 Karolina Szokol1,2,3†
Karolina Szokol1,2,3† Kjell Heuser1,3†
Kjell Heuser1,3† Wannan Tang2,3
Wannan Tang2,3 Vidar Jensen2,3
Vidar Jensen2,3 Rune Enger1,2,3
Rune Enger1,2,3 Peter Bedner4
Peter Bedner4 Christian Steinhäuser4
Christian Steinhäuser4 Erik Taubøll1
Erik Taubøll1 Ole Petter Ottersen5
Ole Petter Ottersen5 Erlend A. Nagelhus1,2,3*
Erlend A. Nagelhus1,2,3*- 1Department of Neurology, Oslo University Hospital, Oslo, Norway
- 2Centre for Molecular Medicine Norway, The Nordic EMBL Partnership, University of Oslo, Oslo, Norway
- 3Letten Centre and GliaLab, Department of Physiology, Institute of Basic Medical Sciences, University of Oslo, Oslo, Norway
- 4Institute of Cellular Neurosciences, University of Bonn, Bonn, Germany
- 5University President’s Office, University of Oslo, Oslo, Norway
Astrocytic endfeet are specialized cell compartments whose important homeostatic roles depend on their enrichment of water and ion channels anchored by the dystrophin associated protein complex (DAPC). This protein complex is known to disassemble in patients with mesial temporal lobe epilepsy and in the latent phase of experimental epilepsies. The mechanistic underpinning of this disassembly is an obvious target of future therapies, but remains unresolved. Here we show in a kainate model of temporal lobe epilepsy that astrocytic endfeet display an enhanced stimulation-evoked Ca2+ signal that outlast the Ca2+ signal in the cell bodies. While the amplitude of this Ca2+ signal is reduced following group I/II metabotropic receptor (mGluR) blockade, the duration is sustained. Based on previous studies it has been hypothesized that the molecular disassembly in astrocytic endfeet is caused by dystrophin cleavage mediated by Ca2+ dependent proteases. Using a newly developed genetically encoded Ca2+ sensor, the present study bolsters this hypothesis by demonstrating long-lasting, enhanced stimulation-evoked Ca2+ signals in astrocytic endfeet.
Introduction
Evidence is accruing that perivascular astrocytic endfeet are highly specialized cell compartments in terms of molecular organization and functional roles (Nagelhus and Ottersen, 2013). Many of the unique features of these processes can be explained by their expression of the brain dystrophin DP71 which orchestrates a molecular assembly that includes the water channel aquaporin-4 (AQP4; Frigeri et al., 2001; Neely et al., 2001; Enger et al., 2012; Waite et al., 2012). The endfoot complement of AQP4 determines the rate by which water accumulates in brain in conditions favoring the development of brain edema (Vajda et al., 2002; Amiry-Moghaddam et al., 2003; Haj-Yasein et al., 2011b). The endfeet are also enriched in the inwardly rectifying K+ channel Kir4.1 (Nagelhus et al., 1999; Higashi et al., 2001). This channel is thought to mediate K+ siphoning in the retina (Kofuji et al., 2000) and contributes to K+ spatial buffering in the CNS at large (Chever et al., 2010; Haj-Yasein et al., 2011a). The unique features of the astrocytic endfeet imply that astrocytes are highly polarized cells, biochemically as well as functionally.
It was recently found that loss of astrocyte polarization is common to several neurological conditions. The endfoot pool of AQP4 drops abruptly after an ischemic insult (Frydenlund et al., 2006; Steiner et al., 2012), and is also strongly reduced in models of Alzheimer’s disease (Yang et al., 2011) and traumatic brain injury (Ren et al., 2013). Similarly, loss of astrocyte polarization—with reductions in AQP4 as well as Kir4.1—has been described in the hippocampus of patients with temporal lobe epilepsy (Schröder et al., 2000; Eid et al., 2005; Heuser et al., 2012). These changes are reproduced in experimental models of epilepsy, including the kainate model (Lee et al., 2012; Alvestad et al., 2013). The loss of Kir4.1 in particular is likely to be pathophysiologically relevant, as glial-conditional Kir4.1 knockout animals display deficient K+ spatial buffering and severe epilepsy (Chever et al., 2010; Haj-Yasein et al., 2011a). Disassembly of endfoot protein complexes emerges as one of several mechanisms whereby astroglia may contribute to hyperexcitability and epileptogenesis (Binder et al., 2012; Binder and Carson, 2013; Crunelli et al., 2014).
The mechanisms underlying the loss of astrocyte polarization in epilepsy have not been resolved. One possible mechanism is that an early injury causes Ca2+ accumulation in endfeet, leading to proteolytic cleavage of the dystrophin associated protein complex (DAPC) at these sites. Such a mechanism is plausible, as astrocytes activated by injury contain calpain (Shields et al., 2000)—a protease that shows affinity to dystrophin and that is activated by Ca2+ (Yoshida et al., 1992).
This hypothesis cannot be tested by conventional Ca2+ imaging, as bulk-loaded synthetic Ca2+ dyes mainly reveal Ca2+ signals at the level of the cell bodies (Reeves et al., 2011). Here we use an approach that allows Ca2+ signals to be monitored in the fine astrocytic processes, including the perivascular endfeet. Specifically, we employed recombinant adeno-associated virus (rAAV) gene delivery of the genetically encoded Ca2+ indicator GCaMP5E (Akerboom et al., 2012) to hippocampal astrocytes in a mouse model of temporal lobe epilepsy. Two-photon Ca2+ imaging of acute hippocampal slices obtained in the epilepsy latent phase revealed elevated stimulation-evoked astrocytic Ca2+ signals, both in endfeet and in astrocytic cell bodies. Indeed, the Ca2+ signals in endfeet outlasted those in cell bodies. The present data point to endfoot Ca2+ signaling as a possible mechanism underpinning the loss of astrocyte polarization in epilepsy.
Material and Methods
Animals
Male C57BL/6N mice of 2–4 months of age (Charles River) were used for all experiments. All procedures were approved by the animal use and care committee of the Institute of Basic Medical Sciences, University of Oslo, and the Centre for Comparative Medicine, Oslo University Hospital.
Plasmid Constructs
The plasmid constructs were generated as described in a separate paper (Tang et al., 2015). In brief, the GCaMP5E DNA sequence was directly taken out from the expression vector pRGCAMP5E (Akerboom et al., 2012) by restriction digest with BamHI and HindIII, and subcloned into the rAAV vector pAAV-6P-SEWB (Shevtsova et al., 2005) with the human SYNAPSIN-1 (SYN) promoter to generate the construct of pAAV-SYN-GCaMP5E. The human GFAP promoter (Hirrlinger et al., 2009) was then inserted with MluI and BamHI into the pAAV-SYN-GCaMP5E vector resulting in the pAAV-GFAP-GCaMP5E construct.
Viral Transduction
rAAVs serotype 1 and 2 were generated as described (Tang et al., 2009), and purified by AVB Sepharose affinity chromatography (Smith et al., 2009). For the virus preparation, the genomic titer was determined by Real-Time PCR (~1.0 × 1012 viral genomes (vg)/ml, TaqMan Assay, Applied Biosystems). For virus infection, adult mice were deeply anesthetized with a mixture of zolazepam (188 mg/kg body weight), tiletamine (188 mg/kg body weight), xylazine (4.5 mg/kg body weight) and fentanyl (26 µg/kg body weight) before viruses were stereotactically injected (Shevtsova et al., 2005) into both hippocampi. Coordinates relative to Bregma were: anteroposterior −2.0 mm, lateral ±1.5 mm, depth 1.5 mm. During each injection, 0.3 µl of purified rAAV (~1.0 × 1012 vg/ml) was delivered.
Intracortical Kainate Injection Model for Mesial TLE
We used deep cortical (juxtahippocampal) kainate injection to elicit an initial status epilepticus (SE). Using this approach, more than 90% of injected animals developed recurrent behavioral seizures after a 5–8 day long latent period. For kainate injections, mice were anesthetized with a mixture of medetomidine (0.3 mg/kg, i.p.) and ketamine (40 mg/kg, i.p.) and kept on a heating blanket. A small craniotomy was performed in a stereotactic frame and kainate (50 nl; 20 mM; Tocris) was injected by a Hamilton pipette (Hamilton Company, NV) at a depth of 1.7 mm at the following coordinates relative to Bregma: anteroposterior −2 mm, lateral +1.5 mm (right). Anesthesia was stopped with atipamezol (300 mg/kg, i.p.) and SE was observed either clinically or by telemetric EEG recording and video monitoring. The animal model has been described in detail in a separate paper (Bedner et al., 2015). The non-injected side served as control for the kainate injected side.
Immunohistochemistry and Confocal Imaging
Virus transduced mice were anesthetized with ~4% isoflurane and intracardially perfused with 1 × phosphate buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4/2H2O, 1.4 mM KH2PO4, pH 7.4, all from Sigma-Aldrich) and 4% paraformaldehyde (PFA, Merck) in PBS prior to decapitation. Brains were removed and fixed in ice-cold 4% PFA/PBS for 2 h, embedded in 2.5% Agarose (Invitrogen) in PBS and sliced on a Vibratome (Leica) into 70 µm sections. Immunostaining was performed with polyclonal rabbit anti-GFP (1:3000, Abcam, #ab6556), chicken anti-GFAP (1:1000, Covance, #PCK-591P), rat anti-CD31 (1:200, BD Biosciences, #550274) and FITC-coupled anti-rabbit, Cy3-coupled anti-chicken, Cy5-coupled anti-rat secondary antibodies (1:200, Jackson Immuno Research, #711095152, #703165155 and #712175153 respectively). Confocal images were acquired on a Zeiss LSM5 PASCAL confocal laser scanning microscopy with 63x/1.4NA oil-immersion objective, equipped with an Argon laser (457, 476, 488, 514 nm) and a Helium Neon laser (543 nm, Carl Zeiss).
Electrophysiology and Two-Photon Ca2+ Imaging
Experiments were performed on hippocampal slices prepared 3–4 weeks after injection of rAAV-GFAP-GCaMP5E and 1, 3 and 7 days after juxtahippocampal, cortical kainate injections. The animals were sacrificed with an overdose of desflurane (Baxter), and brains were removed and cooled in artificial cerebrospinal fluid (ACSF, 0–4°C, bubbled with 95% O2/5% CO2, pH 7.4) containing (in mM): 124 NaCl, 2 KCl, 1.25 KH2PO4, 2 MgSO4, 1 CaCl2, 26 NaHCO3 and 12 glucose. Transverse slices (400 µm) were cut from the dorsal portion of each hippocampus with a Vibratome slicer (Leica) and placed in a humidified interface chamber at 30 ± 1°C and perfused with ACSF containing 2 mM CaCl2. In some experiments the group I/II mGluR antagonist α-methyl-4-carboxyphenylglycine (MCPG; 1 mM, Tocris) or the mGluR5 selective antagonist 2-methyl-6-phenylethynyl pyridine hydrochloride (MPEP; 100 µM, Tocris) were added to the ACSF. Two glass electrodes filled with ACSF and positioned 200–300 µm away from each other in CA1 stratum radiatum served as stimulation and recording electrodes, respectively. Orthodromic synaptic stimulations at 20 Hz for 10 s were delivered and excitatory postsynaptic potentials (fEPSPs) were monitored. Neuronal stimulation-evoked (simultaneous recording while electrical stimulations) astrocytic GCaMP5 fluorescence signals were recorded by a two-photon laser scanning microscope (model “Ultima”, Prairie Technologies), as described previously (Tang et al., 2015). Astrocytic Ca2+ responses on the kainate injected side were compared with those on the non-injected (control) side. Images were recorded with a model “XLPLN 25 × WMP” 1.05NA, water-immersion objective (Olympus, Japan) at 900–910 nm laser pulses. The laser was a model “Chameleon Vision II” (Coherent, Santa Clara, CA). The recording was done either with 1 Hz or 4 Hz frame rate, the images were 512 × 512 px or 256 × 256 px, respectively.
Imaging Analysis
Time-series of fluorescence images were first imported into Fiji ImageJ (Fiji), and regions of interest (ROIs) were manually selected based on morphology. Astrocytic cell bodies were identified by their projecting branches and endfeet by their characteristic circular pattern around transversely cut vessels and elongated, linear appearance along obliquely cut vessels. ROIs over processes were chosen at least 5 µm away from the perimeter of the soma. The relative change in fluorescence (ΔF/F) in each ROI, the individual traces and the histograms were all calculated and plotted by MATLAB (R2011b, MathWorks, Inc.) with custom written scripts. Standard deviation (SD) images were generated from time-lapse image recordings by Fiji ImageJ.
Statistical Analysis
Statistical analyses were performed using Prism (Version 6.0b for Mac OSX, GraphPad Software). One-way ANOVA with Tukey multiple comparisons test was used for comparison of GCaMP5E fluorescence changes in astrocytic somata, processes and endfeet following stimulation of Schaffer collaterals/commissural fibers. Paired t-test was used for comparison before and after wash-in with MCPG and MPEP. P < 0.05 was considered statistically significant.
Results
Viral Transduction Yielded Expression of the Ca2+ Indicator GCaMP5E in Adult Mouse Hippocampal Astrocytes
Injection of the rAAV-GFAP-GCaMP5E construct into the hippocampus yielded robust and selective GCaMP5E expression in hippocampal astrocytes, as revealed by immunolabeling with antibodies against green fluorescent protein (GFP) and glial fibrillary acidic protein (GFAP; Figures 1A,B). Notably, GCaMP5E was expressed within all astrocytic compartments, including the fine astrocytic processes and endfeet adjacent to CD31-immunopositive blood vessels (Figure 1B).
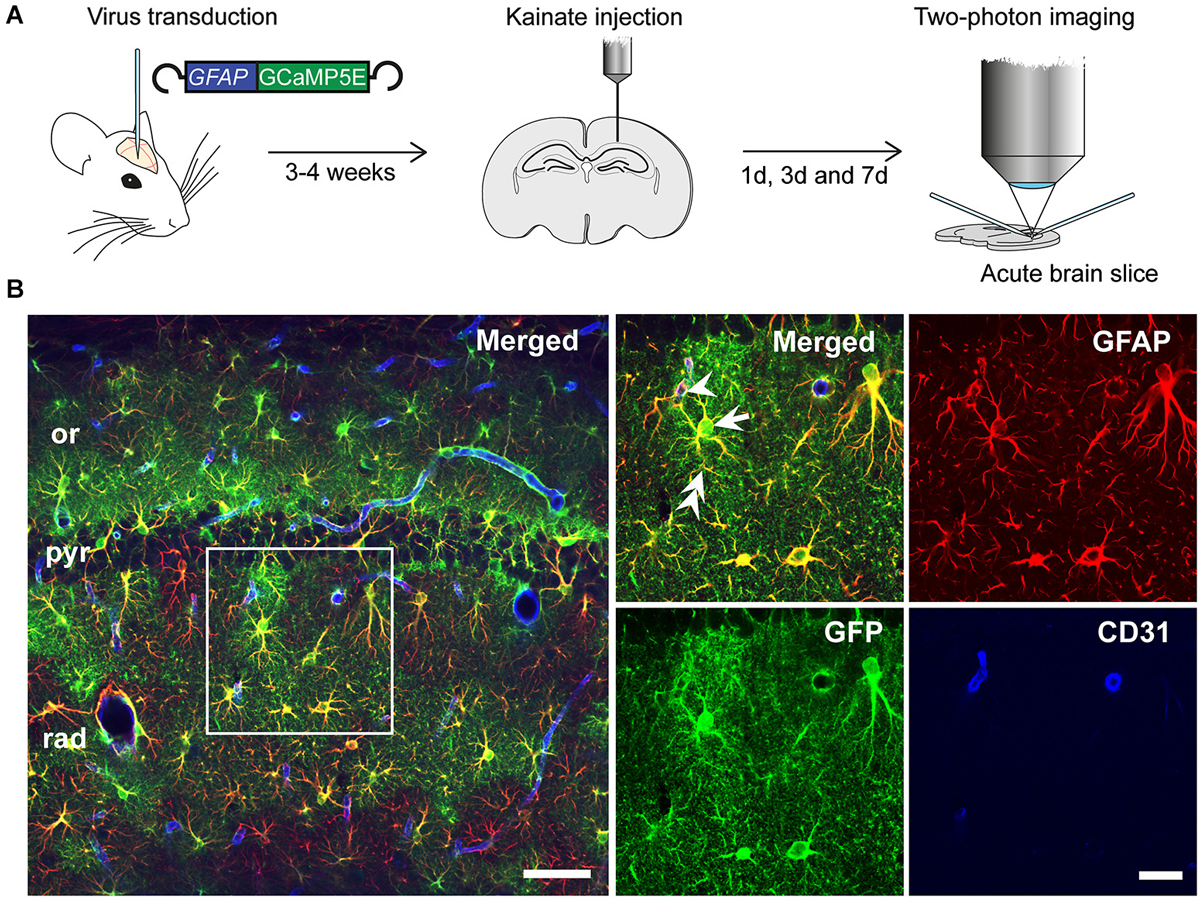
Figure 1. Experimental design. (A) rAAV-GFAP-GCaMP5E virus was injected into both hippocampi 2–3 weeks prior to unilateral, intracortical kainate injection. Acute hippocampal slices were prepared and imaged at 1, 3 and 7 days post kainate injection. (B) Immunofluorescence with green fluorescent protein (GFP) antibodies (green) showed robust GGaMP5E expression in GFAP immunopositive (red) astrocytes. The vascular endothelium was labeled with CD31 antibodies (blue). Arrow: astrocyte soma, double arrow: astrocyte process, arrowhead: endfoot. or, stratum oriens; pyr, stratum pyramidale; rad, stratum radiatum. Scale bars, 50 µm and 20 µm (boxed motif expanded in inset).
Stimulation Induced Ca2+ Signals Within Astrocytic Somata, Processes and Endfeet are Enhanced in the Latent Phase of Epilepsy
Stimulation (20 Hz, 10 s) of Schaffer collateral/commissural fibers (Scc) in acute hippocampal slices from the control side of rAAV-GFAP-GCaMP5E-transduced animals 1 day after intracortical kainate injection elicited brisk Ca2+ signals in the majority of stratum radiatum astrocytes (Figure 2A), as reported for slices from healthy adult mice (Tang et al., 2015). Compared to the control side, the amplitudes of stimulation evoked Ca2+ signals in the kainate injected side were significantly increased in all astrocytic compartments 1 day post injection (Figures 2A,B; soma contralateral 6.8 ± 0.4 vs. ipsilateral 12.0 ± 1.1, P < 0.0001, n = 74 cells, 22 slices, 20 mice and n = 69 cells, 16 slices, 11 mice, respectively; processes 10.7 ± 0.7 vs. 22.2 ± 2.7, P < 0.0001, n = 91 processes, 22 slices, 20 mice and n = 76 processes, 16 slices, 11 mice, respectively; endfeet 7.2 ± 0.7 vs. 14.1 ± 2.1, P = 0.001, n = 36 endfeet, 22 slices, 20 mice vs. 14 endfeet, 16 slices, 11 mice).
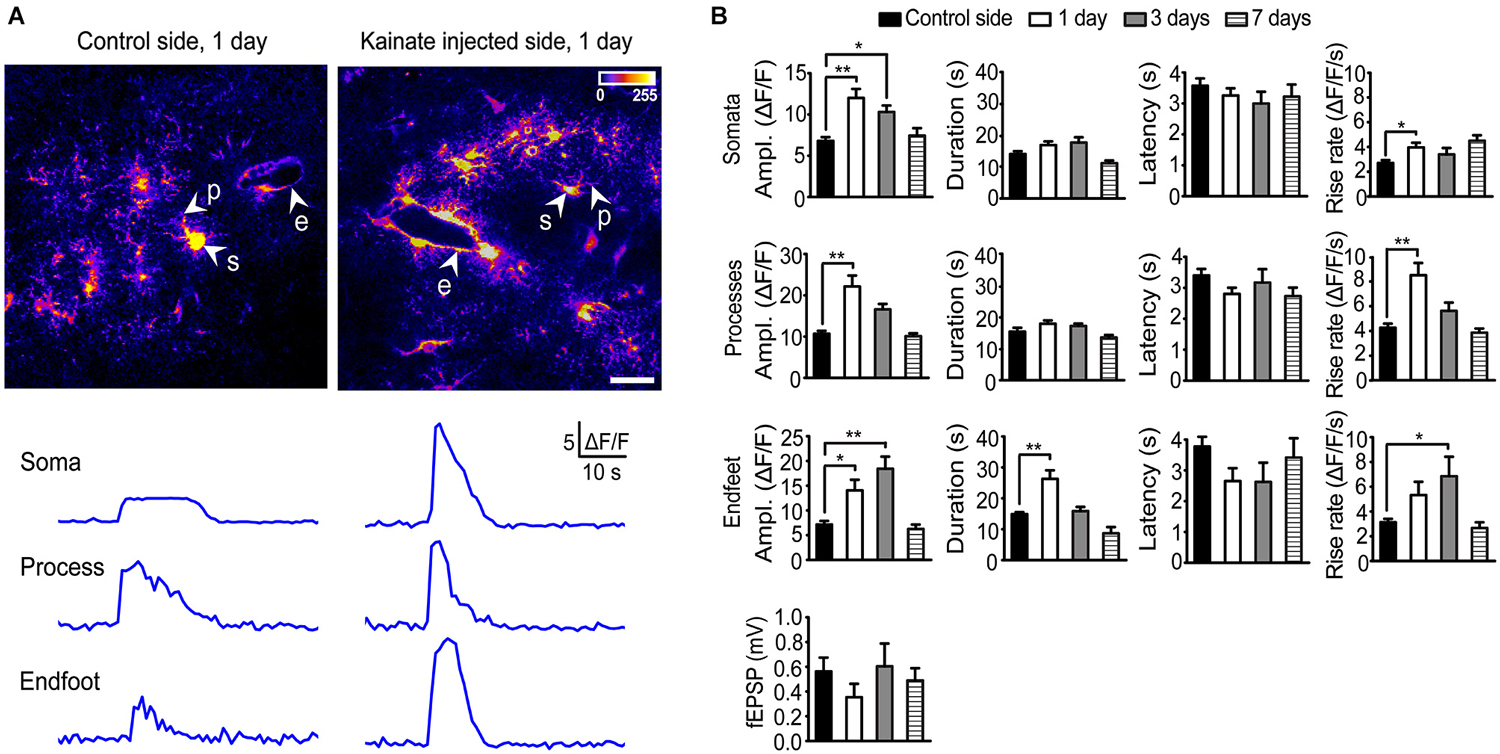
Figure 2. Two-photon imaging of stimulation-evoked astrocytic Ca2+ signals in adult mouse hippocampal slices 1, 3 and 7 days after intracortical kainate injection. (A) Standard deviation (SD) images of GCaMP5E fluorescence intensities from 1 Hz time-lapse recording 1 day after kainate injection. GCaMP5E fluorescent astrocytic somata (s), processes (p), and endfeet (e) are indicated. The fluorescence traces from the astrocytic compartments indicated in the images are shown below. Scale bar, 20 µm. (B) Amplitudes, duration, latency and rise rate of Schaffer collateral/commissural fiber (Scc) stimulation (20 Hz, 10 s) evoked GCaMP5E fluorescence transients in astrocytic somata, processes and endfeet in control (non-injected side; 20 mice, 22 slices), 1 day (11 mice, 16 slices), 3 days (3 mice, 6 slices) and 7 days (6 mice, 9 slices) after kainate injection. Lower panel shows average fEPSP for the different time points. Values are mean ± s.e.m. Asterisk, P < 0.05. Double asterisk, P < 0.0001.
At day 3 the Ca2+ signal amplitudes were still significantly elevated in astrocytic somata and endfeet, but not in processes (soma contralateral 6.8 ± 0.4 vs. ipsilateral 10.3 ± 0.8, P = 0.029, n = 74 cells, 22 slices, 20 mice and n = 39 cells, 6 slices, 3 mice respectively; processes 10.7 ± 0.7 vs. 16.6 ± 1.3, P = 0.12, n = 91 processes, 22 slices, 20 mice and n = 40 processes, 6 slices, 3 mice, respectively; endfeet 7.2 ± 0.7 vs. 18.4 ± 2.4, P < 0.0001, n = 36 endfeet, 22 slices, 20 mice vs. 9 endfeet, 6 slices, 3 mice, respectively). At day 7 the Ca2+ signal amplitudes in all astrocytic compartments had returned to control values. At day 1 post kainate injection the duration of the stimulation evoked Ca2+ signal was significantly increased in the astrocytic endfeet compared to the non-injected side (26.30 ± 2.8 vs. 14.83 ± 0.6, P < 0.0001, 20 endfeet, 16 slices, 11 mice, and n = 36 endfeet, 22 slices, 20 mice, respectively), while no changes were observed in other astrocytic processes or in the cell bodies. The latency from start of Scc stimulation to Ca2+ fluorescence increase was similar in all astrocytic subcompartments and not affected by kainate injection. However, kainate injection increased the Ca2+ signal rise rate in all compartments (for somata and processes at day 1; for endfeet at day 3; Figure 2B).
The Augmented Stimulation Evoked Astrocytic Ca2+ Responses Following Kainate Injection was Dependent on mGluR5
Administration of the group I/II mGluR antagonist MCPG significantly reduced the amplitude of stimulation evoked Ca2+ signals in all astrocyte compartments at day 1 post kainate injection (soma, P = 0.04, 32 somata, 9 slices, 6 mice; processes P = 0.0001, 36 processes, 9 slices, 6 mice; endfeet P = 0.04, 14 endfeet, 9 slices, 6 mice). The duration and latency of the Ca2+ signals were not affected by MCPG, whilst rise rate was significantly reduced only in processes (Figure 3A).
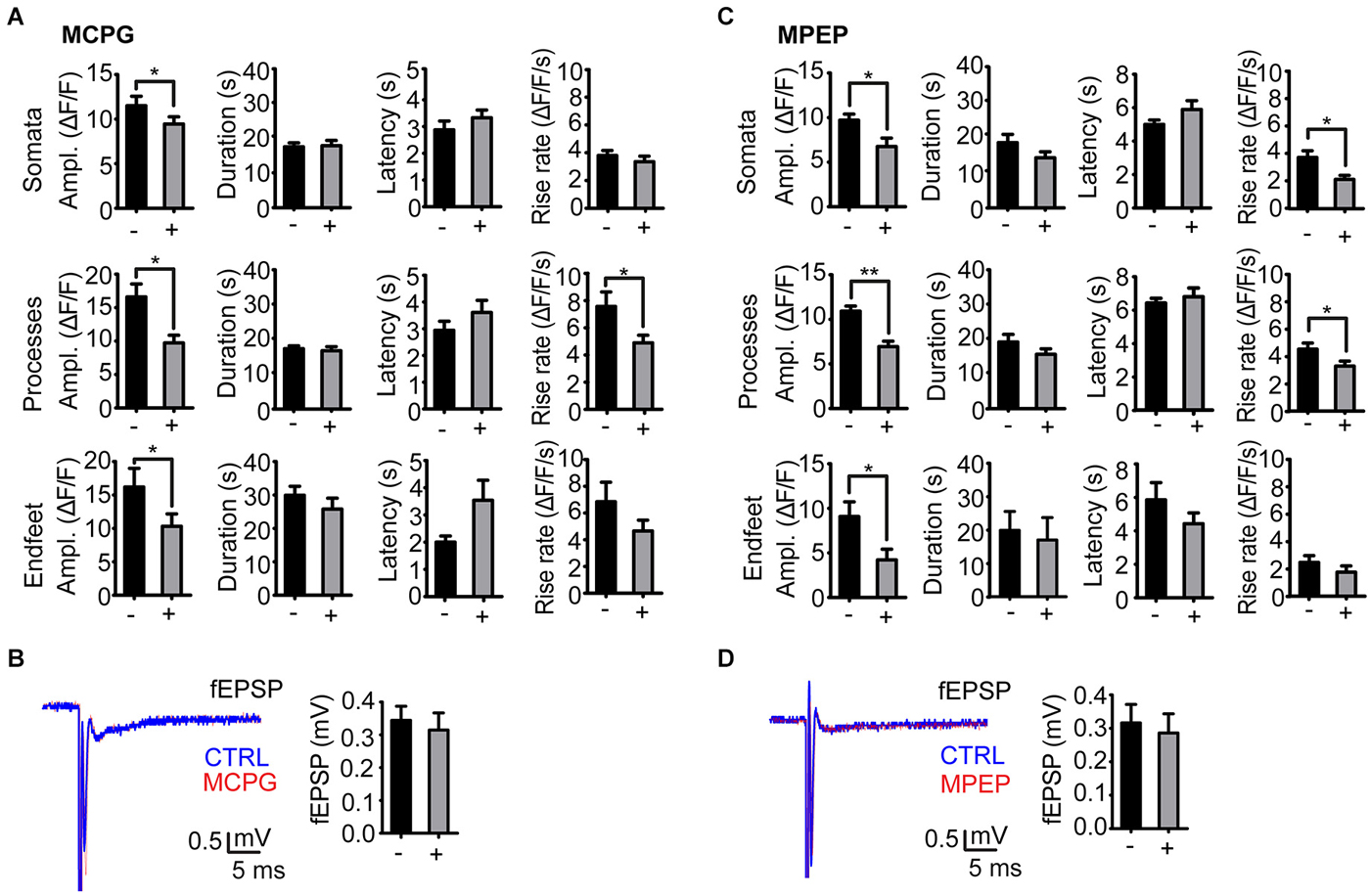
Figure 3. Effect of mGluR antagonists on the stimulation evoked astrocytic Ca2+ signals in the kainate injected side. (A) Amplitude, duration, latency and rise rate of Ca2+ signals in different astrocytic territories before (−) and after (+) administration of the mGluR I/II antagonist MCPG. (B) Representative fEPSP traces before (CTRL; blue) and after (red) MCPG administration and mean fEPSP amplitudes. (C) As in (A), but with MPEP instead of MCPG. (D) As in (B), but with MPEP instead of MCPG. Values are mean ± s.e.m. Asterisk, P < 0.05. Double asterisk, P < 0.0001.
As mGluR5 receptors have been shown to mediate enhanced astrocytic Ca2+ signaling following pilocarpine induced SE (Ding et al., 2007), we applied the mGluR5 selective antagonist MPEP in our model. Similarly to MCPG, administration of MPEP significantly reduced the amplitude of stimulation evoked Ca2+ signals at day 1 after kainate injection in astrocytic somata (P = 0.003, 19 somata, 5 slices, 4 mice), processes (P < 0.0001, 19 processes, 5 slices, 4 mice) and endfeet (P = 0.04, 7 endfeet, 5 slices, 4 mice). MPEP reduced the amplitudes of the Ca2+ transients by 30–40%, i.e., to the level at the non-injected side (Figure 3C). The nonselective antagonist MCPG reduced the Ca2+ transients to the same extent, suggesting that mGluR5 alone is mediating the enhanced Ca2+ signal amplitude in the latent phase. Similarly to MCPG, MPEP did not affect the duration and latency of the Ca2+ signals, and had inconsistent effects on transient rise rate in the three compartments (Figure 3C).
Neither MPEP nor MCPG significantly affected the fEPSP amplitudes (Figures 3B,D).
Discussion
Astrocytes are highly polarized cells, structurally as well as functionally, opening for the possibility of a compartmentation of Ca2+ signaling analogous to that found in neurons. With the advent of genetically encoded Ca2+ sensors this possibility can be experimentally explored. A key question is whether Ca2+ signaling in the astrocytic endfeet could play a role in epileptogenesis, by initiating a sequence of events that lead to disassembly of the DAPC in the endfoot plasma membrane. This complex, known to be critical for K+ and water homeostasis in brain, is lost in patients with mesial temporal lobe epilepsy (Eid et al., 2005; Heuser et al., 2012) and in the latent phase of kainate induced epilepsy (Alvestad et al., 2013).
Here we show that intracortical kainate application leads to a stimulation evoked Ca2+ signal in the endfeet that outlasts the Ca2+ signal in the astrocytic cell bodies. This underlines the idea that endfeet are distinct subcompartments of astroglia (Nagelhus and Ottersen, 2013) and, more specifically, that endfeet serve as diffusion-limited subcellular compartments (Nuriya and Yasui, 2013).
The Ca2+ signal in endfeet is attenuated by blockade of group I/II mGluRs and thus dependent on Ca2+ mobilization from intracellular stores. However, mGluR blockade does not cancel out the difference between endfeet and cell bodies when it comes to the duration of the Ca2+ signal. This suggests that the increased signal duration primarily reflects reduced clearance of Ca2+. An uncoupling of astrocytes could contribute to reduced clearance (Bedner and Steinhäuser, 2013; Bedner et al., 2015).
A disassembly of the DAPC in astrocytic endfeet and the loss of astrocyte polarization that this entails now emerge as a signature event in mesial temporal lobe epilepsy and epilepsy models (Nagelhus and Ottersen, 2013). The disassembly is reflected by a loss of dystrophin DP71, while β-dystroglycan remains (Heuser et al., 2012). Beta-dystroglycan is a member of the DAPC and normally serves to link this complex to extracellular matrix molecules of the pericapillary basal lamina (Neely et al., 2001; Amiry-Moghaddam and Ottersen, 2003).
It has been proposed that the molecular disassembly in astrocytic endfeet is caused by calpain activation (Nagelhus and Ottersen, 2013). Calpain is capable of cleaving DP71, and the expression of this protease is increased in activated astrocytes (Shields et al., 2000). It has not been resolved, however, whether endfeet sustain Ca2+ signals necessary for activation of calpain or any other Ca2+ dependent protease with affinity for dystrophin or dystrophin associated molecules. The present study fills this void and shows that endfeet display Ca2+ signals that even outlast those in the astrocytic cell bodies. The cascade of events underlying the molecular disassembly in endfeet is an obvious target for future therapies.
Author Contributions
KS designed experiments, acquired, analyzed and interpreted data, and wrote the paper; KH conceived the study and supervised experiments, acquired, analyzed and interpreted data, and wrote the paper; WT designed experiments, acquired, analyzed and interpreted data, and wrote the paper; VJ designed and supervised experiments, interpreted data, and wrote the paper; RE analyzed and interpreted data, and wrote the paper; PB designed the animal model, interpreted data and commented upon the manuscript; CS designed the animal model, interpreted data and commented upon the manuscript; ET conceived the study, interpreted data and commented upon the manuscript; OPO conceived the study, interpreted data and wrote the manuscript; EAN conceived the study and supervised experiments, interpreted data and wrote the manuscript. All authors revised the work critically and approved the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by an EMBO short-term fellowship (to WT), by the German Academic Exchange Service (DAAD) mobility program Germany-Norway (to EAN), the Research Council of Norway (NevroNor and FRIMEDBIO Grants to EAN), the European Science Foundation, Euroepinomics, EUROSCORE program (grants to ET and CS), the European Union’s Seventh Framework Programme for research, technological development and demonstration under grant agreement no. 601055 (to EAN), the Letten Foundation and Deutsche Forschungsgemeinschaft (STE 552/3 to CS).
References
Akerboom, J., Chen, T. W., Wardill, T. J., Tian, L., Marvin, J. S., Mutlu, S., et al. (2012). Optimization of a GCaMP calcium indicator for neural activity imaging. J. Neurosci. 32, 13819–13840. doi: 10.1523/JNEUROSCI.2601-12.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Alvestad, S., Hammer, J., Hoddevik, E. H., Skare, O., Sonnewald, U., Amiry-Moghaddam, M., et al. (2013). Mislocalization of AQP4 precedes chronic seizures in the kainate model of temporal lobe epilepsy. Epilepsy Res. 105, 30–41. doi: 10.1016/j.eplepsyres.2013.01.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Amiry-Moghaddam, M., Otsuka, T., Hurn, P. D., Traystman, R. J., Haug, F. M., Froehner, S. C., et al. (2003). An alpha-syntrophin-dependent pool of AQP4 in astroglial end-feet confers bidirectional water flow between blood and brain. Proc. Natl. Acad. Sci. U S A 100, 2106–2111. doi: 10.1073/pnas.0437946100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Amiry-Moghaddam, M., and Ottersen, O. P. (2003). The molecular basis of water transport in the brain. Nat. Rev. Neurosci. 4, 991–1001. doi: 10.1038/nrn1252
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bedner, P., Dupper, A., Hüttmann, K., Müller, J., Herde, M. K., Dublin, P., et al. (2015). Astrocyte uncoupling as a cause of human temporal lobe epilepsy. Brain, in press.
Bedner, P., and Steinhäuser, C. (2013). Altered Kir and gap junction channels in temporal lobe epilepsy. Neurochem. Int. 63, 682–687. doi: 10.1016/j.neuint.2013.01.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Binder, D. K., and Carson, M. J. (2013). Glial cells as primary therapeutic targets for epilepsy. Neurochem. Int. 63, 635–637. doi: 10.1016/j.neuint.2013.09.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Binder, D. K., Nagelhus, E. A., and Ottersen, O. P. (2012). Aquaporin-4 and epilepsy. Glia 60, 1203–1214. doi: 10.1002/glia.22317
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chever, O., Djukic, B., McCarthy, K. D., and Amzica, F. (2010). Implication of Kir4.1 channel in excess potassium clearance: an in vivo study on anesthetized glial-conditional Kir4.1 knock-out mice. J. Neurosci. 30, 15769–15777. doi: 10.1523/JNEUROSCI.2078-10.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Crunelli, V., Carmignoto, G., and Steinhäuser, C. (2014). Astrocytic targets provide new avenues for the therapeutic treatment of epilepsy. Neuroscientist 21, 62–83. doi: 10.1177/1073858414523320
Ding, S., Fellin, T., Zhu, Y., Lee, S. Y., Auberson, Y. P., Meaney, D. F., et al. (2007). Enhanced astrocytic Ca2+ signals contribute to neuronal excitotoxicity after status epilepticus. J. Neurosci. 27, 10674–10684. doi: 10.1523/jneurosci.2001-07.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Eid, T., Lee, T. S., Thomas, M. J., Amiry-Moghaddam, M., Bjørnsen, L. P., Spencer, D. D., et al. (2005). Loss of perivascular aquaporin 4 may underlie deficient water and K+ homeostasis in the human epileptogenic hippocampus. Proc. Natl. Acad. Sci. U S A 102, 1193–1198. doi: 10.1073/pnas.0409308102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Enger, R., Gundersen, G. A., Haj-Yasein, N. N., Eilert-Olsen, M., Thoren, A. E., Vindedal, G. F., et al. (2012). Molecular scaffolds underpinning macroglial polarization: an analysis of retinal Müller cells and brain astrocytes in mouse. Glia 60, 2018–2026. doi: 10.1002/glia.22416
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Frigeri, A., Nicchia, G. P., Nico, B., Quondamatteo, F., Herken, R., Roncali, L., et al. (2001). Aquaporin-4 deficiency in skeletal muscle and brain of dystrophic mdx mice. FASEB J. 15, 90–98. doi: 10.1096/fj.00-0260com
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Frydenlund, D. S., Bhardwaj, A., Otsuka, T., Mylonakou, M. N., Yasumura, T., Davidson, K. G., et al. (2006). Temporary loss of perivascular aquaporin-4 in neocortex after transient middle cerebral artery occlusion in mice. Proc. Natl. Acad. Sci. U S A 103, 13532–13536. doi: 10.1073/pnas.0605796103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Haj-Yasein, N. N., Jensen, V., Vindedal, G. F., Gundersen, G. A., Klungland, A., Ottersen, O. P., et al. (2011a). Evidence that compromised K+ spatial buffering contributes to the epileptogenic effect of mutations in the human Kir4.1 gene (KCNJ10). Glia 59, 1635–1642. doi: 10.1002/glia.21205
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Haj-Yasein, N. N., Vindedal, G. F., Eilert-Olsen, M., Gundersen, G. A., Skare, O., Laake, P., et al. (2011b). Glial-conditional deletion of aquaporin-4 (Aqp4) reduces blood-brain water uptake and confers barrier function on perivascular astrocyte endfeet. Proc. Natl. Acad. Sci. U S A 108, 17815–17820. doi: 10.1073/pnas.1110655108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Heuser, K., Eid, T., Lauritzen, F., Thoren, A. E., Vindedal, G. F., Tauboll, E., et al. (2012). Loss of perivascular Kir4.1 potassium channels in the sclerotic hippocampus of patients with mesial temporal lobe epilepsy. J. Neuropathol. Exp. Neurol. 71, 814–825. doi: 10.1097/NEN.0b013e318267b5af
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Higashi, K., Fujita, A., Inanobe, A., Tanemoto, M., Doi, K., Kubo, T., et al. (2001). An inwardly rectifying K(+) channel, Kir4.1, expressed in astrocytes surrounds synapses and blood vessels in brain. Am. J. Physiol. Cell Physiol. 281, C922–C931.
Hirrlinger, J., Scheller, A., Hirrlinger, P. G., Kellert, B., Tang, W., Wehr, M. C., et al. (2009). Split-cre complementation indicates coincident activity of different genes in vivo. PLoS One 4:e4286. doi: 10.1371/journal.pone.0004286
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kofuji, P., Ceelen, P., Zahs, K. R., Surbeck, L. W., Lester, H. A., and Newman, E. A. (2000). Genetic inactivation of an inwardly rectifying potassium channel (Kir4.1 subunit) in mice: phenotypic impact in retina. J. Neurosci. 20, 5733–5740.
Lee, D. J., Hsu, M. S., Seldin, M. M., Arellano, J. L., and Binder, D. K. (2012). Decreased expression of the glial water channel aquaporin-4 in the intrahippocampal kainic acid model of epileptogenesis. Exp. Neurol. 235, 246–255. doi: 10.1016/j.expneurol.2012.02.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nagelhus, E. A., Horio, Y., Inanobe, A., Fujita, A., Haug, F. M., Nielsen, S., et al. (1999). Immunogold evidence suggests that coupling of K+ siphoning and water transport in rat retinal Müller cells is mediated by a coenrichment of Kir4.1 and AQP4 in specific membrane domains. Glia 26, 47–54. doi: 10.1002/(sici)1098-1136(199903)26:1<47::aid-glia5>3.0.co;2-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nagelhus, E. A., and Ottersen, O. P. (2013). Physiological roles of aquaporin-4 in brain. Physiol. Rev. 93, 1543–1562. doi: 10.1152/physrev.00011.2013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Neely, J. D., Amiry-Moghaddam, M., Ottersen, O. P., Froehner, S. C., Agre, P., and Adams, M. E. (2001). Syntrophin-dependent expression and localization of Aquaporin-4 water channel protein. Proc. Natl. Acad. Sci. U S A 98, 14108–14113. doi: 10.1073/pnas.241508198
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nuriya, M., and Yasui, M. (2013). Endfeet serve as diffusion-limited subcellular compartments in astrocytes. J. Neurosci. 33, 3692–3698. doi: 10.1523/JNEUROSCI.3050-12.2013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Reeves, A. M., Shigetomi, E., and Khakh, B. S. (2011). Bulk loading of calcium indicator dyes to study astrocyte physiology: key limitations and improvements using morphological maps. J. Neurosci. 31, 9353–9358. doi: 10.1523/JNEUROSCI.0127-11.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ren, Z., Iliff, J. J., Yang, L., Yang, J., Chen, X., Chen, M. J., et al. (2013). ‘Hit and Run’ model of closed-skull Traumatic Brain Injury (TBI) reveals complex patterns of post-traumatic AQP4 dysregulation. J. Cereb. Blood Flow Metab. 33, 834–845. doi: 10.1038/jcbfm.2013.30
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schröder, W., Hinterkeuser, S., Seifert, G., Schramm, J., Jabs, R., Wilkin, G. P., et al. (2000). Functional and molecular properties of human astrocytes in acute hippocampal slices obtained from patients with temporal lobe epilepsy. Epilepsia 41(Suppl. 6), S181–S184. doi: 10.1111/j.1528-1157.2000.tb01578.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shevtsova, Z., Malik, J. M., Michel, U., Bähr, M., and Kügler, S. (2005). Promoters and serotypes: targeting of adeno-associated virus vectors for gene transfer in the rat central nervous system in vitro and in vivo. Exp. Physiol. 90, 53–59. doi: 10.1113/expphysiol.2004.028159
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shields, D. C., Schaecher, K. E., Hogan, E. L., and Banik, N. L. (2000). Calpain activity and expression increased in activated glial and inflammatory cells in penumbra of spinal cord injury lesion. J. Neurosci. Res. 61, 146–150. doi: 10.1002/1097-4547(20000715)61:2<146::aid-jnr5>3.3.co;2-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Smith, R. H., Levy, J. R., and Kotin, R. M. (2009). A simplified baculovirus-AAV expression vector system coupled with one-step affinity purification yields high-titer rAAV stocks from insect cells. Mol. Ther. 17, 1888–1896. doi: 10.1038/mt.2009.128
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Steiner, E., Enzmann, G. U., Lin, S., Ghavampour, S., Hannocks, M. J., Zuber, B., et al. (2012). Loss of astrocyte polarization upon transient focal brain ischemia as a possible mechanism to counteract early edema formation. Glia 60, 1646–1659. doi: 10.1002/glia.22383
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tang, W., Ehrlich, I., Wolff, S. B., Michalski, A. M., Wölfl, S., Hasan, M. T., et al. (2009). Faithful expression of multiple proteins via 2A-peptide self-processing: a versatile and reliable method for manipulating brain circuits. J. Neurosci. 29, 8621–8629. doi: 10.1523/JNEUROSCI.0359-09.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tang, W., Szokol, K., Jensen, V., Enger, R., Trivedi, C., Hvalby, Ø., et al. (2015). Stimulation-evoked Ca2+ signals in astrocytic processes at hippocampal CA3-CA1 synapses of adult mice are modulated by glutamate and ATP. J. Neurosci. 35, 3016–3021.
Vajda, Z., Pedersen, M., Füchtbauer, E. M., Wertz, K., Stødkilde-Jørgensen, H., Sulyok, E., et al. (2002). Delayed onset of brain edema and mislocalization of aquaporin-4 in dystrophin-null transgenic mice. Proc. Natl. Acad. Sci. U S A 99, 13131–13136. doi: 10.1073/pnas.192457099
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Waite, A., Brown, S. C., and Blake, D. J. (2012). The dystrophin-glycoprotein complex in brain development and disease. Trends Neurosci. 35, 487–496. doi: 10.1016/j.tins.2012.04.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yang, J., Lunde, L. K., Nuntagij, P., Oguchi, T., Camassa, L. M., Nilsson, L. N., et al. (2011). Loss of astrocyte polarization in the tg-ArcSwe mouse model of Alzheimer’s disease. J. Alzheimers Dis. 27, 711–722. doi: 10.3233/JAD-2011-110725
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: astrocytes, GCaMP, dystrophin, glia, kainate, perivascular
Citation: Szokol K, Heuser K, Tang W, Jensen V, Enger R, Bedner P, Steinhäuser C, Taubøll E, Ottersen OP and Nagelhus EA (2015) Augmentation of Ca2+ signaling in astrocytic endfeet in the latent phase of temporal lobe epilepsy. Front. Cell. Neurosci. 9:49. doi: 10.3389/fncel.2015.00049
Received: 02 December 2014; Accepted: 01 February 2015;
Published online: 25 February 2015.
Edited by:
Luc Leybaert, Ghent University, BelgiumReviewed by:
Vedrana Montana, University of Alabama, USADmitry Lim, Università del Piemonte Orientale “Amedeo Avogadro”, Italy
Copyright © 2015 Szokol, Heuser, Tang, Jensen, Enger, Bedner, Steinhäuser, Taubøll, Ottersen and Nagelhus. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Erlend A. Nagelhus, Letten Centre and GliaLab, Department of Physiology, Institute of Basic Medical Sciences, University of Oslo, Sognsvannsveien 9 0317 Oslo, Norway e-mail:ZS5hLm5hZ2VsaHVzQG1lZGlzaW4udWlvLm5v
† These authors have contributed equally to this work.