 Carolina Koutras
Carolina Koutras Janice E. A. Braun
Janice E. A. Braun- Department of Physiology and Pharmacology, Hotchkiss Brain Institute, Cumming School of Medicine, University of Calgary, Calgary, AB, Canada
Despite a century of intensive investigation the effective treatment of protein aggregation diseases remains elusive. Ordinarily, molecular chaperones ensure that proteins maintain their functional conformation. The appearance of misfolded proteins that aggregate implies the collapse of the cellular chaperone quality control network. That said, the cellular chaperone network is extensive and functional information regarding the detailed action of specific chaperones is not yet available. J proteins (DnaJ/Hsp40) are a family of chaperone cofactors that harness Hsc70 (heat shock cognate protein of 70 kDa) for diverse conformational cellular tasks and, as such, represent novel clinically relevant targets for diseases resulting from the disruption of proteostasis. Here we review incisive reports identifying mutations in individual J protein chaperones and the proteostasis collapse that ensues.
J Proteins: Personal Travel Guides
In neurons, there are significant demands on cellular folding events. Dynamic protein complexes are central to synaptic transmission, a process that occurs with speed, precision and plasticity. Most proteins can exist in more than one conformation and many proteins must change conformation and activity regularly. Rigorous quality control mechanisms operate at the synapse to provide defense against the detrimental effects of functionally impaired proteins and protein complexes. The balance between protecting protein functional integrity and preventing accumulation of misfolded proteins is accomplished by a network of chaperone families including: J proteins (DnaJ/Hsp40), HspA (Hsp70), HspB (small Hsp) HspC (Hsp90), HspD/E (Hsp60/Hsp10), HspH (Hsp110), CCT (TRiC) and numerous regulatory factors (Figure 1). Operating like a switch, J proteins control Hsc70 (70–kDa heat shock cognate protein) by directing and activating Hsc70’s ATPase activity for conformational and refolding work (Kampinga and Craig, 2010; Kakkar et al., 2012). Sequence analysis has highlighted the diversity of the J protein family but has not provided much clarity into the functionality of specific J proteins. Each J protein has a 70 amino acid signature region comprised of four helices, called a J domain. Outside of the J domain, J proteins are structurally divergent, likely the basis for their distinct functional properties. Originally named Hsp40 or DnaJ after the founding members of the family (Georgopoulos et al., 1980; Ohtsuka et al., 1990), we now know that there are 49 J proteins in humans that range in molecular weight from 18–520 kDa and that while some J proteins are induced by heat most members of the J protein family are constitutively expressed (Zhao et al., 2008; Kakkar et al., 2012). The recognition that Hsc70 serves as a central hub and J proteins are the switches for diverse proteostasis events has fueled investigations into understanding the specific role of J proteins.
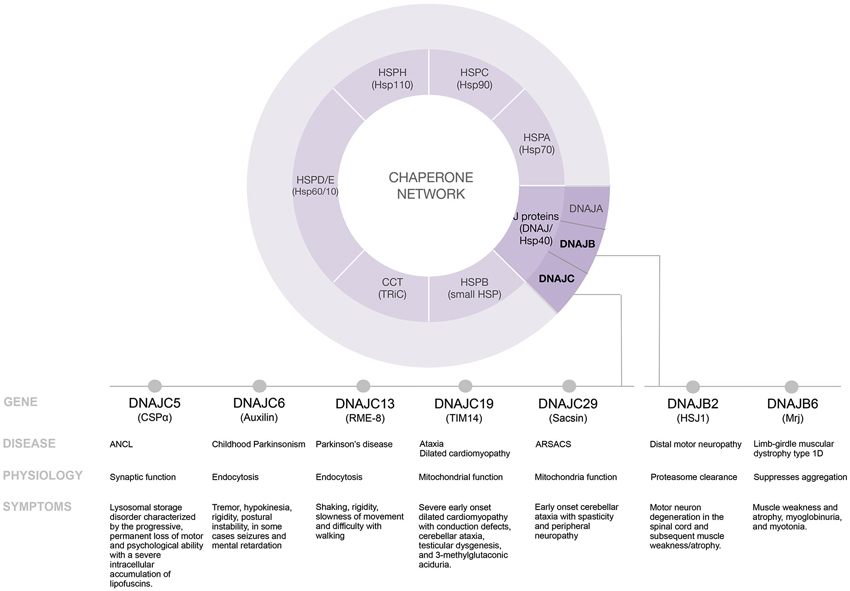
Figure 1. Disease modifying pathways of select J proteins.
Age-Associated Neurodegenerative Disorders
How do folding processes go awry in age? To counteract aging and disease, neurons must routinely regulate protein machinery within tight limits. The quality control machinery constantly protects against the misfolding and aggregation of proteins that is inherent to continuous protein synthesis, generation of folding intermediates and molecular crowding of neural proteins. Neurons which don’t undergo cell division are vulnerable to infectious misfolded aggregated proteins that spread (e.g., prion diseases), mutations that produce toxic proteins characteristic of late onset of neurodegenerative disorders such as Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and Amyotrophic Lateral Sclerosis (ALS) as well as mutations that generate ineffective chaperones. In fact, at the cellular level, pathology may start long before the onset of clinical symptoms associated with failure of the chaperone machinery to meet the demands of misfolded proteins. While it is widely accepted that J proteins maintain and restore protein balance several questions remain regarding the nature of changes to the chaperone network in age related neurodegenerative diseases (Soti and Csermely, 2002; Muchowski and Wacker, 2005; Kim et al., 2013; Labbadia and Morimoto, 2014).
Defining a Trajectory to Understand the Function of Specific J Proteins
The chaperone network is elaborate and elucidating the function of individual J proteins is challenging. Chaperonopathies are a collection of diseases caused by genetically inherited mutations in chaperones. Our growing appreciation that diverse diseases in humans result from select J protein mutations provides new roads to explore the mechanisms of action of individual J proteins. To date, mutations in seven distinct J proteins; DNAJB2, DNAJB6, DNAJC5, DNAJC6, DNAJC13, DNAJC19 and DNAJC29 have been linked to distinct diseases in humans (Table 1). Here we review the information that these recently identified mutations provide regarding individual J protein function and the collapse of proteostasis. Understanding the mechanisms of specific J proteins is key to understanding the proteostasis machinery and will be critically important for J protein-based drug development.
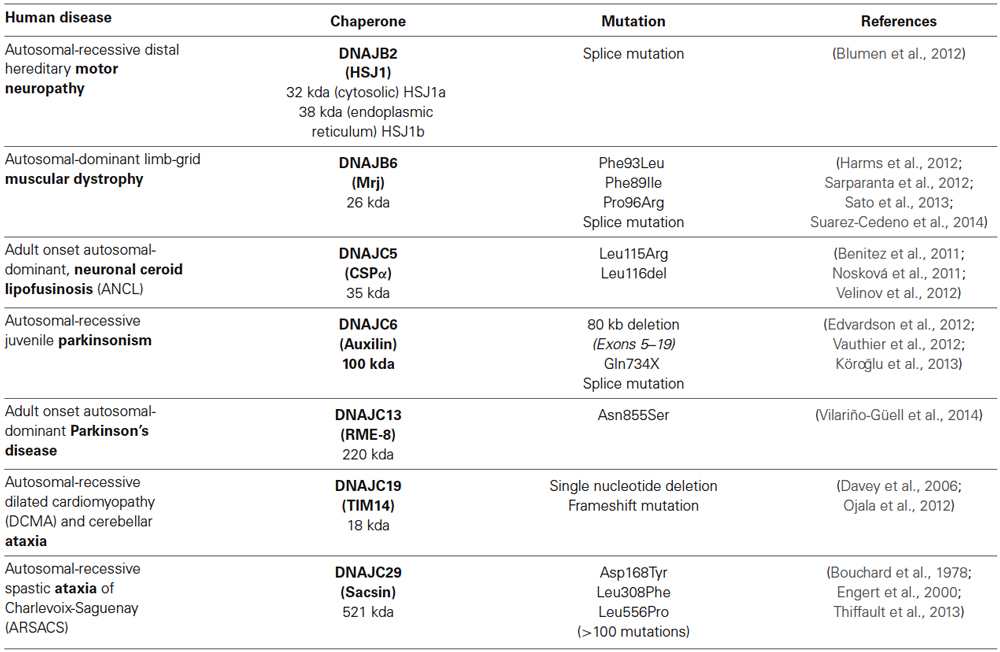
Table 1. J proteins mutations and resulting proteostasis consequences in human diseases.
DNAJB2 (HSJ1)
Mutation of DNAJB2 (HSJ1), a 32/38 kDa J protein, causes recessive distal hereditary motor neuropathy (Blumen et al., 2012). Originally identified in Morocco, individuals show a distal motor weakness, hypotonia, atrophy and paralysis of the lower limbs in young adulthood (age 20) due to progressive degeneration of motor neurons in the spinal cord.
HSJ1 was first identified via expression cloning with antiserum prepared against Alzheimer’s disease brain extracts enriched in helical filaments (Cheetham et al., 1992). It is preferentially expressed in neurons where it is proposed to promote proteasome degradation of proteins tagged with polyubiquitin (Cheetham et al., 1992; Chapple and Cheetham, 2003; Westhoff et al., 2005; Howarth et al., 2007). Alternative splicing produces two isoforms. HSJ1a is a cytosolic 32 kDa form and HSJ1b is a 38 kDa form that is anchored to the cytosolic face of the endoplasmic reticulum via C terminal geranylgeranylation. In addition to its J domain, HSJ1 contains two unique ubiquitin interaction domains that bind ubiquitin to prevent aggregation and direct ubiquitinated/misfolded proteins to the proteasome. Which misfolded proteins are targeted by HSJ1? In cellular models HSJ1a suppresses the aggregation of polyglutamine expanded proteins and mutant superoxide dismutatse1 (SOD1; Chapple et al., 2004; Westhoff et al., 2005; Gao et al., 2011; Blumen et al., 2012). Transgene upregulation of HSJ1a reduces brain huntingtin aggregation in the mouse R6/2 model of Huntington’s disease (Labbadia et al., 2012) and mutant SOD1 aggregation in the mouse SOD1G93A model of ALS (Novoselov et al., 2013). Key questions arise from these findings. While transgenic expression of HSJ1 alone reduces aggregation, it does not fully reverse disease progression and correct life span. Whether reinforcing other J proteins along with HSJ1 will completely circumvent the cascade of degeneration remains an open question. Further, while the splice mutation that give rise to distal motor neuropathy in humans decreases the availability of both HSJ1a and HSJ1b which are broadly expressed in neurons, the mutation causes selective loss of motor neuron function. Obviously, whether HSJ1 has a motor neuron specific or a general neuronal function will be the focus of future investigation.
DNAJB6 (Mrj)
Mutations in DnaJB6 (Mrj; mammalian relative of DnaJ), a 26/36 kDa J protein cause autosomal dominant limb-girdle muscular dystrophy type 1 D (Harms et al., 2012; Sarparanta et al., 2012; Sato et al., 2013; Suarez-Cedeno et al., 2014). Four mutations F96L, F96I, F89I and F93L results in adult or child onset (age 14–68 yrs) limb-girdle muscular dystrophy type 1D which is clinically characterized by elevated serum creatine kinase levels, progressive muscle weakness mainly in the legs. Limb-girdle muscular dystrophies are a heterogeneous group of inherited disorders caused by a number of dominant or recessive mutations that cause myofibrillar myopathy and the DNAJB6 mutations are specifically distinguished by characteristic protein aggregates and autophagic vacuoles (Mitsuhashi and Kang, 2012).
Mrj is a ubiquitous J protein with Hsc70-dependent and -independent activities that is most highly expressed in brain (Chuang et al., 2002; Hageman et al., 2010). Further, expression of Mrj is increased in astrocytes from Parkinson’s disease patients, where it is found to be a component of Lewy bodies (Durrenberger et al., 2009). Alternative splicing produces two isoforms (Hanai and Mashima, 2003). DnaJB6a is 36 kDa and localizes to the nucleus, whereas DnaJB6b is 26 kDa and cytosolic but translocates to the nucleus in response to cellular stress (Andrews et al., 2012). What are Mrj’s targets? Mrj suppresses aggregation and toxicity of several aggregation-prone proteins including: huntingtin, α-synuclein and parkin (Chuang et al., 2002; Durrenberger et al., 2009; Hageman et al., 2010; Kampinga and Craig, 2010; Rose et al., 2011). Possible “client proteins” in addition to disease-causing proteins include: keratin-intermediate filaments (Watson et al., 2007), histone deacetylase (HDAC; Hageman et al., 2010) and the transcription factor, NFATc3 (Dai et al., 2005). Mrj KO is embryonic lethal in mice due to a failure of chorioallantoic attachment in placental development (Hunter et al., 1999). All human mutations causing myopathy are found in a glycine-phenylaline linker region that follows the amino terminal J domain and result in a reduction in the inhibition of protein aggregation (Sarparanta et al., 2012). Furthermore, low expression of Mrj has been linked to breast cancer (Andrews et al., 2012). It remains an open question why mutations in neuronal-enriched Mrj cause myopathy and low levels are linked to cancer rather than neurodegeneration. Further investigation is required to understand these somewhat disparate pieces and develop a clear picture of the neuroprotective actions of Mrj. It is noteworthy that in the wobbler mouse, an ALS model of progressive motor neuron degeneration, DnaJB3, which shares 90% identity with Mrj(DnaJB6), shows reduced expression in the spinal cord prior to loss of motor neurons (Boillée et al., 2002). Overlap in Mrj and DnaJB3 expression as well as possible overlap in DnaJB6/DnaJB3 client proteins remains to be determined.
DNAJC5 (CSPα)
The DnaJC5 gene encodes CSPα, a 35 kDa secretory vesicle protein. Mutations in DnaJC5, cause adult onset, neuronal ceroid lipofusinosis (ANCL), a neurodegenerative disorder characterized by lysosomal accumulation of autofluorescent oxidized lipid and protein waste, called lipofuscin (Benitez et al., 2011; Nosková et al., 2011; Velinov et al., 2012). Deletion of leucine 116 (L116Δ), or mutation of residue 115 from leucine to arginine (L115R) results in ANCL which is clinically characterized by anxiety, depression, speech difficulties, ataxia, myoclonus, involuntary movements, progressive seizures and dementia. Neuronal ceroid lipofuscinoses (NCLs) are a heterogeneous group of inherited lysosomal storage disorders caused by mutations that lead to accumulation of lipofuscin and loss of neurons (Anderson et al., 2013). ANCL is unique among NCLs in that it is adult onset and the only NCL mutations inherited in an autosomal dominant manner.
What is the physiological function of CSPα? CSPα protects against synapse loss, however the precise molecular mechanism underlying neuroprotection is not yet known. In addition to its N terminal J domain, CSPα bears a hydrophobic region followed by the distinctive cysteine string region after which the protein is named. Most of the cysteines are palmitoylated, which anchors CSPα to the synaptic vesicle membrane. CSPα is located on synaptic vesicles (Mastrogiacomo et al., 1994), exocrine vesicles (Braun and Scheller, 1995), and endocrine vesicles (Chamberlain et al., 1996; Brown et al., 1998). At birth, CSPα KO mice appear normal and around postnatal day 20 develop progressive motor deficits and CNS degeneration, followed by early lethality (Fernández-Chacón et al., 2004). The synapse loss in CSPα null mice is activity-dependent and synapses that fire frequently are lost first (Schmitz et al., 2006; Garcia-Junco-Clemente et al., 2010). In Drosophila, CSPα KOs that survive to adulthood are characterized by uncoordinated movements, shaking, and temperature-sensitive paralysis (Zinsmaier et al., 1994). What are CSPα’s targets? Presynaptic targets that demonstrate changes in protein levels early-on in the cascade of neurodegeneration include SNAP25 (Sharma et al., 2011, 2012), dynamin1 (Zhang et al., 2012) and BK channels (Kyle et al., 2013; Ahrendt et al., 2014). Other promising targets include; voltage dependent Ca2+ channels, heterotrimeric G proteins, syntaxin and synaptotagmin (Donnelier and Braun, 2014). Which client proteins are critical for triggering the cascade of events leading to degeneration and which changes are downstream of the primary event remains an open question. In addition to CSPα, CSPβ and CSPγ have been identified in the mammalian genomes (Fernández-Chacón et al., 2004). While brain expresses only CSPα, all CSP isoforms are expressed in testis, whereas the cochlea expresses CSPα and CSPβ (Chandra et al., 2005). In CSPα KO mice, the redundancy of CSPβ in the ribbon synapses protects against neurodegeneration in cochlea, however other J proteins do not protect against the absence of CSPα. Surprisingly, in mice, transgenic expression of α-synuclein abolishes neurodegeneration caused by deletion of CSPα but its protective mechanism has not yet been fully elucidated (Chandra et al., 2005). The L116Δ and L115R mutations that result in ANCL cause CSPα to mislocalize implying both a partial loss of function (at the synaptic vesicle) and a partial gain of function (at the intracellular site of mislocalization). Although progress has been made, mislocalization is not sufficient to explain lysosome dysfunction. There are still gaps in our understanding of the molecular mechanisms underlying CSPα’s neuroprotection with regards to synaptic vesicle release and recycling (Rozas et al., 2012; Sharma et al., 2012).
DNAJC6 (Auxilin)
Mutations in DnaJC6 (auxilin), a 100 kDa nerve-specific J protein cause recessive juvenile parkinsonism characterized by early onset (age 3–18) tremor at rest, bradykinesia, rigidity and postural instability (Edvardson et al., 2012; Vauthier et al., 2012; Köroğlu et al., 2013). The severity and age of onset depends on the extent of the deletion or reduction in expression of auxilin and in some cases includes mental retardation, epilepsy and lack of responsiveness to L-Dopa.
Auxilin is one of the best studied J proteins and its role in uncoating of clathrin from clathrin-coated vesicles is well established (Ungewickell et al., 1995; Eisenberg and Greene, 2007). Neurotransmission requires a rapid continuous recycling of synaptic vesicles. Following fusion, clathrin assembles into a lattice on the presynaptic plasma membrane and deforms plasma membrane into a clathrin coated vesicle that is severed from the membrane by dynamin. After internalization auxilin binds clathrin and Hsc70 and in a J domain-dependent process uncoats clathrin to replenish the pool of synaptic vesicles (Morgan et al., 2001). Unpolymerized clathrin remains in association with Hsc70 until released by nucleotide exchange factors for additional rounds of endocytosis (Morgan et al., 2013). The clathrin binding motif and J domain are located at the C terminus of auxilin and therefore C terminal truncations are expected to render auxilin incapable of uncoating clathrin thereby impairing synaptic transmission (Morgan et al., 2001). Extending on these studies, impaired dopamine receptor recycling may explain the lack of L-Dopa responsiveness observed in some auxilin mutations. Auxilin KO mice have a high rate of postnatal mortality and surviving pups have a low body weight and show recycling and endocytosis defects (Yim et al., 2010). There is a high homology between neural-specific auxilin (DnaJC6) and ubiquitous GAK (cyclin G associated kinase; DnaJC26) with primary differences in the N terminal domain and GAK is thought to partially compensate for auxilin deletion. Interestingly, genome wide association studies have linked DnaJC26 (GAK) to Parkinson’s disease susceptibility (Li et al., 2012).
DNAJC13 (RME-8 Receptor Mediated Endocytosis)
Mutation of DnaJC13 (RME-8 receptor mediated endocytosis 8) a 220 kDa J protein (Asn855Ser) cause adult onset autosomal-dominant Parkinson disease (Vilariño-Güell et al., 2014). Originally discovered in Saskatchewan, onset of disease is between 59 and 85 years and is characterized by slowly progressive tremor, rigidity, bradykinesia and roughly half of the Lewy body inclusions are immunoreactive for DnaJC13. Interestingly, DanJC13 mutation (Ala2057Ser) has also been linked to Tourette syndrome/chronic tic phenotype in patients of European ancestry (Sundaram et al., 2011).
RME-8 was first identified in a screen for endocytotic defects in C. elegans (Zhang et al., 2001) and was subsequently shown to have a role in endocytosis in Drosophila (Chang et al., 2004) and endosomal function in mammals (Girard et al., 2005). Surprisingly, Rme-8 which bears a central J domain, is widely expressed but not especially abundant in brain (Girard et al., 2005). Rme-8 interacts with retromer (Popoff et al., 2009) and WASH complexes (Freeman et al., 2014) and loss of RME-8 disrupts cation independent-mannose-6-phosphate receptor and epidermal growth factor receptor trafficking (Girard and McPherson, 2008; Popoff et al., 2009). There are numerous gaps in our understanding of how disruption of normal Rme-8 function in endosomal recyclying causes Parkinson disease.
DNAJC19 (TIM14 Mitochondrial Import Inner Membrane Translocase Subunit 14)
Mutation of DnaJC19 (Tim14 or Pam 18), an 18 Kda J protein, cause early-childhood-onset (before 3 years) recessive dilated cardiomyopathy and cerebellar ataxia (Davey et al., 2006; Sparkes et al., 2007; Ojala et al., 2012). Originally identified in Alberta Dariusleut Hutterites, DnaJC19 mutations are clinically characterized by raised levels of 3-methylglutaconic acid, a readout of mitochondrial distress, dilated cardiomyopathy, prolongation of the QT interval in the electrocardiogram and cerebellar ataxia (Davey et al., 2006).
DnaJC19 is a transmembrane component of the TIM23 mitochondria import machinery that delivers nuclear encoded proteins to the mitochondrial matrix in an ATP dependent manner (Sinha et al., 2014). DnaJC19 activates mortalin, the mtHsp70 ATPase, and activation is counteracted by MAGMAS, a protein that contains a C-terminal J-like domain that lacks the HPD motif required to recruit and activate the Hsc70 ATPase activity. There are multiple mitochondrial J proteins and our understanding of the differences in function and specificity remains limited. Clearly, DNAJC19 and another mitochondrial J protein, DNAJC15, are not redundant as DnaJC15 does not rescue DnaJC19 mutations that lead to dilated cardiomyopathy and cerebellar ataxia. This underscores the importance of identifying the diverse disease modifying pathways of mitochondrial J proteins.
DNAJC29 (Sacsin)
Mutations in DnaJC29 (sacsin), a 521 kDa J protein, cause early-childhood-onset (age 1–2 years), recessive spastic ataxia of Charlevoix-Saguenay (ARSACS; Bouchard et al., 1978). Originally discovered in Quebec in 1978 (Bouchard et al., 1978), we now know that >100 different mutations in DnaJC29 mutation exist worldwide (Engert et al., 2000; Thiffault et al., 2013). ARSACS is clinically characterized by unsteady gait, spasticity, ataxia, muscle atrophy, due to progressive cerebellar atrophy and peripheral neuropathy. Quebec patients never walk properly and are wheelchair-bound on average by 41 years with life expectancy approximately 51 years. Outside of Quebec, mild ARSACS with onset delayed until later childhood and early 20’s or severe ARSACS with mental retardation is also found (Baets et al., 2010; Thiffault et al., 2013). Deposits of lysosome derived oxidized lipid and protein waste, called lipofuscin, are found in cerebellar cortical neurons or skin (Stevens et al., 2013), somewhat reminiscent of DnaJC5 mutations that cause ANCL.
Sascin is the largest J protein, bearing C termini J domain, an N-terminal ubiquitin-like domain and a higher eukaryotic and prokaryotic nucleotide binding domain (HEPN; Parfitt et al., 2009; Kozlov et al., 2011). Sacsin is predominantly cytosolic with a mitochondria component, it interacts with dynamin-related protein 1, a GTPase required for mitochondrial fission (Kozlov et al., 2011; Girard et al., 2012; Thiffault et al., 2013). Knockdown of sacsin in SH-SY5Y cells results in an overly interconnected and functionally impaired mitochondrial network with mitochondria accumulating in the soma and proximal dendrites (Girard et al., 2012). Sacsin KO mice display age-dependent neurodegeneration of cerebellar Purkinje cells most likely due to defects in mitochondrial proteostasis (Girard et al., 2012). Much work will be needed to untangle the complexities of sacsin mutations and ensuing neurodegeneration.
Future Prospects
Independent threads have recently begun to provide insight into how select J proteins in the protein quality machinery monitor and adjust proteotoxic imbalances in order to maintain neural function. Many knowledge gaps in our understanding of the functionality of specific J proteins exist. It remains to be seen whether identification of further human mutations will yield insights into the biological roles of J proteins. Ultimately, systematic analysis of the mechanisms by which J proteins facilitate proteostasis will enable us to develop novel therapeutic agents and re-purpose drugs currently used for other indications.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ahrendt, E., Kyle, B., Braun, A. P., and Braun, J. E. (2014). Cysteine string protein limits expression of the large conductance, calcium-activated K(+) (BK) channel. PLoS One 9:e86586. doi: 10.1371/journal.pone.0086586
Anderson, G. W., Goebel, H. H., and Simonati, A. (2013). Human pathology in NCL. Biochim. Biophys. Acta 1832, 1807–1826. doi: 10.1016/j.bbadis.2012.11.014
Andrews, J. F., Sykora, L. J., Letostak, T. B., Menezes, M. E., Mitra, A., Barik, S., et al. (2012). Cellular stress stimulates nuclear localization signal (NLS) independent nuclear transport of MRJ. Exp. Cell Res. 318, 1086–1093. doi: 10.1016/j.yexcr.2012.03.024
Baets, J., Deconinck, T., Smets, K., Goossens, D., Van den Bergh, P., Dahan, K., et al. (2010). Mutations in SACS cause atypical and late-onset forms of ARSACS. Neurology 75, 1181–1188. doi: 10.1212/wnl.0b013e3181f4d86c
Benitez, B. A., Alvarado, D., Cai, Y., Mayo, K., Chakraverty, S., Norton, J., et al. (2011). Exome-sequencing confirms DNAJC5 mutations as cause of adult neuronal ceroid-lipofuscinosis. PLoS One 6:e26741. doi: 10.1371/journal.pone.0026741
Blumen, S. C., Astord, S., Robin, V., Vignaud, L., Toumi, N., Cieslik, A., et al. (2012). A rare recessive distal hereditary motor neuropathy with HSJ1 chaperone mutation. Ann. Neurol. 71, 509–519. doi: 10.1002/ana.22684
Boillée, S., Berruti, G., Meccariello, R., Grannec, G., Razan, F., Pierantoni, R., et al. (2002). Early defect in the expression of mouse sperm DNAJ 1, a member of the DNAJ/heat shock protein 40 chaperone protein family, in the spinal cord of the wobbler mouse, a murine model of motoneuronal degeneration. Neuroscience 113, 825–835. doi: 10.1016/s0306-4522(02)00235-x
Bouchard, J. P., Barbeau, A., Bouchard, R., and Bouchard, R. W. (1978). Autosomal recessive spastic ataxia of Charlevoix-Saguenay. Can. J. Neurol. Sci. 5, 61–69.
Braun, J. E., and Scheller, R. H. (1995). Cysteine string protein, a DnaJ family member, is present on diverse secretory vesicles. Neuropharmacology 34, 1361–1369. doi: 10.1016/0028-3908(95)00114-l
Brown, H., Larsson, O., Bränström, R., Yang, S., Leibiger, B., Leibiger, I., et al. (1998). Cysteine string protein (CSP) is an insulin secretory granule-associated protein regulating beta-cell exocytosis. EMBO J. 17, 5048–5058. doi: 10.1093/emboj/17.17.5048
Chamberlain, L. H., Henry, J., and Burgoyne, R. D. (1996). Cysteine string proteins are associated with chromaffin granules. J. Biol. Chem. 271, 19514–19517. doi: 10.1074/jbc.271.32.19514
Chandra, S., Gallardo, G., Fernández-Chacón, R., Schlüter, O. M., and Südhof, T. C. (2005). Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 123, 383–396. doi: 10.1016/j.cell.2005.09.028
Chang, H. C., Hull, M., and Mellman, I. (2004). The J-domain protein Rme-8 interacts with Hsc70 to control clathrin-dependent endocytosis in Drosophila. J. Cell. Biol. 164, 1055–1064. doi: 10.1083/jcb.200311084
Chapple, J. P., and Cheetham, M. E. (2003). The chaperone environment at the cytoplasmic face of the endoplasmic reticulum can modulate rhodopsin processing and inclusion formation. J. Biol. Chem. 278, 19087–19094. doi: 10.1074/jbc.m212349200
Chapple, J. P., van der, S. J., Poopalasundaram, S., and Cheetham, M. E. (2004). Neuronal DnaJ proteins HSJ1a and HSJ1b: a role in linking the Hsp70 chaperone machine to the ubiquitin-proteasome system? Biochem. Soc. Trans. 32, 640–642. doi: 10.1042/bst0320640
Cheetham, M. E., Brion, J. P., and Anderton, B. H. (1992). Human homologues of the bacterial heat-shock protein DnaJ are preferentially expressed in neurons. Biochem. J. 284, 469–476.
Chuang, J. Z., Zhou, H., Zhu, M., Li, S. H., Li, X. J., and Sung, C. H. (2002). Characterization of a brain-enriched chaperone, MRJ, that inhibits huntingtin aggregation and toxicity independently. J. Biol. Chem. 277, 19831–19838. doi: 10.1074/jbc.m109613200
Dai, Y. S., Xu, J., and Molkentin, J. D. (2005). The DnaJ-related factor Mrj interacts with nuclear factor of activated T cells c3 and mediates transcriptional repression through class II histone deacetylase recruitment. Mol. Cell. Biol. 25, 9936–9948. doi: 10.1128/mcb.25.22.9936-9948.2005
Davey, K. M., Parboosingh, J. S., McLeod, D. R., Chan, A., Casey, R., Ferreira, P., et al. (2006). Mutation of DNAJC19, a human homologue of yeast inner mitochondrial membrane co-chaperones, causes DCMA syndrome, a novel autosomal recessive Barth syndrome-like condition. J. Med. Genet. 43, 385–393. doi: 10.1136/jmg.2005.036657
Donnelier, J., and Braun, J. E. (2014). CSPalpha-chaperoning presynaptic proteins. Front. Cell Neurosci. 8:116. doi: 10.3389/fncel.2014.00116
Durrenberger, P. F., Filiou, M. D., Moran, L. B., Michael, G. J., Novoselov, S., Cheetham, M. E., et al. (2009). DnaJB6 is present in the core of Lewy bodies and is highly up-regulated in parkinsonian astrocytes. J. Neurosci. Res. 87, 238–245. doi: 10.1002/jnr.21819
Edvardson, S., Cinnamon, Y., Ta-Shma, A., Shaag, A., Yim, Y. I., Zenvirt, S., et al. (2012). A deleterious mutation in DNAJC6 encoding the neuronal-specific clathrin-uncoating co-chaperone auxilin, is associated with juvenile parkinsonism. PLoS One 7:e36458. doi: 10.1371/journal.pone.0036458
Eisenberg, E., and Greene, L. E. (2007). Multiple roles of auxilin and hsc70 in clathrin-mediated endocytosis. Traffic 8, 640–646. doi: 10.1111/j.1600-0854.2007.00568.x
Engert, J. C., Berube, P., Mercier, J., Dore, C., Lepage, P., Ge, B., et al. (2000). ARSACS, a spastic ataxia common in northeastern Quebec, is caused by mutations in a new gene encoding an 11.5-kb ORF. Nat. Genet. 24, 120–125. doi: 10.1038/72769
Fernández-Chacón, R., Wolfel, M., Nishimune, H., Tabares, L., Schmitz, F., Castellano-Muñoz, M., et al. (2004). The synaptic vesicle protein CSP alpha prevents presynaptic degeneration. Neuron 42, 237–251. doi: 10.1016/s0896-6273(04)00190-4
Freeman, C. L., Hesketh, G., and Seaman, M. N. (2014). RME-8 coordinates the activity of the WASH complex with the function of the retromer SNX dimer to control endosomal tubulation. J. Cell Sci. 127, 2053–2070. doi: 10.1242/jcs.144659
Gao, X. C., Zhou, C. J., Zhou, Z. R., Zhang, Y. H., Zheng, X. M., Song, A. X., et al. (2011). Co-chaperone HSJ1a dually regulates the proteasomal degradation of ataxin-3. PLoS One 6:e19763. doi: 10.1371/journal.pone.0019763
Garcia-Junco-Clemente, P., Cantero, G., Gomez-Sanchez, L., Linares-Clemente, P., Martinez-Lopez, J. A., Lujan, R., et al. (2010). Cysteine string protein-alpha prevents activity-dependent degeneration in GABAergic synapses. J. Neurosci. 30, 7377–7391. doi: 10.1523/jneurosci.0924-10.2010
Georgopoulos, C. P., Lundquist-Heil, A., Yochem, J., and Feiss, M. (1980). Identification of the E. coli. DnaJ gene product. Mol. Gen. Genet. 178, 583–588. doi: 10.1007/BF00337864
Girard, M., Larivière, R., Parfitt, D. A., Deane, E. C., Gaudet, R., Nossova, N., et al. (2012). Mitochondrial dysfunction and Purkinje cell loss in autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS). Proc. Natl. Acad. Sci. U S A 109, 1661–1666. doi: 10.1073/pnas.1113166109
Girard, M., and McPherson, P. S. (2008). RME-8 regulates trafficking of the epidermal growth factor receptor. FEBS Lett. 582, 961–966. doi: 10.1016/j.febslet.2008.02.042
Girard, M., Poupon, V., Blondeau, F., and McPherson, P. S. (2005). The DnaJ-domain protein RME-8 functions in endosomal trafficking. J. Biol. Chem. 280, 40135–40143. doi: 10.1074/jbc.m505036200
Hageman, J., Rujano, M. A., van Waarde, M. A., Kakkar, V., Dirks, R. P., Govorukhina, N., et al. (2010). A DNAJB chaperone subfamily with HDAC-dependent activities suppresses toxic protein aggregation. Mol. Cell 37, 355–369. doi: 10.1016/j.molcel.2010.01.001
Hanai, R., and Mashima, K. (2003). Characterization of two isoforms of a human DnaJ homologue, HSJ2. Mol. Biol. Rep. 30, 149–153. doi: 10.1379/1466-1268(2003)8<8:cotama>2.0.co;2
Harms, M. B., Sommerville, R. B., Allred, P., Bell, S., Ma, D., Cooper, P., et al. (2012). Exome sequencing reveals DNAJB6 mutations in dominantly-inherited myopathy. Ann. Neurol. 71, 407–416. doi: 10.1002/ana.22683
Howarth, J. L., Kelly, S., Keasey, M. P., Glover, C. P., Lee, Y. B., Mitrophanous, K., et al. (2007). Hsp40 molecules that target to the ubiquitin-proteasome system decrease inclusion formation in models of polyglutamine disease. Mol. Ther. 15, 1100–1105. doi: 10.1038/sj.mt.6300163
Hunter, P. J., Swanson, B. J., Haendel, M. A., Lyons, G. E., and Cross, J. C. (1999). Mrj encodes a DnaJ-related co-chaperone that is essential for murine placental development. Development 126, 1247–1258.
Kakkar, V., Prins, L. C., and Kampinga, H. H. (2012). DNAJ proteins and protein aggregation diseases. Curr. Top. Med. Chem. 12, 2479–2490. doi: 10.2174/1568026611212220004
Kampinga, H. H., and Craig, E. A. (2010). The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 11, 579–592. doi: 10.1038/nrm2941
Kim, Y. E., Hipp, M. S., Bracher, A., Hayer-Hartl, M., and Hartl, F. U. (2013). Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 82, 323–355. doi: 10.1146/annurev-biochem-060208-092442
Köroğlu, C., Baysal, L., Cetinkaya, M., Karasoy, H., and Tolun, A. (2013). DNAJC6 is responsible for juvenile parkinsonism with phenotypic variability. Parkinsonism Relat. Disord. 19, 320–324. doi: 10.1016/j.parkreldis.2012.11.006
Kozlov, G., Denisov, A. Y., Girard, M., Dicaire, M. J., Hamlin, J., McPherson, P. S., et al. (2011). Structural basis of defects in the sacsin HEPN domain responsible for autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS). J. Biol. Chem. 286, 20407–20412. doi: 10.1074/jbc.M111.232884
Kyle, B. D., Ahrendt, E., Braun, A. P., and Braun, J. E. (2013). The large conductance, calcium-activated K(+) (BK) channel is regulated by cysteine string protein. Sci. Rep. 3:2447. doi: 10.1038/srep02447
Labbadia, J., and Morimoto, R. I. (2014). Proteostasis and longevity: when does aging really begin? F1000Prime. F1000Prime Rep. 6:7. doi: 10.12703/p6-07
Labbadia, J., Novoselov, S. S., Bett, J. S., Weiss, A., Paganetti, P., Bates, G. P., et al. (2012). Suppression of protein aggregation by chaperone modification of high molecular weight complexes. Brain 135, 1180–1196. doi: 10.1093/brain/aws022
Li, N. N., Chang, X. L., Mao, X. Y., Zhang, J. H., Zhao, D. M., Tan, E. K., et al. (2012). GWAS-linked GAK locus in Parkinson’s disease in Han Chinese and meta-analysis. Hum. Genet. 131, 1089–1093. doi: 10.1007/s00439-011-1133-3
Mastrogiacomo, A., Parsons, S. M., Zampighi, G. A., Jenden, D. J., Umbach, J. A., and Gundersen, C. B. (1994). Cysteine string proteins: a potential link between synaptic vesicles and presynaptic Ca2+ channels. Science 263, 981–982. doi: 10.1126/science.7906056
Mitsuhashi, S., and Kang, P. B. (2012). Update on the genetics of limb girdle muscular dystrophy. Semin. Pediatr. Neurol. 19, 211–218. doi: 10.1016/j.spen.2012.09.008
Morgan, J. R., Jiang, J., Oliphint, P. A., Jin, S., Gimenez, L. E., Busch, D. J., et al. (2013). A role for an Hsp70 nucleotide exchange factor in the regulation of synaptic vesicle endocytosis. J. Neurosci. 33, 8009–8021. doi: 10.1523/jneurosci.4505-12.2013
Morgan, J. R., Prasad, K., Jin, S., Augustine, G. J., and Lafer, E. M. (2001). Uncoating of clathrin-coated vesicles in presynaptic terminals: roles for Hsc70 and auxilin. Neuron 32, 289–300. doi: 10.1016/s0896-6273(01)00467-6
Muchowski, P. J., and Wacker, J. L. (2005). Modulation of neurodegeneration by molecular chaperones. Nat. Rev. Neurosci. 6, 11–22. doi: 10.1038/nrn1587
Nosková, L., Stranecky, V., Hartmannova, H., Pristoupilova, A., Baresova, V., Ivanek, R., et al. (2011). Mutations in DNAJC5, encoding cysteine-string protein alpha, cause autosomal-dominant adult-onset neuronal ceroid lipofuscinosis. Am. J. Hum. Genet. 89, 241–252. doi: 10.1016/j.ajhg.2011.07.003
Novoselov, S. S., Mustill, W. J., Gray, A. L., Dick, J. R., Kanuga, N., Kalmar, B., et al. (2013). Molecular chaperone mediated late-stage neuroprotection in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. PLoS One 8:e73944. doi: 10.1371/journal.pone.0073944
Ohtsuka, K., Masuda, A., Nakai, A., and Nagata, K. (1990). A novel 40-kDa protein induced by heat shock and other stresses in mammalian and avian cells. Biochem. Biophys. Res. Commun. 166, 642–647. doi: 10.1016/0006-291x(90)90857-j
Ojala, T., Polinati, P., Manninen, T., Hiippala, A., Rajantie, J., Karikoski, R., et al. (2012). New mutation of mitochondrial DNAJC19 causing dilated and noncompaction cardiomyopathy, anemia, ataxia and male genital anomalies. Pediatr. Res. 72, 432–437. doi: 10.1038/pr.2012.92
Parfitt, D. A., Michael, G. J., Vermeulen, E. G., Prodromou, N. V., Webb, T. R., Gallo, J. M., et al. (2009). The ataxia protein sacsin is a functional co-chaperone that protects against polyglutamine-expanded ataxin-1. Hum. Mol. Genet. 18, 1556–1565. doi: 10.1093/hmg/ddp067
Popoff, V., Mardones, G. A., Bai, S. K., Chambon, V., Tenza, D., Burgos, P. V., et al. (2009). Analysis of articulation between clathrin and retromer in retrograde sorting on early endosomes. Traffic 10, 1868–1880. doi: 10.1111/j.1600-0854.2009.00993.x
Rose, J. M., Novoselov, S. S., Robinson, P. A., and Cheetham, M. E. (2011). Molecular chaperone-mediated rescue of mitophagy by a Parkin RING1 domain mutant. Hum. Mol. Genet. 20, 16–27. doi: 10.1093/hmg/ddQ528
Rozas, J. L., Gomez-Sanchez, L., Mircheski, J., Linares-Clemente, P., Nieto-Gonzalez, J. L., Vazquez, M. E., et al. (2012). Motorneurons require cysteine string protein-alpha to maintain the readily releasable vesicular pool and synaptic vesicle recycling. Neuron 74, 151–165. doi: 10.1016/j.neuron.2012.02.019
Sarparanta, J., Jonson, P. H., Golzio, C., Sandell, S., Luque, H., Screen, M., et al. (2012). Mutations affecting the cytoplasmic functions of the co-chaperone DNAJB6 cause limb-girdle muscular dystrophy. Nat. Genet. 44, 450–452. doi: 10.1038/ng.1103
Sato, T., Hayashi, Y. K., Oya, Y., Kondo, T., Sugie, K., Kaneda, D., et al. (2013). DNAJB6 myopathy in an Asian cohort and cytoplasmic/nuclear inclusions. Neuromuscul. Disord. 23, 269–276. doi: 10.1016/j.nmd.2012.12.010
Schmitz, F., Tabares, L., Khimich, D., Strenzke, N., de la Villa-Polo, P., Castellano-Munoz, M., et al. (2006). CSPalpha-deficiency causes massive and rapid photoreceptor degeneration. Proc. Natl. Acad. Sci. U S A 103, 2926–2931. doi: 10.1073/pnas.0510060103
Sharma, M., Burre, J., and Sudhof, T. C. (2011). CSPalpha promotes SNARE-complex assembly by chaperoning SNAP-25 during synaptic activity. Nat. Cell Biol. 13, 30–39. doi: 10.1038/ncb2131
Sharma, M., Burre, J., Bronk, P., Zhang, Y., Xu, W., and Sudhof, T. C. (2012). CSPalpha knockout causes neurodegeneration by impairing SNAP-25 function. EMBO J. 31, 829–841. doi: 10.1038/emboj.2011.467
Sinha, D., Srivastava, S., Krishna, L., and D’Silva, P. (2014). Unraveling the intricate organization of Mammalian mitochondrial presequence translocases: existence of multiple translocases for maintenance of mitochondrial function. Mol. Cell. Biol. 34, 1757–1775. doi: 10.1128/MCB.01527-13
Soti, C., and Csermely, P. (2002). Chaperones come of age. Cell Stress Chaperones 7, 186–190. doi: 10.1379/1466-1268(2002)007%3C0186:CCOA%3E2.0.CO;2
Sparkes, R., Patton, D., and Bernier, F. (2007). Cardiac features of a novel autosomal recessive dilated cardiomyopathic syndrome due to defective importation of mitochondrial protein. Cardiol. Young 17, 215–217. doi: 10.1017/s1047951107000042
Stevens, J. C., Murphy, S. M., Davagnanam, I., Phadke, R., Anderson, G., Nethisinghe, S., et al. (2013). The ARSACS phenotype can include supranuclear gaze palsy and skin lipofuscin deposits. J. Neurol. Neurosurg. Psychiatry 84, 114–116. doi: 10.1136/jnnp-2012-303634
Suarez-Cedeno, G., Winder, T., and Milone, M. (2014). DNAJB6 myopathy: a vacuolar myopathy with childhood onset. Muscle Nerve 49, 607–610. doi: 10.1002/mus.24106
Sundaram, S. K., Huq, A. M., Sun, Z., Yu, W., Bennett, L., Wilson, B. J., et al. (2011). Exome sequencing of a pedigree with Tourette syndrome or chronic tic disorder. Ann. Neurol. 69, 901–904. doi: 10.1002/ana.22398
Thiffault, I., Dicaire, M. J., Tetreault, M., Huang, K. N., Demers-Lamarche, J., Bernard, G., et al. (2013). Diversity of ARSACS mutations in French-Canadians. Can. J. Neurol. Sci. 40, 61–66.
Ungewickell, E., Ungewickell, H., Holstein, S. E. H., Linder, R., Prasad, K., Barouch, W., et al. (1995). Role of auxilin in uncoating clathrin-coated vesicles. Nature 378, 632–635. doi: 10.1038/378632a0
Vauthier, V., Jaillard, S., Journel, H., Dubourg, C., Jockers, R., and Dam, J. (2012). Homozygous deletion of an 80 kb region comprising part of DNAJC6 and LEPR genes on chromosome 1P31.3 is associated with early onset obesity, mental retardation and epilepsy. Mol. Genet. Metab. 106, 345–350. doi: 10.1016/j.ymgme.2012.04.026
Velinov, M., Dolzhanskaya, N., Gonzalez, M., Powell, E., Konidari, I., Hulme, W., et al. (2012). Mutations in the gene DNAJC5 cause autosomal dominant kufs disease in a proportion of cases: study of the parry family and 8 other families. PLoS One 7:e29729. doi: 10.1371/journal.pone.0029729
Vilariño-Güell, C., Rajput, A., Milnerwood, A. J., Shah, B., Szu-Tu, C., Trinh, J., et al. (2014). DNAJC13 mutations in Parkinson disease. Hum. Mol. Genet. 23, 1794–1801. doi: 10.1093/hmg/ddt570
Watson, E. D., Geary-Joo, C., Hughes, M., and Cross, J. C. (2007). The Mrj co-chaperone mediates keratin turnover and prevents the formation of toxic inclusion bodies in trophoblast cells of the placenta. Development 134, 1809–1817. doi: 10.1242/dev.02843
Westhoff, B., Chapple, J. P., van der, S. J., Hohfeld, J., and Cheetham, M. E. (2005). HSJ1 is a neuronal shuttling factor for the sorting of chaperone clients to the proteasome. Curr. Biol. 15, 1058–1064. doi: 10.1016/j.cub.2005.04.058
Yim, Y. I., Sun, T., Wu, L. G., Raimondi, A., De, C. P., Eisenberg, E., et al. (2010). Endocytosis and clathrin-uncoating defects at synapses of auxilin knockout mice. Proc. Natl. Acad. Sci. U S A 107, 4412–4417. doi: 10.1073/pnas.1000738107
Zhang, Y., Grant, B., and Hirsh, D. (2001). RME-8, a conserved J-domain protein, is required for endocytosis in Caenorhabditis elegans. Mol. Biol. Cell 12, 2011–2021. doi: 10.1091/mbc.12.7.2011
Zhang, Y. Q., Henderson, M. X., Colangelo, C. M., Ginsberg, S. D., Bruce, C., Wu, T., et al. (2012). Identification of CSPalpha clients reveals a role in dynamin 1 regulation. Neuron 74, 136–150. doi: 10.1016/j.neuron.2012.01.029
Zhao, X., Braun, A. P., and Braun, J. E. (2008). Biological roles of neural J proteins. Cell. Mol. Life. Sci. 65, 2385–2396. doi: 10.1007/s00018-008-8089-z
Keywords: sacsin, Tim14, Rme-8, auxilin, CSPα, HSJ1, Mrj, Hsp40
Citation: Koutras C and Braun JEA (2014) J protein mutations and resulting proteostasis collapse. Front. Cell. Neurosci. 8:191. doi: 10.3389/fncel.2014.00191
Received: 08 May 2014; Accepted: 21 June 2014;
Published online: 08 July 2014.
Edited by:
Pier Giorgio Mastroberardino, Erasmus MC University Medical Center Rotterdam, NetherlandsReviewed by:
Daniel Kaganovich, Hebrew University of Jerusalem, IsraelEileen M. Lafer, University of Texas Health Science Center at San Antonio, USA
Copyright © 2014 Koutras and Braun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Janice E. A. Braun, Department of Physiology and Pharmacology, Hotchkiss Brain Institute, Cumming School of Medicine, University of Calgary, 3330 Hospital Dr. N.W., Calgary, AB T2N 4N1, Canada e-mail:YnJhdW5qQHVjYWxnYXJ5LmNh