 Jamie Maguire
Jamie Maguire- Department of Neuroscience, Tufts University School of Medicine, Boston, MA, USA
GABAergic neurotransmission is highly plastic, undergoing dynamic alterations in response to changes in the environment, such as following both acute and chronic stress. Stress-induced plasticity of GABAergic inhibition is thought to contribute to changes in neuronal excitability associated with stress, which is particularly relevant for stress-related disorders and seizure susceptibility. Here we review the literature demonstrating several mechanisms altering GABAergic inhibition associated with stress, including brain region-specific alterations in GABAA receptor (GABAAR) subunit expression, changes in chloride homeostasis, and plasticity at GABAergic synapses. Alterations in the expression of specific GABAAR subunits have been documented in multiple brain regions associated with acute or chronic stress. In addition, recent work demonstrates stress-induced alterations in GABAergic inhibition resulting from plasticity in intracellular chloride levels. Acute and chronic stress-induced dephosphorylation and downregulation of the K+/Cl− co-transporter, KCC2, has been implicated in compromising GABAergic control of corticotropin-releasing hormone (CRH) neurons necessary for mounting the physiological response to stress. Acute stress also unmasks the capacity for both long-term potentiation and long-term depression, in distinct temporal windows, at GABAergic synapses on parvocellular neuroendocrine cells (PNCs) in the paraventricular nucleus (PVN) of the hypothalamus. This review highlights the complexity in the plasticity of GABAergic neurotransmission associated with stress and the relationship to neuronal excitability, including alterations in GABAAR expression, synaptic plasticity at GABAergic synapses, and changes in chloride homeostasis.
Alterations in neuronal excitability and seizure susceptibility associated with stress have largely been attributed to changes in GABAergic inhibition. Here we review the literature describing stress-induced alterations in neuronal excitability and seizure susceptibility associated with stress and the role of GABAergic neurotransmission. Alterations in the expression of GABAARs and the functional consequences on GABAergic inhibition following both acute and chronic stress in multiple brain regions are discussed. In addition, recent evidence pointing to synaptic plasticity at GABAergic synapses and alterations in intracellular chloride levels resulting in compromised GABAergic inhibition following stress is also highlighted.
Stress-Induced Changes in Excitability
Anecdotally, it is widely accepted that stress is a precipitating factor for seizures (for review see Maguire and Salpekar, 2013). Patients with epilepsy frequently self-report that stress exacerbates their seizures (Neugebauer et al., 1994; Frucht et al., 2000; Haut et al., 2003, 2007; Nakken et al., 2005; Sperling et al., 2008) and increased cortisol levels are positively correlated with seizure frequency in patients with epilepsy (Galimberti et al., 2005) (for review see Lai and Trimble, 1997; Maguire and Salpekar, 2013). However, the relationship between stress and seizure susceptibility is complex. Broadly speaking, acute stress is thought to be anticonvulsant; whereas, chronic stress is thought to increase seizure susceptibility. This section will review the evidence in animal models documenting changes in seizure susceptibility associated with both acute and chronic stress.
Acute Stress
Swim stress (Soubrie et al., 1980; Pericic et al., 2000, 2001; Reddy and Rogawski, 2002), acute cold stress (de Lima and Rae, 1991), and acute restraint stress (de Lima and Rae, 1991) have been shown to increase seizure threshold (for review see Joels, 2009). The anticonvulsant actions of acute stress are thought to be mediated by the production of stress-derived neurosteroids (for review see Rogawski and Reddy, 2004). In response to stress, deoxycorticosterone (DOC) is released which can be metabolized into the neuroactive derivative, allotetrahydrodeoxycorticosterone (THDOC) which has been demonstrated to exert anticonvulsant actions. Further, DOC itself has also been shown to exhibit anticonvulsant properties, a process which requires neurosteroidogenesis (Reddy and Rogawski, 2002) (for review see Rogawski and Reddy, 2004). The anticonvulsant actions of stress-derived neurosteroids are thought to be mediated by their actions on GABAARs (for review see Rogawski and Reddy, 2004), which will be discussed more thoroughly in a later section (Stress, seizure susceptibility, and GABAARs). In addition, recent studies also demonstrate a role for glucocorticoid receptors in the anticonvulsant action of acute stress on seizure susceptibility (Maggio and Segal, 2012), suggesting the involvement of multiple pathways regulating excitability in response to acute stress.
These data clearly demonstrate the anticonvulsant effects of acute stress, which is contrary to the notion that stress is associated with increased seizure susceptibility and is a trigger for seizures in patients with epilepsy (Minter, 1979) (for review see Maguire and Salpekar, 2013). Although acute stress may have anticonvulsant effects, chronic stress appears to increase seizure susceptibility in animal models, a topic which will be covered in more detail in the following section.
Chronic Stress
Not many controlled studies have been undertaken to elucidate the impact of chronic stress on seizure susceptibility. However, the few studies that have been conducted suggest that chronic social isolation or restraint stress increase susceptibility to seizures induced with bicuculline, picrotoxin, kainic acid, and kindling (Matsumoto et al., 2003; Chadda and Devaud, 2004; Jones et al., 2013). Numerous factors likely mediate the effects of chronic stress on neuronal excitability and seizure susceptibility, including effects on synaptic transmission, adult neurogenesis, and hippocampal remodeling (for review see McEwen, 1999, 2000). The hippocampus is a particularly vulnerable region to the adverse effects of stress, which may mediate the increased neuronal excitability and seizure susceptibility associated with chronic stress (for review see McEwen, 1999, 2000). Furthermore, chronic social isolation stress has been shown to decrease the production of neurosteroids (Serra et al., 2000; Dong et al., 2001), which have anticonvulsant properties (for review see Rogawski and Reddy, 2004), and, thereby, may also influence neuronal excitability. Furthermore, early life stress has also been demonstrated to increase the excitability of principal neurons in the hippocampus and increase seizure susceptibility (for review see Koe et al., 2009), suggesting long-term effects of chronic stress on network excitability.
In addition to stress, there is also a relationship between psychiatric disorders and epilepsy (for review see Jones and O'Brien, 2013). Individuals with psychiatric disorders have a greater risk for developing epilepsy (Forsgren and Nystrom, 1990; Hesdorffer et al., 2000, 2006) and is associated with a poorer outcome (Hitiris et al., 2007; Kanner et al., 2009; Petrovski et al., 2010) (for review see Jones and O'Brien, 2013). These studies suggest that chronic anxiety and other stress-related disorders may increase the susceptibility for seizures. Further, this evidence suggests a proconvulsant role for chronic stress and highlights the complex relationship between stress and seizure susceptibility.
Stress, Seizure Susceptibility, and GABAARs
GABAARs are the primary site of neurosteroid action and likely mediate their anticonvulsant effects (for review see Reddy, 2003). THDOC is produced in response to acute stress, increasing to physiologically-relevant levels which can act on GABAARs (Purdy et al., 1991; Barbaccia et al., 1996a). Neurosteroids act preferentially at δ-containing GABAARs (Wohlfarth et al., 2002), but at higher concentrations can also act on different GABAAR subtypes (Stell et al., 2003; Belelli et al., 2009). In addition to the direct, positive allosteric modulation of GABAARs, neurosteroids can also alter the expression of GABAARs (for review see Maguire and Mody, 2009; Mody and Maguire, 2011). In fact, alterations in the expression of GABAARs associated with both acute and chronic stress may underlie changes in neuronal excitability and seizure susceptibility which will be discussed in more detail below.
Stress-Induced Alterations in GABAergic Inhibition
Many studies examining the impact of stress on GABAergic signaling have relied on the expression of GABA synthesizing enzymes and the binding of GABA ligands. Fewer follow up studies have focused on changes in specific GABAAR subtypes in specific brain regions and the impact on GABAergic inhibition. Many of these studies have focused on stress-induced changes in GABA in the hippocampus in relation to changes in neuronal excitability (for review see Joels, 2009); however, recent interest has also focused on the role of GABA within the stress neurocircuitry (for review see Gunn et al., 2011), particularly the PVN of the hypothalamus.
Acute Stress
In addition to the direct modulatory effects of acute stress-derived neurosteroids on GABAergic inhibition, acute stress has also been proposed to alter GABAergic inhibition via changes in GABA synthesis, release, and the expression of specific GABAAR subunits (for review see Maguire and Mody, 2009; Mody and Maguire, 2011). Changes in GABA synthesis following acute stress have been suggested from alterations in the expression of GABA (Yoneda et al., 1983; Otero Losada, 1988; Acosta et al., 1993) as well as glutamic acid decarboxylase (GAD) (Yoneda et al., 1983; Otero Losada, 1988; Maroulakou and Stylianopoulou, 1991; Acosta et al., 1993; Bowers et al., 1998), the enzyme responsible for the synthesis of GABA (Table 1). GABA levels are decreased in the striatum following acute cold stress (Acosta et al., 1993) and decreased in the olfactory bulb following acute immobilization stress (Otero Losada, 1988). In contrast, GABA levels are increased in the striatum and hypothalamus following acute immobilization stress (Yoneda et al., 1983). GAD expression has been shown to be increased in numerous brain regions following acute thermal stress (Maroulakou and Stylianopoulou, 1991) and acute immobilization/restraint stress (Yoneda et al., 1983; Bowers et al., 1998).
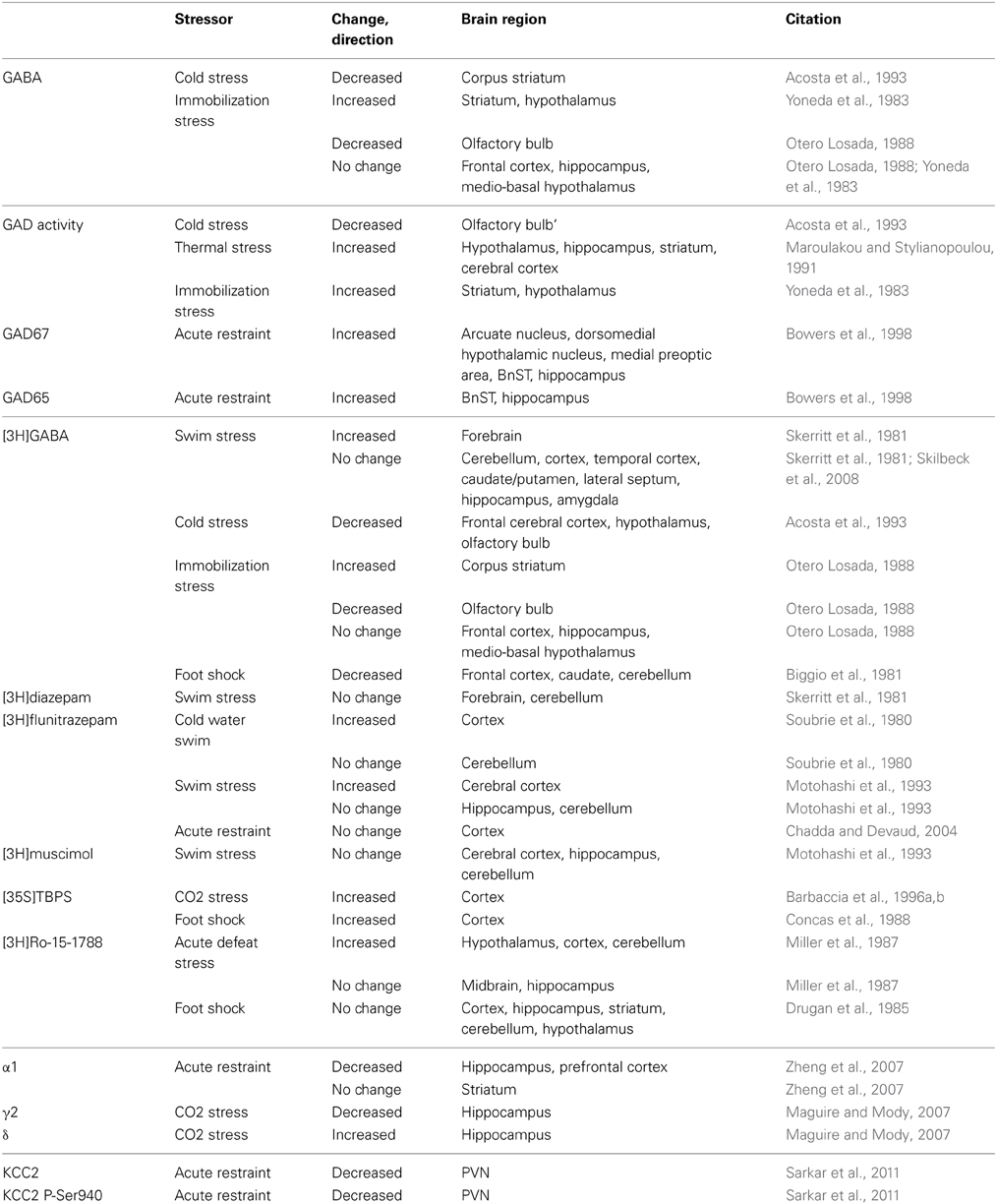
Table 1. Acute stress-induced changes related to GABA.
There are also changes in the expression of GABAARs following acute stress, evident from changes in the binding of radio-labeled GABA ligands. The binding of [3H]GABA is increased in the forebrain following acute swim stress (Skerritt et al., 1981) and in the striatum following acute immobilization stress (Otero Losada, 1988). In contrast, [3H]GABA binding is decreased in the cortex, hypothalamus and olfactory bulb following acute cold stress (Acosta et al., 1993) and decreased in the olfactory bulb following acute immobilization stress (Otero Losada, 1988). [3H]flunitrazepam binding is increased in the cortex following swim stress (Soubrie et al., 1980; Motohashi et al., 1993; Chadda and Devaud, 2004) and t-[35S]butylbicyclophosphorothionate ([35S]TBPS) binding is increased in the cortex following either CO2 or foot shock stress (Concas et al., 1988; Barbaccia et al., 1996b). The binding of [3H]Ro-15-1788 is decreased in the hypothalamus, cortex, and cerebellum following acute defeat stress (Miller et al., 1987). However, no changes were observed in [3H]diazepam (Skerritt et al., 1981) or [3H]muscimol binding (Motohashi et al., 1993) (Table 1). These data are summarized in Table 1 and demonstrate the complexity in the plasticity of GABAARs associated with acute stress (for review see Skilbeck et al., 2010). In addition, changes in specific GABAAR subunits have been described following acute stress.
Table 1 summarizes changes brain region-specific changes in the expression of specific GABAAR subunits associated with several different models of acute stress. Expression of the GABAAR α1 and γ2 subunits are decreased in the hippocampus and prefrontal cortex following acute restraint stress or CO2 stress (Maguire and Mody, 2007; Zheng et al., 2007). In contrast, the GABAAR δ subunit expression is increased following acute stress in the hippocampus following CO2 stress (Maguire and Mody, 2007) (Table 1). Functional alterations in GABAergic inhibition following acute stress have also been documented in the hippocampus. Following acute restraint stress, there is an increase in the frequency of spontaneous inhibitory post-synaptic currents (sIPSCs) in CA1 pyramidal cells (Hu et al., 2010). In addition, there is an increase in the tonic GABAergic inhibition recorded in dentate gyrus granule cells (DGGCs) following acute CO2 inhalation stress, consistent with the increased expression of the GABAAR δ subunit (Maguire and Mody, 2007). Interestingly, some of the alterations in GABAAR subunit expression can be mimicked by treatment with THDOC (Maguire and Mody, 2007), suggesting a role for neurosteroids in stress-induced GABAAR plasticity.
Alterations in GABAergic inhibition have also been observed in other brain regions following acute stress. The frequency, but not amplitude, of sIPSCs is increased in pyramidal neurons in the prefrontal cortex following acute stress (inescapable shock). Interestingly, these changes were prevented if the animal was able to exert some control over the stress (escapable shock) (Varela et al., 2012). The frequency of sIPSCs is also increased in the PVN following high frequency stimulation in slices from mice subjected to acute restraint stress (Inoue et al., 2013), which is dependent upon glucocorticoid receptor activation and retrograde opioid signaling (Wamsteeker Cusulin et al., 2013). The increased frequency of sIPSCs in the PVN following acute stress may be due to the increased burst firing of GABAergic interneurons in the peri-PVN area (Shin et al., 2011). These data demonstrate alterations in GABAergic inhibition in several different brain regions following acute stress which may involve both pre- and post-synaptic mechanisms.
Chronic Stress
Alterations in GABAergic inhibition also occur following chronic stress, although these changes appear to be unique from those observed following acute stress. Changes in GABA synthesis following chronic stress have been suggested from alterations in the expression of GABA, GAD65, and GAD67 (Table 2). A reduction in the concentration of GABA was observed in the cortex, hypothalamus, and olfactory bulb following chronic cold stress (Acosta et al., 1993). GAD expression is decreased in the striatum and olfactory bulb following chronic cold stress (Acosta et al., 1993); whereas, GAD65 and GAD67 expression are increased in numerous brain regions following chronic intermittent stress (Bowers et al., 1998) and unchanged following repeated swim stress (Montpied et al., 1993). Changes in the expression of GABAARs following chronic stress have also been suggested from changes in the binding of radio-labeled GABA ligands. [3H]GABA binding is decreased in the hypothalamus following chronic cold stress (Acosta et al., 1993). The binding of [3H]flunitrazepam is decreased in the frontal cortex following chronic foot shock stress and increased following chronic immobilization stress (Braestrup et al., 1979). No change in [3H]flunitrazepam or [3H]muscimol binding was observed following repeated swim stress (Braestrup et al., 1979; Motohashi et al., 1993). These data are summarized in Table 2. Changes in specific GABAAR subunits have also been described following chronic stress.
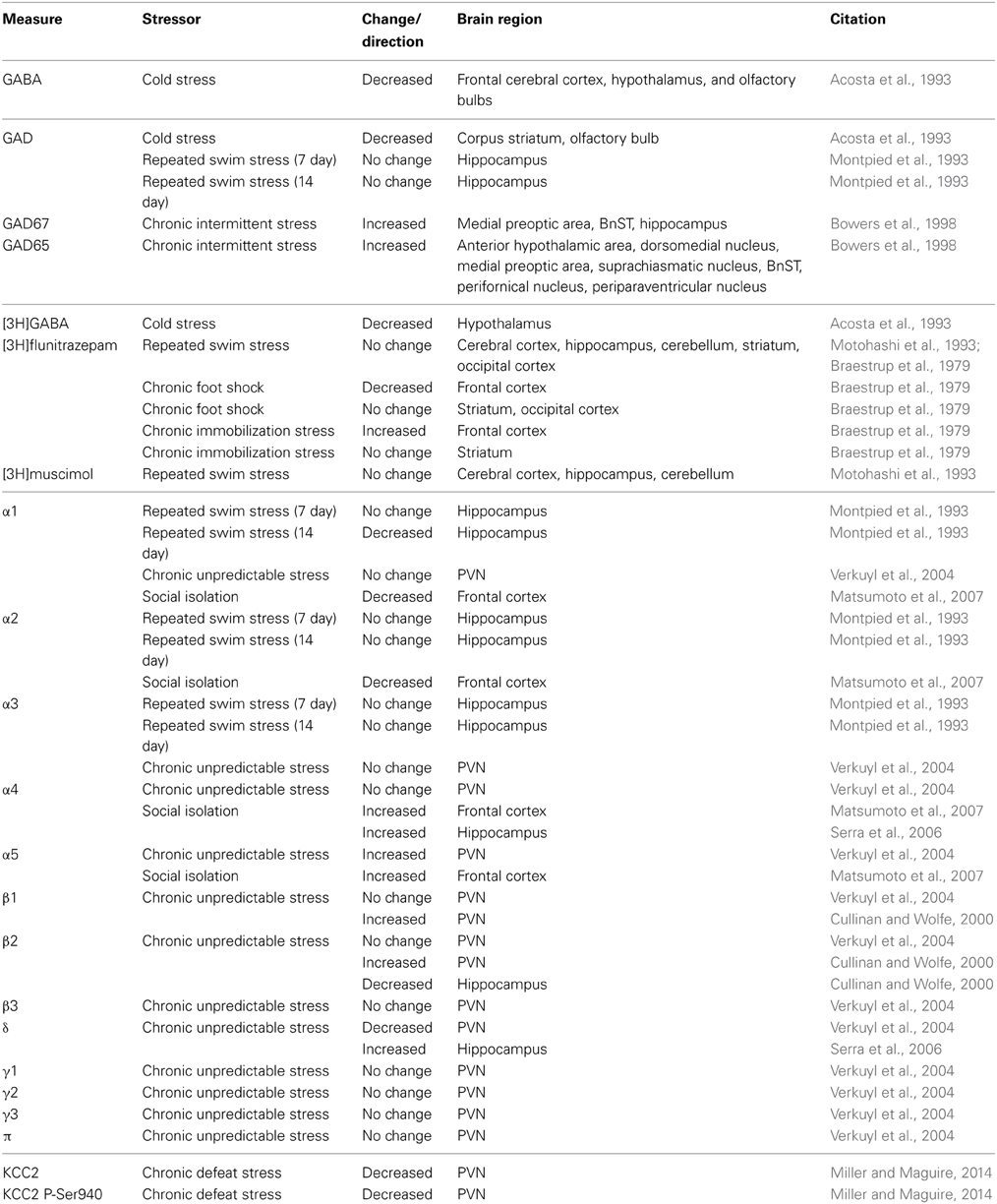
Table 2. Chronic stress-induced changes related to GABA.
A decrease in the expression of the GABAAR α1 subunit has been observed in the hippocampus following repeated swim stress with no change observed in α2 or α3 (Montpied et al., 1993). Similarly, a decrease in the expression of the GABAAR β 2 subunit was observed in the hippocampus following chronic unpredictable stress (Cullinan and Wolfe, 2000). An upregulation of α5 and β 1 subunits have been observed in the PVN following chronic unpredictable stress (Cullinan and Wolfe, 2000; Verkuyl et al., 2004); whereas, a decrease in the expression of the δ subunit was observed in the PVN with no change in α1, α3, α4, γ1, γ2, γ3, or π expression (Montpied et al., 1993; Verkuyl et al., 2004). Chronic social isolation stress results in a decrease in α1 and α2 expression and an increase in α4 and α5 expression in the frontal cortex (Matsumoto et al., 2007). Similarly, an increase in α4 and δ subunit expression has been observed in the hippocampus following chronic social isolation stress (Serra et al., 2006). These changes are summarized in Table 2, highlighting the brain region-specific alterations in GABAARs following chronic stress. Consistent with changes in GABAAR subunit expression, functional changes in GABAergic inhibition have been observed following chronic stress. An increase in the frequency of sIPSCs has been observed in CA1 pyramidal neurons following chronic restraint stress (Hu et al., 2010), which is mediated by glucocorticoid receptor activation (Hu et al., 2010). In contrast, a decrease in the frequency of sIPSCs has been observed following chronic mild stress in DGGCs (Holm et al., 2011), which is consistent with a decrease in the expression of the α1/α2 and γ2 subunits (Matsumoto et al., 2007). Consistent with an upregulation of the GABAAR α4, α5 and δ subunit (Serra et al., 2006; Matsumoto et al., 2007), an increase in tonic GABAergic inhibition has been measured in DGGCs following chronic stress (Serra et al., 2008; Holm et al., 2011). In the PVN, a decrease in the frequency of sIPSCs has been observed (Verkuyl et al., 2004), which can be mimicked with exogenous corticosterone (Verkuyl et al., 2005) and reversed with adrenalectomy (Verkuyl and Joels, 2003). These data demonstrate the brain region-specific plasticity in GABAergic neurotransmission associated with chronic stress, which may underlie changes in neuronal excitability but also changes in sensitivity to pharmacological compounds.
Impaired GABAergic inhibition following chronic stress may also result from a decrease in neurosteroid synthesis. A decrease in the production of 3α,5α-tetrahydroprogesterone (3α,5α-THP; allopregnanolone) has been observed following chronic stress (Serra et al., 2000; Dong et al., 2001; Pinna et al., 2003; Matsumoto et al., 2007). Decreased levels of neurosteroids may limit the allosteric modulation of GABAARs as well as potentially contribute to the observed changes in GABAAR subunit expression (for review see Maguire and Mody, 2009). The decreased production of endogenous positive modulators of GABAARs, combined with alterations in GABAAR subunit expression and decreased binding of GABA ligands, indicates altered GABAergic signaling in multiple brain regions following chronic stress.
Stress-Induced Alterations in Chloride Homeostasis
Effective GABAergic inhibition requires the maintenance of the chloride gradient, which is accomplished by the K+/Cl− co-transporter, KCC2, in the adult brain (Rivera et al., 1999, 2005; Payne et al., 2003). Recent studies have begun to investigate the impact of changes in chloride homeostasis on GABAergic inhibition under both physiological and pathological conditions, including following acute and chronic stress.
Acute Stress
Our lab recently demonstrated dynamic changes in GABAergic inhibition in CRH neurons in the PVN following acute restraint stress. CRH neurons are at the apex of control of the hypothalamic-pituitary-adrenal (HPA) axis, which mediates the body's physiological response to stress. Following acute restraint stress, KCC2 is dephosphorylated at residue Ser 940 and downregulated in the PVN (Sarkar et al., 2011) (Table 1). Although functional deficits in KCC2 transport have not directly been measured following acute stress, shifts in EGABA and compromised GABAergic control of CRH neurons (Hewitt et al., 2009; Sarkar et al., 2011) are thought to result from the dephosphorylation and downregulation of KCC2, leading to excitatory actions of GABA (Sarkar et al., 2011). The changes in KCC2 and GABAergic inhibition following acute stress are unique to these CRH neurons and we believe are part of the signaling cascade required to mount a rapid, all-or-none response to stress.
Chronic Stress
Alterations in KCC2 in the PVN, such as those observed following acute stress, persist following chronic social defeat stress (Miller and Maguire, 2014). The dephosphorylation and downregulation of KCC2 in the PVN is accompanied by stress-induced elevations in corticosterone throughout the chronic social defeat stress paradigm (Miller and Maguire, 2014). These data are consistent with the role of dephosphorylation and downregulation of KCC2 in the PVN in mounting the physiological response to stress. However, deficits in KCC2 transporter function have not been directly measured following chronic stress and future studies are required to determine the significance of these changes on neuronal excitability. Interestingly, the ability of neurosteroids to potentiate GABAergic inhibition and limit the activity of PNCs is reduced following early life stress due to compromised GABAergic inhibition associated with a shift in EGABA in PNCs (Gunn et al., 2013). Thus, it appears that chronic stress impairs the GABAergic control of PNCs, via downregulation of KCC2, which has significant implications for KCC2 as a therapeutic target. Stress-induced changes in chloride plasticity in other brain regions and the impact on neuronal excitability and seizure susceptibility remains to be explored, but are necessary steps given the interest in targeting KCC2 for therapeutics.
Plasticity in the GABAergic Control of the HPA Axis
In addition to changes in GABAergic neurotransmission resulting from changes in GABAAR subunit expression and/or chloride plasticity, elegant studies have demonstrated stress-induced synaptic plasticity at GABAergic synapses following acute stress. Acute restraint stress has been demonstrated to unmask the capacity for activity-dependent long-term potentiation at GABAergic synapses (LTPGABA) on PNCs in the PVN, a process which involves the activation of β-adrenergic receptors and an upregulation of mGluR1 receptors (Inoue et al., 2013). This potentiation of GABAergic inhibition has been proposed to overwhelm the chloride extrusion mechanisms (Inoue and Bains, 2014), accomplished by KCC2, which accounts for the collapse in the chloride gradient following stress (Hewitt et al., 2009; Sarkar et al., 2011). Interestingly, there is temporal specificity in the plasticity of GABAergic inhibition on PNCs. At 90 min post-restraint stress, GABAergic synapses on PNCs exhibit long-term depression (LTDGABA), mediated by the actions of glucocorticoids (Wamsteeker Cusulin et al., 2013) which is thought to limit the stress response. These data demonstrate the bidirectional plasticity in GABAergic inhibition on PNCs following acute stress and highlight the importance of synaptic plasticity and the dynamic impact on GABAergic neurotransmission in the regulation of the HPA axis.
Concluding Remarks
Here we review the role of stress in the plasticity of GABAergic inhibition. In addition to changes in GABAAR subunit expression, recent evidence demonstrates a greater complexity in the plasticity of GABAergic neurotransmission associated with stress, involving changes in chloride homeostasis and synaptic plasticity at GABAergic synapses. These data suggest that there may be additional targets, other than GABAARs, for managing the impact of stress on GABAergic neurotransmission which has implications for stress-related disorders and seizure susceptibility.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
Jamie Maguire was supported by NIH Grant R01 NS073574 and a Research Grant from the Epilepsy Foundation.
References
Acosta, G. B., Otero Losada, M. E., and Rubio, M. C. (1993). Area-dependent changes in GABAergic function after acute and chronic cold stress. Neurosci. Lett. 154, 175–178. doi: 10.1016/0304-3940(93)90200-5
Barbaccia, M. L., Concas, A., Roscetti, G., Bolacchi, F., Mostallino, M. C., Purdy, R. H., et al. (1996a). Stress-induced increase in brain neuroactive steroids: antagonism by abecarnil. Pharmacol. Biochem. Behav. 54, 205–210. doi: 10.1016/0091-3057(95)02133-7
Barbaccia, M. L., Roscetti, G., Trabucchi, M., Mostallino, M. C., Concas, A., Purdy, R. H., et al. (1996b). Time-dependent changes in rat brain neuroactive steroid concentrations and GABA(A) receptor function after acute stress. Neuroendocrinology 63, 166–172. doi: 10.1159/000126953
Belelli, D., Harrison, N. L., Maguire, J., Macdonald, R. L., Walker, M. C., and Cope, D. W. (2009). Extrasynaptic GABAA receptors: form, pharmacology, and function. J. Neurosci. 29, 12757–12763. doi: 10.1523/JNEUROSCI.3340-09.2009
Biggio, G., Corda, M. G., Concas, A., Demontis, G., Rossetti, Z., and Gessa, G. L. (1981). Rapid changes in GABA binding induced by stress in different areas of the rat brain. Brain Res. 229, 363–369. doi: 10.1016/0006-8993(81)91000-3
Bowers, G., Cullinan, W. E., and Herman, J. P. (1998). Region-specific regulation of glutamic acid decarboxylase (GAD) mRNA expression in central stress circuits. J. Neurosci. 18, 5938–5947.
Braestrup, C., Nielsen, M., Nielsen, E. B., and Lyon, M. (1979). Benzodiazepine receptors in the brain as affected by different experimental stresses: the changes are small and not undirectional. Psychopharmacology (Berl.). 65, 273–277. doi: 10.1007/BF00492215
Chadda, R., and Devaud, L. L. (2004). Sex differences in effects of mild chronic stress on seizure risk and GABAA receptors in rats. Pharmacol. Biochem. Behav. 78, 495–504. doi: 10.1016/j.pbb.2004.03.022
Concas, A., Serra, M., Atsoggiu, T., and Biggio, G. (1988). Foot-shock stress and anxiogenic +|-Carbolines increase t-[35S]Butylbicyclophosphorothionate binding in the rat cerebral cortex, an effect opposite to anxiolytics and +|-Aminobutyric Acid Mimetics. J. Neurochem. 51, 1868–1876. doi: 10.1111/j.1471-4159.1988.tb01170.x
Cullinan, W. E., and Wolfe, T. J. (2000). Chronic stress regulates levels of mRNA transcripts encoding beta subunits of the GABA(A) receptor in the rat stress axis. Brain Res. 887, 118–124. doi: 10.1016/S0006-8993(00)03000-6
de Lima, T. C. M., and Rae, G. A. (1991). Effects of cold-restraint and swim stress on convulsions induced by pentylenetetrazol and electroshock: influence of naloxone pretreatment. Pharmacol. Biochem. Behav. 40, 297–300. doi: 10.1016/0091-3057(91)90556-H
Dong, E., Matsumoto, K., Uzunova, V., Sugaya, I., Takahata, H., Nomura, H., et al. (2001). Brain 5alpha-dihydroprogesterone and allopregnanolone synthesis in a mouse model of protracted social isolation. Proc. Natl. Acad. Sci. U.S.A. 98, 2849–2854. doi: 10.1073/pnas.051628598
Drugan, R. C., McIntyre, T. D., Alpern, H. P., and Maier, S. F. (1985). Coping and seizure susceptibility: control over shock protects against bicuculline-induced seizures. Brain Res. 342, 9–17. doi: 10.1016/0006-8993(85)91347-2
Forsgren, L., and Nystrom, L. (1990). An incident case-referent study of epileptic seizures in adults. Epilepsy Res. 6, 66–81. doi: 10.1016/0920-1211(90)90010-S
Frucht, M. M., Quigg, M., Schwaner, C., and Fountain, N. B. (2000). Distribution of seizure precipitants among epilepsy syndromes. Epilepsia 41, 1534–1539. doi: 10.1111/j.1499-1654.2000.001534.x
Galimberti, C. A., Magri, F., Copello, F., Arbasino, C., Cravello, L., Casu, M., et al. (2005). Seizure frequency and cortisol and dehydroepiandrosterone sulfate (DHEAS) levels in women with epilepsy receiving antiepileptic drug treatment. Epilepsia 46, 517–523. doi: 10.1111/j.0013-9580.2005.59704.x
Gunn, B. G., Brown, A. R., Lambert, J. J., and Belelli, D. (2011). Neurosteroids and GABA(A) receptor interactions: a focus on stress. Front. Neurosci. 5:131. doi: 10.3389/fnins.2011.00131
Gunn, B. G., Cunningham, L., Cooper, M. A., Corteen, N. L., Seifi, M., Swinny, J. D., et al. (2013). Dysfunctional astrocytic and synaptic regulation of hypothalamic glutamatergic transmission in a mouse model of early-life adversity: relevance to neurosteroids and programming of the stress response. J. Neurosci. 33, 19534–19554. doi: 10.1523/JNEUROSCI.1337-13.2013
Haut, S. R., Hall, C. B., Masur, J., and Lipton, R. B. (2007). Seizure occurrence: precipitants and prediction. Neurology 69, 1905–1910. doi: 10.1212/01.wnl.0000278112.48285.84
Haut, S. R., Vouyiouklis, M., and Shinnar, S. (2003). Stress and epilepsy: a patient perception survey. Epilepsy Behav. 4, 511–514. doi: 10.1016/S1525-5050(03)00182-3
Hesdorffer, D. C., Hauser, W. A., Annegers, J. F., and Cascino, G. (2000). Major depression is a risk factor for seizures in older adults. Ann. Neurol. 47, 246–249. doi: 10.1002/1531-8249(200002)47:2<246::AID-ANA17>3.0.CO;2-E
Hesdorffer, D. C., Hauser, W. A., Olafsson, E., Ludvigsson, P., and Kjartansson, O. (2006). Depression and suicide attempt as risk factors for incident unprovoked seizures. Ann. Neurol. 59, 35–41. doi: 10.1002/ana.20685
Hewitt, S. A., Wamsteeker, J. I., Kurz, E. U., and Bains, J. S. (2009). Altered chloride homeostasis removes synaptic inhibitory constraint of the stress axis. Nat. Neurosci. 12, 438–443. doi: 10.1038/nn.2274
Hitiris, N., Mohanraj, R., Norrie, J., Sills, G. J., and Brodie, M. J. (2007). Predictors of pharmacoresistant epilepsy. Epilepsy Res. 75, 192–196. doi: 10.1016/j.eplepsyres.2007.06.003
Holm, M. M., Nieto-Gonzalez, J. L., Vardya, I., Henningsen, K., Jayatissa, M. N., Wiborg, O., et al. (2011). Hippocampal GABAergic dysfunction in a rat chronic mild stress model of depression. Hippocampus 21, 422–433. doi: 10.1002/hipo.20758
Hu, W., Zhang, M., Czeh, B., Flugge, G., and Zhang, W. (2010). Stress impairs GABAergic network function in the hippocampus by activating nongenomic glucocorticoid receptors and affecting the integrity of the parvalbumin-expressing neuronal network. Neuropsychopharmacology 35, 1693–1707. doi: 10.1038/npp.2010.31
Inoue, W., Baimoukhametova, D. V., Fuzesi, T., Cusulin, J. I., Koblinger, K., Whelan, P. J., et al. (2013). Noradrenaline is a stress-associated metaplastic signal at GABA synapses. Nat. Neurosci. 16, 605–612. doi: 10.1038/nn.3373
Inoue, W., and Bains, J. S. (2014). Beyond inhibition: GABA synapses tune the neuroendocrine stress axis. Bioessays 36, 561–569. doi: 10.1002/bies.201300178
Joels, M. (2009). Stress, the hippocampus, and epilepsy. Epilepsia 50, 586–597. doi: 10.1111/j.1528-1167.2008.01902.x
Jones, N. C., Lee, H. E., Yang, M., Rees, S. M., Morris, M. J., O'Brien, T. J., et al. (2013). Repeatedly stressed rats have enhanced vulnerability to amygdala kindling epileptogenesis. Psychoneuroendocrinology. 38, 263–270. doi: 10.1016/j.psyneuen.2012.06.005
Jones, N. C., and O'Brien, T. J. (2013). Stress, epilepsy, and psychiatric comorbidity: how can animal models inform the clinic? Epilepsy Behav. 26, 363–369. doi: 10.1016/j.yebeh.2012.09.002
Kanner, A. M., Byrne, R., Chicharro, A., Wuu, J., and Frey, M. (2009). A lifetime psychiatric history predicts a worse seizure outcome following temporal lobectomy. Neurology 72, 793–799. doi: 10.1212/01.wnl.0000343850.85763.9c
Koe, A. S., Jones, N. C., and Salzberg, M. R. (2009). Early life stress as an influence on limbic epilepsy: an hypothesis whose time has come? Front. Behav. Neurosci. 3:24. doi: 10.3389/neuro.08.024.2009
Lai, C. W., and Trimble, M. R. (1997). Stress and epilepsy. J. Epilepsy 10, 177–186. doi: 10.1016/S0896-6974(97)00017-0
Maggio, N., and Segal, M. (2012). Stress and corticosteroid modulation of seizures and synaptic inhibition in the hippocampus. Exp. Neurol. 234, 200–207. doi: 10.1016/j.expneurol.2011.12.035
Maguire, J., and Mody, I. (2007). Neurosteroid synthesis-mediated regulation of GABA(A) receptors: relevance to the ovarian cycle and stress. J. Neurosci. 27, 2155–2162. doi: 10.1523/JNEUROSCI.4945-06.2007
Maguire, J., and Mody, I. (2009). Steroid hormone fluctuations and GABA(A)R plasticity. Psychoneuroendocrinology. 34, S84–S90. doi: 10.1016/j.psyneuen.2009.06.019
Maguire, J., and Salpekar, J. A. (2013). Stress, seizures, and hypothalamic-pituitary-adrenal axis targets for the treatment of epilepsy. Epilepsy Behav. 26, 352–362. doi: 10.1016/j.yebeh.2012.09.040
Maroulakou, I. G., and Stylianopoulou, F. (1991). The effects of adrenalectomy and thermal stress on glutamic acid decarboxylase activity in different regions of the rat brain. Neurochem. Res. 16, 1265–1268. doi: 10.1007/BF00966656
Matsumoto, K., Nomura, H., Murakami, Y., Taki, K., Takahata, H., and Watanabe, H. (2003). Long-term social isolation enhances picrotoxin seizure susceptibility in mice: up-regulatory role of endogenous brain allopregnanolone in GABAergic systems. Pharmacol. Biochem. Behav. 75, 831–835. doi: 10.1016/S0091-3057(03)00169-2
Matsumoto, K., Puia, G., Dong, E., and Pinna, G. (2007). GABA(A) receptor neurotransmission dysfunction in a mouse model of social isolation-induced stress: possible insights into a non-serotonergic mechanism of action of SSRIs in mood and anxiety disorders. Stress 10, 3–12. doi: 10.1080/10253890701200997
McEwen, B. S. (1999). Stress and hippocampal plasticity. Annu. Rev. Neurosci. 22, 105–122. doi: 10.1146/annurev.neuro.22.1.105
McEwen, B. S. (2000). Effects of adverse experiences for brain structure and function. Biol. Psychiatry 48, 721–731. doi: 10.1016/S0006-3223(00)00964-1
Miller, L. G., Thompson, M. L., Greenblatt, D. J., Deutsch, S. I., Shader, R. I., and Paul, S. M. (1987). Rapid increase in brain benzodiazepine receptor binding following defeat stress in mice. Brain Res. 414, 395–400. doi: 10.1016/0006-8993(87)90023-0
Miller, S., and Maguire, J. (2014). Deficits in KCC2 and activation of the HPA axis lead to depression-like behavior following social defeat. Horm. Stud. 2, 1–10. doi: 10.7243/2052-8000-2-2
Minter, R. E. (1979). Can emotions precipitate seizures–a review of the question. J. Fam. Pract. 8, 55–59.
Mody, I., and Maguire, J. (2011). The reciprocal regulation of stress hormones and GABA(A) receptors. Front. Cell. Neurosci. 6:4. doi: 10.3389/fncel.2012.00004
Montpied, P., Weizman, A., Weizman, R., Kook, K. A., Morrow, A. L., and Paul, S. M. (1993). Repeated swim-stress reduces GABAA receptor +|subunit mRNAs in the mouse hippocampus. Mol. Brain Res. 18, 267–272. doi: 10.1016/0169-328X(93)90199-Y
Motohashi, N., Okamoto, Y., Osada, M., and Yamawaki, S. (1993). Acute swim stress increases benzodiazepine receptors, but not GABAA or GABAB receptors, in the rat cerebral cortex. Neurochem. Int. 23, 327–330. doi: 10.1016/0197-0186(93)90076-H
Nakken, K. O., Solaas, M. H., Kjeldsen, M. J., Friis, M. L., Pellock, J. M., and Corey, L. A. (2005). Which seizure-precipitating factors do patients with epilepsy most frequently report? Epilepsy Behav. 6, 85–89. doi: 10.1016/j.yebeh.2004.11.003
Neugebauer, R., Paik, M., Hauser, W. A., Nadel, E., Leppik, I., and Susser, M. (1994). Stressful life events and seizure frequency in patients with epilepsy. Epilepsia 35, 336–343. doi: 10.1111/j.1528-1157.1994.tb02441.x
Otero Losada, M. E. (1988). Changes in central GABAergic function following acute and repeated stress. Br. J. Pharmacol. 93, 483–490. doi: 10.1111/j.1476-5381.1988.tb10302.x
Payne, J. A., Rivera, C., Voipio, J., and Kaila, K. (2003). Cation-chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci. 26, 199–206. doi: 10.1016/S0166-2236(03)00068-7
Pericic, D., Jazvinscak, M., Svob, D., and Mirkovic, K. (2001). Swim stress alters the behavioural response of mice to GABA-related and some GABA-unrelated convulsants. Epilepsy Res. 43, 145–152. doi: 10.1016/S0920-1211(00)00194-7
Pericic, D., Svob, D., Jazvinscak, M., and Mirkovic, K. (2000). Anticonvulsive effect of swim stress in mice. Pharmacol. Biochem. Behav. 66, 879–886. doi: 10.1016/S0091-3057(00)00267-7
Petrovski, S., Szoeke, C. E. I., Jones, N. C., Salzberg, M. R., Sheffield, L. J., Huggins, R. M., et al. (2010). Neuropsychiatric symptomatology predicts seizure recurrence in newly treated patients. Neurology 75, 1015–1021. doi: 10.1212/WNL.0b013e3181f25b16
Pinna, G., Dong, E., Matsumoto, K., Costa, E., and Guidotti, A. (2003). In socially isolated mice, the reversal of brain allopregnanolone down-regulation mediates the anti-aggressive action of fluoxetine. Proc. Natl. Acad. Sci. U.S.A. 100, 2035–2040. doi: 10.1073/pnas.0337642100
Purdy, R. H., Morrow, A. L., Moore, P. H., and Paul, S. M. (1991). Stress-induced elevations of gamma-aminobutyric-acid type-a receptor-active steroids in the rat-brain. Proc. Natl. Acad. Sci. U.S.A. 88, 4553–4557. doi: 10.1073/pnas.88.10.4553
Reddy, D. S. (2003). Is there a physiological role for the neurosteroid THDOC in stress-sensitive conditions? Trends Pharmacol. Sci. 24, 103–106. doi: 10.1016/S0165-6147(03)00023-3
Reddy, D. S., and Rogawski, M. A. (2002). Stress-induced deoxycorticosterone-derived neurosteroids modulate GABA(A) receptor function and seizure susceptibility. J. Neurosci. 22, 3795–3805.
Rivera, C., Voipio, J., and Kaila, K. (2005). Two developmental switches in GABAergic signalling: the K+-Cl- cotransporter KCC2 and carbonic anhydrase CAVII. J. Physiol. 562, 27–36. doi: 10.1113/jphysiol.2004.077495
Rivera, C., Voipio, J., Payne, J. A., Ruusuvuori, E., Lahtinen, H., Lamsa, K., et al. (1999). The K+/Cl- co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397, 251–255. doi: 10.1038/16697
Rogawski, M. A., and Reddy, D. S. (2004). Neurosteroids: Endogenous Modulators of Seizure Susceptibility. Epilepsy: Scientific Foundations of Clinical Practice (New York, NY: Marcel Dekker), 319–355.
Sarkar, J., Wakefield, S., Mackenzie, G., Moss, S. J., and Maguire, J. (2011). Neurosteroidogenesis is required for the physiological response to stress: role of neurosteroid-sensitive GABAA receptors. J. Neurosci. 31, 18198–18210. doi: 10.1523/JNEUROSCI.2560-11.2011
Serra, M., Mostallino, M. C., Talani, G., Pisu, M. G., Carta, M., Mura, M. L., et al. (2006). Social isolation-induced increase in alpha and delta subunit gene expression is associated with a greater efficacy of ethanol on steroidogenesis and GABA receptor function. J. Neurochem. 98, 122–133. doi: 10.1111/j.1471-4159.2006.03850.x
Serra, M., Pisu, M. G., Littera, M., Papi, G., Sanna, E., Tuveri, F., et al. (2000). Social isolation-induced decreases in both the abundance of neuroactive steroids and GABA(A) receptor function in rat brain. J. Neurochem. 75, 732–740. doi: 10.1046/j.1471-4159.2000.0750732.x
Serra, M., Pisu, M. G., Mostallino, M. C., Sanna, E., and Biggio, G. (2008). Changes in neuroactive steroid content during social isolation stress modulate GABAA receptor plasticity and function. Brain Res. Rev. 57, 520–530. doi: 10.1016/j.brainresrev.2007.06.029
Shin, S. Y., Han, T. H., Lee, S. Y., Han, S. K., Park, J. B., Erdelyi, F., et al. (2011). Direct Corticosteroid modulation of GABAergic neurons in the anterior hypothalamic area of GAD65-eGFP mice. Korean J. Physiol. Pharmacol. 15, 163–169. doi: 10.4196/kjpp.2011.15.3.163
Skerritt, J. H., Trisdikoon, P., and Johnston, G. A. (1981). Increased GABA binding in mouse brain following acute swim stress. Brain Res. 215, 398–403. doi: 10.1016/0006-8993(81)90524-2
Skilbeck, K. J., Hinton, T., and Johnston, G. A. R. (2008). Sex-differences and stress: Effects on regional high and low affinity [3H]GABA binding. Neurochem. Int. 52, 1212–1219. doi: 10.1016/j.neuint.2008.01.001
Skilbeck, K. J., Johnston, G. A., and Hinton, T. (2010). Stress and GABA receptors. J. Neurochem. 112, 1115–1130. doi: 10.1111/j.1471-4159.2009.06539.x
Soubrie, P., Thiebot, M. H., Jobert, A., Montastruc, J. L., Hery, F., and Hamon, M. (1980). Decreased convulsant potency of picrotoxin and pentetrazol and enhanced [3H]flunitrazepam cortical binding following stressful manipulations in rats. Brain Res. 189, 505–517. doi: 10.1016/0006-8993(80)90109-2
Sperling, M. R., Schilling, C. A., Glosser, D., Tracy, J. I., and Asadi-Pooya, A. A. (2008). Self-perception of seizure precipitants and their relation to anxiety level, depression, and health locus of control in epilepsy. Seizure 17, 302–307. doi: 10.1016/j.seizure.2007.09.003
Stell, B. M., Brickley, S. G., Tang, C. Y., Farrant, M., and Mody, I. (2003). Neuroactive steroids reduce neuronal excitability by selectively enhancing tonic inhibition mediated by delta subunit-containing GABA(A) receptors. Proc. Natl. Acad. Sci. U.S.A. 100, 14439–14444. doi: 10.1073/pnas.2435457100
Varela, J. A., Wang, J., Christianson, J. P., Maier, S. F., and Cooper, D. C. (2012). Control over stress, but not stress per se increases prefrontal cortical pyramidal neuron excitability. J. Neurosci. 32, 12848–12853. doi: 10.1523/JNEUROSCI.2669-12.2012
Verkuyl, J. M., Hemby, S. E., and Joels, M. (2004). Chronic stress attenuates GABAergic inhibition and alters gene expression of parvocellular neurons in rat hypothalamus. Eur. J. Neurosci. 20, 1665–1673. doi: 10.1111/j.1460-9568.2004.03568.x
Verkuyl, J. M., and Joels, M. (2003). Effect of adrenalectomy on miniature inhibitory postsynaptic currents in the paraventricular nucleus of the hypothalamus. J. Neurophysiol. 89, 237–245. doi: 10.1152/jn.00401.2002
Verkuyl, J. M., Karst, H., and Joels, M. (2005). GABAergic transmission in the rat paraventricular nucleus of the hypothalamus is suppressed by corticosterone and stress. Eur. J. Neurosci. 21, 113–121. doi: 10.1111/j.1460-9568.2004.03846.x
Wamsteeker Cusulin, J. I., Fuzesi, T., Inoue, W., and Bains, J. S. (2013). Glucocorticoid feedback uncovers retrograde opioid signaling at hypothalamic synapses. Nat. Neurosci. 16, 596–604. doi: 10.1038/nn.3374
Wohlfarth, K. M., Bianchi, M. T., and Macdonald, R. L. (2002). Enhanced neurosteroid potentiation of ternary GABA(A) receptors containing the delta subunit. J. Neurosci. 22, 1541–1549.
Yoneda, Y., Kanmori, K., Ida, S., and Kuriyama, K. (1983). Stress-induced alterations in metabolism of gamma-aminobutyric acid in rat brain. J. Neurochem. 40, 350–356. doi: 10.1111/j.1471-4159.1983.tb11289.x
Keywords: GABA, neurosteroids, stress, KCC2, GABAA receptors, GABAAR, THDOC
Citation: Maguire J (2014) Stress-induced plasticity of GABAergic inhibition. Front. Cell. Neurosci. 8:157. doi: 10.3389/fncel.2014.00157
Received: 30 December 2013; Paper pending published: 21 January 2014;
Accepted: 19 May 2014; Published online: 06 June 2014.
Edited by:
Andrea Barberis, Fondazione Istituto Italiano di Tecnologia, ItalyReviewed by:
Yehezkel Ben-Ari, Institut National de la Santé et de la Recherche Médicale, FranceAmiel Rosenkranz, RFUMS - Chicago Medical School, USA
Copyright © 2014 Maguire. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jamie Maguire, Department of Neuroscience, Tufts University School of Medicine, 136 Harrison Ave., SC205, Boston, MA 02111, USA e-mail:SmFtaWUuTWFndWlyZUB0dWZ0cy5lZHU=