 Hansen Wang
Hansen WangIntroduction
Fragile X mental retardation protein (FMRP) is a RNA binding protein, the absence of which due to silencing of the FMR1 gene causes fragile X syndrome, an X-linked neurodevelopmental disorder (Bassell and Warren, 2008; Bhakar et al., 2012; Santoro et al., 2012). FMRP regulates the transport, stability and translation of its mRNA targets. Loss of FMRP alters translational control and receptor-mediated signaling pathways, leading to aberrant dendritic spine dynamics, synaptic dysfunction and behavioral phenotypes in fragile X syndrome (Wang et al., 2012; Sidorov et al., 2013; Suhl et al., 2014). Since the identification of FMR1 as the causative gene for fragile X syndrome in 1991, studies have mainly focused on understanding the function of FMRP. More and more potential FMRP mRNA targets and interacting proteins have been identified in the mammalian brain, supporting the critical roles of FMRP in neurodevelopment, synaptic plasticity and other neurological disorders apart from fragile X syndrome (Wang et al., 2008b, 2012; Pasciuto and Bagni, 2014a; Suhl et al., 2014).
Fragile X syndrome, the most common monogenic cause of autism spectrum disorders (ASDs), has been leading the way for better understanding of autism and other neurodevelopmental disorders (Belmonte and Bourgeron, 2006; Bhakar et al., 2012; Banerjee et al., 2014; Cook et al., 2014). Clinically, a substantial proportion of children with fragile X syndrome meets diagnostic criteria for ASDs (Budimirovic and Kaufmann, 2011). Genetically and biologically, many of the neuronal targets of FMRP overlap with genes associated with ASDs, suggesting the common pathways that are dysregulated and might be potential therapeutic targets for these neurodevelopmental disorders (Auerbach et al., 2011; Zoghbi and Bear, 2012; Darnell and Klann, 2013). Interestingly, studies in recent years have further revealed that FMRP regulates a multitude of synaptic proteins and components of signaling pathways that not only affect neurodevelopment, but also contribute to the pathogenesis of neurodegenerative diseases such as Alzheimer disease (AD), the leading cause for dementia in the elderly (Malter et al., 2010; Sokol et al., 2011; Westmark et al., 2011; Hamilton et al., 2014). FMRP may play a pivotal role in the association between neurodevelopmental and neurodegenerative disorders across lifespan.
FMRP and AD Pathogenesis
AD is pathologically characterized by the presence of plaques comprised of β amyloid (Aβ) and neurofibrillary tangles (NFTs) containing hyperphosphorylated tau protein (Selkoe, 2011; Holtzman et al., 2012; Ubhi and Masliah, 2013; Bloom, 2014). A considerable amount of evidence suggests that soluble Aβ oligomers are the predominant neurotoxic species of Aβ, with Aβ 42 fragment as the particularly potent form (Klyubin et al., 2012; Masters and Selkoe, 2012; Klein, 2013). Aβ oligomers exert the toxic effects by binding to their receptors on neuronal synapses, disrupting normal synaptic signaling pathways, which further leads to synaptic damage accompanied by neuronal loss (Benilova et al., 2012; Sheng et al., 2012; Pozueta et al., 2013; Wang et al., 2013; Tu et al., 2014).
FMRP in Aβ Mediated Synaptic Toxicity
A growing number of synaptic proteins have been proposed as potential Aβ receptors or coreceptors, which are believed to mediate Aβ induced synaptic dysfunction (Karran et al., 2011; Paula-Lima et al., 2013; Pozueta et al., 2013; Overk and Masliah, 2014). Those receptors include, but are not limited to, NMDARs, mGluR5, AMPARs, cellular prion protein (PrPC), PSD-95, and EphB2 (Lacor et al., 2004; Lauren et al., 2009; Cisse et al., 2011; Larson and Lesne, 2012; Mucke and Selkoe, 2012; Um et al., 2013; Tu et al., 2014). In fact, some of Aβ receptors (NMDARs, mGluR5, and PSD-95) and their associated scaffolding proteins and adhesion molecules such as SAPAP, Shank, Homer, and SynGAP1, are those whose mRNAs are FMRP targets (Darnell and Klann, 2013; Santini and Klann, 2014), suggesting that FMRP might be involved in initiating toxic effects of Aβ oligomers through regulating Aβ receptors (Figure 1A).
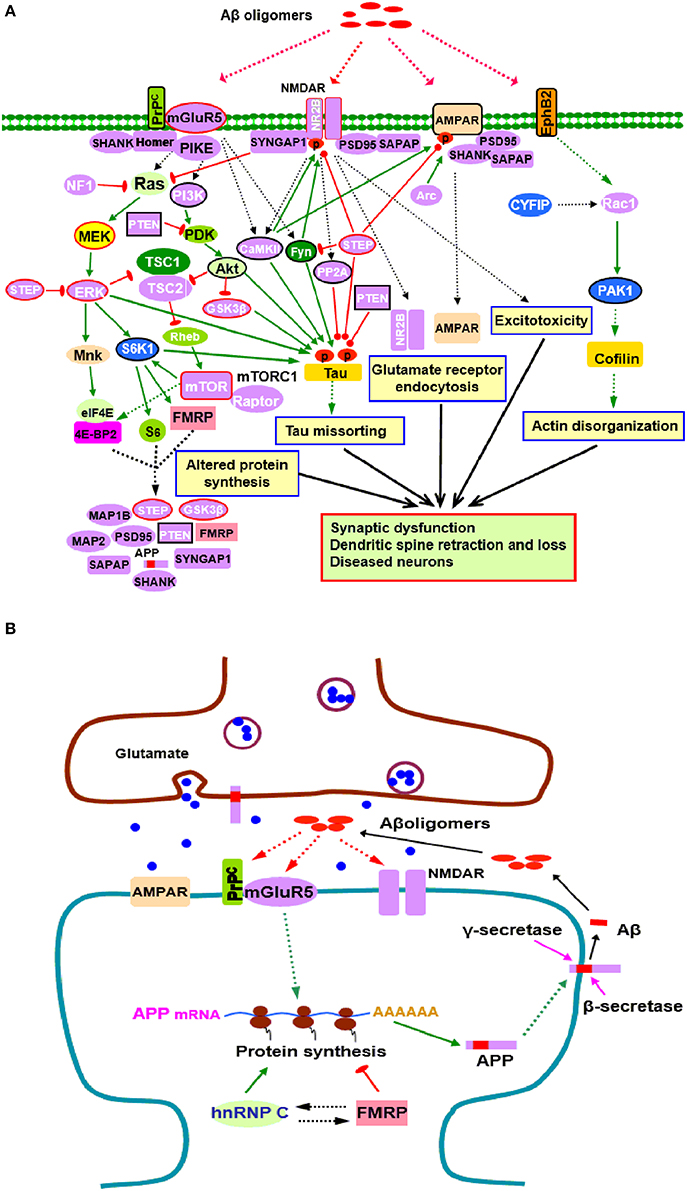
Figure 1. Potential roles of FMRP in the pathogenesis of AD. (A) FMRP might be involved in oligomeric Aβ induced neurotoxicity. At pathological concentrations, Aβ oligomers may interact with multiple neuronal synaptic receptors such as mGluR5-PrPC, NMDARs, AMPARs, and EphB2, triggering a series of toxic synaptic events which may involve FMRP and eventually lead to synaptic dysfunction and neuronal loss. These events include: Aberrant activation of PI3K-Akt-mTORC1 and MEK-ERK signaling pathways linked to cap-dependent translation result in altered synthesis of synaptic proteins; Oligomeric Aβ exposure disrupts the balance between tau kinase (GSK3β, CaMKII, Akt, Fyn, and ERK1/2) and phosphatase (PP2A, STEP, and PTEN) activities, inducing tau hyperphosphorylation and aggregation; Stimulating EphB2-Rac1/PAK1 signaling by Aβ oligomers induces cofilin phosphorylation and actin depolymerization, leading to actin network disorganization; Binding of Aβ oligomers to PrPC-mGluR5 activates Fyn kinase which phosphorylates not only tau, but also NR2B subunit of NMDARs, enhancing NMDAR activity and causing excitotoxicity; STEP is also activated, inactivates Fyn, and dephosphorylates AMPARs and NMDARs, resulting in endocytosis of glutamate receptors, a cellular process involves Arc, PSD-95, SAPAP, and other synaptic proteins. Purple proteins are those whose mRNAs are FMRP targets (Darnell and Klann, 2013; Pasciuto and Bagni, 2014b; Santini and Klann, 2014); the blue ones are the interacting proteins of FMRP (Pasciuto and Bagni, 2014a). Proteins with red lines around them indicate those that have been successfully manipulated either pharmacologically or genetically to reverse molecular, cellular and/or behavioral phenotypes in animal models of AD (Zhang et al., 2010; Malinow, 2012; Caccamo et al., 2014; Feld et al., 2014; Hamilton et al., 2014; Llorens-Martin et al., 2014) as well as ASDs (Goebel-Goody et al., 2012; Guo et al., 2012; Won et al., 2012; Darnell and Klann, 2013; Osterweil et al., 2013; Wang and Doering, 2013; Wang, 2014). Proteins with black lines around them are the ones that have been reported to be potential targets for AD therapy (Griffin et al., 2005; Lafay-Chebassier et al., 2005; Ma et al., 2008; Cisse et al., 2011; Moriguchi, 2011; Chang et al., 2012; Gross and Bassell, 2014; Nygaard et al., 2014; Sontag and Sontag, 2014). (B) FMRP in APP synthesis. Aβ oligomers stimulate dendritic APP synthesis through PrPC-mGluR5 mediated protein translation dependent pathway, providing template for secretase cleavage to produce Aβ and other metabolites. A positive feedback may exist whereby production of APP results in increased substrate for amyloidogenic processing and release of Aβ, which then acitivates mGluR5 to further stimulate APP translation. In this process, FMRP competes with the other RNA binding protein hnRNP C to modulate APP translation. FMRP is a repressor of APP translation, whereas hnRNP C acts as an enhancer. The rate of APP synthesis is directly influenced by the relative association of each RNA binding protein (Lee et al., 2010). In signaling pathways, arrows indicate positive (green) or inhibitory (red) consequence on downstream components, but they do not necessarily represent direct interactions.
Aβ induced synaptic dysfunction/loss is a complicated process involving multiple signaling pathways and biological events (Benilova et al., 2012; Tu et al., 2014). FMRP may be implicated in this process through selective regulation of components in those signaling pathways perturbed by Aβ oligomers (Figure 1A). Binding of glutamate receptors (NMDARs, mGluR5, and AMPARs) by Aβ oligomers impairs glutamatergic neurotransmission and triggers aberrant activation of downstream pathways, including PI3K-Akt-mTOR and MEK-ERK signaling pathways (Benilova et al., 2012; Pozueta et al., 2013) which are coupled to cap-dependent translation (Darnell and Klann, 2013; Santini and Klann, 2014). FMRP may regulate the activity of these translational control pathways directly since several components (PIKE, PI3K, mTOR, S6K1, and ERK) and negative regulators (PTEN, NF1, and STEP) of the pathways are FMRP targets (Figure 1A). FMRP also plays a critical role in regulating the balance of cap-dependent translation of its target mRNAs (Darnell and Klann, 2013; Sidorov et al., 2013; Santini and Klann, 2014). Dysregulation of the translational control pathways caused by Aβ oligomers will affect the protein products of FMRP targets, including Aβ precursor protein (APP), ARC, STEP, PTEN, GSK3β and other molecules which are closely linked to AD or other neurodegenerative disorders (Figure 1A), further associating FMRP with neurodegeneration.
Aβ induced synaptic degeneration also involves surface removal and endocytosis of glutmate receptors (NMDARs and AMPARs) (Um et al., 2012; Boehm, 2013; Tu et al., 2014). The FMRP targets, such as the scaffolding protein PSD-95 and SAPAP3, immediate-early gene product Arc, protein kinase CaMKII and tyrosine phosphatase STEP, are key determinants of NMDAR or AMPAR endocytosis. PSD-95 and SAPAP3 tethers the receptors to signaling proteins and neuronal cytoskeleton (Wang et al., 2010; Chen et al., 2011); Arc is a component of AMPAR endocytosis machinery and regulates the endocytosis rate (Chowdhury et al., 2006; Waung et al., 2008); CaMKII and STEP mediate surface expression or internalization of NMDARs and AMPARs through phosphorylation/dephosphorylation of relative receptor subunits (Wang et al., 2008b; Zhang et al., 2008; Lisman et al., 2012), supporting possible involvement of FMRP in glutmate receptor endocytosis (Figure 1A). The actin disorganization contributes to AD pathogenesis (Maloney and Bamburg, 2007; Bamburg and Bloom, 2009; Penzes and Vanleeuwen, 2011). The Rac1/PAK1 signaling downstream of EphB2 is involved in actin cytokeleton remodeling; CYFIP1/2 are linked to Rac1. Binding of EphB2 by Aβ oligomers stimulates PAK1, which further induces cofilin phosphorylation to mediate actin depolymerization, thereby inducing actin network disorganization, dendritic spine shrinkage and loss (Ma et al., 2012; Abekhoukh and Bardoni, 2014; Cisse and Checler, 2014). In this signaling pathway, Rac1 is a FMRP target, and CYFIP1/2 and PAK interact with FMRP (Figure 1A). Thus, FMRP is likely to play a role in actin disorganization in AD pathogenesis.
The presence of hyperphosphorylated tau enriched NFTs is a classical AD pathological hallmark. Tau, a microtubule associated protein (MAP), becomes hyperphosphorylated and disassociated from microtubules under pathological conditions, subsequently forming soluble aggregates, insoluble filaments, and eventually NFTs in affected brain regions (Medina and Avila, 2014; Zempel and Mandelkow, 2014). Studies have shown that Aβ-induced synaptic loss and toxicity are tau dependent (Vossel et al., 2010; Roberson et al., 2011). There is a causal association between oligomeric Aβ exposure and tau phosphorylation (Larson et al., 2012; Boehm, 2013). Tau phosphorylation is regulated by a balance between tau kinase and phosphatase activities. Disruption of this balance by Aβ exposure is suggested to cause abnormal tau phosphorylation and thereby contributes to tau aggregation (Wang et al., 2007; Martin et al., 2013a,b). Of the tau protein kinases, GSK3β, CaMKII, ERK1/2, and S6K1 are known to be FMRP targets (Figure 1A). The decrease in the levels or activity of protein phosphatase(s) that dephosphorylate tau also contributes to AD pathology (Braithwaite et al., 2012; Martin et al., 2013a); among these phosphatases, PP2A, STEP, and PTEN are FMRP targets (Figure 1A). FMRP thus, might be involved in tau pathology through regulating those tau kinases and phosphatases. Hyperphosphorylated tau sequesters normal tau, and the other two major MAPs (MAP1 and MAP2), causing disruption of microtubules and misfolding of tau (Zempel and Mandelkow, 2014). Notably, both MAP1A and MAP2 are FMRP targets, further implicating FMRP in neurofibrillary degeneration (Figure 1A).
FMRP in Aβ Stimuiated APP Synthesis
Aβ is produced by the sequential proteolytic cleavage of APP by β - and γ-secretases via amyloidogenic pathway (Masters and Selkoe, 2012). The expression of APP can be upregulated upon mGluR5 stimulation. FMRP, which is also regulated by mGluR5 (Ronesi and Huber, 2008; Wang et al., 2008a; Wang and Zhuo, 2012), binds to and represses the translation of APP mRNA due to mGluR5 activation (Westmark and Malter, 2007; Westmark, 2013).
The mGluR5 links FMRP with APP. It is known that mGluR5 acts as a coreceptor for Aβ oligomers bound to PrPC (Um et al., 2013). Aβ oligomers can stimulate APP synthesis through the mGluR5 and protein translation dependent pathway which involves FMRP, providing template for secretase cleavage to produce Aβ and other metabolites (Westmark and Malter, 2007; Westmark, 2013). Aβ oligomer interactions with mGluR5-PrPC may function to accelerate Aβ production through the FMRP dependent signaling pathway, suggesting that a positive feedback loop may exist in AD, whereby translation of APP results in increased substrate for amyloidogenic processing and generation of Aβ which then stimulates mGluR5 signaling to induce further synaptic synthesis of APP (Westmark and Malter, 2007; Westmark, 2013) (Figure 1B). Thus, in addition to the established role in fragile X syndrome and autism, FMRP likely contributes directly to AD pathogenesis through mGluR5 dependent APP production.
FMRP Targets and AD Therapy
As discussed above, a number of signaling pathways, including PI3K-Akt-mTORC1, MEK-ERK and PAK1 pathways, have been found to be involved in the neurodegenerative progression of AD. Therapies for AD might require the development of drugs targeting these aberrant signaling pathways, among which several key signaling proteins such as PI3K, mTOR, ERK and PAK1, are targets of FMRP. In addition, the FMRP targeted Aβ oligomer receptors including mGluR5 and NMDARs could be ideal therapeutic targets for AD (Figure 1A). Particularly, pharmacological inhibition or genetic deletion of mGluR5 was recently found to rescue learning deficits, or reduce Aβ oligomers and plaques in AD mice (Um et al., 2013; Hamilton et al., 2014).
Tau plays crucial roles in the neuronal cytoskeleton stabilization and is an important target for AD therapies (Gotz et al., 2012; Himmelstein et al., 2012; Giacobini and Gold, 2013). Interventions focused on preventing or reducing tau hyperphosphorylation and mislocalization may provide additional strategies for treatment of AD. The therapeutic tactics include Tau kinase inhibitors and phosphatase activators (Giacobini and Gold, 2013; Zempel and Mandelkow, 2014). The relevant kinases and phosphatases could be the FMRP targets such as GSK3β, ERK, S6K1, PP2A, PTEN, and STEP (Figure 1A). Although the tau based treatments are encouraging, additional work are undoubtedly needed to optimize each treatment for further development of safe and effective therapies.
Therefore, FMRP targeted signaling molecules not only provide therapeutic strategies for fragile X syndrome and other ASDs (Darnell and Klann, 2013; Santini and Klann, 2014), but may serve as potential targets for treatment of AD. Indeed, many components of altered signaling pathways in AD overlap with those in ASDs. A number of signaling proteins targeted by FMRP have been successfully manipulated either pharmacologically or genetically to reverse molecular, cellular and/or behavioral phenotypes in animal models of both ASDs and AD (Figure 1A). FMRP thus, acts as a molecular link between ASDs and AD through the common signaling pathways among the diseases. Developing novel therapies directed at FMRP targets may benefit both neurodevelopmental and neurodegenerative disorders.
Future Perspectives
It is now known that FMRP controls signaling pathways that could be associated with both neurodevelopmental and neurodegenerative disorders. FMRP not only regulates gene expression at the translational level, but also interacts with a multitude of proteins at both presynaptic and postsynaptic sites (Pasciuto and Bagni, 2014a; Myrick et al., 2015). However, so far no systemic proteomic analysis of FMRP interactome in brain has been reported. Additionally, although many of the interacting proteins of FMRP such as transactive response DNA-binding protein-43 (TDP-43), survival of motor neuron 1 (SMN1) and CYFIP are known to be linked to neurological disorders (Abekhoukh and Bardoni, 2014; Pasciuto and Bagni, 2014a), the significance of the protein-protein interaction to individual proteins and diseases still need to be further characterized.
The AD animal or cell models are powerful tools for investigating the pathogenesis of the neurodegenerative disease. It will be useful to set up the transgenic AD mice/Aβ-treated primary neuronal culture which either lack or overexpress FMRP for further evaluating the pathological role of FMRP in AD. FMRP regulates specific mRNA/protein targets at different developmental stages and in different brain areas. Future studies will need to provide detailed information on FMRP mRNA targets and FMRP interactome in relevant brain areas at specific developmental stages of AD animal models. The information will greatly help to further elucidate the pathogenesis of this neurodegenerative disease and develop relative therapeutic strategies.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Hansen Wang was supported by the National Natural Science Foundation of China (NSFC, No.30200152) for Rett syndrome studies and the Fragile X Research Foundation of Canada.
Abbreviations
4E-BP2, eIF4E-binding protein 2; AD, Alzheimer disease; Aβ, β amyloid; AMPAR, α-amino-3-hydroxyl-4-isoxazole propionic acid receptors; APP, amyloid precursor protein; Arc, activity-regulated cytoskeleton-associated protein; CYFIP, cytoplasmic FMRP interacting protein; eIF4E, eukaryotic initiation factor 4E; EphB2, ephrin type-B receptor 2; ERK, extracellular signal related kinase; FMRP, fragile X mental retardation protein; GABA, gamma aminobutyric acid; GSK3β, glycogen synthase kinase-3β; hnRNP C, heterogeneous nuclear ribonucleoprotein C; MAP1A/2, microtubule associated protein 1A/2; MEK, mitogen-activated protein kinase/ERK kinase; mGluR5, metabotropic glutamate receptor 5; Mnk, mitogen-activated protein kinase interacting kinase; mTOR, mammalian target of rapamycin; mTORC1, mammalian target of rapamycin complex 1; NF1, neurofibromatosis 1; NMDAR, N-methyl-d-aspartate receptors; PAK1, p21-activated kinase 1; PI3K, phosphatidylinositol 3-kinase; PIKE, PI3K enhancer; PP2A, protein phosphatase 2A; PrPC, cellular prion protein; PSD-95, postsynaptic density 95; PTEN, Phosphatase and tensin homolog; Raptor, regulatory-associated protein of mTOR; S6K1, p70 ribosomal kinase 1; SAPAP, SAP90/PSD-95–associated protein; SHANK, Src homology 3 (SH3) and multiple ankyrin repeat domains protein; STEP, striatal-enriched protein tyrosine phosphatase; SYNGAP1, synaptic Ras guanosine triphosphatase (GTPase)–activating protein 1; TSC1/2, tuberous sclerosis complex 1/2.
References
Abekhoukh, S., and Bardoni, B. (2014). CYFIP family proteins between autism and intellectual disability: links with Fragile X syndrome. Front. Cell. Neurosci. 8:81. doi: 10.3389/fncel.2014.00081
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Auerbach, B. D., Osterweil, E. K., and Bear, M. F. (2011). Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature 480, 63–68. doi: 10.1038/nature10658
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bamburg, J. R., and Bloom, G. S. (2009). Cytoskeletal pathologies of Alzheimer disease. Cell Motil. Cytoskeleton 66, 635–649. doi: 10.1002/cm.20388
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Banerjee, S., Riordan, M., and Bhat, M. A. (2014). Genetic aspects of autism spectrum disorders: insights from animal models. Front. Cell. Neurosci. 8:58. doi: 10.3389/fncel.2014.00058
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bassell, G. J., and Warren, S. T. (2008). Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron 60, 201–214. doi: 10.1016/j.neuron.2008.10.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Belmonte, M. K., and Bourgeron, T. (2006). Fragile X syndrome and autism at the intersection of genetic and neural networks. Nat Neurosci 9, 1221–1225. doi: 10.1038/nn1765
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Benilova, I., Karran, E., and De Strooper, B. (2012). The toxic Abeta oligomer and Alzheimer's disease: an emperor in need of clothes. Nat. Neurosci. 15, 349–357. doi: 10.1038/nn.3028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bhakar, A. L., Dolen, G., and Bear, M. F. (2012). The pathophysiology of fragile X. (and what it teaches us about synapses). Annu. Rev. Neurosci. 35, 417–443. doi: 10.1146/annurev-neuro-060909-153138
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bloom, G. S. (2014). Amyloid-beta and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 71, 505–508. doi: 10.1001/jamaneurol.2013.5847
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Boehm, J. (2013). A ‘danse macabre’: tau and Fyn in STEP with amyloid beta to facilitate induction of synaptic depression and excitotoxicity. Eur. J. Neurosci. 37, 1925–1930. doi: 10.1111/ejn.12251
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Braithwaite, S. P., Stock, J. B., Lombroso, P. J., and Nairn, A. C. (2012). Protein phosphatases and Alzheimer's disease. Prog. Mol. Biol. Transl. Sci. 106, 343–379. doi: 10.1016/B978-0-12-396456-4.00012-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Budimirovic, D. B., and Kaufmann, W. E. (2011). What can we learn about autism from studying fragile X syndrome? Dev. Neurosci. 33, 379–394. doi: 10.1159/000330213
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Caccamo, A., De Pinto, V., Messina, A., Branca, C., and Oddo, S. (2014). Genetic reduction of mammalian target of rapamycin ameliorates Alzheimer's disease-like cognitive and pathological deficits by restoring hippocampal gene expression signature. J. Neurosci. 34, 7988–7998. doi: 10.1523/JNEUROSCI.0777-14.2014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chang, P. K., Verbich, D., and McKinney, R. A. (2012). AMPA receptors as drug targets in neurological disease–advantages, caveats, and future outlook. Eur. J. Neurosci. 35, 1908–1916. doi: 10.1111/j.1460-9568.2012.08165.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, X., Nelson, C. D., Li, X., Winters, C. A., Azzam, R., Sousa, A. A., et al. (2011). PSD-95 is required to sustain the molecular organization of the postsynaptic density. J. Neurosci. 31, 6329–6338. doi: 10.1523/JNEUROSCI.5968-10.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chowdhury, S., Shepherd, J. D., Okuno, H., Lyford, G., Petralia, R. S., Plath, N., et al. (2006). Arc/Arg3.1 interacts with the endocytic machinery to regulate AMPA receptor trafficking. Neuron 52, 445–459. doi: 10.1016/j.neuron.2006.08.033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cisse, M., and Checler, F. (2014). Eph receptors: new players in Alzheimer's disease pathogenesis. Neurobiol. Dis. 73C, 137–149. doi: 10.1016/j.nbd.2014.08.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cisse, M., Halabisky, B., Harris, J., Devidze, N., Dubal, D. B., Sun, B., et al. (2011). Reversing EphB2 depletion rescues cognitive functions in Alzheimer model. Nature 469, 47–52. doi: 10.1038/nature09635
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cook, D., Nuro, E., and Murai, K. K. (2014). Increasing our understanding of human cognition through the study of Fragile X Syndrome. Dev. Neurobiol. 74, 147–177. doi: 10.1002/dneu.22096
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Darnell, J. C., and Klann, E. (2013). The translation of translational control by FMRP: therapeutic targets for FXS. Nat. Neurosci. 16, 1530–1536. doi: 10.1038/nn.3379
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Feld, M., Krawczyk, M. C., Sol Fustinana, M., Blake, M. G., Baratti, C. M., Romano, A., et al. (2014). Decrease of ERK/MAPK overactivation in prefrontal cortex reverses early memory deficit in a mouse model of Alzheimer's disease. J. Alzheimers Dis. 40, 69–82. doi: 10.3233/JAD-131076
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Giacobini, E., and Gold, G. (2013). Alzheimer disease therapy–moving from amyloid-beta to tau. Nat. Rev. Neurol. 9, 677–686. doi: 10.1038/nrneurol.2013.223
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Goebel-Goody, S. M., Wilson-Wallis, E. D., Royston, S., Tagliatela, S. M., Naegele, J. R., and Lombroso, P. J. (2012). Genetic manipulation of STEP reverses behavioral abnormalities in a fragile X syndrome mouse model. Genes Brain Behav. 11, 586–600. doi: 10.1111/j.1601-183X.2012.00781.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gotz, J., Ittner, A., and Ittner, L. M. (2012). Tau-targeted treatment strategies in Alzheimer's disease. Br. J. Pharmacol. 165, 1246–1259. doi: 10.1111/j.1476-5381.2011.01713.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Griffin, R. J., Moloney, A., Kelliher, M., Johnston, J. A., Ravid, R., Dockery, P., et al. (2005). Activation of Akt/PKB, increased phosphorylation of Akt substrates and loss and altered distribution of Akt and PTEN are features of Alzheimer's disease pathology. J. Neurochem. 93, 105–117. doi: 10.1111/j.1471-4159.2004.02949.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gross, C., and Bassell, G. J. (2014). Neuron-specific regulation of class I PI3K catalytic subunits and their dysfunction in brain disorders. Front. Mol. Neurosci. 7:12. doi: 10.3389/fnmol.2014.00012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guo, W., Murthy, A. C., Zhang, L., Johnson, E. B., Schaller, E. G., Allan, A. M., et al. (2012). Inhibition of GSK3beta improves hippocampus-dependent learning and rescues neurogenesis in a mouse model of fragile X syndrome. Hum. Mol. Genet. 21, 681–691. doi: 10.1093/hmg/ddr501
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hamilton, A., Esseltine, J. L., DeVries, R. A., Cregan, S. P., and Ferguson, S. S. (2014). Metabotropic glutamate receptor 5 knockout reduces cognitive impairment and pathogenesis in a mouse model of Alzheimer's disease. Mol. Brain 7:40. doi: 10.1186/1756-6606-7-40
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Himmelstein, D. S., Ward, S. M., Lancia, J. K., Patterson, K. R., and Binder, L. I. (2012). Tau as a therapeutic target in neurodegenerative disease. Pharmacol. Ther. 136, 8–22. doi: 10.1016/j.pharmthera.2012.07.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Holtzman, D. M., Mandelkow, E., and Selkoe, D. J. (2012). Alzheimer disease in 2020. Cold Spring Harb. Perspect. Med. 2:a011585. doi: 10.1101/cshperspect.a011585
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Karran, E., Mercken, M., and de Strooper, B. (2011). The amyloid cascade hypothesis for Alzheimer's disease: an appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 10, 698–712. doi: 10.1038/nrd3505
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Klein, W. L. (2013). Synaptotoxic amyloid-beta oligomers: a molecular basis for the cause, diagnosis, and treatment of Alzheimer's disease? J. Alzheimers Dis. 33(Suppl. 1), S49–S65. doi: 10.3233/JAD-2012-129039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Klyubin, I., Cullen, W. K., Hu, N. W., and Rowan, M. J. (2012). Alzheimer's disease Abeta assemblies mediating rapid disruption of synaptic plasticity and memory. Mol. Brain 5:25. doi: 10.1186/1756-6606-5-25
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lacor, P. N., Buniel, M. C., Chang, L., Fernandez, S. J., Gong, Y., Viola, K. L., et al. (2004). Synaptic targeting by Alzheimer's-related amyloid beta oligomers. J. Neurosci. 24, 10191–10200. doi: 10.1523/JNEUROSCI.3432-04.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lafay-Chebassier, C., Paccalin, M., Page, G., Barc-Pain, S., Perault-Pochat, M. C., Gil, R., et al. (2005). mTOR/p70S6k signalling alteration by Abeta exposure as well as in APP-PS1 transgenic models and in patients with Alzheimer's disease. J. Neurochem. 94, 215–225. doi: 10.1111/j.1471-4159.2005.03187.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Larson, M. E., and Lesne, S. E. (2012). Soluble Abeta oligomer production and toxicity. J. Neurochem. 120(Suppl. 1), 125–139. doi: 10.1111/j.1471-4159.2011.07478.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Larson, M., Sherman, M. A., Amar, F., Nuvolone, M., Schneider, J. A., Bennett, D. A., et al. (2012). The complex PrP(c)-Fyn couples human oligomeric Abeta with pathological tau changes in Alzheimer's disease. J. Neurosci. 32, 16857a–16871a. doi: 10.1523/JNEUROSCI.1858-12.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lauren, J., Gimbel, D. A., Nygaard, H. B., Gilbert, J. W., and Strittmatter, S. M. (2009). Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 457, 1128–1132. doi: 10.1038/nature07761
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, E. K., Kim, H. H., Kuwano, Y., Abdelmohsen, K., Srikantan, S., Subaran, S. S., et al. (2010). hnRNP C promotes APP translation by competing with FMRP for APP mRNA recruitment to P bodies. Nat. Struct. Mol. Biol. 17, 732–739. doi: 10.1038/nsmb.1815
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lisman, J., Yasuda, R., and Raghavachari, S. (2012). Mechanisms of CaMKII action in long-term potentiation. Nat. Rev. Neurosci. 13, 169–182. doi: 10.1038/nrn3192.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Llorens-Martin, M., Jurado, J., Hernandez, F., and Avila, J. (2014). GSK-3beta, a pivotal kinase in Alzheimer disease. Front. Mol. Neurosci. 7:46. doi: 10.3389/fnmol.2014.00046
Ma, Q. L., Yang, F., Calon, F., Ubeda, O. J., Hansen, J. E., Weisbart, R. H., et al. (2008). p21-activated kinase-aberrant activation and translocation in Alzheimer disease pathogenesis. J. Biol. Chem. 283, 14132–14143. doi: 10.1074/jbc.M708034200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ma, Q. L., Yang, F., Frautschy, S. A., and Cole, G. M. (2012). PAK in Alzheimer disease, Huntington disease and X-linked mental retardation. Cell. Logist. 2, 117–125. doi: 10.4161/cl.21602
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Malinow, R. (2012). New developments on the role of NMDA receptors in Alzheimer's disease. Curr. Opin. Neurobiol. 22, 559–563. doi: 10.1016/j.conb.2011.09.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maloney, M. T., and Bamburg, J. R. (2007). Cofilin-mediated neurodegeneration in Alzheimer's disease and other amyloidopathies. Mol. Neurobiol. 35, 21–44. doi: 10.1007/BF02700622
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Malter, J. S., Ray, B. C., Westmark, P. R., and Westmark, C. J. (2010). Fragile X Syndrome and Alzheimer's disease: another story about APP and beta-amyloid. Curr. Alzheimer Res. 7, 200–206. doi: 10.2174/156720510791050957
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Martin, L., Latypova, X., Wilson, C. M., Magnaudeix, A., Perrin, M. L., and Terro, F. (2013a). Tau protein phosphatases in Alzheimer's disease: the leading role of PP2A. Ageing Res. Rev. 12, 39–49. doi: 10.1016/j.arr.2012.06.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Martin, L., Latypova, X., Wilson, C. M., Magnaudeix, A., Perrin, M. L., Yardin, C., et al. (2013b). Tau protein kinases: involvement in Alzheimer's disease. Ageing Res. Rev. 12, 289–309. doi: 10.1016/j.arr.2012.06.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Masters, C. L., and Selkoe, D. J. (2012). Biochemistry of amyloid beta-protein and amyloid deposits in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2:a006262. doi: 10.1101/cshperspect.a006262
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Medina, M., and Avila, J. (2014). New perspectives on the role of tau in Alzheimer's disease. Implications for therapy. Biochem. Pharmacol. 88, 540–547. doi: 10.1016/j.bcp.2014.01.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moriguchi, S. (2011). Pharmacological study on Alzheimer's drugs targeting calcium/calmodulin-dependent protein kinase II. J. Pharmacol. Sci. 117, 6–11. doi: 10.1254/jphs.11R06CP
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mucke, L., and Selkoe, D. J. (2012). Neurotoxicity of amyloid beta-protein: synaptic and network dysfunction. Cold Spring Harb. Perspect. Med. 2:a006338. doi: 10.1101/cshperspect.a006338
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Myrick, L. K., Deng, P. Y., Hashimoto, H., Oh, Y. M., Cho, Y., Poidevin, M. J., et al. (2015). Independent role for presynaptic FMRP revealed by an FMR1 missense mutation associated with intellectual disability and seizures. Proc. Natl. Acad. Sci. U.S.A. 112, 949–956. doi: 10.1073/pnas.1423094112
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nygaard, H. B., van Dyck, C. H., and Strittmatter, S. M. (2014). Fyn kinase inhibition as a novel therapy for Alzheimer's disease. Alzheimers Res. Ther. 6, 8. doi: 10.1186/alzrt238
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Osterweil, E. K., Chuang, S. C., Chubykin, A. A., Sidorov, M., Bianchi, R., Wong, R. K., et al. (2013). Lovastatin corrects excess protein synthesis and prevents epileptogenesis in a mouse model of fragile X syndrome. Neuron 77, 243–250. doi: 10.1016/j.neuron.2012.01.034
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Overk, C. R., and Masliah, E. (2014). Pathogenesis of synaptic degeneration in Alzheimer's disease and Lewy body disease. Biochem. Pharmacol. 88, 508–516. doi: 10.1016/j.bcp.2014.01.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pasciuto, E., and Bagni, C. (2014a). SnapShot: FMRP interacting proteins. Cell 159, 218–e211. doi: 10.1016/j.cell.2014.08.036
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pasciuto, E., and Bagni, C. (2014b). SnapShot: FMRP mRNA targets and diseases. Cell 158, 1446–e1441. doi: 10.1016/j.cell.2014.08.035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Paula-Lima, A. C., Brito-Moreira, J., and Ferreira, S. T. (2013). Deregulation of excitatory neurotransmission underlying synapse failure in Alzheimer's disease. J. Neurochem. 126, 191–202. doi: 10.1111/jnc.12304
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Penzes, P., and Vanleeuwen, J. E. (2011). Impaired regulation of synaptic actin cytoskeleton in Alzheimer's disease. Brain Res. Rev. 67, 184–192. doi: 10.1016/j.brainresrev.2011.01.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pozueta, J., Lefort, R., and Shelanski, M. L. (2013). Synaptic changes in Alzheimer's disease and its models. Neuroscience 251, 51–65. doi: 10.1016/j.neuroscience.2012.05.050
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Roberson, E. D., Halabisky, B., Yoo, J. W., Yao, J., Chin, J., Yan, F., et al. (2011). Amyloid-beta/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer's disease. J. Neurosci. 31, 700–711. doi: 10.1523/JNEUROSCI.4152-10.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ronesi, J. A., and Huber, K. M. (2008). Metabotropic glutamate receptors and fragile x mental retardation protein: partners in translational regulation at the synapse. Sci. Signal. 1:pe6. doi: 10.1126/stke.15pe6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Santini, E., and Klann, E. (2014). Reciprocal signaling between translational control pathways and synaptic proteins in autism spectrum disorders. Sci. Signal. 7:re10. doi: 10.1126/scisignal.2005832
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Santoro, M. R., Bray, S. M., and Warren, S. T. (2012). Molecular mechanisms of fragile X syndrome: a twenty-year perspective. Annu. Rev. Pathol. 7, 219–245. doi: 10.1146/annurev-pathol-011811-132457
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Selkoe, D. J. (2011). Alzheimer's disease. Cold Spring Harb. Perspect. Biol. 3:a004457. doi: 10.1101/cshperspect.a004457
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sheng, M., Sabatini, B. L., and Sudhof, T. C. (2012). Synapses and Alzheimer's disease. Cold Spring Harb. Perspect. Biol. 4:a005777. doi: 10.1101/cshperspect.a005777
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sidorov, M. S., Auerbach, B. D., and Bear, M. F. (2013). Fragile X mental retardation protein and synaptic plasticity. Mol. Brain 6:15. doi: 10.1186/1756-6606-6-15
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sokol, D. K., Maloney, B., Long, J. M., Ray, B., and Lahiri, D. K. (2011). Autism, Alzheimer disease, and fragile X: APP, FMRP, and mGluR5 are molecular links. Neurology 76, 1344–1352. doi: 10.1212/WNL.0b013e3182166dc7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sontag, J. M., and Sontag, E. (2014). Protein phosphatase 2A dysfunction in Alzheimer's disease. Front. Mol. Neurosci. 7:16. doi: 10.3389/fnmol.2014.00016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Suhl, J. A., Chopra, P., Anderson, B. R., Bassell, G. J., and Warren, S. T. (2014). Analysis of FMRP mRNA target datasets reveals highly associated mRNAs mediated by G-quadruplex structures formed via clustered WGGA sequences. Hum. Mol. Genet. 23, 5479–5491. doi: 10.1093/hmg/ddu272
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tu, S., Okamoto, S., Lipton, S. A., and Xu, H. (2014). Oligomeric Abeta-induced synaptic dysfunction in Alzheimer's disease. Mol. Neurodegener. 9:48. doi: 10.1186/1750-1326-9-48
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ubhi, K., and Masliah, E. (2013). Alzheimer's disease: recent advances and future perspectives. J. Alzheimers Dis. 33(Suppl. 1), S185–S194. doi: 10.3233/JAD-2012-129028.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Um, J. W., Kaufman, A. C., Kostylev, M., Heiss, J. K., Stagi, M., Takahashi, H., et al. (2013). Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer abeta oligomer bound to cellular prion protein. Neuron 79, 887–902. doi: 10.1016/j.neuron.2013.06.036
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Um, J. W., Nygaard, H. B., Heiss, J. K., Kostylev, M. A., Stagi, M., Vortmeyer, A., et al. (2012). Alzheimer amyloid-beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 15, 1227–1235. doi: 10.1038/nn.3178
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vossel, K. A., Zhang, K., Brodbeck, J., Daub, A. C., Sharma, P., Finkbeiner, S., et al. (2010). Tau reduction prevents Abeta-induced defects in axonal transport. Science 330, 198. doi: 10.1126/science.1194653
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, H. (2014). Lipid rafts: a signaling platform linking cholesterol metabolism to synaptic deficits in autism spectrum disorders. Front. Behav. Neurosci. 8:104. doi: 10.3389/fnbeh.2014.00104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, H., and Doering, L. C. (2013). Reversing autism by targeting downstream mTOR signaling. Front. Cell. Neurosci. 7:28. doi: 10.3389/fncel.2013.00028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, H., Kim, S. S., and Zhuo, M. (2010). Roles of fragile X mental retardation protein in dopaminergic stimulation-induced synapse-associated protein synthesis and subsequent alpha-amino-3-hydroxyl-5-methyl-4-isoxazole-4-propionate. (AMPA). receptor internalization. J. Biol. Chem. 285, 21888–21901. doi: 10.1074/jbc.M110.116293
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, H., Ren, C. H., Gunawardana, C. G., and Schmitt-Ulms, G. (2013). Overcoming barriers and thresholds - signaling of oligomeric Abeta through the prion protein to Fyn. Mol. Neurodegener. 8:24. doi: 10.1186/1750-1326-8-24
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, H., Wu, L. J., Kim, S. S., Lee, F. J., Gong, B., Toyoda, H., et al. (2008b). FMRP acts as a key messenger for dopamine modulation in the forebrain. Neuron 59, 634–647. doi: 10.1016/j.neuron.2008.06.027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, H., Wu, L. J., Zhang, F., and Zhuo, M. (2008a). Roles of calcium-stimulated adenylyl cyclase and calmodulin-dependent protein kinase IV in the regulation of FMRP by group I metabotropic glutamate receptors. J. Neurosci. 28, 4385–4397. doi: 10.1523/JNEUROSCI.0646-08.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, H., and Zhuo, M. (2012). Group I metabotropic glutamate receptor-mediated gene transcription and implications for synaptic plasticity and diseases. Front. Pharmacol. 3:189. doi: 10.3389/fphar.2012.00189
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, J. Z., Grundke-Iqbal, I., and Iqbal, K. (2007). Kinases and phosphatases and tau sites involved in Alzheimer neurofibrillary degeneration. Eur. J. Neurosci. 25, 59–68. doi: 10.1111/j.1460-9568.2006.05226.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, T., Bray, S. M., and Warren, S. T. (2012). New perspectives on the biology of fragile X syndrome. Curr. Opin. Genet. Dev. 22, 256–263. doi: 10.1016/j.gde.2012.02.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Waung, M. W., Pfeiffer, B. E., Nosyreva, E. D., Ronesi, J. A., and Huber, K. M. (2008). Rapid translation of Arc/Arg3.1 selectively mediates mGluR-dependent LTD through persistent increases in AMPAR endocytosis rate. Neuron 59, 84–97. doi: 10.1016/j.neuron.2008.05.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Westmark, C. J. (2013). What's hAPPening at synapses? The role of amyloid beta-protein precursor and beta-amyloid in neurological disorders. Mol. Psychiatry 18, 425–434. doi: 10.1038/mp.2012.122
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Westmark, C. J., and Malter, J. S. (2007). FMRP mediates mGluR5-dependent translation of amyloid precursor protein. PLoS Biol. 5:e52. doi: 10.1371/journal.pbio.0050052
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Westmark, C. J., Westmark, P. R., O'Riordan, K. J., Ray, B. C., Hervey, C. M., Salamat, M. S., et al. (2011). Reversal of fragile X phenotypes by manipulation of AbetaPP/Abeta levels in Fmr1KO mice. PLoS ONE 6:e26549. doi: 10.1371/journal.pone.0026549
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Won, H., Lee, H. R., Gee, H. Y., Mah, W., Kim, J. I., Lee, J., et al. (2012). Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 486, 261–265. doi: 10.1038/nature11208
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zempel, H., and Mandelkow, E. (2014). Lost after translation: missorting of Tau protein and consequences for Alzheimer disease. Trends Neurosci. 37, 721–732. doi: 10.1016/j.tins.2014.08.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, Y., Kurup, P., Xu, J., Carty, N., Fernandez, S. M., Nygaard, H. B., et al. (2010). Genetic reduction of striatal-enriched tyrosine phosphatase. (STEP). reverses cognitive and cellular deficits in an Alzheimer's disease mouse model. Proc. Natl. Acad. Sci. U.S.A. 107, 19014–19019. doi: 10.1073/pnas.1013543107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, Y., Venkitaramani, D. V., Gladding, C. M., Kurup, P., Molnar, E., Collingridge, G. L., et al. (2008). The tyrosine phosphatase STEP mediates AMPA receptor endocytosis after metabotropic glutamate receptor stimulation. J. Neurosci. 28, 10561–10566. doi: 10.1523/JNEUROSCI.2666-08.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zoghbi, H. Y., and Bear, M. F. (2012). Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb. Perspect. Biol. 4:a009886. doi: 10.1101/cshperspect.a009886
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: FMRP, Alzheimer disease, fragile X syndrome, autism, β amyloid, neurodegeneration, tau, synaptic plasticity
Citation: Wang H (2015) Fragile X mental retardation protein: from autism to neurodegenerative disease. Front. Cell. Neurosci. 9:43. doi: 10.3389/fncel.2015.00043
Received: 04 January 2015; Paper pending published: 22 January 2015;
Accepted: 28 January 2015; Published online: 12 February 2015.
Edited by:
Rakez Kayed, University of Texas Medical Branch, USAReviewed by:
Diana Laura Castillo-Carranza, University of Texas Medical Branch, USAIndra Chandrasekar, Sanford Research, USA
Copyright © 2015 Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence:aGFuc2VuLndhbmdAdXRvcm9udG8uY2E=