 Luca Rosario La Rosa1
Luca Rosario La Rosa1 Lorena Perrone2
Lorena Perrone2 Morten Schallburg Nielsen3
Morten Schallburg Nielsen3 Pietro Calissano4
Pietro Calissano4 Olav Michael Andersen3,5
Olav Michael Andersen3,5 Carmela Matrone3*
Carmela Matrone3*- 1Institute of Cellular Biology and Neurobiology, National Council of Research of Rome, Rome, Italy
- 2Laboratory CiMoTheMA EA 3808, Faculty of Medicine and Pharmacy, University of Poitiers, Poitiers, France
- 3Department of Biomedicine, Aarhus University, Aarhus, Denmark
- 4European Brain Research Institute, Rome, Italy
- 5Danish Research Institute of Translational Neuroscience (DANDRITE), Nordic-EMBL Partnership, Aarhus, Denmark
The intracellular transport and localization of amyloid precursor protein (APP) are critical determinants of APP processing and β-amyloid peptide production, thus crucially important for the pathophysiology of Alzheimer’s disease (AD). Notably, the C-terminal Y682ENPTY687 domain of APP binds to specific adaptors controlling APP trafficking and sorting in neurons. Mutation on the Y682 residue to glycine (Y682G) leads to altered APP sorting in hippocampal neurons that favors its accumulation in intracellular compartments and the release of soluble APPα. Such alterations induce premature aging and learning and cognitive deficits in APP Y682G mutant mice (APPYG/YG). Here, we report that Y682G mutation affects formation of the APP complex with sortilin-related receptor (SorLA), resulting in endo-lysosomal dysfunctions and neuronal degeneration. Moreover, disruption of the APP/SorLA complex changes the trafficking pathway of SorLA, with its consequent increase in secretion outside neurons. Mutations in the SorLA gene are a prognostic factor in AD, and changes in SorLA levels in cerebrospinal fluid are predictive of AD in humans. These results might open new possibilities in comprehending the role played by SorLA in its interaction with APP and in the progression of neuronal degeneration. In addition, they further underline the crucial role played by Y682 residue in controlling APP trafficking in neurons.
Introduction
Amyloid precursor protein (APP) has been extensively studied for its role as the precursor of β-amyloid peptide (Aβ) in Alzheimer’s disease (AD). However, its physiological function still remains largely enigmatic.
Amyloid precursor protein is a transmembrane protein that is actively sorted among numerous compartments in the cell. After its synthesis, the newly translated APP polypeptide undergoes several post-translational modifications in the Golgi apparatus, including N- and O-glycosylation, sulfation, phosphorylation, and palmitoylation (Guo et al., 2012a; Jiang et al., 2014). Mature APP is then trafficked to the cell surface. Once APP reaches the cell surface, it is rapidly internalized and subsequently trafficked through endocytic and recycling compartments back to the cell surface or degraded in the lysosome (Guo et al., 2012a; Jiang et al., 2014).
Amyloid precursor protein trafficking in neurons is largely controlled by the Y682ENPTY687 domain via its specific binding to numerous cytosolic adaptor proteins (Russo et al., 2002; Tarr et al., 2002; Zhou et al., 2004, 2009; Tamayev et al., 2009; Muller and Zheng, 2012). In particular (tyrosine-682), Y682 modulates the interaction with adaptor proteins through its phosphorylation and dephosphorylation, suggesting that this residue functions as a switch that activates certain APP signaling pathways (Russo et al., 2002; Tarr et al., 2002; Zhou et al., 2004, 2009; Tamayev et al., 2009; Muller and Zheng, 2012). Relevantly, the trafficking of APP affects two competitive enzymatic APP processing pathways, mediated by α- and β-secretases. Notably, an imbalance to the β-secretase-mediated cleavage of APP leads to the accumulation of neurotoxic fragments, including Aβ peptide, which is considered a major cause of AD (Guo et al., 2012a; Jiang et al., 2014).
We previously reported that substituting Y682 with a glycine in knock-in animals (APPYG/YG) promotes a premature, age-dependent decline in cognition, learning, and locomotor performance (Matrone et al., 2012). The Y682G mutation induces a redistribution of APP inside neurons and its concomitant accumulation in intracellular compartments (Barbagallo et al., 2010; Matrone et al., 2011; Matrone, 2013). Additionally, the lysosome area is enlarged, and the number of lysosomes is significantly decreased in dorsal root ganglia and fibroblasts from APPYG/YG mice (Basso and Matrone, 2013; Matrone, 2013).
We believe that Y682 plays a crucial role in APP trafficking by possibly controlling APP binding to adaptors and consequently preventing alteration in its cleavage and sorting, finally resulting in neuronal degeneration and death.
Notably, aberrant APP trafficking and deficits in the endo-lysosomal system have been largely described as early causes of neurodegeneration and AD (Nixon et al., 2005; Yu et al., 2005; Lee and Landreth, 2010; Nixon and Yang, 2011). Additionally, aging, a major risk factor for AD, is also associated with defects in autophagocytosis (Cuervo, 2008; Salminen and Kaarniranta, 2009).
Here, we report that Y682G mutation induces alternative APP trafficking toward late endosomes (LEs) and lysosomes, ensuing functional alterations of the lysosomal system. Additionally, sortilin-related receptor (SorLA), which controls the trafficking of APP between the trans-Golgi network (TGN) and early endosomes (EEs) by interacting with specific cytosolic adaptors (Willnow and Andersen, 2013), fails to bind mutated APP and is largely secreted outside neurons. Notably, defects in the binding of SorLA and its interacting adaptors are considered important risk factors for AD (Guo et al., 2012b).
These results underline the relevance of residue Y682 in controlling APP trafficking and sorting in neurons by likely interacting with SorLA. In addition, they further emphasize the role of SorLA as a potential target protein to monitor APP activity in neurons. Finally, they point to the importance of identification of further adaptors to better understand the molecular events described here.
Results
Mutated APP is Sorted to the Lysosome and Late Endosome
To analyze the role of Y682 in controlling APP trafficking and sorting, we first determined the cellular compartment where mutated APP is preferentially localized using hippocampal neurons from APPYG/YG and control mice.
We previously reported the accumulation of APP in intracellular compartments and alterations in the number and size of lysosomes from APPYG/YG neurons (Matrone et al., 2011, 2012). Caster et al. recently reported that APP mutation on Y682 preferentially accumulated in lysosomes from HeLa cells (Caster and Kahn, 2013). Therefore, we began our analysis by focusing on the endo-lysosomal compartments of hippocampal neurons from wildtype (WT) and APPYG/YG mice.
We performed confocal microscopy of neurons that were stained with antibodies against APP and either lysosomal-associated membrane protein 1 (Lamp1) or Rab7 (Figure 1). We found an augmented overlap between the localization of APP (red) and Lamp1 (green) in APPYG/YG neurons (YG) compared with WT cells (Figure 1I). We also detected an increase in the co-localization between APP and Rab7, a marker of the LE (Figure 1R).
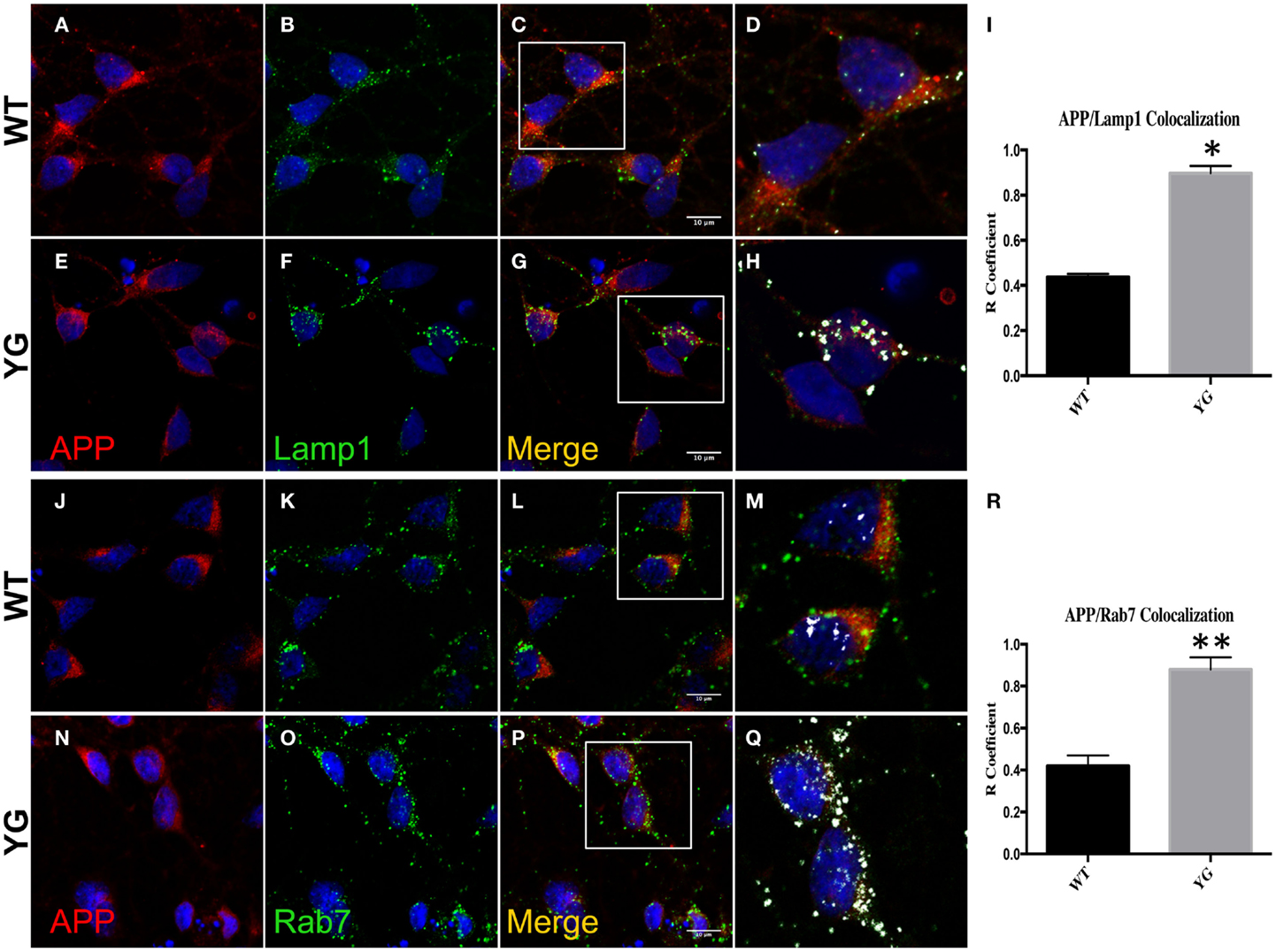
Figure 1. Amyloid precursor protein increases in LAMP1- and Rab7-positive vesicles. (A–G) Confocal microscopy analysis of double-staining of mouse anti-APP [red; (A,E)] and rabbit anti-Lamp1 [green; (B,F)] in WT and APPYG/YG (YG) hippocampal neurons [(A–D) and (E–H), respectively; 63× objective). Scale bars = 10 μm. The panels are representative of five different experiments performed in duplicate. [(C,G) and (D,H)] Co-localization analysis of WT and APPYG/YG hippocampal neurons. The analysis was performed using Zen/Zeiss software. Notice that the area of overlap between APP and Lamp1 immunostaining (white) was significantly increased in APPYG/YG mice (H) compared with WT (D). (D,H) High magnification of (C,G). The (R) coefficient (Pearson’s coefficient) was used for the quantitative and comparative analyses (I). The data are expressed as mean ± SEM. n = 10. *p < 0.05. (J–P) Confocal microscopy analysis of double-staining for mouse anti-APP [red; (J,N)] and rabbit anti-Rab7 [green; (K,O)] in WT and APPYG/YG (YG) hippocampal neurons [(J–M) and (N–Q), respectively; 63× objective]. Scale bars = 10 μm. The panels are representative of four different experiments performed in duplicate. [(L,P) and (M,Q)] Co-localization analysis of WT and APPYG/YG hippocampal neurons. The analysis was performed using Zen software. Notice that the area of overlap between APP and Rab7 immunostaining (white) was significantly increased in APPYG/YG mice (Q) compared with WT (M). (M,Q) High magnification of (L,P). The (R) coefficient (Pearson’s coefficient) was used for the quantitative and comparative analyses (R). The data are expressed as mean ± SEM. n = 8. *p < 0.05.
This increase in APP localization inside Lamp1- and Rab7-positive vesicles suggested that the Y682G mutation disturbed the recycling of APP to the plasma membrane (PM), triggering its accumulation in endo-lysosomal compartments.
We next examined whether APP trafficking to the EE and Golgi compartment was also altered in APPYG/YG neurons. Interestingly, immunostaining for EEA1, a marker of EEs, indicated a reduction of co-localization with mutated APP in hippocampal neurons (Figures 2A–I). Similarly, APP localization in Golgi compartments that were positive for (Figures 2M,Q,R) Giantin was also reduced by the Y682G mutation (Figures 2J–R).
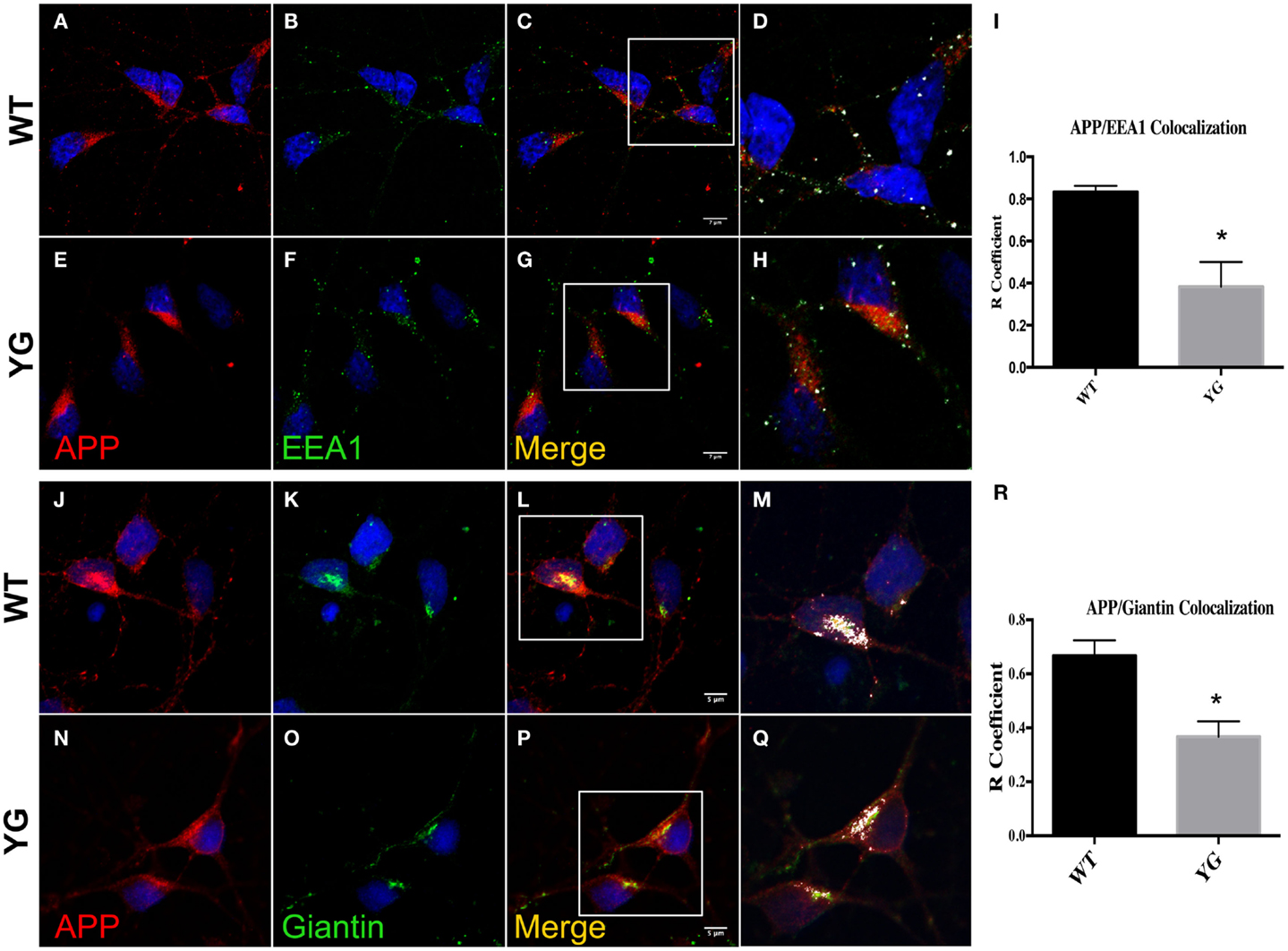
Figure 2. Amyloid precursor protein decreases in Golgi compartment and early endosomes. (A–G) Confocal microscopy analysis of double-staining for rabbit anti-APP red; (A,E)] and mouse anti-EEA1 [green; (B,F)] in WT and APPYG/YG (YG) hippocampal neurons [(A–D) and (E–H), respectively; 63× objective]. Scale bars = 7 μm. The panels are representative of three different experiments performed in duplicate. (C,G) and (D,H) Co-localization analysis of WT and APPYG/YG hippocampal neurons. The analysis was performed using Zen software. Notice that the area of overlap between APP and EEA1 immunostaining (white) was significantly decreased in APPYG/YG mice (H) compared with WT (D). The R coefficient (Pearson’s coefficient) was used for the quantitative and comparative analyses (I). The data are expressed as mean ± SEM. n = 6. **p < 0.001. (J,P) Confocal microscopy analysis of double-staining for rabbit anti-APP [red; (J,N)] and mouse anti-giantin [green; (K,O)] in WT and APPYG/YG (YG) hippocampal neurons [(J–L) and (N–P), respectively; 63× objective]. Scale bars = 5 μm. The panels are representative of five different experiments performed in duplicate. (L,P) and (M,Q) Co-localization analysis of WT and APPYG/YG hippocampal neurons. The analysis was performed using Zen software. The (R) coefficient (Pearson’s coefficient) was used for the quantitative and comparative analyses (R). The data are expressed as mean ± SEM. n = 10. **p < 0.01.
Y682 Mutation Affects Lysosomal Activity in Hippocampal YG Neurons
We next investigated whether the observed accumulation of APP in Lamp1-positive vesicles leads to a defect in lysosomal activity. To test for lysosomal activity, we used western blotting (WB) to detect the conversion of immature to mature cathepsin D (CD), a lysosomal protease that is used as an indirect marker of lysosomal function (Luzio et al., 2007). The WB analysis of homogenates from WT and APPYG/YG neurons showed a significant decrease in the level of the two CD active fragments, which migrate at 32 and 14 kDa (CDm; Figures 3A,B). Furthermore, the enzymatic activity of CD and cathepsin B (CB) confirmed a significant reduction of lysosomal activity in neurons isolated from APPYG/YG mice (Figure 3C). These data clearly suggest impairment in the lysosomal machinery and a key role played by the Y682ENPTY687 domain in controlling APP trafficking in neurons and preventing lysosomal dysfunction. These results are also interesting when considering that a reduction of CD activity has been previously reported in brains from AD patients (Yang et al., 2011).
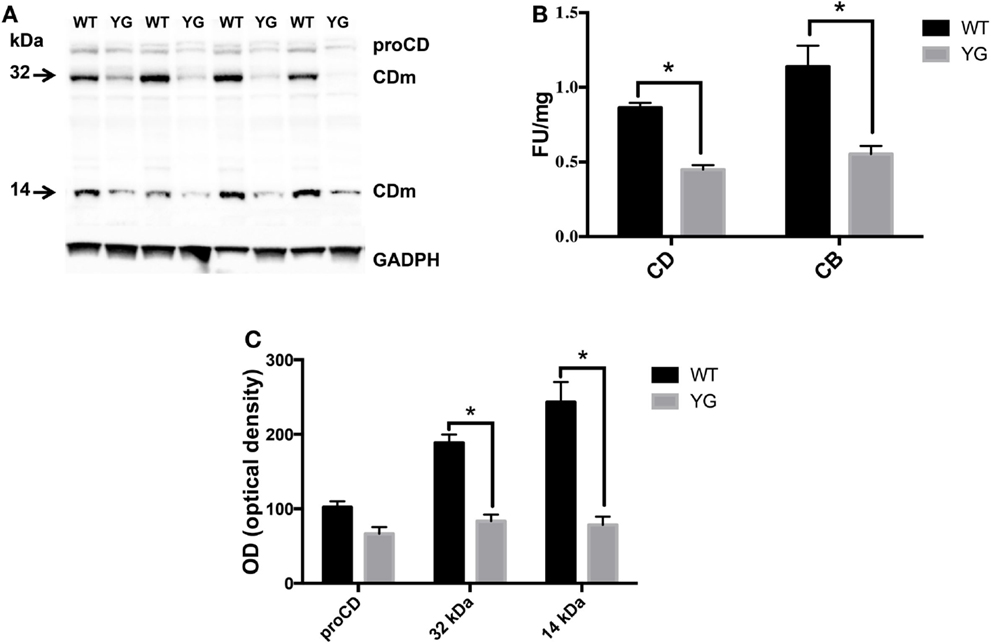
Figure 3. Lysosome activity is impaired in APPYG/YG hippocampal neurons. (A) Western blot analysis of cathepsin D in hippocampi from WT and APPYG/YG (YG) mice and (B) the corresponding densitometric analysis. The data were normalized to the corresponding GADPH values. The optical density (OD) is expressed as a percentage (mean ± SEM) of the proCD values from WT samples. n = 3. *p < 0.05. (C) Cathepsin D (CD) and cathepsin B (CB) cysteine protease activity in WT and APPYG/YG (YG) hippocampal neurons. The samples were collected and analyzed according to the procedure suggested by the manufacturer and described in the Section “Materials and Methods.” The relative activity is expressed as fluorescent units (FU)/mg protein. The data are expressed as mean ± SEM. n = 3. *p < 0.05.
Y682G Mutation Affects APP Binding to SorLA and Increases SorLA Secretion Outside Neurons
The Y682ENPTY687 motif within the APP cytoplasmic domain plays a crucial role in the sorting and trafficking of APP through its binding to specific adaptor proteins. The recruitment of specific adaptors to APP is linked to its routing throughout the endomembrane system and its phosphorylation state, particularly the Y682 residue (Russo et al., 2002; Tarr et al., 2002; Zhou et al., 2004, 2009; Tamayev et al., 2009; Muller and Zheng, 2012).
The Vps10p-D receptor, SorLA, is a neuronal protein that shuttles from the PM to endosomes and TGN and regulates the trafficking and processing of APP (Andersen et al., 2005). SorLA confines APP to the Golgi apparatus and affects its transition to the cell surface, a step that is crucial for conversion to both soluble APPα (sAPPα) and sAPPβ/Aβ (Andersen et al., 2005, 2006; Dodson et al., 2008; Fjorback et al., 2012). We observed the enhancement of mutated APP trafficking to Rab7-positive vesicles and sought to determine whether Y682G substitution impairs the binding of APP to SorLA.
Confocal microscopy of hippocampal neurons that were immunostained for APP and SorLA showed a strong co-localization between SorLA and APP in WT cells (Figures 4A–D). However, such co-localization was significantly reduced in APPYG/YG neurons (Figures 4E–H). Interestingly, confocal microscopy analysis indicated a decrease in SorLA in the cell body and an increase along the neurites of Y682G neurons (Figure 4).
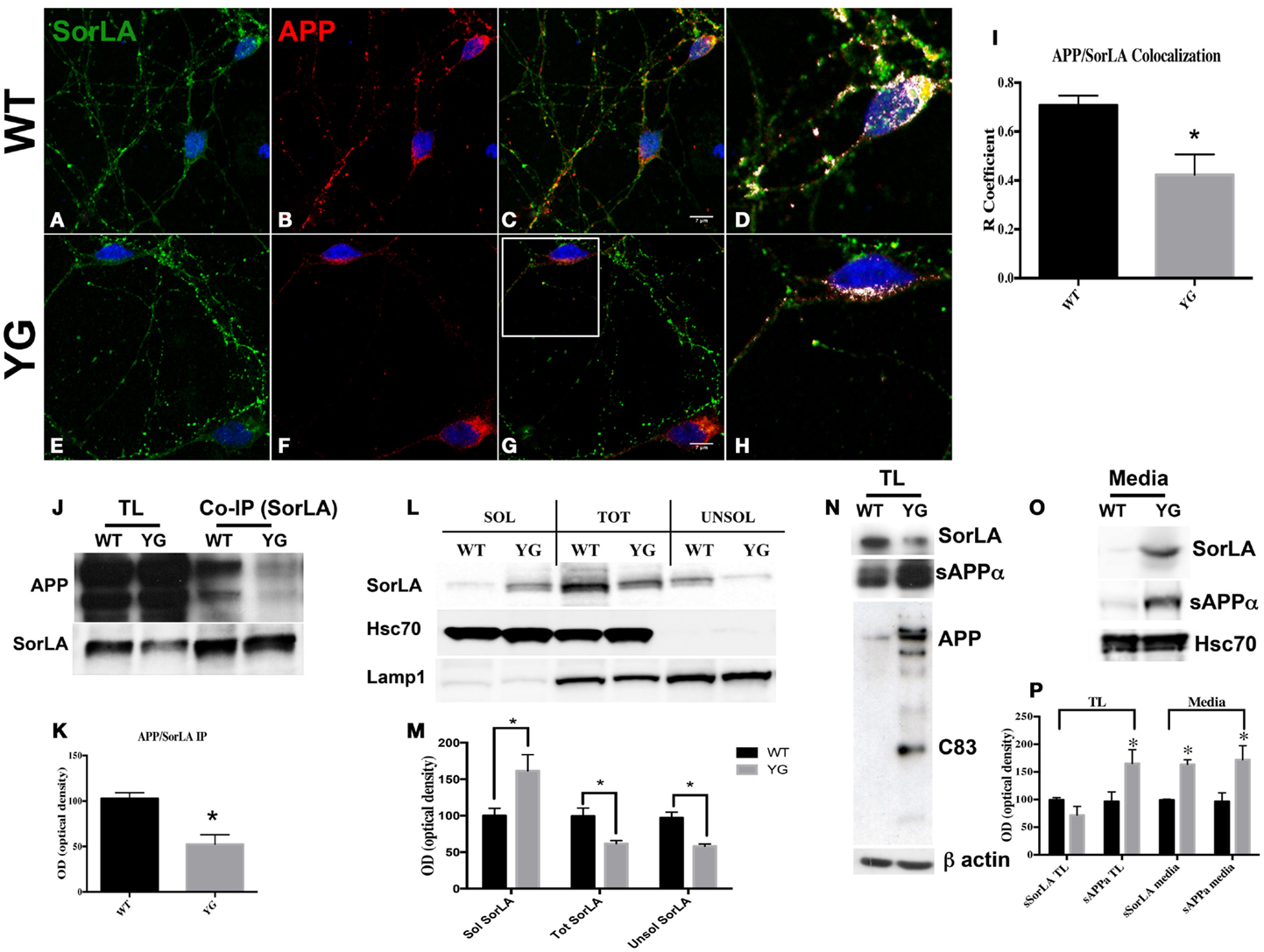
Figure 4. Y682G mutation prevents binding of APP to SorLA and increases SorLA secretion. (A–H) Confocal microscopy analysis of double-staining for rabbit anti-SorLA [green; (A,E)] and mouse anti-APP [red; (B,F)] in WT and APPYG/YG (YG) hippocampal neurons [(A–C) and (E–G), respectively; 63× objective]. Scale bars = 7 μm. (D,H) high magnification of the (C) and (G), respectively. Co-localization analysis was performed using Zen software (I). The panels are representative of seven different experiments. (G) Quantitative analysis was performed by comparing the R coefficient (Pearson’s coefficient) between WT and APPYG/YG (YG) neurons. n = 7. *p < 0.05. (J,K). Protein samples from hippocampi from either WT or APPYG/YG (YG) mice were immunoprecipitated with mouse anti-α-SorLA antibody (co-IP SorLA) and analyzed with rabbit anti-APP. TL, total lysate; IP, immunoprecipitate. The same membrane was stripped and analyzed with mouse anti-SorLA. (L,M) SorLA WB analysis in the soluble and insoluble fractions from hippocampal tissues from WT and APPYG/YG (YG) mice. As positive and negative controls for the fractionations, we used HSC70 protein and Lamp1, respectively. The optical density analysis is reported below. Data from the soluble (Sol) and insoluble (Unsol) fractions were normalized to the corresponding HSC70 and Lamp1 values and are expressed as a percentage of WT (n = 4). *p < 0.05 vs. WT (Newman–Keuls test). (N) WB analysis of APP, sAPPα, and sSorLA from hippocampal primary neuronal cultures of WT and APPYG/YG (YG) mice (O) Medium collected from APPYG/YG (YG) neurons and the corresponding control (WT) samples were analyzed by WB for sAPPα and sSorLA (see Materials and Methods). WB is representative of three different experiments. (P) Densitometric analysis of (N,O). Black histogram: WT; Gray histogram: YG. (N = 3; Newman–Keuls test) *p < 0.05 vs. WT.
Consistent with the reduced co-localization, co-immuno precipitation (co-IP) experiments from the hippocampi of WT and APPYG/YG mice showed a reduced complex formation between APP and SorLA in APPYG/YG neurons (Figures 4J,K).
The way in which Y682 mutation disturbs the binding of SorLA to APP is still unknown. One plausible mechanism is that some adaptor proteins may fail to bind APP or SorLA, resulting in alterations of the APP/SorLA complex.
Sortilin-related receptor has been largely described as a potential tool that is predictive of the progression of AD in humans. In fact, the levels of SorLA appeared to be reduced in AD brain tissues and increased in cerebrospinal fluid (Ikeuchi et al., 2010; Guo et al., 2012b). Therefore, we hypothesized that the lack of formation of the APP/SorLA complex might result in the missorting of SorLA and its secretion outside neurons. To test this hypothesis, we analyzed the protein levels of SorLA in both soluble and insoluble hippocampal fractions from APPYG/YG mice and relative controls (Figures 4L,M). As expected, SorLA levels in the soluble fraction from APPYG/YG tissue was significantly increased, whereas its levels appeared to be reduced in the corresponding insoluble fraction (Figures 4L,M). The increased secretion of SorLA from APPYG/YG neurons was further analyzed in the medium of primary hippocampal neurons. Also here, the amount of SorLA released from APPYG/YG neurons into the medium was greatly increased when compared to the control samples (Figures 4O,P). Such increase was paralleled by an intensification of sAPPα amount in the medium of APPYG/YG neurons. Consistently, the corresponding SorLA intracellular levels appeared significantly reduced (Figure 4N). Notably and consistent with the evidence that Y682G mutation induces a preferential α-secretase-mediated APP cleavage (Basso and Matrone, 2013), WB analysis performed with anti-sAPPα antibody showed an increase in sAPPα in APPYG/YG neurons (Figure 4N).
Discussion
Altogether, our data further elucidate the role played by the Y682ENPTY687 domain in controlling APP trafficking and sorting in neurons. Our data support a model whereby alterations in the binding of APP to SorLA likely leads to an aberrant misrouting of APP in hippocampal neurons, resulting in APP accumulation in endo-lysosomal compartments and autophagic defects (see Box 1).
Box 1. Y682G mutation alters APP trafficking and leads to an increase in SorLA secretion.
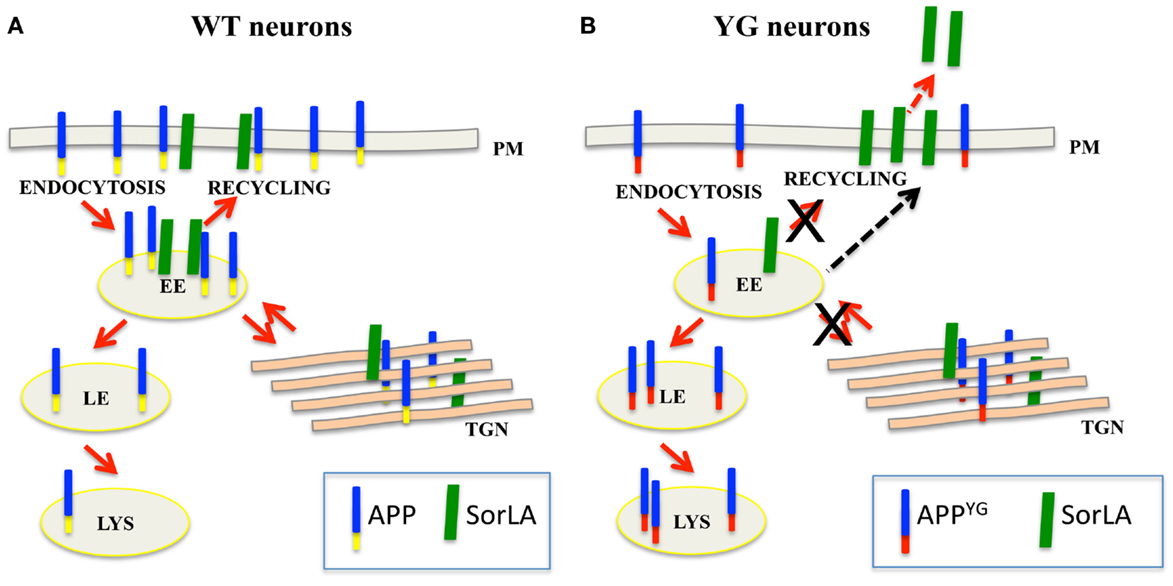
(A) In normal neurons, SorLA (green) controls APP (blue–yellow) trafficking from early endosome (EE) back to the trans-Golgi network (TGN) or plasma membrane (PM). (B) Because of the Y682G mutation on the Y682ENPTY687 domain of APP (YG), SorLA is no longer able to interact with mutated APP (blue–red); consequently, mutated APP mislocalizes to the late endosome (LE) and lysosome (LS) where it is likely processed to generate CTF peptides (Basso and Matrone, 2013). The accumulation of APP in the late endosome and lysosomes induces alterations in lysosomal activity and leads to the neuronal defects that were previously reported in APPYG/YG mice (YG) (Matrone, 2013). As a result of the lack of binding to APP, SorLA is trafficked to the plasma membrane where it is rapidly cleaved and secreted outside neurons.
The intracellular portion of APP, which encompasses the Y682ENPTY687 sequence of AICD, has been reported to interact with several different adaptors. Y682ENPTY687 plays a crucial role in modulating the binding and unbinding of APP to specific adaptor proteins via phosphorylation and dephosphorylation of the Y682 residues. When Y682 is phosphorylated, it creates docking sites for cytosolic proteins, such as Shc, Grb2, and Grb7, and inhibits the binding of others, such as the Fe65 family. Differently, when Y682 is dephosphorylated, the binding of proteins containing a phosphotyrosine-binding (PTB) domain is impaired (Russo et al., 2002; Tarr et al., 2002; Zhou et al., 2004, 2009; Tamayev et al., 2009; Muller and Zheng, 2012).
Here, we report that when Y682 residue is mutated (Y682G mutation), APP is misrouted to the LEs/lysosomes, triggering lysosomal defects and contributing to progressive and premature aging and the neurodegenerative phenotype previously reported in YG knock-in mice (Matrone et al., 2012).
In addition, we provide clear evidence of the role played by Y682 in the interaction of APP with SorLA, and we speculate on the possibility that the lack of a proper interaction between SorLA and APP could lead to the misrouting of APP in neurons and consequently to the lysosomal defects previously observed in APPYG/YG mice (Matrone et al., 2012). Interestingly, the lack of APP and SorLA interaction also results in increased secretion of SorLA into the soluble fraction of APPYG/YG brains and into the medium of cultured hippocampal APPYG/YG neurons.
The role of SorLA in controlling APP trafficking has been previously clarified (Andersen et al., 2005). SorLA directly binds APP and controls APP trafficking from EEs back to the TGN or PM, thus preventing β- and α-secretase-mediated APP cleavage.
The complexity in the APP and SorLA interaction has been previously predicted in the SorLA-null mouse model and in several SorLA-overexpressing cell lines (Andersen et al., 2005; Spoelgen et al., 2006; Schmidt et al., 2012; Mehmedbasic et al., 2015). All these studies have allowed the identification of numerous putative SorLA binding sites on both the N- and C-terminal sequences of APP depending on the cellular context, pointing to the possibility that APP interacts with SorLA at multiple sites and that such interaction probably requires the additional collaboration of other adaptors (Andersen et al., 2005; Spoelgen et al., 2006; Schmidt et al., 2012; Mehmedbasic et al., 2015).
Accordingly, we report here that the alteration in APP binding to SorLA (and likely to other adaptors) leads to APP accumulation in LEs and lysosomes and to autophagic defects, which are considered a major risk factor for AD and dementia during aging (Cuervo, 2008; Salminen and Kaarniranta, 2009).
In addition, evidence from pathophysiological and genetic studies clearly suggests that SorLA plays a crucial role in AD-related processes. Several studies of brain or cerebrospinal fluid specimens have reported alterations in the levels of SorLA in the brains of some individuals with sporadic AD (Ma et al., 2009; Ikeuchi et al., 2010). Moreover, several mutations of the SorLA gene have been associated with AD (Rogaeva et al., 2007; Alexopoulos et al., 2011; Caglayan et al., 2014).
Based on the critical phenotype previously observed in APPYG/YG mice, which resulted in premature aging and cognitive and learning deficits (Matrone et al., 2011, 2012), and the potential role of SorLA as a predictive factor of neurodegeneration in AD patients (Rogaeva et al., 2007; Alexopoulos et al., 2011; Caglayan et al., 2014), we believe that these results strongly underscore the relevance of the correct APP binding to adaptor proteins, via Y682ENPTY687 domain, as a future potential strategy to prevent neurodegeneration in vivo.
Materials and Methods
Animal Model
Male homozygous APP Y682G mice (APPYG/YG) and WT controls (C57BL/6J, approximately 30–40 days of age) were used in this study. The APPYG/YG mice were a generous gift from Prof. D’Adamio and have been previously described in detail (Barbagallo et al., 2010, 2011; Matrone et al., 2011, 2012; Basso and Matrone, 2013; Matrone, 2013). The mice from both lines were bred and group-housed in the Lab Animal Centre of Taconic (Ejby, DK) at an ambient temperature of 22–23°C and on a 12/12 h dark/light cycle (lights on 7 a.m.).
All of the mice were anesthetized by 2-bromo-2-chloro-1,1,1-trifluoroethane inhalation and euthanized in accordance with international guidelines on the ethical use of animals (European Communities Council Directive of November 24, 1986; 86/609/EEC) and Danish guidelines. The brains were quickly removed from the skulls, and the hippocampi were dissected on ice according to the procedure previously described by La Rosa et al. (2013).
We previously reported that neuronal decline in APPYG/YG mice starts to be evident at 3 months (Matrone et al., 2012), for this reason all of the experiments reported herein were performed in 3-month-old APPYG/YG and control mice.
Immunoprecipitation
For the IP reactions, protein samples were added to Dynabeads-Protein G (30 μg/100 μl) according to the procedure described by the manufacturer (Invitrogen, Naerum, Denmark) and eluted with 0.1 M citrate buffer (pH 2.3).
For the IP analysis, we used anti-mouse anti-SorLA from Abcam [mouse monoclonal (3B6B11) to SorLA]. The WB analysis was performed using rabbit anti-APP antibody (clone Y188) from Abcam.
Western Blot Analysis
Whole mouse brain was sonicated in a buffer containing 20 mM Tris-base pH 7.4, 250 mM sucrose, 1 mM EDTA, 1 mM EGTA plus protease (Roche, Complete) and phosphatase inhibitors.
To obtain soluble and insoluble fractions, the lysates were first spun at 1,000 × g for 15 min and then for an additional 4 h at 100,000 × g. The soluble fraction was separated from the pellet and analyzed by WB. Equal amounts (30 μg) of proteins were separated on 4–12% Bis-Tris SDS-PAGE gels (Invitrogen, DK), blotted onto nitrocellulose membranes (Amersham, DK), and incubated overnight with the appropriate primary antibody. We used rabbit anti-cathepsin D and rabbit anti-Lamp1 from Santa Cruz (Santa Cruz, CA, USA, DK); mouse anti-SorLA (clone 3B6B11) and rabbit anti-APP (clone Y188) from Abcam (Abcam, UK).
Media collected from APPYG/YG neurons and the correspondent control samples were centrifuged at 10,000 × g for 30 min to remove cell debris, and the supernatant was further centrifuged at 100,000 × g for 12 h at 4°C. The pellets were solubilized in 2% SDS-loading buffer and analyzed by WB for sAPPα and sSorLA. To detect α-secretase-mediated APP cleaved peptides, we used anti α-APP (MyBioSource, clone MBS533522). An antibody against the N-terminal domain of SorLA (sc33822) from Santa Cruz Biotechnology (Santa Cruz, CA, USA) was used to assess the amount of SorLA released into the medium and retained in neurons.
Primary Hippocampal Neuronal Cultures
Hippocampal neurons were prepared from embryonic day 17/18 (E17/18) mice as previously reported (Matrone et al., 2008, 2009). The hippocampi were dissected in Hank’s Balanced Salt Solution buffered with HEPES and dissociated via trypsin/ethylenediaminetetraacetic acid treatment. A total of 106 cells were plated on 3.5-cm dishes that were precoated with poly-l-lysine. After 2 days of culture in neurobasal medium that contained B-27 supplement and Glutamax, cytosine arabinofuranoside was added to reduce glial proliferation.
Confocal Microscopy and Co-Localization Analysis
The neurons were fixed for 20 min in phosphate-buffered saline (PBS) that contained 4% formaldehyde, permeabilized with 0.05% Triton (5–10 min for 20°C), and processed for double-labeling with the appropriate antibodies. Secondary antibodies coupled to Alexa dyes (488 or 594) were obtained from Abcam (Cambridge, UK). The nuclei were visualized by staining with DAPI (1 μg/ml; Sigma Brøndby, Denmark). Digital images were obtained with a Zeiss LSM confocal lsm780 system using 63× oil NA 1.3 objective. Quantification of the co-localization experiments was performed using Zen 2009 software. Pearson coefficients (R coefficients) were used as co-localization coefficients.
For the immunofluorescence analysis, we used rabbit anti-Lamp1 (sc199992), rabbit anti-APP (clone Y188), mouse anti-Rab7 (Rab7-117), mouse anti-EEA1 [anti-EEA1 antibody (1G11); EE marker], and mouse anti-giantin (clone 9B6), from Abcam. For the experiments reported in Figures 4A–H,N,O we used anti α-APP (MyBioSource, clone MBS533522) and anti-SorLA (sc33822) from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Measurement of Cathepsin D and B Activity
Enzymatic CD and CB activity was examined in hippocampal tissues from WT and YG mice using a CD or CB activity assay kit (Abcam). Briefly, tissues were washed twice in ice-cold PBS and then homogenized in extraction buffer. After 10 min incubation on ice, the extract was centrifuged at 10,000 × g for 5 min, and 50 μl of the supernatant was mixed with an equal volume of 2× reaction buffer and 2 μl substrate (GKPIFFRLK[Dnp]-DR-NH2-MCA substrate for CD or 10 mM Ac-RR-AFC substrate for CB) in a 96-well microplate. The plates were kept in the dark at 37°C for 1 h, and fluorescence was recorded using a FLUOstar Optima plate reader (BMG Labtech, Milan, Italy). The protein concentration was determined by the BCA method (Bio-Rad Laboratories, Hercules, CA, USA) at 328 and 460 nm excitation and emission wavelengths, respectively, for CD activity or 400 and 505 nm excitation and emission wavelengths for CB activity.
Statistical Analysis
The statistical analysis was performed using two-tailed t-tests, and multiple comparisons were made using one-way, two-way, or two-way repeated-measures analysis of variance (ANOVA) followed by a post hoc test using GraphPad Prism 6 software (San Diego, CA, USA). Statistical significance was accepted at the 95% confidence level (p < 0.05).
Author Contributions
LLR and CM designed the study and performed the experiments; MN, OA, and CM interpreted and analyzed the data; MN, LP, and PC provided critical discussion about biological significance; OA and CM wrote the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was funded by the Lundbeck Foundation (grant no. R108 and R151 to CM) and Danish Council 2014 to CM. Ciotti M. T., Graziani A. (National Council of Research), Johansen M. G., and Kamakh F. (Department of Biomedicine, AU) are thanked for their excellent technical assistance. We are grateful to Servillo Felicio for help with the statistical analysis and IT support. We thank BioMed Proofreading LLC for English editing.
Abbreviations
Aβ, amyloid beta; APP, amyloid precursor protein; CTF, carboxyl terminal fragment; EEA1, early endosome-associated protein 1; Lamp1, lysosomal-associated membrane protein 1; sAPPα, soluble amyloid precursor protein alpha; sAPPβ, soluble amyloid precursor protein beta; IF, immunofluorescence; IP, immunoprecipitation; SorLA, sortilin-related receptor; TL, total lysate; WB, western blot; WT, wildtype; YG, mutated amyloid precursor protein: APP Y682G.
References
Alexopoulos, P., Guo, L. H., Kratzer, M., Westerteicher, C., Kurz, A., and Perneczky, R. (2011). Impact of SORL1 single nucleotide polymorphisms on Alzheimer’s disease cerebrospinal fluid markers. Dement. Geriatr. Cogn. Disord. 32, 164–170. doi:10.1159/000332017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Andersen, O. M., Reiche, J., Schmidt, V., Gotthardt, M., Spoelgen, R., Behlke, J., et al. (2005). Neuronal sorting protein-related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proc. Natl. Acad. Sci. U.S.A. 102, 13461–13466. doi:10.1073/pnas.0503689102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Andersen, O. M., Schmidt, V., Spoelgen, R., Gliemann, J., Behlke, J., Galatis, D., et al. (2006). Molecular dissection of the interaction between amyloid precursor protein and its neuronal trafficking receptor SorLA/LR11. Biochemistry 45, 2618–2628. doi:10.1021/bi052120v
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Barbagallo, A. P., Wang, Z., Zheng, H., and D’Adamio, L. (2011). A single tyrosine residue in the amyloid precursor protein intracellular domain is essential for developmental function. J. Biol. Chem. 286, 8717–8721. doi:10.1074/jbc.C111.219873
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Barbagallo, A. P., Weldon, R., Tamayev, R., Zhou, D., Giliberto, L., Foreman, O., et al. (2010). Tyr(682) in the intracellular domain of APP regulates amyloidogenic APP processing in vivo. PLoS ONE 5:e15503. doi:10.1371/journal.pone.0015503
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Basso, E., and Matrone, C. (2013). NGF and APP interplay: focus on YENPTY motif of amyloid precursor protein and Y682 residue. Cell Biol. Res. Ther. 2:2. doi:10.4172/2324-9293.1000106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Caglayan, S., Takagi-Niidome, S., Liao, F., Carlo, A. S., Schmidt, V., Burgert, T., et al. (2014). Lysosomal sorting of amyloid-beta by the SORLA receptor is impaired by a familial Alzheimer’s disease mutation. Sci. Transl. Med. 6, 223ra20. doi:10.1126/scitranslmed.3007747
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Caster, A. H., and Kahn, R. A. (2013). Recruitment of the Mint3 adaptor is necessary for export of the amyloid precursor protein (APP) from the Golgi complex. J. Biol. Chem. 288, 28567–28580. doi:10.1074/jbc.M113.481101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cuervo, A. M. (2008). Autophagy and aging: keeping that old broom working. Trends Genet. 24, 604–612. doi:10.1016/j.tig.2008.10.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dodson, S. E., Andersen, O. M., Karmali, V., Fritz, J. J., Cheng, D., Peng, J., et al. (2008). Loss of LR11/SORLA enhances early pathology in a mouse model of amyloidosis: evidence for a proximal role in Alzheimer’s disease. J. Neurosci. 28, 12877–12886. doi:10.1523/JNEUROSCI.4582-08.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fjorback, A. W., Seaman, M., Gustafsen, C., Mehmedbasic, A., Gokool, S., Wu, C., et al. (2012). Retromer binds the FANSHY sorting motif in SorLA to regulate amyloid precursor protein sorting and processing. J. Neurosci. 32, 1467–1480. doi:10.1523/JNEUROSCI.2272-11.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guo, Q., Wang, Z., Li, H., Wiese, M., and Zheng, H. (2012a). APP physiological and pathophysiological functions: insights from animal models. Cell Res. 22, 78–89. doi:10.1038/cr.2011.116
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guo, L. H., Westerteicher, C., Wang, X. H., Kratzer, M., Tsolakidou, A., Jiang, M., et al. (2012b). SORL1 genetic variants and cerebrospinal fluid biomarkers of Alzheimer’s disease. Eur. Arch. Psychiatry Clin. Neurosci. 262, 529–534. doi:10.1007/s00406-012-0295-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ikeuchi, T., Hirayama, S., Miida, T., Fukamachi, I., Tokutake, T., Ebinuma, H., et al. (2010). Increased levels of soluble LR11 in cerebrospinal fluid of patients with Alzheimer disease. Dement. Geriatr. Cogn. Disord. 30, 28–32. doi:10.1159/000315539
Jiang, S., Li, Y., Zhang, X., Bu, G., Xu, H., and Zhang, Y. W. (2014). Trafficking regulation of proteins in Alzheimer’s disease. Mol. Neurodegener. 9, 6. doi:10.1186/1750-1326-9-6
La Rosa, L. R., Matrone, C., Ferraina, C., Panico, M. B., Piccirilli, S., Di Certo, M. G., et al. (2013). Age-related changes of hippocampal synaptic plasticity in AbetaPP-null mice are restored by NGF through p75NTR. J. Alzheimers Dis. 33, 265–272. doi:10.3233/JAD-2012-112108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, C. Y., and Landreth, G. E. (2010). The role of microglia in amyloid clearance from the AD brain. J. Neural Transm. 117, 949–960. doi:10.1007/s00702-010-0433-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Luzio, J. P., Pryor, P. R., and Bright, N. A. (2007). Lysosomes: fusion and function. Nat. Rev. Mol. Cell Biol. 8, 622–632. doi:10.1038/nrm2217
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ma, Q. L., Galasko, D. R., Ringman, J. M., Vinters, H. V., Edland, S. D., Pomakian, J., et al. (2009). Reduction of SorLA/LR11, a sorting protein limiting beta-amyloid production, in Alzheimer disease cerebrospinal fluid. Arch. Neurol. 66, 448–457. doi:10.1001/archneurol.2009.22
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Matrone, C. (2013). A new molecular explanation for age-related neurodegeneration: the Tyr682 residue of amyloid precursor protein. Bioessays 35, 847–852. doi:10.1002/bies.201300041
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Matrone, C., Barbagallo, A. P., La Rosa, L. R., Florenzano, F., Ciotti, M. T., Mercanti, D., et al. (2011). APP is phosphorylated by TrkA and regulates NGF/TrkA signaling. J. Neurosci. 31, 11756–11761. doi:10.1523/JNEUROSCI.1960-11.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Matrone, C., Ciotti, M. T., Mercanti, D., Marolda, R., and Calissano, P. (2008). NGF and BDNF signaling control amyloidogenic route and Abeta production in hippocampal neurons. Proc. Natl. Acad. Sci. U.S.A. 105, 13139–13144. doi:10.1073/pnas.0806133105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Matrone, C., Luvisetto, S., La Rosa, L. R., Tamayev, R., Pignataro, A., Canu, N., et al. (2012). Tyr682 in the Abeta-precursor protein intracellular domain regulates synaptic connectivity, cholinergic function, and cognitive performance. Aging Cell 11, 1084–1093. doi:10.1111/acel.12009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Matrone, C., Marolda, R., Ciafre, S., Ciotti, M. T., Mercanti, D., and Calissano, P. (2009). Tyrosine kinase nerve growth factor receptor switches from prosurvival to proapoptotic activity via Abeta-mediated phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 106, 11358–11363. doi:10.1073/pnas.0904998106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mehmedbasic, A., Christensen, S. K., Nilsson, J., Ruetschi, U., Gustafsen, C., Poulsen, A. S., et al. (2015). SorLA complement-type repeat domains protect the amyloid precursor protein against processing. J. Biol. Chem. 290, 3359–3376. doi:10.1074/jbc.M114.619940
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Muller, U. C., and Zheng, H. (2012). Physiological functions of APP family proteins. Cold Spring Harb. Perspect. Med. 2, a006288. doi:10.1101/cshperspect.a006288
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nixon, R. A., Wegiel, J., Kumar, A., Yu, W. H., Peterhoff, C., Cataldo, A., et al. (2005). Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 64, 113–122.
Nixon, R. A., and Yang, D. S. (2011). Autophagy failure in Alzheimer’s disease – locating the primary defect. Neurobiol. Dis. 43, 38–45. doi:10.1016/j.nbd.2011.01.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rogaeva, E., Meng, Y., Lee, J. H., Gu, Y., Kawarai, T., Zou, F., et al. (2007). The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat. Genet. 39, 168–177. doi:10.1038/ng1943
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Russo, C., Dolcini, V., Salis, S., Venezia, V., Zambrano, N., Russo, T., et al. (2002). Signal transduction through tyrosine-phosphorylated C-terminal fragments of amyloid precursor protein via an enhanced interaction with Shc/Grb2 adaptor proteins in reactive astrocytes of Alzheimer’s disease brain. J. Biol. Chem. 277, 35282–35288. doi:10.1074/jbc.M110785200
Salminen, A., and Kaarniranta, K. (2009). Regulation of the aging process by autophagy. Trends Mol. Med. 15, 217–224. doi:10.1016/j.molmed.2009.03.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schmidt, V., Baum, K., Lao, A., Rateitschak, K., Schmitz, Y., Teichmann, A., et al. (2012). Quantitative modelling of amyloidogenic processing and its influence by SORLA in Alzheimer’s disease. EMBO J. 31, 187–200. doi:10.1038/emboj.2011.352
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Spoelgen, R., von Arnim, C. A., Thomas, A. V., Peltan, I. D., Koker, M., Deng, A., et al. (2006). Interaction of the cytosolic domains of sorLA/LR11 with the amyloid precursor protein (APP) and beta-secretase beta-site APP-cleaving enzyme. J. Neurosci. 26, 418–428. doi:10.1523/JNEUROSCI.3882-05.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tamayev, R., Zhou, D., and D’Adamio, L. (2009). The interactome of the amyloid beta precursor protein family members is shaped by phosphorylation of their intracellular domains. Mol. Neurodegener. 4, 28. doi:10.1186/1750-1326-4-28
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tarr, P. E., Roncarati, R., Pelicci, G., Pelicci, P. G., and D’Adamio, L. (2002). Tyrosine phosphorylation of the beta-amyloid precursor protein cytoplasmic tail promotes interaction with Shc. J. Biol. Chem. 277, 16798–16804. doi:10.1074/jbc.M110286200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Willnow, T. E., and Andersen, O. M. (2013). Sorting receptor SORLA – a trafficking path to avoid Alzheimer disease. J. Cell. Sci. 126(Pt 13), 2751–2760. doi:10.1242/jcs.125393
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yang, D. S., Stavrides, P., Mohan, P. S., Kaushik, S., Kumar, A., Ohno, M., et al. (2011). Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer’s disease ameliorates amyloid pathologies and memory deficits. Brain 134(Pt 1), 258–277. doi:10.1093/brain/awq341
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yu, W. H., Cuervo, A. M., Kumar, A., Peterhoff, C. M., Schmidt, S. D., Lee, J. H., et al. (2005). Macroautophagy – a novel Beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J. Cell Biol. 171, 87–98. doi:10.1083/jcb.200505082
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhou, D., Noviello, C., D’Ambrosio, C., Scaloni, A., and D’Adamio, L. (2004). Growth factor receptor-bound protein 2 interaction with the tyrosine-phosphorylated tail of amyloid beta precursor protein is mediated by its Src homology 2 domain. J. Biol. Chem. 279, 25374–25380. doi:10.1074/jbc.M400488200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhou, D., Zambrano, N., Russo, T., and D’Adamio, L. (2009). Phosphorylation of a tyrosine in the amyloid-beta protein precursor intracellular domain inhibits Fe65 binding and signaling. J. Alzheimers Dis. 16, 301–307. doi:10.3233/JAD-2009-0970
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: APP, Y682G mutation, SorLA, Alzheimer’s disease, neuronal trafficking, lysosomes, endosome, Abeta
Citation: La Rosa LR, Perrone L, Nielsen MS, Calissano P, Andersen OM and Matrone C (2015) Y682G mutation of amyloid precursor protein promotes endo-lysosomal dysfunction by disrupting APP–SorLA interaction. Front. Cell. Neurosci. 9:109. doi: 10.3389/fncel.2015.00109
Received: 08 December 2014; Accepted: 10 March 2015;
Published online: 07 April 2015.
Edited by:
Francesco Moccia, University of Pavia, ItalyReviewed by:
Gunnar Keppler Gouras, Lund University, SwedenPaul M. Salvaterra, Beckman Research Institute of the City of Hope, USA
Copyright: © 2015 La Rosa, Perrone, Nielsen, Calissano, Andersen and Matrone. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carmela Matrone, Department of Biomedicine, University of Aarhus, BartholinAllé 6, Aarhus 8000, Denmark e-mail:bWF0cm9uZUBiaW9tZWQuYXUuZGs=