 Marko Uutela1
Marko Uutela1 Jesse Lindholm2
Jesse Lindholm2 Tomi Rantamäki2
Tomi Rantamäki2 Juzoh Umemori2
Juzoh Umemori2 Kerri Hunter1
Kerri Hunter1 Vootele Võikar2
Vootele Võikar2 Maija L. Castrén1,3*
Maija L. Castrén1,3*- 1Institute of Biomedicine/Physiology, University of Helsinki, Helsinki, Finland
- 2Neuroscience Center, University of Helsinki, Helsinki, Finland
- 3Department of Child Neurology, Hospital for Children and Adolescents, University Hospital of Helsinki, Helsinki, Finland
Fluoxetine is used as a therapeutic agent for autism spectrum disorder (ASD), including Fragile X syndrome (FXS). The treatment often associates with disruptive behaviors such as agitation and disinhibited behaviors in FXS. To identify mechanisms that increase the risk to poor treatment outcome, we investigated the behavioral and cellular effects of fluoxetine on adult Fmr1 knockout (KO) mice, a mouse model for FXS. We found that fluoxetine reduced anxiety-like behavior of both wild-type and Fmr1 KO mice seen as shortened latency to enter the center area in the open field test. In Fmr1 KO mice, fluoxetine normalized locomotor hyperactivity but abnormally increased exploratory activity. Reduced brain-derived neurotrophic factor (BDNF) and increased TrkB receptor expression levels in the hippocampus of Fmr1 KO mice associated with inappropriate coping responses under stressful condition and abolished antidepressant activity of fluoxetine. Fluoxetine response in the cell proliferation was also missing in the hippocampus of Fmr1 KO mice when compared with wild-type controls. The postnatal mRNA expression of serotonin transporter (SERT) was reduced in the thalamic nuclei of Fmr1 KO mice during the time of transient innervation of somatosensory neurons suggesting that developmental changes of SERT expression were involved in the differential cellular and behavioral responses to fluoxetine in wild-type and Fmr1 mice. The results indicate that changes of BDNF/TrkB signaling contribute to differential behavioral responses to fluoxetine among individuals with ASD.
Introduction
Fragile X syndrome (FXS) is a common inherited cause of intellectual disability and a well characterized form of autism spectrum disease (ASD). The behavioral phenotype of FXS includes hyperactivity, difficulties with regulation of attention, and many features that are associated with infantile autism, including motor stereotypies, poor eye contact, social avoidance, perseverative and self-injurious behavior, and delayed speech development (Hagerman et al., 2010). It has been estimated that approximately 30% of males with FXS meet the diagnostic criteria for autism (Brown et al., 1986; Hernandez et al., 2009; Hagerman et al., 2010). Perseveration in speech and behavior in FXS resemble obsessive and compulsive behavior. Obsessive thoughts and behavior are sometimes problems for FXS individuals. FXS is caused by a loss of functional FMR1 protein (FMRP), an RNA-binding protein that interacts with many pre- and postsynaptic transcripts and regulates their translation (Darnell et al., 2011). The absence of FMRP leads to aberrances in local synaptic connections, membrane excitability, and circuit activity (Bassel and Warren, 2008; Gibson et al., 2008). Alterations of neural progenitor cell proliferation and differentiation both in developing and adult brain contribute to the pathophysiology of FXS (Castrén et al., 2005; Luo et al., 2010). Several studies indicate that brain-derived neurotrophic factor (BDNF) and its tropomyosin-related kinase B (TrkB) receptors are involved in the plasticity changes in FXS (Uutela et al., 2012; Castrén and Castrén, 2014) as well as in autism (Perry et al., 2001; Miyazaki et al., 2004; Connolly et al., 2006; Correia et al., 2011; Garcia et al., 2012).
Selective serotonin reuptake inhibitors (SSRIs) such as fluoxetine are often prescribed medications for ASD (Aman et al., 2005; Oswald and Sonenklar, 2007). Although some studies suggest that fluoxetine may be beneficial for core features of ASD in adults (Williams et al., 2013) and in individual cases and subgroups of children with autism (DeLong et al., 1998, 2002; Hollander et al., 2012), a recent meta-analysis indicates that there is not enough evidence to support the use of SSRIs in autism (Williams et al., 2013). In addition, the possible side-effects of the drug treatment are a main concern in clinics. Treatment with fluoxetine has been shown to be of benefit to some FXS individuals with autism, social anxiety, or selective mutism (Hagerman et al., 1994). However, fluoxetine may not be suitable to all individuals with FXS and it can cause mood changes, restlessness, and aggression (Hagerman et al., 2009).
Fluoxetine acts primarily as an inhibitor of serotonin transporter (SERT) and blocks serotonin uptake from the synaptic cleft into presynaptic vesicles in the central nervous system (Wong et al., 1974). Fluoxetine also inhibits a number of ion channels and may suppress excitotoxicity (Kim et al., 2013b). The mechanisms of fluoxetine action involve multiple molecular pathways, including the activation of serotonergic receptors (Banasr et al., 2004), the cAMP-CREB signaling pathway (Warner-Schmidt and Duman, 2006), and signaling pathways associated with BDNF and TrkB (Duman and Monteggia, 2006; Warner-Schmidt and Duman, 2006). The clinical antidepressant effects of fluoxetine have been shown to be mediated via changes in neurogenesis and neuronal elimination (Sairanen et al., 2005; Duman and Monteggia, 2006). In the present study, we investigated behavioral and cellular effects of long-term fluoxetine treatment on adult Fmr1 knockout (KO) mice, a mouse model for FXS, and examined the contribution of BDNF and TrkB to fluoxetine responses in FXS.
Materials and Methods
Animals
Fmr1 KO mice (B6.129P2-Fmr1tm1/Cgr/J) purchased from Jackson Laboratory (Bar Harbor, ME, USA) and maintained on the C57BL/6JOlaHsd substrain in the Animal Centre of University of Helsinki were used for the behavioral studies. Male mice at the age of 3–4 months were used. Each experimental group contained four mice. Group-housed mice were maintained under 12-h light–dark cycle (lights on from 06.00 to 18.00 h) with food and water available ad libitum. The behavioral experiments were carried out during light phase (between 09.00 and 16.00 h). Fmr1-KO mice and their WT littermates used at postnatal day 7–8 (P7-8) were on inbred FVB background (Bakker et al., 1994). Animal experiments were performed in accordance with the guidelines of the National Institutes of Health Guide for the Care and Use of Laboratory Animals and European Economic Community Council Directive. All animal procedures were approved by the Experimental Animal Ethics Committee of Finland.
Fluoxetine Administration and Cell Birth Studies
Fluoxetine was administered via drinking water (0.10 mg/ml, about 10 mg/kg/day, Orion Pharma, Finland) and control mice received water without fluoxetine. Mice received an intraperitoneal injection of bromodeoxyuridine (BrdU, Sigma-Aldrich) at a dose of 75 mg/kg four times every 2 h (300 mg/kg total) starting 24 h before sacrifice for studies investigating the proliferation and short term survival of newborn cells in the hippocampus. Hippocampi were dissected after cervical dislocation in CO2 anesthesia. The BrdU labeling was detected as described previously (Wu and Castrén, 2009). Briefly, deoxyribonucleic acid was extracted from the hippocampi, denatured, and dot-blotted onto membrane. The BrdU incorporation was detected by immunostaining with mouse BrdU-specific monoclonal primary antibody (Roche, 1-299-964).
Behavioral Testing
Behavioral testing was performed between 9:00 AM and 4:00 PM by experimenters who were blinded to the genotypes at the time of testing.
Open field test
The mice were released in the corner of novel open field arena (30 × 30 cm, Med Associates, St. Albans, VT, USA) surrounded by frames with infra-red light barriers for detection of animal’s position. Horizontal and vertical activity was recorded for 30 min (light intensity ~150 lx). Peripheral zone was defined as a 6 cm-wide corridor along the wall.
Forced swim test
Mice were placed in a clear, 21°C water-filled cylinder (diameter, 20 cm; depth, 13 cm) for 6 min and the immobility time of the mice was measured between 2 and 6 min.
BDNF ELISA
For the BDNF expression studies, hippocampi were collected from mice sacrificed by cervical dislocation followed by anesthesia with CO2. Samples were frozen on dry ice, and stored at –70°C until use. The BDNF expression was determined using BDNF ELISA (Quantikine human BDNF kit, R&D Systems) as described previously (Louhivuori et al., 2011).
Western Analysis
The samples were homogenized and processed in a lysis buffer for Western analysis as previously described (Castrén et al., 2002). The protein concentration of the supernatant samples was determined using Biorad DC protein assay. The protein extracts (60 μg) were electrophoresed on 7.5% sodium dodecyl sulfate polyacrylamide minigels and transferred to 0.2 mm nitrocellulose membranes (Schleicher & Schuell) for 1 h at 400 mA. The membranes were washed 10 min in TBS, pH 7.4 (0.1 M Tris, 0.15 M NaCl) and blocked in 5% non-fat dry milk, in TBS with 0.1% Tween 20 (TBST) for 1.5 h. The incubation with rabbit anti-TrkB (1:1000, sc-11, Santa Cruz Biotechnology) at +4°C overnight was followed by washes in TBST and incubation with horseradish-peroxidase-conjugated secondary antibody (1:10000, Bio-Rad Laboratories) for 1.5 h at room temperature. Detection was performed using the enhanced chemiluminescence kit (ECL++ kit, Amersham Biosciences) and Fuji LAS-3000 camera (Tamro Medlabs, Vantaa, Finland). Data were analyzed using NIH Image J software.
In Situ Hybridization
Mouse brains at P7-8 were fixed in 4% paraformaldehyde (PFA) in phosphate buffered saline (PBS) overnight and processed for frozen sectioning. Brains were washed twice in PBS after fixation and then soaked in cryoprotective solution (30% sucrose in PBS). Brains were mounted in Tissue-Tek®(Sakura Finetek, Zoeterwoude, Netherlands), frozen on dry ice, and stored at –80°C until cut. Brains were cut in 12 μm thick sections and collected onto Superfrost® Plus microscope slides (Menzel GmbH & Co. KG, Braunschweig, Germany) with MICROM HM 550 cryostat (MICROM International GmbH, Walldorf, Germany) and the slides were stored at –80°C until use.
In situ hybridization with the oligonucleotide probes was performed as described by Wisden and Morris (1994). Oligonucleotides complementary to mouse SERT (5′-ATG AGG TAG TAG AGC GCC CAG GCT ATG ATG GTG TT-3′) were 3′ labeled with [α33P]-dATP (3000/mmol; Amerham Biosciences) to a specific activity of 6–7 × 10-7cpm/pmol using terminal deoxynucleotidyl transferase (Finnzymes). Hybridization was performed overnight (42°C) on postfixed sections in the presence of 1 × 106 cpm/ml labeled probe in buffer containing 50% formamide, 4× standard saline citrate (SSC; 1×SSC: 150 mM NaCl, 15 mM sodium citrate), 10% dextran sulfate and 10 mM dithiothreitol. After overnight hybridization at 42°C, the sections were dipped into 1×SSC and then sequentially for 3 min each at room temperature in 1×SSC, 0.1×SSC, 70% ethanol, and 94% ethanol. Microscope slides were exposed to film (Fuji medical X-ray film super RX) for three weeks. 14C standard scale was included to every film. Hybridization signal intensities were quantified from films scanned with Fujifilm FLA-5100 scanning device.
Neural Progenitor Cultures
Neural progenitors were propagated from the wall of lateral ventricles of wild-type and Fmr1 KO pups as previously described (Castrén et al., 2005). Cells were grown as free-floating aggregates referred to as neurospheres in Dulbecco’s modified Eagle’s medium F-12 nutrient mixture (DMEM/F-12) media containing B27 supplement (both from Gibco, Life Technologies Ltd.), L-glutamine (2 mM), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES, 15 mM), penicillin (100 U/ml), and streptomycin (100 U/ml) (all from Sigma-Aldrich), in the presence of basic fibroblastic growth factor (10 ng/ml) and epidermal growth factor (20 ng/ml) (both from PeproTech) in a 5% CO2-humidified incubator at +37oC. The culture medium was refreshed and growth factors were added three times per week. The cells were passaged by manual trituration at approximately two weeks intervals. Neuronal progenitor cells from WT and Fmr1 KO mice were plated at a concentration of 100000 cells/10 ml plate and grown as neurospheres for 5 days. Medium was changed and growth factors last added 5 h prior to the start of treatments. Cells were treated with 1 μM fluoxetine in parallel with corresponding non-treated controls for 48 h. The cells were then collected as cell pellets and stored at –70°C until further use.
RNA Extraction and Real-Time Quantitative PCR
Total RNA was extracted from frozen cells by using QIAzol (Qiagen, Valencia, CA, USA) and treated with DNaseI (Thermo Fisher Scientific Inc., Rockford, IL, USA) according to the manufacturer’s instruction. We used 2–4 μg of total RNA to synthesize cDNA using the Maxima First Strand cDNA Synthesis Kit (Thermo Fisher Scientific Inc., Rockford, IL, USA). Real-time quantitative PCR was performed using the Maxima SYBR Green qPCR Master Mix (Thermo Fisher Scientific Inc., Rockford, IL, USA) and the CFX96 TouchTM detection system (Bio-Rad, Hercules, CA, USA). The primers described previously (Karpova et al., 2009) were used to amplify specific cDNA regions of transcripts: the coding region in the exon IX of the Bdnf gene for the total Bdnf mRNA (5′-GAAGGCTGCAGGGGCATAGACAAA-3′ and 5′-TACACAGGAAGTGTCTATCCTTATG-3′); the exon IV (5′-ACCGAAGTATGAAATAACCATAGTAAG-3′) and (5′-TGTTTACTTTGACAAGTAGTGACTGAA-3′), Gapdh (5′-GGTGAAGGTCGGTGTGAACGG-3′ and 5′-ATGTAGTTGAGGTCAATGAAGGG-3′) as a housekeeping control gene. Ct and quantitative values were calculated from each sample using CFX ManagerTM software (Bio-Rad, Hercules, CA, USA) and the quantitative values were normalized to the control Gapdh levels.
Data Analysis
Data obtained from behavioral tests were analyzed with Statview software (SAS, Cary, NC, USA), unless specified otherwise, a two-way repeated-measures analysis of variance (ANOVA) followed by Fishers’s protected least significance post hoc test.
Immunoblot bands were quantified using NIH ImageJ software. All the data are presented as means ±SEM. Statistical analyses were performed using GraphPad Prism 4.0 for Windows (GraphPad Software, San Diego, CA, USA). Quantitative analysis of signals on X-ray films was performed with AIDA Image Analyzer (version 3.44.035, Raytest Isotopenmessgeräte GmbH, Straubenhardt, Germany) software. Brightness and contrast were optimized before measuring.
For comparison between two groups, Student’s t-test was used. Two-way ANOVA was used to reveal main effect and interaction between the factors followed by Bonferroni post hoc test. The criterion for significance was set to P < 0.05.
Results
Behavioral Responses of Fmr1 KO Mice to Fluoxetine in the Open Field Test
We observed a significant Fmr1 KO genotype and fluoxetine treatment interaction (two-way ANOVA: F(1,33) = 2,294; P < 0.05) in the locomotor activity in the open field test. Fmr1 KO mice were hyperactive when compared with non-treated mice (P < 0.05) and treatment with fluoxetine reduced the motor activity of Fmr1 KO mice to wild-type levels (P = 0.295; Figure 1A). There was a main effect of fluoxetine treatment (two-way ANOVA: F(1,12) = 6.948; P < 0.05) but no effects of the mouse genotype (two-way ANOVA: F(1,12) = 0.695; P > 0.05) on the latency to enter the center arena of the open field. Fluoxetine reduced significantly the latency in both wild-type (P < 0.05) and Fmr1 KO mice (P < 0.05; Figure 1B). In addition, a significant genotype × treatment effect in the exploratory activity and unconditioned anxiety-related behavior (two-way ANOVA: F(1,12) = 5.863; 0.05) was found. Fluoxetine increased the time that Fmr1 KO mice spent in the central square (P < 0.05; Figure 1C) and appropriately decreased the time that the transgenic mice stayed along the perimeter (P < 0.05; data not shown) without having any effects on this behavior in wild-type mice. As shown in Figure 1D, particularly the resting time in center was increased (P < 0.05) in Fmr1 KO mice by fluoxetine treatment. No genotype or fluoxetine effects were found on the total resting time (two-way ANOVA: F(1,12) = 0.875; P > 0.05 and F(1,12) = 2.353; P > 0.05, respectively).
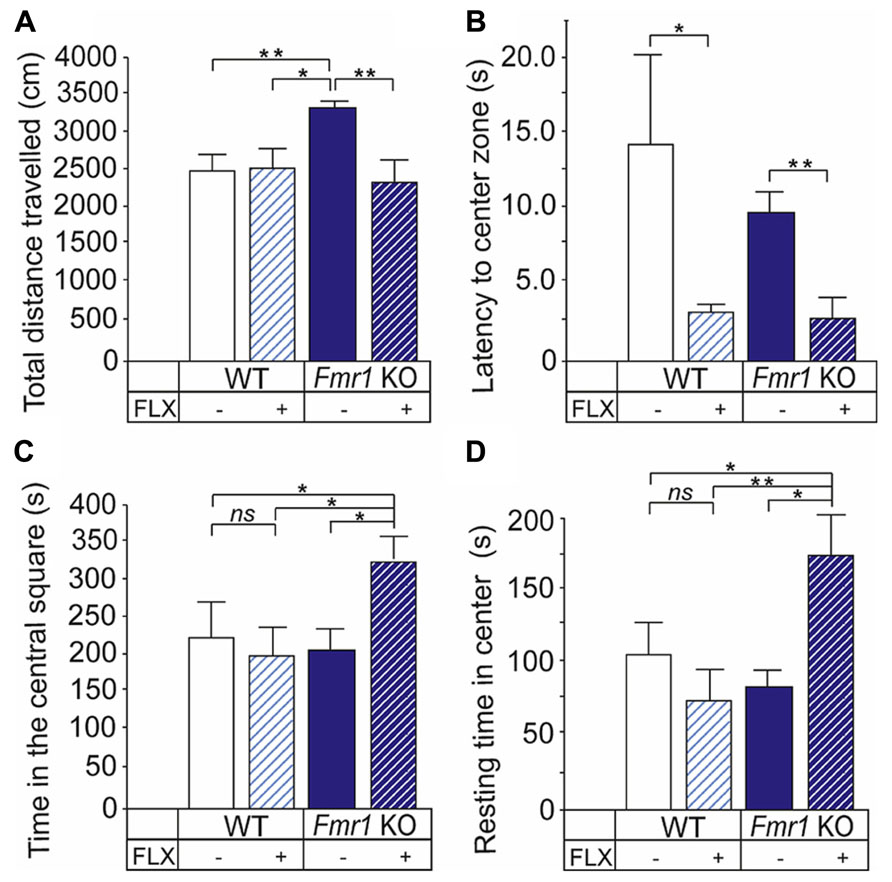
FIGURE 1. Altered behavioral responses to fluoxetine of Fmr1 KO mice in the open field test. (A) Fmr1 KO mice (Fmr1 KO) show locomotor hyperactivity seen as longer distance traveled by the mice when compared with wild-type controls (WT). The locomotor activity of Fmr1 KO mice was decreased to the level of WT mice Fluoxetine reduced the latency to enter the center area of the open field in both WT and Fmr1 KO mice. (C) Fluoxetine increased the time that Fmr1 KO mice spent in the central square but did not have any effects on this behavior in WT mice. (D) The resting time in center was increased by fluoxetine in Fmr1 KO mice. Error bars indicate means ± SEM. *P < 0.05, **P < 0.01.
Behavioral Responses of Fmr1 KO Mice to Fluoxetine in the Forced Swim Test
We investigated the antidepressant effects of fluoxetine on the phenotype of Fmr1 KO mice by submitting the mice to the forced swim test, which estimates behavioral despair under stressful and inescapable conditions, and it is widely used screening test of antidepressant drugs to assess their antidepressant activity (Porsolt et al., 1977; Cryan et al., 2002; Prut and Belzung, 2003). In this test, wild-type mice respond to antidepressants by reducing their immobility time (Porsolt et al., 1977). We found a significant Fmr1 KO genotype and long-term fluoxetine treatment interaction (two-way ANOVA: F(1,12) = 11,211; P < 0.01). The swimming immobility of Fmr1 KO mice was decreased (P < 0.001) when compared with WT littermates without any treatment (Figure 2A). Fluoxetine administration reduced (P < 0.01) immobility time of wild-type mice but had no effect on this immobility score of Fmr1 KO mice (Figure 2A).
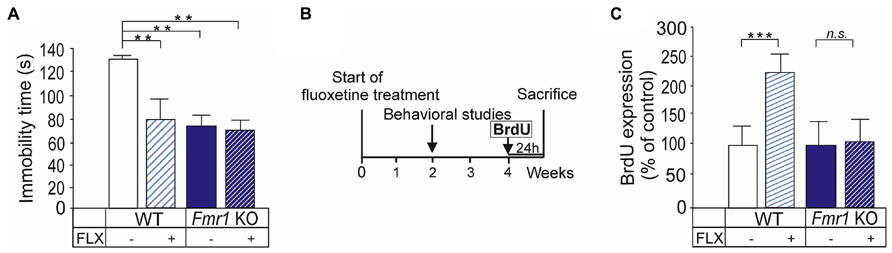
FIGURE 2. Altered fluoxetine responses on mobility in the forced swim test and proliferation of hippocampal cells. (A) Fmr1 KO mice showed abnormal coping responses under stressful condition in the forced swim test and the immobility time of Fmr1 KO mice was reduced when compared with wild-type controls. Fluoxetine reduced immobility of wild-type mice but not that of Fmr1 KO mice. (B) The effects of long-term fluoxetine treatment were examined on hippocampal cell proliferation analyzed by incorporation of BrdU in newborn cells. (C) Fluoxetine increased significantly hippocampal cell proliferation in wild type but not in Fmr1 KO mice after fluoxetine treatment. Error bars indicate means ± SEM. **P < 0.01, ***P < 0.001.
Effects of Fluoxetine on Cell Proliferation in the Hippocampus of Fmr1 KO Mice
The stimulatory effect of fluoxetine on progenitor cell proliferation is implicated to its therapeutic effects. We assessed the short-term effect of fluoxetine on cell proliferation by the BrdU staining in the hippocampus 24 h after intraperitoneal BrdU injections (Figure 2B). As shown in Figure 2C, treatment with fluoxetine increased the BrdU staining 2.2-fold in wild-type mice but did not have any effects on the BrdU expression in the hippocampus of Fmr1 KO mice (ANOVA: F(3,14) = 5,117; P < 0.05). The data indicate that the normal response to fluoxetine on proliferation rate was missing in the absence of FMRP.
Responses to Fluoxetine in the Expression of BDNF and TrkB in the Absence of FMRP
BDNF/TrkB signaling is implicated in the fluoxetine effects and chronic, but not acute, fluoxetine treatment increase BDNF in the rodent brain (Nibuya et al., 1995; Duman and Monteggia, 2006). The expression of BDNF was reduced in the hippocampus of Fmr1 KO mice when compared with wild-type controls (Figure 3A) as shown previously in older Fmr1 KO mice (Uutela et al., 2012). Treatment with fluoxetine did not have any significant effects on the BDNF protein expression in the hippocampus of wild-type or Fmr1 KO mice in our experimental setting (Figure 3A). The expression of TrkB receptors was increased in the hippocampus of the Fmr1 KO mice when compared with wild-type controls (P < 0.05), suggesting a role for TrkB in altered fluoxetine responses in FXS (Figure 3B). There was a tendency toward increased TrkB protein in the wild-type hippocampus after fluoxetine treatment and the expression of TrkB protein remained higher in the hippocampus of Fmr1 KO than in wild-type controls after treatment but the effects of fluoxetine on the TrkB protein expression did not reach the level of significance (Figure 3B).
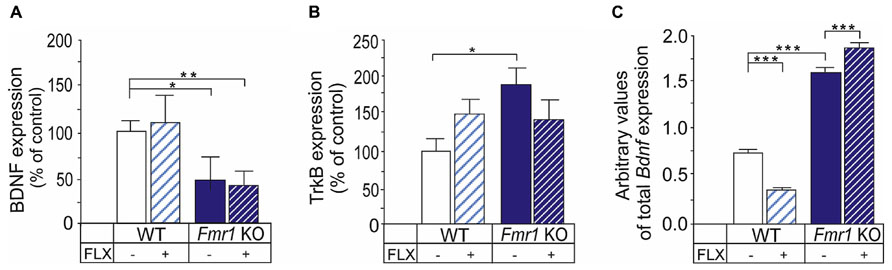
FIGURE 3. Effects of fluoxetine on the BDNF and TrkB protein expression in the hippocampus of Fmr1 KO mice. (A) The BDNF protein expression was reduced in the hippocampus of Fmr1 KO mice (Fmr1 KO) when compared to that of wild-type mice (WT). The expression levels were not changed significantly after treatment with fluoxetine under our experimental conditions. (B) The TrkB receptor protein was significantly increased in the hippocampus of Fmr1 KO mice when compared to WT controls. Fluoxetine did not have any significant effects on the TrkB expression in WT or Fmr1 KO mice. (C) The expression of Bdnf mRNA was increased in undifferentiated neural progenitors derived from Fmr1 KO mice when compared with WT controls and the responses to fluoxetine treatment were different in WT and transgenic progenitors. Error bars indicate means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
Our previous studies have shown that the dendritic targeting and expression of Bdnf mRNA are increased in cortical and hippocampal neurons of Fmr1 KO (Louhivuori et al., 2011). We examined responses to fluoxetine on Bdnf mRNA levels in undifferentiated cortical progenitors derived from Fmr1 KO mice. We found that the basal expression level of the total Bdnf mRNA in progenitors lacking FMRP was significantly higher than that in wild-type progenitors (genotype, F(1,20) = 1148.5; P < 2.0e–16; Figure 3C). A two-way ANOVA showed that there was an interaction between genotype and drug treatment (genotype × treatment interaction, F(1,20) = 100.0, P = 3.16e–09), and fluoxetine treatment reduced total Bdnf mRNA in wild-type progenitor cultures whereas the expression was increased by fluoxetine in cultures derived from Fmr1 KO mice. The expression of exon IV transcripts correlated with that of total Bdnf mRNA (data not shown), but its large variation due to a low expression level suggested that promoter IV-driven Bdnf transcription was not utilized significantly in proliferating undifferentiated neural progenitors.
SERT Expression in Postnatal Brain of Fmr1 KO Mouse
The early development of serotonergic system has important functions in cortical maturation and plasticity (Vitalis and Parnavelas, 2003). Changes in the SERT expression during early brain development induce long-lasting behavioral alterations that associate with changes of responses to fluoxetine and expression of BDNF and TrkB (Karpova et al., 2009; Kiryanova et al., 2013). Transient SERT expression mediates innervation and the uptake of serotonin by axons and terminals of thalamic sensory neurons at P1–P10 before the total maturation of serotonergic system (Lebrand et al., 1996). We examined the SERT mRNA expression in the sensory relay nuclei of the thalamus of Fmr1 KO mice at P7-8. The SERT expression was slightly but significantly reduced (90% of control, P < 0.003) in the medial geniculate nucleus (MGN) of the auditory relay in Fmr1 KO mice when compared with wild-type controls (Figures 4A, B). Signal intensities in the dorsal lateral geniculate nucleus (dLGN) of the visual in Fmr1-KO mouse relay showed a tendency to decreased levels and the ratio of the SERT mRNA expression in dLGN to that in the ventrobasal nucleus (VB) of the somatosensory relay (90% of control, P < 0.048) was significantly reduced when compared with wild-type controls (Figures 4C, D) suggesting dysregulation of serotonin-dependent developmental processes in FXS.
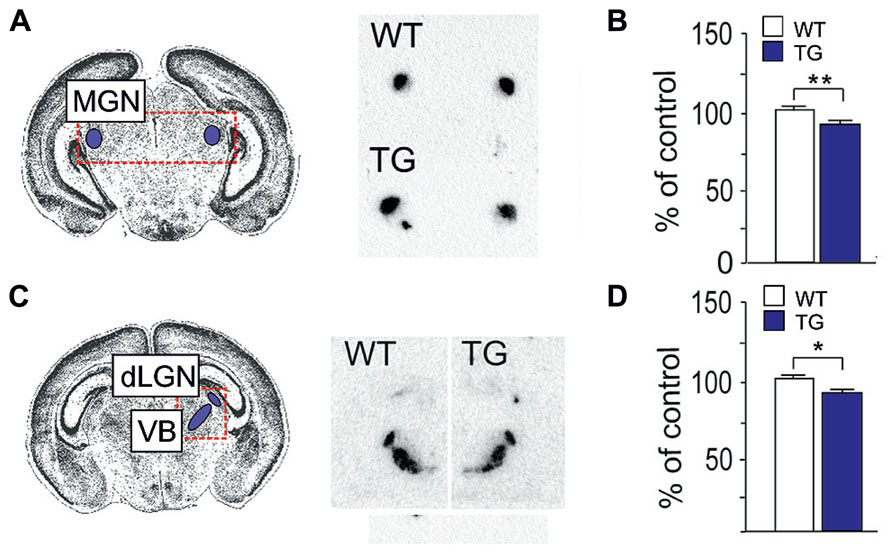
FIGURE 4. Expression of SERT mRNA in the thalamic nuclei of Fmr1 KO mice at P7-8. (A) The location of the medial geniculate nucleus (MGN) in the Nissl-stained brain section and radioactive in situ hybridization with SERT showing the nucleus in the brain sections of a wild-type (WT) and Fmr1 KO transgenic (TG) mouse. (B) A bar graph representing the decrease in the signal intensity of the SERT expression in the MGN of TG mice when compared with that of WT mice. (C) The location of the dorsal lateral geniculate nucleus (dLGN) and ventrobasal nucleus (VB) in the Nissl-stained brain section and the in situ SERT hybridization signal in the nuclei in the brain sections of a WT and TG mouse. (D) A bar graph representing the ratio of the signal intensities of the SERT expression in the dLGN to that in the VB of WT and TG mice. n(WT) = 7, n(TG) = 7. Error bars indicate means ± SEM. *P < 0.05, **P < 0.01.
Discussion
Fmr1 KO Mice as a Model for Autism for Fluoxetine Studies
ASD consists of a range of complex neurodevelopmental disorders, characterized by aberrant reciprocal social interactions, impaired communication, and stereotyped repetitive behaviors with narrow restricted interests. ASD varies in character and severity. The clinical phenotypes reflect heterogeneity of genetic/epigenetic/environmental factors which may contribute to alterations in developmental processes and neuronal plasticity that associate with defects in synapse and neuronal network function in autism (Hughes, 2009). A genetic association among autism and the TrkB gene (Correia et al., 2011), abnormal blood BDNF levels in children with autism (Nelson et al., 2001; Miyazaki et al., 2004; Connolly et al., 2006; Iughetti et al., 2011), and increased BDNF protein expression in postmortem brain tissue of autistic individuals (Perry et al., 2001; Garcia et al., 2012) suggests that BDNF/TrkB signaling plays a role in the pathophysiology of autism. FXS is the cause of autism in 2–6% of all children diagnosed with autism and the syndrome is the best characterized form of ASD (Dölen and Bear, 2009). FXS is caused by a loss of functional FMRP and Fmr1 KO mice recapitulate the main features of the human FXS (Hagerman et al., 1994). Studies of Fmr1 KO mice have revealed that BDNF/TrkB signaling is involved in the alterations of neurogenesis and synapse function in FXS (Louhivuori et al., 2011; Uutela et al., 2012). Here, we show aberrant behavioral and cellular responses to fluoxetine in Fmr1 KO mice. We show that the aberrant responses associate with alterations in the expression of BDNF and TrkB receptors. Furthermore, a reduced transient SERT mRNA expression in the thalamic nuclei of Fmr1 KO mice suggests developmental changes in the maturation of the serotonin system that can have long-lasting effects on the behavior.
Alterations of Behavioral Effects of Fluoxetine in FXS Mice
We observed that fluoxetine reduced the latency of Fmr1 KO and wild-type mice to enter the center area in the open field test indicating reduced anxiety in both mouse groups. Fluoxetine normalized the locomotor hyperactivity characteristics of Fmr1 KO mice (Bakker et al., 1994; Peier et al., 2000; Spencer et al., 2005; Mineur et al., 2006) and increased the exploratory activity of these mice, seen as longer time that the mice stayed in the center of the open field when compared to that of fluoxetine-treated wild-type mice. Fluoxetine did not display this type of anxiolytic effect in wild-type mice. The behavioral response to fluoxetine in Fmr1 KO mice may correlate with disinhibited behaviors and agitation which are known side-effects of fluoxetine treatment in FXS individuals. In the forced swim test, Fmr1 KO mice showed reduced behavioral despair under the stressful condition when compared with wild-type mice. Fluoxetine had no effects on the immobility time of Fmr1 KO mice suggesting that the normal antidepressant effect of fluoxetine was missing in the absence of FMRP.
The Absence of FMRP Affects cellular Responses to Fluoxetine
We found that the aberrant behavioral responses to fluoxetine in Fmr1 KO mice correlated with alterations of cellular responses. Fluoxetine did not increase the proliferation of hippocampal cells in Fmr1 KO mice like is normally seen in wild-type mice. The responses in cell proliferation and neurogenesis are implicated particularly in the antidepressant effects of fluoxetine. The forced swim test is used to assess the antidepressant activity of drugs, and defects in cell proliferation responses in Fmr1 KO mice are consistent with the unresponsiveness to fluoxetine in the forced swim test. BDNF/TrkB signaling plays an essential role for the antidepressant effects of fluoxetine (Saarelainen et al., 2003; Monteggia et al., 2007; Ibarguen-Vargas et al., 2009). Behavioral effects of fluoxetine are blunted in animals with reduced BDNF expression in the central nervous system (Saarelainen et al., 2003; Ibarguen-Vargas et al., 2009). The expression of BDNF shows age-dependent changes in murine brain and temporal alterations of BDNF expression have been found in Fmr1 KO mouse brain. The reduced expression of hippocampal BDNF protein in the Fmr1 KO male mice at the age of 3–4 months in the present study is in agreement with an enhanced age-dependent decay of BDNF expression in the absence of FMRP (Uutela et al., 2012) and unresponsiveness to fluoxetine in the forced swim test. However, previous studies have revealed that the expression of BDNF protein is increased in the hippocampus of young Fmr1 KO mice (Louhivuori et al., 2011; Uutela et al., 2012). We found previously an increased expression and dendritic targeting of Bdnf mRNAs in neurons of Fmr1 KO mice (Louhivuori et al., 2011). Here, we showed that the Bdnf mRNA expression is increased in FMRP-deficient neural progenitors which express normal levels of BDNF protein (Louhivuori et al., 2011) and that the fluoxetine responses are also affected on mRNA levels in these cells.
The dynamic alterations of BDNF expression levels in Fmr1 KO mice contribute to a behavioral phenotype that differs from the phenotype of Bdnf +/- mice with reduced BDNF expression (Uutela et al., 2012). Bdnf +/- mice are indistinguishable from wild-type mice in behavioral tests investigating anxiety, fear-associated learning, behavioral despair, and spatial learning (Kernie et al., 2000; MacQueen et al., 2001). In early adulthood, Bdnf +/- mice show aggressiveness that has been linked with dysfunction of serotonergic neurons (Lyons et al., 1999). Defects in associative learning and reduced startle responses at higher intensities are consistent findings in Fmr1 KO mice but not seen in Bdnf +/- mice (Uutela et al., 2012), whereas locomotor hyperactivity is characteristics of Fmr1 KO mice that may be seen in Bdnf +/- mice when stressed (Kernie et al., 2000; Rios et al., 2001). Reduced non-social but increased social anxiety have been reported in Fmr1 KO mice (Bakker et al., 1994; Peier et al., 2000; Mineur et al., 2002; Spencer et al., 2005; Mineur et al., 2006) but the anxiety phenotype of Fmr1 KO mice has not been consistent in all studies (Van Dam et al., 2000; Mineur et al., 2002; Nielsen et al., 2002; Zhao et al., 2005; Bernardet and Crusio, 2006).
Reduced behavioral despair in adult Fmr1 KO mice in the forced swim test was associated with increased hippocampal TrkB receptors. Similarly, mice with overexpression of TrkB in neurons show reduced behavioral despair (Koponen et al., 2005). BDNF and TrkB are implicated in learning and memory processes, including acquisition of fear learning within amygdala (Rattiner et al., 2004). Overexpression of TrkB in transgenic mice reduces anxiety and the increased TrkB expression in Fmr1 KO mice was likely linked with the reduced anxiety. Neuronal release of BDNF can alter anxiety-like behaviors in mice (Berton et al., 2006; Chen et al., 2006) and age-dependent changes in the expression of BDNF observed in the brain of Fmr1 KO mice (Uutela et al., 2012) could at least partially explain the alterations seen in the anxiety phenotype in different studies. Fluoxetine displayed an abnormal anxiolytic effect that did not associate with any significant changes in the TrkB expression in the hippocampus of Fmr1 KO mice in our experimental setting and further studies are needed to explore the regulation and functional responses of TrkB receptors after treatment with fluoxetine in different experimental conditions in FXS mice.
There is evidence that BDNF signaling is critical for the normal development and function of central serotonergic neurons (Lyons et al., 1999). Reduced levels of endogenous BDNF cause alterations of serotonergic receptor expression but brain serotonin levels and fiber density are normal in Bdnf +/- mice at early age. We found that the postnatal SERT mRNA expression was reduced in the thalamic nuclei of Fmr1 KO mice during the time of transient innervation of somatosensory neurons indicating developmental changes in the serotonergic system that contribute to alterations of BDNF/TrkB signaling and behavioral responses in adult Fmr1 KO mice. Previously, defects of AMPA receptor GluR1 subtype surface insertion have been shown after inhibition of 5-HT2A receptor also indicating defects in serotonergic system in FXS (Xu et al., 2012). Changes in the SERT mRNA expression in the sensory thalamic nuclei during postnatal period are consistent with alterations of developmental plasticity in both visual and auditory systems of Fmr1 KO mice (Dolen et al., 2007; Kim et al., 2013a). Temporal and spatial changes of serotonin expression during early development may cause long-lasting behavioral alterations in FXS. Indeed, targeting SERT expression by fluoxetine during postnatal development results in reduced behavioral despair in adult mice as seen in Fmr1 KO mice (Karpova et al., 2009).
Fluoxetine Treatment in ASD
Fluoxetine is often used to treat individuals with ASD (Aman et al., 2005; Oswald and Sonenklar, 2007) and its effects have been evaluated in several clinical studies. A recently published meta-analysis does not support the use of SSRIs in autism (Williams et al., 2013). However, positive effects of fluoxetine on core autistic symptoms have been shown in individual cases and subgroups of autistic children and in adults with ASD (DeLong et al., 1998, 2002; Makkonen et al., 2011; Hollander et al., 2012). Anxiety and obsessive–compulsive symptoms which associate with autism can be ameliorated by fluoxetine in adult ASD (Buchsbaum et al., 2001; Hollander et al., 2012). In children, beneficial effects have been particularly shown in language impairment. Relatively few side-effects were observed over a 12-week fluoxetine treatment period (Hollander et al., 2012) but long-time consequences of fluoxetine treatment on human brain maturation are not known.
Improved understanding of distinct molecular mechanisms linked to SSRI action in ASD could facilitate optimal pharmacological intervention of individuals with ASD. Dysregulated serotonergic signaling in autism is supported by platelet hyperserotonemia in some of ASD individuals (Piven et al., 1991). Furthermore, linkage studies have identified ASD candidate genes in serotonergic pathways, including the gene that encodes SERT (SLC6A4) (Devlin et al., 2005; Brune et al., 2006). Chronic treatment with fluoxetine enhances serotonergic transmission that may activate mechanisms involved in regulation of intracortical inhibitory–excitatory balance and reduced γ -aminobutyric acid (GABA) signaling is reported by fluoxetine treatment (Maya-Vetencourt et al., 2008).
In the present study, we examined effects of long-term fluoxetine treatment in FXS that represents a monogenic cause of ASD. We observed aberrances of behavioral fluoxetine responses which correlated with alterations of BDNF and TrkB expression. Alterations of both excitatory and inhibitory neurotransmission are implicated in FXS and the outcome of fluoxetine treatment on function of neuronal circuits in FXS is difficult to predict. Enhanced explorative activity of Fmr1 KO mice after fluoxetine treatment is in agreement with activation seen as restlessness, mood changes, and disinhibited behaviors in about 20% of individuals with FXS (Hagerman et al., 1994). The present study suggests that molecular mechanisms underlying ASD may associate with developmental changes that influence fluoxetine responses. Further studies are needed to investigate genetic and epigenetic factors which modulate responses to fluoxetine in ASD more in detail.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Outi Nikkilä for help in mouse genotyping and Dr. Eero Castrén for providing facilities for animal studies. Behavioral studies were performed in Mouse Behavioral Unit that is supported by Biocenter Finland. This study received financial support from the Academy of Finland, Arvo and Lea Ylppö Foundation, and Finnish Brain Research Foundation.
References
Aman, M. G., Lam, K. S., and Van Bourgondien, M. E. (2005). Medication patterns in patients with autism: temporal, regional, and demographic influences. J. Child Adolesc. Psychopharmacol. 15, 116–126. doi: 10.1089/cap.2005.15.116
Bakker, C. E., Verheij, C., Willemsen, R., Vanderhelm, R., Oerlemans, F., Vermey, M., et al. (1994). Fmr1 knockout mice: a model to study fragile X mental retardation. Cell 78, 23–33. doi: 10.1016/0092-8674(94)90569-X
Banasr, M., Hery, M., Printemps, R., and Daszuta, A. (2004). Serotonin-induced increases in adult cell proliferation and neurogenesis are mediated through different and common 5-HT receptor subtypes in the dentate gyrus and the subventricular zone. Neuropsychopharmacology 29, 450–460. doi: 10.1038/sj.npp.1300320
Bassel, G. J., and Warren, S. T. (2008). Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron 60, 201–214. doi: 10.1016/j.neuron.2008.10.004
Bernardet, M., and Crusio, W. E. (2006). Fmr1 KO mice as a possible model of autistic features. Sci. World J. 6, 1164–1176.
Berton, O., Mcclung, C. A., Dileone, R. J., Krishnan, V., Renthal, W., Russo, S. J., et al. (2006). Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science 311, 864–868. doi: 10.1126/science.1120972
Brown, W. T., Jenkins, E. C., Cohen, I. L., Fisch, G. S., Wolf-Schein, E. G., Gross, A., et al. (1986). Fragile X and autism: a multicenter survey. Am. J. Med. Genet. 23, 341–352. doi: 10.1002/ajmg.1320230126
Brune, C., Kim, S., Salt, J., Leventhal, B., Lord, C., and Cook, E. J. (2006). 5-HTTLPR genotype-specific phenotype in children and adolescents with autism. Am. J. Psychiatry 163, 2148–2156. doi: 10.1176/appi.ajp.163.12.2148
Buchsbaum, M. S., Hollander, E., Haznedar, M. M., Tang, C., Spiegel-Cohen, J., Wei, T. C., et al. (2001). Effect of fluoxetine on regional cerebral metabolism in autistic spectrum disorders: a pilot study. Int. J. Neuropsychopharmacol. 4, 119–125. doi: 10.1017/S1461145701002280
Castrén, M., Lampinen, K. E., Miettinen, R., Koponen, E., Sipola, I., Bakker, C. E., et al. (2002). BDNF regulates the expression of fragile X mental retardation protein mRNA in the hippocampus. Neurobiol. Dis. 11, 221–229. doi: 10.1006/nbdi.2002.0544
Castrén, M., Tervonen T., Kärkkäinen V., Heinonen S., Castrén E., Larsson K., et al. (2005). Altered neuronal differentiation of neural stem cells in fragile X syndrome. Proc. Natl. Acad. Sci. U.S.A. 102, 17408–17449. doi: 10.1073/pnas.0508995102
Castrén, M. L., and Castrén, E. (2014). BDNF in fragile X syndrome. Neuropharmacology 76, 729–736. doi: 10.1016/j.neuropharm.2013.05.018
Chen, Z. Y., Jing, D., Bath, K. G., Ieraci, A., Khan, T., Siao, C. J., et al. (2006). Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science 314, 140–143. doi: 10.1126/science.1129663
Connolly, A., Chez, M., Streif, E. M., Keeling, R. M., Golumbek, P. T., Kwon, J. M., et al. (2006). Brain-derived neurotrophic factor and autoantibodies to neural antigens in sera of children with autistic spectrum disorders, Landau-Kleffner syndrome, and epilepsy. Biol. Psychiatry 59, 354–363. doi: 10.1016/j.biopsych.2005.07.004
Correia, C. T., Coutinho, A. M., Sequeira, A. F., Sousa, I. G., Venda, L. L., Almeida, J. P., et al. (2011). Increased BDNF levels and NTRK2 gene association suggest a disruption of BDNF/TRKB signaling in autism. Genes Brain Behav. 9, 841–848. doi: 10.1111/j.1601-183X.2010.00627.x
Cryan, J. F., Markou, A., and Lucki, I. (2002). Assessing antidepressant activity in rodents: recent developments and future needs. Trends Pharmacol. Sci. 23, 238–245. doi: 10.1016/S0165-6147(02)02017-5
Darnell, J. C., Van Driesche, S. J., Zhang, C., Hung, K. Y., Mele, A., Fraser, C. E., et al. (2011). FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261. doi: 10.1016/j.cell.2011.06.013
DeLong, G. R., Ritch, C. R., and Burch, S. (2002). Fluoxetine response in children with autistic spectrum disorders: correlation with familial major affective disorder and intellectual achievement. Dev. Med. Child Neurol. 44, 652–659. doi: 10.1111/j.1469-8749.2002.tb00266.x
DeLong, G. R., Teague, L. A., and Mcswain Kamran, M. (1998). Effects of fluoxetine treatment in young children with idiopathic autism. Dev. Med. Child Neurol. 40, 551–562. doi: 10.1111/j.1469-8749.1998.tb15414.x
Devlin, B., Cook, E. J., Coon, H., Dawson, G., Grigorenko, E., Mcmahon, W., et al. (2005). Autism and the serotonin transporter: the long and short of it. Mol. Psychiatry 10, 1110–1116. doi: 10.1038/sj.mp.4001724
Dolen, G., Osterweil, E., Rao, B. S.S ,Smith, G. B., Auerbach, B. D., Chattarji, S., et al. (2007). Correction of fragile X syndrome in mice. Neuron 56, 955–962. doi: 10.1016/j.neuron.2007.12.001
Duman, R. S., and Monteggia, L. M. (2006). A neurotrophic model for stress-related mood disorders. Biol. Psychiatry 59, 1116–1127. doi: 10.1016/j.biopsych.2006.02.013
Dölen, G., and Bear, M. F. (2009). Fragile X syndrome and autism: from disease model to therapeutic targets. J. Neurodevelop. Disord. 1, 133–140. doi: 10.1007/s11689-009-9015-x
Garcia, K., Yu, G., Nicolini, C., Michalski, B., Garzon, D., Chiu, V. S., et al. (2012). Altered balance of proteolytic isoforms of pro-brain-derived neurotrophic factor in autism. J. Neuropathol. Exp. Neurol. 71, 289–297. doi: 10.1097/NEN.0b013e31824b27e4
Gibson, J. R., Bartley, A. F., Hays, S. A., and Huber, K. M. (2008). Imbalance of neocortical excitation and inhibition and altered UP states reflect network hyperexcitability in the mouse model of fragile X syndrome. J. Neurophysiol. 100, 2615–2626. doi: 10.1152/jn.90752.2008
Hagerman, R., Berry-Kravis, E., Kaufmann, W., Ono, M., Tartaglia, N., Lachiewicz, A., et al. (2009). Advances in the treatment of fragile X syndrome. Pediatrics 123, 378–390. doi: 10.1542/peds.2008-0317
Hagerman, R., Fulton, M. J., Leaman, A., Riddle, J., Hagerman, K., and Sobesky, W. (1994). A survey of fluoxetine therapy in fragile syndrome. Dev. Brain Dysfunct. 7, 155–164.
Hagerman, R., Hoem, G., and Hagerman, P. (2010). Fragile X and autism: intertwined at the molecular level leading to targeted treatments. Mol. Autism 1, 1–14. doi: 10.1186/2040-2392-1-12
Hernandez, R. N., Feinberg, R. L., Vaurio, R., Passanante, N. M., Thompson, R. E., and Kaufmann, W. E. (2009). Autism spectrum disorder in fragile X syndrome: a longitudinal evaluation. Am. J. Med. Genet. 149A, 1125–1137. doi: 10.1002/ajmg.a.32848
Hollander, E., Soorya, L., Chaplin, W., Anagnostou, E., Taylor, B. P., Ferretti, C. J., et al. (2012). A double-blind placebo-controlled trial of fluoxetine for repetitive behaviors and global severity in adult autism spectrum disorders. Am. J. Psychiatry 169, 292–299. doi: 10.1176/appi.ajp.2011.10050764
Hughes, J. (2009). Update on autism: a review of 1300 reports published in 2008. Epilepsy Behav. 16, 569–589. doi: 10.1016/j.yebeh.2009.09.023
Ibarguen-Vargas, Y., Surget, A., Vourc’h, P., Leman, S., Andres, C. R., Gardier, A. M., et al. (2009). Deficit in BDNF does not increase vulnerability to stress but dampens antidepressant-like effects in the unpredictable chronic mild stress. Behav. Brain Res. 202, 245–251. doi: 10.1016/j.bbr.2009.03.040
Iughetti, L., Casarosa, E., Predieri, B., Patianna, V., and Luisi, S. (2011). Plasma brain-derived neurotrophic factor concentrations in children and adolescents. Neuropeptides 45, 205–211. doi: 10.1016/j.npep.2011.02.002
Karpova, N., Lindholm, J., Pruunsild, P., Timmusk, T., and Castrén, E. (2009). Long-lasting behavioural and molecular alterations induced by early postnatal fluoxetine exposure are restored by chronic fluoxetine treatment in adult mice. Eur. Neuropsychopharmacol. 19, 97–108. doi: 10.1016/j.euroneuro.2008.09.002
Kernie, S., Liebl, D. J., and Parada, F. B. (2000). BDNF regulates eating behavior and locomotor activity in mice. EMBO J. 19, 1290–1300. doi: 10.1093/emboj/19.6.1290
Kim, H., Gibboni, R., Kirkhart, C., and Bao, S. (2013a). Impaired critical period plasticity in primary auditory cortex of fragile X model mice. J. Neurosci. 33, 15686–15692. doi: 10.1523/JNEUROSCI.3246-12.2013
Kim, H. J., Kim, T. H., Choi, S. J., Hong, Y. J., Yang, J. S., Sung, K. W., et al. (2013b). Fluoxetine suppresses synaptically induced [Ca2+]i spikes and excitotoxicity in cultured rat hippocampal neurons. Brain Res. 1490, 23–34. doi: 10.1016/j.brainres.2012.10.062
Kiryanova, V., McAllister, B., and Dyck, R. (2013). Long-term outcomes of developmental exposure to fluoxetine: a review of the animal literature. Dev. Neurosci. 35, 437–439. doi: 10.1159/000355709
Koponen, E., Rantamäki, T., Voikar, V., Saarelainen, T., MacDonald, E., and Castrén, E. (2005). Enhanced BDNF signaling is associated with an antidepressant-like behavioral response and changes in brain monoamines. Cell. Mol. Neurobiol. 25, 973–980. doi: 10.1007/s10571-005-8468-z
Lebrand, C., Cases, O., Adelbrecht, C., Doye, A., Alvarez, C., El Mestikawy, S., et al. (1996). Transient uptake and storage of serotonin in developing thalamic neurons. Neuron 17, 823–835. doi: 10.1016/S0896-6273(00)80215-9
Louhivuori, V., Vicario, A., Uutela, M., Rantamäki, T., Louhivuori, L. M., Castrén, E., et al. (2011). BDNF and TrkB in neuronal differentiation of Fmr1-knockout mouse. Neurobiol. Dis. 41, 469–480. doi: 10.1016/j.nbd.2010.10.018
Luo, Y., Shan, G., Guo, W., Smrt, R. D., Johnson, E. B., Li, X., et al. (2010). Fragile x mental retardation protein regulates proliferation and differentiation of adult neural stem/progenitor cells. PLoS Genet. 6:e1000898. doi: 10.1371/journal.pgen.1000898
Lyons, W. E., Maumonas, L. A., Ricaurte, G. A., Coppola, V., Reid, S. W., Bora, S. H., et al. (1999). Brain-derived neurotrophic factor-deficient mice develop aggressiveness and hyperphagia in conjunction with brain serotonergic abnormalities. Proc. Natl. Acad. Sci. U.S.A. 96, 15239–15244. doi: 10.1073/pnas.96.26.15239
MacQueen, G., Ramakrishnan, K., Croll, S. D., Siuciak, J. A., Yu, G., Young, L. T., et al. (2001). Performance of heterozygous brain-derived neurotrophic factor knockout mice on behavioral analogues of anxiety, nociception, and depression. Behav. Neurosci. 115, 1145–1153. doi: 10.1037/0735-7044.115.5.1145
Makkonen, I., Riikonen, R., Kuikka, J. T., Kokki, H., Bressler, J., Marshall, C., et al. (2011). Brain derived neurotrophic factor and serotonin transporter binding as markers of clinical response to fluoxetine therapy in children with autism. J. Pediatr. Neurol. 9, 1–8. doi: 10.3233/JPN-2010-0446
Maya-Vetencourt, J., Sale, A., Viegi, A., Baroncelli, L., De Pasquale, R., O’Leary, O., et al. (2008). The antidepressant fluoxetine restores plasticity in the adult visual cortex. Science 320, 385–388. doi: 10.1126/science.1150516
Mineur, Y. S., Huynh, L. X., and Crusio, W. E. (2006). Social behavior deficits in the Fmr1 mutant mouse. Behav. Brain Res. 168, 172–175. doi: 10.1016/j.bbr.2005.11.004
Mineur, Y. S., Sluyter, F., De Wit, S., Oostra, B. A., and Crusio, W. E. (2002). Behavioral and neuroanatomical characterization of the Fmr1 knockout mouse. Hippocampus 12, 39–46. doi: 10.1002/hipo.10005
Miyazaki, K., Narita, N., Sakuta, R., Miyahara, T., Naruse, H., Okado, N., et al. (2004). Serum neurotrophin concentrations in autism and mental retardation: a pilot study. Brain Dev. 26, 292–295. doi: 10.1016/S0387-7604(03)00168-2
Monteggia, L. M., Luikart, B., Barrot, M., Theobold, D., Malkovska, I., Nef, S., et al. (2007). Brain-derived neurotrophic factor conditional knockouts show gender differences in depression-related behaviors. Biol. Psychiatry 15, 187–197. doi: 10.1016/j.biopsych.2006.03.021
Nelson, K., Grether, J. K., Croen, L. A., Dambrosia, J. M., Dickens, B. F., Jelliffe, L. L., et al. (2001). Neuropeptides and neurotrophins in neonatal blood of children with autism or mental retardation. Ann. Neurol. 49, 597–606. doi: 10.1002/ana.1024
Nibuya, M., Morinobu, S., and Duman, R. S. (1995). Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J. Neurosci. 15, 7539–7547.
Nielsen, D. M., Derber, W. J., McClellan, D. A., and Crnic, L. S. (2002). Alterations in the auditory startle response in Fmr1 targeted mutant mouse models of fragile X syndrome. Brain Res. 927. 8–17. doi: 10.1016/S0006-8993(01)03309-1
Oswald, D. P., and Sonenklar, N. A. (2007). Medication use among children with autism spectrum disorders. J. Child Adolesc. Psychopharmacol. 17, 348–355. doi: 10.1089/cap.2006.17303
Peier, A. M., McIlwain, K. L., Kenneson, A., Warren, S. T., Paylor, R., and Nelson, D. L. (2000). (Over)correction of FMR1 deficiency with YAC transgenics: behavioral and physical features. Hum. Mol. Genet. 9, 1145–1159. doi: 10.1093/hmg/9.8.1145
Perry, E. K., Lee, M. L. W., Martin-Ruiz, C. M., Court, J. A., Volsen, S. G., Merrit, J., et al. (2001). Cholinergic activity in autism: abnormalities in the cerebral cortex and basal forebrain. Am. J. Psychiatry 158, 1058–1066. doi: 10.1176/appi.ajp.158.7.1058
Piven, J., Tsai, G., Nehme, E., Coyle, J., Chase, G., and Folstein, S. (1991). Platelet serotonin, a possible marker for familial autism. J. Autism Dev. Disord. 21, 51–59. doi: 10.1007/BF02206997
Porsolt, R. D., Le Pichon, M., and Jalfre, M. (1977). Depression: a new animal model sensitive to antidepressant treatments. Nature 266, 730–732. doi: 10.1038/266730a0
Prut, L., and Belzung, C. (2003). The open field as a paradigm to measure the effects of drugs on anxiety-like behaviors: a review. Eur. J. Pharmacol. 463, 3–33. doi: 10.1016/S0014-2999(03)01272-X
Rattiner, L. M., Davis, M., French, C. T., and Ressler, K. J. (2004). Brain-derived neurotrophic factor and tyrosine kinase receptor B involvement in amygdala-dependent fear conditioning. J. Neurosci. 19, 4796–4806. doi: 10.1523/JNEUROSCI.5654-03.2004
Rios, M., Fan, G., Fekete, C., Kelly, J., Bates, B., Kuehn, R., et al. (2001). Conditional deletion of brain-derived neurotrophic factor in the postnatal brain leads to obesity and hyperactivity. Mol. Endocrinol. 15, 1748–1757. doi: 10.1210/mend.15.10.0706
Saarelainen, T., Hendolin, P., Lucas, G., Koponen, E., Sairanen, M., MacDonald, E., et al. (2003). Activation of the TrkB neurotrophin receptor is induced by antidepressant drugs and is required for antidepressant-induced behavioural effects. J. Neurosci. 23, 349–357.
Sairanen, M., Lucas, G., Ernfors, P., Castrén, M., and Castrén, E. (2005). BDNF signaling and antidepressant drugs differentially but coordinatedly regulate neurogenesis, survival and turnover of new-born neurons in the adult dentate gyrus. J. Neurosci. 25, 1089–1094. doi: 10.1523/JNEUROSCI.3741-04.2005
Spencer, C. M., Alekseyenko, O., Serysheva, E., Yuva-Paylor, L. A., and Paylor, R. (2005). Altered anxiety-related and social behaviors in the Fmr1 knockout mouse model of fragile X syndrome. Genes Brain Behav. 4, 420–430. doi: 10.1111/j.1601-183X.2005.00123.x
Uutela, M., Lindholm, J., Louhivuori, V., Wei, H., Louhivuori, L. M., Pertovaara, A., et al. (2012). Reduction of BDNF expression in Fmr1 knockout mice worsens cognitive deficits but improves hyperactivity and sensorimotor deficits. Genes Brain Behav. 5, 513–523. doi: 10.1111/j.1601-183X.2012.00784.x
Van Dam, D., D’Hooge, R., Hauben, E., Reyniers, E., Gantois, I., Bakker, C. E., et al. (2000). Spatial learning, contextual fear conditioning and conditioned emotional response in Fmr1 knockout mice. Behav. Brain Res. 117, 127–136. doi: 10.1016/S0166-4328(00)00296-5
Warner-Schmidt, J. L., and Duman, R. S. (2006). Hippocampal neurogenesis: opposing effects of stress and antidepressant treatment. Hippocampus 16, 239–249. doi: 10.1002/hipo.20156
Williams, K., Brignell, A., Randall, M., Silove, N., and Hazell, P. (2013). Selective serotonin reuptake inhibitors (SSRIs) for autism spectrum disorders (ASD). Cochrane Database. Syst. Rev. 20, CD004677. doi: 10.1002/14651858.CD004677.pub3
Wisden, W., and Morris, B. (1994). “In situ hybridization with synthetic oligonucleotide probes,” in In Situ Protocols for Neurobiology, eds W. Wisden and B. Morris (London: Academic Press), 1–34.
Vitalis, T., and Parnavelas, J. G. (2003). The role of serotonin in early cortical development. Dev. Neurosci. 25, 245–256. doi: 10.1159/000072272
Wong, D. T., Horng, J. S., Bymaster, F. P., Hauser, K. L., and Molloy, B. B. (1974). A selective inhibitor of serotonin uptake: Lilly 110140, 3-(p-trifluoromethylphenoxy)-N-methyl-3 phenylpropylamine. Life Sci. 15, 471–479. doi: 10.1016/0024-3205(74)90345-2
Wu, X., and Castrén, E. (2009). Co-treatment with diazepam prevents the effects of fluoxetine on the proliferation and survival of hippocampal dentate granule cells. Biol. Psychiatry 66, 5–8. doi: 10.1016/j.biopsych.2009.01.023
Xu, Z., Yang, Q., Ma, L., Liu, S., Chen, G., Wu, Y., et al. (2012). Deficits in LTP induction by 5-HT2A receptor antagonist in a mouse model for fragile X syndrome. PLoS ONE 7:e48741. doi: 10.1371/journal.pone.0048741
Keywords: behavior, autism, BDNF, neurogenesis, TrkB receptors
Citation: Uutela M, Lindholm J, Rantamäki T, Umemori J, Hunter K, Võikar V and Castrén ML (2014) Distinctive behavioral and cellular responses to fluoxetine in the mouse model for Fragile X syndrome. Front. Cell. Neurosci. 8:150. doi: 10.3389/fncel.2014.00150
Received: 17 January 2014; Accepted: 09 May 2014;
Published online: 28 May 2014.
Edited by:
Laurie Doering, McMaster University, CanadaReviewed by:
Osborne F. Almeida, Max Planck Institute of Psychiatry, GermanyMasami Kojima, National Institute of Advanced Science and Technology, Japan
Copyright © 2014 Uutela, Lindholm, Rantamäki, Umemori, Hunter, Võikar and Castrén. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maija L. Castrén, Institute of Biomedicine/Physiology, University of Helsinki, P. O. Box 63 (Haartmaninkatu 8), FIN-00014 Helsinki, Finland e-mail:bWFpamEuY2FzdHJlbkBoZWxzaW5raS5maQ==