 Julia Margulis1,2,3
Julia Margulis1,2,3 Steven Finkbeiner1,2,3,4*
Steven Finkbeiner1,2,3,4*
- 1Gladstone Institute of Neurological Disease, J. David Gladstone Institutes, San Francisco, CA, USA
- 2Department of Neurology, University of California at San Francisco, San Francisco, CA, USA
- 3Department of Physiology, University of California at San Francisco, San Francisco, CA, USA
- 4Taube/Koret Center for Huntington’s Disease Research, San Francisco, CA, USA
Selective neuronal loss is a hallmark of neurodegenerative diseases, including Huntington’s disease (HD). Although mutant huntingtin, the protein responsible for HD, is expressed ubiquitously, a subpopulation of neurons in the striatum is the first to succumb. In this review, we examine evidence that protein quality control pathways, including the ubiquitin proteasome system, autophagy, and chaperones, are significantly altered in striatal neurons. These alterations may increase the susceptibility of striatal neurons to mutant huntingtin-mediated toxicity. This novel view of HD pathogenesis has profound therapeutic implications: protein homeostasis pathways in the striatum may be valuable targets for treating HD and other misfolded protein disorders.
Huntington’s Disease
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disorder caused by a mutation in the gene encoding the huntingtin (Htt) protein. The mutation is an expansion of CAG repeats that encodes a homomeric polyglutamine stretch in the first exon of Htt. Alleles with 35 repeats put an individual at risk for HD; 40 or more invariably lead to disease. Although mutant Htt (mHtt) is expressed ubiquitously, the key pathological hallmark of HD is the selective loss of striatal medium spiny neurons (MSNs) that express enkephalin and γ-aminobutyric acid (GABA; Graveland et al., 1985). As HD progresses, degeneration also occurs in the cortex and later the globus pallidus and thalamus (Vonsattel et al., 1985; Vonsattel and DiFiglia, 1998).
Huntington’s disease arises from the abnormal accumulation of mHtt. In HD mouse models and human patients, the appearance of visible mHtt aggregates called inclusion bodies (IBs) correlates with the onset of behavioral deficits (Davies et al., 1997). IB formation is restricted anatomically despite ubiquitous expression of mHtt. Many mechanisms attempt to explain selective striatal degeneration – including differential Htt expression, mitochondrial dysfunction, and neurotrophic factor expression – but none accounts for the regional selectivity of IBs.
IB Formation is an Indicator of Cellular Proteostasis
Inclusion body formation indicates a mismatch between the production and clearance of aggregation-prone protein. The mechanism of IB formation in HD is unclear, but the length of the polyQ repeat region correlates with the number of IBs in diseased brains (Vonsattel et al., 1985; Becher et al., 1998). While the role IBs play in cellular toxicity has been controversial, evidence suggests that IB formation can be dissociated from neurodegeneration (Klement et al., 1998; Saudou et al., 1998; Kim et al., 1999) and is a coping response to mHtt rather than a direct source of toxicity (Arrasate et al., 2004). In addition to aggregated mHtt, IBs contain ubiquitin, molecular chaperones, and proteasome subunits, suggesting that cells have insufficient capacity to clear misfolded mHtt (Sieradzan et al., 1999; Stenoien et al., 1999; Waelter et al., 2001; Mitra and Finkbeiner, 2008). Further evidence suggests that cells can degrade IBs even after they form: Yamamoto et al. (2000) generated an inducible HD mouse model in which they terminated mHtt production after IBs and behavioral deficits arose. Turning off mHtt production caused IBs to disappear and reversed the behavioral deficits (Yamamoto et al., 2000). Although this study demonstrated that IB formation is reversible, it did not address whether IBs are cleared all at once or dissolved gradually. Later work showed that IBs in mHtt-expressing neurons disappear abruptly (Arrasate et al., 2004; Miller et al., 2010), suggesting that neurons can spontaneously and rapidly metabolize IBs. Autophagy and the ubiquitin-proteasome system (UPS) have been implicated in this metabolism. Thus, the balance between the formation and clearance of IBs can provide insight into the efficiency of proteostasis pathways.
IB Formation is Cell-Selective
In HD brains, IBs localize within the nucleus and, more commonly, the neuropil of striatal and cerebral cortical neurons. Other subcortical structures, such as the globus pallidus and the thalamus, exhibit fewer IBs (DiFiglia et al., 1997; Maat-Schieman et al., 1999; Sieradzan et al., 1999). Within the human striatum, IBs are only present in 1–4% of neurons, but are more prevalent in the cerebral cortex, which exhibits less cell death in HD (Gutekunst et al., 1999; Sieradzan et al., 1999). Furthermore, few IBs form in the most vulnerable striatal neurons; only 4% of MSNs exhibit IBs, while 50% of NADPH-positive striatal neurons do (Kuemmerle et al., 1999). Greater neuronal death in the striatum does not explain this differential distribution, as both low- and high-grade cases exhibit similar numbers of striatal IBs (Gutekunst et al., 1999).
Differential mHtt expression within the cortex and striatum may account for different IB levels in these regions. Indeed, in a neuronal culture system, the rate of IB formation was tightly correlated with mHtt levels (Arrasate et al., 2004; Miller et al., 2010). In immunolabeling studies, Htt levels were relatively low in striatal neurons but were uniformly high in cortical pyramidal neurons (Ferrante et al., 1997; Fusco et al., 1999; Sieradzan and Mann, 2001; Gourfinkel-An et al., 2004). However, even when striatal and cortical neurons expressed mHtt equally, cortical neurons formed IBs more readily (Tagawa et al., 2004; Arrasate and Finkbeiner, 2012). Thus, intrinsic differences in how cell types handle misfolded proteins contribute to differences in IB formation.
Proteostasis in the Striatum
Neurons are postmitotic cells that require consistently functional proteostasis pathways. While dividing cells can simply dilute misfolded or aggregated proteins through division and growth, neurons rely on intracellular protein quality control pathways, such as degradation, to maintain protein quality (Eden et al., 2011). In addition, as neurons survive throughout an organism’s lifetime, their proteostasis mechanisms must withstand stressors over time. Misfolded proteins, such as mHtt, stress the proteostasis system, which can dysregulate protein quality control mechanisms and lead to cell death. Striatal MSNs are particularly vulnerable to degeneration and cell death even though mHtt is expressed ubiquitously. Here, we review evidence that striatal MSNs have global changes in proteostasis that render them unable to manage protein misfolding.
Ubiquitin Proteasome System
The UPS degrades misfolded and mutated intracellular proteins. Proteins targeted for degradation are ubiquitinated (i.e., tagged with a polyubiquitin chain; Pickart and Fushman, 2004) and delivered to the proteasome where they are unfolded and hydrolyzed (Goldberg, 2003; Pickart and Cohen, 2004). Originally, mHtt IBs were thought to clog the proteasome (Bence et al., 2001). Later work showed that proteasome function was inhibited prior to IB formation and that IB formation actually improved UPS flux (Bennett et al., 2005; Mitra et al., 2009). These findings suggested that diffuse mHtt – mHtt protein outside of a visible IB – impairs proteasome function. IBs may sequester this diffuse population of protein. Subsequently, Hipp et al. (2012) showed that mHtt does not directly block the proteasome. Instead, they found that misfolded mHtt overwhelmed the chaperone system, leading to misfolding of metastable proteins and increased substrate load which in turn overwhelmed the UPS (Hipp et al., 2012).
Mutant Htt has differential effects on UPS function in the striatum compared to other brain regions. Levels of a single ubiquitin-activating enzyme, Ube1, are lower in the striatum and cortex than in the cerebellum in CAG140Q knock-in mice (Wade et al., 2014). Conversely, a number of other UPS-associated proteins are upregulated in the striatum and downregulated in the cortex of R6/2 HD model mice (Liu et al., 2007). This upregulation suggests that striatal neurons have an increased need for UPS function, which may make the striatum more susceptible to UPS stressors. Indeed, age-dependent reduction in proteasomal function was shown to be exacerbated in the striatum (Zhou et al., 2003). In addition, global knockout of Parkin, an E3 ubiquitin ligase, resulted in mitochondrial respiration defects and increased oxidative stress in the striatum (Damiano et al., 2014; Figure 1).
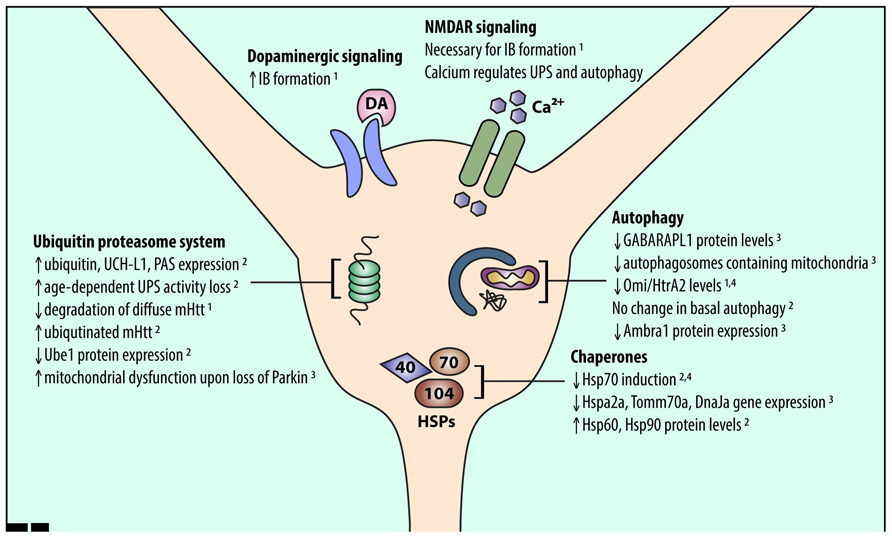
FIGURE 1. An overview of alterations in striatal proteostasis pathways. Protein quality control in neurons is accomplished through three major pathways: the UPS, chaperones and the heat shock response, and autophagy. Recent work indicates that striatal neurons may express and induce the proteins involved in these pathways differently than other cell types. Listed findings were performed in HD and wild-type model systems as follows: (1) primary striatal neuron, (2) HD mouse striatum, (3) wild-type mouse striatum, (4) human HD striatum.
Recent work also indicates that UPS activity may be lower in the striatum than in the cortex. Tsvetkov et al. (2013) demonstrated that diffuse mHtt is degraded more rapidly in cortical than in striatal neurons. This difference in degradation rate may be due to the UPS, as diffuse mHtt is ubiquitinated (Jana et al., 2001; Waelter et al., 2001; Steffan et al., 2004) and ubiquitinated mHtt accumulates upon proteasomal inhibition in many HD models (Wyttenbach et al., 2000; Jana et al., 2001; Waelter et al., 2001; Lunkes et al., 2002; Zhou et al., 2003). In addition, incubation of mHtt with mouse striatal lysates (compared to cortical or cerebellar lysates) resulted in more ubiquitinated mHtt, pointing to reduced clearance of ubiquitinated mHtt (Wade et al., 2014). Thus, diffuse mHtt may be degraded differently in striatal neurons due to basal differences in striatal UPS function.
Protein Chaperone Network
The protein chaperone network, which includes the heat shock proteins (HSPs), controls cellular protein folding. Since HSPs prevent misfolded proteins from aggregating, target proteins for degradation, and refold misfolded proteins (Sõti et al., 2005; Muchowski and Wacker, 2005; Westerheide and Morimoto, 2005), they may protect against neurodegenerative disease.
Recently, gene expression data from the Allen Brain Institute revealed many chaperone genes that are expressed at different levels in the striatum and cortex, including Hspa2, DnaJa, various Hsp90 co-chaperones, and Tomm70a (Tebbenkamp and Borchelt, 2010). Many of these genes were downregulated in striatum compared to cortex, suggesting reduced capacity for proteostasis stress in striatum. In addition, mHtt expression upregulated Hsp70 in cerebellar neurons, which are largely spared in HD, but not in striatal neurons. Therefore, vulnerable cell populations likely cannot sufficiently upregulate their chaperone system to manage misfolded mHtt (Tagawa et al., 2007). Moreover, in HD mouse models, insufficient activation of HSPs and the heat shock response (HSR) in the striatum was associated with altered chromatin architecture, which reduced access to HSP promoters (Labbadia et al., 2011). The HSR may also be inhibited by proteins that form β-sheets (Olzscha et al., 2011), as mHtt likely does (Thakur and Wetzel, 2002; Poirier et al., 2005). Thus, mHtt misfolding in the striatum may encourage β-sheet-containing mHtt aggregates to form, which inhibit HSPs and further prevent the cell from eliminating mHtt.
Conversely, other work identified HSPs that were upregulated in the striatum and downregulated in the cortex of R6/2 mice (Liu et al., 2007; Figure 1). Thus, understanding changes in chaperone protein levels rather than gene expression may help unravel their role in striatal-selective degeneration. Altered gene expression may also not be the only way protein levels are regulated in the brain. Recent studies show that mRNA expression of ribosomal proteins varies across brain regions (Kondrashov et al., 2011; Jackson, 2014), which may explain why striatal chaperone gene and protein expression are not correlated. Further studies are needed to fully unravel HSP network function in striatal neurons.
Autophagy
Macroautophagy (hereafter referred to as autophagy) sequesters long-lived proteins, organelles, or parasites within double-membrane autophagosomes (Rubinsztein et al., 2007), which fuse with lysosomes to degrade the sequestered contents. In many cellular and in vivo HD models, upregulating autophagy reduces IBs (Qin et al., 2003; Ravikumar et al., 2004; Shibata et al., 2006; Tsvetkov et al., 2010). Thus, autophagy likely regulates IB formation and clearance.
Autophagy-related protein expression varies across brain regions. Le Grand et al. (2013) showed that GABARAPL1, an Atg8 subfamily protein, is highly expressed in the cortex compared to striatum. In another study, wild-type mouse cortex exhibited more mitochondria-containing autophagosomes than did wild-type striatum (Diedrich et al., 2011). Finally, levels of Ambra1, a member of the autophagy core complex, were increased in mouse striatal interneurons compared to MSNs (Sepe et al., 2014). These data indicate that basal levels of autophagy may be lower in susceptible striatal neurons.
Misfolded and aggregated mHtt may impair autophagy induction in striatal neurons. For example, mHtt expression reduces the expression of Omi/HtrA2 in cultured striatal neurons and in human HD striatum (Inagaki et al., 2008). Omi/HtrA2, a mitochondrial chaperone and protease (Clausen et al., 2002), regulates autophagy and mitophagy (Li et al., 2010; Cilenti et al., 2014). Thus, reduced expression of autophagy-related proteins and reduced induction of autophagy may make striatal neurons more vulnerable to mHtt (Figure 1). Interestingly, basal autophagy was similar in the striatum and cortex in a recent study of BACHD mice (Baldo et al., 2013), suggesting that further investigations are needed to compare autophagy induction in different neuronal populations.
Synaptic Activity Regulates Proteostasis
Cell non-autonomous pathways, such as neuronal signaling and synaptic activity, may also affect striatal proteostasis. Neuronal activity can affect levels of ubiquitinated proteins in the post-synaptic density (Ehlers, 2003) and the subcellular localization and biochemical composition of proteasomes (Bingol and Schuman, 2006; Tai et al., 2010). Neuronal stimulation can also induce autophagy (Shehata et al., 2012; Otabe et al., 2014).
The striatum receives significant dopaminergic and excitatory glutamatergic inputs from the substantia nigra and cerebral cortex, respectively. Excitotoxicity caused by glutamatergic signaling via N-methyl-D-aspartate receptors (NMDARs) may contribute to striatal-selective degeneration in HD (Levine et al., 1999; Zeron et al., 2002). This increased sensitivity to NMDAR activation may also affect striatal proteostasis mechanisms. Okamoto et al. (2009) showed that NMDAR extrasynaptic activity is necessary for mHtt IB formation. Dopaminergic input to the striatum, which potentiates glutamate excitotoxicity (Cepeda et al., 1998; Tang et al., 2007), also affects proteostasis. Dopamine can increase IB formation in primary neuron cultures and cell lines (Charvin et al., 2005; Robinson et al., 2008), suggesting that projections from the cortex and substantia nigra to the striatum may promote striatal susceptibility in HD by altering striatal proteostasis mechanisms (Figure 1).
In addition to their role as glutamate receptors, NMDARs also regulate calcium influx. Calcium dyshomeostasis can induce excitotoxicity and may cause cell death in HD models (Bezprozvanny and Hayden, 2004; Tang et al., 2005). Striatal mitochondria were found to have reduced calcium buffering capacity, and expression of calcium binding proteins in HD mouse striatal neurons was reduced, suggesting that calcium dyshomeostasis is involved in striatal-selective degeneration (Thomas, 2006; Oliveira and Gonçalves, 2009). Studies also indicate that calcium signaling can affect proteostasis. Calcium and Ca2+/calmodulin-dependent protein kinase II (CaMKII) can regulate UPS function and autophagy in neurons (Djakovic et al., 2009; Bingol et al., 2010; Decuypere et al., 2011). Thus, differences in striatal calcium handling may alter proteostasis capacity and induction. Overall, understanding how NMDAR, dopamine, and calcium signaling affect proteostasis will divulge cell non-autonomous mechanisms that may explain the regional selectivity of IB formation in HD.
Therapeutic Implications
Targeting cellular proteostasis pathways may be therapeutically beneficial in HD. Table 1 contains a summary of proteostasis targets tested in HD models.
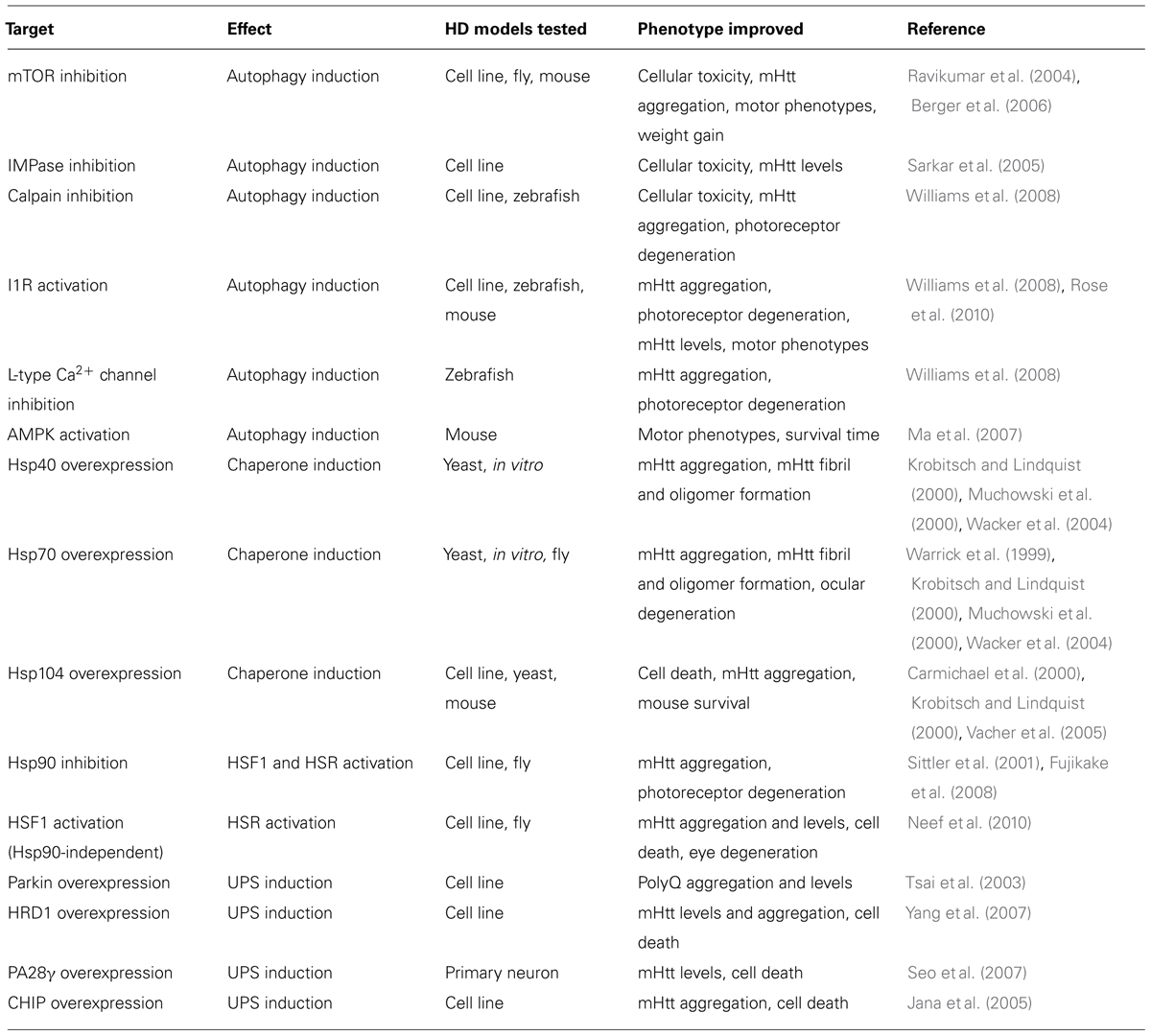
TABLE 1. Proteostasis targets tested in HD models.
Activating the UPS pathway is an intriguing therapeutic strategy. Overexpressing specific E3 ubiquitin ligase enzymes, such as Parkin and HrdI, increased clearance of mHtt by the UPS (Tsai et al., 2003; Yang et al., 2007). Overexpressing CHIP, a co-chaperone and a ubiquitin ligase, also reduced mHtt aggregation and cell death in vitro (Jana et al., 2005). Alternatively, UPS function can be induced by endogenously activating the 20S proteasome via PA700, PA200, or PA28 proteasome activators (Huang and Figueiredo-Pereira, 2010). Indeed, activating PA28γ improved cell viability in striatal neurons expressing mHtt (Seo et al., 2007) but did not improve motor phenotypes or pathology in the R6/2 mouse model (Bett et al., 2006). These results indicate that differences between in vitro and in vivo models of HD must be considered before developing effective UPS-targeting therapies.
Manipulating chaperone function may also be therapeutically effective. For example, Hsp40 and Hsp70 can reduce mHtt-dependent aggregation and toxicity (Warrick et al., 1999; Krobitsch and Lindquist, 2000; Muchowski et al., 2000; Wacker et al., 2004), while Hsp104 can reduce mHtt-induced aggregation and cell death (Carmichael et al., 2000; Krobitsch and Lindquist, 2000; Vacher et al., 2005). Furthermore, activating heat shock factor 1 (HSF1) activity, which regulates HSP expression, can suppress mHtt levels and IB formation, reduce Drosophila photoreceptor degeneration, and prolong lifespan of R6/2 mice (Sittler et al., 2001; Fujimoto et al., 2005; Fujikake et al., 2008; Neef et al., 2010, 2011). Chemical chaperones were also shown to be neuroprotective in HD mouse models (Tanaka et al., 2004; Gardian et al., 2005). A Phase 2 clinical trial of one such chaperone, phenylbutyrate, was completed in 2007 and demonstrated that phenylbutyrate was well tolerated in HD patients (Hersch, 2008). In 2014, the metal “chaperone” PBT2, which promoted degradation of extracellular β-amyloid by transporting metal ions into cells (Crouch et al., 2011) was examined in a Phase 2 clinical trial for HD where it was also shown to be well tolerated and had a minor positive effect on cognition (Prana Biotechnology). Further investigations must determine if results obtained in chaperone overexpression-based systems are translatable to more physiological HD models.
Finally, upregulating autophagy can ameliorate symptoms and pathology in many HD models. Inducing mammalian target of rapamycin (mTOR)-dependent autophagy reduced neurodegeneration in a fly HD model and improved behavior and motor performance in mouse HD models (Ravikumar et al., 2004; Berger et al., 2006; Sarkar et al., 2009). Inducing autophagy independently of mTOR also reduced mHtt aggregation and toxicity in various models (Sarkar et al., 2005; Ma et al., 2007; Zhang et al., 2007; Williams et al., 2008; Rose et al., 2010; Tsvetkov et al., 2010). The compounds identified in these studies act via inhibition of calpain or inositol monophosphatase (IMPase), activation of the imidazoline type 1 receptor (I1R) or AMP-activated protein kinase (AMPK), and antagonism of L-type Ca2+ channels. While autophagy is a promising therapeutic target, the degree of autophagy induction must be optimized if overactive autophagy is detrimental, as seen in some circumstances (Chakrabarti et al., 2009).
Over the past decade, most therapies tested in HD clinical trials have either targeted dopamine or NMDA signaling (Bonelli and Hofmann, 2007). As discussed above, both dopaminergic and NMDA signaling can affect striatal proteostasis; however, it is unclear whether the few compounds that have some effect on HD do so via proteostasis pathways. As most of these compounds do not markedly influence HD progression, it is likely that direct targeting of proteostasis pathways will be necessary to achieve clinical success.
Protein homeostasis has an important role in striatal-selective neurodegeneration in HD, and it is a strategic focus of therapeutic efforts. Since obvious symptoms of HD do not often develop until the fourth or fifth decade of life (Kieburtz et al., 1994), proteostasis pathways likely manage misfolded mHtt fairly well for a long time. Thus, future studies may find that only modestly increasing proteostasis function can stall disease indefinitely.
Concluding Remarks
The gene responsible for HD was identified in 1993. Since then, the characteristic pathology of HD has been puzzling. If mHtt expression is ubiquitous, why do MSNs degenerate first? Although many hypotheses have emerged, the regional selectivity of IB formation indicates that differences in striatal proteostasis capacity are responsible for the selective degeneration of MSNs. Recent evidence supports this claim, demonstrating that components of autophagy, the UPS, and chaperone systems are expressed or regulated differently in striatal neurons than in other brain regions. Thus, targeting proteostasis pathways specifically in the striatum may uncover new treatments for HD.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We apologize to our colleagues whose work we were unable to include due to space limitations. We thank members of the Finkbeiner laboratory for helpful discussions and Gary Howard, Crystal Herron, and Celeste Brennecka for editorial assistance. This work was made possible with support from the Larry L. Hillblom Center for the Biology of Aging Graduate Fellowship (to Julia Margulis), the National Institutes of Health (NIH) grants 3R01 NS039074, U24 NS078370, 2R01 NS045091, R01 NS083990 (to Steven Finkbeiner) from the National Institutes of Aging and by the Roddenberry Stem Cell Program (to Steven Finkbeiner), The Taube/Koret Center for Neurodegenerative Disease (Steven Finkbeiner), the Hellman Family Foundation Alzheimer’s Disease Research Program (Steven Finkbeiner), and the Keck Foundation (Steven Finkbeiner).
References
Arrasate, M., and Finkbeiner, S. (2012). Protein aggregates in Huntington’s disease. Exp. Neurol. 238, 1–11. doi: 10.1016/j.expneurol.2011.12.013
Arrasate, M., Mitra, S., Schweitzer, E. S., Segal, M. R., and Finkbeiner, S. (2004). Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431, 805–810. doi: 10.1038/nature02998
Baldo, B., Soylu, R., and Petersén, Å. (2013). Maintenance of basal levels of autophagy in Huntington’s disease mouse models displaying metabolic dysfunction. PLoS ONE 8:e83050. doi: 10.1371/journal.pone.0083050
Becher, M. W., Kotzuk, J. A., Sharp, A. H., Davies, S. W., Bates, G. P., Price, D. L.,et al. (1998). Intranuclear neuronal inclusions in Huntington’s disease and dentatorubral and pallidoluysian atrophy: correlation between the density of inclusions and IT15 CAG triplet repeat length. Neurobiol. Dis. 4, 387–397. doi: 10.1006/nbdi.1998.0168
Bence, N. F., Sampat, R. M., and Kopito, R. R. (2001). Impairment of the ubiquitin-proteasome system by protein aggregation. Science 292, 1552–1555. doi: 10.1126/science.292.5521.1552
Bennett, E. J., Bence, N. F., Jayakumar, R., and Kopito, R. R. (2005). Global impairment of the ubiquitin-proteasome system by nuclear or cytoplasmic protein aggregates precedes inclusion body formation. Mol. Cell 17, 351–365. doi: 10.1016/j.molcel.2004.12.021
Berger, Z., Ravikumar, B., Menzies, F. M., Oroz, L. G., Underwood, B. R., Pangalos, M. N.,et al. (2006). Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum. Mol. Genet. 15, 433–442. doi: 10.1093/hmg/ddi458
Bett, J. S., Goellner, G. M., Woodman, B., Pratt, G., Rechsteiner, M., and Bates, G. P. (2006). Proteasome impairment does not contribute to pathogenesis in R6/2 Huntington’s disease mice: exclusion of proteasome activator REGgamma as a therapeutic target. Hum. Mol. Genet. 15, 33–44. doi: 10.1093/hmg/ddi423
Bezprozvanny, I., and Hayden, M. R. (2004). Deranged neuronal calcium signaling and Huntington disease. Biochem. Biophys. Res. Commun. 322, 1310–1317. doi: 10.1016/j.bbrc.2004.08.035
Bingol, B., and Schuman, E. M. (2006). Activity-dependent dynamics and sequestration of proteasomes in dendritic spines. Nat. Cell Biol. 441, 1144–1148. doi: 10.1038/nature04769
Bingol, B., Wang, C.-F., Arnott, D., Cheng, D., Peng, J., and Sheng, M. (2010). Autophosphorylated CaMKIIalpha acts as a scaffold to recruit proteasomes to dendritic spines. Cell 140, 567–578. doi: 10.1016/j.cell.2010.01.024
Bonelli, R. M., and Hofmann, P. (2007). A systematic review of the treatment studies in Huntington’s disease since 1990. Expert Opin. Pharmacother. 8, 141–153. doi: 10.1517/14656566.8.2.141
Carmichael, J., Chatellier, J., Woolfson, A., Milstein, C., Fersht, A. R., and Rubinsztein, D. C. (2000). Bacterial and yeast chaperones reduce both aggregate formation and cell death in mammalian cell models of Huntington’s disease. Proc. Natl. Acad. Sci. U.S.A. 97, 9701–9705. doi: 10.1073/pnas.170280697
Cepeda, C., Colwell, C. S., Itri, J. N., Gruen, E., and Levine, M. S. (1998). Dopaminergic modulation of early signs of excitotoxicity in visualized rat neostriatal neurons. Eur. J. Neurosci. 10, 3491–3497. doi: 10.1046/j.1460-9568.1998.00357.x
Chakrabarti, L., Eng, J., Ivanov, N., Garden, G. A., and La Spada, A. R. (2009). Autophagy activation and enhanced mitophagy characterize the Purkinje cells of pcd mice prior to neuronal death. Mol. Brain 2, 24. doi: 10.1186/1756-6606-2-24
Charvin, D., Vanhoutte, P., Pages, C., Borrelli, E., Borelli, E., and Caboche, J. (2005). Unraveling a role for dopamine in Huntington’s disease: the dual role of reactive oxygen species and D2 receptor stimulation. Proc. Natl. Acad. Sci. U.S.A. 102, 12218–12223. doi: 10.1073/pnas.0502698102
Cilenti, L., Ambivero, C. T., Ward, N., Alnemri, E. S., Germain, D., and Zervos, A. S. (2014). Inactivation of Omi/HtrA2 protease leads to the deregulation of mitochondrial Mulan E3 ubiquitin ligase and increased mitophagy. Biochim. Biophys. Acta 1843, 1295–1307. doi: 10.1016/j.bbamcr.2014.03.027
Clausen, T., Southan, C., and Ehrmann, M. (2002). The HtrA family of proteases: implications for protein composition and cell fate. Mol. Cell 10, 443–455. doi: 10.1016/S1097-2765(02)00658-5
Crouch, P. J., Savva, M. S., Hung, L. W., Donnelly, P. S., Mot, A. I., Parker, S. J.,et al. (2011). The Alzheimer’s therapeutic PBT2 promotes amyloid-β degradation and GSK3 phosphorylation via a metal chaperone activity. J. Neurochem. 119, 220–230. doi: 10.1111/j.1471-4159.2011.07402.x
Damiano, M., Gautier, C. A., Bulteau, A.-L., Ferrando-Miguel, R., Gouarne, C., Paoli, M. G.,et al. (2014). Tissue- and cell-specific mitochondrial defect in parkin-deficient mice. PLoS ONE 9:e99898. doi: 10.1371/journal.pone.0099898
Davies, S. W., Turmaine, M., Cozens, B. A., DiFiglia, M., Sharp, A. H., Ross, C. A.,et al. (1997). Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90, 537–548. doi: 10.1016/S0092-8674(00)80513-9
Decuypere, J.-P., Bultynck, G., and Parys, J. B. (2011). A dual role for Ca(2+) in autophagy regulation. Cell Calcium 50, 242–250. doi: 10.1016/j.ceca.2011.04.001
Diedrich, M., Kitada, T., Nebrich, G., Koppelstaetter, A., Shen, J., Zabel, C.,et al. (2011). Brain region specific mitophagy capacity could contribute to selective neuronal vulnerability in Parkinson’s disease. Proteome Sci. 9, 59. doi: 10.1186/1477-5956-9-59
DiFiglia, M., Sapp, E., Chase, K. O., Davies, S. W., Bates, G. P., Vonsattel, J. P.,et al. (1997). Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277, 1990–1993. doi: 10.1126/science.277.5334.1990
Djakovic, S. N., Schwarz, L. A., Barylko, B., DeMartino, G. N., and Patrick, G. N. (2009). Regulation of the proteasome by neuronal activity and calcium/calmodulin-dependent protein kinase II. J. Biol. Chem. 284, 26655–26665. doi: 10.1074/jbc.M109.021956
Eden, E., Geva-Zatorsky, N., Issaeva, I., Cohen, A., Dekel, E., Danon, T.,et al. (2011). Proteome half-life dynamics in living human cells. Science 331, 764–768. doi: 10.1126/science.1199784
Ehlers, M. D. (2003). Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat. Neurosci. 6, 231–242. doi: 10.1038/nn1013
Ferrante, R. J., Gutekunst, C. A., Persichetti, F., McNeil, S. M., Kowall, N. W., Gusella, J. F.,et al. (1997). Heterogeneous topographic and cellular distribution of huntingtin expression in the normal human neostriatum. J. Neurosci. 17, 3052–3063.
Fujikake, N., Nagai, Y., Popiel, H. A., Okamoto, Y., Yamaguchi, M., and Toda, T. (2008). Heat shock transcription factor 1-activating compounds suppress polyglutamine-induced neurodegeneration through induction of multiple molecular chaperones. J. Biol. Chem. 283, 26188–26197. doi: 10.1074/jbc.M710521200
Fujimoto, M., Takaki, E., Hayashi, T., Kitaura, Y., Tanaka, Y., Inouye, S.,et al. (2005). Active HSF1 significantly suppresses polyglutamine aggregate formation in cellular and mouse models. J. Biol. Chem. 280, 34908–34916. doi: 10.1074/jbc.M506288200
Fusco, F. R., Chen, Q., Lamoreaux, W. J., Figueredo-Cardenas, G., Jiao, Y., Coffman, J. A.,et al. (1999). Cellular localization of huntingtin in striatal and cortical neurons in rats: lack of correlation with neuronal vulnerability in Huntington’s disease. J. Neurosci. 19, 1189–1202.
Gardian, G., Browne, S. E., Choi, D.-K., Klivenyi, P., Gregorio, J., Kubilus, J. K.,et al. (2005). Neuroprotective effects of phenylbutyrate in the N171-82Q transgenic mouse model of Huntington’s disease. J. Biol. Chem. 280, 556–563. doi: 10.1074/jbc.M410210200
Goldberg, A. L. (2003). Protein degradation and protection against misfolded or damaged proteins. Nature 426, 895–899. doi: 10.1038/nature02263
Gourfinkel-An, I., Cancel, G., Trottier, Y., Devys, D., Tora, L., Lutz, Y.,et al. (2004). Differential distribution of the normal and mutated forms of huntingtin in the human brain. Ann. Neurol. 42, 712–719. doi: 10.1002/ana.410420507
Graveland, G. A., Williams, R. S., and DiFiglia, M. (1985). Evidence for degenerative and regenerative changes in neostriatal spiny neurons in Huntington’s disease. Science 227, 770–773. doi: 10.1126/science.3155875
Gutekunst, C. A., Li, S. H., Yi, H., Mulroy, J. S., Kuemmerle, S., Jones, R.,et al. (1999). Nuclear and neuropil aggregates in Huntington’s disease: relationship to neuropathology. J. Neurosci. 19, 2522–2534.
Hersch, S. M. (2008). PHEND-HD: a safety, tolerability, and biomarker study of phenylbutyrate in symptomatic HD. Neurotherapeutics 5, 363. doi: 10.1016/j.nurt.2007.10.058
Hipp, M. S., Patel, C. N., Bersuker, K., Riley, B. E., Kaiser, S. E., Shaler, T. A.,et al. (2012). Indirect inhibition of 26S proteasome activity in a cellular model of Huntington’s disease. J. Cell Biol. 196, 573–587. doi: 10.1083/jcb.201110093
Huang, Q., and Figueiredo-Pereira, M. E. (2010). Ubiquitin/proteasome pathway impairment in neurodegeneration: therapeutic implications. Apoptosis 15, 1292–1311. doi: 10.1007/s10495-010-0466-z
Inagaki, R., Tagawa, K., Qi, M.-L., Enokido, Y., Ito, H., Tamura, T.,et al. (2008). Omi / HtrA2 is relevant to the selective vulnerability of striatal neurons in Huntington’s disease. Eur. J. Neurosci. 28, 30–40. doi: 10.1111/j.1460-9568.2008.06323.x
Jackson, W. S. (2014). Selective vulnerability to neurodegenerative disease: the curious case of prion protein. Dis. Model. Mech. 7, 21–29. doi: 10.1242/dmm.012146
Jana, N. R., Dikshit, P., Goswami, A., Kotliarova, S., Murata, S., Tanaka, K.,et al. (2005). Co-chaperone CHIP associates with expanded polyglutamine protein and promotes their degradation by proteasomes. J. Biol. Chem. 280, 11635–11640. doi: 10.1074/jbc.M412042200
Jana, N. R., Zemskov, E. A., Wang, G. H., and Nukina, N. (2001). Altered proteasomal function due to the expression of polyglutamine-expanded truncated N-terminal huntingtin induces apoptosis by caspase activation through mitochondrial cytochrome c release. Hum. Mol. Genet. 10, 1049–1059. doi: 10.1093/hmg/10.10.1049
Kieburtz, K., MacDonald, M., Shih, C., Feigin, A., Steinberg, K., Bordwell, K.,et al. (1994). Trinucleotide repeat length and progression of illness in Huntington’s disease. J. Med. Genet. 31, 872–874. doi: 10.1136/jmg.31.11.872
Kim, M., Lee, H. S., LaForet, G., McIntyre, C., Martin, E. J., Chang, P.,et al. (1999). Mutant huntingtin expression in clonal striatal cells: dissociation of inclusion formation and neuronal survival by caspase inhibition. J. Neurosci. 19, 964–973.
Klement, I. A., Skinner, P. J., Kaytor, M. D., Yi, H., Hersch, S. M., Clark, H. B.,et al. (1998). Ataxin-1 nuclear localization and aggregation. Cell 95, 41–53. doi: 10.1016/S0092-8674(00)81781-X
Kondrashov, N., Pusic, A., Stumpf, C. R., Shimizu, K., Hsieh, A. C., Xue, S.,et al. (2011). Ribosome-mediated specificity in Hox mRNA translation and vertebrate tissue patterning. Cell 145, 383–397. doi: 10.1016/j.cell.2011.03.028
Krobitsch, S., and Lindquist, S. (2000). Aggregation of huntingtin in yeast varies with the length of the polyglutamine expansion and the expression of chaperone proteins. Proc. Natl. Acad. Sci. U.S.A. 97, 1589–1594. doi: 10.1073/pnas.97.4.1589
Kuemmerle, S., Gutekunst, C. A., Klein, A. M., Li, X. J., Li, S. H., Beal, M. F.,et al. (1999). Huntington aggregates may not predict neuronal death in Huntington’s disease. Ann. Neurol. 46, 842–849. doi: 10.1002/1531-8249(199912)46:6<842::AID-ANA6>3.0.CO;2-O
Labbadia, J., Cunliffe, H., Weiss, A., Katsyuba, E., Sathasivam, K., Seredenina, T.,et al. (2011). Altered chromatin architecture underlies progressive impairment of the heat shock response in mouse models of Huntington disease. J. Clin. Invest. 121, 3306. doi: 10.1172/JCI57413DS1
Le Grand, J. N., Bon, K., Fraichard, A., Zhang, J., Jouvenot, M., Risold, P.-Y.,et al. (2013). Specific distribution of the autophagic protein GABARAPL1/GEC1 in the developing and adult mouse brain and identification of neuronal populations expressing GABARAPL1/GEC1. PLoS ONE 8:e63133. doi: 10.1371/journal.pone.0063133.t001
Levine, M. S., Klapstein, G. J., Koppel, A., Gruen, E., Cepeda, C., Vargas, M. E.,et al. (1999). Enhanced sensitivity to N-methyl-D-aspartate receptor activation in transgenic and knockin mouse models of Huntington’s disease. J. Neurosci. Res. 58, 515–532. doi: 10.1002/(SICI)1097-4547(19991115)58:4<515::AID-JNR5>3.0.CO;2-F
Li, B., Hu, Q., Wang, H., Man, N., Ren, H., Wen, L.,et al. (2010). Omi/HtrA2 is a positive regulator of autophagy that facilitates the degradation of mutant proteins involved in neurodegenerative diseases. Cell Death Differ. 17, 1773–1784. doi: 10.1038/cdd.2010.55
Liu, X., Miller, B. R., Rebec, G. V., and Clemmer, D. E. (2007). Protein expression in the striatum and cortex regions of the brain for a mouse model of Huntington’s disease. J. Proteome Res. 6, 3134–3142. doi: 10.1021/pr070092s
Lunkes, A., Lindenberg, K. S., Ben-Haïem, L., Weber, C., Devys, D., Landwehrmeyer, G. B.,et al. (2002). Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Mol. Cell 10, 259–269. doi: 10.1016/S1097-2765(02)00602-0
Ma, T. C., Buescher, J. L., Oatis, B., Funk, J. A., Nash, A. J., Carrier, R. L.,et al. (2007). Metformin therapy in a transgenic mouse model of Huntington’s disease. Neurosci. Lett. 411, 98–103. doi: 10.1016/j.neulet.2006.10.039
Maat-Schieman, M. L., Dorsman, J. C., Smoor, M. A., Siesling, S., Van Duinen, S. G., Verschuuren, J. J.,et al. (1999). Distribution of inclusions in neuronal nuclei and dystrophic neurites in Huntington disease brain. J. Neuropathol. Exp. Neurol. 58, 129–137. doi: 10.1097/00005072-199902000-00003
Miller, J., Arrasate, M., Shaby, B. A., Mitra, S., Masliah, E., and Finkbeiner, S. (2010). Quantitative relationships between huntingtin levels, polyglutamine length, inclusion body formation, and neuronal death provide novel insight into huntington’s disease molecular pathogenesis. J. Neurosci. 30, 10541–10550. doi: 10.1523/JNEUROSCI.0146-10.2010
Mitra, S., and Finkbeiner, S. (2008). The ubiquitin-proteasome pathway in Huntington’s disease. ScientificWorldJournal 8, 421–433. doi: 10.1100/tsw.2008.60
Mitra, S., Tsvetkov, A. S., and Finkbeiner, S. (2009). Single neuron ubiquitin-proteasome dynamics accompanying inclusion body formation in huntington disease. J. Biol. Chem. 284, 4398–4403. doi: 10.1074/jbc.M806269200
Muchowski, P. J., Schaffar, G., Sittler, A., Wanker, E. E., Hayer-Hartl, M. K., and Hartl, F. U. (2000). Hsp70 and hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc. Natl. Acad. Sci. U.S.A. 97, 7841–7846. doi: 10.1073/pnas.140202897
Muchowski, P. J., and Wacker, J. L. (2005). Modulation of neurodegeneration by molecular chaperones. Nat. Rev. Neurosci. 6, 11–22. doi: 10.1038/nrn1587
Neef, D. W., Jaeger, A. M., and Thiele, D. J. (2011). Heat shock transcription factor 1 as a therapeutic target in neurodegenerative diseases. Nat. Rev. Drug Discov. 10, 930–944. doi: 10.1038/nrd3453
Neef, D. W., Turski, M. L., and Thiele, D. J. (2010). Modulation of heat shock transcription factor 1 as a therapeutic target for small molecule intervention in neurodegenerative disease. PLoS Biol. 8:e1000291. doi: 10.1371/journal.pbio.1000291
Okamoto, S.-I., Pouladi, M. A., Talantova, M., Yao, D., Xia, P., Ehrnhoefer, D. E.,et al. (2009). Balance between synaptic versus extrasynaptic NMDA receptor activity influences inclusions and neurotoxicity of mutant huntingtin. Nat. Med. 15, 1407–1413. doi: 10.1038/nm.2056
Oliveira, J. M. A., and Gonçalves, J. (2009). In situ mitochondrial Ca2+ buffering differences of intact neurons and astrocytes from cortex and striatum. J. Biol. Chem. 284, 5010–5020. doi: 10.1074/jbc.M807459200
Olzscha, H., Schermann, S. M., Woerner, A. C., Pinkert, S., Hecht, M. H., Tartaglia, G. G.,et al. (2011). Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell 144, 67–78. doi: 10.1016/j.cell.2010.11.050
Otabe, H., Nibuya, M., Shimazaki, K., Toda, H., Suzuki, G., Nomura, S.,et al. (2014). Electroconvulsive seizures enhance autophagy signaling in rat hippocampus. Prog. Neuropsychopharmacol. Biol. Psychiatry 50, 37–43. doi: 10.1016/j.pnpbp.2013.11.012
Pickart, C. M., and Cohen, R. E. (2004). Proteasomes and their kin: proteases in the machine age. Nat. Rev. Mol. Cell Biol. 5, 177–187. doi: 10.1038/nrm1336
Pickart, C. M., and Fushman, D. (2004). Polyubiquitin chains: polymeric protein signals. Curr. Opin. Chem. Biol. 8, 610–616. doi: 10.1016/j.cbpa.2004.09.009
Poirier, M. A., Jiang, H., and Ross, C. A. (2005). A structure-based analysis of huntingtin mutant polyglutamine aggregation and toxicity: evidence for a compact beta-sheet structure. Hum. Mol. Genet. 14, 765–774. doi: 10.1093/hmg/ddi071
Qin, Z.-H., Wang, Y., Kegel, K. B., Kazantsev, A., Apostol, B. L., Thompson, L. M.,et al. (2003). Autophagy regulates the processing of amino terminal huntingtin fragments. Hum. Mol. Genet. 12, 3231–3244. doi: 10.1093/hmg/ddg346
Ravikumar, B., Vacher, C., Berger, Z., Davies, J. E., Luo, S., Oroz, L. G.,et al. (2004). Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 36, 585–595. doi: 10.1038/ng1362
Robinson, P., Lebel, M., and Cyr, M. (2008). Dopamine D1 receptor-mediated aggregation of N-terminal fragments of mutant huntingtin and cell death in a neuroblastoma cell line. Neuroscience 153, 762–772. doi: 10.1016/j.neuroscience.2008.02.052
Rose, C., Menzies, F. M., Renna, M., Acevedo-Arozena, A., Corrochano, S., Sadiq, O.,et al. (2010). Rilmenidine attenuates toxicity of polyglutamine expansions in a mouse model of Huntington’s disease. Hum. Mol. Genet. 19, 2144–2153. doi: 10.1093/hmg/ddq093
Rubinsztein, D. C., Gestwicki, J. E., Murphy, L. O., and Klionsky, D. J. (2007). Potential therapeutic applications of autophagy. Nat. Rev. Drug Discov. 6, 304–312. doi: 10.1038/nrd2272
Sarkar, S., Floto, R. A., Berger, Z., Imarisio, S., Cordenier, A., Pasco, M.,et al. (2005). Lithium induces autophagy by inhibiting inositol monophosphatase. J. Cell Biol. 170, 1101–1111. doi: 10.1083/jcb.200504035
Sarkar, S., Ravikumar, B., Floto, R. A., and Rubinsztein, D. C. (2009). Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ. 16, 46–56. doi: 10.1038/cdd.2008.110
Saudou, F., Finkbeiner, S., Devys, D., and Greenberg, M. E. (1998). Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 95, 55–66. doi: 10.1016/S0092-8674(00)81782-1
Seo, H., Sonntag, K.-C., Kim, W., Cattaneo, E., and Isacson, O. (2007). Proteasome activator enhances survival of Huntington’s disease neuronal model cells. PLoS ONE 2:e238. doi: 10.1371/journal.pone.0000238.g004
Sepe, S., Nardacci, R., Fanelli, F., Rosso, P., Bernardi, C., Cecconi, F.,et al. (2014). Expression of Ambra1 in mouse brain during physiological and Alzheimer type aging. Neurobiol. Aging 35, 96–108. doi: 10.1016/j.neurobiolaging.2013.07.001
Shehata, M., Matsumura, H., Okubo-Suzuki, R., Ohkawa, N., and Inokuchi, K. (2012). Neuronal stimulation induces autophagy in hippocampal neurons that is involved in AMPA receptor degradation after chemical long-term depression. J. Neurosci. 32, 10413–10422. doi: 10.1523/JNEUROSCI.4533-11.2012
Shibata, M., Lu, T., Furuya, T., Degterev, A., Mizushima, N., Yoshimori, T.,et al. (2006). Regulation of intracellular accumulation of mutant Huntingtin by Beclin 1. J. Biol. Chem. 281, 14474–14485. doi: 10.1074/jbc.M600364200
Sieradzan, K. A., and Mann, D. (2001). The selective vulnerability of nerve cells in Huntington’s disease. Neuropathol. Appl. Neurobiol. 27, 1–21. doi: 10.1046/j.0305-1846.2001.00299.x
Sieradzan, K. A., Mechan, A. O., Jones, L., Wanker, E. E., Nukina, N., and Mann, D. M. (1999). Huntington’s disease intranuclear inclusions contain truncated, ubiquitinated huntingtin protein. Exp. Neurol. 156, 92–99. doi: 10.1006/exnr.1998.7005
Sittler, A., Lurz, R., Lueder, G., Priller, J., Lehrach, H., Hayer-Hartl, M. K.,et al. (2001). Geldanamycin activates a heat shock response and inhibits huntingtin aggregation in a cell culture model of Huntington’s disease. Hum. Mol. Genet. 10, 1307–1315. doi: 10.1093/hmg/10.12.1307
Sõti, C., Nagy, E., Giricz, Z., Vígh, L., Csermely, P., and Ferdinandy, P. (2005). Heat shock proteins as emerging therapeutic targets. Br. J. Pharmacol. 146, 769–780. doi: 10.1038/sj.bjp.0706396
Steffan, J. S., Agrawal, N., Pallos, J., Rockabrand, E., Trotman, L. C., Slepko, N.,et al. (2004). SUMO modification of Huntingtin and Huntington’s disease pathology. Science 304, 100–104. doi: 10.1126/science.1092194
Stenoien, D. L., Cummings, C. J., Adams, H. P., Mancini, M. G., Patel, K., DeMartino, G. N.,et al. (1999). Polyglutamine-expanded androgen receptors form aggregates that sequester heat shock proteins, proteasome components and SRC-1, and are suppressed by the HDJ-2 chaperone. Hum. Mol. Genet. 8, 731–741. doi: 10.1093/hmg/8.5.731
Tagawa, K., Hoshino, M., Okuda, T., Ueda, H., Hayashi, H., Engemann, S.,et al. (2004). Distinct aggregation and cell death patterns among different types of primary neurons induced by mutant huntingtin protein. J. Neurochem. 89, 974–987. doi: 10.1111/j.1471-4159.2004.02372.x
Tagawa, K., Marubuchi, S., Qi, M.-L., Enokido, Y., Tamura, T., Inagaki, R.,et al. (2007). The induction levels of heat shock protein 70 differentiate the vulnerabilities to mutant huntingtin among neuronal subtypes. J. Neurosci. 27, 868–880. doi: 10.1523/JNEUROSCI.4522-06.2007
Tai, H.-C., Besche, H., Goldberg, A. L., and Schuman, E. M. (2010). Characterization of the brain 26S proteasome and its interacting proteins. Front. Mol. Neurosci. 3:12. doi: 10.3389/fnmol.2010.00012
Tanaka, M., Machida, Y., Niu, S., Ikeda, T., Jana, N. R., Doi, H.,et al. (2004). Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat. Med. 10, 148–154. doi: 10.1038/nm985
Tang, T.-S., Chen, X., Liu, J., and Bezprozvanny, I. (2007). Dopaminergic signaling and striatal neurodegeneration in Huntington’s disease. J. Neurosci. 27, 7899–7910. doi: 10.1523/JNEUROSCI.1396-07.2007
Tang, T.-S., Slow, E., Lupu, V., Stavrovskaya, I. G., Sugimori, M., Llinás, R.,et al. (2005). Disturbed Ca2+ signaling and apoptosis of medium spiny neurons in Huntington’s disease. Proc. Natl. Acad. Sci. U.S.A. 102, 2602–2607. doi: 10.1073/pnas.0409402102
Tebbenkamp, A. T. N., and Borchelt, D. R. (2010). Analysis of chaperone mRNA expression in the adult mouse brain by meta analysis of the allen brain atlas. PLoS ONE 5:e13675. doi: 10.1371/journal.pone.0013675
Thakur, A. K., and Wetzel, R. (2002). Mutational analysis of the structural organization of polyglutamine aggregates. Proc. Natl. Acad. Sci. U.S.A. 99, 17014–17019. doi: 10.1073/pnas.252523899
Thomas, E. A. (2006). Striatal specificity of gene expression dysregulation in Huntington’s disease. J. Neurosci. Res. 84, 1151–1164. doi: 10.1002/jnr.21046
Tsai, Y. C., Fishman, P. S., Thakor, N. V., and Oyler, G. A. (2003). Parkin facilitates the elimination of expanded polyglutamine proteins and leads to preservation of proteasome function. J. Biol. Chem. 278, 22044–22055. doi: 10.1074/jbc.M212235200
Tsvetkov, A. S., Arrasate, M., Barmada, S., Ando, D. M., Sharma, P., Shaby, B. A.,et al. (2013). Proteostasis of polyglutamine varies among neurons and predicts neurodegeneration. Nat. Chem. Biol. 9, 586–592. doi: 10.1038/nchembio.1308
Tsvetkov, A. S., Miller, J., Arrasate, M., Wong, J. S., Pleiss, M. A., and Finkbeiner, S. (2010). A small-molecule scaffold induces autophagy in primary neurons and protects against toxicity in a Huntington disease model. Proc. Natl. Acad. Sci. U.S.A. 107, 16982–16987. doi: 10.1073/pnas.1004498107
Vacher, C., Oroz, L. G., and Rubinsztein, D. C. (2005). Overexpression of yeast hsp104 reduces polyglutamine aggregation and prolongs survival of a transgenic mouse model of Huntington’s disease. Hum. Mol. Genet. 14, 3425–3433. doi: 10.1093/hmg/ddi372
Vonsattel, J. P., and DiFiglia, M. (1998). Huntington disease. J. Neuropathol. Exp. Neurol. 57, 369–384. doi: 10.1097/00005072-199805000-00001
Vonsattel, J. P., Myers, R. H., Stevens, T. J., Ferrante, R. J., Bird, E. D., and Richardson, E. P. (1985). Neuropathological classification of Huntington’s disease. J. Neuropathol. Exp. Neurol. 44, 559–577. doi: 10.1097/00005072-198511000-00003
Wacker, J. L., Zareie, M. H., Fong, H., Sarikaya, M., and Muchowski, P. J. (2004). Hsp70 and Hsp40 attenuate formation of spherical and annular polyglutamine oligomers by partitioning monomer. Nat. Struct. Mol. Biol. 11, 1215–1222. doi: 10.1038/nsmb860
Wade, B. E., Wang, C.-E., Yan, S., Bhat, K., Huang, B., Li, S.,et al. (2014). Ubiquitin-activating enzyme activity contributes to differential accumulation of mutant huntingtin in brain and peripheral tissues. J. Neurosci. 34, 8411–8422. doi: 10.1523/JNEUROSCI.0775-14.2014
Waelter, S., Boeddrich, A., Lurz, R., Scherzinger, E., Lueder, G., Lehrach, H.,et al. (2001). Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol. Biol. Cell 12, 1393–1407. doi: 10.1091/mbc.12.5.1393
Warrick, J. M., Chan, H. Y. E., Gray-Board, G. L., Chai, Y., Paulson, H. L., and Bonini, N. M. (1999). Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat. Genet. 23, 425–428. doi: 10.1038/70532
Westerheide, S. D., and Morimoto, R. I. (2005). Heat shock response modulators as therapeutic tools for diseases of protein conformation. J. Biol. Chem. 280, 33097–33100. doi: 10.1074/jbc.R500010200
Williams, A., Sarkar, S., Cuddon, P., Ttofi, E. K., Saiki, S., Siddiqi, F. H.,et al. (2008). Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat. Chem. Biol. 4, 295–305. doi: 10.1038/nchembio.79
Wyttenbach, A., Carmichael, J., Swartz, J., Furlong, R. A., Narain, Y., Rankin, J.,et al. (2000). Effects of heat shock, heat shock protein 40 (HDJ-2), and proteasome inhibition on protein aggregation in cellular models of Huntington’s disease. Proc. Natl. Acad. Sci. U.S.A. 97, 2898–2903. doi: 10.1073/pnas.97.6.2898
Yamamoto, A., Lucas, J. J., and Hen, R. (2000). Reversal of neuropathology and motor dysfunction in a conditional model of Huntington’s disease. Cell 101, 57–66. doi: 10.1016/S0092-8674(00)80623-6
Yang, H., Zhong, X., Ballar, P., Luo, S., Shen, Y., Rubinsztein, D. C.,et al. (2007). Ubiquitin ligase Hrd1 enhances the degradation and suppresses the toxicity of polyglutamine-expanded huntingtin. Exp. Cell Res. 313, 538–550. doi: 10.1016/j.yexcr.2006.10.031
Zeron, M. M., Hansson, O., Chen, N., Wellington, C. L., Leavitt, B. R., Brundin, P.,et al. (2002). Increased sensitivity to N-methyl-D-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington’s disease. Neuron 33, 849–860. doi: 10.1016/S0896-6273(02)00615-3
Zhang, L., Yu, J., Pan, H., Hu, P., Hao, Y., Cai, W.,et al. (2007). Small molecule regulators of autophagy identified by an image-based high-throughput screen. Proc. Natl. Acad. Sci. U.S.A. 104, 19023–19028. doi: 10.1073/pnas.0709695104
Keywords: striatum, proteostasis, autophagy, proteasome, Huntington’s disease
Received: 01 April 2014; Accepted: 17 July 2014;
Published online: 07 August 2014.
Citation: Margulis J and Finkbeiner S (2014) Proteostasis in striatal cells and selective neurodegeneration in Huntington’s disease. Front. Cell. Neurosci. 8:218. doi: 10.3389/fncel.2014.00218
Edited by:
Marie-Christin Pauly, University Medical Center Freiburg, GermanyReviewed by:
Marie-Christin Pauly, University Medical Center Freiburg, GermanyWilliam Mobley, University of California at San Diego, USA
Copyright © 2014 Margulis and Finkbeiner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Steven Finkbeiner, Gladstone Institute of Neurological Disease, J. David Gladstone Institutes, 1650 Owens Street, San Francisco, CA 94158, USA e-mail:c2ZpbmtiZWluZXJAZ2xhZHN0b25lLnVjc2YuZWR1