 Valentine Lahmy
Valentine Lahmy Romain Long3,4
Romain Long3,4 Didier Morin
Didier Morin Tangui Maurice
Tangui Maurice- 1Inserm U 710, University of Montpellier 2, Montpellier, France
- 2Amylgen, Montferrier-sur-Lez, France
- 3Inserm U 955, Team 03, Créteil, France
- 4Faculty of Medicine, Université Paris-Est, Unité Mixte de Recherche S955, Université Paris-Est Créteil Val-de-Marne, Créteil, France
Alzheimer’s disease (AD), the most prevalent dementia in the elderly, is characterized by progressive synaptic and neuronal loss. Mitochondrial dysfunctions have been consistently reported as an early event in AD and appear before Aβ deposition and memory decline. In order to define a new neuroprotectant strategy in AD targeting mitochondrial alterations, we develop tetrahydro-N,N-dimethyl-2,2-diphenyl-3-furanmethanamine (ANAVEX2-73, AE37), a mixed muscarinic receptor ligand and a sigma-1 receptor (σ1R) agonist. We previously reported that ANAVEX2-73 shows anti-amnesic and neuroprotective activities in mice injected intracerebroventricular (ICV) with oligomeric amyloid-β25–35 peptide (Aβ25–35). The σ1R is present at mitochondria-associated endoplasmic reticulum (ER) membranes, where it acts as a sensor/modulator of ER stress responses and local Ca2+ exchanges with the mitochondria. We therefore evaluated the effect of ANAVEX2-73 and PRE-084, a reference σ1R agonist, on preservation of mitochondrial integrity in Aβ25–35-injected mice. In isolated mitochondria from hippocampus preparations of Aβ25–35 injected animals, we measured respiration rates, complex activities, lipid peroxidation, Bax/Bcl-2 ratios and cytochrome c release into the cytosol. Five days after Aβ25–35 injection, mitochondrial respiration in mouse hippocampus was altered. ANAVEX2-73 (0.01–1 mg/kg IP) restored normal respiration and PRE-084 (0.5–1 mg/kg IP) increased respiration rates. Both compounds prevented Aβ25–35-induced increases in lipid peroxidation levels, Bax/Bcl-2 ratio and cytochrome c release into the cytosol, all indicators of increased toxicity. ANAVEX2-73 and PRE-084 efficiently prevented the mitochondrial respiratory dysfunction and resulting oxidative stress and apoptosis. The σ1R, targeted selectively or non-selectively, therefore appears as a valuable target for protection against mitochondrial damages in AD.
Introduction
Alzheimer’s disease (AD), the most common form of dementia in the elderly is defined histologically by the presence of the two hallmarks: extracellular senile plaques constituted with amyloid-β (Aβ) proteins and intracellular inclusions of hyperphosphorylated microtubule-associated Tau protein, refered as neurofibrillary tangles (Selkoe, 2004). Clinically, the disease is characterized by progressive cognitive decline associated with synaptic and neuronal loss in regions critical for memory process, e.g., hippocampus, entorhinal and frontal cortex (Crews and Masliah, 2010). Mitochondria, which are the main energy provider in the cell, are particularly enriched in synapses where energy is critical for synaptic transmission (Verstreken et al., 2005). Mitochondrial dysfunctions have been consistently reported as an early event in AD physiopathology and appear before Aβ deposition and memory deficits in AD patients and transgenic mice (Maurer et al., 2000; Caspersen et al., 2005; Mosconi et al., 2008). Indeed, during AD pathogenesis, Aβ oligomers accumulate in mitochondria, resulting in disrupted energy metabolism, increased oxidative stress and apoptosis (Lustbader et al., 2004; Caspersen et al., 2005; Manczak et al., 2006). Studies in AD patients and transgenic mice have reported consistent decreases in tricarboxylic acid cycle enzymes and cytochrome c oxidase activities (Yates et al., 1990; Maurer et al., 2000; Caspersen et al., 2005; Leuner et al., 2007). Moreover, studies in isolated mitochondria from transgenic mice brains or in isolated mitochondria exposed to Aβ oligomers showed decreased respiration rates and ATP production and increased oxidative stress, suggesting Aβ-related defects in mitochondrial respiratory chain (Casley et al., 2002a; Aleardi et al., 2005; Caspersen et al., 2005; Clementi et al., 2005; Leuner et al., 2007). Disruption in mitochondrial respiratory chain results in increased production of oxidative stress by mitochondria, a key element in AD physiopathology (Smith et al., 1996; Leuner et al., 2012). One other aspect of mitochondrial failure is the opening of mitochondrial permeability transition pore (mPTP) which can be triggered by different effectors, e.g., mitochondrial Ca2+ overload and oxidative stress (Kroemer and Reed, 2000). mPTP opening could also be triggered by the translocation to the mitochondrial outer membrane of the cytosolic proapototic members of the Bcl-2 family proteins. Among them is the proapototic protein Bax which, in proapoptotic conditions, translocates to mitochondria, dimerizes and constitutes a pore in the mitochondrial membrane and/or contributes to the formation of mPTP (Jürgensmeier et al., 1998; Marzo et al., 1998; Narita et al., 1998). Bax activity could be antagonized by heterodimerization with antiapoptotic Bcl-2 family proteins, including Bcl-2 itself (Hanada et al., 1995; Yin et al., 1995; Yang et al., 1997). mPTP opening allows liberation of mitochondrial protein, such as cytochrome c, which promotes apoptosis, binding to the apoptosis protease activation factor (APAf-1) and forming a complex indicated as “apoptosome” (Liu et al., 1996; Yang et al., 1997).
In the present study, we used the intracerebroventricular (ICV) injection of Aβ25–35 fragment in oligomeric form in mice as a rapid, standardized pharmacological model of AD toxicity, suitable to investigate the impact of mitochondrial alteration. Indeed, ICV Aβ25–35 injection results in neuroinflammation and reactive gliosis, pro-apoptotic caspase activity, oxidative stress, reduction in the number of neurons measured in hippocampal pyramidal cell layers, loss of cholinergic neurons, and memory deficits (Maurice et al., 1996; Delobette et al., 1997; Meunier et al., 2006; Villard et al., 2009, 2011; Zussy et al., 2011). In vitro evidences have been accumulated suggesting that the peptide also altered mitochondrial physiology, after direct application on isolated mitochondria or in mitochondria isolated from cultured neurons exposed to Aβ25–35 (Canevari et al., 1999; Casley et al., 2002a,b; Aleardi et al., 2005; Clementi et al., 2005; Dong et al., 2010; Ren et al., 2011). Isolated mitochondria from rat brain subjected to subchronic ICV injection of Aβ25–35 or Aβ1–40 produced significantly more H2O2 compared to control mice after three days of treatment, suggesting mitochondrial dysfunction in this model (Kaminsky and Kosenko, 2008). However, to our knowledge, the functionality of isolated mitochondria has never been explored ex vivo after ICV administration of Aβ25–35 in rodents.
Tetrahydro-N,N-dimethyl-2,2-diphenyl-3-furanmethanamine hydrochloride (ANAVEX2-73, AE37) is a novel ligand, acting at muscarinic acetylcholine (mAChR) and σ1 receptors (σ1R) with affinities in the low micromolar range (Espallergues et al., 2007). We previously reported that this compound had anti-amnesic and neuroproective potential in the AD model in mice induced by the ICV injection of Aβ25–35 peptide. In particular, ANAVEX2-73 attenuated cellular loss, Aβ1–42 seeding, Tau hyperphosphorylation and learning and memory deficits observed several days after Aβ25–35 injection (Villard et al., 2009, 2011; Lahmy et al., 2013). The drug acts synergistically on its two main targets, as an antagonist of M2 mAChR and as an agonist of σ1R. The σ1R is an endoplasmic reticulum (ER)-resident molecular chaperone (Hayashi and Su, 2007), also expressed at the mitochondrial membrane (Klouz et al., 2002), particularly at mitochondria-associated ER membranes (MAM). Its activity regulates Ca2+ exchange from ER to mitochondria (Hayashi and Su, 2007), known to be critical for mitochondrial bioenergetics (Cárdenas et al., 2010). Moreover, the σ1R is involved in regulation of the mitochondrial membrane potential, reactive oxygen species (ROS) production and apoptosis (Tsai et al., 2009; Meunier and Hayashi, 2010) and σ1R agonists have been shown to protect mitochondrial function in vivo against ischemic damage (Klouz et al., 2008; Tagashira et al., 2013).
In this study, we analyzed whether the neuroprotective activity of ANAVEX2-73 involves mitochondrial protection. We analyzed the respiratory activity in mitochondria isolated from the mouse hippocampus, five days after ICV injection of oligomerized Aβ25–35. We measured respiration rates of freshly extracted mitochondria and the activity of the respiratory chain complexes I to IV. We then analyzed biochemical markers of mitochondrial damage, including Bax, Bcl-2 protein expression and cytochrome c release from the mitochondria into the cytosol. PRE-084 was used as a reference σ1R agonist.
Material and Methods
Animals
Male Swiss OF-1 mice (Depré, St Doulchard, France), aged 7–9 weeks and weighing 32 ± 2 g were used. They were housed in plastic cages in groups with free access to food and water, except during behavioral experiments. They were kept in a regulated environment (23 ± 1°C, 50–60% humidity) under a 12 h light/dark cycle (light on at 8:00 am). Behavioral experiments were carried out between 10:00 am and 4:00 pm, in an experimental room within the animal facility. All animal procedures were conducted in adherence with the 2010/63 EU Directive.
Drugs and Administration Procedures
The amyloid-β[25–35] (Aβ25–35) and scrambled Aβ25–35 (Sc. Aβ) peptides were purchased from Genepep (Saint Jean-de-Védas, France). They were solubilized in sterile distilled water at a concentration of 3 mg/ml and stored at −20°C until use. Before injection, peptides were aggregated by incubation at 37°C for 4 days (Maurice et al., 1996). This procedure is currently standardized in the laboratory and previous analyses using electronic microscopy, IR spectroscopy, and photon correlation spectroscopy techniques showed that the aggregated Aβ25–35 solution is mainly composed of a mixture of soluble oligomeric amyloid species, which sizes extended from 52.8 to 295.3 nm (98%) (Zussy et al., 2011, 2013). They were administered intracerebroventricularly (ICV) in a final volume of 3 µl per mouse, as previously described (Maurice et al., 1996; Meunier et al., 2006). Tetrahydro-N,N-dimethyl-2,2-diphenyl-3-furanmethanamine hydrochloride (ANAVEX2-73, AE37) was provided by Dr Alexandre Vamvakides (Anavex Life Science, Pallini, Greece). PRE-084 was a gift from Dr Tsung-Ping Su (NIDA/NIH, Baltimore, MD, USA). 2-(3,4-Dichlorophenyl)-N-(2-dimethylaminoethyl)-N-methylethanamine dihydrobromide (BD1047) was from Sigma-Aldrich (St Quentin-Fallavier, France). Drugs were solubilized in physiological saline at the concentration of 5 mg/ml and then diluted to the appropriate concentrations. They were injected intraperitoneally (IP) in a volume of 100 µl/20 g body weight. Drugs were injected once, 20 min before the Aβ25–35 peptide, also injected once. Animals were used 1, 3, 5, or 7 days after injections, as indicated in the figure legends.
Lipid Peroxidation
Mice were killed by decapitation and brains were rapidly removed, the hippocampus dissected out, weighed, and kept in liquid nitrogen until assayed. After thawing, the hippocampus was homogenized in cold methanol (1/10 w/v), centrifuged at 1,000 g for 5 min and the supernatant collected. Homogenate was added to a solution containing 1 mM FeSO4, 0.25 M H2SO4, 1 mM xylenol orange, and incubated for 30 min at room temperature. Absorbance was measured at 580 nm (A5801), and 10 ml of 1 mM cumene hydroperoxide (CHP; Sigma-Aldrich) was added to the sample and incubated for 30 min at room temperature, to determine the maximal oxidation level. Absorbance was measured at 580 nm (A5802). The level of lipid peroxidation was determined as CHP equivalents according to: CHP eq. 1/4 A5801/A5802 [CHP (nmol)] dilution, and expressed as CHP eq. per wet tissue weight.
Respiration Rates
Five days after peptide injection, mice were sacrificed, and their hippocampus rapidly removed on ice and homogenized in 10 ml of ice-cold homogenization buffer (220 mM mannitol, 70 mM sucrose, 10 mM 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES; Sigma-Aldrich), 2 mM ethylene glycol tetraacetic acid (EGTA; Sigma-Aldrich), pH 7.4) using a Teflon potter homogenizer. Homogenate was centrifuged at 1,000 g for 5 min. The supernatant was then centrifuged at 10,000 g for 10 min. Pellet was collected and resuspended in 50 µl of ice-cold isolation buffer (220 mM mannitol, 70 mM sucrose, 10 mM HEPES, 0.01 mM EGTA, pH 7.4). Mitochondrial protein content was determined by spectrophotometry. O2 consumption was measured with a Clarke type oxygen electrode (Hansatech, Cergy, France). Mitochondria (0.8 mg/ml) were loaded in the chamber filled up with respiration buffer maintained at 30°C (50 mM sucrose, 100 mM KCl, 10 mM HEPES, 5 mM KH2PO4, 0.5 mM MgCl2, pH 7.4). State 2 respiration was measured after addition of 5 mM pyruvate-malate and state 3 respiration was triggered by addition of 0.5 mM adenosine diphosphate (ADP; Sigma-Aldrich). State 4 respiration was then measured after addition of 4 µM carboxyatractyloside (CAT; Sigma-Aldrich), a blocker of the adenosine trisphosphate (ATP)-ADP carrier, and uncoupled state with 1 µM tyrphostine (uncoupling agent). The mean of two measures of respiration rates for the four states were calculated for each sample. The control ratio was calculated as the ratio between state 3 and state 4 respiratory rates.
In a series of experiment, the effects of direct application of the drugs (ANAVEX2-73 and PRE-084, in the 10−8 to 10−4 M concentration range) were examined. Isolated mitochondria (0.8 mg/ml) were loaded in the chamber. Drugs were applied and after 5 min, the respiration was examined.
Complex Activities
Five days after peptide injection, mice were sacrificed, and their hippocampus rapidly removed on ice. Mitochondria were isolated in the same way as for respiration rates. Mitochondrial protein content was determined using a BCA assay (Pierce Biotechnology, Rockford, IL, USA). Mitochondria were frozen/thawed four times to break mitochondrial membranes. Complex activities were measured at 30°C, using a Jasco V-530 spectrophotometer (Jasco, Nantes, France). Complex activities are calculated as nmol/min/mg of proteins and expressed as percentage of Sc. Aβ-treated group data.
Complex I (NADH ubiquinone reductase)
The oxidation of nicotinamide adenine dinucleotide (NADH; Sigma-Aldrich) by complex I was measured using decylubiquinone (DUQ; Sigma-Aldrich), an ubiquinone analog, as electron acceptor. 200 µM NADH and 100 µM DUQ were added to the assay medium (25 mM KH2PO4, 5 mM MgCl2, 250 µM KCN, 1 mg/ml bovine serum albumin (BSA; Sigma-Aldrich), pH 7.4) in a final volume of 1.5 ml. Enzyme activity was measured by initiating the reaction with 0.15 mg of mitochondrial protein. The decrease in absorbance due to NADH oxidation was measured at 340 nm.
Complex II (succinate dehydrogenase)
The activity of complex II was measured indirectly by record of the reduction of 2,6-dichlorophenolindo-phenol (DCIP; Sigma-Aldrich) following the oxidation of succinate. 6 mM succinate was incubated with 0.05 mg of mitochondrial proteins for 5 min at 30°C in the assay medium (10 mM KH2PO4, 2 mM ethylenediaminetetraacetic acid (EDTA; Sigma-Aldrich), 2 µM rotenone, 1 mg/ml BSA) in a final volume of 1.5 ml. 100 µM DUQ and 80 µM DCIP were then added and the decrease in absorbance at 600 nm, corresponding to the reduction of DCIP, was measured.
Complex III (ubiquinol cytochrome c reductase)
The oxidation of DUQH2 by complex III was determined using cytochrome c(III) as an electron acceptor. 0.25 mg of mitochondrial protein was added in the assay medium (10 mM KH2PO4, 2 mM EDTA, 1 mg/ml BSA, 2 µM rotenone, 250 µM KCN) in a final volume of 1.5 ml. The reaction was started with the addition of 100 µM DUQH2 (Sigma-Aldrich) and the increase in absorbance at 550 nm, corresponding to reduction of cytochrome c was measured. Because cytochrome c(III) could also be reduced by DUQH2 independently of complex III, the reduction of cytochrome c(III) was measured in the presence of 1 µM antimycin (an inhibitor of complex III activity) and deducted from the total reduction measured before.
Complex IV (cytochrome c oxidase)
0.025 mg of mitochondrial protein were incubated for 1 min at 30°C in the assay medium (10 mM KH2PO4, 10 mM EDTA, 2 mM MgCl2, 1 mg/ml BSA) in a final volume of 1.5 ml. The reaction was started when 33 µM of cytochrome c(II) was added to the assay medium and the decreased in absorbance at 550 nm, corresponding to oxidation of cytochrome c(II), was measured.
Western Blotting
Mice were sacrificed at indicated days after injections and the hippocampus rapidly dissected on ice and kept at −80°C until used. For cytochrome c release experiments, the hippocampus were homogenized with a glass Dounce homogenizer in ice-cold homogenization buffer (250 µM sucrose, 10 mM HEPES, pH 7.4), including a protease and phosphatase inhibitor cocktail (Roche Diagnostics, Meylan, France) in a final volume of 250 µl. Homogenates were centrifuged at 600 g for 5 min and the supernatant collected and centrifuged again at 10,300 g for 20 min. The supernatant, corresponding to the cytosolic fraction (C), and the pellet, corresponding to the crude mitochondrial fraction (M), were separated. The mitochondrial fraction was resuspended in 50 µl of ice-cold isolation buffer (250 mM mannitol, 5 mM HEPES, 0.5 mM EGTA, pH 7.4).
For Bax and Bcl-2 experiments, the hippocampus were homogenized by sonication in an ice-cold lysis buffer (125 mM Tris HCl pH 6.8, 4% sodium dodecyl sulfate (SDS; Sigma-Aldrich), 20% glycerol) including a protease and phosphatase inhibitor cocktail (Roche Diagnostics, Meylan, France). Homogenates were heated at 70°C for 10 min and centrifuged at 16,000 g for 30 min. The supernatant was collected.
For all samples, protein concentration was determined using a BCA assay (Pierce Biotechnology, Rockford, IL, USA) according to the manufacturer’s instructions.
Proteins, 20 µg per lane, were resolved on a 12% SDS-polyacrylamid gel and transfered to a polyvinylidene fluoride (PVDF) membrane (GE Healthcare, Orsay, France). After 1 h blocking in 5% non-fat dry milk in a 20 mM Tris-buffered saline pH 7.5 buffer containing 0.1% Tween-20 (TBS-T), membranes were incubated overnight at 4°C with the following primary antibodies: mouse anti-cytochrome c (CytC, dilution 1/1000; BioLegend, San Diego, CA, USA), mouse anti-oxphos-complex IV subunit I (Oxphos, 1/1000; Invitrogen Life Technologies, St Aubin, France), rabbit anti-Bax (1/2000; Cell Signaling Technology, Ozyme, St Quentin-en-Yvelines, France), mouse anti-Bcl-2 antibody (1/1000; Santa Cruz Biotechnology; Heidelberg, Germany). After brief washes, membranes were incubated for 1 h at room temperature with corresponding secondary antibody: goat anti-mouse IgG peroxidase conjugate (1/2000; Sigma-Aldrich) or goat anti-rabbit IgG peroxidase conjugate (1/2000; Sigma-Aldrich). The immunoreactive bands were visualized with the enhanced chemiluminescence reagent (ECL, Millipore, Molsheim, France) using an Odyssey® Fc fluorescent imaging system (Li-Cor, Eurobio, Courtaboeuf, France). Then, membranes were stripped using the Restore Western Blot Stripping Buffer (Pierce Biotechnology) and reprobed with anti-β-tubulin antibody (1/2000) (Sigma-Aldrich). The intensity of peroxidase activity was quantified using the Odyssey® Fc software (Li-Cor).
Statistical Analyses
Data were expressed as mean ± S.E.M. The number of animals is indicated in the figure legends. Data were analyzed using one-way ANOVA (F values), followed by the Dunnett’s post-hoc multiple comparison test. For reading clarity, ANOVA values were all reported in the figure legends. The level of statistical significance was p < 0.05.
Results
ICV administration of Aβ25–35 in mice induced a robust (+50–70%) increase in lipid peroxidation level in the hippocampus of the animals, starting at 5–7 days after injection (Figure 1A). The IP pretreatment with ANAVEX2-73, in the 0.01–1 mg/kg dose-range dose-dependently prevented the Aβ25–35-induced increase in lipid peroxidation with highly significant effect measured at 0.3 and 1 mg/kg (Figure 1B). The reference σ1R agonist PRE-084 similarly blocked Aβ25–35-induced increase in lipid peroxidation at 0.5 and 1 mg/kg (Figure 1C). Note that both drug effects at the highest doses were completely blocked by the reference σ1R antagonist BD1047, confirming the σ1R selectivity (Figures 1B,C). These initial observations confirmed that ICV injection of Aβ25–35 induces a marked oxidative stress in mice and that the two compounds act as anti-oxidant drugs. The prominent role of mitochondrial dysfunction in ROS generation and oxidative stress led us to analyze the impact of the drug treatments on mitochondrial respiration.
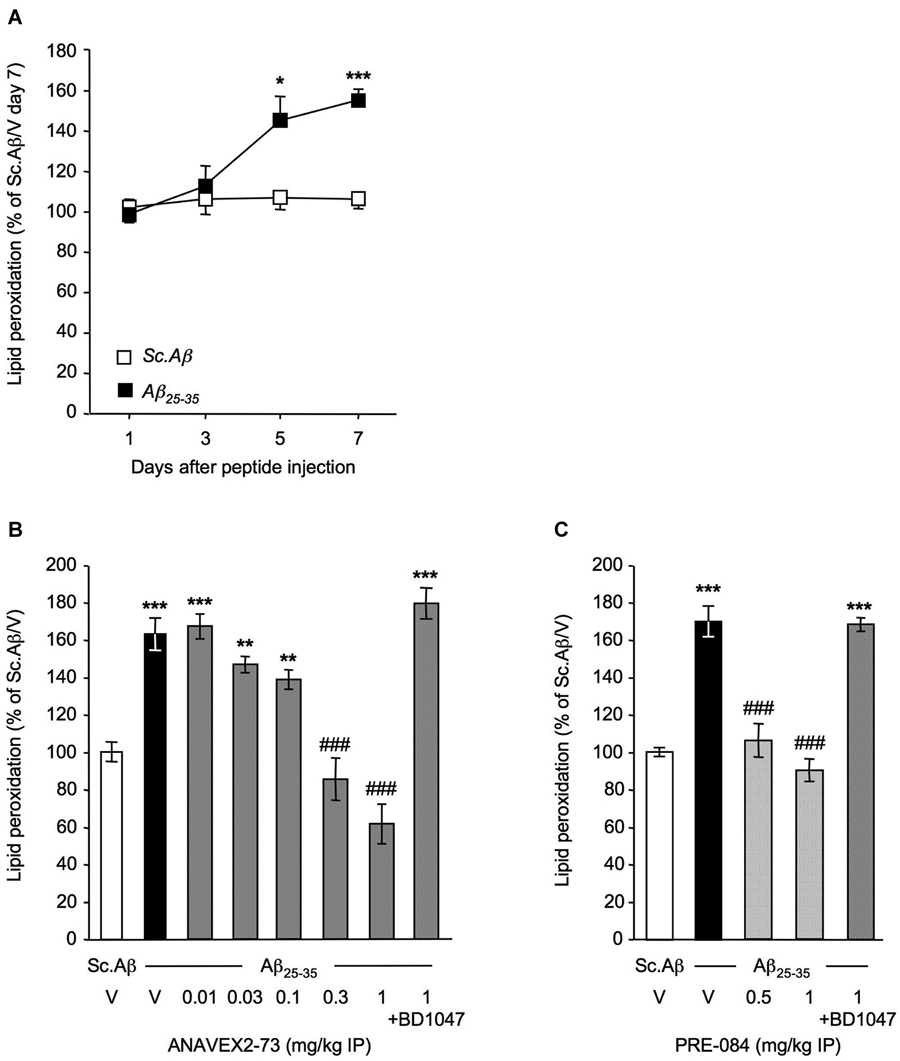
Figure 1. ANAVEX2-73 and PRE-084 prevented oxidative stress in Aβ25–35-treated mice. Mice were administered ICV with Sc. Aβ or Aβ25–35 peptide (9 nmol) and sacrificed after 1, 3, 5 or 7 days for lipid peroxidation measures (A). n = 6 per group, *p < 0.05, ***p < 0.001 vs. the (Sc. Aβ + V)-treated group at the same timepoint; t-test. Mice were then administered IP with (B) ANAVEX2-73 (0.01–1 mg/kg) or (C) PRE-084 (0.5–1 mg/kg) 20 min before Aβ25–35 (9 nmol). In two groups, BD1047 (10 mg/kg) was administered simultaneously with the highest dose of each agonist. Mice were sacrificed after 7 days for lipid peroxidation measures. One-way ANOVA: F(7,70) = 22.5, p < 0.0001, n = 6–14 per group in (B); F(4,69) = 31.9, p < 0.0001, n = 6–22 in (C). **p < 0.01, ***p < 0.001 vs. the (Sc. Aβ+V)-treated group; ###p < 0.001 vs. the (Aβ25–35+V)-treated group; Dunnett’s test.
O2 consumption was analyzed in mice, five days after Sc. Aβ and Aβ25–35 injection, and following ANAVEX2-73 or PRE-084 treatment (Figure 2). The Aβ25–35 ICV treatment failed to affect state 2 or state 4 O2 consumption (Figure 2A), but significantly altered by −23% and −22% the state 3 or uncoupled state O2 consumption, respectively (Figure 2B). As a consequence the peptide injection significantly altered the respiratory control ratio by −17% (Figure 2C). The ANAVEX2-73 treatment (0.3 mg/kg IP) significantly prevented the Aβ25–35-induced decreases in O2 consumption (Figure 2B). However, since the drug increased in fact significantly O2 consumption at all states (Figure 2A), there was no significant impact on the respiratory control ratio (Figure 2C). A similar profile was observed with PRE-084 (0.5 mg/kg IP) in Aβ25–35-treated mice (Figures 2A,B) but the increase in state 3 O2 consumption was more marked resulting in a restoration of the respiratory control ratio value, i.e., a significant prevention of the Aβ25–35-induced alteration (Figure 2C). Note that neither ANAVEX2-73 nor PRE-084 affected O2 consumption in Sc. Aβ-treated animals. In addition, we analyzed whether the drugs have a direct effect on mitochondrial respiration. The drugs were both applied at increasing concentrations, from 10−8 M to 10−4 M, on isolated mitochondria and respiration analyzed. State 3 O2 consumption and respiratory control ratio are presented (Figures 3A,B). The drugs affected both parameters only at the highest concentrations tested.
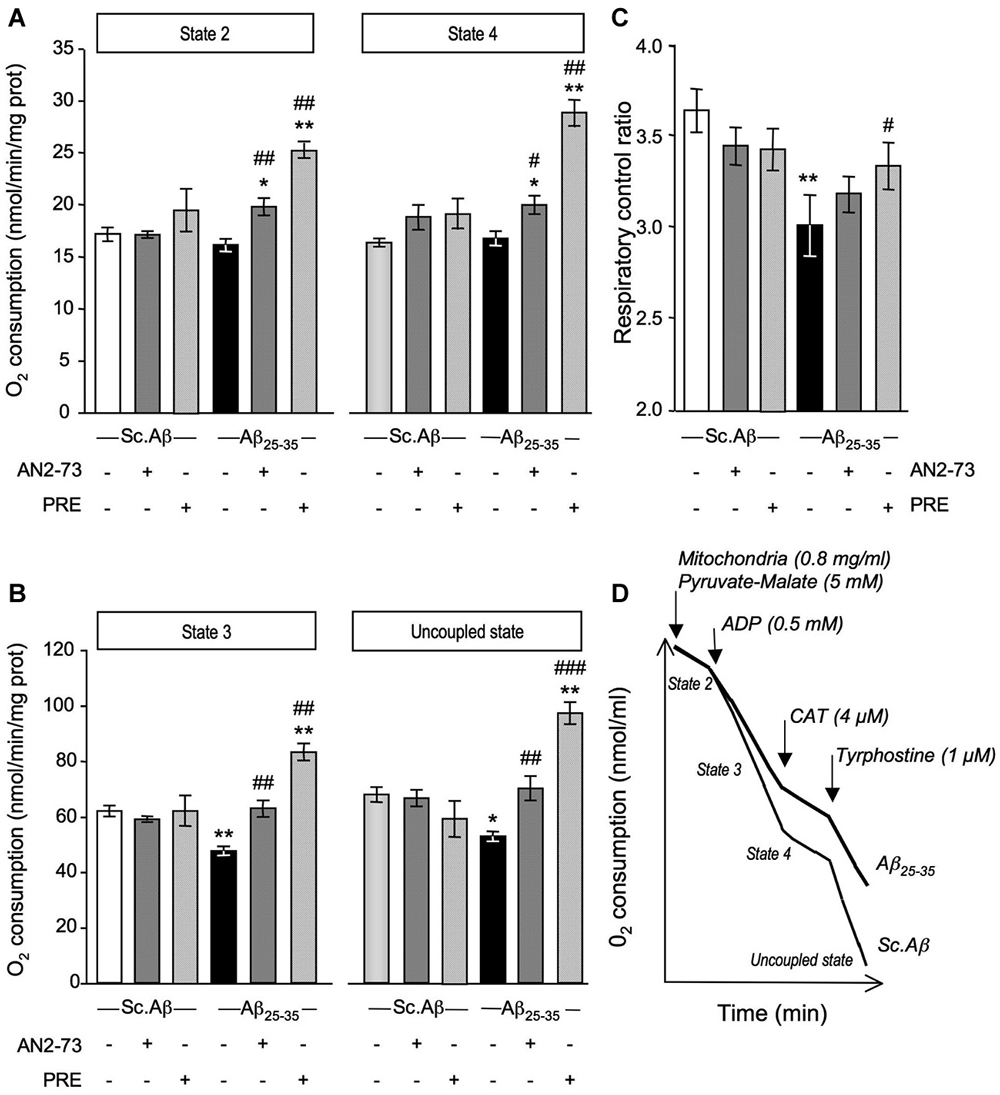
Figure 2. ANAVEX2-73 (AN2-73) and PRE-084 (PRE) protected against Aβ25–35-induced alteration of mitochondrial respiration in mice. Mice were treated with ANAVEX2-73 (0.3 mg/kg IP), PRE-084 (0.5 mg/kg IP) or vehicle, before injection of Sc. Aβ or Aβ25–35 (9 nmol ICV). (A–C) Mitochondria (0.8 mg/ml) were loaded in the chamber with appropriate buffer at 30°C. State 2 respiration was activated by addition of pyruvate-malate (5 mM). State 3 respiration was induced with ADP. State 4 respiration was provoked by the addition of carboxyatractyloside (CAT), a blocker of ATP-ADP carrier. Finally, addition of tyrphostine, an uncoupling agent, activated the uncoupled respiration. The respiratory control ratio is the state 3/state 4 ratio. n = 4–8 per group, F(5,38) = 7.11, p < 0.0001 for state 2 and F(5,38) = 8.43, p < 0.0001 for state 4 in (A); F(5,38) = 13.1, p < 0.0001 for state 3 and F(5,38)= 8.43, p < 0.0001 for state 3 in (B); F(5,38) = 2.58, p < 0.05 in (C). *p < 0.05, **p < 0.01 vs. Sc. Aβ/Veh; #p < 0.05, ##p < 0.01, ###p < 0.001 vs. Aβ25–35/Veh. Dunnett’s test. (D) Typical trace of O2 consumption for Sc. Aβ and Aβ25–35-injected mice.
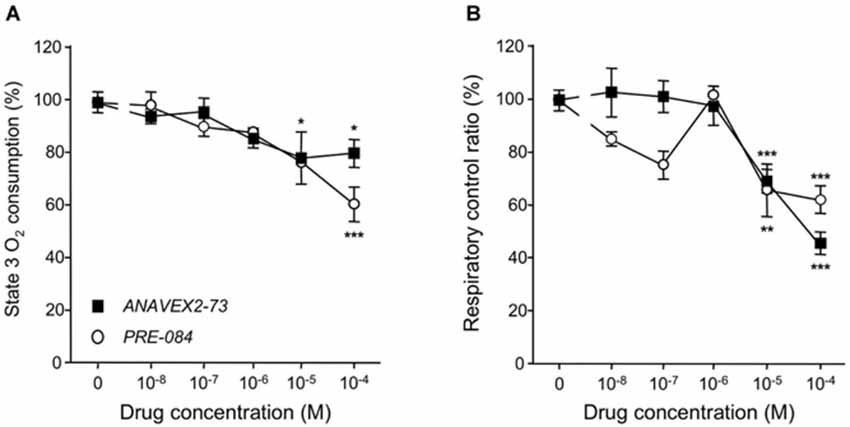
Figure 3. Direct application of ANAVEX2-73 or PRE-084 affected mitochondrial respiration only at high concentrations. Mitochondria (0.8 mg/ml) were loaded in the chamber with appropriate buffer at 30°C. The drugs (10−8 to 10−4 M) were bath applied 5 min before the repspiration measures. State 2 respiration was activated by addition of pyruvate-malate (5 mM). State 3 respiration was induced with ADP. State 4 respiration was provoked by the addition of carboxyatractyloside (CAT), a blocker of ATP-ADP carrier. The respiratory control ratio is the state 3/state 4 ratio. n = 4–8 per group (n = 14 for the no-drug group), F(5,38) = 3.08, p < 0.05 for ANAVEX2-73 and F(5,31) = 5.19, p < 0.01 for PRE-084 in (A); F(5,38) = 22.1, p < 0.0001 for ANAVEX2-73 and F(5,31)= 7.53, p < 0.001 for PRE-084 in (B). *p < 0.05, **p < 0.01, ***p < 0.001 vs. no-drug data; Dunnett’s test.
We analyzed the nature of the Aβ25–35-induced alteration of respiration (Figure 4). The measure of each complex activity showed that only the complex IV activity was significantly decreased by −14% (Figure 4A). The ANAVEX2-73 treatment prevented the Aβ25–35-induced decrease in complex IV activity (Figure 4B).
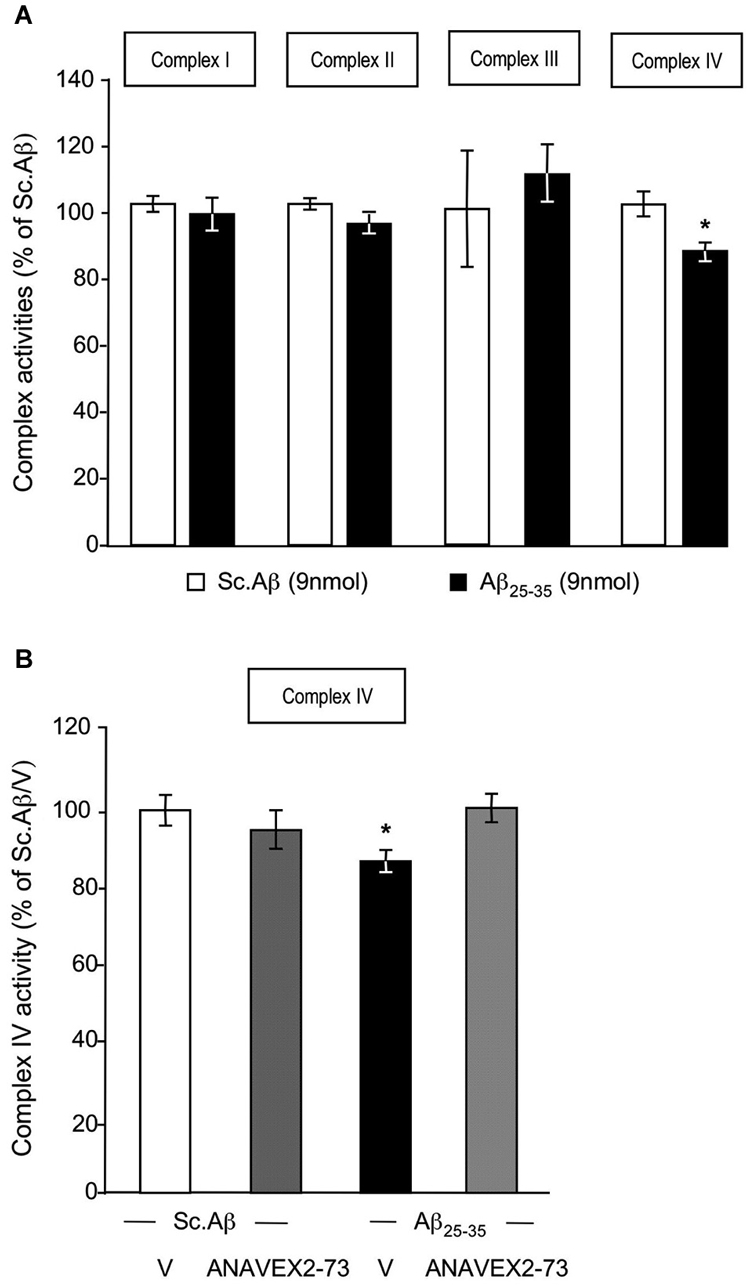
Figure 4. ANAVEX2-73 protected against Aβ25–35-induced alteration of complex IV activity. Mitochondria were loaded in the spectrophotometer cuvette with appropriate buffer at 30°C and variations of absorption were recorded after addition of specific substrates. (A) Effect of Aβ25–35 ICV injection on complex activities (% of Sc. Aβ) n = 8–9 per group, * p < 0.05 vs. Sc. Aβ Student’s t-test. (B) Mice were treated with ANAVEX2-73 (0.3 mg/kg IP) or vehicle, before injection of Sc. Aβ or Aβ25–35 (9 nmol ICV). Complex IV activity was measured after 7 days (% of Sc. Aβ). n = 5–9 per group, F(3,26) = 3.00, p < 0.05. * p < 0.05 vs. Sc. Aβ; Dunnett’s test.
Among the different biochemical markers that can be examined to evidence the damage resulting from impaired mitochondrial functionality, we first addressed cytochrome c release from the mitochondrial membrane into the cytosol (Figure 5A). The time-course analysis showed that significant cytochrome c release can be evidenced at day 5 after the peptide injection (Figure 5B). At this timepoint, both the ANAVEX2-73 treatment (Figure 5C) and PRE-084 treatment (Figure 5D) completely prevented the Aβ25–35-induced cytochrome c release in the mouse hippocampus.
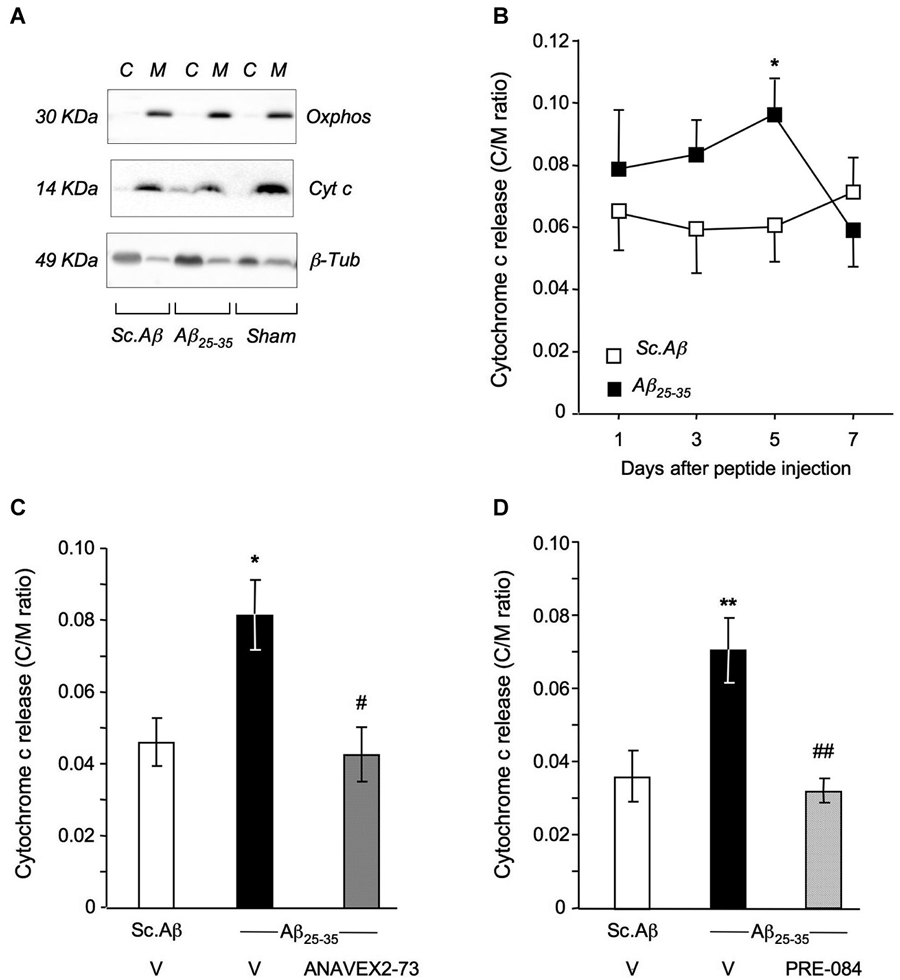
Figure 5. ANAVEX2-73 and PRE-084 prevented Aβ25–35-induced cytochrome c release. (A) Typical blots of mitochondrial (M) and cytosolic (C) fractions with cytochrome c, oxphos and β-tubulin antibodies. (B) Cytochrome c content (cytosol to mitochondrial content ratios). Mice were injected with Sc. Aβ or Aβ25–35 (9 nmol ICV) and sacrificed at indicated days. n = 9–12 per group. * p < 0.05 vs. Sc. Aβ. Student’s t-test. (C, D) Mice were treated with ANAVEX2-73 (0.3 mg/kg IP) or PRE-084 (0.5 mg/kg IP) before Aβ25–35 and sacrificed after 5 days. n = 8–17, F(2,37) = 5.96, p < 0.01 in (C); n = 8–14, F(2,33) = 8.19, p < 0.01 in (D). *p < 0.05, **p < 0.01 vs. Sc. Aβ, #p < 0.05, ##p < 0.01 vs. Aβ25–35; Dunnett’s test.
We then measured Bax and Bcl-2 protein expression, by a western blot approach, in order to confirm that the impact of the treatment on mitochondrial damage readily transfer into anti-apoptosis protection (Figure 6). The Aβ25–35 treatment resulted, between 1 to 7 days after injection, in a progressive but non-significant increase in Bax level in the hippocampus (Figure 6A), accompanied by a trend to diminution of Bcl-2 level (Figure 6B). As a consequence, the Bax/Bcl-2 ratio increased, and significantly at days 5 and 7 after the peptide injection (Figure 5C). ANAVEX2-73 was tested at 0.1–1 mg/kg. A dose-dependent decrease in Bax level was measured at the highest dose tested (Figure 6D). The drug impacted marginally Bcl-2 levels (Figure 6E), but the data examined as Bax/Bcl-2 ratio clearly showed a beneficial effect of the compound, particularly significant at 0.3 mg/kg (Figure 6F).
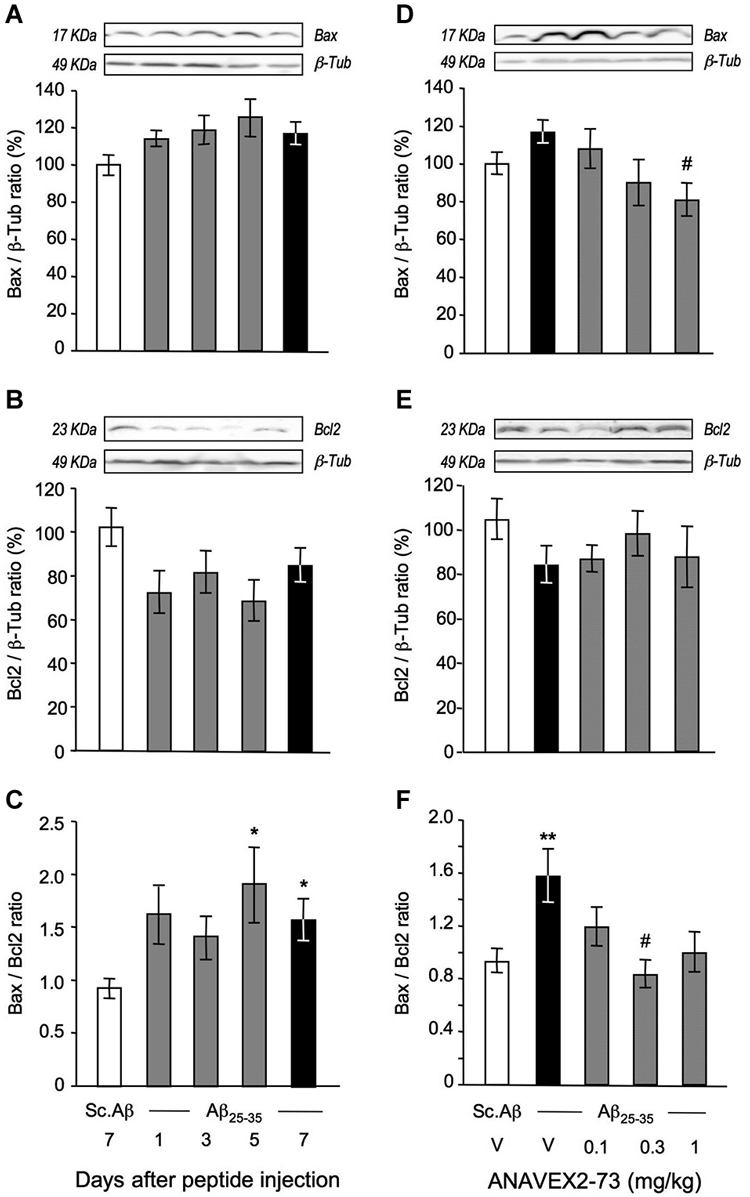
Figure 6. ANAVEX2-73 protected against Aβ25–35-induced increase in Bax/Bcl-2 ratio in the mouse hippocampus. (A–C) Mice were sacrificed at 1, 3, 5 and 7 days after Aβ25–35 injection and Bax and Bcl-2 contents determined by western blots. (D–F) Effect of ANAVEX2-73 (0.1–1 mg/kg IP) on Bax and Bcl-2 levels, 7 days after Aβ25–35 injection. Typical blots are represented above the bar graphs. Mice were injected with ANAVEX2-73 or vehicle before Aβ25–35 peptide (9 nmol). n = 5–15 per groups. F(4,44) = 1.98, p > 0.05 in (A) ; F(4,44) = 1.95, p > 0.05 in (B) ; F(4,44) = 2.66, p < 0.05 in (C); F(4,49)= 3.01, p < 0.05 in (D), F(4,49) = 0.89, p > 0.05 in (E), F(4,49) = 3.09, p < 0.05 in (F). *p < 0.05, **p < 0.01 vs. Sc. Aβ; #p < 0.05 vs. Aβ25–35; Dunnett’s test.
Discussion
In this study, we showed that mitochondrial functionality was altered several days after an ICV injection of the Aβ25–35 peptide in mouse. It was shown by reduced respiration rates, decreased complex IV activity and increased oxidative stress and proapoptotic markers. Five days after a single injection of oligomeric Aβ25–35 peptide, respiration was reduced in isolated mitochondria from mouse hippocampus, as compared with control Sc. Aβ-treated mice. Basal respiration (State 2 and State 4) was unaffected by Aβ25–35, but ADP-stimulated respiration (State 3) was significantly decreased. Reduced State 3 respiration resulted in a decreased control ratio in Aβ25–35-injected mice. Uncoupled state was also altered, suggesting a defect in respiratory chain complex I-IV rather than a defect in ATP synthase complex activity. The reduced respiratory rates and control ratio observed in this in vivo model are coherent with previous studies exploring the impact of Aβ peptides, and particularly Aβ25–35, on mitochondrial respiration in vitro. Application of Aβ25–35 on isolated mitochondria decreased respiration rates (Casley et al., 2002a; Aleardi et al., 2005; Clementi et al., 2005). Interestingly, in these studies, basal respiration (State 4) was also altered after Aβ25–35 treatment. In the same way, mitochondria isolated from cultured neurons and exposed to Aβ25–35 showed decreased ATP production, suggesting a reduced mitochondrial respiration (Casley et al., 2002b; Dong et al., 2010). In search for an explanation to this decreased mitochondrial respiration after Aβ25–35, we measured the respiratory chain complex I-IV activities. We found that complex IV activity was significantly reduced 5 days after Aβ25–35. In most of the in vitro studies, application of Aβ25–35 on cultured neurons or directly on isolated mitochondria resulted in reduced activities of complexes I and IV (Casley et al., 2002a,b; Aleardi et al., 2005; Clementi et al., 2005; Dong et al., 2010). This appears to be a difference between in vitro and in vivo studies with the Aβ25–35 peptide. It could explain the decreased state 4 respiration observed in vitro. But, consistent with our results, are data in transgenic mice expressing human mutant amyloid precursor protein (hAPPm). Aβ peptide has been found to accumulate in mitochondria and respiratory rates and complex IV activity were also decreased in these animals (Caspersen et al., 2005; Rhein et al., 2009; Du et al., 2010). Moreover, Canevari et al. (1999) described a specific and concentration-dependent decrease in complex IV activity after Aβ25–35 application in isolated mitochondria. Disruption of complex IV activity induced by Aβ peptides could be due to a direct interaction between Aβ and the enzymatic subunits (Crouch et al., 2006; Hernandez-Zimbron et al., 2012) but also to the direct induction of oxidative stress by Aβ peptides, and particularly Aβ25–35. Indeed, membrane lipid peroxidation induced by ROS overproduction might alter complex IV subunits and disrupt complex IV activity (Paradies et al., 1998; Bobba et al., 2013), as we confirmed the marked increase in oxidative stress in mouse hippocampus, already described in this model (Meunier et al., 2006). The release of cytochrome c from mitochondria into the cytosol that was increased 5 days after Aβ25–35 injection can also contribute to the dysfunction of the mitochondrial respiratory chain and thus to the oxidative stress. Concomitantly to the cytochrome c release, the ratio Bax/Bcl-2 was significantly increased in mouse hippocampus, 5 and 7 days after Aβ25–35 injection and this is likely related to the cellular response to the high level of oxidative stress and decreased mitochondrial respiration (Mattson et al., 2008).
The aim of the study was to establish whether the localization of the σ1R on mitochondria (Klouz et al., 2002) and more precisely at MAM (Hayashi and Su, 2007) is related to its particular neuroprotective activity. Indeed, both ANAVEX2-73 and PRE-084, prevented Aβ25–35-induced oxidative stress and increase in pro-apototic markers. The results are coherent with previously reported observations, since the σ1R has been involved in regulation of oxidative stress (Pal et al., 2012; Tsai et al., 2012). Moreover, σ1R agonists prevented oxidative stress-induced apoptosis in vitro through upregulation of the anti-apoptotic Bcl-2 protein expression (Meunier and Hayashi, 2010). The σ1R activation also prevented Bax increase and caspase-3 activation, linked to cytochrome c release, after glutamate insult in retinal ganglional cells (Tchedre and Yorio, 2008). Another study also showed a decreased caspase-3 activation after PRE-084 treatment in a Huntington’s disease model (Hyrskyluoto et al., 2013). ANAVEX2-73 and PRE-084 both fully reversed state 3 and uncoupled state respiration deficits. Surprisingly both drugs also increased basal respiration (state 2 and state 4) and PRE-084, but not ANAVEX2-73 increased state 3 and uncoupled state respiration, compared to control mice. These differences in ANAVEX2-73 and PRE-084 effects could be explained by the higher affinity of the later for the σ1 receptor. Although state 3 respiration raised to the control level after ANAVEX2-73 treatment, CR was not different from the Aβ25–35 group because state 4 respiration also increased. However, the high increased of both state 3 and state 4 respiration after PRE-084 treatment increased the control ratio to the control level. Such increased respiration after stimulation of σ1R has never been described and is not observed in Sc. Aβ-treated control mice. An attempt to explain this phenomenon is challenging. The increased state 3 and uncoupled state respiration rates observed after PRE-084 treatment were clearly not an artifact or due to experimental conditions as all the samples were prepared in strictly the same way and measures realized randomly among treatment groups and in several experimental sessions. Few studies have examined the involvement of σ1R in mitochondrial respiration. BHDP is a selective ligand with nanomolar affinity for the σ1 receptor (Klouz et al., 2003). A three days pretreatment with BHDP in rats prevented the decrease in CR in mitochondria isolated from rat liver after ischemia-reperfusion (Klouz et al., 2008). We previously suggested that protective effect on mitochondrial function of σ1 agonist could be attributable to the presence of σ1R to the mitochondrial membrane (Klouz et al., 2002). Indeed, σ1R are present in lipid rafts and modify their composition and particularly the ganglioside content (Hayashi and Su, 2005). In mitochondrial membrane, lipid rafts microdomains, and particularly the glycosphingolipid GD3, are involved in alteration of mitochondrial membrane potential and apoptosis (Malorni et al., 2007). Therefore, an explanation of increased mitochondrial respiration by σ1 ligand could be through the capacity of the protein to modulate lipid rafts gangliosids at mitochondrial membrane. Another explanation could be the chaperone activity of the σ1 protein on InsP3 receptor at the ER membrane (Hayashi and Su, 2007). The InsP3 receptor stabilization allows proper Ca2+ exchange between ER and mitochondria, which are critical for mitochondrial bioenergetics (Cárdenas et al., 2010). Cutamesine (SA4503), another selective σ1 agonist, increased ATP production in cardiomyocytes, even without pathological conditions, suggesting an increase in mitochondrial respiration after σ1R stimulation (Tagashira et al., 2013). The increased mitochondrial bioenergetics after σ1 agonist treatment is largely attribuable to ER Ca2+ mobilization into mitochondria, because treatment with xestospongin C, an antagonist of the InsP3 receptor abolished the effects of the σ1 agonist on ATP production. However, in our model, this mechanism did not seem to be involved in the σ1 protein-induced increased mitochondrial respiration because the agonist was injected only once, five days before the mitochondrial isolation. Moreover, Tsai et al. (2009) described Ca2+-independent perturbations of mitochondrial functions in hippocampal neurons after σ1 receptor knockdown with siRNA. To note, another interesting consequence of σ1R activity is the modulation of different genes expression. Some of them are involved in oxidative stress regulation, e.g., NAD(P)H quinone oxidoreductase (Nqo1), Superoxide dismutase (SOD), Atf-4 and Prdx6 (Pal et al., 2012; Tsai et al., 2012). It could be worth investigating whether the PRE-084-induced increased respiratory efficacy could be due to the modification of expression of specific genes.
In conclusion, we provided here the clear evidence that the neuroprotective activity of σ1 receptor agonists, including the mixed muscarinic ligand/σ1 agonist ANAVEX2-73, involves mitochondrial protection in AD models. Indeed, the drug protected mitochondria against Aβ25–35 insult. Mitochondrial dysfunction in AD is a critical hallmark of the pathology, as mitochondria are, at the same time, a target and promoter of the physiopathological processus (for review, see Eckert et al., 2010; Selfridge et al., 2013). Mitochondrial dysfunction and, notably, resulting oxidative stress are responsible for the increased Aβ production (Dyrks et al., 1992; Leuner et al., 2007) and Tau hyperphosphorylation (Melov et al., 2007), the two biochemical signatures of AD. Thus, by preserving mitochondrial function ANAVEX2-73 could expectedly be more than a symptomatic drug and block the physiopathological amplification of the pathology.
Conflict of Interest Statement
Valentine Lahmy and Vanessa Villard are employees of Amylgen. Tangui Maurice is a member of the scientific advisory board of Anavex Life Sciences and scientific director of Amylgen. The companies had no role in the present research, except providing drug and funding. Other authors declare no conflict of interest.
Acknowledgments
This work was supported by collaboration contracts between Anavex Life Sciences (New York, NY, USA) and Amylgen (Montferrier-sur-Lez, France) and between Amylgen and the University of Montpellier (France). The companies had no role in study design or analysis. Valentine Lahmy is recipient of a CIFRE PhD funding (ANRT, Paris, France).
References
Aleardi, A. M., Benard, G., Augereau, O., Malgat, M., Talbot, J. C., Mazat, J. P., et al. (2005). Gradual alteration of mitochondrial structure and function by beta-amyloids: importance of membrane viscosity changes, energy deprivation, reactive oxygen species production and cytochrome c release. J. Bioenerg. Biomembr. 37, 207–225. doi: 10.1007/s10863-005-6631-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bobba, A., Amadoro, G., Valenti, D., Corsetti, V., Lassandro, R., and Atlante, A. (2013). Mitochondrial respiratory chain Complexes I and IV are impaired by β-amyloid via direct interaction and through Complex I-dependent ROS production, respectively. Mitochondrion 13, 298–311. doi: 10.1016/j.mito.2013.03.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Canevari, L., Clark, J. B., and Bates, T. E. (1999). β-Amyloid fragment 25–35 selectively decreases complex IV activity in isolated mitochondria. FEBS Lett. 457, 131–134. doi: 10.1016/S0014-5793(99)01028-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cárdenas, C., Miller, R. A., Smith, I., Bui, T., Molgó, J., Müller, M., et al. (2010). Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 142, 270–283. doi: 10.1016/j.cell.2010.06.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Casley, C. S., Canevari, L., Land, J. M., Clark, J. B., and Sharpe, M. A. (2002b). β-Amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J. Neurochem. 80, 91–100. doi: 10.1046/j.0022-3042.2001.00681.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Casley, C. S., Land, J. M., Sharpe, M. A., Clark, J. B., Duchen, M. R., and Canevari, L. (2002a). β-Amyloid fragment 25–35 causes mitochondrial dysfunction in primary cortical neurons. Neurobiol. Dis. 10, 258–267. doi: 10.1006/nbdi.2002.0516
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Caspersen, C., Wang, N., Yao, J., Sosunov, A., Chen, X., Lustbader, J. W., et al. (2005). Mitochondrial Aβ: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 19, 2040–2041. doi: 10.1096/fj.05-3735fje
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Clementi, M. E., Marini, S., Coletta, M., Orsini, F., Giardina, B., and Misiti, F. (2005). Aβ31–35 and Aβ25–35 fragments of amyloid beta-protein induce cellular death through apoptotic signals: role of the redox state of methionine-35. FEBS Lett. 579, 2913–2918. doi: 10.1016/j.febslet.2005.04.041
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Crews, L., and Masliah, E. (2010). Molecular mechanisms of neurodegeneration in Alzheimer’s disease. Hum. Mol. Genet. 19, R12–R20. doi: 10.1093/hmg/ddq160
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Crouch, P. J., Barnham, K. J., Duce, J. A., Blake, R. E., Masters, C. L., and Trounce, I. A. (2006). Copper-dependent inhibition of cytochrome c oxidase by Aβ1–42 requires reduced methionine at residue 35 of the Aβ peptide. J. Neurochem. 99, 226–236. doi: 10.1111/j.1471-4159.2006.04050.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Delobette, S., Privat, A., and Maurice, T. (1997). In vitro aggregation facilities β-amyloid peptide-(25-35)-induced amnesia in the rat. Eur. J. Pharmacol. 319, 1–4. doi: 10.1016/s0014-2999(96)00922-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dong, W., Huang, F., Fan, W., Cheng, S., Chen, Y., Zhang, W., et al. (2010). Differential effects of melatonin on amyloid-β peptide 25–35-induced mitochondrial dysfunction in hippocampal neurons at different stages of culture. J. Pineal Res. 48, 117–125. doi: 10.1111/j.1600-079x.2009.00734.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Du, H., Guo, L., Yan, S., Sosunov, A. A., McKhann, G. M., and Yan, S. S. (2010). Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc. Natl. Acad. Sci. U S A 107, 18670–18675. doi: 10.1073/pnas.1006586107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dyrks, T., Dyrks, E., Hartmann, T., Masters, C., and Beyreuther, K. (1992). Amyloidogenicity of βA4 and βA4-bearing amyloid protein precursor fragments by metal-catalyzed oxidation. J. Biol. Chem. 267, 18210–18217.
Eckert, A., Schulz, K. L., Rhein, V., and Götz, J. (2010). Convergence of amyloid-β and tau pathologies on mitochondria in vivo. Mol. Neurobiol. 41, 107–114. doi: 10.1007/s12035-010-8109-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Espallergues, J., Lapalud, P., Christopoulos, A., Avlani, V. A., Sexton, P. M., Vamvakides, A., et al. (2007). Involvement of the sigma1 (σ1) receptor in the anti-amnesic, but not antidepressant-like, effects of the aminotetrahydrofuran derivative ANAVEX1–41. Br. J. Pharmacol. 152, 267–279. doi: 10.1038/sj.bjp.0707386
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hanada, M., Aimé-Sempé, C., Sato, T., and Reed, J. C. (1995). Structure-function analysis of Bcl-2 protein. Identification of conserved domains important for homodimerization with Bcl-2 and heterodimerization with Bax. J. Biol. Chem. 270, 11962–11969. doi: 10.1074/jbc.270.20.11962
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hayashi, T., and Su, T. P. (2005). The potential role of σ1 receptors in lipid transport and lipid raft reconstitution in the brain: implication for drug abuse. Life Sci. 77, 1612–1624. doi: 10.1016/j.lfs.2005.05.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hayashi, T., and Su, T. P. (2007). Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca2+ signaling and cell survival. Cell 131, 596–610. doi: 10.1016/j.cell.2007.08.036
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hernandez-Zimbron, L. F., Luna-Muñoz, J., Mena, R., Vazquez-Ramirez, R., Kubli-Garfias, C., Cribbs, D. H., et al. (2012). Amyloid-β peptide binds to cytochrome C oxidase subunit 1. PLoS One 7:e42344. doi: 10.1371/journal.pone.0042344
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hyrskyluoto, A., Pulli, I., Törnqvist, K., Ho, T. H., Korhonen, L., and Lindholm, D. (2013). Sigma1 receptor agonist PRE084 is protective against mutant huntingtin-induced cell degeneration: involvement of calpastatin and the NF-κB pathway. Cell Death Dis. 4:e646. doi: 10.1038/cddis.2013.170
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jürgensmeier, J. M., Xie, Z., Deveraux, Q., Ellerby, L., Bredesen, D., and Reed, J. C. (1998). Bax directly induces release of cytochrome c from isolated mitochondria. Proc. Natl. Acad. Sci. USA 95, 4997–5002. doi: 10.1073/pnas.95.9.4997
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kaminsky, Y. G., and Kosenko, E. A. (2008). Effects of amyloid-β peptides on hydrogen peroxide-metabolizing enzymes in rat brain in vivo. Free Radic. Res. 42, 564–573. doi: 10.1080/10715760802159057
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Klouz, A., Saïd, D. B., Ferchichi, H., Kourda, N., Ouanes, L., Lakhal, M., et al. (2008). Protection of cellular and mitochondrial functions against liver ischemia by N-benzyl-N’-(2-hydroxy-3,4-dimethoxybenzyl)-piperazine (BHDP), a σ1 ligand. Eur. J. Pharmacol. 578, 292–299. doi: 10.1016/j.ejphar.2007.09.038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Klouz, A., Sapena, R., Liu, J., Maurice, T., Tillement, J. P., Papadopoulos, V., et al. (2002). Evidence for σ1-like receptors in isolated rat liver mitochondrial membranes. Br. J. Pharmacol. 135, 1607–1615. doi: 10.1038/sj.bjp.0704626
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Klouz, A., Tillement, J. P., Boussard, M. F., Wierzbicki, M., Berezowski, V., Cecchelli, R., et al. (2003). [3H]BHDP as a novel and selective ligand for σ1 receptors in liver mitochondria and brain synaptosomes of the rat. FEBS Lett. 553, 157–162. doi: 10.1016/s0014-5793(03)01011-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kroemer, G., and Reed, J. C. (2000). Mitochondrial control of cell death. Nat. Med. 6, 513–519. doi: 10.1038/74994
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lahmy, V., Meunier, J., Malmström, S., Naert, G., Givalois, L., Kim, S. H., et al. (2013). Blockade of Tau hyperphosphorylation and Aβ1–42 generation by the aminotetrahydrofuran derivative ANAVEX2–73, a mixed muscarinic and σ1 receptor agonist, in a nontransgenic mouse model of Alzheimer’s disease. Neuropsychopharmacology 38, 1706–1723. doi: 10.1038/npp.2013.70
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Leuner, K., Hauptmann, S., Abdel-Kader, R., Scherping, I., Keil, U., Strosznajder, J. B., et al. (2007). Mitochondrial dysfunction: the first domino in brain aging and Alzheimer’s disease?. Antioxid. Redox Signal. 9, 1659–1675. doi: 10.1089/ars.2007.1763
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Leuner, K., Müller, W. E., and Reichert, A. S. (2012). From mitochondrial dysfunction to amyloid β formation: novel insights into the pathogenesis of Alzheimer’s disease. Mol. Neurobiol. 46, 186–193. doi: 10.1007/s12035-012-8307-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, X., Kim, C. N., Yang, J., Jemmerson, R., and Wang, X. (1996). Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 86, 147–157. doi: 10.1016/s0092-8674(00)80085-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lustbader, J. W., Cirilli, M., Lin, C., Xu, H. W., Takuma, K., Wang, N., et al. (2004). ABAD directly links Aβ to mitochondrial toxicity in Alzheimer’s disease. Science 304, 448–452. doi: 10.1126/science.1091230
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Malorni, W., Giammarioli, A. M., Garofalo, T., and Sorice, M. (2007). Dynamics of lipid raft components during lymphocyte apoptosis: the paradigmatic role of GD3. Apoptosis 12, 941–949. doi: 10.1007/s10495-007-0757-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Manczak, M., Anekonda, T. S., Henson, E., Park, B. S., Quinn, J., and Reddy, P. H. (2006). Mitochondria are a direct site of a beta accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 15, 1437–1449. doi: 10.1093/hmg/ddl066
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marzo, I., Brenner, C., Zamzami, N., Jürgensmeier, J. M., Susin, S. A., Vieira, H. L., et al. (1998). Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science 281, 2027–2031. doi: 10.1126/science.281.5385.2027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mattson, M. P., Gleichmann, M., and Cheng, A. (2008). Mitochondria in neuroplasticity and neurological disorders. Neuron 60, 748–766. doi: 10.1016/j.neuron.2008.10.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maurer, I., Zierz, S., and Möller, H. J. (2000). A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol. Aging 21, 455–462. doi: 10.1016/s0197-4580(00)00112-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maurice, T., Lockhart, B. P., and Privat, A. (1996). Amnesia induced in mice by centrally administered β-amyloid peptides involves cholinergic dysfunction. Brain Res. 706, 181–193. doi: 10.1016/0006-8993(95)01032-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Melov, S., Adlard, P. A., Morten, K., Johnson, F., Golden, T. R., Hinerfeld, D., et al. (2007). Mitochondrial oxidative stress causes hyperphosphorylation of tau. PLoS One 2:e536. doi: 10.1371/journal.pone.0000536
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Meunier, J., and Hayashi, T. (2010). Sigma-1 receptors regulate Bcl-2 expression by reactive oxygen species-dependent transcriptional regulation of nuclear factor κB. J. Pharmacol. Exp. Ther. 332, 388–397. doi: 10.1124/jpet.109.160960
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Meunier, J., Ieni, J., and Maurice, T. (2006). The anti-amnesic and neuroprotective effects of donepezil against amyloid β25–35 peptide-induced toxicity in mice involve an interaction with the σ1 receptor. Br. J. Pharmacol. 149, 998–1012. doi: 10.1038/sj.bjp.0706927
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mosconi, L., Pupi, A., and De Leon, M. J. (2008). Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease. Ann. N Y Acad. Sci. 1147, 180–195. doi: 10.1196/annals.1427.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Narita, M., Shimizu, S., Ito, T., Chittenden, T., Lutz, R. J., Matsuda, H., et al. (1998). Bax interacts with the permeability transition pore to induce permeability transition and cytochrome c release in isolated mitochondria. Proc. Natl. Acad. Sci. U S A 95, 14681–14686. doi: 10.1073/pnas.95.25.14681
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pal, A., Fontanilla, D., Gopalakrishnan, A., Chae, Y. K., Markley, J. L., and Ruoho, A. E. (2012). The σ1 receptor protects against cellular oxidative stress and activates antioxidant response elements. Eur. J. Pharmacol. 682, 12–20. doi: 10.1016/j.ejphar.2012.01.030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Paradies, G., Ruggiero, F. M., Petrosillo, G., and Quagliariello, E. (1998). Peroxidative damage to cardiac mitochondria: cytochrome oxidase and cardiolipin alterations. FEBS Lett. 424, 155–158. doi: 10.1016/s0014-5793(98)00161-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ren, R., Zhang, Y., Li, B., Wu, Y., and Li, B. (2011). Effect of β-amyloid (25–35) on mitochondrial function and expression of mitochondrial permeability transition pore proteins in rat hippocampal neurons. J. Cell. Biochem. 112, 1450–1457. doi: 10.1002/jcb.23062
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rhein, V., Song, X., Wiesner, A., Ittner, L. M., Baysang, G., Meier, F., et al. (2009). Amyloid-β and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc. Natl. Acad. Sci. U S A 106, 20057–20062. doi: 10.1073/pnas.0905529106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Selfridge, J. E., E, L., Lu, J., and Swerdlow, R. H. (2013). Role of mitochondrial homeostasis and dynamics in Alzheimer’s disease. Neurobiol. Dis. 51, 3–12. doi: 10.1016/j.nbd.2011.12.057
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Selkoe, D. J. (2004). Cell biology of protein misfolding: the examples of Alzheimer’s and Parkinson’s diseases. Nat. Cell Biol. 6, 1054–1061. doi: 10.1038/ncb1104-1054
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Smith, M. A., Perry, G., Richey, P. L., Sayre, L. M., Anderson, V. E., Beal, M. F., et al. (1996). Oxidative damage in Alzheimer’s. Nature 382, 120–121. doi: 10.1038/382120b0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tagashira, H., Zhang, C., Lu, Y. M., Hasegawa, H., Kanai, H., Han, F., et al. (2013). Stimulation of σ1-receptor restores abnormal mitochondrial Ca²+ mobilization and ATP production following cardiac hypertrophy. Biochim. Biophys. Acta 1830, 3082–3094. doi: 10.1016/j.bbagen.2012.12.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tchedre, K. T., and Yorio, T. (2008). σ1 receptors protect RGC-5 cells from apoptosis by regulating intracellular calcium, Bax levels and caspase-3 activation. Invest. Ophthalmol. Vis. Sci. 49, 2577–2588. doi: 10.1167/iovs.07-1101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tsai, S. Y., Hayashi, T., Harvey, B. K., Wang, Y., Wu, W. W., Shen, R. F., et al. (2009). σ1 Receptors regulate hippocampal dendritic spine formation via a free radical-sensitive mechanism involving Rac1xGTP pathway. Proc. Natl. Acad. Sci. U S A 106, 22468–22473. doi: 10.1073/pnas.0909089106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tsai, S. Y., Rothman, R. K., and Su, T. P. (2012). Insights into the σ1 receptor chaperone’s cellular functions: a microarray report. Synapse 66, 42–51. doi: 10.1002/syn.20984
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Verstreken, P., Ly, C. V., Venken, K. J., Koh, T. W., Zhou, Y., and Bellen, H. J. (2005). Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron 47, 365–378. doi: 10.1016/j.neuron.2005.06.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Villard, V., Espallergues, J., Keller, E., Alkam, T., Nitta, A., Yamada, K., et al. (2009). Antiamnesic and neuroprotective effects of the aminotetrahydrofuran derivative ANAVEX1–41 against amyloid β25–35-induced toxicity in mice. Neuropsychopharmacology 34, 1552–1566. doi: 10.1038/npp.2008.212
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Villard, V., Espallergues, J., Keller, E., Vamvakides, A., and Maurice, T. (2011). Anti-amnesic and neuroprotective potentials of the mixed muscarinic receptor/sigma1 (σ1) ligand ANAVEX2–73, a novel aminotetrahydrofuran derivative. J. Psychopharmacol. 25, 1101–1117. doi: 10.1177/0269881110379286
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yang, J., Liu, X., Bhalla, K., Kim, C. N., Ibrado, A. M., Cai, J., et al. (1997). Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science 275, 1129–1132. doi: 10.1126/science.275.5303.1129
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yates, C. M., Butterworth, J., Tennant, M. C., and Gordon, A. (1990). Enzyme activities in relation to pH and lactate in postmortem brain in Alzheimer-type and other dementias. J. Neurochem. 55, 1624–1630. doi: 10.1111/j.1471-4159.1990.tb04948.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yin, X. M., Oltvai, Z. N., and Korsmeyer, S. J. (1995). Heterodimerization with Bax is required for Bcl-2 to repress cell death. Curr. Top. Microbiol. Immunol. 194, 331–338. doi: 10.1007/978-3-642-79275-5_38
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zussy, C., Brureau, A., Delair, B., Marchal, S., Keller, E., Ixart, G., et al. (2011). Time-course and regional analyses of the physiopathological changes induced after cerebral injection of an amyloid β fragment in rats. Am. J. Pathol. 179, 315–334. doi: 10.1016/j.ajpath.2011.03.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zussy, C., Brureau, A., Keller, E., Marchal, S., Blayo, C., Delair, B., et al. (2013). Alzheimer’s disease related markers, cellular toxicity and behavioral deficits induced six weeks after oligomeric amyloid-β peptide injection in rats. PLoS One 8:e53117. doi: 10.1371/journal.pone.0053117
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: Alzheimer’s disease, mitochondrial damages, ANAVEX2-73, sigma-1 receptor, cytoprotection
Citation: Lahmy V, Long R, Morin D, Villard V and Maurice T (2015) Mitochondrial protection by the mixed muscarinic/σ1 ligand ANAVEX2-73, a tetrahydrofuran derivative, in Aβ25–35 peptide-injected mice, a nontransgenic Alzheimer’s disease model. Front. Cell. Neurosci. 8:463. doi: 10.3389/fncel.2014.00463
Received: 29 October 2014; Accepted: 20 December 2014;
Published online: 20 January 2015.
Edited by:
Fabio Blandini, National Institute of Neurology C. Mondino Foundation, ItalyReviewed by:
Rafael Linden, Federal University of Rio de Janeiro, BrazilMohan Pabba, Center for Addiction and Mental Health, Canada
Copyright © 2015 Lahmy, Long, Morin, Villard and Maurice. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tangui Maurice, Inserm U 710, University of Montpellier, cc 105, place Eugène Bataillon, 34095 Montpellier cedex 5, France e-mail:bWF1cmljZUB1bml2LW1vbnRwMi5mcg==