 Vedrana Tadic
Vedrana Tadic Tino Prell
Tino Prell Julian Grosskreutz
Julian Grosskreutz- Hans Berger Department of Neurology, Jena University Hospital, Jena, Germany
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease characterized by progressive loss of upper and lower motor neurons. Although the etiology remains unclear, disturbances in calcium homoeostasis and protein folding are essential features of neurodegeneration in this disorder. Here, we review recent research findings on the interaction between endoplasmic reticulum (ER) and mitochondria, and its effect on calcium signaling and oxidative stress. We further provide insights into studies, providing evidence that structures of the ER mitochondria calcium cycle serve as a promising targets for therapeutic approaches for treatment of ALS.
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disorder characterized by progressive degeneration of the upper (spasticity, dysphagia, dysarthria) and lower motor neurons (atrophy, fasciculations). Approximately 90% of ALS patients have sporadic ALS (sALS) which is the most prevalent form and about 10% have the inherited or familial form of ALS (fALS). The latter form is believed to be due to several genes including SOD1, TARDBP, FUS, OPTN, and VCP. In addition, a hexanucleotide (GGGGCC) repeat expansion in the first intron of the C9ORF72 gene (DeJesus-Hernandez et al., 2011; Renton et al., 2011) has lately been demonstrated as being associated with ALS. However, the etiology of the disease is still unclear, although recent studies indicate that calcium (Ca2+) disturbances, ER stress, and mitochondrial dysfunction are involved in the pathogenesis of ALS (Grosskreutz et al., 2010; Lautenschlaeger et al., 2012). Other mechanisms possibly involved in ALS-related pathophysiology comprise: oxidative stress, protein aggregation, dysregulated endosomal trafficking, impaired axonal transport, neuroinflammation, and dysregulated transcription and RNA processing (Ferraiuolo et al., 2011). Several properties of motor neurons make them more vulnerable than other neuronal groups. Motor neurons express high levels of Ca2+–permeable α-amino-5-methyl-3-hydroxisoxazolone-4-propionate (AMPA) receptors that lack the GluR2 subunit which makes them more vulnerable to excitotoxicity and dysregulation of intracellular Ca2+ homeostasis (Williams et al., 1997). Also, low levels of Ca2+ -buffering proteins contributes greatly to this vulnerability (Ince et al., 1993). Because of high metabolic demands, motor neurons are largely dependent on optimal mitochondrial function, a robust cytoskeleton and an axonal transport mechanism. Despite all the above facts, there remain numerous unanswered questions in ALS related to selectivity and specificity of the cellular targets of motor neuron degeneration and cell-specific aspects of mitochondrial Ca2+ signaling. This review focuses on crosstalk between ER, mitochondria, oxidative stress and calcium.
ALS Genes and Encoded Proteins – Role in Pathophyisology
Cu/Zn superoxide dismutase 1 (SOD 1)
About 20% of fALS patients carry a mutation in the SOD1 gene. Indeed, more than 170 different SOD1 mutations have been described in ALS families (http://alsod.iop.kcl.ac.uk/). Generally, SOD1 mutations have not been linked to decreased SOD activity (Kostrominova, 2010; Fischer et al., 2011), instead mutant SOD1 likely acts through a combination of several mechanisms, including protein misfolding, mitochondrial dysfunction, oxidative damage, cytoskeletal abnormalities, and defective axonal transport, excitotoxicity, in addition to inadequate growth factor signaling and inflammation (Cozzolino et al., 2008).
Fused in sarcoma (FUS) and TAR DNA-binding protein (TDP-43)
Mutations in the gene encoding fused in sarcoma/translocated in liporsarcoma (FUS/TLS or FUS) are linked to 4% of fALS cases. Mutations in TARDBP (which encodes TDP-43) account for 4% of fALS and a smaller percentage of sALS. The toxicity of TDP-43 and FUS/TSL proteins is linked to their altered intracellular localization. Both TDP-43 and FUS/TLS are mainly localized in cell nuclei where they control gene transcription and pre-mRNA (Buratti et al., 2001; Winton et al., 2008; Kwiatkowski et al., 2009). In the presence of mutations or stress, these proteins accumulate in the cytosol (Liu-Yesucevitz et al., 2010; Dewey et al., 2012; Daigle et al., 2013; Walker et al., 2013; Watanabe et al., 2013). FUS triggers ER stress and causes fragmentation of the Golgi apparatus in patients with fALS (Farg et al., 2013). In NSC34 transfected cells, wild type human TDP-43 caused ER stress (Suzuki et al., 2011). In the same cell line, mutant TDP-43 induced mitochondrial dysfunction and probably caused oxidative stress (Duan et al., 2010). Experiments with TDP-43 mutation in zebrafish resulted in impairment of neuromuscular junctions (Armstrong and Drapeau, 2013).
Chromosome 9 open reading frame 72 (C9ORF72)
Large expansions of a non-coding GGGGCC-repeat in the first intron of the C9ORF72 gene are accountable for 40% of fALS. C9ORF72 hexanucleotide repeats form highly stable RNA G-quadruplexes, which probably influence RNA transcription, splicing, translation and transport (Fratta et al., 2012). C9ORF72 pathology is characterized by intracellular inclusions, however the major proteins forming these inclusions have not yet been elucidated (Mori et al., 2013).
Other genes
Mutations in the valosin–containing protein (VCP) are responsible for 1–2% of fALS cases. In mice, overexpression of mutant VCP produces ubiquitin- and TDP-43-positive inclusions, suggesting that TDP-43 plays a role in VCP-induced disease (Rodriguez-Ortiz et al., 2013). Mutant VCP also impact mitochondria, such as via a decrease in ATP production related to mitochondrial uncoupling (Bartolome et al., 2013).
Another mutated gene found in patients with ALS comprises OPTN that encodes the protein optineurin which regulates membrane trafficking, protein secretion, cell division and host defense against pathogens (Kachaner et al., 2012). Wild-type optineurin suppresses nuclear factor-kappa B (NF-κB) activity, but the ALS-causing mutant optineurin is unable to suppress NF-κB activity. Therefore, there is an indication that inappropriate NF-κB activation is the pathogenic mechanism underlying optineurin mutation-related ALS (Akizuki et al., 2013). In two patients carrying mutation in OPTN, was shown that loss of function rather than proteinopathy itself resulted in the formation of TDP-43 inclusions in neuronal and glial cytoplasm, and Golgi apparatus fragmentation (Kamada et al., 2014).
Vesicle-associated membrane protein (VAMP)-associated protein B (VAPB) is usually ER-resident and is integral to its structure, protein transport, lipid metabolism, and the UPR. VAPB toxicity is probably mediated by impaired Ca2+ homeostasis and ER stress (Langou et al., 2010; De Vos et al., 2012; Morotz et al., 2012).
The ERMCC and Calcium Disturbances in ALS
The ER and mitochondria form a highly dynamic interconnected network that is involved in the generation of Ca2+ signals. Ca2+ release from ER is controlled by ryanodine receptors (RyRs, Ca2+-gated Ca2+ channels) (Meissner, 2002; Lanner et al., 2010), the inositol 1,4,5-triphosphate receptor-gated channels (IP3Rs), and the translocon (Taylor and Tovey, 2010). Restocking of the ER with Ca2+ is executed by the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA; Wuytack et al., 2002; Verkhratsky, 2005; Lipskaia et al., 2009). Ultimately, the plasma membrane Na+/Ca2+ exchanger and Ca2+ ATPase remove Ca2+ from the cell (Rhodes and Sanderson, 2009; Figure 1).
Mitochondria take up Ca2+ via a Ca2+-sensitive electrogenic carrier, the mitochondrial uniporter (mUP) which is gated by cytosolic Ca2+ in a biphasic-dependent manner (Gunter and Sheu, 2009). Ca2+ uptake into mitochondria is facilitated by Ca2+/calmodulin. However, sustained cytosolic Ca2+ levels inactivate the uniporter, preventing further Ca2+ uptake (Moreau et al., 2006). Accumulated Ca2+ in the mitochondria can slowly be ejected back into the cytosol through Na+/Ca2+ and 2H+/Ca2+ exchangers (Pivovarova and Andrews, 2010; Figure 1). Once intramitochondrial Ca2+ rises above a certain threshold, the voltage- and Ca2+-dependent high-conductance channel in the inner membrane, known as the mitochondrial permeability transition pore (mPTP), opens, leading to cell death either by apoptosis or necrosis (Leung and Halestrap, 2008; Martin, 2010b). Mitochondria contain similar low Ca2+ levels as resting cells, but accumulate a considerable amount during stimulated Ca2+ entry, which affects numerous cellular processes such as cellular energy metabolism, synaptic transmission and excitability, intracellular signaling, generation of ROS, and activation of apoptosis (Chinopoulos and Adam-Vizi, 2010; Starkov, 2010).
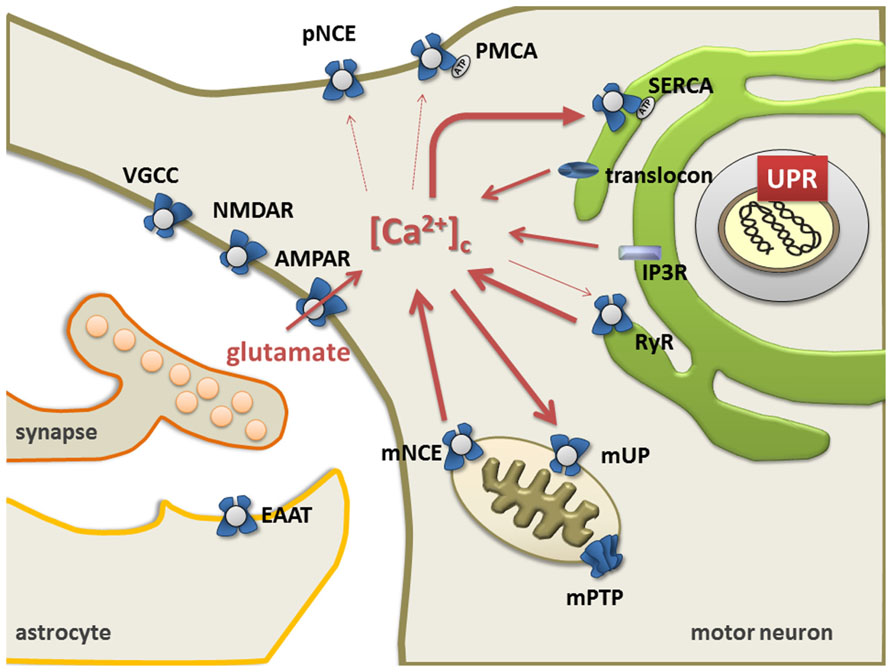
FIGURE 1. The endoplasmic reticulum mitochondria Ca2+cycle (ERMCC). Ca2+can enter cytosol through: the AMPA receptor, the ryanodine receptor (RyR) at the ER membranes, the opening of the inositol 1,4,5 trisphosphate receptor (IP3R), the translocon at the ER membrane, and/or the plasmalemmal voltage gated Ca2+channels (VGCC). Triggered by physiological activity of AMPA receptors with pathologically increased Ca2+-permeability in ALS, a chronic shift of Ca2+ from the ER to the mitochondria (i.e., through Ca2+-induced Ca2+release through RyR and mitochondrial uptake through the uniporter mUP) causes depletion of ER Ca2+levels with protein misfolding (UPR) and chronic mitochondrial Ca2+overload. Cytosolic Ca2+clearance is facilitated by the plasma membrane Ca2+ATPase, the plasmalemmal Na+/Ca2+exchanger (NCE), the sarco/endoplasmicreticulum Ca2+ATPase (SERCA), and the Golgi apparatus. Astrocytes control the level of persisting glutamate at the glutamatergic synapse through glutamate transporters (EAAT), but also exert life-supporting functions in motor neurons (i.e., BDNF, IGF, VEGF). (NMDAR = NMDA receptors, VGCC = voltage gated Ca2+channels, Na/K = Na+/K+pump, pNCE = plasmalemmal Na+/Ca2+ exchanger, PMCA = plasmalemmal Ca2+ATPase, mNCE = mitochondrial Na+/Ca2+ exchanger, SERCA = sarco-endoplasmic Ca2+ATPase). Modified picture taken from (Prell et al., 2013).
Several studies have previously investigated abnormalities of Ca2+ homeostasis, ER and mitochondria as well as excitotoxicity in motor neurons in ALS (Grosskreutz et al., 2010; Lautenschlager et al., 2013). Based on the models described by Berridge (2002), a persistent shift of Ca2+ from the ER to mitochondria (i.e., through Ca2+induced Ca2+ release via RyR and mitochondrial uptake through mUP) was postulated. This could be triggered by the physiological activity of AMPA receptors together with a pathologically increased Ca2+-permeability (Grosskreutz et al., 2010). This in turn, leads to a depletion of Ca2+ levels in the ER, resulting in protein folding dysfunction and chronic mitochondrial Ca2+ overload. Both protein misfolding and Ca2+ overload can then induce apoptosis through Bcl-2 dependent mechanisms (Grosskreutz et al., 2010). Since Ca2+ appears to be shuttled back and forth between the ER and the mitochondrial compartment, the process has been termed the ER–mitochondria Ca2+ cycle (ERMCC, Figure 1; Grosskreutz et al., 2010).
Impact of ER Stress on Mitochondria
Recent studies indicate that ER stress is involved in the pathogenesis of familial and sporadic ALS (Ilieva et al., 2007; Atkin et al., 2008; Walker, 2010; Lautenschlaeger et al., 2012; Prell et al., 2012). ER stress occurs when ER Ca2+ content is depleted (Verkhratsky, 2005) and misfolded proteins accumulate in the ER. To cope with ER stress, cells activate the unfolded protein response (UPR; Figure 2). The UPR mediates the (1) upregulation of genes encoding ER-resident chaperones, (2) down-regulation of general protein synthesis in order to reduce the ER protein load, and, (3) degradation of misfolded proteins by the proteasome (Kozutsumi et al., 1988; Yoshida et al., 2001; Dudek et al., 2009; Figure 2). On a cellular level, ER stress is transduced by three proximal sensors of the UPR: the double-stranded RNA-activated protein kinase (PKR)-like ER kinase (PERK), the basic leucine-zipper transcription factor 6 (ATF6) and the inositol requiring enzyme 1 (IRE1) (Prell et al., 2013; Figure 2). When protein misfolding can no longer be compensated for, the prolonged UPR triggers apoptosis by the caspase pathways (Nakagawa et al., 2000; Figure 2).
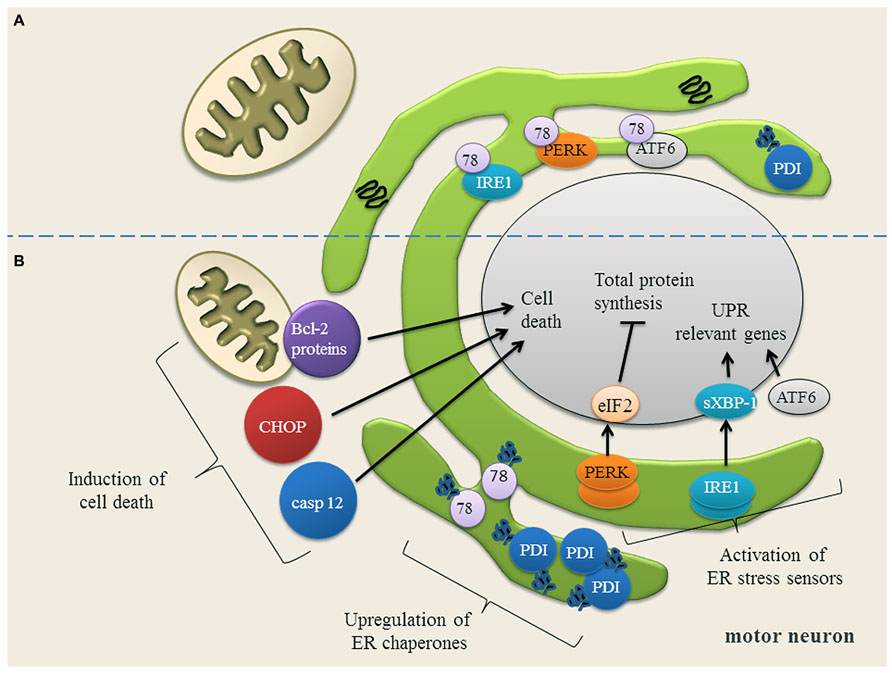
FIGURE 2. (A) Motor neuron in a physiological state and under ER stress conditions. In motor neurons, most proteins in the ER are physiologically properly folded. The ER chaperones Grp78 (78) and protein disulfide isomerase (PDI) are moderately expressed and assist protein folding. The unfolded protein response (UPR) sensor proteins (IRE1, PERK, ATF6) are bound to Grp78 and are therefore in an inactive state. (B) Upon ER stress, Grp78 dissociates from IRE1, PERK and ATF6 resulting in the activation and up-regulation of these three UPR sensor proteins. IRE1 splices XBP-1 mRNA (sXBP-1) which up-regulates the expression of UPR relevant genes. ATF6 is transported to Golgi apparatus, where it is cleaved. The cytosolic domain of ATF6 translocates to the nucleus to activate gene expression. PERK phosphorylates eIF2α, thereby down-regulating the total protein synthesis to prevent an overload of the ER. Once the expression of ER chaperones Grp78 and PDI is induced, they bind to the misfolded proteins and try to assist their folding. If restoring of ER homeostasis fails, cell death proteins transcription factor C/EBP homologous protein (CHOP) and caspase 12 (casp 12) are induced and the balance of Bcl-2 family members is disturbed, anti-apoptotic Bcl-2 family members are down-regulated, while pro-apoptotic Bcl-2 family members are up-regulated. Picture modified from (Lautenschlaeger et al., 2012).
ER stress can affect mitochondria because both organelles are functionally and morphologically connected by several pathways (Vannuvel et al., 2013). In particular, the contact between ER and mitochondria is essential for coordination of the Ca2+ transfer (Rowland and Voeltz, 2012). The proteins B-cell lymphoma 2 (Bcl-2), Bcl-2-associated X protein (Bax/Bak) and the Bcl-2-interacting killer (BIK) can enhance Ca2+ transfer from ER to mitochondria and the ensuing Ca2+ accumulation activates apoptosis via cytochrome C (Germain et al., 2002; Nutt et al., 2002; Fannjiang et al., 2004; Kong et al., 2005). The PERK/ATF4 pathway can induce Lon protease that controls the assembly and/or the degradation of cytochrome C (Margineantu et al., 2002; Venkatesh et al., 2012). Bid, which is a pro-apoptotic BH3-only protein can be cleaved upon ER stress, which subsequently activates caspase-2 or caspase-8 leading to apoptosis (Upton et al., 2008; Uchibayashi et al., 2011). Recruitment of the dynamin-related protein 1 mediates the fission of the outer mitochondrial membrane (Breckenridge et al., 2003).
Therapeutic Strategies Focused on ER Stress
Several therapeutic strategies aim to target the ER (Table 1; Figure 4). For instance, Salubrinal is a substance that reduces ER stress by activating the UPR. UPR activation is mediated via phosphorylation of elF2α and activation of PERK (Boyce et al., 2005). Salubrinal prevented neuronal cell death triggered by several ER stress inducers (Smith et al., 2005; Reijonen et al., 2008). Moreover, Salubrinal, Guanabenz and Phenazine have all been shown to reduce ER stress in worms and zebrafish expressing mutated TDP-43 (Vaccaro et al., 2013). In SOD1G93A mice, Salubrinal decreased muscle strength loss and extended survival (Saxena et al., 2009).
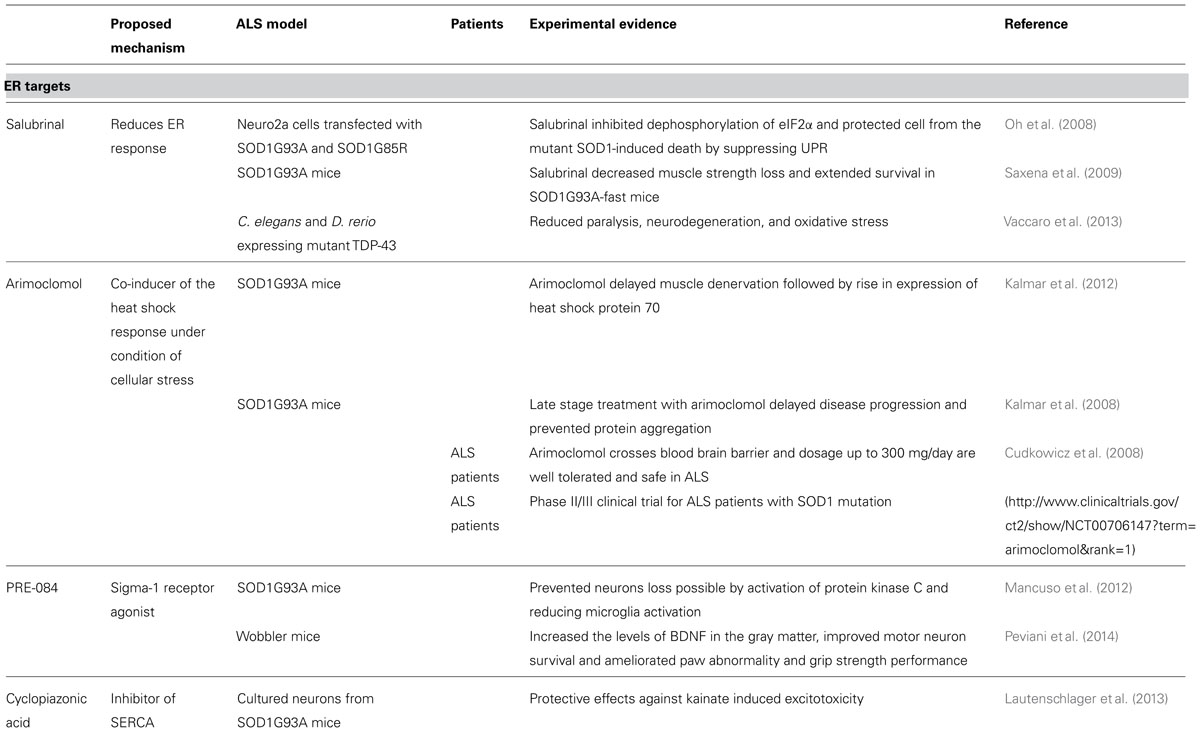
TABLE 1. Substances targeting ER.
Another approach to target the ER is to encourage the natural cellular protein-folding machinery via activation of the heat shock transcription factor 1 (Hsf1). Hsf1 is the master activator of chaperone protein gene expression (Neef et al., 2011). Overexpression of human molecular chaperone hHSJ1a in vivo mediated late-stage neuroprotection in the SOD1G93A mouse model, probably through a combination of chaperone, co-chaperone and pro-ubiquitylation activity on SOD1 (Novoselov et al., 2013). Arimoclomol, a hydroxylamine derivate and a co-inducer of the heat shock response delayed muscle denervation in the SOD1G93A mouse followed by a rise in expression of the heat shock protein 70 (Kalmar et al., 2012). The therapeutic potential of this drug is under investigation in a phase II/III clinical trial for ALS patients with SOD1 mutations (http://www.clinicaltrials.gov/ct2/show/NCT00706147?term = arimoclomol&rank=1). Other available pharmaceuticals that up-regulate heat shock response and that may be used to treat ALS include Celastrol and 17-AAG (Kalmar et al., 2014).
Accumulation of misfolded proteins may also be targeted by small molecule regulators of autophagy such as antipsychotics (fluspirilene, trifluoperazine, pimozide) and calcium-channel modulators (nicardipine, niguldipine, amiodarone; Sarkar et al., 2007; Zhang et al., 2007).
The Sigma-1 receptors have also gained attention in the recent past. The receptors are a chaperone proteins residing at the mitochondrion-associated ER membrane, where they affect mitochondrial Ca2+ influx by stabilizing IP3R and acting as inter-organelle signaling modulators of Ca2+ homeostatasis, ER stress and apoptosis. The Sigma-1 receptor agonist PRE-084 prevented neurons loss in SOD1G93A transgenic mice, probably by the activation of protein kinase C and reducing microglia activation (Mancuso et al., 2012). Neuroprotective effects of PRE-084 have also been demonstrated in the wobbler mouse model not linked to SOD1 mutation that is characterized by progressive neural atrophy shortly after birth (Peviani et al., 2014). Another agonist of Sigma-1 receptor SA4503 prevented SOD1G93A–induced neurotoxicity in NSC34 cells and extended survival of SOD1G93A mice (Ono et al., 2014). Pharmacological manipulation of the Sigma-1 receptor may increase availability of growth factors, as well as the modulation of astrocytosis and of macrophage/microglia as part of the mechanism involved in Sigma 1 receptor-mediated neuroprotection (Peviani et al., 2014).
Treatment with Geldanamycin, an inducer of heat shock response, successfully blocked protein aggregation but not Ca2+ dysregulation or loss of mitochondrial membrane potential (ΔΨ) in murine motor neurons expressing human SOD1G93A (Tradewell et al., 2011). This implies chaperone-based therapies would possibly require co-therapy targeting other important mechanisms of toxicity.
Cross-Talk Between Calcium, Mitochondria, and Reactive Oxygen Species Signaling
Mitochondrial Dysfunction
Mitochondria are central for energy metabolism and have been well studied in relation to ALS pathogenesis (von Lewinski and Keller, 2005; Cozzolino and Carri, 2012; Jaiswal, 2013). The levels of mutated mitochondrial DNA (mtDNA) were higher in ALS patients but the amount of mtDNA was reduced compared to controls. This reduction correlated well with a decrease of a mitochondrial marker, citrate synthase activity and with the activities of respiratory chain complexes I + III, II + III, and IV, suggesting a loss of mitochondria in ALS spinal cords (Wiedemann et al., 2002). Activity of cytochrome C oxidase in mitochondria is reduced in the spinal cord of sALS patients (Borthwick et al., 1999) and ALS spinal neurons show varied and reduced mtDNA gene copy numbers and increased mtDNA gene deletions (Keeney and Bennett, 2010). Oxidative stress, protein nitration and aggregation, and excitotoxicity participate in the process of motor neuron degeneration caused by mutated SOD1 (Martin et al., 2007). One of the pathological hallmarks of ALS is aggregation of ubiquitinated proteins in motor neurons (Stieber et al., 2000; Figure 3). SOD1, FUS, TDP-43, OPTN, and UBQLN2 have been identified as forming aggregates. Whether there is a causal relationship between misfolded proteins and mitochondrial dysfunction for novel mutations is still largely unknown, but there is considerable body of literature describing SOD1 and more recently TDP-43. Mutant SOD1 forms insoluble aggregates in mitochondria at the surface of the outer membrane (Figure 3). Further, there is direct connection between mutated SOD1 and impaired mitochondrial function (Liu et al., 2004; Pasinelli et al., 2004; Pickles et al., 2013). Bcl-2 has been identified as an interacting partner of mutated SOD1 because SOD1 induces mitochondrial morphological changes and impairs mitochondrial membrane integrity only in the presence of Bcl-2. This leads to the release of cytochrome C, ultimately leading to cell death (Pedrini et al., 2010).
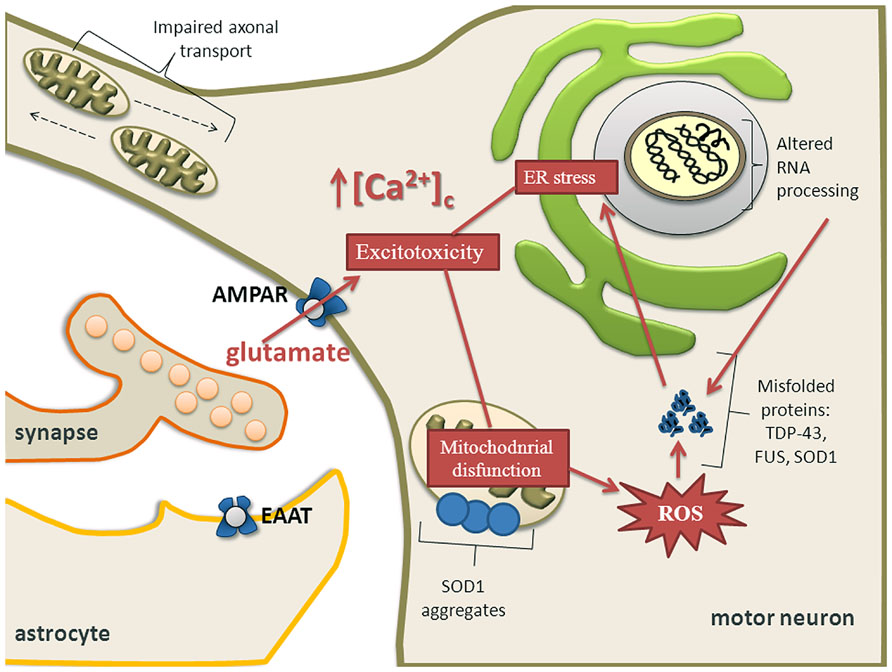
FIGURE 3. Calcium dysregulation, ER stress and mitochondrial impairment are major components of excitotoxicity in motor neurons. Mitochondrial dysfunction causes activation of proteolytic and ROS generating-generating enzyme systems. Mutant SOD1 forms insoluble aggregates in mitochondria at the surface of the outer membrane. Motor neuron might also undergo transcriptional dysregulation and abnormal RNA processing which together with depleted ER Ca2+ levels and overproduction of ROS contribute to aberrant protein folding. Aberrant proteins form aggregates leading to ER stress and ultimately activating apoptotic pathways, especially when the unfolded protein response is exhausted. Impaired axonal transport may also contribute to an energy deficit in the distal axon and the dying back axonopathy that is observed in ALS.
Degenerating mitochondrial vacuoles have been reported in presymptomatic mice expressing mutant SOD1 in previous studies (Wong et al., 1995; Kong and Xu, 1998). However, mitochondrial disturbances are not restricted to SOD1 mutations. In patients with ALS, dense conglomerates of mitochondria have been found in the anterior horn of lumbar and spinal cord and proximal axons (Hirano et al., 1984; Sasaki and Iwata, 1996). It has been demonstrated that neuronal Ca2+, mitochondrial volume and a number of synaptic vesicles are increased in ALS patients (Siklos et al., 1996). In addition, overexpression of TDP-43 causes mitochondrial dysfunction and induces mitophagy (Hong et al., 2012) and oxidative injury in NSC34 cell line (Duan et al., 2010; Lu et al., 2012). In a yeast model, TDP-43 aggregates around mitochondria and there is an inverse correlation between respiratory activity and toxicity of the mutant protein (Braun et al., 2011). Overexpression of wild-type TDP-43 resulted in reduced mitochondrial length and density in neurites of primary motor neurons and conversely, suppression of TDP-43 resulted in significantly increased mitochondrial length and density in neurites (Wang et al., 2013). Neuronal mitochondrial transport and morphological abnormalities occur in vivo in SOD1 and TDP-43 ALS (Figure 3) mouse models but show differences in temporal and spatial manifestation. This implies that different molecular mechanisms may be involved (Magrane et al., 2013).
Mitochondrial Calcium Dynamics
Ca2+ regulation of mitochondrial metabolism
Ca2+ plays a central role in cell signaling at numerous levels. The tricarboxylic acid cycle consists of a series of reactions that produce energy through the breakdown of proteins, fatty acids and carbohydrates. Ca2+ within mitochondria regulates the most important task of the organelle: ATP production by oxidative phosphorylation. The physiological increase of mitochondrial Ca2+ stimulates the adenine nucleotide transporter (Mildaziene et al., 1995) and synthesis of ATP complex V (Das and Harris, 1990). Moreover, mitochondrial Ca2+ increase activates three matrix dehydrogenases: isocitrate dehydrogenase, α-ketogluterate dehydrogenase and pyruvate dehydrogenase (McCormack and Denton, 1979, 1993; McCormack et al., 1990). All three dehydrogenases enhance the reaction rate of many of the steps in the tricarboxylic acid cycle and therefore increase flux throughout the pathway, raising ATP production (Jouaville et al., 1999). Furthermore, it has been shown that motor neurons have an insufficient mitochondrial capacity to buffer large Ca2+ elevations which is partly due to a reduced mitochondrial density per volume compared to non-motor neurons (Grosskreutz et al., 2007). Mitochondrial disfunction and impaired Ca2+ homeostasis largely contribute to selective vulnerability of motor neurons (Jaiswal et al., 2009; Jaiswal and Keller, 2009).
Ca2+ overload and activation of permeability transition pore
One main mediator of mitochondrial function or dysfunction in neurons is the mPTP, which is a Ca2+ dependent high-conductance channel in the inner membrane of mitochondria (Brenner and Moulin, 2012). The mPTP comprises of the voltage-dependent anion channel (VDAC), the adenine nucleotide translocator (ANT) and cyclophilin D. Since VDAC and ANT are not essential for functioning of mPTP regulator (Juhaszova et al., 2008), the soluble matrix protein cyclophilin D received special attention (Giorgio et al., 2010). The mPTP opening is promoted by binding of cyclophilin D to the inner mitochondrial membrane (Di Lisa and Bernardi, 2009) and is favored by Ca2+ overload, ROS, inorganic phosphate and mitochondrial depolarization (Crompton, 1999; Brustovetsky et al., 2002; Bernardi et al., 2006). Binding of cyclophilin D to the inner mitochondrial membrane can be prevented by Cyclosporine A. Opening the mPTP causes a release of cytochrome C, which subsequently leads to apoptotic cell death (Liu et al., 1996; Crompton et al., 1998). High concentrations of cyclophylin D were found in swollen mitochondria in the SOD1 animal model. Modifying mPTP through different genetic and pharmacological manipulations has been shown to be protective in animal models of ALS. Genetic ablation of cyclophilin D delayed disease onset and extended the lifespan in the ALS mouse model (Martin et al., 2009). However, in another study, deleting cyclophilin D in the SOD1 mouse model did not lead to prolongation of survival, although it improved mitochondrial buffering capacity and attenuated mitochondrial damage (Parone et al., 2013). Therefore the role of cyclophylin D as a potential therapeutic is not fully understood.
Therapeutic strategies focused on mitochondrial Ca2+ dynamics
Because there is growing evidence for mitochondrial dysfunction in ALS, mitochondria are promising therapeutic targets (Table 2; Figure 4). However, studies targeting mitochondria have failed so far. The mPTP modulator Olesoxime (TRO19622) had a neuroprotective effect in motor neuron cell culture and in ALS rodents (Bordet et al., 2007; Martin, 2010a; Sunyach et al., 2012), but failed in a phase III clinical trial (http://www.als.net/ALS-Research/Olesoxime/ALS-Topics/). Further, Dexpramipexole, which reduces mPTP opening and increases cellular energy supply, did not have significant effects on survival and disease progression in a recent clinical trial (Cudkowicz et al., 2013).
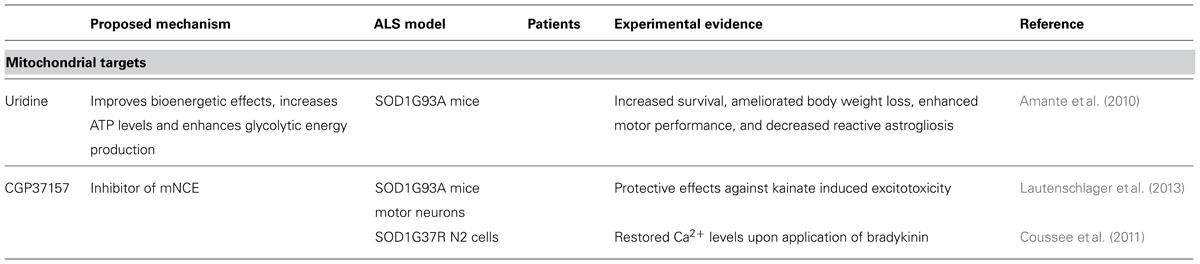
TABLE 2. Substances targeting mitochondria.
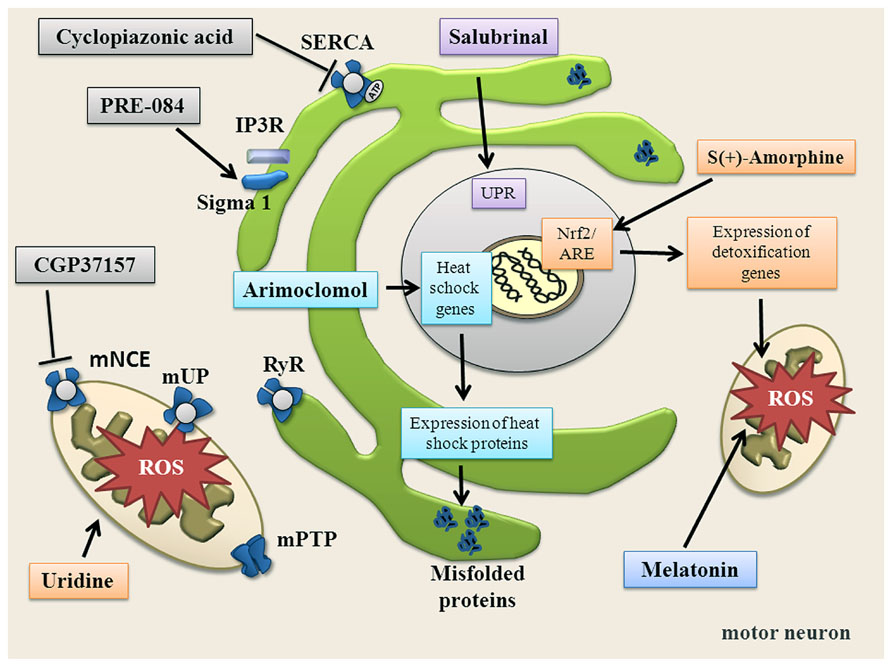
FIGURE 4. Summary of substances targeting ER, mitochondria, oxidative stress and altered Ca2+ homeostasis. Salubrinal increases the unfolded protein response (UPR) via phosphorylation of elF2α and activation of PERK. Arimoclomol induces heat shock response. PRE-084 activates Sigma-1 receptor. Uridine increases bioenergetic effects, increases ATP and enhances glycolytic energy production. CGP37157 blocks mNCE. Cyclopiazonic acid blocks SERCA. S(+)-Apomorphine activates the Nrf2/ARE pathway. Melatonin directly scavenges free radicals.
Minocycline and creatine, compounds that improve mitochondrial function have also failed in human trials (Shefner et al., 2004; Gordon et al., 2007). Other medications targeting mitochondria such as coenzyme Q and cylosporin A have been studied, but all trials in humans were negative (Appel et al., 1988; Kaufmann et al., 2009). Uridine, a pyrimidine nucleoside, extended survival in SOD1G93A mice, probably by improving bioenergetic effects, increasing ATP levels, and enhancing glycolytic energy production (Amante et al., 2010). However, it has not been tested in ALS patients yet.
Melatonin recently gained interest because of its ability to decrease cytochrome C release and caspase-3 activation. It delayed disease onset in the SOD1G93A mice model. Besides its mitochondria stabilization effects in ALS, melatonin attenuated the activation of astrocytes and microglia (Zhang et al., 2013).
CGP37157, which is able to cross blood-brain barrier (Gonzalez-Lafuente et al., 2012), blocks the mNCE. It showed protective effects against kainate induced excitotoxicity in SOD1G93A mice motor neurons (Lautenschlager et al., 2013), restored calcium levels in SOD1G37R N2 cells (Coussee et al., 2011) and protected rat hippocampal slices upon veratridine-induced sodium and calcium overload (Gonzalez-Lafuente et al., 2012).
The transport of ADP, ATP and inorganic phosphates across mitochondrial membranes is regulated by the VDAC at the outer mitochondrial membrane. VDAC is regulated by Bcl-2 and both can form toxic complexes with mutated SOD1 (Pasinelli et al., 2004; Arbel and Shoshan-Barmatz, 2010; Israelson et al., 2010; Pedrini et al., 2010). Interactions between VDAC1, Bcl-2 and mutated SOD1 inhibits the conductance of VDAC1, leading to cell death (Israelson et al., 2010; Pedrini et al., 2010). It was demonstrated that small SOD1-like therapeutic peptides specifically block the formation in symptomatic SOD1G93A mice by restoring mitochondrial ADP permeability (Tan et al., 2013).
Human TDP-43 caused mitochondrial morphologic abnormality and decrease of mitochondrial complex I activity, and mitochondrial transmembrane potential in human TDP-43 stably tranfected NSC-34 cells. Dimethoxy curcumin was able to ameliorate mitochondrial dysfunction in the same experiment, which makes this drug interesting as a potential therapeutic for TDP-43 linked ALS (Lu et al., 2012).
Calcium and Mitochondrial ROS
Mitochondria are the main sites of ROS formation as by-products of ATP production (Coyle and Puttfarcken, 1993; Brand, 2010). However, mitochondrial Ca2+ overload and abnormal oxidative phosphorylation increase ROS production and oxidative stress (Carriedo et al., 2000; Mattiazzi et al., 2002; Murphy, 2009; Figure 3). Many of the stated mitochondrial respiratory abnormalities have been linked to reduced activity of mitochondrial complexes I and IV (Mattiazzi et al., 2002; Rizzardini et al., 2006; Son et al., 2008; Coussee et al., 2011).
Reactive oxygen species and oxidative stress
Moderate levels of ROS and reactive nitrogen species (RNS) promote cellular proliferation, regulation and survival. Typical ROS are free radical species such as superoxide (), hydroxyl radicals (•OH) and non-radical species like hydrogen peroxide (H2O2). The respiratory chain complexes I and III are the primary mitochondrial sources of Oxidative stress has been implicated as a pathological mechanism of both fALS and sALS (Ferrante et al., 1997). ROS has been detected in the spinal cord and cerebrospinal fluid of sALS patients (Tohgi et al., 1999). Increased H2O2 and oxidative damage to protein and DNA were observed in mutated SOD1 transgenic mice (Liu et al., 1999). Many ALS causing genes and genes modifiers are known to influence ROS production (Carter et al., 2009). SOD1 mutation induces oxidative modifications of several proteins in ALS: SOD1, translationally controlled tumor protein (TCTP), ubiquitin carboxyl-terminal hydrolase-L1 (UCH-L1) and probably alphaB-crystallin. These oxidative modifications lead to structural alteration and a decline of protein activity (Poon et al., 2005). ROS also directly influences transcriptional factors such as NF-kB, activator protein 1 (AP-1), and HIF-1α which are all involved in the regulation of gene expression and maintaining cellular homeostasis (Haddad, 2002).
ROS dysregulation of these factors is observed in ALS pathology (Iaccarino et al., 2011; Moreau et al., 2011) as the involvement of protein disulfide isomerase (PDI) family members plays an important role in oxidative folding of human secretory proteins (Rutkevich et al., 2010; Figure 2). PDI’s enzymatic activity can be inactivated by oxidation and S-nitrosylation of their active site thiol groups. In motor neurons of patients with ALS, PDI was widely distributed and aggregated (Atkin et al., 2008). Therefore it was assumed that PDI is inactivated due to S-nitrosylation in the affected neurons, which causes protein misfolding in ALS (Honjo et al., 2011). Studies of genetics, model organisms, and patient’s tissue samples support PDI upregulation triggered by ER stress and post-translational inhibition of PDI due to S-nitrosylation (Atkin et al., 2008; Walker and Atkin, 2011).
Therapeutic strategies focused on reactive oxygen species
Design of novel antioxidant strategies to selectively target oxidative stress and redox imbalance might be an important approach (Table 3; Figure 4). However, the antioxidant treatment therapy in ALS has not been successful so far (Traynor et al., 2006; Bedlack et al., 2007; Beghi et al., 2011). SOD1 is tightly connected with the nuclear erythroid 2-related-factor 2 (Nrf2). Nrf2 is a transcriptional factor and main expression regulator of many antioxidant/detoxification genes via its interaction with the antioxidant response element (ARE). Because it helps neuronal cells to cope with toxic effect of oxidative stress, pharmacological targeting of Nrf2/ARE pathway was proposed as a tool against neurodegeneration in ALS (Petri et al., 2012; Milani et al., 2013; Figure 4). S(+9)-Apomorphine, a CNS penetrating activator of the Nrf2/ARE pathway was able to reduce pathological oxidative stress and improved survival in fibroblasts of ALS patients, and also slowed disease progression in SOD1G93mice (Mead et al., 2013). On the other hand, Guo et al. (2013) reported a rather modest impact of Nrf2 on the course of disease in SOD1G93A mice.
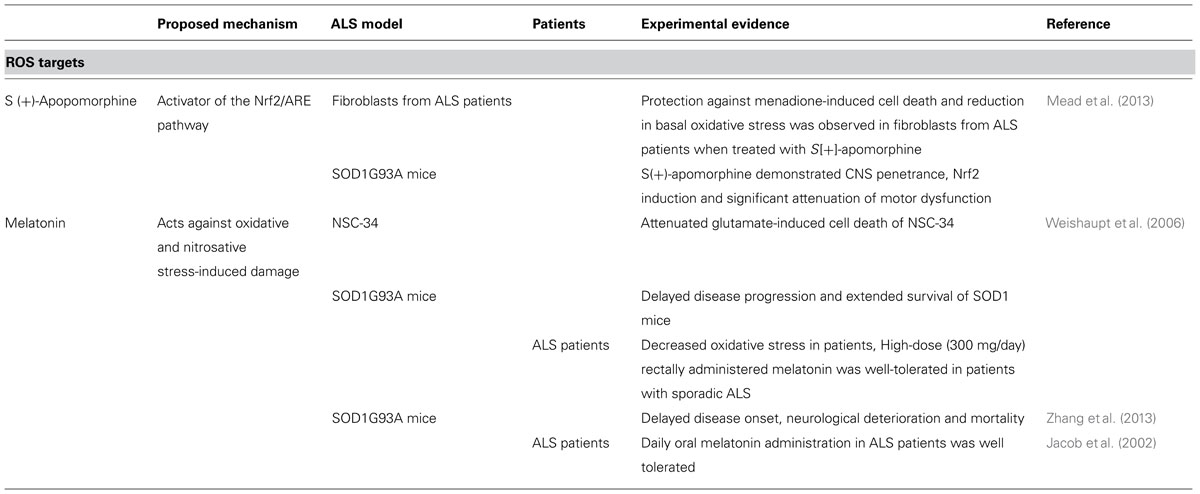
TABLE 3. Substances targeting ROS.
Beneficial effects have been reported in NSC-34 cells for melatonin, a hormone which acts against oxidative and nitrosative stress-induced damage (Weishaupt et al., 2006). In several studies, treatment with melatonin prolonged survival in the SOD1G93A mice (Weishaupt et al., 2006; Zhang et al., 2013), however, Dardiotis et al. (2013) showed contrary results in the same mouse model, possibly because melatonin exacerbated neurodegeneration.
In patients with ALS, high doses of melatonin were well tolerated (Jacob et al., 2002; Weishaupt et al., 2006) and it was reported that circulating serum protein carbonyls, which are oxidative stress markers, were decreased in melatonin treated ALS patients (Weishaupt et al., 2006).
Diacetylbis(N(4)-methylthiosemicarbazonato) copper(II), that inhibits the action of peroxynitrite on SOD1 and ensues nitration of cellular proteins, significantly delays onset of paralysis, prolongs lifespan and prevents accumulation of TDP-43 in the spinal cord of SOD1G93A mice. Therefore, it represents a potential neuroprotective agent targeting multiple disease pathways in ALS (Soon et al., 2011). In the spinal cord of ALS patients, metallothioneins (Zn modulators and anti-oxidant reaction inducers), were seriously reduced (Hozumi et al., 2008). It was demonstrated that metallothionein-III prevents the loss of motor neurons and prolongs the life span of ALS mice (Hashimoto et al., 2011; Hozumi, 2013).
Calcium Homeostasis and ROS
Ca2+ plays a role in ROS production and, vice versa, the redox state can modulate Ca2+ signaling. Components of ROS homeostasis are regulated by Ca2+-dependent pathways. Ca2+ stimulates NO synthesis, inhibits complex IV, and leads to ROS production at the complex III (Feissner et al., 2009). Depending on the targeted protein, the type and concentration of ROS and the duration of exposure, interactions between Ca2+ and ROS signaling can be stimulating or inhibiting.
Ryanodine receptors are stimulated by oxidation
Ryanodine receptors (RyR) belong to a class of intracellular Ca2+ channels and are important mediators of Ca2+ induced Ca2+ release in excitable cells such as muscles and neurons (Fabiato, 1983; McPherson et al., 1991). RyR are opened by Ca2+ itself which may induce propagated Ca2+release on the ER surface. Ca2+ entry through AMPA receptors caused RyR mediated Ca2+-induced Ca2+ release from the ER in embryonic motor neurons in co-culture with neonatal Schwan cell (Jahn et al., 2006). In embryonic motor neurons, Ca2+-induced Ca2+ release was shown to contribute greatly to AMPA receptor stimulation induced Ca2+-induced Ca2+ release through RyR and Ca2+ dysregulation (Grosskreutz et al., 2004). RyR form tetramers in the sarcoplasmic reticulum (SR) and ER membranes (Xu et al., 1998; Fill and Copello, 2002). The reversible oxidation of endogenous SH groups opens the channel and releases Ca2+ from SR (Abramson and Salama, 1989; Xu et al., 1998). Since sulfhydryl oxidation of reactive thiols is involved in the gating of the Ca2+ release channel, RyR represents an important target in oxidative cell damage (Liu et al., 1994; Zable et al., 1997; Marengo et al., 1998). Activity of the RyR channel complex is regulated as a response to changes in transmembrane redox potential (Feng et al., 2000). When Ca2+ channel activators lower the redox potential of the RyR, the thiol groups get oxidized and the channel opens. When Ca2+ channel inhibitors increase the redox potential of the RyR, the disulfides are reduced and the channel closes (Feng et al., 2000). ROS, such as and H2O2, can activate the channel by direct oxidation of redox-sensing thiols (Rowe et al., 1983; Boraso and Williams, 1994; Zima et al., 2004). The endogenous ligand of RyR, FKBP12, stabilizes RyR in the absence of activation and prevents Ca2+ leakage from the ER. The concentration of FKBP12 was decreased in ALS patients indicating the importance of equilibrium between FKBP12 and RyR in neurodegeneration (Kihira et al., 2005).
IP3R are stimulated by oxidation
A second receptor that induces the release of Ca2+ from the ER is the IP3R. Ca2+ overload in the ER discharges IP3R spontaneously (Missiaen et al., 1991; Rooney et al., 1991). The most important ligands that modulate IP3R channel activity are InsP3 and Ca2+. At low concentrations Ca2+ activates the channel, whereas at higher concentrations, Ca2+ inhibits the channel (Foskett et al., 2007). IP3R can be directly activated by oxidative agents, such as (Suzuki and Ford, 1992) and H2O2 (Wesson and Elliott, 1995). Thimerosal, a sulfhydryl-oxidizing agent, stimulates IP3R channels isolated from rat cerebellum and incorporated into artificial lipid bilayer (Thrower et al., 1996) and HeLa cells (Bootman et al., 1992). Overexpression of the IP3R2 shortened the lifespan in SOD1G93A mice, which implicates the importance of ER Ca2+ release by IP3Rs and that impaired function of this receptor can be destructive in ALS (Staats et al., 2012a). IP3-gated Ca2+ seems to be a key regulator of TDP-43 nucleoplasmic shuttling and proteostasis. Pathologic TDP-43 aggregation disturbs Ca2+-dependent TDP-43 shuttling, indicating pharmacological manipulation of IP3R as a target in TDP-43 induced neurodegeneration in vivo (Kim et al., 2012).
Phospholipase C delta 1 (PLCδ1) increases InsP3 formation which releases calcium from ER through IP3R. The expression of PLCδ1 is increased in ALS mouse spinal cord and neurons. Genetic ablation of PLCδ1 prevented shrinkage of motor neurons in ALS mice, suggesting that PLCδ1 is also a candidate for new targets in ALS research (Staats et al., 2013).
SERCA and plasma membrane Ca2+-ATPase are inhibited by oxidation
SERCA transfers Ca2+ from the cell cytosol to the lumen of the SR (MacLennan et al., 1997). SERCA is very sensitive to redox state but contrary to RyR and IP3R, oxidation inhibits SERCA activity (Kaplan et al., 2003). SERCA is reversibly regulated through NO-dependent S-glutathiolation of specific cysteine residues (Adachi et al., 2004), where irreversible sulfonylation reduces SERCA (Ying et al., 2008). Thiol oxidizing agents inhibit and glutathione stimulate SERCA (Scherer and Deamer, 1986). Amino acid peroxides selectively oxidize cysteine residues of SERCA and inactivate the pump (Dremina et al., 2007). O2•- and H2O2/•OH have been shown to inhibit Ca2+ uptake into the sarcoplasmic reticulum (Rowe et al., 1983; Kukreja et al., 1988; Xu et al., 1997). H2O2/•OH directly interfere with the ATP binding site. Since the Ca2+ uptake into ER is coupled to the ATP hydrolysis, restriction of ATPase activity decreases Ca2+ uptake (Xu et al., 1997). Ca2+ transport and ATPase activity of plasmalemmal Ca2+ ATPase can be inhibited by ROS due to oxidation of SH groups and peroxidation of membrane phospholipids (Kaneko et al., 1989). In SOD1G93A motor neurons, ER Ca2+ uptake by SERCA was shown to be increased (Lautenschlager et al., 2013). Ca2+ handling is reshaped during disease progression in the SOD1G93A mouse model. Increased plasma membrane extrusion upon mitochondrial failure likely indicates a compensatory mechanism in the disease. This study puts the focus on further investigations of mitochondrial and plasma membrane Ca2+ transporters such as plasmalemmal Ca2+ ATPase and plasma membrane Na+/Ca2+ exchanger (Fuchs et al., 2013).
Therapeutic strategies targeting RyR, IP3R, and SERCA
Blocking RyR using dantrolene has provided protection of motor neurons exposed to a brief excitotoxic insult in vitro, but did not show a protective effect in SOD1G93A mice. This indicates that Ca2+ release through RyRs have a modest role in SOD1 mice (Staats et al., 2012b). Inhibiting SERCA by cyclopiazonic acid showed protective effects against kainate induced excitotoxicity in SOD1G93A cultured motor neurons (Lautenschlager et al., 2013). Although there are not many studies targeting ERMCC Ca2+ channels, these could be valuable targets for further investigation.
Concluding Remarks
Riluzole is currently the only approved drug for ALS, but at best it only slows disease progression for some months. It is crucial to understand disease pathophysiology and to recognize the major upstream events that lead to motor neuron death. Disturbances of Ca2+ homeostasis and ER function are well-known features of motor neuron degeneration in ALS. Dysregulation in between the ERMCC is therefore characterized accumulation of misfolded proteins, oxidative stress and motor neuron death. Therapeutic drugs aiming to stabilize the ERMCC, reduce ER stress and support the UPR may be effective in a wide range of neuron diseases. However, genetic and the majority of pharmacologic strategies to protect ER and mitochondria against exitotoxicity have been unsuccessful. Nevertheless, these negative results, added to the many failed trials in the past, raise the question of the suitability of our experimental models, which are mostly murine. Perhaps we should focus on new tools such as induced pluripotent stem cells taken from ALS patients and derived into motor neurons. They could generate more suitible models.
Newly discovered genes that cause ALS may also offer new therapeutics for ALS. These strategies are currently underway. The SOD1G93A mice model has been extensively investigated so far, but there is an urgent requirement for additional models of ALS such as TDP-43, FUS and VCP. Moreover, the drugs that failed in clinical trials could still prove to play a valuble role as part of a combination strategy with other molecules in the future, such as drugs that operate in distinct or overlapping pathways. Developing of “smart drugs”, such as Arimoclomol that enhance protein folding capacity only under conditions of cellular stress, may also be good direction in drug development.
Finally a significant point comprises establishing improved pharmacokinetic profiles. The safety properties and most efficient dose of the drug in humans have to be adequately established prior to phase III trials. Taken together, the key for success is in basic and clinical researchers continuing to work together.
Author Contributions
All authors contributed in the conception and design of the present review, as well as in drafting and revising the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by BMBF (the Bundesministerium für Bildung und Forschung) grants to Julian Grosskreutz in the framework of the E-RARE program (PYRAMID) and JPND program (SOPHIA) of the European Union. We thank N. Kroegel for language editing. The authors report no conflicts of interests.
Abbreviations:
ALS, amyotrophic lateral sclerosis; AMPA, α-amino-5-methyl-3-hydroxisoxazolone-4-propionate; ANT, adenine nucleotide translocator; AP-1, activator protein 1; ARE, antioxidant response element; ATF6, basic leucine-zipper transcription factor 6; Bax/Bak, Bcl-2-associated X protein; Bcl-2, B-cell lymphoma 2 protein; BIK, Bcl-2 interacting killer protein; C9ORF72, chromosome 9 open reading frame 72; CHOP, transcription factor C/EBP homologous protein; eIF2α, eukaryotic initiation factor-2; ER, endoplasmic reticulum; ERMCC, endoplasmic reticulum mitochondria calcium cycle; fALS, familial amyotrophic lateral sclerosis; FUS/TSL, fused in sarcoma/translated in liposarcoma; HIF-1α, hypoxia-induced factor; Hsf1, heat shock transcription factor 1; InsP3, inositol 1,4,5-trisphosphate; IP3R, inositol 1,4,5-triphosphate receptor-gated channel; IRE1, inositol-requiring enzyme 1; mNCE, mitochondrial sodium calcium exchanger; mPTP, mitochondrial permeability transition pore; mUP, mitochondrial uniporter; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NO, nitric oxide; Nrf2, erythroid 2-related-factor 2; OPTN, optineurin; PDI, protein disulfide isomerase; PERK, the double-stranded RNA-activated protein kinase (PKR)-like ER kinase; PLCδ1, phospolipase C delta 1; ROS, reactive oxygen species; RyR, ryanodine receptors; sALS, sporadic amyotrophic lateral sclerosis; SERCA, sarco/endoplasmic reticulum Ca2+ ATPase; SOD1, Cu/Zn superoxide dismutase type 1; SR, sarcoplasmic reticulum; TARDBP, TAR DNA binding protein; TCTP, translationally controlled tumor protein; TDP-43, transactive response DNA binding protein 43 kDa; UBQLN2, ubiquilin-2; UCH-L1, ubiquitin carboxy-terminal hydrolase L1; UPR, unfolded protein response; VAPB, vesicle-associated membrane protein (VAMP)-associated protein B; VCP, valosin-containing protein; VDAC, voltage-dependent anion channel.
References
Abramson, J. J., and Salama, G. (1989). Critical sulfhydryls regulate calcium release from sarcoplasmic reticulum. J. Bioenerg. Biomembr. 21, 283–294. doi: 10.1007/BF00812073
Adachi, T., Weisbrod, R. M., Pimentel, D. R., Ying, J., Sharov, V. S., Schoneich, C., et al. (2004). S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat. Med. 10, 1200–1207. doi: 10.1038/nm1119
Akizuki, M., Yamashita, H., Uemura, K., Maruyama, H., Kawakami, H., Ito, H., et al. (2013). Optineurin suppression causes neuronal cell death via NF-kappaB pathway. J. Neurochem. 126, 699–704. doi: 10.1111/jnc.12326
Amante, D. J., Kim, J., Carreiro, S. T., Cooper, A. C., Jones, S. W., Li, T., et al. (2010). Uridine ameliorates the pathological phenotype in transgenic G93A-ALS mice. Amyotroph. Lateral Scler. 11, 520–530. doi: 10.3109/17482968.2010.491867
Appel, S. H., Stewart, S. S., Appel, V., Harati, Y., Mietlowski, W., Weiss, W., et al. (1988). A double-blind study of the effectiveness of cyclosporine in amyotrophic lateral sclerosis. Arch. Neurol. 45, 381–386. doi: 10.1001/archneur.1988.00520280027011
Arbel, N., and Shoshan-Barmatz, V. (2010). Voltage-dependent anion channel 1-based peptides interact with Bcl-2 to prevent antiapoptotic activity. J. Biol. Chem. 285, 6053–6062. doi: 10.1074/jbc.M109.082990
Armstrong, G. A., and Drapeau, P. (2013). Calcium channel agonists protect against neuromuscular dysfunction in a genetic model of TDP-43 mutation in ALS. J. Neurosci. 33, 1741–1752. doi: 10.1523/JNEUROSCI.4003-12.2013
Atkin, J. D., Farg, M. A., Walker, A. K., McLean, C., Tomas, D., and Horne, M. K. (2008). Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol. Dis. 30, 400–407. doi: 10.1016/j.nbd.2008.02.009
Bartolome, F., Wu, H. C., Burchell, V. S., Preza, E., Wray, S., Mahoney, C. J., et al. (2013). Pathogenic VCP mutations induce mitochondrial uncoupling and reduced ATP levels. Neuron 78, 57–64. doi: 10.1016/j.neuron.2013.02.028
Bedlack, R. S., Traynor, B. J., and Cudkowicz, M. E. (2007). Emerging disease-modifying therapies for the treatment of motor neuron disease/amyotropic lateral sclerosis. Expert. Opin. Emerg. Drugs 12, 229–252. doi: 10.1517/14728214.12.2.229
Beghi, E., Chio, A., Couratier, P., Esteban, J., Hardiman, O., Logroscino, G., et al. (2011). The epidemiology and treatment of ALS: focus on the heterogeneity of the disease and critical appraisal of therapeutic trials. Amyotroph. Lateral Scler. 12, 1–10. doi: 10.3109/17482968.2010.502940
Bernardi, P., Krauskopf, A., Basso, E., Petronilli, V., Blachly-Dyson, E., Di Lisa, F., et al. (2006). The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 273, 2077–2099. doi: 10.1111/j.1742-4658.2006.05213.x
Berridge, M. J. (2002). The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium 32, 235–249. doi: 10.1016/S0143416002001823
Bootman, M. D., Taylor, C. W., and Berridge, M. J. (1992). The thiol reagent, thimerosal, evokes Ca2+ spikes in HeLa cells by sensitizing the inositol 1,4,5-trisphosphate receptor. J. Biol. Chem. 267, 25113–25119.
Boraso, A., and Williams, A. J. (1994). Modification of the gating of the cardiac sarcoplasmic reticulum Ca(2+)-release channel by H2O2 and dithiothreitol. Am. J. Physiol. 267, H1010-H1016.
Bordet, T., Buisson, B., Michaud, M., Drouot, C., Galea, P., Delaage, P., et al. (2007). Identification and characterization of cholest-4-en-3-one, oxime (TRO19622), a novel drug candidate for amyotrophic lateral sclerosis. J. Pharmacol. Exp. Ther. 322, 709–720. doi: 10.1124/jpet.107.123000
Borthwick, G. M., Johnson, M. A., Ince, P. G., Shaw, P. J., and Turnbull, D. M. (1999). Mitochondrial enzyme activity in amyotrophic lateral sclerosis: implications for the role of mitochondria in neuronal cell death. Ann. Neurol. 46, 787–790. doi: 10.1002/1531-8249(199911)46:5<787::AID-ANA17>3.0.CO;2-8
Boyce, M., Bryant, K. F., Jousse, C., Long, K., Harding, H. P., Scheuner, D., et al. (2005). A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science 307, 935–939. doi: 10.1126/science.1101902
Brand, M. D. (2010). The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 45, 466–472. doi: 10.1016/j.exger.2010.01.003
Braun, R. J., Sommer, C., Carmona-Gutierrez, D., Khoury, C. M., Ring, J., Buttner, S., et al. (2011). Neurotoxic 43-kDa TAR DNA-binding protein (TDP-43) triggers mitochondrion-dependent programmed cell death in yeast. J. Biol. Chem. 286, 19958–19972. doi: 10.1074/jbc.M110.194852
Breckenridge, D. G., Stojanovic, M., Marcellus, R. C., and Shore, G. C. (2003). Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J. Cell Biol. 160, 1115–1127. doi: 10.1083/jcb.200212059
Brenner, C., and Moulin, M. (2012). Physiological roles of the permeability transition pore. Circ. Res. 111, 1237–1247. doi: 10.1161/CIRCRESAHA.112.265942
Brustovetsky, N., Brustovetsky, T., Jemmerson, R., and Dubinsky, J. M. (2002). Calcium-induced cytochrome c release from CNS mitochondria is associated with the permeability transition and rupture of the outer membrane. J. Neurochem. 80, 207–218. doi: 10.1046/j.0022-3042.2001.00671.x
Buratti, E., Dork, T., Zuccato, E., Pagani, F., Romano, M., and Baralle, F. E. (2001). Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J. 20, 1774–1784. doi: 10.1093/emboj/20.7.1774
Carriedo, S. G., Sensi, S. L., Yin, H. Z., and Weiss, J. H. (2000). AMPA exposures induce mitochondrial Ca(2+) overload and ROS generation in spinal motor neurons in vitro. J. Neurosci. 20, 240–250.
Carter, B. J., Anklesaria, P., Choi, S., and Engelhardt, J. F. (2009). Redox modifier genes and pathways in amyotrophic lateral sclerosis. Antioxid. Redox. Signal. 11, 1569–1586. doi: 10.1089/ars.2008.2414
Chinopoulos, C., and Adam-Vizi, V. (2010). Mitochondrial Ca2+ sequestration and precipitation revisited. FEBS J. 277, 3637-3651. doi: 10.1111/j.1742-4658.2010.07755.x
Coussee, E., De Smet, P., Bogaert, E., Elens, I., Van Damme, P., Willems, P., et al. (2011). G37R SOD1 mutant alters mitochondrial complex I activity, Ca(2+) uptake and ATP production. Cell Calcium 49, 217–225. doi: 10.1016/j.ceca.2011.02.004
Coyle, J. T., and Puttfarcken, P. (1993). Oxidative stress, glutamate, and neurodegenerative disorders. Science 262, 689–695. doi: 10.1126/science.7901908
Cozzolino, M., and Carri, M. T. (2012). Mitochondrial dysfunction in ALS. Prog. Neurobiol. 97, 54–66. doi: 10.1016/j.pneurobio.2011.06.003
Cozzolino, M., Ferri, A., and Carri, M. T. (2008). Amyotrophic lateral sclerosis: from current developments in the laboratory to clinical implications. Antioxid. Redox. Signal. 10, 405–443. doi: 10.1089/ars.2007.1760
Crompton, M. (1999). The mitochondrial permeability transition pore and its role in cell death. Biochem. J. 341(Pt 2), 233–249. doi: 10.1042/0264-6021:3410233
Crompton, M., Virji, S., and Ward, J. M. (1998). Cyclophilin-D binds strongly to complexes of the voltage-dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore. Eur. J. Biochem. 258, 729–735. doi: 10.1046/j.1432-1327.1998.2580729.x
Cudkowicz, M. E., Shefner, J. M., Simpson, E., Grasso, D., Yu, H., Zhang, H., et al. (2008). Arimoclomol at dosages up to 300 mg/day is well tolerated and safe in amyotrophic lateral sclerosis. Muscle Nerve 38, 837–844. doi: 10.1002/mus.21059
Cudkowicz, M. E., van den Berg, L. H., Shefner, J. M., Mitsumoto, H., Mora, J. S., Ludolph, A., et al. (2013). Dexpramipexole versus placebo for patients with amyotrophic lateral sclerosis (EMPOWER): a randomised, double-blind, phase 3 trial. Lancet Neurol. 12, 1059–1067. doi: 10.1016/S1474-4422(13)70221-7
Daigle, J. G., Lanson, N. A. Jr., Smith, R. B., Casci, I., Maltare, A., Monaghan, J., et al. (2013). RNA-binding ability of FUS regulates neurodegeneration, cytoplasmic mislocalization and incorporation into stress granules associated with FUS carrying ALS-linked mutations. Hum. Mol. Genet. 22, 1193–1205. doi: 10.1093/hmg/dds526
Dardiotis, E., Panayiotou, E., Feldman, M. L., Hadjisavvas, A., Malas, S., Vonta, I., et al. (2013). Intraperitoneal melatonin is not neuroprotective in the G93ASOD1 transgenic mouse model of familial ALS and may exacerbate neurodegeneration. Neurosci. Lett. 548, 170–175. doi: 10.1016/j.neulet.2013.05.058
Das, A. M., and Harris, D. A. (1990). Control of mitochondrial ATP synthase in heart cells: inactive to active transitions caused by beating or positive inotropic agents. Cardiovasc. Res. 24, 411–417. doi: 10.1093/cvr/24.5.411
De Vos, K. J., Morotz, G. M., Stoica, R., Tudor, E. L., Lau, K. F., Ackerley, S., et al. (2012). VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum. Mol. Genet. 21, 1299–1311. doi: 10.1093/hmg/ddr559
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
Dewey, C. M., Cenik, B., Sephton, C. F., Johnson, B. A., Herz, J., and Yu, G. (2012). TDP-43 aggregation in neurodegeneration: are stress granules the key? Brain Res. 1462, 16–25. doi: 10.1016/j.brainres.2012.02.032
Di Lisa, F., and Bernardi, P. (2009). A CaPful of mechanisms regulating the mitochondrial permeability transition. J. Mol. Cell Cardiol. 46, 775–780. doi: 10.1016/j.yjmcc.2009.03.006
Dremina, E. S., Sharov, V. S., Davies, M. J., and Schoneich, C. (2007). Oxidation and inactivation of SERCA by selective reaction of cysteine residues with amino acid peroxides. Chem. Res. Toxicol. 20, 1462–1469. doi: 10.1021/tx700108w
Duan, W., Li, X., Shi, J., Guo, Y., Li, Z., and Li, C. (2010). Mutant TAR DNA-binding protein-43 induces oxidative injury in motor neuron-like cell. Neuroscience 169, 1621–1629. doi: 10.1016/j.neuroscience.2010.06.018
Dudek, J., Benedix, J., Cappel, S., Greiner, M., Jalal, C., Muller, L., et al. (2009). Functions and pathologies of BiP and its interaction partners. Cell Mol. Life Sci. 66, 1556–1569. doi: 10.1007/s00018-009-8745-y
Fabiato, A. (1983). Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am. J. Physiol. 245, C1-C14.
Fannjiang, Y., Cheng, W. C., Lee, S. J., Qi, B., Pevsner, J., McCaffery, J. M., et al. (2004). Mitochondrial fission proteins regulate programmed cell death in yeast. Genes Dev. 18, 2785–2797. doi: 10.1101/gad.1247904
Farg, M. A., Soo, K. Y., Warraich, S. T., Sundaramoorthy, V., Blair, I. P., and Atkin, J. D. (2013). Ataxin-2 interacts with FUS and intermediate-length polyglutamine expansions enhance FUS-related pathology in amyotrophic lateral sclerosis. Hum. Mol. Genet. 22, 717–728. doi: 10.1093/hmg/dds479
Feissner, R. F., Skalska, J., Gaum, W. E., and Sheu, S. S. (2009). Crosstalk signaling between mitochondrial Ca2+ and ROS. Front Biosci. 14:1197–1218. doi: 10.2741/3303
Feng, W., Liu, G., Allen, P. D., and Pessah, I. N. (2000). Transmembrane redox sensor of ryanodine receptor complex. J. Biol. Chem. 275, 35902–35907. doi: 10.1074/jbc.C000523200
Ferraiuolo, L., Kirby, J., Grierson, A. J., Sendtner, M., and Shaw, P. J. (2011). Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 7, 616–630. doi: 10.1038/nrneurol.2011.152
Ferrante, R. J., Browne, S. E., Shinobu, L. A., Bowling, A. C., Baik, M. J., MacGarvey, U., et al. (1997). Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J. Neurochem. 69, 2064–2074. doi: 10.1046/j.1471-4159.1997.69052064.x
Fill, M., and Copello, J. A. (2002). Ryanodine receptor calcium release channels. Physiol. Rev. 82, 893–922.
Fischer, L. R., Igoudjil, A., Magrane, J., Li, Y., Hansen, J. M., Manfredi, G., et al. (2011). SOD1 targeted to the mitochondrial intermembrane space prevents motor neuropathy in the Sod1 knockout mouse. Brain 134, 196–209. doi: 10.1093/brain/awq314
Foskett, J. K., White, C., Cheung, K. H., and Mak, D. O. (2007). Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 87, 593–658. doi: 10.1152/physrev.00035.2006
Fratta, P., Mizielinska, S., Nicoll, A. J., Zloh, M., Fisher, E. M., Parkinson, G., et al. (2012). C9orf72 hexanucleotide repeat associated with amyotrophic lateral sclerosis and frontotemporal dementia forms RNA G-quadruplexes. Sci. Rep. 2, 1016. doi: 10.1038/srep01016
Fuchs, A., Kutterer, S., Muhling, T., Duda, J., Schutz, B., Liss, B., et al. (2013). Selective mitochondrial Ca2+ uptake deficit in disease endstage vulnerable motoneurons of the SOD1G93A mouse model of amyotrophic lateral sclerosis. J. Physiol. 591, 2723–2745. doi: 10.1113/jphysiol.2012.247981
Germain, M., Mathai, J. P., and Shore, G. C. (2002). BH-3-only BIK functions at the endoplasmic reticulum to stimulate cytochrome c release from mitochondria. J. Biol. Chem. 277, 18053–18060. doi: 10.1074/jbc.M201235200
Giorgio, V., Soriano, M. E., Basso, E., Bisetto, E., Lippe, G., Forte, M. A., et al. (2010). Cyclophilin D in mitochondrial pathophysiology. Biochim. Biophys. Acta 1797, 1113–1118. doi: 10.1016/j.bbabio.2009.12.006
Gonzalez-Lafuente, L., Egea, J., Leon, R., Martinez-Sanz, F. J., Monjas, L., Perez, C., et al. (2012). Benzothiazepine CGP37157 and its isosteric 2’-methyl analogue provide neuroprotection and block cell calcium entry. ACS Chem. Neurosci. 3, 519–529. doi: 10.1021/cn300009e
Gordon, P. H., Moore, D. H., Miller, R. G., Florence, J. M., Verheijde, J. L., Doorish, C., et al. (2007). Efficacy of minocycline in patients with amyotrophic lateral sclerosis: a phase III randomised trial. Lancet Neurol. 6, 1045–1053. doi: 10.1016/S1474-4422(07)70270-3
Grosskreutz, J., Haastert, K., Dewil, M., Van Damme, P., Callewaert, G., Robberecht, W., et al. (2007). Role of mitochondria in kainate-induced fast Ca(2+) transients in cultured spinal motor neurons. Cell Calcium 42, 59–69. doi: 10.1016/j.ceca.2006.11.010
Grosskreutz, J., Jaeckel, M., Laderer, C., Haastert, K., Claus, P., Grothe, C., et al. (2004). Calcium-induced calcium release as mechanism of AMPA-mediated excitotoxicity in motoneurons. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 5, 106.
Grosskreutz, J., Van Den, B. L., and Keller, B. U. (2010). Calcium dysregulation in amyotrophic lateral sclerosis. Cell Calcium 47, 165–174. doi: 10.1016/j.ceca.2009.12.002
Gunter, T. E., and Sheu, S. S. (2009). Characteristics and possible functions of mitochondrial Ca(2+) transport mechanisms. Biochim. Biophys. Acta 1787, 1291–1308. doi: 10.1016/j.bbabio.2008.12.011
Guo, Y., Zhang, Y., Wen, D., Duan, W., An, T., Shi, P., et al. (2013). The modest impact of transcription factor Nrf2 on the course of disease in an ALS animal model. Lab. Invest. 93, 825–833. doi: 10.1038/labinvest.2013.73
Haddad, J. J. (2002). Antioxidant and prooxidant mechanisms in the regulation of redox(y)-sensitive transcription factors. Cell Signal. 14, 879–897. doi: 10.1016/S0898-6568(02)00053-0
Hashimoto, K., Hayashi, Y., Watabe, K., Inuzuka, T., and Hozumi, I. (2011). Metallothionein-III prevents neuronal death and prolongs life span in amyotrophic lateral sclerosis model mice. Neuroscience 189, 293–298. doi: 10.1016/j.neuroscience.2011.05.034
Hirano, A., Nakano, I., Kurland L. T., Mulder D. W., Holley P. W., and Saccomanno, G. (1984). Fine structural study of neurofibrillary changes in a family with amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 43, 571–480.
Hong, K., Li, Y., Duan, W., Guo, Y., Jiang, H., Li, W., et al. (2012). Full-length TDP-43 and its C-terminal fragments activate mitophagy in NSC34 cell line. Neurosci. Lett. 530, 144–149. doi: 10.1016/j.neulet.2012.10.003
Honjo, Y., Kaneko, S., Ito, H., Horibe, T., Nagashima, M., Nakamura, M., et al. (2011). Protein disulfide isomerase-immunopositive inclusions in patients with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 14, 408–413. doi: 10.3109/17482968.2011.594055
Hozumi, I. (2013). Roles and Therapeutic potential of metallothioneinsin neurodegenerative diseases. Curr. Pharm. Biotechnol. 9, 294–298.
Hozumi, I., Yamada, M., Uchida, Y., Ozawa, K., Takahashi, H., and Inuzuka, T. (2008). The expression of metallothioneins is diminished in the spinal cords of patients with sporadic ALS. Amyotroph. Lateral Scler. 9, 294–298. doi: 10.1080/17482960801934312
Iaccarino, C., Mura, M. E., Esposito, S., Carta, F., Sanna, G., Turrini, F., et al. (2011). Bcl2-A1 interacts with pro-caspase-3: implications for amyotrophic lateral sclerosis. Neurobiol. Dis. 43, 642–650. doi: 10.1016/j.nbd.2011.05.013
Ilieva, E. V., Ayala, V., Jove, M., Dalfo, E., Cacabelos, D., Povedano, M., et al. (2007). Oxidative and endoplasmic reticulum stress interplay in sporadic amyotrophic lateral sclerosis. Brain 130, 3111–3123. doi: 10.1093/brain/awm190
Ince, P., Stout, N., Shaw, P., Slade, J., Hunziker, W., Heizmann, C. W., et al. (1993). Parvalbumin and calbindin D-28k in the human motor system and in motor neuron disease. Neuropathol. Appl. Neurobiol. 19, 291–299. doi: 10.1111/j.1365-2990.1993.tb00443.x
Israelson, A., Arbel, N., Da Cruz, S., Ilieva, H., Yamanaka, K., Shoshan-Barmatz, V., et al. (2010). Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron 67, 575–587. doi: 10.1016/j.neuron.2010.07.019
Jacob, S., Poeggeler, B., Weishaupt, J. H., Siren, A. L., Hardeland, R., Bahr, M., et al. (2002). Melatonin as a candidate compound for neuroprotection in amyotrophic lateral sclerosis (ALS): high tolerability of daily oral melatonin administration in ALS patients. J. Pineal Res. 33, 186–187. doi: 10.1034/j.1600-079X.2002.02943.x
Jahn, K., Grosskreutz, J., Haastert, K., Ziegler, E., Schlesinger, F., Grothe, C., et al. (2006). Temporospatial coupling of networked synaptic activation of AMPA-type glutamate receptor channels and calcium transients in cultured motoneurons. Neuroscience 142, 1019–1029. doi: 10.1016/j.neuroscience.2006.07.034
Jaiswal, M. K. (2013). Calcium, mitochondria, and the pathogenesis of ALS: the good, the bad, and the ugly. Front. Cell. Neurosci. 7:199. doi: 10.3389/fncel.2013.00199
Jaiswal, M. K., and Keller, B. U. (2009). Cu/Zn superoxide dismutase typical for familial amyotrophic lateral sclerosis increases the vulnerability of mitochondria and perturbs Ca2+ homeostasis in SOD1G93A mice. Mol. Pharmacol. 75, 478–489. doi: 10.1124/mol.108.050831
Jaiswal, M. K., Zech, W. D., Goos, M., Leutbecher, C., Ferri, A., Zippelius, A., et al. (2009). Impairment of mitochondrial calcium handling in a mtSOD1 cell culture model of motoneuron disease. BMC Neurosci. 10:64. doi: 10.1186/1471-2202-10-64
Jouaville, L. S., Pinton, P., Bastianutto, C., Rutter, G. A., and Rizzuto, R. (1999). Regulation of mitochondrial ATP synthesis by calcium: evidence for a long-term metabolic priming. Proc. Natl. Acad. Sci. U.S.A. 96, 13807–13812. doi: 10.1073/pnas.96.24.13807
Juhaszova, M., Wang, S., Zorov, D. B., Nuss, H. B., Gleichmann, M., Mattson, M. P., et al. (2008). The identity and regulation of the mitochondrial permeability transition pore: where the known meets the unknown. Ann. N. Y. Acad. Sci. 1123, 197–212. doi: 10.1196/annals.1420.023
Kachaner, D., Genin, P., Laplantine, E., and Weil, R. (2012). Toward an integrative view of Optineurin functions. Cell Cycle 11, 2808–2818. doi: 10.4161/cc.20946
Kalmar, B., Edet-Amana, E., and Greensmith, L. (2012). Treatment with a coinducer of the heat shock response delays muscle denervation in the SOD1-G93A mouse model of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 13, 378–392. doi: 10.3109/17482968.2012.660953
Kalmar, B., Lu, C. H., and Greensmith, L. (2014). The role of heat shock proteins in amyotrophic lateral sclerosis: the therapeutic potential of Arimoclomol. Pharmacol. Ther. 141, 40–54. doi: 10.1016/j.pharmthera.2013.08.003
Kalmar, B., Novoselov, S., Gray, A., Cheetham, M. E., Margulis, B., and Greensmith, L. (2008). Late stage treatment with arimoclomol delays disease progression and prevents protein aggregation in the SOD1 mouse model of ALS. J. Neurochem. 107, 339–350. doi: 10.1111/j.1471-4159.2008.05595.x
Kamada, M., Izumi, Y., Ayaki, T., Nakamura, M., Kagawa, S., Kudo, E., et al. (2014). Clinicopathologic features of autosomal recessive amyotrophic lateral sclerosis associated with optineurin mutation. Neuropathology 34, 64–70. doi: 10.1111/neup.12051
Kaneko, M., Beamish, R. E., and Dhalla, N. S. (1989). Depression of heart sarcolemmal Ca2+-pump activity by oxygen free radicals. Am. J. Physiol. 256, H368-H374.
Kaplan, P., Babusikova, E., Lehotsky, J., and Dobrota, D. (2003). Free radical-induced protein modification and inhibition of Ca2+-ATPase of cardiac sarcoplasmic reticulum. Mol. Cell Biochem. 248, 41–47. doi: 10.1023/A:1024145212616
Kaufmann, P., Thompson, J. L., Levy, G., Buchsbaum, R., Shefner, J., Krivickas, L. S., et al. (2009). Phase II trial of CoQ10 for ALS finds insufficient evidence to justify phase III. Ann. Neurol. 66, 235–244. doi: 10.1002/ana.21743
Keeney, P. M., and Bennett, J. P. Jr. (2010). ALS spinal neurons show varied and reduced mtDNA gene copy numbers and increased mtDNA gene deletions. Mol. Neurodegener. 5, 21. doi: 10.1186/1750-1326-5-21
Kihira, T., Utunomiya, H., and Kondo, T. (2005). Expression of FKBP12 and ryanodine receptors (RyRs) in the spinal cord of MND patients. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 6, 94–99. doi: 10.1080/14660820510034442
Kim, S. H., Zhan, L., Hanson, K. A., and Tibbetts, R. S. (2012). High-content RNAi screening identifies the Type 1 inositol triphosphate receptor as a modifier of TDP-43 localization and neurotoxicity. Hum. Mol. Genet. 21, 4845–4856. doi: 10.1093/hmg/dds321
Kong, D., Xu, L., Yu, Y., Zhu, W., Andrews, D. W., Yoon, Y., et al. (2005). Regulation of Ca2+-induced permeability transition by Bcl-2 is antagonized by Drpl and hFis1. Mol. Cell Biochem. 272, 187–199. doi: 10.1007/s11010-005-7323-3
Kong, J., and Xu, Z. (1998). Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J. Neurosci. 18, 3241–3250.
Kostrominova, T. Y. (2010). Advanced age-related denervation and fiber-type grouping in skeletal muscle of SOD1 knockout mice. Free Radic. Biol. Med. 49, 1582-1593. doi: 10.1016/j.freeradbiomed.2010.08.022
Kozutsumi, Y., Segal, M., Normington, K., Gething, M. J., and Sambrook, J. (1988). The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature 332, 462–464. doi: 10.1038/332462a0
Kukreja, R. C., Okabe, E., Schrier, G. M., and Hess, M. L. (1988). Oxygen radical-mediated lipid peroxidation and inhibition of Ca2+-ATPase activity of cardiac sarcoplasmic reticulum. Arch. Biochem. Biophys. 261, 447–457. doi: 10.1016/0003-9861(88)90361-X
Kwiatkowski, T. J. Jr., Bosco, D. A., Leclerc, A. L., Tamrazian, E., Vanderburg, C. R., Russ, C., et al. (2009). Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208. doi: 10.1126/science.1166066
Langou, K., Moumen, A., Pellegrino, C., Aebischer, J., Medina, I., Aebischer, P., et al. (2010). AAV-mediated expression of wild-type and ALS-linked mutant VAPB selectively triggers death of motoneurons through a Ca2+-dependent ER-associated pathway. J. Neurochem. 114, 795–809. doi: 10.1111/j.1471-4159.2010.06806.x
Lanner, J. T., Georgiou, D. K., Joshi, A. D., and Hamilton, S. L. (2010). Ryanodine receptors: structure, expression, molecular details, and function in calcium release. Cold Spring Harb. Perspect. Biol. 2, a003996. doi: 10.1101/cshperspect.a003996
Lautenschlaeger, J., Prell, T., and Grosskreutz, J. (2012). Endoplasmic reticulum stress and the ER mitochondrial calcium cycle in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 13, 166–177. doi: 10.3109/17482968.2011.641569
Lautenschlager, J., Prell, T., Ruhmer, J., Weidemann, L., Witte, O. W., and Grosskreutz, J. (2013). Overexpression of human mutated G93A SOD1 changes dynamics of the ER mitochondria calcium cycle specifically in mouse embryonic motor neurons. Exp. Neurol. 13, 166–167. doi: 10.1016/j.expneurol.2013.03.027
Leung, A. W., and Halestrap, A. P. (2008). Recent progress in elucidating the molecular mechanism of the mitochondrial permeability transition pore. Biochim. Biophys. Acta 1777, 946–952. doi: 10.1016/j.bbabio.2008.03.009
Lipskaia, L., Hulot, J. S., and Lompre, A. M. (2009). Role of sarco/endoplasmic reticulum calcium content and calcium ATPase activity in the control of cell growth and proliferation. Pflugers Arch. 457, 673–685. doi: 10.1007/s00424-007-0428-7
Liu, D., Wen, J., Liu, J., and Li, L. (1999). The roles of free radicals in amyotrophic lateral sclerosis: reactive oxygen species and elevated oxidation of protein, DNA, and membrane phospholipids. FASEB J. 13, 2318–2328.
Liu, G., Abramson, J. J., Zable, A. C., and Pessah, I. N. (1994). Direct evidence for the existence and functional role of hyperreactive sulfhydryls on the ryanodine receptor-triadin complex selectively labeled by the coumarin maleimide 7-diethylamino-3-(4’-maleimidylphenyl)-4-methylcoumarin. Mol. Pharmacol. 45, 189–200.
Liu, J., Lillo, C., Jonsson, P. A., Vande, V. C., Ward, C. M., Miller, T. M., et al. (2004). Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron 43, 5–17. doi: 10.1016/j.neuron.2004.06.016
Liu, X., Kim, C. N., Yang, J., Jemmerson, R., and Wang, X. (1996). Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 86, 147–157. doi: 10.1016/S0092-8674(00)80085-9
Liu-Yesucevitz, L., Bilgutay, A., Zhang, Y. J., Vanderweyde, T., Citro, A., Mehta, T., et al. (2010). Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS ONE 5:e13250. doi: 10.1371/journal.pone.0013250
Lu, J., Duan, W., Guo, Y., Jiang, H., Li, Z., Huang, J., et al. (2012). Mitochondrial dysfunction in human TDP-43 transfected NSC34 cell lines and the protective effect of dimethoxy curcumin. Brain Res. Bull. 89, 185–190. doi: 10.1016/j.brainresbull.2012.09.005
MacLennan, D. H., Rice, W. J., and Green, N. M. (1997). The mechanism of Ca2+ transport by sarco(endo)plasmic reticulum Ca2+-ATPases. J. Biol. Chem. 272, 28815–28818. doi: 10.1074/jbc.272.46.28815
Magrane, J., Cortez, C., Gan, W., and Manfredi, G. (2013). Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum. Mol. Genet. 23, 1413–1424. doi: 10.1093/hmg/ddt528
Mancuso, R., Olivan, S., Rando, A., Casas, C., Osta, R., and Navarro, X. (2012). Sigma-1R agonist improves motor function and motoneuron survival in ALS mice. Neurotherapeutics 9, 814–826. doi: 10.1007/s13311-012-0140-y
Marengo, J. J., Hidalgo, C., and Bull, R. (1998). Sulfhydryl oxidation modifies the calcium dependence of ryanodine-sensitive calcium channels of excitable cells. Biophys. J. 74, 1263–1277. doi: 10.1016/S0006-3495(98)77840-3
Margineantu, D. H., Gregory, C. W., Sundell, L., Sherwood, S. W., Beechem, J. M., and Capaldi, R. A. (2002). Cell cycle dependent morphology changes and associated mitochondrial DNA redistribution in mitochondria of human cell lines. Mitochondrion 1, 425–435. doi: 10.1016/S1567-7249(02)00006-5
Martin, L. J. (2010a). Olesoxime, a cholesterol-like neuroprotectant for the potential treatment of amyotrophic lateral sclerosis. IDrugs 13, 568–580.
Martin, L. J. (2010b). The mitochondrial permeability transition pore: a molecular target for amyotrophic lateral sclerosis therapy. Biochim. Biophys. Acta 1802, 186–197. doi: 10.1016/j.bbadis.2009.07.009
Martin, L. J., Gertz, B., Pan, Y., Price, A. C., Molkentin, J. D., and Chang, Q. (2009). The mitochondrial permeability transition pore in motor neurons: involvement in the pathobiology of ALS mice. Exp. Neurol. 218, 333–346. doi: 10.1016/j.expneurol.2009.02.015
Martin, L. J., Liu, Z., Chen, K., Price, A. C., Pan, Y., Swaby, J. A., et al. (2007). Motor neuron degeneration in amyotrophic lateral sclerosis mutant superoxide dismutase-1 transgenic mice: mechanisms of mitochondriopathy and cell death. J. Comp. Neurol. 500, 20–46. doi: 10.1002/cne.21160
Mattiazzi, M., D’Aurelio, M., Gajewski, C. D., Martushova, K., Kiaei, M., Beal, M. F., et al. (2002). Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J. Biol. Chem. 277, 29626–29633. doi: 10.1074/jbc.M203065200
McCormack, J. G., and Denton, R. M. (1979). The effects of calcium ions and adenine nucleotides on the activity of pig heart 2-oxoglutarate dehydrogenase complex. Biochem. J. 180, 533–544.
McCormack, J. G., and Denton, R. M. (1993). Mitochondrial Ca2+ transport and the role of intramitochondrial Ca2+ in the regulation of energy metabolism. Dev. Neurosci. 15, 165–173. doi: 10.1159/000111332
McCormack, J. G., Halestrap, A. P., and Denton, R. M. (1990). Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol. Rev. 70, 391–425.
McPherson, P. S., Kim, Y. K., Valdivia, H., Knudson, C. M., Takekura, H., Franzini-Armstrong, C., et al. (1991). The brain ryanodine receptor: a caffeine-sensitive calcium release channel. Neuron 7, 17–25. doi: 10.1016/0896-6273(91)90070-G
Mead, R. J., Higginbottom, A., Allen, S. P., Kirby, J., Bennett, E., Barber, S. C., et al. (2013). S[+] Apomorphine is a CNS penetrating activator of the Nrf2-ARE pathway with activity in mouse and patient fibroblast models of amyotrophic lateral sclerosis. Free Radic. Biol. Med. 61C, 438-452. doi: 10.1016/j.freeradbiomed.2013.04.018
Meissner, G. (2002). Regulation of mammalian ryanodine receptors. Front. Biosci. 7:d2072-d2080. doi: 10.2741/meissner Source: PubMed
Milani, P., Ambrosi, G., Gammoh, O., Blandini, F., and Cereda, C. (2013). SOD1 and DJ-1 converge at Nrf2 pathway: a clue for antioxidant therapeutic potential in neurodegeneration. Oxid. Med. Cell Longev. 2013, 836760. doi: 10.1155/2013/836760
Mildaziene, V., Baniene, R., Nauciene, Z., Bakker, B. M., Brown, G. C., Westerhoff, H. V., et al. (1995). Calcium indirectly increases the control exerted by the adenine nucleotide translocator over 2-oxoglutarate oxidation in rat heart mitochondria. Arch. Biochem. Biophys. 324, 130–134. doi: 10.1006/abbi.1995.9918
Missiaen, L., Taylor, C. W., and Berridge, M. J. (1991). Spontaneous calcium release from inositol trisphosphate-sensitive calcium stores. Nature 352, 241–244. doi: 10.1038/352241a0
Moreau, B., Nelson, C., and Parekh, A. B. (2006). Biphasic regulation of mitochondrial Ca2+ uptake by cytosolic Ca2+ concentration. Curr. Biol. 16, 1672–1677. doi: 10.1016/j.cub.2006.06.059
Moreau, C., Gosset, P., Kluza, J., Brunaud-Danel, V., Lassalle, P., Marchetti, P., et al. (2011). Deregulation of the hypoxia inducible factor-1alpha pathway in monocytes from sporadic amyotrophic lateral sclerosis patients. Neuroscience 172, 110–117. doi: 10.1016/j.neuroscience.2010.10.040
Mori, K., Weng, S. M., Arzberger, T., May, S., Rentzsch, K., Kremmer, E., et al. (2013). The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339, 1335–1338. doi: 10.1126/science.1232927
Morotz, G. M., De Vos, K. J., Vagnoni, A., Ackerley, S., Shaw, C. E., and Miller, C. C. (2012). Amyotrophic lateral sclerosis-associated mutant VAPBP56S perturbs calcium homeostasis to disrupt axonal transport of mitochondria. Hum. Mol. Genet. 21, 1979–1988. doi: 10.1093/hmg/dds011
Murphy, M. P. (2009). How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13. doi: 10.1042/BJ20081386
Nakagawa, T., Zhu, H., Morishima, N., Li, E., Xu, J., Yankner, B. A., et al. (2000). Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 403, 98–103. doi: 10.1038/47513
Neef, D. W., Jaeger, A. M., and Thiele, D. J. (2011). Heat shock transcription factor 1 as a therapeutic target in neurodegenerative diseases. Nat. Rev. Drug Discov. 10, 930–944. doi: 10.1038/nrd3453
Novoselov, S. S., Mustill, W. J., Gray, A. L., Dick, J. R., Kanuga, N., Kalmar, B., et al. (2013). Molecular chaperone mediated late-stage neuroprotection in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. PLoS ONE 8:e73944. doi: 10.1371/journal.pone.0073944
Nutt, L. K., Pataer, A., Pahler, J., Fang, B., Roth, J., McConkey, D. J., et al. (2002). Bax and Bak promote apoptosis by modulating endoplasmic reticular and mitochondrial Ca2+ stores. J. Biol. Chem. 277, 9219–9225. doi: 10.1074/jbc.M106817200
Oh, Y. K., Shin, K. S., Yuan, J., and Kang, S. J. (2008). Superoxide dismutase 1 mutants related to amyotrophic lateral sclerosis induce endoplasmic stress in neuro2a cells. J. Neurochem. 104, 993–1005. doi: 10.1111/j.1471-4159.2007.05053.x
Ono, Y., Tanaka, H., Takata, M., Nagahara, Y., Noda, Y., Tsuruma, K., et al. (2014). SA4503, a sigma-1 receptor agonist, suppresses motor neuron damage in in vitro and in vivo amyotrophic lateral sclerosis models. Neurosci. Lett. 559, 174–178. doi: 10.1016/j.neulet.2013.12.005
Parone, P. A., Da Cruz, S., Han, J. S., McAlonis-Downes, M., Vetto, A. P., Lee, S. K., et al. (2013). Enhancing mitochondrial calcium buffering capacity reduces aggregation of misfolded SOD1 and motor neuron cell death without extending survival in mouse models of inherited amyotrophic lateral sclerosis. J. Neurosci. 33, 4657–4671. doi: 10.1523/JNEUROSCI.1119-12.2013
Pasinelli, P., Belford, M. E., Lennon, N., Bacskai, B. J., Hyman, B. T., Trotti, D., et al. (2004). Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron 43, 19–30. doi: 10.1016/j.neuron.2004.06.021
Pedrini, S., Sau, D., Guareschi, S., Bogush, M., Brown, R. H. Jr., Naniche, N., et al. (2010). ALS-linked mutant SOD1 damages mitochondria by promoting conformational changes in Bcl-2. Hum. Mol. Genet. 19, 2974–2986. doi: 10.1093/hmg/ddq202
Petri, S., Korner, S., and Kiaei, M. (2012). Nrf2/ARE signaling pathway: key mediator in oxidative stress and potential therapeutic target in ALS. Neurol. Res. Int. 2012, 878030. doi: 10.1155/2012/878030
Peviani, M., Salvaneschi, E., Bontempi, L., Petese, A., Manzo, A., Rossi, D., et al. (2014). Neuroprotective effects of the Sigma-1 receptor (S1R) agonist PRE-084, in a mouse model of motor neuron disease not linked to SOD1 mutation. Neurobiol. Dis. 62, 218–232. doi: 10.1016/j.nbd.2013.10.010
Pickles, S., Destroismaisons, L., Peyrard, S. L., Cadot, S., Rouleau, G. A., Brown, R. H., et al. (2013). Mitochondrial damage revealed by immunoselection for ALS-linked misfolded SOD1. Hum. Mol. Genet. 22, 3947–3959. doi: 10.1093/hmg/ddt249
Pivovarova, N. B., and Andrews, S. B. (2010). Calcium-dependent mitochondrial function and dysfunction in neurons. FEBS J. 277, 3622–3636. doi: 10.1111/j.1742-4658.2010.07754.x
Poon, H. F., Hensley, K., Thongboonkerd, V., Merchant, M. L., Lynn, B. C., Pierce, W.M., et al. (2005). Redox proteomics analysis of oxidatively modified proteins in G93A-SOD1 transgenic mice - a model of familial amyotrophic lateral sclerosis. Free Radic. Biol. Med. 39, 453–462. doi: 10.1016/j.freeradbiomed.2005.03.030
Prell, T., Lautenschlager, J., and Grosskreutz, J. (2013). Calcium-dependent protein folding in amyotrophic lateral sclerosis. Cell Calcium 54, 132–143. doi: 10.1016/j.ceca.2013.05.007
Prell, T., Lautenschlager, J., Witte, O. W., Carri, M. T., and Grosskreutz, J. (2012). The unfolded protein response in models of human mutant G93A amyotrophic lateral sclerosis. Eur. J. Neurosci. 35, 652–660. doi: 10.1111/j.1460-9568.2012.08008.x
Reijonen, S., Putkonen, N., Norremolle, A., Lindholm, D., and Korhonen, L. (2008). Inhibition of endoplasmic reticulum stress counteracts neuronal cell death and protein aggregation caused by N-terminal mutant huntingtin proteins. Exp. Cell Res. 314, 950–960. doi: 10.1016/j.yexcr.2007.12.025
Renton, A. E., Majounie, E., Waite, A., Simon-Sanchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
Rhodes, J. D., and Sanderson, J. (2009). The mechanisms of calcium homeostasis and signalling in the lens. Exp. Eye Res. 88, 226–234. doi: 10.1016/j.exer.2008.10.025
Rizzardini, M., Lupi, M., Mangolini, A., Babetto, E., Ubezio, P., and Cantoni, L. (2006). Neurodegeneration induced by complex I inhibition in a cellular model of familial amyotrophic lateral sclerosis. Brain Res. Bull. 69, 465–474. doi: 10.1016/j.brainresbull.2006.02.013
Rodriguez-Ortiz, C. J., Hoshino, H., Cheng, D., Liu-Yescevitz, L., Blurton-Jones, M., Wolozin, B., et al. (2013). Neuronal-specific overexpression of a mutant valosin-containing protein associated with IBMPFD promotes aberrant ubiquitin and TDP-43 accumulation and cognitive dysfunction in transgenic mice. Am. J. Pathol. 183, 504–515. doi: 10.1016/j.ajpath.2013.04.014
Rooney, T. A., Renard, D. C., Sass, E. J., and Thomas, A. P. (1991). Oscillatory cytosolic calcium waves independent of stimulated inositol 1,4,5-trisphosphate formation in hepatocytes. J. Biol. Chem. 266, 12272–12282.
Rowe, G. T., Manson, N. H., Caplan, M., and Hess, M. L. (1983). Hydrogen peroxide and hydroxyl radical mediation of activated leukocyte depression of cardiac sarcoplasmic reticulum. Participation of the cyclooxygenase pathway. Circ. Res. 53, 584–591. doi: 10.1161/01.RES.53.5.584
Rowland, A. A., and Voeltz, G. K. (2012). Endoplasmic reticulum-mitochondria contacts: function of the junction. Nat. Rev. Mol. Cell Biol. 13, 607–625. doi: 10.1038/nrm3440
Rutkevich, L. A., Cohen-Doyle, M. F., Brockmeier, U., and Williams, D. B. (2010). Functional relationship between protein disulfide isomerase family members during the oxidative folding of human secretory proteins. Mol. Biol. Cell 21, 3093–3105. doi: 10.1091/mbc.E10-04-0356
Sarkar, S., Perlstein, E. O., Imarisio, S., Pineau, S., Cordenier, A., Maglathlin, R. L., et al. (2007). Small molecules enhance autophagy and reduce toxicity in Huntington’s disease models. Nat. Chem. Biol. 3, 331–338. doi: 10.1038/nchembio883
Sasaki, S., and Iwata, M. (1996). Ultrastructural study of synapses in the anterior horn neurons of patients with amyotrophic lateral sclerosis. Neurosci. Lett. 204, 53–56. doi: 10.1016/0304-3940(96)12314-4
Saxena, S., Cabuy, E., and Caroni, P. (2009). A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat. Neurosci. 12, 627–636. doi: 10.1038/nn.2297
Scherer, N. M., and Deamer, D. W. (1986). Oxidative stress impairs the function of sarcoplasmic reticulum by oxidation of sulfhydryl groups in the Ca2+-ATPase. Arch. Biochem. Biophys. 246, 589–601. doi: 10.1016/0003-9861(86)90314-0
Shefner, J. M., Cudkowicz, M. E., Schoenfeld, D., Conrad, T., Taft, J., Chilton, M., et al. (2004). A clinical trial of creatine in ALS. Neurology 63, 1656–1661. doi: 10.1212/01.WNL.0000142992.81995.F0
Siklos, L., Engelhardt, J., Harati, Y., Smith, R. G., Joo, F., and Appel, S. H. (1996). Ultrastructural evidence for altered calcium in motor nerve terminals in amyotropic lateral sclerosis. Ann. Neurol. 39, 203–216. doi: 10.1002/ana.410390210
Smith, W. W., Jiang, H., Pei, Z., Tanaka, Y., Morita, H., Sawa, A., et al. (2005). Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum. Mol. Genet. 14, 3801–3811. doi: 10.1093/hmg/ddi396
Son, M., Leary, S. C., Romain, N., Pierrel, F., Winge, D. R., Haller, R. G., et al. (2008). Isolated cytochrome c oxidase deficiency in G93A SOD1 mice overexpressing CCS protein. J. Biol. Chem. 283, 12267–12275. doi: 10.1074/jbc.M708523200
Soon, C. P., Donnelly, P. S., Turner, B. J., Hung, L. W., Crouch, P. J., Sherratt, N. A., et al. (2011). Diacetylbis(N(4)-methylthiosemicarbazonato) copper(II) (CuII(atsm)) protects against peroxynitrite-induced nitrosative damage and prolongs survival in amyotrophic lateral sclerosis mouse model. J. Biol. Chem. 286, 44035–44044. doi: 10.1074/jbc.M111.274407
Staats, K. A., Bogaert, E., Hersmus, N., Jaspers, T., Luyten, T., Bultynck, G., et al. (2012a). Neuronal overexpression of IP(3) receptor 2 is detrimental in mutant SOD1 mice. Biochem. Biophys. Res. Commun. 429, 210–213. doi: 10.1016/j.bbrc.2012.10.094
Staats, K. A., Van Rillaer, M., Scheveneels, W., Verbesselt, R., Van Damme, P., Robberecht, W., et al. (2012b). Dantrolene is neuroprotective in vitro, but does not affect survival in SOD1(G(9)(3)A) mice. Neuroscience 220, 26–31. doi: 10.1016/j.neuroscience.2012.06.050
Staats, K. A., Van Helleputte, L., Jones, A. R., Bento-Abreu, A., Van Hoecke, A., Shatunov, A., et al. (2013). Genetic ablation of phospholipase C delta 1 increases survival in SOD1(G93A) mice. Neurobiol. Dis. 60, 11–17. doi: 10.1016/j.nbd.2013.08.006
Starkov, A. A. (2010). The molecular identity of the mitochondrial Ca2+ sequestration system. FEBS J. 277, 3652–3663. doi: 10.1111/j.1742-4658.2010.07756.x
Stieber, A., Gonatas, J. O., and Gonatas, N. K. (2000). Aggregation of ubiquitin and a mutant ALS-linked SOD1 protein correlate with disease progression and fragmentation of the Golgi apparatus. J. Neurol. Sci. 173, 53–62. doi: 10.1016/S0022-510X(99)00300-7
Sunyach, C., Michaud, M., Arnoux, T., Bernard-Marissal, N., Aebischer, J., Latyszenok, V., et al. (2012). Olesoxime delays muscle denervation, astrogliosis, microglial activation and motoneuron death in an ALS mouse model. Neuropharmacology 62, 2346–2352. doi: 10.1016/j.neuropharm.2012.02.013
Suzuki, H., Lee, K., and Matsuoka, M. (2011). TDP-43-induced death is associated with altered regulation of BIM and Bcl-xL and attenuated by caspase-mediated TDP-43 cleavage. J. Biol. Chem. 286, 13171–13183. doi: 10.1074/jbc.M110.197483
Suzuki, Y. J., and Ford, G. D. (1992). Superoxide stimulates IP3-induced Ca2+ release from vascular smooth muscle sarcoplasmic reticulum. Am. J. Physiol. 262, H114-H116.
Tan, W., Naniche, N., Bogush, A., Pedrini, S., Trotti, D., and Pasinelli, P. (2013). Small peptides against the mutant SOD1/Bcl-2 toxic mitochondrial complex restore mitochondrial function and cell viability in mutant SOD1-mediated ALS. J. Neurosci. 33, 11588–11598. doi: 10.1523/JNEUROSCI.5385-12.2013
Taylor, C. W., and Tovey, S. C. (2010). IP(3) receptors: toward understanding their activation. Cold Spring Harb. Perspect. Biol. 2, a004010. doi: 10.1101/cshperspect.a004010
Thrower, E. C., Duclohier, H., Lea, E. J., Molle, G., and Dawson, A. P. (1996). The inositol 1,4,5-trisphosphate-gated Ca2+ channel: effect of the protein thiol reagent thimerosal on channel activity. Biochem. J. 318(Pt 1), 61–66.
Tohgi, H., Abe, T., Yamazaki, K., Murata, T., Ishizaki, E., and Isobe, C. (1999). Increase in oxidized NO products and reduction in oxidized glutathione in cerebrospinal fluid from patients with sporadic form of amyotrophic lateral sclerosis. Neurosci. Lett. 260, 204–206. doi: 10.1016/S0304-3940(98)00986-0
Tradewell, M. L., Cooper, L. A., Minotti, S., and Durham, H. D. (2011). Calcium dysregulation, mitochondrial pathology and protein aggregation in a culture model of amyotrophic lateral sclerosis: mechanistic relationship and differential sensitivity to intervention. Neurobiol. Dis. 42, 265–275. doi: 10.1016/j.nbd.2011.01.016
Traynor, B. J., Bruijn, L., Conwit, R., Beal, F., O’Neill, G., Fagan, S. C., et al. (2006). Neuroprotective agents for clinical trials in ALS: a systematic assessment. Neurology 67, 20–27. doi: 10.1212/01.wnl.0000223353.34006.54
Uchibayashi, R., Tsuruma, K., Inokuchi, Y., Shimazawa, M., and Hara, H. (2011). Involvement of Bid and caspase-2 in endoplasmic reticulum stress- and oxidative stress-induced retinal ganglion cell death. J. Neurosci. Res. 89, 1783–1794. doi: 10.1002/jnr.22691
Upton, J. P., Austgen, K., Nishino, M., Coakley, K. M., Hagen, A., Han, D., et al. (2008). Caspase-2 cleavage of BID is a critical apoptotic signal downstream of endoplasmic reticulum stress. Mol. Cell Biol. 28, 3943–3951. doi: 10.1128/MCB.00013-08
Vaccaro, A., Patten, S. A., Aggad, D., Julien, C., Maios, C., Kabashi, E., et al. (2013). Pharmacological reduction of ER stress protects against TDP-43 neuronal toxicity in vivo. Neurobiol. Dis. 55, 64–75. doi: 10.1016/j.nbd.2013.03.015
Vannuvel, K., Renard, P., Raes, M., and Arnould, T. (2013). Functional and morphological impact of ER stress on mitochondria. J. Cell. Physiol. 228, 1802–1818. doi: 10.1002/jcp.24360
Venkatesh, S., Lee, J., Singh, K., Lee, I., and Suzuki, C. K. (2012). Multitasking in the mitochondrion by the ATP-dependent Lon protease. Biochim. Biophys. Acta 1823, 56–66. doi: 10.1016/j.bbamcr.2011.11.003
Verkhratsky, A. (2005). Physiology and pathophysiology of the calcium store in the endoplasmic reticulum of neurons. Physiol. Rev. 85, 201–279. doi: 10.1152/physrev.00004.2004
von Lewinski, F., and Keller, B. U. (2005). Ca2+, mitochnodria and selective motoneruon vulnerability: implications for ALS. Trends Neurosci. 28, 494–500. doi: 10.1016/j.tins.2005.07.001
Walker, A. K. (2010). Protein disulfide isomerase and the endoplasmic reticulum in amyotrophic lateral sclerosis. J. Neurosci. 30, 3865–3867. doi: 10.1523/JNEUROSCI.0408-10.2010
Walker, A. K., and Atkin, J. D. (2011). Mechanisms of neuroprotection by protein disulphide isomerase in amyotrophic lateral sclerosis. Neurol. Res. Int. 2011, 317340. doi: 10.1155/2011/317340
Walker, A. K., Soo, K. Y., Sundaramoorthy, V., Parakh, S., Ma, Y., Farg, M. A., et al. (2013). ALS-associated TDP-43 induces endoplasmic reticulum stress, which drives cytoplasmic TDP-43 accumulation and stress granule formation. PLoS ONE 8:e81170. doi: 10.1371/journal.pone.0081170
Wang, W., Li, L., Lin, W. L., Dickson, D. W., Petrucelli, L., Zhang, T., et al. (2013). The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum. Mol. Genet. 22, 4706–4719. doi: 10.1093/hmg/ddt319
Watanabe, S., Kaneko, K., and Yamanaka, K. (2013). Accelerated disease onset with stabilized familial amyotrophic lateral sclerosis (ALS)-linked mutant TDP-43 proteins. J. Biol. Chem. 288, 3641–3654. doi: 10.1074/jbc.M112.433615
Weishaupt, J. H., Bartels, C., Polking, E., Dietrich, J., Rohde, G., Poeggeler, B., et al. (2006). Reduced oxidative damage in ALS by high-dose enteral melatonin treatment. J. Pineal Res. 41, 313–323. doi: 10.1111/j.1600-079X.2006.00377.x
Wesson, D. E., and Elliott, S. J. (1995). The H2O2-generating enzyme, xanthine oxidase, decreases luminal Ca2+ content of the IP3-sensitive Ca2+ store in vascular endothelial cells. Microcirculation 2, 195–203. doi: 10.3109/10739689509146767
Wiedemann, F. R., Manfredi, G., Mawrin, C., Beal, M. F., and Schon, E. A. (2002). Mitochondrial DNA and respiratory chain function in spinal cords of ALS patients. J. Neurochem. 80, 616–625. doi: 10.1046/j.0022-3042.2001.00731.x
Williams, T. L., Day, N. C., Ince, P. G., Kamboj, R. K., and Shaw, P. J. (1997). Calcium-permeable alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors: a molecular determinant of selective vulnerability in amyotrophic lateral sclerosis. Ann. Neurol. 42, 200–207. doi: 10.1002/ana.410420211
Winton, M. J., Igaz, L. M., Wong, M. M., Kwong, L. K., Trojanowski, J. Q., and Lee, V. M. (2008). Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J. Biol. Chem. 283, 13302–13309. doi: 10.1074/jbc.M800342200
Wong, P. C., Pardo, C. A., Borchelt, D. R., Lee, M. K., Copeland, N. G., Jenkins, N. A., et al. (1995). An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 14, 1105–1116. doi: 10.1016/0896-6273(95)90259-7
Wuytack, F., Raeymaekers, L., and Missiaen, L. (2002). Molecular physiology of the SERCA and SPCA pumps. Cell Calcium 32, 279–305. doi: 10.1016/S0143416002001847
Xu, K. Y., Zweier, J. L., and Becker, L. C. (1997). Hydroxyl radical inhibits sarcoplasmic reticulum Ca(2+)-ATPase function by direct attack on the ATP binding site. Circ. Res. 80, 76–81. doi: 10.1161/01.RES.80.1.76
Xu, L., Eu, J. P., Meissner, G., and Stamler,J. S. (1998). Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science 279, 234–237. doi: 10.1126/science.279.5348.234
Ying, J., Sharov, V., Xu, S., Jiang, B., Gerrity, R., Schoneich, C., et al. (2008). Cysteine-674 oxidation and degradation of sarcoplasmic reticulum Ca(2+) ATPase in diabetic pig aorta. Free Radic. Biol. Med. 45, 756-762. doi: 10.1016/j.freeradbiomed.2008.05.029
Yoshida, H., Matsui, T., Yamamoto, A., Okada, T., and Mori, K. (2001). XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891. doi: 10.1016/S0092-8674(01)00611-0
Zable, A. C., Favero, T. G., and Abramson, J. J. (1997). Glutathione modulates ryanodine receptor from skeletal muscle sarcoplasmic reticulum. Evidence for redox regulation of the Ca2+ release mechanism. J. Biol. Chem. 272, 7069–7077. doi: 10.1074/jbc.272.11.7069
Zhang, L., Yu, J., Pan, H., Hu, P., Hao, Y., Cai, W., et al. (2007). Small molecule regulators of autophagy identified by an image-based high-throughput screen. Proc. Natl. Acad. Sci. U.S.A. 104, 19023–19028. doi: 10.1073/pnas.0709695104
Zhang, Y., Cook, A., Kim, J., Baranov, S. V., Jiang, J., Smith, K., et al. (2013). Melatonin inhibits the caspase-1/cytochrome c/caspase-3 cell death pathway, inhibits MT1 receptor loss and delays disease progression in a mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 55, 26–35. doi: 10.1016/j.nbd.2013.03.008
Keywords: amyotrophic lateral sclerosis, ER stress, protein misfolding, calcium dysregulation, SOD1, TDP-43, mitochondria, oxidative stress
Citation: Tadic V, Prell T, Lautenschlaeger J and Grosskreutz J (2014) The ER mitochondria calcium cycle and ER stress response as therapeutic targets in amyotrophic lateral sclerosis. Front. Cell. Neurosci. 8:147. doi: 10.3389/fncel.2014.00147
Received: 27 February 2014; Accepted: 07 May 2014;
Published online: 30 May 2014.
Edited by:
Manoj Kumar Jaiswal, Center for Neuroscience and Regenerative Medicine, USAReviewed by:
Tibor Kristian, University of Maryland School of Medicine, USAAlexej Verkhratsky, University of Manchester, UK
Pavle R. Andjus, University of Belgrade, Serbia
Anthony Robert White, The University of Melbourne, Australia
Copyright © 2014 Tadic, Prell, Lautenschlaeger and Grosskreutz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vedrana Tadic, Hans Berger Department of Neurology, Jena University Hospital, Erlanger Allee 101, 07747 Jena, Germany e-mail:dmVkcmFuYS50YWRpY0BtZWQudW5pLWplbmEuZGU=