 Alexandre Henriques1,2†
Alexandre Henriques1,2† Stefan Kastner3†Eva Chatzikonstantinou3Claudia Pitzer3Christian Plaas3Friederike Kirsch3
Stefan Kastner3†Eva Chatzikonstantinou3Claudia Pitzer3Christian Plaas3Friederike Kirsch3 Oliver Wafzig3Carola Krüger3Robert Spoelgen3
Oliver Wafzig3Carola Krüger3Robert Spoelgen3 Jose-Luis Gonzalez De Aguilar1,2Norbert Gretz4
Jose-Luis Gonzalez De Aguilar1,2Norbert Gretz4 Armin Schneider3*
Armin Schneider3*- 1INSERM, U1118, Mécanismes Centraux et Péripheriques de la Neurodégénérescence, Strasbourg, France
- 2UMRS1118, Fédération de Médecine Translationnelle de Strasbourg, Université de Strasbourg, France
- 3Sygnis Bioscience GmbH & Co KG, Heidelberg, Germany
- 4Medical Research Center, Medical Faculty Mannheim, University of Heidelberg, Mannheim, Germany
Background: Amyotrophic lateral sclerosis (ALS) is an incurable fatal motoneuron disease with a lifetime risk of approximately 1:400. It is characterized by progressive weakness, muscle wasting, and death ensuing 3–5 years after diagnosis. Granulocyte-colony stimulating factor (G-CSF) is a drug candidate for ALS, with evidence for efficacy from animal studies and interesting data from pilot clinical trials. To gain insight into the disease mechanisms and mode of action of G-CSF, we performed gene expression profiling on isolated lumbar motoneurons from SOD1G93A mice, the most frequently studied animal model for ALS, with and without G-CSF treatment.
Results: Motoneurons from SOD1G93A mice present a distinct gene expression profile in comparison to controls already at an early disease stage (11 weeks of age), when treatment was initiated. The degree of deregulation increases at a time where motor symptoms are obvious (15 weeks of age). Upon G-CSF treatment, transcriptomic deregulations of SOD1G93A motoneurons were notably restored. Discriminant analysis revealed that SOD1 mice treated with G-CSF has a transcriptom close to presymptomatic SOD1 mice or wild type mice. Some interesting genes modulated by G-CSF treatment relate to neuromuscular function such as CCR4-NOT or Prss12.
Conclusions: Our data suggest that G-CSF is able to re-adjust gene expression in symptomatic SOD1G93A motoneurons. This provides further arguments for G-CSF as a promising drug candidate for ALS.
Introduction
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disease that results in progressive loss of motoneurons, motor weakness, and death within 1–5 years after disease onset. Although more than 130 years have passed since the disease was first described by Charcot its etiology is still enigmatic, precluding a truly rational drug development. Motoneurons in the spinal cord and the motor unit are the common endpoint in all hypotheses concerning pathophysiology (Gordon, 2013; Turner et al., 2013). The transgenic SOD-1G93A mice are a model for ALS and recapitulate most of the ALS pathology, including loss of motor units. The first evidence of stress on the motor axis in this model occurs early before appearance of motor deficit (Frey et al., 2000).
G-CSF was primarily described as a hematopoietic factor able to stimulate the proliferation and differentiation of myeloid precursors. It is used for the treatment of chemotherapy-induced neutropenia and for bone marrow transplants (Kobbe et al., 2009; Yang et al., 2012). G-CSF has more recently been uncovered as a neurotrophic factor able to rescue motoneurons from apoptosis, protect motor functions and enhance overall survival of SOD1G93A mice through the preservation of motor units (Pitzer et al., 2008; Henriques et al., 2010a,b, 2011).
Pilot clinical studies have investigated the potential of G-CSF as a drug candidate in ALS. Administrations were mainly subcutaneous with limited duration (unique or repeated injection for up to 5 days) and enrollment ranged from 10 to 39 patients. Adverse effects were minor and the drug was well tolerated in all studies. Effects of G-CSF on disease progression are unclear since all studies suffer from small cohort sizes. One study concluded that G-CSF treatment did not bring benefit for patients (Nefussy et al., 2010), while others either reported slower disease progression upon treatment (Martinez et al., 2009; Zhang et al., 2009), reduced damage of white matter tracts (Duning et al., 2011) or lower level of pro-inflammatory markers in the cerebrospinal fluid (Chio et al., 2011).
Effects of G-CSF treatment in SOD-1G93A mice have been examined at the clinical, electrophysiological, and histological level. Here we sought to determine if and what changes on the gene expression level in alpha motoneurons would be related to G-CSF treatment. Therefore, our objective was to (1) define changes on the mRNA level that occur in spinal motoneurons in an animal model of ALS, the SOD1G93A mice and (2) characterize the effects of G-CSF on detected changes in the transcriptome of those motoneurons. We combined laser microdissection (LMD), RNA amplification, and gene expression profiling on DNA arrays to study these cell-type specific expression changes.
Materials and Methods
ALS Model
We used mice transgenic for the SOD1G93A mutation (Gurney et al., 1994) on a C57/bl6 background (B6.Cg-Tg(SOD1-G93A)1Gur/J strain; Jackson laboratory, Bar Harbor, Maine, USA), harboring the high copy number of the mutant allele human SOD1. The hemizygous line was maintained by mating transgenic males with C57BL/6 wild-type females. Transgenic littermate females were used in all experiments. They were either treated with vehicle (250 mM sorbitol, 0.004% Tween-80 and 10 mM sodium acetate buffer) or with G-CSF (filgrastim dissolved in vehicle, 30 μg/kg body weight/day) with an osmotic, subcutaneously implanted, paravertebrally located minipump (Alzet Minipump, model 2004; ALZET Osmotic Pumps, Cupertino, CA, USA) (Pitzer et al., 2008). Wild type littermates served as control group (group 3). Each mouse was housed under standard conditions in cages together with 4-6 littermates and fed ad libitum, in a room that also housed male mice, thus resulting in likely estrous cycle synchronization. All animal experiments were approved by the Regierungspräsidium Karlsruhe (Karlsruhe, Germany).
Tissue Isolation
All kinds of surgical instruments (forceps, scissors) were cleaned with RNase Zap spray or 100 % ethanol before use. Directions for handling RNA or tissue for RNA isolation (e.g., gloves, etc.) were followed. For anesthetizing purpose, a mixture of ketamine (120 mg/kg boday mass) and xylasine (Rompun, 16 mg/kg body mass was injected intraperitoneally. After complete absence of peripheral nociceptive reflexes the animals were perfused transcardially via the left ventricle with ice-cold HBSS. The pump was run at 10 ml/min for 3 min. Care was taken not to injure any blood vessel during the preparation. The perfused mouse was fixed onto a metal platform cooled by wet ice to avoid RNA degradation. After removal of the skin, cranial bones and vertebrae were carefully removed dorsally and spinal cords were dissected from the remaining opened cranium and vertebral canal. The thoracic position T9 was marked with a blue dye spot. Tissue was frozen on dry ice for about 5–10 min and stored at −80°C.
Laser Microdissection
Spinal cords were cryosectioned into 8 μm coronal sections, starting at the lumbar position L2. Thirty sections per mice were collected. Sections taken for microdissection were 100 μm apart. The tissue on the POL-membrane of a frame slide was overlaid with thionin staining solution so that the whole sample was completely covered. After 20 s ultraPURE™ water was poured onto the sample to wash away excess thionin. The frame slide with the tissue sample on the POL-membrane was mounted onto the microscope. Samples were collected into the cap of a 0.65 ml LMD tube filled with 35 μl of buffer RLT (Qiagen RNeasy® Micro Kit). Using the program “Leica Laser Microdissection V 5.0,” cells were marked and then automatically cut by a moving laser beam (power 40–50, speed 4, specimen/balance 11, offset 45). A total of 300 motoneurons per mouse spinal cord have been sampled by laser microdissection (Supplementary Figure 1), from a total of 35 mice (week 11 wt: n = 6; SOD n = 8); week 15 [wt n = 7; SOD (vehicle): n = 7; SOD (G-CSF treated): n = 7)], and subjected amplified RNA to array hybridization. Cutting was done at 40x magnification. Experimenters involved in laser microdissection were blind to the identity of the animal.
RNA Amplification
Each RNA sample was linearly amplified by two consecutive rounds of T7 polymerase-driven amplification. The precipitated total RNA of each LMD sample was reverse transcribed with a first-strand synthesis mix comprising 10 U Superscript III (Invitrogen, Karlsruhe, Germany), 1 U Superasin (Ambion, Huntingdon, UK) and 0.5 pmol of T7-T18V primer by incubating for 2 h at 50°C after second-strand synthesis with 6 U DNA polymerase I, 2 h at 22°C with 0.2 U RNaseH, and 15 min at 16°C with 4 U T4 DNA polymerase I (Invitrogen). The T7 promoter tagged cDNA template was purified by standard phenol extraction and ethanol precipitation. This cDNA was used for first round amplified antisense RNA production applying a MegaScript kit (Ambion) and purified with an RNeasy Mini Kit (Qiagen) according to the manufacturer's instructions. Second round amplification was performed accordingly but using T7-tagged random primer for first strand cDNA synthesis and T18V primer for second-strand synthesis, yielding amplified sense RNA. UV measurements at 280 and 260 nm and 2100 Bioanalyzer runs using the RNA-6000 Nano-LabChip (Agilent, Böblingen, Germany) were used to ensure the quality and quantity of the sense RNA produced. Only samples showing RNA integrity numbers (RIN) above 8 were used. The yield varied from 40 to 80 μg.
Array Analyses
The arrays were performed with the GeneChip® mouse genome 430 2.0 array from Affymetrix (Affymetrix, Santa Clara, CA, USA). Expression console software (Affymetrix, Santa Clara, CA, USA) was used to normalize arrays with the gcrma algorithm (Harr and Schlotterer, 2006) and to perform quality control. The database was filtered with intensity and standard deviation. We removed genes when one of the following conditions was not met: less than 4 arrays had intensity higher than the centile 20% of all genes or the standard deviation was lower than centile 30% of all genes.
Discriminant analysis has been performed with JMP 11.0.0 (SAS Institute, Cary, North Carolina) with genes presenting a minimum standard deviation greater than 1.2 in all individuals. Class prediction has been computed using a non-parametric k-Nearest Neighbors algorithm.
Differential gene expression among experimental groups has been assessed by ANOVA followed by a post-hoc Tukey's Honest, with genes presenting a minimum fold-change of 1.5-fold. We considered a p-value lower than 0.05 with a FDR lower than 10% as significant difference. Analyses were performed using BRB-ArrayTools developed by Dr. Richard Simon and BRB-ArrayTools Development Team (http://linus.nci.nih.gov).
WEB-based Gene SeT AnaLysis Toolkit was used to detect enrichment of biological functions (WebGestalt, http://bioinfo.vanderbilt.edu/webgestalt/) (Zhang et al., 2005; Wang et al., 2013).
Real-Time Quantitative PCR
One μg total RNA used for the microarray was used to synthesize cDNA using superscript II reverse transcriptase (Invitrogen) and oligo-dT primers, accordingly to manufacturer's recommendation. Quantitative reverse transcription-polymerase chain reaction was performed using the Lightcycler system with SYBR-Green reagent (Roche). Cycling conditions were as follows: 10 min at 95°C; 5 s at 95°C, 10 s at the annealing temperature of 64°C, 30 s at 72°C for 50 cycles and 10 min at 95°C. Relative quantification were calculated after normalization to the reference gene cyclophilin B.
Results
Design of the Experiment
A first group of SOD1G93A and WT mice was included in the study at week 11 of age when SOD1G93A mice present no signs of motor dysfunction but subtle signs of denervation detectable by electromyography (Pitzer et al., 2008). The second cohort of mice was treated with G-CSF or vehicle from week 11 to week 15. At the time of study completion, SOD1G93A mice presented clear motor impairment and motoneuron degeneration is documented (Pitzer et al., 2008; Henriques et al., 2011).
During laser microdissection, there is a risk of collecting cells surrounding motoneurons and therefore analyzing non-neuronal transcripts (Supplementary Figure 1A, Perrin et al., 2005; Bandyopadhyay et al., 2013). To assess the proportion of contaminating RNA, we have studied the expression values of highly specific markers for astrocytes, oligodendrocytes, microglia, endothelial cells, and neurons (Cahoy et al., 2008). All experimental groups show high expression of the specific neuronal markers, while markers for glial and endothelial cells are present at very low levels or not detected (Supplementary Figure 1B). These data suggest that contamination with RNA from other cells than motoneurons is minimal and irrelevant for the following analyses.
The design of the present study should be informative on genes altered in motoneurons of SOD1G93A mice from the clinically non-symptomatic to an early symptomatic stage, and give insight into genes influenced by G-CSF treatment (Figure 1A). Results were analyzed by unsupervised and supervised approaches (Figure 1B), and biological functions for relevant genes analyzed based on gene ontology.
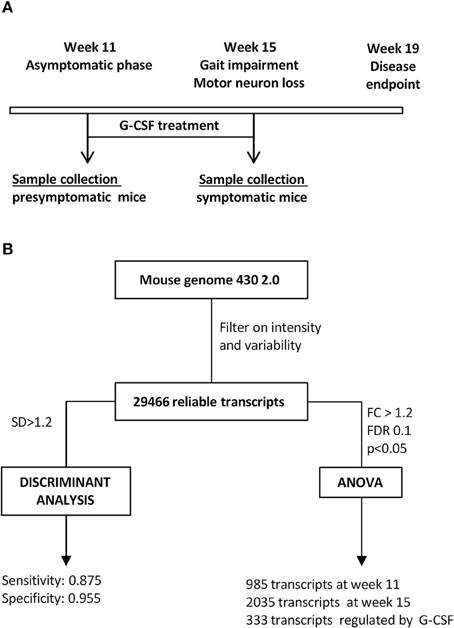
Figure 1. Study design. (A) Time course of the study. Given is the disease course in SOD1G93A mice, time of treatment and time of sacrifice for all experimental groups. (B) Flow chart for analytical steps for analysis.
Gene Regulation in SOD1G93A Motoneurons
At 11 weeks of age, when SOD1G93A mice do not show motor symptoms despite early electromyographic changes we detected 985 transcripts with a significantly altered expression, of which 725 genes were upregulated and 260 dowregulated (Table 1). At 15 weeks of age, when motor symptoms are obvious and muscular denervation signs are prominent by electromyography, the number of significantly deregulated transcripts was two times higher, with 790 genes upregulated and 1245 dowregulated (Table 2). Functional annotation using gene ontology revealed that metabolic process, reponse to stimulus/stress and nucleic acid binding were the main biological and molecular functions altered in SOD-1 motoneurons (Supplementary Figure 2).
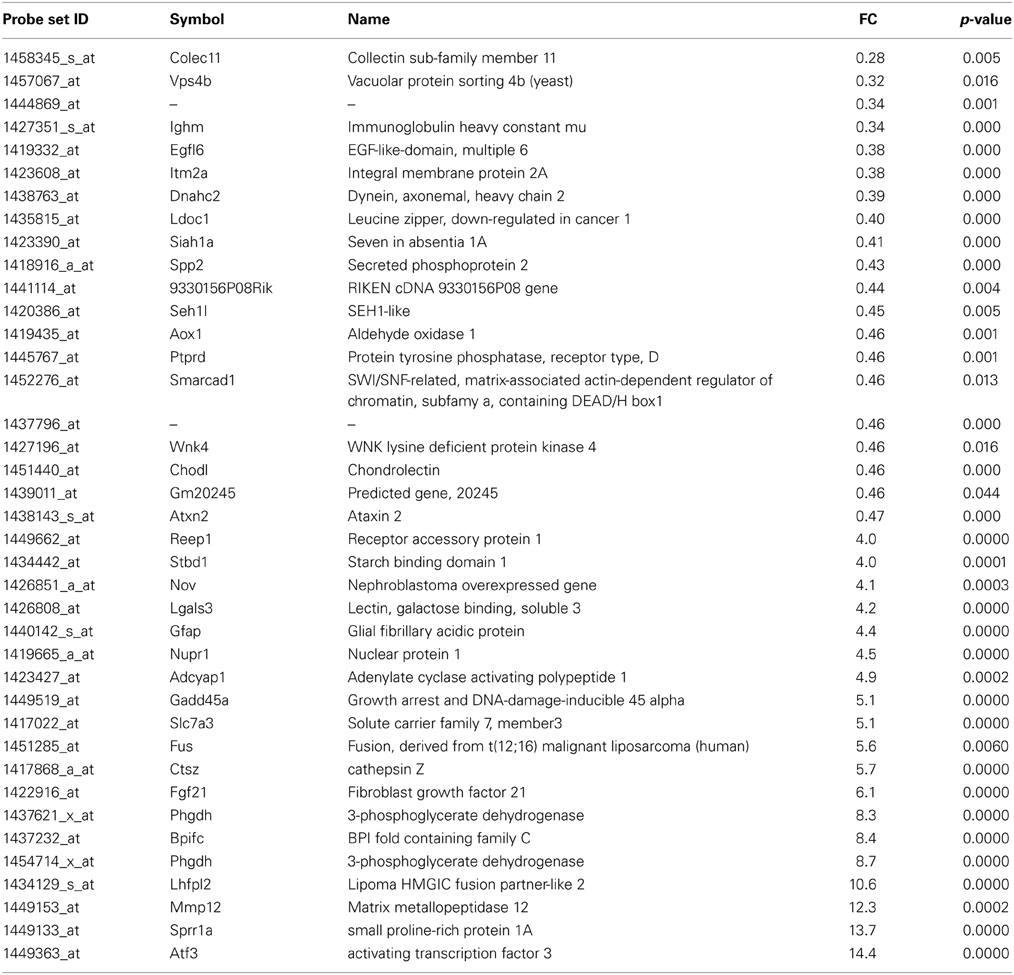
Table 1. List of the main transcripts whose expression are altered in SOD1G93A mice at 11 week of age.
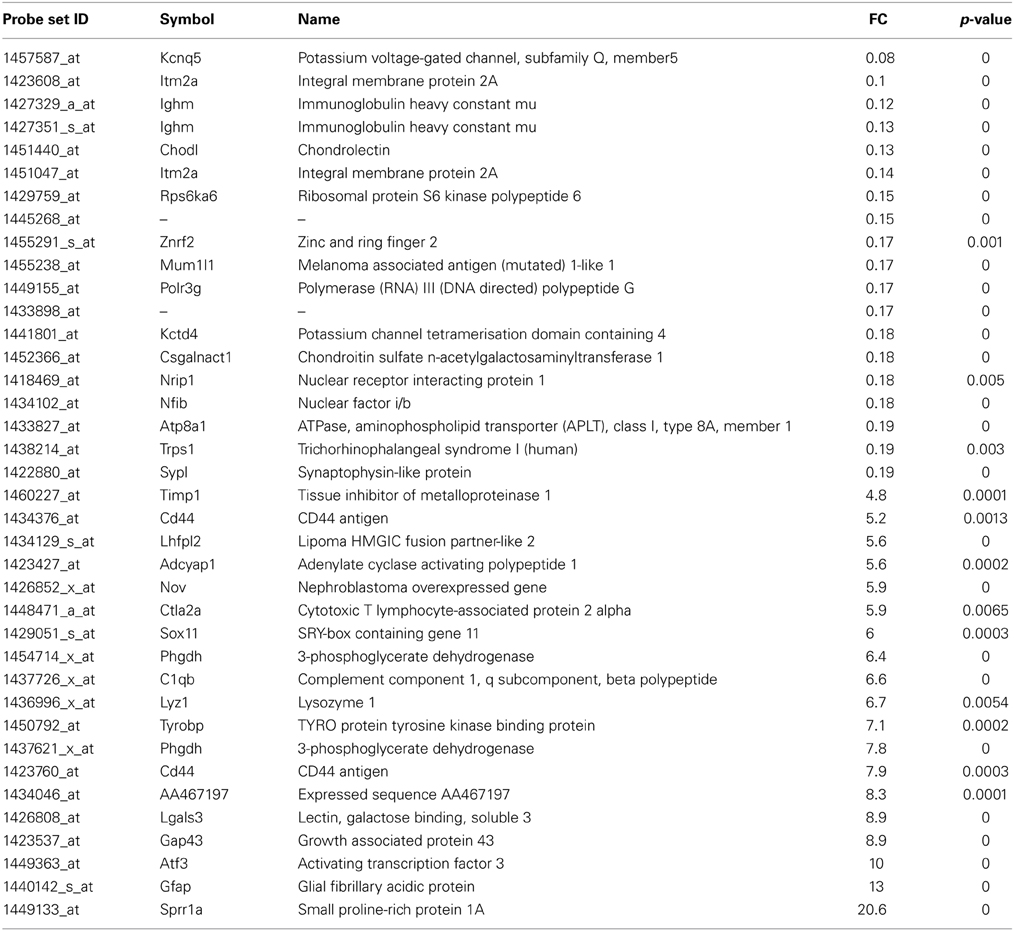
Table 2. List of the main transcripts whose expression are altered in SOD1G93A mice at 15 week of age.
At the presymptomatic disease stage (week 11), genes which were most downregulated were the collectin sub-family member 11 (colec11) and vacuolar protein sorting 4b (vpsb4) (Table 1). Expression of colec11 has been reported in neurons and is involved in innate immunity (Motomura et al., 2008; Ohtani et al., 2012). vpsb4 is known to interact with the pro-apoptotic pathway (Cui et al., 2014) and the autophagosome in neurodegenerative conditions (Han et al., 2012). The most strongly upregulated genes were related to response to stress and injury, such as activating transcription factor 3 (atf3) and small proline-rich repeat protein 1A (sprr1A). Upregulation of atf3 has already been noted in motoneurons after axotomy (Tsujino et al., 2000) and in response to deficiency in neurotrophic factors (Hyatt Sachs et al., 2007). Sprr1A was the second most stimulated gene and, similar to atf3, its expression is enhanced in motoneurons upon axotomy in order to promote axonal growth (Bonilla et al., 2002; Li and Strittmatter, 2003; Starkey et al., 2009). Interestingly, the fus (fused in sarcoma) gene was strongly upregulated in the presymtomatic disease stage. Fus protein is a RNA-binding protein and has important functions in RNA metabolism (e.g. splicing, formation of stress granule, transport and local translation). Mutations in fus are found in familial forms of ALS, and inclusions of the protein are present in patients with frontotemporal dementia, a syndrome that is connected to ALS (Ling et al., 2013).
At the symptomatic disease stage, the most overexpressed genes were mainly associated with motoneuron stress response and support of axonal regeneration, similar to the presymptomatic disease stage (Table 2). Beside sprr1A and atf3, the growth associated protein 43 (gap43) shows increased expression (9-fold) which is involved in axonal regeneration after motoneuron injury (Chen et al., 2010; Grasselli et al., 2011). Interestingly, the transcription factor responsible for sprr1A expression in motoneurons, sox11, is strongly upregulated at this disease stage. Potassium voltage-gated channel, subfamily Q, member 5 (kcnq5) was the most downregulated gene (downregulated 14-fold) at the symptomatic disease stage. Its function in motoneurons is not described, but should be related to the control of resting and release properties of synapses (Huang and Trussell, 2011). The expression of chondrolectin (chodl) was also strongly repressed in motoneurons of SOD1G93A mice. Interestingly, this protein promotes motor axon growth and growth cone interactions, and its downregulation is associated with motoneuron degeneration in an animal model of spinal muscular atrophy (Zhong et al., 2012; Sleigh et al., 2014). As shown in Supplementary Table 1, 83 transcripts were commonly deregulated at both asymptomatic and symptomatic disease stages. We noted deregulation at the two disease stages for several growth factors (FGF21, VGF, TGFa), and genes related to DNA/RNA metabolism (wars, eef1a1, eif4e).
G-CSF Partially Restores the Motoneuron Transcriptional Signature in SOD1G93A Mice
In order to better understand the effects of G-CSF in the SOD1G93A mice, we have monitored transcriptional changes in motoneurons at a symptomatic disease stage upon a 4 weeks treatment (from week 11 to week 15). We performed a discriminant analysis with transcripts showing a standard deviation greater than 1.2 across all samples (734 transcripts).
As shown in a canonical plot, control groups are close to each other and SOD1G93A mice are distant from non-transgenic animals (Figure 2A). The SOD1 mice treated with G-CSF (green dots in Figure 2) tended to have a transcriptomic signature different from the sympatomatic SOD1 mice (red dots in Figure 2), that was closer to non-symptomatic mice. This observation was further confirmed by class prediction using a non-parametric k-Nearest Neighbors algorithm, used to predict the class of unknown samples. The algorithm was built with the signatures of all samples with the exclusion of mice treated with G-CSF. There were 4 defined classes: wild type 11 weeks, SOD-1G93A 11 weeks (presymptomatic), wild type 15 weeks and SOD-1G93A mice 15 weeks (symptomatic). SOD-1G93A mice treated with G-CSF were considered as unknown and the algorithm sorted them into the defined classes based on their transcriptome signature. The mean percent of correct classification reached 96% of efficacy for the defined classes, with a sensitivity above 0.875 and a specificity of 0.955.
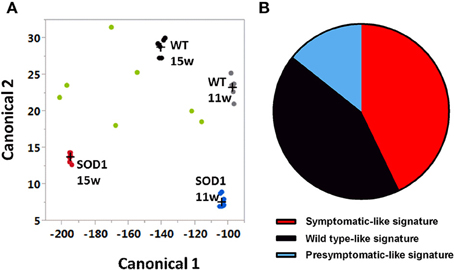
Figure 2. Canonical plot shows dispersion of treated animals off the non-treated groups. Multivariate analyses have been conducted with genes having a standard deviation greater than 1.2 (734 transcripts). (A) Canonical plot shows that symptomatic SOD-1 mice (red dots) are distant from other groups, including asymptomatic SOD1 mice (blue dots) and when considered as unknown, SOD-1 mice treated with G-CSF (green dots) show a strong dispersion and tend to be close to wild types at 11 weeks (gray dots) or 15 weeks of age (black dots). (B) After G-CSF treatment, discriminant analysis classified only three samples of SOD1 motorneurons as symptomatic motor neurons. One sample was considered has presymptomatic and three as wild type motor neurons.
The group treated with G-CSF was composed by seven SOD-1G93A mice. Three out of seven were sorted as symptomatic SOD-1G93A, one as asymptomatic SOD-1G93A 11 weeks, and three as wild type 15 weeks (Figure 2B). These data suggest that G-CSF can modulate the gene expression in motoneurons of 15 weeks old SOD-1G93A mice and can prevent disease-related changes to approach the transcriptomic signature of healthy mice or asymptomatic SOD1G93A animals.
Genes Regulated by G-CSF in Motoneurons of SOD1G93A Mice
Next, we focused on the identity of differentially expressed genes in motoneurons after G-CSF treatment. G-CSF had an effect on the regulation of 463 transcripts in total, and was able to adjust the expression of 187 transcripts that were either up- or downregulated in SOD1G93A mice compared to controls (Tables 3, 4, Figure 3).
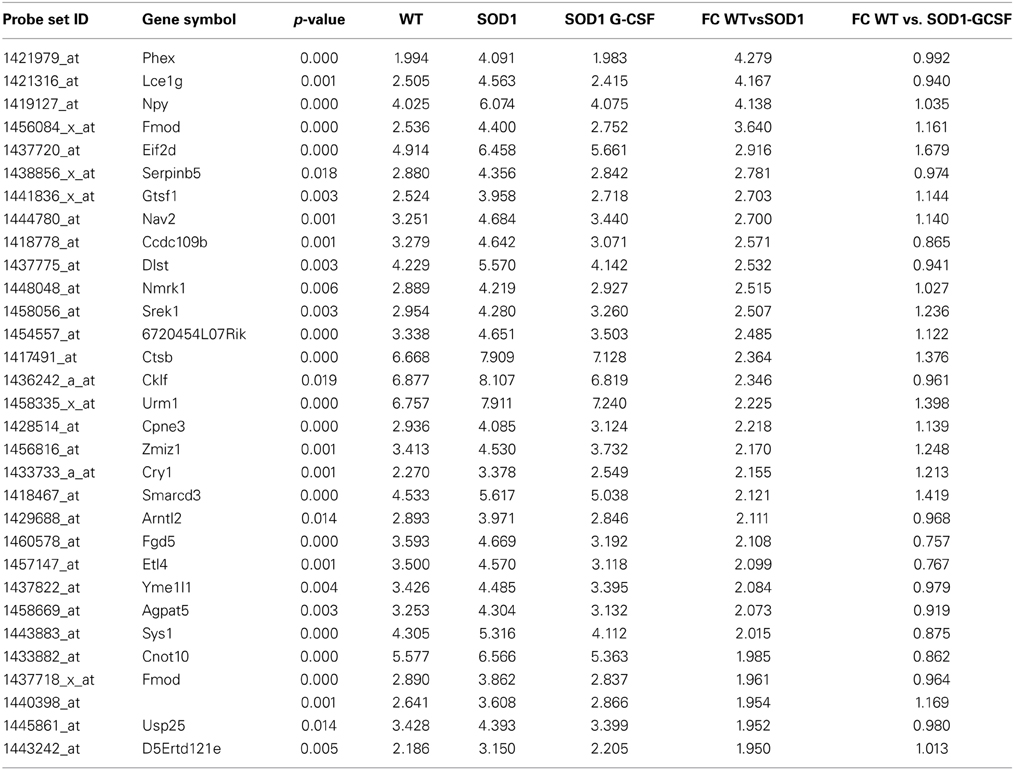
Table 3. List of the main transcripts whose expression is downregulated by G-CSF treatment in SOD1G93A mice at 15 week of age.
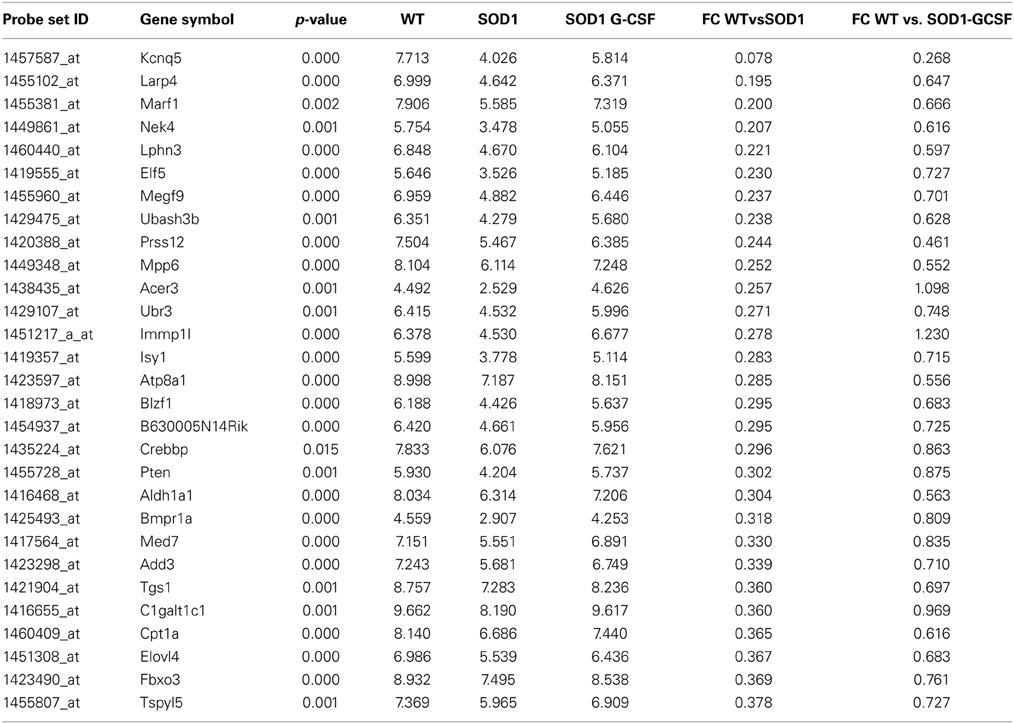
Table 4. List of the main transcripts whose expression is upregulated by G-CSF treatment in SOD1G93A mice at 15 week of age.
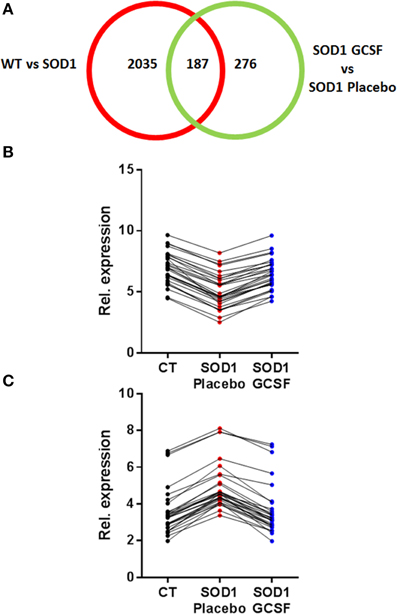
Figure 3. G-CSF treatment modulates transgene-related gene expression changes. (A) Chart showing the number of differentially regulated genes after G-CSF treatment in the motoneurons of SOD1 mice. (B–C) Given is the relative expression of the 30 most significantly modified transcripts re-adjusted by G-CSF treatment and previously found downregulated (B) or upregulated (C) in symptomatic SOD1 mice.
Two of the most downregulated transcripts in SOD1G93A mice, kcnq5 and larp4, had their expression enhanced by a factor 3 upon G-CSF treatment as compared to placebo group. Larp4 has no known specific functions in neurons but is known to regulates mRNA stability (Yang et al., 2011). G-CSF prevented the upregulation of NPY, a well-known neuropeptide involved in energetic metabolism and food intake but also expressed by neurons in response to stress such as spinal cord inflammation (Taylor et al., 2014). Upregulation of cklf, a cytokine with lymphocyte chemoattractive properties, was also inhibited by treatment (Han et al., 2001). Interestingly, Cpt1a and elovl4, two transcripts directly associated with lipid metabolism, were downregulated in SOD1G93A motoneurons, and upregulated by G-CSF. Cpt1a controls the entrance of fatty acids into mitochondria and is therefore the rate limiting enzyme for fatty acid oxidation, and Elovl4 elongates long fatty acids without distinction on their saturation levels, suggesting that G-CSF could enhance the processing of fatty acids in spinal cord. Additionally, G-CSF modulated the expression of 251 previously unaltered transcripts. The effect of G-CSF was prominent on gene ontology biological functions deregulated in SOD1G93A mice, including metabolic process, and response to stimulus (Supplementary Figure 2).
In order to assess the reliability of the analysis, we selected several genes for validation by quantitative real time PCR (qPCR). Those genes were selected because of their altered expression in SOD1G93A mice at 15 weeks of age and by a significant effect of G-CSF on their regulation. An additional criterion was their potential role in the neurodegenerative process of ALS based on the literature. We chose to study the regulation of CNOT10, CTSB, Prss12 and BMPR1a. CNOT10 is a member of the CCR4-NOT complex that acts as a chaperon platform (Collart and Panasenko, 2012). The CCR4-NOT complex controls and promotes neuromuscular junction formation in drosophila (Pradhan et al., 2012). Prss12, also known as motopsin, is a protease specific to neurons and its expression is downregulated upon stress, such as axotomy, in motoneurons (Numajiri et al., 2009). Its regulation is therefore seen as a marker of denervation. We chose ctsb, as meta-analysis of transcriptomic studies in ALS have shown its expression systematically altered, both in human patients and SOD1G93A mice (Saris et al., 2013). Ctsb codes for cathepsin B, a protein associated with neuroinflammation, motoneuron degeneration, and tissue destruction (Kikuchi et al., 2003). BMPR1a is a receptor serine/threonine kinase and regulates apoptosis and cell differentiation (Mishina et al., 2004). In the central nervous system, BMP signaling is required during formation of large-transmitting synapses (Xiao et al., 2013), build-up of neuromuscular junctions (Higashi-Kovtun et al., 2010) and is stimulated upon spinal cord and sciatic nerve injury (Tsujii et al., 2009; Sahni et al., 2010).
Relative expressions for these transcripts were highly similar to the relative abundance derived from microarray analyses (Figure 4). In particular, upregulation of CNOT10 present in SOD1G93A mice is completely reversed upon G-CSF treatment. The regulation of bmpr1a showed a trend to the wild type value but this was not significant in the qPCR analysis.
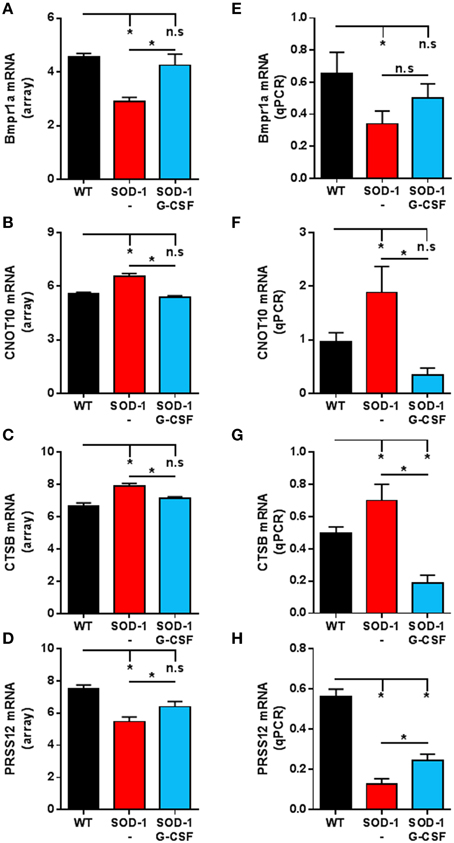
Figure 4. Validation of the expression of selected transcripts by qPCR. (A–D) Expression levels from microarray analyses of Bmpr1a (A), CNOT10 (B), CTSB (C) and PRSS12 (D) at the symptomatic disease stage. (E–H) Expression levels derived from quantitative PCR of Bmpr1a (E), CNOT10 (F), CTSB (G) and PRSS12 (H) at the symptomatic disease stage. Data are presented as mean ± standard variation of the mean. *p < 0.05 (ANOVA, followed by multicomparison t-test).
Taken together, these data demonstrate that G-CSF promotes transcriptional changes in motoneurons in symptomatic SOD1G93A mice and can correct the alterations of many disease related gene expression changes.
Discussion
ALS is a fatal condition characterized by the constant death of motoneurons. Although the triggering factors for ALS remain unknown, the step that induces symptoms is the disruption of the motor units, with the dismantlement of neuromuscular junctions and motoneuron stress and death. The study of global changes in the transcriptome of specific cells in the motor axis is a promising approach to address the complexity of ALS (Henriques and Gonzalez De Aguilar, 2011).
Transcriptomic Changes of Motoneurons from SOD1G93A Mice
Here, we studied the transcriptome of motoneurons of SOD1G93A mice, at two disease stages and after a pharmacological treatment with G-CSF, a trophic factor with neuroprotective properties (Pitzer et al., 2008; Henriques et al., 2010a,b, 2011). Our aim was to characterize the transcriptomic reponse of motor neurons to a neuroprotective treatment based on G-CSF.
In 2005, a first study was published describing the transcriptome of laser microdissected motoneurons of SOD1G93A mice at three different disease stages (Perrin et al., 2005). The conclusion of the authors was that few changes occurred at presymptomatic (28 deregulated transcripts) and early symptomatic (148 deregulated transcripts) stages, and that symptomatic stages were characterized by few expression changes in cell-death-associated genes. They found deregulation of related to energetic metabolism, cell growth and/or maintenance (Perrin et al., 2005). Many of these genes also showed an altered expression in our study. In 2007, another group published data on the transcriptome of dissected motoneurons from SOD1G93A mice, and reported dysregulations on genes related to transcription and nuclear proteins, protein synthesis, metabolism and cell cycle/growth (Ferraiuolo et al., 2007). Both studies identified lower deregulation events than our study. This difference can most likely be explained by higher statistical power in our study as both studies used fewer animals (n = 3/group) than our study (at least n = 6 per group). Higher degree of contaminating RNA from motoneuron-surrounding cells could also account for the differences between studies.
Based on our results, the transcriptome of isolated motoneurons of SOD1G93A mice is characterized by the deregulation of genes, starting at presymptomatic disease stages, related to response to stress, to regulation of transcription and to metabolic processes.
Response to stress is stimulated early on, at presymptomatic disease stage. Trophic signaling seems also to be stimulated, with the upregulation of the trophic factors such as fgf21 and vgf at presymptomatic and symptomatic disease stages. FGF21 belongs to the fibroblast growth factor family, known to counteract motoneuron death after axotomy-induced apoptosis (Cuevas et al., 1995), but also shown to regulate activation of astrocytes in animal models of ALS (Cassina et al., 2005). Additionally, motoneurons of SOD1G93A show an upregulation of transcription factors related to stress response, that are also closely connected to trophic factors, such as ATF3 (Averill et al., 2004).
During laser-microdissection of motoneurons, small amount of astrocyte RNA may have been captured. Although this contamination is minimal in our study, genes that are expressed at low abundance in motoneurons but at high levels in glial cells could be detected as differentially regulated, provided that the difference between two groups is very strong. An indication for that could be the detection of GFAP among the deregulatated transcripts in our study (upregulated in SOD1 mice). Presence of transcripts of glial origin in studies working on laser-captured motoneurons has been described previously (Perrin et al., 2005; Ferraiuolo et al., 2007). Therefore, it is possible that the alterations in the transcriptome linked to energetic/lipid metabolism arise from astrocytes surrounding motoneurons. G-CSF was indeed interestingly able to stimulate the expression of cpt1a in SOD1G93A mice, which is surprising since fatty acids are not the main energetic source for neurons. It is therefore possible that upregulation of cpt1a takes place in astrocytes and not in motoneurons, presumably to sustain higher energetic needs for neurons. Indeed, during energetic stress, astrocytes are able to convert lipid to ketone bodies that will be used as energetic fuel by surrounding neurons (Guzman and Blazquez, 2004). Interestingly, recent studies have shown increased ketone bodies in the cerebrospinal fluid of patients and fatty acid and ketone body supplementations have neuroprotective effects on motoneurons in animal models of ALS (Blasco et al., 2010; Schmitt et al., 2014). Energetic metabolism represents one promising field of research in ALS. Patients present with hypermetabolism, higher energetic needs and a higher level of circulating lipids is associated with better survival (Schmitt et al., 2014). Two recent clinical reports strongly suggest benefit after high lipid/caloric diets in ALS patients with a strong effect on survival (Dorst et al., 2013; Wills et al., 2014).
We found deregulation of several genes associated to RNA metabolism, such as Wars, Eef1a1 and Eif4e. Growing evidence suggests a role for defect RNA processing in motoneuron death. ALS patients present pathological inclusion of TDP-43 and FUS, two proteins that closely interact with RNA metabolism (Ling et al., 2013). Mutations in TARDBP (coding for TDP-43) and FUS are described in ALS patients with a family history but also in patients with fronto-temporal dementia (FTD) with a high risk (around 30%) to develop ALS. Expanded repeats in the gene C9orf72 are largely present in sporadic and familial ALS and FTD cases. Interestingly, whose expanded repeats are believed to induce RNA toxicity through a novel negative gain-of-function. Our results therefore provide interesting new clues when related to most recent pathophysiological hypotheses in ALS research.
G-CSF Action on Motoneurons
Our transcriptomic study describes for the first time the transcriptomic changes in motoneurons after a neuroprotective/ -regenerative treatment. We chose to perform our study with SOD1G93A mice at 15 week of age with an administration of G-CSF for 4 weeks. At this age, motor symptom are clearly established in untreated animals (Pitzer et al., 2008; Henriques et al., 2011) and G-CSF has a significant beneficial effect on motor functions and preserves motoneuron counts (Pitzer et al., 2008). In our study, the altered transcriptome of SOD1G93A alpha motoneurons has been modified by G-CSF and the expressions of 187 genes were partially restored. Genes involved in the maintenance of the motor-units, such as Prss12 and CNOT10, were modulated upon treatment, but effect of G-CSF was also observed on other different molecular functions such as neuroinflammation, RNA processing, and energetic metabolism. Completely unsupervised analyses identified more than half of treated mice as asymptomatic based on their transcriptomic profile, confirming the positive effect of the treatment on disease progression.
Modification of the transcriptom was most likely due to a direct effect of G-CSF on the motor neurons. Motoneurons express the receptor of G-CSF in basal condition and can upregulate it after an acute stress in neonatal age (Pitzer et al., 2008; Henriques et al., 2010a). We have also demonstrated that G-CSF can rescue motoneurons in a pure apoptotic condition, induced by sciatic nerve axotomy (Henriques et al., 2010a) and promotes reinnervation after sciatic nerve crush (Henriques et al., 2011). However, we cannot formally conclude whether all or most of the observed changes are due to direct actions of G-CSF on motoneurons or reflect improved health status of the alpha motoneurons by indirect treatment effects. Indeed, G-CSF was first described as a hematopoietic growth factor involved in the proliferation and differentiation of neutrophils. Despite an abundant literature on the involvement of neuroinflammation in ALS (Philips and Robberecht, 2011), there is no evidence that neutrophils have a role in the initiation or progression of ALS, neither in animal model nor in ALS patients. Additionally, a recent report suggests that G-CSF might be involved in response to muscle damage, which is also a characteristic of ALS(Kruger et al., 2014).
Conclusion
G-CSF was able to re-adjust the expression of genes important for motoneuron survival and function but also genes from other physiological pathways. The global changes occurring after treatment strengthens the notion of multimodal activity of G-CSF for preservation of motor units in ALS. Our results complement the existing data on characterization of G-CSF's action in mouse ALS models (Pitzer et al., 2008; Henriques et al., 2010a, 2011; Naumenko et al., 2011; Pollari et al., 2011) on the gene expression level. This increases confidence in G-CSF as a promising drug candidate for ALS.
Author Contributions
Armin Schneider designed the study. Stefan Kastner, Eva Chatzikonstantinou, Friederike Kirsch, Carola Krüger, Christian Plaas performed animal experiments, histological work, laser microdissection, RNA preparation and amplification. Norbert Gretz performed array hybridizations. Alexandre Henriques performed data analysis. Armin Schneider, Alexandre Henriques, Stefan Kastner, Claudia Pitzer, Carola Krüger, Robert Spoelgen, Jose-Luis Gonzalez De Aguilar and Armin Schneider were involved in data evaluation and discussions. Alexandre Henriques and Armin Schneider wrote the manuscript. All authors read and approved the final manuscript.
Conflict of Interest Statement
Some authors are employees of Sygnis Bioscience. Armin Schneider is inventor on patent applications claiming the use of G-CSF for the treatment of neurodegenerative conditions. The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to acknowledge the excellent technical assistance of Ulrike Bolz, Frank Herzog, and Gisela Eisenhardt. This work was supported by funds from European Community's Health Seventh Framework Program under grant agreement no. 259867 (FP7/2007-2013). Alexandre Henriques is a research fellow receiving funds from FP7/2007-2013. Jose-Luis Gonzalez De Aguilar is recipient of a “Chaire d'Excellence INSERM/Université de Strasbourg.”
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fncel.2014.00464/abstract
Supplementary Figure 1. Laser microdissection and neuronal specificity. (A) Representative pictures of spinal cord section before and after laser capture. Sections are stained with thionin. (B) Given is the relative expression of genes that are accepted as specific markers of astrocytes (n = 6 markers), neurons (n = 7 markers), oligodendrocytes (n = 7), microglia (n = 4) and endothelial cells (n = 4).
Supplementary Figure 2. Biological and molecular functions of genes altered in SOD1G93A mice. Given are the main biological (A) and molecular (B) functions of genes altered in SOD1G93A motoneurons at presymptomatic and symptomatic disease stages, and re-adjusted after G-CSF treatment.
Availability of Supporting Data. Microarray data files have been uploaded to the Gene Expression Omnibus (GEO) database repository, under the accession number GSE60856 (www.ncbi.nlm.nih.gov/geo/).
References
Averill, S., Michael, G. J., Shortland, P. J., Leavesley, R. C., King, V. R., and Bradbury, E. J. (2004). NGF and GDNF ameliorate the increase in ATF3 expression which occurs in dorsal root ganglion cells in response to peripheral nerve injury. Eur. J. Neurosci. 19, 1437–1445. doi: 10.1111/j.1460-9568.2004.03241.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bandyopadhyay, U., Cotney, J., Nagy, M., Oh, S., Leng, J., Mahajan, M., et al. (2013). RNA-Seq profiling of spinal cord motor neurons from a presymptomatic SOD1 ALS mouse. PLoS ONE 8:e53575. doi: 10.1371/journal.pone.0053575
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Blasco, H., Corcia, P., Moreau, C., Veau, S., Fournier, C., Vourc'h, P., et al. (2010). 1H-NMR-based metabolomic profiling of CSF in early amyotrophic lateral sclerosis. PLoS ONE 5:e13223. doi: 10.1371/journal.pone.0013223
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bonilla, I. E., Tanabe, K., and Strittmatter, S. M. (2002). Small proline-rich repeat protein 1A is expressed by axotomized neurons and promotes axonal outgrowth. J. Neurosci. 22, 1303–1315.
Cahoy, J. D., Emery, B., Kaushal, A., Foo, L. C., Zamanian, J. L., Christopherson, K. S., et al. (2008). A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J. Neurosci. 28, 264–278. doi: 10.1523/JNEUROSCI.4178-07.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cassina, P., Pehar, M., Vargas, M. R., Castellanos, R., Barbeito, A. G., Estevez, A. G., et al. (2005). Astrocyte activation by fibroblast growth factor-1 and motor neuron apoptosis: implications for amyotrophic lateral sclerosis. J. Neurochem. 93, 38–46. doi: 10.1111/j.1471-4159.2004.02984.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, L. J., Ren, Y. H., Liu, L., Zhang, X. Q., Zhao, Y., Wu, W. T., et al. (2010). Upregulated expression of GAP-43 mRNA and protein in anterior horn motoneurons of the spinal cord after brachial plexus injury. Arch. Med. Res. 41, 513–518. doi: 10.1016/j.arcmed.2010.10.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chio, A., Mora, G., La Bella, V., Caponnetto, C., Mancardi, G., Sabatelli, M., et al. (2011). Repeated courses of granulocyte colony-stimulating factor in amyotrophic lateral sclerosis: clinical and biological results from a prospective multicenter study. Muscle Nerve 43, 189–195. doi: 10.1002/mus.21851
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Collart, M. A., and Panasenko, O. O. (2012). The Ccr4–not complex. Gene 492, 42–53. doi: 10.1016/j.gene.2011.09.033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cuevas, P., Carceller, F., and Gimenez-Gallego, G. (1995). Acidic fibroblast growth factor prevents post-axotomy neuronal death of the newborn rat facial nerve. Neurosci. Lett. 197, 183–186. doi: 10.1016/0304-3940(95)11926-N
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cui, G., Wang, Y., Yu, S., Yang, L., Li, B., Wang, W., et al. (2014). The expression changes of vacuolar protein sorting 4B (VPS4B) following middle cerebral artery occlusion (MCAO) in adult rats brain hippocampus. Cell. Mol. Neurobiol. 34, 83–94. doi: 10.1007/s10571-013-9989-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dorst, J., Cypionka, J., and Ludolph, A. C. (2013). High-caloric food supplements in the treatment of amyotrophic lateral sclerosis: a prospective interventional study. Amyotroph. Lateral Scler. Frontotemporal Degener. 14, 533–536. doi: 10.3109/21678421.2013.823999
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Duning, T., Schiffbauer, H., Warnecke, T., Mohammadi, S., Floel, A., Kolpatzik, K., et al. (2011). G-CSF prevents the progression of structural disintegration of white matter tracts in amyotrophic lateral sclerosis: a pilot trial. PLoS ONE 6:e17770. doi: 10.1371/journal.pone.0017770
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ferraiuolo, L., Heath, P. R., Holden, H., Kasher, P., Kirby, J., and Shaw, P. J. (2007). Microarray analysis of the cellular pathways involved in the adaptation to and progression of motor neuron injury in the SOD1G93A mouse model of familial ALS. J. Neurosci. 27, 9201–9219. doi: 10.1523/JNEUROSCI.1470-07.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Frey, D., Schneider, C., Xu, L., Borg, J., Spooren, W., and Caroni, P. (2000). Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J. Neurosci. 20, 2534–2542.
Gordon, P. H. (2013). Amyotrophic lateral sclerosis: an update for 2013 clinical features, pathophysiology, management and therapeutic trials. Aging Disease 4, 295–310. doi: 10.14336/AD.2013.0400295
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Grasselli, G., Mandolesi, G., Strata, P., and Cesare, P. (2011). Impaired sprouting and axonal atrophy in cerebellar climbing fibres following in vivo silencing of the growth-associated protein GAP-43. PLoS ONE 6:e20791. doi: 10.1371/journal.pone.0020791
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gurney, M. E., Pu, H., Chiu, A. Y., Dal Canto, M. C., Polchow, C. Y., Alexander, D. D., et al. (1994). Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 264, 1772–1775. doi: 10.1126/science.8209258
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guzman, M., and Blazquez, C. (2004). Ketone body synthesis in the brain: possible neuroprotective effects. Prostaglandins Leukot. Essent. Fatty Acids 70, 287–292. doi: 10.1016/j.plefa.2003.05.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Han, J. H., Ryu, H. H., Jun, M. H., Jang, D. J., and Lee, J. A. (2012). The functional analysis of the CHMP2B missense mutation associated with neurodegenerative diseases in the endo-lysosomal pathway. Biochem. Biophys. Res. Commun. 421, 544–549. doi: 10.1016/j.bbrc.2012.04.041
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Han, W., Lou, Y., Tang, J., Zhang, Y., Chen, Y., Li, Y., et al. (2001). Molecular cloning and characterization of chemokine-like factor 1 (CKLF1), a novel human cytokine with unique structure and potential chemotactic activity. Biochem. J. 357(Pt 1), 127–135. doi: 10.1042/0264-6021:3570127
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Harr, B., and Schlotterer, C. (2006). Comparison of algorithms for the analysis of Affymetrix microarray data as evaluated by co-expression of genes in known operons. Nucleic Acids Res. 34:e8. doi: 10.1093/nar/gnj010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Henriques, A., and Gonzalez De Aguilar, J. L. (2011). Can transcriptomics cut the gordian knot of amyotrophic lateral sclerosis? Curr. Genomics 12, 506–515. doi: 10.2174/138920211797904043
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Henriques, A., Pitzer, C., Dittgen, T., Klugmann, M., Dupuis, L., and Schneider, A. (2011). CNS-targeted viral delivery of G-CSF in an animal model for ALS: improved efficacy and preservation of the neuromuscular unit. Mol. Therapy 19, 284–292. doi: 10.1038/mt.2010.271
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Henriques, A., Pitzer, C., Dupuis, L., and Schneider, A. (2010a). G-CSF protects motoneurons against axotomy-induced apoptotic death in neonatal mice. BMC Neurosci. 11:25. doi: 10.1186/1471-2202-11-25
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Henriques, A., Pitzer, C., and Schneider, A. (2010b). Neurotrophic growth factors for the treatment of amyotrophic lateral sclerosis: where do we stand? Front. Neurosci. 4:32. doi: 10.3389/fnins.2010.00032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Higashi-Kovtun, M. E., Mosca, T. J., Dickman, D. K., Meinertzhagen, I. A., and Schwarz, T. L. (2010). Importin-beta11 regulates synaptic phosphorylated mothers against decapentaplegic, and thereby influences synaptic development and function at the Drosophila neuromuscular junction. J. Neurosci. 30, 5253–5268. doi: 10.1523/JNEUROSCI.3739-09.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Huang, H., and Trussell, L. O. (2011). KCNQ5 channels control resting properties and release probability of a synapse. Nat. Neurosci. 14, 840–847. doi: 10.1038/nn.2830
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hyatt Sachs, H., Schreiber, R. C., Shoemaker, S. E., Sabe, A., Reed, E., and Zigmond, R. E. (2007). Activating transcription factor 3 induction in sympathetic neurons after axotomy: response to decreased neurotrophin availability. Neuroscience 150, 887–897. doi: 10.1016/j.neuroscience.2007.10.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kikuchi, H., Yamada, T., Furuya, H., Doh-ura, K., Ohyagi, Y., Iwaki, T., et al. (2003). Involvement of cathepsin B in the motor neuron degeneration of amyotrophic lateral sclerosis. Acta Neuropathol. 105, 462–468. doi: 10.1007/s00401-002-0667-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kobbe, G., Bruns, I., Fenk, R., Czibere, A., and Haas, R. (2009). Pegfilgrastim for PBSC mobilization and autologous haematopoietic SCT. Bone Marrow Transplant. 43, 669–677. doi: 10.1038/bmt.2009.59
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kruger, K., Pilat, C., Schild, M., Lindner, N., Frech, T., Muders, K., et al. (2014). Progenitor cell mobilization after exercise is related to systemic levels of G-CSF and muscle damage. Scand. J. Med. Sci. Sports doi: 10.1111/sms.12320
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, S., and Strittmatter, S. M. (2003). Delayed systemic Nogo-66 receptor antagonist promotes recovery from spinal cord injury. J. Neurosci. 23, 4219–4227.
Ling, S. C., Polymenidou, M., and Cleveland, D. W. (2013). Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416–438. doi: 10.1016/j.neuron.2013.07.033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Martinez, H. R., Gonzalez-Garza, M. T., Moreno-Cuevas, J. E., Caro, E., Gutierrez-Jimenez, E., and Segura, J. J. (2009). Stem-cell transplantation into the frontal motor cortex in amyotrophic lateral sclerosis patients. Cytotherapy 11, 26–34. doi: 10.1080/14653240802644651
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mishina, Y., Starbuck, M. W., Gentile, M. A., Fukuda, T., Kasparcova, V., Seedor, J. G., et al. (2004). Bone morphogenetic protein type IA receptor signaling regulates postnatal osteoblast function and bone remodeling. J. Biol. Chem. 279, 27560–27566. doi: 10.1074/jbc.M404222200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Motomura, W., Yoshizaki, T., Ohtani, K., Okumura, T., Fukuda, M., Fukuzawa, J., et al. (2008). Immunolocalization of a novel collectin CL-K1 in murine tissues. J. Histochem. Cytochem. 56, 243–252. doi: 10.1369/jhc.7A7312.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Naumenko, N., Pollari, E., Kurronen, A., Giniatullina, R., Shakirzyanova, A., Magga, J., et al. (2011). Gender-Specific mechanism of synaptic impairment and its prevention by GCSF in a mouse model of ALS. Front. Cellular Neurosci. 5:26. doi: 10.3389/fncel.2011.00026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nefussy, B., Artamonov, I., Deutsch, V., Naparstek, E., Nagler, A., and Drory, V. E. (2010). Recombinant human granulocyte-colony stimulating factor administration for treating amyotrophic lateral sclerosis: a pilot study. Amyotroph. Lateral Scler. 11, 187–193. doi: 10.3109/17482960902933809
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Numajiri, T., Mitsui, S., Hisa, Y., Ishida, T., Nishino, K., and Yamaguchi, N. (2009). The expression of a motoneuron-specific serine protease, motopsin (PRSS12), after facial nerve axotomy in mice. J. Plast. Reconstr. Aesthet. Surg. 59, 393–397. doi: 10.1016/j.bjps.2005.04.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ohtani, K., Suzuki, Y., and Wakamiya, N. (2012). Biological functions of the novel collectins CL-L1, CL-K1, and CL-P1. J. Biomed. Biotechnol. 2012:493945. doi: 10.1155/2012/493945
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Perrin, F. E., Boisset, G., Docquier, M., Schaad, O., Descombes, P., and Kato, A. C. (2005). No widespread induction of cell death genes occurs in pure motoneurons in an amyotrophic lateral sclerosis mouse model. Hum. Mol. Genet. 14, 3309–3320. doi: 10.1093/hmg/ddi357
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Philips, T., and Robberecht, W. (2011). Neuroinflammation in amyotrophic lateral sclerosis: role of glial activation in motor neuron disease. Lancet Neurol. 10, 253–263. doi: 10.1016/S1474-4422(11)70015-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pitzer, C., Kruger, C., Plaas, C., Kirsch, F., Dittgen, T., Muller, R., et al. (2008). Granulocyte-colony stimulating factor improves outcome in a mouse model of amyotrophic lateral sclerosis. Brain 131(Pt 12), 3335–3347. doi: 10.1093/brain/awn243
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pollari, E., Savchenko, E., Jaronen, M., Kanninen, K., Malm, T., Wojciechowski, S., et al. (2011). Granulocyte colony stimulating factor attenuates inflammation in a mouse model of amyotrophic lateral sclerosis. J. Neuroinflammation 8:74. doi: 10.1186/1742-2094-8-74
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pradhan, S. J., Nesler, K. R., Rosen, S. F., Kato, Y., Nakamura, A., Ramaswami, M., et al. (2012). The conserved P body component HPat/Pat1 negatively regulates synaptic terminal growth at the larval Drosophila neuromuscular junction. J, Cell Sci. 125(Pt 24), 6105–6116. doi: 10.1242/jcs.113043
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sahni, V., Mukhopadhyay, A., Tysseling, V., Hebert, A., Birch, D., McGuire, T. L., et al. (2010). BMPR1a and BMPR1b signaling exert opposing effects on gliosis after spinal cord injury. J. Neurosci. 30, 1839–1855. doi: 10.1523/JNEUROSCI.4459-09.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Saris, C. G., Groen, E. J., Koekkoek, J. A., Veldink, J. H., and van den Berg, L. H. (2013). Meta-analysis of gene expression profiling in amyotrophic lateral sclerosis: a comparison between transgenic mouse models and human patients. Amyotroph. Lateral Scler. Frontotemporal Degener. 14, 177–189. doi: 10.3109/21678421.2012.729842
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schmitt, F., Hussain, G., Dupuis, L., Loeffler, J. P., and Henriques, A. (2014). A plural role for lipids in motor neuron diseases: energy, signaling and structure. Front. Cell. Neurosci. 8:25. doi: 10.3389/fncel.2014.00025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sleigh, J. N., Barreiro-Iglesias, A., Oliver, P. L., Biba, A., Becker, T., Davies, K. E., et al. (2014). Chondrolectin affects cell survival and neuronal outgrowth in in vitro and in vivo models of spinal muscular atrophy. Hum. Mol. Genet., 23, 855–869. doi: 10.1093/hmg/ddt477
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Starkey, M. L., Davies, M., Yip, P. K., Carter, L. M., Wong, D. J., McMahon, S. B., et al. (2009). Expression of the regeneration-associated protein SPRR1A in primary sensory neurons and spinal cord of the adult mouse following peripheral and central injury. J. Comp. Neurol. 513, 51–68. doi: 10.1002/cne.21944
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Taylor, B. K., Fu, W., Kuphal, K. E., Stiller, C. O., Winter, M. K., Chen, W., et al. (2014). Inflammation enhances Y1 receptor signaling, neuropeptide Y-mediated inhibition of hyperalgesia, and substance P release from primary afferent neurons. Neuroscience 256, 178–194. doi: 10.1016/j.neuroscience.2013.10.054
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tsujii, M., Akeda, K., Iino, T., and Uchida, A. (2009). Are BMPs involved in normal nerve and following transection?: a pilot study. Clin. Orthop. Relat. Res. 467, 3183–3189. doi: 10.1007/s11999-009-1009-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tsujino, H., Kondo, E., Fukuoka, T., Dai, Y., Tokunaga, A., Miki, K., et al. (2000). Activating transcription factor 3 (ATF3) induction by axotomy in sensory and motoneurons: a novel neuronal marker of nerve injury. Mol. Cell. Neurosci. 15, 170–182. doi: 10.1006/mcne.1999.0814
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Turner, M. R., Bowser, R., Bruijn, L., Dupuis, L., Ludolph, A., McGrath, M., et al. (2013). Mechanisms, models and biomarkers in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Frontotemporal Degener. 14, 19–32. doi: 10.3109/21678421.2013.778554
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, J., Duncan, D., Shi, Z., and Zhang, B. (2013). WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013. Nucleic Acids Res. 41, W77–W83. doi: 10.1093/nar/gkt439
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wills, A. M., Hubbard, J., Macklin, E. A., Glass, J., Tandan, R., Simpson, E. P., et al. (2014). Hypercaloric enteral nutrition in patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled phase 2 trial. Lancet. 383, 2065–2072. doi: 10.1016/S0140-6736(14)60222-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xiao, L., Michalski, N., Kronander, E., Gjoni, E., Genoud, C., Knott, G., et al. (2013). BMP signaling specifies the development of a large and fast CNS synapse. Nat. Neurosci. 16, 856–864. doi: 10.1038/nn.3414
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yang, B. B., Savin, M. A., and Green, M. (2012). Prevention of chemotherapy-induced neutropenia with pegfilgrastim: pharmacokinetics and patient outcomes. Chemotherapy 58, 387–398. doi: 10.1159/000345626
Yang, R., Gaidamakov, S. A., Xie, J., Lee, J., Martino, L., Kozlov, G., et al. (2011). La-related protein 4 binds poly(A), interacts with the poly(A)-binding protein MLLE domain via a variant PAM2w motif, and can promote mRNA stability. Mol. Cell. Biol. 31, 542–556. doi: 10.1128/MCB.01162-10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, B., Kirov, S., and Snoddy, J. (2005). WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 33, W741–W748. doi: 10.1093/nar/gki475
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, Y., Wang, L., Fu, Y., Song, H., Zhao, H., Deng, M., et al. (2009). Preliminary investigation of effect of granulocyte colony stimulating factor on amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 10, 430–431. doi: 10.3109/17482960802588059
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhong, Z., Ohnmacht, J., Reimer, M. M., Bach, I., Becker, T., and Becker, C. G. (2012). Chondrolectin mediates growth cone interactions of motor axons with an intermediate target. J. Neurosci. 32, 4426–4439. doi: 10.1523/JNEUROSCI.5179-11.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: motoneuron, ALS, G-CSF, neurodegeneration, mouse model, gene expression, laser microdissection
Citation: Henriques A, Kastner S, Chatzikonstantinou E, Pitzer C, Plaas C, Kirsch F, Wafzig O, Krüger C, Spoelgen R, Gonzalez De Aguilar J-L, Gretz N and Schneider A (2015) Gene expression changes in spinal motoneurons of the SOD1G93A transgenic model for ALS after treatment with G-CSF. Front. Cell. Neurosci. 8:464. doi: 10.3389/fncel.2014.00464
Received: 21 October 2014; Accepted: 20 December 2014;
Published online: 20 January 2015.
Edited by:
Shawn Hayley, Carleton University, CanadaReviewed by:
Rafael Linden, Federal University of Rio de Janeiro, BrazilFlorence Evelyne PERRIN, University of Montpellier, France
Copyright © 2015 Henriques, Kastner, Chatzikonstantinou, Pitzer, Plaas, Kirsch, Wafzig, Krüger, Spoelgen, Gonzalez De Aguilar, Gretz and Schneider. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Armin Schneider, Sygnis Bioscience, Im Neuenheimer Feld 515, 69120 Heidelberg, Germany e-mail:c2NobmVpZGVyQHN5Z25pcy5kZQ==
†These authors have contributed equally to this work.