 Arthur P. H. de Jong
Arthur P. H. de Jong Diasynou Fioravante
Diasynou Fioravante- 1Department of Neurobiology, Harvard Medical School, Boston, MA, USA
- 2Department of Neurobiology, Physiology and Behavior, Center for Neuroscience, University of California Davis, Davis, CA, USA
The complex manner in which patterns of presynaptic neural activity are translated into short-term plasticity (STP) suggests the existence of multiple presynaptic calcium (Ca2+) sensors, which regulate the amplitude and time-course of STP and are the focus of this review. We describe two canonical Ca2+-binding protein domains (C2 domains and EF-hands) and define criteria that need to be met for a protein to qualify as a Ca2+ sensor mediating STP. With these criteria in mind, we discuss various forms of STP and identify established and putative Ca2+ sensors. We find that despite the multitude of proposed sensors, only three are well established in STP: Munc13, protein kinase C (PKC) and synaptotagmin-7. For putative sensors, we pinpoint open questions and potential pitfalls. Finally, we discuss how the molecular properties and modes of action of Ca2+ sensors can explain their differential involvement in STP and shape net synaptic output.
Introduction
Synaptic transmission is initiated by action potential-evoked influx of calcium (Ca2+) into the presynaptic terminal, which triggers fusion of vesicles by binding to a specialized Ca2+ sensor. Bursts of action potentials lead to the buildup of residual Ca2+ ([Ca2+]residual) in the terminal, which outlives neuronal activity, and induce multiple forms of short-term presynaptic plasticity (STP), including facilitation, depression, augmentation and post-tetanic potentiation (PTP) (reviewed in Fioravante and Regehr, 2011). STP plays a crucial role in synaptic computations and shapes the properties of microcircuits (reviewed in Abbott and Regehr, 2004; Regehr, 2012).
The dynamics of some forms of STP are dictated by the kinetics of [Ca2+]residual (Delaney et al., 1989; Kamiya and Zucker, 1994) and can be explained by changes in vesicular release probability (Katz and Miledi, 1968; Zucker and Stockbridge, 1983) or by depletion of the readily releasable pool (RRP) of vesicles (Bailey and Chen, 1988; Liu and Tsien, 1995; von Gersdorff and Matthews, 1997). However, at several synapses the magnitude of facilitation is higher than can be explained by [Ca2+]residual alone, and both facilitation and PTP decay slower than the [Ca2+]residual signal (Regehr et al., 1994; Atluri and Regehr, 1996; Brager et al., 2003; Felmy et al., 2003; Fioravante et al., 2011; Figure 1). Furthermore, many types of STP rely on the regulation of steps upstream of vesicle fusion (Dittman and Regehr, 1998; Wang and Kaczmarek, 1998), including RRP refilling and Ca2+ influx through voltage-gated Ca2+ channels (VGCCs; Stevens and Wesseling, 1998; Xu and Wu, 2005; Mochida et al., 2008; Müller et al., 2008; Leal et al., 2012). These events are strongly Ca2+-dependent, and thus Ca2+ sensors must be activated to induce and sustain STP. The Ca2+ sensors that mediate STP are the topic of this mini-review. First, we will discuss the molecular structure and function of two Ca2+-binding domains employed by Ca2+ sensors: C2 domains and EF-hands. Subsequently, we will define the criteria for establishing Ca2+ sensors for STP, and, guided by these criteria, discuss a body of recent literature on well accepted and putative sensors that regulate STP.
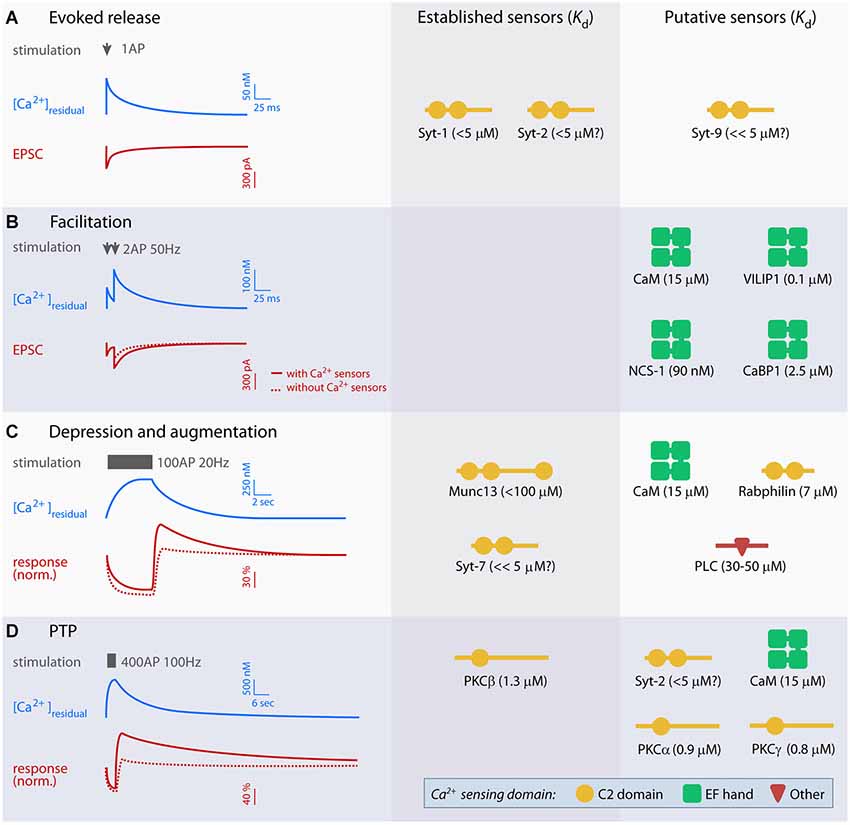
Figure 1. Overview of established and putative presynaptic Ca2+ sensors in evoked release and short-term plasticity (STP). Left panel displays idealized traces of [Ca2+]residual and excitatory postsynaptic currents (EPSCs; A and B) or baseline-normalized responses (C and D) during synaptic plasticity based on experiments at parallel fiber synapses, mossy fiber-CA3 synapses, the crayfish neuromuscular junction and the calyx of Held. Typical stimulation paradigms used to elicit various forms of STP are indicated in gray. Scale bars are approximate, but note that the amplitude and kinetics of the Ca2+ signal and STP vary significantly between preparations. Right panels show established and putative Ca2+ sensors for evoked release (A) and each form of STP (B–D), and their Ca2+ dissociation constant (Kd). Kd values were obtained from: syt-1 C2AB (with PIP2) (van den Bogaart et al., 2012), free calmodulin (CaM; Xia and Storm, 2005), visin-like protein (VILIP-1) (myristroylated) (Li et al., 2011), neuronal calcium sensor 1 (NCS-1) (myristroylated) and CaBP1 (Aravind et al., 2008), Munc13 C2B (Shin et al., 2010), Rabphilin C2B (Ubach et al., 1999), PLCδ1 (Grobler and Hurley, 1998) PKCα, -β and γ (Torrecillas et al., 2004). The Kd values of syt-2, -7 and -9 have not been measured directly, but indirect measurements suggest that syt-2 is similar to syt-1, whereas syt-7 and 9 may have lower Kd (Sugita et al., 2002).
Ca2+-Binding Motifs
C2 Domains
The best described Ca2+ sensors in the context of synapses are C2 domains, which are found in many signal transduction and membrane trafficking proteins (Rizo and Südhof, 1998). C2 domains consist of ~130 amino acids that form a compact β-sandwich of two 4-stranded β-sheets. Three loops connecting the β-sheets at the top of the domain contain 4–5 highly conserved aspartates that coordinate the binding of 2 to 3 Ca2+ ions (Shao et al., 1996; Ubach et al., 1998; Fernandez et al., 2001). The Ca2+-binding properties of C2 domains have been described in detail in synaptotagmin (syt), which acts as the Ca2+ sensor for synchronous vesicle fusion at most synapses (Pang and Südhof, 2010). Mutations that interfere with Ca2+ binding on syt-1 alter the Ca2+-sensitivity of vesicle fusion (Nishiki and Augustine, 2004; Shin et al., 2009; Kochubey and Schneggenburger, 2011; Kochubey et al., 2011). Analogous mutation analyses of other C2 domains showed similar effects on Ca2+ binding (Shin et al., 2010; Fioravante et al., 2014; Liu et al., 2014). Some C2 domains naturally lack these aspartate residues and cannot bind Ca2+ (e.g., Pappa et al., 1998). Ca2+ binding increases the affinity of C2 domains for phospholipids (Brose et al., 1992; Fernandez et al., 2001), thus recruiting the domain to the plasma membrane. In addition, it may trigger a conformational change that increases association with effector proteins (for instance syt-1 binding to SNAREs (Bai et al., 2004)) or exposes a domain within the protein (e.g., the MUN domain of Munc13 (Shin et al., 2010)). Many C2 domains display a steep increase in Ca2+ affinity in the presence of phosphatidylinositol 4,5-biphosphate (PIP2; van den Bogaart et al., 2012), which helps localize the domain to the PIP2-enriched active zone (Rohrbough and Broadie, 2005).
EF-Hands
The EF-hand is the most common Ca2+-binding motif, with diverse cellular functions including cytoplasmic Ca2+ buffering and signal transduction (Skelton et al., 1994; Schaub and Heizmann, 2008; Schwaller, 2009). The motif consists of two α-helices connected by a linker of 12 amino acids (Lewit-Bentley and Réty, 2000). Six residues within this linker coordinate binding to a single Ca2+ ion, and their mutation abolishes Ca2+ binding (Maune et al., 1992). Examples of EF-hand-containing proteins with proposed Ca2+-sensing roles in STP include calmodulin (CaM), neuronal calcium sensor 1 (NCS-1) and visin-like proteins (VILIPs).
CaM is the prototypical EF-hand protein that interacts with numerous effector proteins in a Ca2+-dependent manner (Xia and Storm, 2005). Important presynaptic effectors are CaM-dependent kinase II (CaMKII), myosin light chain kinase (MLCK), adenylyl cyclase, the protein phosphatase calcineurin, Munc13, VGCCs and Ca2+-activated potassium channels, all of which regulate presynaptic function (de Jong and Verhage, 2009; Adelman et al., 2012). Because the Ca2+ affinity of CaM is differentially regulated by its binding partners, different CaM-protein complexes vary in their Ca2+ sensitivity (Olwin and Storm, 1985; Xia and Storm, 2005) and could therefore be differentially engaged during various forms of STP. Direct assessment of the role of CaM as a Ca2+ sensor for STP has proven difficult because manipulations of CaM levels alter expression of >200 genes (Pang et al., 2010) and rescue experiments in neuronal preparations with Ca2+-binding mutants of CaM have not been conducted thus far.
Definition of a Ca2+ Sensor for STP
With a plethora of C2- and EF-hand-containing proteins in the presynaptic terminal, there are numerous candidate Ca2+ sensors for STP. We propose that in order to qualify as a sensor for STP, a protein must fulfill the following three criteria:
1. Ca2+ must bind directly to the protein. An obvious requirement for a Ca2+ sensor is that it must bind Ca2+. Some EF-hands and C2 domains lack the Ca2+-coordinating residues and cannot bind Ca2+. Therefore, Ca2+ binding must be experimentally established for each protein.
2. Protein must be part of, or directly modulate, vesicle availability or the vesicle release machinery. Changes in availability and/or fusogenicity of synaptic vesicles and in presynaptic Ca2+ influx shape STP (Dutta Roy et al., 2014). A Ca2+ sensor for STP must therefore directly affect vesicle availability (recruitment, docking, priming) and/or the vesicle fusion machinery, including VGCCs and SM proteins (for a discussion of release machinery, see Südhof, 2013). This definition includes enzymes like kinases, which directly regulate the properties of these components. For the purpose of this review, we do not consider Ca2+ buffers (e.g., parvalbumin) and pumps, which indirectly affect STP by changing the spatiotemporal distribution of free Ca2+ through binding or extrusion (Müller et al., 2007; Scullin and Partridge, 2010), or components of the endocytotic machinery, which can affect vesicle or release site availability after prolonged episodes of exocytosis (Wilkinson and Lin, 2004; Hosoi et al., 2009).
3. Mutations that interfere with Ca2+binding affect STP. Even if a protein satisfies criteria 1 and 2, it is not a Ca2+ sensor for STP unless Ca2+ binding is required for the protein’s function in STP. For instance, whether Ca2+ binding to Doc2 is required for spontaneous release is debated and the role of Doc2 as a Ca2+ sensor for spontaneous release remains unclear (Groffen et al., 2010; Pang et al., 2011). Therefore, it is necessary to show that mutation of the Ca2+ binding site abolishes function (for example using a knockout/rescue or knockin approach) in order to conclude that a protein is a Ca2+ sensor mediating STP. It could even be argued that a requirement for Ca2+ binding during plasticity must be demonstrated in order to establish a protein as a Ca2+ sensor, but the technology for this type of experiments is currently lacking.
Ca2+ Sensors in STP
Facilitation
At synapses with low initial release probability, brief bursts of activity can induce transient facilitation of release, which relies on increased release probability due to elevated [Ca2+]residual (Katz and Miledi, 1968; Kamiya and Zucker, 1994; Regehr et al., 1994). However, this mechanism alone cannot fully explain the magnitude of facilitation at all synapses (Atluri and Regehr, 1996; Felmy et al., 2003), and additional Ca2+-dependent processes have been suggested (Zucker and Regehr, 2002), including the existence of a yet unidentified presynaptic Ca2+ sensor distinct from syt-1 (Bain and Quastel, 1992; Saraswati et al., 2007). Enhancement of Ca2+ currents is an attractive mechanism to mediate facilitation, and the capability of Ca2+/CaM to modulate overexpressed VGCCs during strong depolarization has been studied extensively (Catterall et al., 2013). Ca2+/CaM binds to a regulatory domain of Cav2.1, the VGCC that mediates the P/Q type Ca2+ current driving synaptic transmission in most synapses. In heterologous cell lines, this interaction leads to enhancement of Ca2+ currents, which depends on Ca2+ binding to CaM (Lee et al., 1999; DeMaria et al., 2001). Several EF-hand-containing proteins including VILIPs, CaBPs and NCS-1 (collectively named neuronal Ca2+ sensors, or nCaS) also modulate Ca2+ influx through VGCCs (Few et al., 2005; Lautermilch et al., 2005; Burgoyne, 2007; Dason et al., 2012; Catterall et al., 2013) and may affect facilitation in a manner that depends on the nCaS binding domain of VGCCs (Tsujimoto et al., 2002; Sippy et al., 2003; Mochida et al., 2008; Leal et al., 2012). For none of these protein functions, however, has a Ca2+ binding requirement been established, and some of them may actually be independent of Ca2+ (Few et al., 2005). In addition, due to the lack of suitable genetic models, most experiments rely on overexpression of exogenous proteins (Mochida et al., 2003). Whether nCaS are specifically involved in the regulation of STP, or the altered STP is a consequence of altered basal synaptic properties, remains controversial (Dason et al., 2012).
Depression and Recovery from Depression
Prolonged high-frequency stimulation leads to transient decrease in presynaptic strength, which can be due to depletion of the RRP (Elmqvist and Quastel, 1965; Liu and Tsien, 1995; Schneggenburger et al., 2002) and activity-dependent decrease in Ca2+ influx (Forsythe et al., 1998; Xu and Wu, 2005) (for a complete review of known mechanisms of depression, see Regehr, 2012). CaM, CaBP1 and NCS-1 have been proposed as putative Ca2+ sensors to mediate the latter effect (Xu and Wu, 2005; Catterall and Few, 2008; Mochida et al., 2008). Depression can be slowed by Ca2+-dependent replenishment of the RRP (Stevens and Wesseling, 1998; Wang and Kaczmarek, 1998). The vesicle priming factor Munc13 acts as a Ca2+ sensor to determine the rate of depression, via its C2B and CaM-binding domains. Ca2+ binding to the C2B domain of Munc13 activates its MUN domain that promotes assembly of the machinery responsible for vesicle fusion, thereby increasing refilling of the RRP (Shin et al., 2010; Ma et al., 2011). Indeed, Munc13 knockout neurons expressing a variant of the protein with mutated Ca2+-coordinating aspartates display increased synaptic depression without affecting initial release probability (Shin et al., 2010). In addition, Munc13 binds Ca2+/CaM, and this interaction also accelerates RRP refilling (Junge et al., 2004; Lipstein et al., 2012, 2013). In line with this observation, CaM inhibitors slow the RRP refilling rate (Sakaba and Neher, 2001; Hosoi et al., 2007). Although a Ca2+-binding CaM mutant has not been studied in this context, the CaM/Munc13 interaction is strongly Ca2+-dependent (Junge et al., 2004; Dimova et al., 2006; Lipstein et al., 2012), thus making the Ca2+/CaM-Munc13 complex a likely Ca2+-sensor for STP.
Synaptotagmin-7 has also been identified as a sensor that regulates depression, operating via its two Ca2+-binding C2 domains (Liu et al., 2014). At the zebrafish neuromuscular junction, syt-7 regulates desynchronized release (Wen et al., 2010), but its function in mammalian neurons has been debated (Maximov et al., 2008; Bacaj et al., 2013; Liu et al., 2014). A recent study showed that in syt-7 knockout mice, initial release probability is unaffected but the rate of vesicle replenishment during and after bursts of activity is significantly reduced (Liu et al., 2014). This phenotype is rescued by wild-type syt-7 but not by syt-7 carrying mutations of the Ca2+ binding sites, demonstrating that syt-7 is a Ca2+ sensor that mediates RRP refilling. Syt-7 also probably interacts with Ca2+/CaM (Liu et al., 2014), but the functional significance of this complex remains to be identified.
In contrast to the proteins discussed above that promote recovery from depression, rabphilin is thought to slow down recovery from depression (Deák et al., 2006). Rabphilin is a synaptic vesicle protein with two Ca2+-sensing C2 domains (Yamaguchi et al., 1993; Ubach et al., 1999; Coudevylle et al., 2008), but whether Ca2+ binding is required for its role in STP has not been determined.
Augmentation and PTP
Augmentation and PTP are two closely related forms of STP that require prolonged high-frequency stimulation (Magleby, 1973; Magleby and Zengel, 1976a; Stevens and Wesseling, 1999; Habets and Borst, 2005; Korogod et al., 2005). For augmentation, varying stimulus duration increases the peak amplitude of the enhancement without significantly affecting the time course of decay (Magleby, 1979). The mechanisms underlying augmentation are not well understood and changes in both release probability and Ca2+-dependent replenishment of the RRP have been proposed (Magleby and Zengel, 1976b; Stevens and Wesseling, 1999; Rosenmund et al., 2002; Kalkstein and Magleby, 2004). Munc13 and syt-7 have been suggested as Ca2+ sensors for augmentation (Shin et al., 2010; Lipstein et al., 2013; Liu et al., 2014), but since both sensors affect depression as well, dissociation of their roles in synaptic depression vs. augmentation has not been possible. Various phospholipase C (PLC) isoforms could also act as Ca2+ sensors because they require binding of a Ca2+ ion for activation of their catalytic domain (Grobler and Hurley, 1998; Rebecchi and Pentyala, 2000). Pharmacological studies suggest that PLC activation is required for augmentation (Rosenmund et al., 2002) but not PTP (Genc et al., 2014). PLC hydrolyses PIP2 to diacylglycerol, which could lead to potentiation of synaptic transmission via Munc13 and protein kinase C (PKC; de Jong and Verhage, 2009).
PTP typically lasts longer than augmentation and shows a progressive increase in the time course of decay with increased duration and frequency of stimulation (Magleby, 1979; Korogod et al., 2005). Pharmacological (e.g., Alle et al., 2001; Brager et al., 2002; Beierlein et al., 2007; Korogod et al., 2007) and genetic (Fioravante et al., 2011, 2012, 2014; Chu et al., 2014) studies at several synapses have firmly established the requirement for PKC in PTP. Three PKC isoforms (α, β and γ) possess a C2 domain and bind Ca2+ with low micromolar affinity (Torrecillas et al., 2004; Newton, 2010; Figure 1). PKCs enhance release through phosphorylation of effectors, including components of the vesicular release machinery such as Munc18 (Wierda et al., 2007; de Jong and Verhage, 2009; Genc et al., 2014). Mutations of the Ca2+-coordinating aspartates in the C2 domain of PKCβ abolish its ability to support PTP, without affecting basal synaptic function (Fioravante et al., 2014).
PKCβ is probably not the only Ca2+ sensor for PTP. At the immature calyx of Held, PTP depends on PKCγ (Chu et al., 2014). Moreover, at the parallel fiber-Purkinje cell synapse in the cerebellum, PKCα can readily support PTP in the absence of PKCβ and γ (Fioravante et al., 2012). It remains to be tested whether Ca2+ binding to PKCα and γ is necessary for PTP and whether all PKC isoforms act through Munc18 phosphorylation. Finally, pharmacological studies suggest that Ca2+/CaM, acting via MLCK, makes a small contribution to PTP at immature, but not functionally mature, synapses (Lee et al., 2008; Fioravante et al., 2011).
Tetanic stimulation enhances not only evoked responses (i.e., PTP) but also spontaneous events in a Ca2+-dependent manner. The frequency (Zengel and Magleby, 1981; Zucker and Lara-Estrella, 1983; Eliot et al., 1994; Habets and Borst, 2005), and at some synapses also the amplitude (He et al., 2009), of spontaneous events increase after tetanization. Because of similarities in the time course of these effects with PTP, a common mechanism has been speculated (Zengel and Magleby, 1981). However, the effects of [Ca2+]residual on spontaneous transmission were recently shown to be independent of PKC (Xue and Wu, 2010; Fioravante et al., 2011; but see Brager et al., 2003) and the increase in amplitude requires syt-2 (He et al., 2009). The Ca2+ sensors remain unknown.
Differential Engagement of Ca2+ Sensors and Implications for STP
Different patterns of neuronal activity result in variable Ca2+ signals stretching over an order of magnitude (Figure 1). Diverse sensors are therefore needed to translate the Ca2+ signals into distinct forms of STP. Factors such as Ca2+ affinity, specific (sub-)cellular expression and mechanisms of action contribute to the specialization of sensors for different forms of STP. For example, NCS-1 has high affinity for Ca2+ and localizes at the plasma membrane (O’Callaghan et al., 2002; Burgoyne, 2007) where it could rapidly respond to local Ca2+ signals. PKCβ, on the other hand, has lower Ca2+ affinity, is cytoplasmic at rest (Newton, 2010) and likely has to phosphorylate more than one substrates to induce plasticity; therefore, sustained, global Ca2+increases are likely required for its activation, in agreement with the prolonged stimulation requirement for PTP (Habets and Borst, 2005; Korogod et al., 2005). Even for the same sensor, Ca2+ affinity can vary as a result of effector binding, phospholipid binding, and post-translational modifications (Xia and Storm, 2005; Li et al., 2011; van den Bogaart et al., 2012). Finally, specific expression patterns of Ca2+ sensors could help explain why identical activation regimes do not always lead to the same STP across synapses or during development (Rosenmund et al., 2002; Chu et al., 2014).
Most synapses exhibit multiple forms of STP and the net synaptic output reflects the interaction between these different forms (de Jong and Verhage, 2009). It is therefore likely that different Ca2+ sensors interact, and might even compete (Chu et al., 2014), during STP. The dynamics of these interactions should be considered when building computational models of STP. Traditionally, such models combine use-dependent depletion and Ca2+-dependent facilitation to explain synaptic output (Tsodyks et al., 1998; Fuhrmann et al., 2002; Pfister et al., 2010). Introduction of additional components such as vesicle replenishment, which are engaged under conditions that activate the corresponding Ca2+ sensors, more accurately reflects our understanding of the underlying biology and allows better prediction of synaptic and network behavior (Hennig, 2013).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Drs. P.S. Kaeser, M. Verhage, M. Thanawala, W. Regehr and E. Antzoulatos for critically reading the manuscript. This work was funded by the Netherlands Organization for Scientific Research (NWO, 825.12.028) to Arthur P. H. de Jong and UC Davis College of Biological Sciences Dean’s start-up award to Diasynou Fioravante.
References
Abbott, L. F., and Regehr, W. G. (2004). Synaptic computation. Nature 431, 796–803. doi: 10.10.1038/nature03010
Adelman, J. P., Maylie, J., and Sah, P. (2012). Small-Conductance Ca2+-Activated K+ channels: form and function. Annu. Rev. Physiol. 74, 245–269. doi: 10.1146/annurev-physiol-020911-153336
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Alle, H., Jonas, P., and Geiger, J. R. (2001). PTP and LTP at a hippocampal mossy fiber-interneuron synapse. Proc. Natl. Acad. Sci. U S A 98, 14708–14713. doi: 10.1073/pnas.251610898
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Aravind, P., Chandra, K., Reddy, P. P., Jeromin, A., Chary, K. V. R., and Sharma, Y. (2008). Regulatory and structural EF-hand motifs of neuronal calcium sensor-1: Mg2+ modulates Ca2+ binding, Ca2+-induced conformational changes and equilibrium unfolding transitions. J. Mol. Biol. 376, 1100–1115. doi: 10.1016/j.jmb.2007.12.033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Atluri, P. P., and Regehr, W. G. (1996). Determinants of the time course of facilitation at the granule cell to Purkinje cell synapse. J. Neurosci. 16, 5661–5671.
Bacaj, T., Wu, D., Yang, X., Morishita, W., Zhou, P., Xu, W., et al. (2013). Synaptotagmin-1 and synaptotagmin-7 trigger synchronous and asynchronous phases of neurotransmitter release. Neuron 80, 947–959. doi: 10.1016/j.neuron.2013.10.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bai, J., Wang, C.-T., Richards, D. A., Jackson, M. B., and Chapman, E. R. (2004). Fusion Pore dynamics are regulated by Synaptotagmin•t-SNARE interactions. Neuron 41, 929–942. doi: 10.1016/s0896-6273(04)00117-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bailey, C. H., and Chen, M. (1988). Morphological basis of short-term habituation in Aplysia. J. Neurosci. 8, 2452–2459.
Bain, A. I., and Quastel, D. M. (1992). Multiplicative and additiva Ca(2+)-dependent components of facilitation at mouse endplates. J. Physiol. 455, 383–405.
Beierlein, M., Fioravante, D., and Regehr, W. G. (2007). Differential expression of posttetanic potentiation and retrograde signaling mediate target-dependent short-term synaptic plasticity. Neuron 54, 949–959. doi: 10.1016/j.neuron.2007.06.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brager, D. H., Cai, X., and Thompson, S. M. (2003). Activity-dependent activation of presynaptic protein kinase C mediates post-tetanic potentiation. Nat. Neurosci. 6, 551–552. doi: 10.1038/nn1067
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brager, D. H., Capogna, M., and Thompson, S. M. (2002). Short-term synaptic plasticity, simulation of nerve terminal dynamics, and the effects of protein kinase C activation in rat hippocampus. J. Physiol. 541, 545–559. doi: 10.1113/jphysiol.2001.015842
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brose, N., Petrenko, A. G., Südhof, T. C., and Jahn, R. (1992). Synaptotagmin: a calcium sensor on the synaptic vesicle surface. Science 256, 1021–1025. doi: 10.1126/science.1589771
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Burgoyne, R. D. (2007). Neuronal calcium sensor proteins: generating diversity in neuronal Ca2+ signalling. Nat. Rev. Neurosci. 8, 182–193. doi: 10.1038/nrn2093
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Catterall, W. A., and Few, A. P. (2008). Calcium channel regulation and presynaptic plasticity. Neuron 59, 882–901. doi: 10.1016/j.neuron.2008.09.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Catterall, W. A., Leal, K., and Nanou, E. (2013). Calcium channels and short-term synaptic plasticity. J. Biol. Chem. 288, 10742–10749. doi: 10.1074/jbc.R112.411645
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chu, Y., Fioravante, D., Leitges, M., and Regehr, W. G. (2014). Calcium-dependent PKC isoforms have specialized roles in short-term synaptic plasticity. Neuron 82, 859–871. doi: 10.1016/j.neuron.2014.04.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Coudevylle, N., Montaville, P., Leonov, A., Zweckstetter, M., and Becker, S. (2008). Structural determinants for Ca2+ and phosphatidylinositol 4,5-bisphosphate binding by the C2A domain of rabphilin-3A. J. Biol. Chem. 283, 35918–35928. doi: 10.1074/jbc.M804094200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dason, J. S., Romero-Pozuelo, J., Atwood, H. L., and Ferrús, A. (2012). Multiple roles for Frequenin/NCS-1 in synaptic function and development. Mol. Neurobiol. 45, 388–402. doi: 10.1007/s12035-012-8250-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Deák, F., Shin, O.-H., Tang, J., Hanson, P., Ubach, J., Jahn, R., et al. (2006). Rabphilin regulates SNARE-dependent re-priming of synaptic vesicles for fusion. EMBO J. 25, 2856–2866. doi: 10.1038/sj.emboj.7601165
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
de Jong, A. P. H., and Verhage, M. (2009). Presynaptic signal transduction pathways that modulate synaptic transmission. Curr. Opin. Neurobiol. 19, 245–253. doi: 10.1016/j.conb.2009.06.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Delaney, K. R., Zucker, R. S., and Tank, D. W. (1989). Calcium in motor nerve terminals associated with posttetanic potentiation. J. Neurosci. 9, 3558–3567.
DeMaria, C. D., Soong, T. W., Alseikhan, B. A., Alvania, R. S., and Yue, D. T. (2001). Calmodulin bifurcates the local Ca2+ signal that modulates P/Q-type Ca2+ channels. Nature 411, 484–489. doi: 10.1038/35078091
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dimova, K., Kawabe, H., Betz, A., Brose, N., and Jahn, O. (2006). Characterization of the Munc13-calmodulin interaction by photoaffinity labeling. Biochim. Biophys. Acta 1763, 1256–1265. doi: 10.1016/j.bbamcr.2006.09.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dittman, J. S., and Regehr, W. G. (1998). Calcium dependence and recovery kinetics of presynaptic depression at the climbing fiber to Purkinje cell synapse. J. Neurosci. 18, 6147–6162.
Dutta Roy, R., Stefan, M. I., and Rosenmund, C. (2014). Biophysical properties of presynaptic short-term plasticity in hippocampal neurons: insights from electrophysiology, imaging and mechanistic models. Front. Cell. Neurosci. 8:141. doi: 10.3389/fncel.2014.00141
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Eliot, L. S., Kandel, E. R., and Hawkins, R. D. (1994). Modulation of spontaneous transmitter release during depression and posttetanic potentiation of Aplysia sensory-motor neuron synapses isolated in culture. J. Neurosci. 14, 3280–3292.
Elmqvist, D., and Quastel, D. M. (1965). A quantitative study of end-plate potentials in isolated human muscle. J. Physiol. 178, 505–529.
Felmy, F., Neher, E., and Schneggenburger, R. (2003). Probing the intracellular calcium sensitivity of transmitter release during synaptic facilitation. Neuron 37, 801–811. doi: 10.1016/s0896-6273(03)00085-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fernandez, I., Araç, D., Ubach, J., Gerber, S. H., Shin, O., Gao, Y., et al. (2001). Three-dimensional structure of the synaptotagmin 1 C2B-domain: synaptotagmin 1 as a phospholipid binding machine. Neuron 32, 1057–1069. doi: 10.1016/s0896-6273(01)00548-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Few, A. P., Lautermilch, N. J., Westenbroek, R. E., Scheuer, T., and Catterall, W. A. (2005). Differential regulation of CaV2.1 channels by calcium-binding protein 1 and visinin-like protein-2 requires N-terminal myristoylation. J. Neurosci. 25, 7071–7080. doi: 10.1523/jneurosci.0452-05.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fioravante, D., Chu, Y., de Jong, A. P., Leitges, M., Kaeser, P. S., and Regehr, W. G. (2014). Protein kinase C is a calcium sensor for presynaptic short-term plasticity. Elife 3:e03011. doi: 10.7554/elife.03011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fioravante, D., Chu, Y., Myoga, M. H., Leitges, M., and Regehr, W. G. (2011). Calcium-dependent isoforms of protein kinase C mediate posttetanic potentiation at the calyx of held. Neuron 70, 1005–1019. doi: 10.1016/j.neuron.2011.04.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fioravante, D., Myoga, M. H., Leitges, M., and Regehr, W. G. (2012). Adaptive regulation maintains posttetanic potentiation at cerebellar granule cell synapses in the absence of calcium-dependent PKC. J. Neurosci. 32, 13004–13009. doi: 10.1523/jneurosci.0683-12.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fioravante, D., and Regehr, W. G. (2011). Short-term forms of presynaptic plasticity. Curr. Opin. Neurobiol. 21, 269–274. doi: 10.1016/j.conb.2011.02.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Forsythe, I. D., Tsujimoto, T., Barnes-Davies, M., Cuttle, M. F., and Takahashi, T. (1998). Inactivation of presynaptic calcium current contributes to synaptic depression at a fast central synapse. Neuron 20, 797–807. doi: 10.1016/s0896-6273(00)81017-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fuhrmann, G., Segev, I., Markram, H., and Tsodyks, M. (2002). Coding of temporal information by activity-dependent synapses. J. Neurophysiol. 87, 140–148.
Genc, O., Kochubey, O., Toonen, R. F., Verhage, M., and Schneggenburger, R. (2014). Munc18–1 is a dynamically regulated PKC target during short-term enhancement of transmitter release. Elife 3:e01715. doi: 10.7554/eLife.01715
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Grobler, J. A., and Hurley, J. H. (1998). Catalysis by phospholipase C delta1 requires that Ca2+ bind to the catalytic domain, but not the C2 domain. Biochemistry 37, 5020–5028. doi: 10.1021/bi972952w
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Groffen, A. J., Martens, S., Díez Arazola, R., Cornelisse, L. N., Lozovaya, N., de Jong, A. P. H., et al. (2010). Doc2b is a high-affinity Ca2+ sensor for spontaneous neurotransmitter release. Science 327, 1614–1618. doi: 10.1126/science.1183765
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Habets, R. L. P., and Borst, J. G. G. (2005). Post-tetanic potentiation in the rat calyx of Held synapse. J. Physiol. 564, 173–187. doi: 10.1113/jphysiol.2004.079160
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
He, L., Xue, L., Xu, J., McNeil, B. D., Bai, L., Melicoff, E., et al. (2009). Compound vesicle fusion increases quantal size and potentiates synaptic transmission. Nature 459, 93–97. doi: 10.1038/nature07860
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hennig, M. H. (2013). Theoretical models of synaptic short term plasticity. Front. Comput. Neurosci. 7:45. doi: 10.3389/fncom.2013.00154
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hosoi, N., Holt, M., and Sakaba, T. (2009). Calcium dependence of exo- and endocytotic coupling at a glutamatergic synapse. Neuron 63, 216–229. doi: 10.1016/j.neuron.2009.06.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hosoi, N., Sakaba, T., and Neher, E. (2007). Quantitative analysis of calcium-dependent vesicle recruitment and its functional role at the calyx of Held synapse. J. Neurosci. 27, 14286–14298. doi: 10.1523/jneurosci.4122-07.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Junge, H. J., Rhee, J.-S., Jahn, O., Varoqueaux, F., Spiess, J., Waxham, M. N., et al. (2004). Calmodulin and Munc13 form a Ca2+ sensor/effector complex that controls short-term synaptic plasticity. Cell 118, 389–401. doi: 10.1016/j.cell.2004.06.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kalkstein, J. M., and Magleby, K. L. (2004). Augmentation increases vesicular release probability in the presence of masking depression at the Frog neuromuscular junction. J. Neurosci. 24, 11391–11403. doi: 10.1523/jneurosci.2756-04.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kamiya, H., and Zucker, R. S. (1994). Residual Ca2 + and short-term synaptic plasticity. Nature 371, 603–606. doi: 10.1038/371603a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Katz, B., and Miledi, R. (1968). The role of calcium in neuromuscular facilitation. J. Physiol. 195, 481–492.
Kochubey, O., Lou, X., and Schneggenburger, R. (2011). Regulation of transmitter release by Ca2+ and synaptotagmin: insights from a large CNS synapse. Trends Neurosci. 34, 237–246. doi: 10.1016/j.tins.2011.02.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kochubey, O., and Schneggenburger, R. (2011). Synaptotagmin increases the dynamic range of synapses by driving Ca2+-evoked release and by clamping a near-linear remaining Ca2+ sensor. Neuron 69, 736–748. doi: 10.1016/j.neuron.2011.01.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Korogod, N., Lou, X., and Schneggenburger, R. (2005). Presynaptic Ca2+ requirements and developmental regulation of posttetanic potentiation at the calyx of held. J. Neurosci. 25, 5127–5137. doi: 10.1523/jneurosci.1295-05.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Korogod, N., Lou, X., and Schneggenburger, R. (2007). Posttetanic potentiation critically depends on an enhanced Ca2+ sensitivity of vesicle fusion mediated by presynaptic PKC. Proc. Natl. Acad. Sci. U S A 104, 15923–15928. doi: 10.1073/pnas.0704603104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lautermilch, N. J., Few, A. P., Scheuer, T., and Catterall, W. A. (2005). Modulation of CaV2.1 channels by the neuronal calcium-binding protein visinin-like protein-2. J. Neurosci. 25, 7062–7070. doi: 10.1523/jneurosci.0447-05.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Leal, K., Mochida, S., Scheuer, T., and Catterall, W. A. (2012). Fine-tuning synaptic plasticity by modulation of Ca(V)2.1 channels with Ca2+ sensor proteins. Proc. Natl. Acad. Sci. U S A 109, 17069–17074. doi: 10.1073/pnas.1215172109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, J. S., Kim, M.-H., Ho, W.-K., and Lee, S.-H. (2008). Presynaptic release probability and readily releasable pool size are regulated by two independent mechanisms during posttetanic potentiation at the calyx of held synapse. J. Neurosci. 28, 7945–7953. doi: 10.1523/JNEUROSCI.2165-08.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, A., Wong, S., Gallagher, D., Li, B., Storm, D., Scheuer, T., et al. (1999). Ca2+/calmodulin binds to and modulates P/Q-type calcium channels. Nature 399, 155–159. doi: 10.1038/20194
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lewit-Bentley, A., and Réty, S. (2000). EF-hand calcium-binding proteins. Curr. Opin. Struct. Biol. 10, 637–643. doi: 10.1016/S0959-440X(00)00142-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, C., Pan, W., Braunewell, K. H., and Ames, J. B. (2011). Structural analysis of Mg2+ and Ca2+ binding, myristoylation and dimerization of the neuronal calcium sensor and visinin-like protein 1 (VILIP-1). J. Biol. Chem. 286, 6354–6366. doi: 10.1074/jbc.M110.173724
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lipstein, N., Sakaba, T., Cooper, B. H., Lin, K.-H., Strenzke, N., Ashery, U., et al. (2013). Dynamic control of synaptic vesicle replenishment and short-term plasticity by Ca(2+)-calmodulin-Munc13–1 signaling. Neuron 79, 82–96. doi: 10.1016/j.neuron.2013.05.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lipstein, N., Schaks, S., Dimova, K., Kalkhof, S., Ihling, C., Kölbel, K., et al. (2012). Nonconserved Ca(2+)/calmodulin binding sites in Munc13s differentially control synaptic short-term plasticity. Mol. Cell. Biol. 32, 4628–4641. doi: 10.1128/MCB.00933-12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, H., Bai, H., Hui, E., Yang, L., Evans, C. S., Wang, Z., et al. (2014). Synaptotagmin 7 functions as a Ca2+-sensor for synaptic vesicle replenishment. Elife 3:e01524. doi: 10.7554/elife.01524
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, G., and Tsien, R. W. (1995). Properties of synaptic transmission at single hippocampal synaptic boutons. Nature 375, 404–408. doi: 10.1038/375404a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ma, C., Li, W., Xu, Y., and Rizo, J. (2011). Munc13 mediates the transition from the closed syntaxin-Munc18 complex to the SNARE complex. Nat. Struct. Mol. Biol. 18, 542–549. doi: 10.1038/nsmb.2047
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Magleby, K. L. (1973). The effect of repetitive stimulation on facilitation of transmitter release at the frog neuromuscular junction. J. Physiol. 234, 327–352.
Magleby, K. L. (1979). Facilitation, augmentation and potentiation of transmitter release. Prog. Brain Res. 49, 175–182. doi: 10.1016/s0079-6123(08)64631-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Magleby, K. L., and Zengel, J. E. (1976a). Augmentation: a process that acts to increase transmitter release at the frog neuromuscular junction. J. Physiol. 257, 449–470.
Magleby, K. L., and Zengel, J. E. (1976b). Long term changes in augmentation, potentiation and depression of transmitter release as a function of repeated synaptic activity at the frog neuromuscular junction. J. Physiol. 257, 471–494.
Maune, J. F., Klee, C. B., and Beckingham, K. (1992). Ca2+ binding and conformational change in two series of point mutations to the individual Ca(2+)-binding sites of calmodulin. J. Biol. Chem. 267, 5286–5295.
Maximov, A., Lao, Y., Li, H., Chen, X., Rizo, J., Sorensen, J. B., et al. (2008). Genetic analysis of synaptotagmin-7 function in synaptic vesicle exocytosis. Proc. Natl. Acad. Sci. U S A 105, 3986–3991. doi: 10.1073/pnas.0712372105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mochida, S., Few, A. P., Scheuer, T., and Catterall, W. A. (2008). Regulation of Presynaptic CaV2.1 Channels by Ca2+ sensor proteins mediates short-term synaptic plasticity. Neuron 57, 210–216. doi: 10.1016/j.neuron.2007.11.036
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mochida, S., Westenbroek, R. E., Yokoyama, C. T., Itoh, K., and Catterall, W. A. (2003). Subtype-selective reconstitution of synaptic transmission in sympathetic ganglion neurons by expression of exogenous calcium channels. Proc. Natl. Acad. Sci. U S A 100, 2813–2818. doi: 10.1073/pnas.262787299
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Müller, M., Felmy, F., and Schneggenburger, R. (2008). A limited contribution of Ca2+ current facilitation to paired-pulse facilitation of transmitter release at the rat calyx of held. J. Physiol. 586, 5503–5520. doi: 10.1113/jphysiol.2008.155838
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Müller, M., Felmy, F., Schwaller, B., and Schneggenburger, R. (2007). Parvalbumin is a mobile presynaptic Ca2+ buffer in the calyx of held that accelerates the decay of Ca2+ and short-term facilitation. J. Neurosci. 27, 2261–2271. doi: 10.1523/jneurosci.5582-06.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Newton, A. C. (2010). Protein kinase C: poised to signal. Am. J. Physiol. Endocrinol. Metab. 298, E395–E402. doi: 10.1152/ajpendo.00477.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nishiki, T., and Augustine, G. J. (2004). Dual roles of the C2B domain of synaptotagmin I in synchronizing Ca2+-dependent neurotransmitter release. J. Neurosci. 24, 8542–8550. doi: 10.1523/jneurosci.2545-04.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
O’Callaghan, D. W., Ivings, L., Weiss, J. L., Ashby, M. C., Tepikin, A. V., and Burgoyne, R. D. (2002). Differential use of Myristoyl groups on neuronal calcium sensor proteins as a determinant of spatio-temporal aspects of Ca2+ signal transduction. J. Biol. Chem. 277, 14227–14237. doi: 10.1074/jbc.m111750200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Olwin, B. B., and Storm, D. R. (1985). Calcium binding to complexes of calmodulin and calmodulin binding proteins. Biochemistry 24, 8081–8086. doi: 10.1021/bi00348a037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pang, Z. P., Bacaj, T., Yang, X., Zhou, P., Xu, W., and Südhof, T. C. (2011). Doc2 supports spontaneous synaptic transmission by a Ca(2+)-independent mechanism. Neuron 70, 244–251. doi: 10.1016/j.neuron.2011.03.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pang, Z. P., and Südhof, T. C. (2010). Cell biology of Ca2+-triggered exocytosis. Curr. Opin. Cell Biol. 22, 496–505. doi: 10.1016/j.ceb.2010.05.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pang, Z. P., Xu, W., Cao, P., and Südhof, T. C. (2010). Calmodulin suppresses Synaptotagmin-2 transcription in cortical neurons. J. Biol. Chem. 285, 33930–33939. doi: 10.1074/jbc.M110.150151
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pappa, H., Murray-Rust, J., Dekker, L. V., Parker, P. J., and McDonald, N. Q. (1998). Crystal structure of the C2 domain from protein kinase C-delta. Structure 6, 885–894. doi: 10.1016/s0969-2126(98)00090-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pfister, J.-P., Dayan, P., and Lengyel, M. (2010). Synapses with short-term plasticity are optimal estimators of presynaptic membrane potentials. Nat. Neurosci. 13, 1271–1275. doi: 10.1038/nn.2640
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rebecchi, M. J., and Pentyala, S. N. (2000). Structure, function and control of phosphoinositide-specific Phospholipase C. Physiol. Rev. 80, 1291–1335.
Regehr, W. G. (2012). Short-term presynaptic plasticity. Cold Spring Harb. Perspect. Biol. 4:a005702. doi: 10.1101/cshperspect.a005702
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Regehr, W. G., Delaney, K. R., and Tank, D. W. (1994). The role of presynaptic calcium in short-term enhancement at the hippocampal mossy fiber synapse. J. Neurosci. 14, 523–537.
Rizo, J., and Südhof, T. C. (1998). C2-domains, structure and function of a universal Ca2+-binding domain. J. Biol. Chem. 273, 15879–15882. doi: 10.1074/jbc.273.26.15879
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rohrbough, J., and Broadie, K. (2005). Lipid regulation of the synaptic vesicle cycle. Nat. Rev. Neurosci. 6, 139–150. doi: 10.1038/nrn1608
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rosenmund, C., Sigler, A., Augustin, I., Reim, K., Brose, N., and Rhee, J.-S. (2002). Differential control of vesicle priming and short-term plasticity by Munc13 isoforms. Neuron 33, 411–424. doi: 10.1016/s0896-6273(02)00568-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sakaba, T., and Neher, E. (2001). Calmodulin mediates rapid recruitment of fast-releasing synaptic vesicles at a calyx-type synapse. Neuron 32, 1119–1131. doi: 10.1016/s0896-6273(01)00543-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Saraswati, S., Adolfsen, B., and Littleton, J. T. (2007). Characterization of the role of the Synaptotagmin family as calcium sensors in facilitation and asynchronous neurotransmitter release. Proc. Natl. Acad. Sci. U S A 104, 14122–14127. doi: 10.1073/pnas.0706711104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schaub, M. C., and Heizmann, C. W. (2008). Calcium, troponin, calmodulin, S100 proteins: from myocardial basics to new therapeutic strategies. Biochem. Biophys. Res. Commun. 369, 247–264. doi: 10.1016/j.bbrc.2007.10.082
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schneggenburger, R., Sakaba, T., and Neher, E. (2002). Vesicle pools and short-term synaptic depression: lessons from a large synapse. Trends Neurosci. 25, 206–212. doi: 10.1016/s0166-2236(02)02139-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schwaller, B. (2009). The continuing disappearance of “pure” Ca2+ buffers. Cell. Mol. Life Sci. 66, 275–300. doi: 10.1007/s00018-008-8564-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Scullin, C. S., and Partridge, L. D. (2010). Contributions of SERCA pump and ryanodine-sensitive stores to presynaptic residual Ca2+. Cell Calcium 47, 326–338. doi: 10.1016/j.ceca.2010.01.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shao, X., Davletov, B. A., Sutton, R. B., Südhof, T. C., and Rizo, J. (1996). Bipartite Ca2+-binding motif in C2 domains of synaptotagmin and protein kinase C. Science 273, 248–251. doi: 10.1126/science.273.5272.248
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shin, O.-H., Lu, J., Rhee, J.-S., Tomchick, D. R., Pang, Z. P., Wojcik, S. M., et al. (2010). Munc13 C2B domain is an activity-dependent Ca2+ regulator of synaptic exocytosis. Nat. Struct. Mol. Biol. 17, 280–288. doi: 10.1038/nsmb.1758
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shin, O.-H., Xu, J., Rizo, J., and Sudhof, T. C. (2009). Differential but convergent functions of Ca2+ binding to synaptotagmin-1 C2 domains mediate neurotransmitter release. Proc. Natl. Acad. Sci. U S A 106, 16469–16474. doi: 10.1073/pnas.0908798106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sippy, T., Cruz-Martín, A., Jeromin, A., and Schweizer, F. E. (2003). Acute changes in short-term plasticity at synapses with elevated levels of neuronal calcium sensor-1. Nat. Neurosci. 6, 1031–1038. doi: 10.1038/nn1117
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Skelton, N. J., Kördel, J., Akke, M., Forsén, S., and Chazin, W. J. (1994). Signal transduction versus buffering activity in Ca(2+)-binding proteins. Nat. Struct. Biol. 1, 239–245. doi: 10.1038/nsb0494-239
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stevens, C. F., and Wesseling, J. F. (1998). Activity-dependent modulation of the rate at which synaptic vesicles become available to undergo exocytosis. Neuron 21, 415–424. doi: 10.1016/s0896-6273(00)80550-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stevens, C. F., and Wesseling, J. F. (1999). Augmentation is a potentiation of the Exocytotic process. Neuron 22, 139–146. doi: 10.1016/s0896-6273(00)80685-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Südhof, T. C. (2013). Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron 80, 675–690. doi: 10.1016/j.neuron.2013.10.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sugita, S., Shin, O.-H., Han, W., Lao, Y., and Südhof, T. C. (2002). Synaptotagmins form a hierarchy of exocytotic Ca2+ sensors with distinct Ca2+ affinities. EMBO J. 21, 270–280. doi: 10.1093/emboj/21.3.270
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Torrecillas, A., Laynez, J., Menéndez, M., Corbalán-García, S., and Gómez-Fernández, J. C. (2004). Calorimetric study of the interaction of the C2 domains of classical protein kinase C isoenzymes with Ca2+ and phospholipids. Biochemistry 43, 11727–11739. doi: 10.1021/bi0489659
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tsodyks, M., Pawelzik, K., and Markram, H. (1998). Neural networks with dynamic synapses. Neural Comput. 10, 821–835. doi: 10.1162/089976698300017502
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tsujimoto, T., Jeromin, A., Saitoh, N., Roder, J. C., and Takahashi, T. (2002). Neuronal calcium sensor 1 and activity-dependent facilitation of P/Q-type calcium currents at presynaptic nerve terminals. Science 295, 2276–2279. doi: 10.1126/science.1068278
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ubach, J., García, J., Nittler, M. P., Südhof, T. C., and Rizo, J. (1999). Structure of the Janus-faced C2B domain of rabphilin. Nat. Cell Biol. 1, 106–112. doi: 10.1038/10076
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ubach, J., Zhang, X., Shao, X., Südhof, T. C., and Rizo, J. (1998). Ca2+ binding to synaptotagmin: how many Ca2+ ions bind to the tip of a C2-domain? EMBO J. 17, 3921–3930. doi: 10.1093/emboj/17.14.3921
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
van den Bogaart, G., Meyenberg, K., Diederichsen, U., and Jahn, R. (2012). Phosphatidylinositol 4,5-bisphosphate increases Ca2+ affinity of synaptotagmin-1 by 40-fold. J. Biol. Chem. 287, 16447–16453. doi: 10.1074/jbc.M112.343418
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
von Gersdorff, H., and Matthews, G. (1997). Depletion and Replenishment of vesicle pools at a ribbon-type synaptic terminal. J. Neurosci. 17, 1919–1927.
Wang, L. Y., and Kaczmarek, L. K. (1998). High-frequency firing helps replenish the readily releasable pool of synaptic vesicles. Nature 394, 384–388. doi: 10.1038/28645
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wen, H., Linhoff, M. W., McGinley, M. J., Li, G.-L., Corson, G. M., Mandel, G., et al. (2010). Distinct roles for two synaptotagmin isoforms in synchronous and asynchronous transmitter release at zebrafish neuromuscular junction. Proc. Natl. Acad. Sci. U S A 107, 13906–13911. doi: 10.1073/pnas.1008598107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wierda, K. D. B., Toonen, R. F. G., de Wit, H., Brussaard, A. B., and Verhage, M. (2007). Interdependence of PKC-dependent and PKC-independent pathways for presynaptic plasticity. Neuron 54, 275–290. doi: 10.1016/j.neuron.2007.04.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wilkinson, R. S., and Lin, M. Y. (2004). Endocytosis and synaptic plasticity: might the tail wag the dog? Trends Neurosci. 27, 171–174. doi: 10.1016/j.tins.2004.01.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xia, Z., and Storm, D. R. (2005). The role of calmodulin as a signal integrator for synaptic plasticity. Nat. Rev. Neurosci. 6, 267–276. doi: 10.1038/nrn1647
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xu, J., and Wu, L.-G. (2005). The decrease in the presynaptic calcium current is a major cause of short-term depression at a calyx-type synapse. Neuron 46, 633–645. doi: 10.1016/j.neuron.2005.03.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xue, L., and Wu, L.-G. (2010). Post-tetanic potentiation is caused by two signalling mechanisms affecting quantal size and quantal content. J. Physiol. 588, 4987–4994. doi: 10.1113/jphysiol.2010.196964
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yamaguchi, T., Shirataki, H., Kishida, S., Miyazaki, M., Nishikawa, J., Wada, K., et al. (1993). Two functionally different domains of rabphilin-3A, Rab3A p25/smg p25A-binding and phospholipid- and Ca(2+)-binding domains. J. Biol. Chem. 268, 27164–27170.
Zengel, J. E., and Magleby, K. L. (1981). Changes in miniature endplate potential frequency during repetitive nerve stimulation in the presence of Ca2+, Ba2+ and Sr2+ at the frog neuromuscular junction. J. Gen. Physiol. 77, 503–529. doi: 10.1085/jgp.77.5.503
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zucker, R. S., and Lara-Estrella, L. O. (1983). Post-tetanic decay of evoked and spontaneous transmitter release and a residual-calcium model of synaptic facilitation at crayfish neuromuscular junctions. J. Gen. Physiol. 81, 355–372. doi: 10.1085/jgp.81.3.355
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zucker, R. S., and Regehr, W. G. (2002). Short-term synaptic plasticity. Annu. Rev. Physiol. 64, 355–405. doi: 10.1146/annurev.physiol.64.092501.114547
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: C2 domain, protein kinase C, Munc13, synaptotagmin, calmodulin, post-tetanic potentiation, residual calcium, short-term plasticity
Citation: de Jong APH and Fioravante D (2014) Translating neuronal activity at the synapse: presynaptic calcium sensors in short-term plasticity. Front. Cell. Neurosci. 8:356. doi: 10.3389/fncel.2014.00356
Received: 18 September 2014; Accepted: 09 October 2014;
Published online: 29 October 2014.
Edited by:
Philippe Isope, Centre National pour la Recherche Scientifique, FranceReviewed by:
Marco Canepari, Institut National de la Santé et de la Recherche Médicale, FranceErwin Neher, Max Planck Institute for Biophysical Chemistry, Germany
Copyright © 2014 de Jong and Fioravante. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Arthur P. H. de Jong, Department of Neurobiology, Harvard Medical School, 220 Longwood Ave, Boston, MA 02115, USA e-mail:YXJ0aHVyX2RlX2pvbmdAaG1zLmhhcnZhcmQuZWR1;
Diasynou Fioravante, Department of Neurobiology, Physiology and Behavior, Center for Neuroscience, University of California Davis, 1544 Newton Court, Davis, CA 95618, USA e-mail:ZGZpb3JhdmFudGVAdWNkYXZpcy5lZHU=