 Alison X. Xie
Alison X. Xie Jeremy Petravicz
Jeremy Petravicz Ken D. McCarthy
Ken D. McCarthy- 1Department of Pharmacology, School of Medicine, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA
- 2Department of Brain and Cognitive Sciences, Picower Institute for Learning and Memory, Massachusetts Institute of Technology, Cambridge, MA, USA
Astrocytes are the predominant glial type in the central nervous system and play important roles in assisting neuronal function and network activity. Astrocytes exhibit complex signaling systems that are essential for their normal function and the homeostasis of the neural network. Altered signaling in astrocytes is closely associated with neurological and psychiatric diseases, suggesting tremendous therapeutic potential of these cells. To further understand astrocyte function in health and disease, it is important to study astrocytic signaling in vivo. In this review, we discuss molecular tools that enable the selective manipulation of astrocytic signaling, including the tools to selectively activate and inactivate astrocyte signaling in vivo. Lastly, we highlight a few tools in development that present strong potential for advancing our understanding of the role of astrocytes in physiology, behavior, and pathology.
Introduction
G protein coupled receptors (GPCRs) are the primary molecules through which non-excitable cells transduce information from external cues to biological responses. There are four major families of GPCRs that are distinguished by their composition, ability to activate intracellular signaling cascades, and the functional consequences associated with their activation. The importance of GPCRs is underscored by the fact that these molecules are the most common targeted class of proteins of therapeutic agents. Astrocytes express each of the major classes of GPCRs (Porter and McCarthy, 1997) clearly demonstrating that these cells are dynamically coupled to the activity of their surrounding cellular and chemical milieu. It is likely that astrocyte GPCRs are activated by neurotransmitters released from neurons synaptically as well as through volume transmission. It is also likely that neighboring non-neuronal cells including microglia, vascular endothelial cells, astrocytes, and other resident CNS cells release molecules that activate astrocyte GPCRs and modulate astrocyte activity. In certain situations, low levels of ambient neurotransmitters might tonically activate astrocyte GPCRs. However, it is likely that in most cases astrocyte GPCRs are spatially restricted to discrete signaling domains that are activated with different temporal characteristics dependent on the source of the signal and biological response being affected. Layered onto this signaling complexity is the morphological complexity of astrocytes; the fine processes of an individual astrocyte within the CA1 region of the hippocampus can associate with ~100,000 synapses (Bushong et al., 2002). This being the case, different regions of an individual astrocyte are likely responding simultaneously to local signals (from neurons or other cell types) with different functional outcomes. Collectively, this information underscores how difficult it is to replicate in vitro or in situ the complicated pattern of GPCR activation that normally occurs in vivo.
By far, the emphasis in astrocyte GPCR signaling activity has been on the regulation of Ca2+. This is not surprising given that Ca2+ fluxes play a very important role in regulating biological processes and Ca2+ is the only signaling molecule that we can readily monitor selectively in astrocytes in complex tissue such as brain. Consequently, we know a lot about astrocyte Ca2+ responses following the activation of Gq-GPCRs and the consequences of increasing astrocyte Ca2+ by a number of different approaches. Two important points need to be kept in mind when considering findings in this area. First, most investigations linking increases in astrocyte Ca2+ with functional responses use pharmacological methods to increase astrocyte Ca2+ and consequently findings may not reflect physiological responses. Second, while the field has focused on the role of astrocyte Ca2+ in functional responses, the activation of Gq-coupled GPCRs leads to the modulation of a broad set of signaling cascades beyond changes in Ca2+; the variety of effector proteins in Gαq “interactome” may affect astrocyte Ca2+ responses as well as play important roles in physiological responses to Gq-GPCR activation (Figure 1; Sanchez-Fernandez et al., 2014).
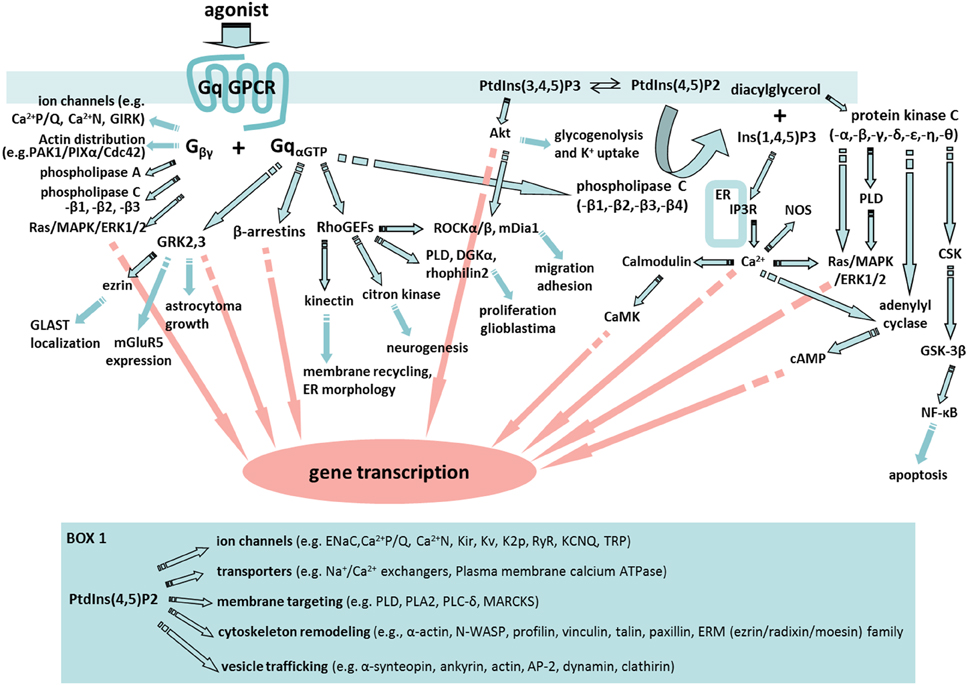
Figure 1. A fabric of signaling cascades can be activated by astrocyte Gq-GPCRs. Upon Gq-GPCR activation, Gαq subunits are known to interact with G protein-coupled receptor kinases (GRKs), β-arrestins, Rho family of guanine exchange factors (RhoGEFs) and phospholipase C (PLC) in astrocytes or astroglia. In addition, Gβγ subunits are able to regulate ion channel properties, as well as to interact with signaling molecules including cdc42, PAK-PIXα, phospholipase A (PLA), PLC, and Ras/MAPK/ERK1/2. Many of the key signaling molecules regulated by Gβγ are expressed in astrocytes or astroglia. Therefore, Gq-GPCR activation in astrocytes is likely to activate an entire fabric of downstream signaling pathways that may include (1) GRKs-mediated glutamate transporter (GLAST) localization (Nijboer et al., 2013), gene transcription (Atkinson et al., 2009), metabotropic glutamate receptor 5 (mGluR5) expression (Sorensen and Conn, 2003), and astrocytoma growth (Woerner et al., 2012); (2) β-arrestin-mediated gene silencing (McLennan et al., 2008; Miyatake et al., 2009; Zhu and Reiser, 2014); (3) RhoGEFs-mediated activation of Kinectin (Santama et al., 2004), citron kinase (Ackman et al., 2007), phospholipase D (PLD) (Burkhardt et al., 2014), diacylglycerol kinase (DGKa) (Kefas et al., 2013), Rhophilin2 (Vedrenne and Hauri, 2006; Danussi et al., 2013), Rock (Holtje et al., 2005; Lau et al., 2011), and mDia1 (Shinohara et al., 2012); (4) PLC regulation of PtdIns(3,4,5)P3 (PIP3)/protein kinase B (PKB or Akt) pathway (DiNuzzo et al., 2013; Kong et al., 2013), PtdIns(4,5)P2(PIP2)/diacylglycerol (DAG)/protein kinase C (PKC) pathway, and PIP2/Ins(1,4,5)P3(IP3) pathway. Moreover, PKC activation in astrocytes (Wang et al., 2002) engages PLD (Servitja et al., 2003), c-src tyrosine kinase (CSK) (Jo et al., 2014), glycogen synthase kinase (GSK) (Sanchez et al., 2003), and cAMP signaling. Many of these signaling pathways are known to trigger cellular responses that are important for astrocyte function including gene transcription and cell migration. Box 1. Selected PIP2-induced signaling pathways in which key molecules are expressed by astrocytes. These signaling molecules include: Epithelial sodium channel (ENaC) (Miller and Loewy, 2013), PLA2 (Ha et al., 2014), Myristoylated alanine-rich C-kinase substrate (MARCKS) (Vitkovic et al., 2005), Wiskott–Aldrich syndrome protein (WASP) (Murk et al., 2013), profilin (Molotkov et al., 2013), vinculin, talin, and paxillin (Kalman and Szabo, 2001), ERM protein family (Persson et al., 2010), α1-syntrophin (Masaki et al., 2010), ankyrin (Lee et al., 2012), adipocyte protein 2 (AP2) (Rossello et al., 2012), and clathirin (Pascual-Lucas et al., 2014).
Molecular Tools for Selective Activation of Astrocytes In Vivo
Temporal Control of Astrocyte Activation In Vivo Using Optogenetics
Optogenetics is an extremely powerful tool for activating and inactivating neuronal circuits in vivo with high temporal resolution. Optogenetic regulation of neuronal activity generally occurs through the flux of ions that either depolarize or hyperpolarize neurons with high temporal and spatial resolution (Schoenenberger et al., 2011; Lin, 2012). More recently, a number of investigators have used this technology to activate astrocyte signaling in situ and in vivo (Gourine et al., 2010; Perea et al., 2014; Yamashita et al., 2014). It is important to understand that while optogenetic activation of astrocytes can lead to changes in ion fluxes across plasma and intracellular compartment membranes, these fluxes do not remotely reflect the changes in signaling that occur following the activation of astrocyte GPCRs, the primary mode for activating astrocyte signaling (Agulhon et al., 2012; Sanchez-Fernandez et al., 2014).
Multiple variants of channelrhodopsin (ChR2) have been expressed in astrocytes, primarily to elicit Ca2+ responses (Table 1). In vivo, activation of the ChR2 variant ChR2 (H134R) (Nagel et al., 2005) in astrocytes in the ventral surface of the medulla oblongata was shown to lead to increases in intracellular Ca2+ and alteration in respiratory activity of rats (Gourine et al., 2010). This same variant of ChR2 was employed to illustrate the ability of astrocyte Ca2+ elevations to alter the firing and orientation responses of mouse primary visual cortex excitatory and inhibitory neurons in vivo (Perea et al., 2014). Most recently, Beppu et al. (2014) created mice expressing the ChR variant ChR2 (C128S) or an optogenetic proton pump (ArchT). Activation of these optogenetic tools lead to, respectively, acidification or alkalization of astrocytes which modulated glutamate release and ischemic damage in vivo. The remainder of studies using ChR2 or its variants have been performed using culture or slice preparations, but provide valuable insights into the mechanisms of their ability to activate astrocytes signaling (Figueiredo et al., 2014). A second variant, CatCh (Ca2+ translocating ChR, which has improved Ca2+ permeability) (Kleinlogel et al., 2011) when expressed in cultured astrocytes, was found to also increase Ca2+ but with varying reliability compared to ChR2 (H134R) (Li et al., 2012; Figueiredo et al., 2014). A non-ChR2 light activated channel, the light-gated Ca2+-permeable ionotropic GluR6 glutamate receptor (LiGluR), has also been used in cultured astrocytes, with again varying results (Li et al., 2012).
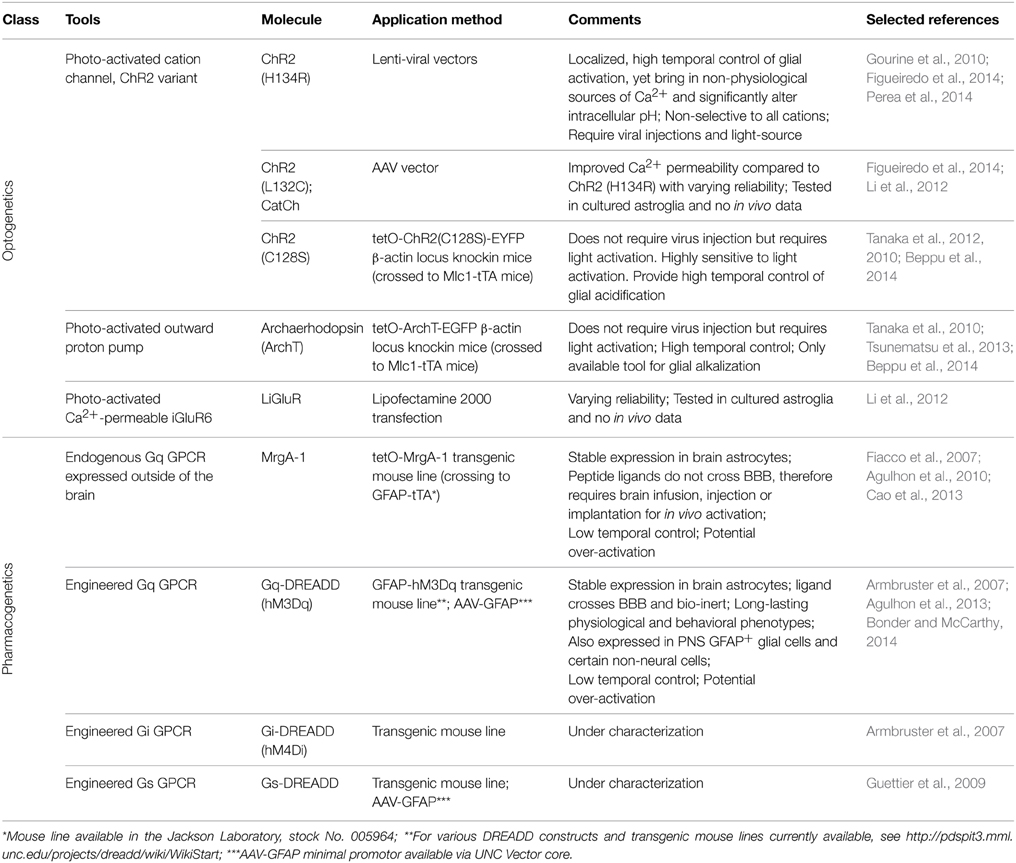
Table 1. Tools for selectively activation of astrocyte signaling in vivo.
The advantages of using optogenetics to activate astrocytes are the ability to temporally and spatially control the extent of activation. However, there are a number of important caveats involved in the use of optogenetics to activate astrocyte signaling cascades. First, it is unresolved as to the source of Ca2+ when using these optogenetic actuators in astrocytes. Extracellular Ca2+ entry through ChR2 variants seems the most logical mechanism, but a recent comparative study found that intracellular stores are the primary source (Figueiredo et al., 2014). These finding are at odds with studies from other laboratories (Li et al., 2012), and previous research from the same authors (Gourine et al., 2010). Current evidence indicates that both external and internal Ca2+ sources are involved in the ChR2 induced Ca2+ signal. Second, ChR2 and its variants are non-selective cation channels, allowing the entry of Na+ K+, H+, and Ca2+ upon activation (Nagel et al., 2003). ChR2 expressed in the cultured astroglia cell line GL261 display significant influx of Na+ and Ca2+ across the cell membrane based upon fluorescent indicator imaging (Ono et al., 2014). Additionally, significant alterations in intracellular pH were also observed in the same study, which has been corroborated in the recent study by Beppu et al. (2014). Interestingly, the ChR2 (H134R) used in the two current in vivo studies of astrocyte function was engineered to increase its ability to depolarize cultured HEK293 and neuronal cells over wild type ChR2 (Nagel et al., 2005; Beppu et al., 2014). Astrocytes process intracellular changes in Na+, Ca2+, and protons in different ways that may affect several different astrocyte functions. For example, increases in intracellular sodium and membrane depolarization, which is known to occur with ChR2-stimulation (Gourine et al., 2010), could alter glutamate reuptake efficacy (Djukic et al., 2007; Unichenko et al., 2012; Verkhratsky et al., 2013). Alterations to intracellular pH in astrocytes are important in the regulation of ischemic damage in vivo (Benesova et al., 2009; Beppu et al., 2014) and gap junction connectivity (Duffy et al., 2004). It is not unreasonable to hypothesize that ChR2 activation leads to membrane depolarization of astrocytes and interferes with a number of transport systems, most importantly glutamate, which may lead to effects on neuronal activity. In addition, to use optogenetics in vivo it is necessary to use a viral vector to deliver the optogenetic construct and may require the insertion of a light probe to activate the optogenetic channel in deeper brain regions. It seems very likely that these insults will lead to pathological changes that alter astrocyte responses. Overall, while optogenetic tools are great tools to activate or silence neurons, using optogenetics to increase astrocyte Ca2+ bypasses the majority of signaling pathways that are activated by GPCRs, as well as result in aberrant ion fluxes that likely never occur under physiological conditions (Fiacco et al., 2007; Agulhon et al., 2012; Wang et al., 2013).
Selective Activation of Astrocyte Gq-GPCR Signaling with MrgA-1
The first experimental system that enabled the specific activation of endogenous astrocyte signaling cascades in complex tissue was the Mas-related gene A1 (MrgA-1) transgenic mouse line (Fiacco et al., 2007) (Table 1). MrgA-1 is a Gq-GPCR that is normally expressed in a subsets of nociceptive sensory neurons (Dong et al., 2001) but not in the brain. The ligand used to activate MrgA-1, the peptide FMRF, does not activate endogenous brain Gq-GPCRs (Fiacco et al., 2007). To achieve cell specificity in the brain, MrgA-1 expression was controlled by using a tetracycline-controlled inducible expression system (tetO system), which requires a second transgene, the transcription transactivator (tTA). By crossing tetO-MrgA-1 transgenic mouse to GFAP-tTA transgenic mouse, GFAP+ glia become the only cells in the brain that express both tTA and tetO-MrgA-1, therefore the only CNS cells to express MrgA-1 receptor (Fiacco et al., 2007). As a native GPCR, MrgA-1 activation triggers the entire fabric of signaling cascades normally activated by endogenous astrocyte Gq-GPCRs; an important component that is absent when selectively increasing specific signaling molecules such as IP3 and Ca2+. With respect to Ca2+, MrgA-1 activation leads to a similar spatial and temporal response as endogenous Gq-GPCRs in hippocampal astrocytes (Fiacco et al., 2007).
MrgA-1 transgenic mice were used to prepare brain slices to test the gliotransmission hypothesis at hippocampal CA3-CA1 synapses (Fiacco et al., 2007; Agulhon et al., 2010; Wang et al., 2012, 2013; Devaraju et al., 2013). Initial reports showed that bath application of FMRFa induced widespread Ca2+ elevations in stratum radiatum astrocytes from MrgA-1 mice, while CA1 neuronal Ca2+ activity, excitatory synaptic transmission and short- or long-term excitatory synaptic plasticity in CA3-CA1 synapses were not affected (Fiacco et al., 2007; Agulhon et al., 2010). Later Wang et al. reported that although MrgA-1 mediated astrocyte activation did not change neuronal excitability and miniature excitatory synaptic currents (mEPSCs) in neurons near the surface of hippocampal slices, both bath and microinjection of FMRFa led to a transient hyperpolarization and decreased mEPSC frequency in neurons below 80 μm depth in the slices (Wang et al., 2012, 2013). These studies suggested that selective activation of Gq-GPCR signaling in astrocytes increased activity of the Na+/K+ ATPase, resulting in a reduction of extracellular K+ which consequently hyperpolarized neurons and suppressed excitatory transmission (Wang et al., 2012). The Gq-GPCR activated change in [K+] was hard to detect in the superficial layer of the slices, where the constant bath perfusion buffered the effects (Wang et al., 2012). The K+ removal hypothesis was supported by an independent study from Devaraju et al. who found that both Schaffer Collaterals stimulation and selective stimulation of astrocytic MrgA-1s potentiated inward K+ current and glutamate uptake in hippocampal astrocytes (Devaraju et al., 2013). These data suggest that astrocytic Gq-GPCR activation may regulate neuronal excitability and modulate neuronal network activity indirectly rather than inducing the release of gliotransmitters.
The MrgA-1 mouse line is rarely used for studying astrocyte function in vivo because the peptide agonists do not effectively cross blood brain barrier. Recently, Cao et al. used MrgA-1 mouse line to study the role of astrocytic activation in behavior by implanting infusion cannula or osmotic pumps into the brain of MrgA-1 mouse (Cao et al., 2013). Brain infusion of the peptide agonist of MrgA-1 mice induced antidepressant-like effect in forced swimming test and reversed depression-like behavior in MrgA-1 mice suggesting that astrocytic Gq-GPCR signaling is capable of modulating depressive-like behaviors (Cao et al., 2013).
Pharmacogenetic Activation of Astrocytic Signaling In Vivo Using DREADDs
In 2007, a new family of engineered GPCRs, Designer Receptor Exclusively Activated by Designer Drugs (DREADD) were developed (Armbruster et al., 2007) and have become the best option for activating GPCR signaling in specific cell populations in vivo (Rogan and Roth, 2011) (Table 1). A significant advantage of DREADDs compared to MrgA-1 is that the ligand of DREADDs, clozapine N-oxide (CNO), crosses BBB (Bender et al., 1994), therefore enabling non-invasive manipulation of receptor activity via peripheral injections (e.g., intraperitoneal or intravenous injections) and even via drinking water (Jain et al., 2013). The M3 muscarinic cholinergic receptor (M3AChR) was engineered through directed molecular evolution (Dong et al., 2010) that led to a striking decrease the affinity of this receptor for its native agonist (acetylcholine) as well as to a large increase in affinity for CNO. In addition, DREADDs do not exhibit constitutive activity and CNO is pharmacologically inert in the absence of DREADDs (Armbruster et al., 2007; Nichols and Roth, 2009; Dong et al., 2010). Consequently, mice expressing DREADDs do not exhibit a phenotype in the absence of CNO and CNO does not lead to a phenotype in wild type mice (Alexander et al., 2009; Guettier et al., 2009; Agulhon et al., 2013). Since their development, DREADDs have been extensively used to chronically and acutely activate (Gq-DREADDs) and silence (Gi-DREADDs) specific subsets of neurons in vivo (Wess et al., 2013).
Gq-DREADD was introduced into astrocyte research studies with the development of GFAP-Gq-DREADD mice for specifically activating GFAP+ glial Gq-GPCR signaling in vivo (Agulhon et al., 2013). Gq-DREADD expression was regulated by the 2.2 Kb human GFAP promotor fragment; a hemagglutinin (HA) tag was added to the N-terminus of the Gq-DREADD for highly specific antibody staining. Extensive immunostaining studies demonstrated that the expression of Gq-DREADD was restricted to GFAP+ glia in the CNS and PNS (Agulhon et al., 2013). Bath application of CNO in situ or i.p. injection of CNO in vivo led to Ca2+ increases in brain astrocytes, without affecting Ca2+ in nearby neurons; CNO induced Ca2+ increases occurred throughout astrocytes including their fine processes within the neuropil. The development of this model enabled, for the first time, examination of the behavioral and physiological consequences of specifically activating Gq-GPCR signaling in GFAP+ glia. CNO administration to GFAP-Gq-DREADD transgenic mice revealed robust and unexpected behavioral and physiological phenotypes that were absent in litter mate controls; phenotypic changes include robust increases in heart rate and blood pressure, saliva formation, a decrease in body temperature, and increased sedation in the presence of a GABA receptor agonist (Agulhon et al., 2013). These findings suggest that GFAP+ glia have the potential for modulating a number of important physiological processes.
In addition to GFAP-Gq-DREADD mice, transgenic mouse lines expressing Gs- and Gi-DREADD specifically in GFAP+ glia were developed and are currently under characterization in the McCarthy laboratory. GFAP-DREADD transgenic mice offer the best system to non-invasively and simultaneously activate widely distributed astrocyte populations. Other systems for manipulating astrocytic activity in vivo, including MrgA-1 transgenic mice and optogenetics, requires direct application of ligand/light to brain tissue and thus have spatial limitations with regard to cells being activated at a given time. Region-specific expression DREADD can be achieved via viral delivery (Bull et al., 2014). Once the expression pattern is established, one can activate a subset of astrocytes acutely or chronically in dose-dependent manner, and behavior and physiological outcome can be measured from free-moving, awake mice. For the first time in astrocyte research, we can now test the contribution of astrocytes GPCR signaling in physiology and behavior, as well as verify the previously known astrocytic function in intact animals.
There are caveats associated with using pharmacogenetic systems to study the role of astrocyte Gq-GPCR signaling in vivo. First, engineered GPCRs are driven by an exogenous promoter system and consequently the levels of expression are likely to be different than that of endogenous astrocytic GPCRs. While this does not appear to lead to markedly different Ca2+ responses compared to endogenous receptors, it has not been confirmed that the engineered GPCR signaling cascades are regulated in a similar manner. Second, it is impossible to mimic the temporal and spatial characteristics of in vivo GPCR activation, a caveat associated with all pharmacological stimulation. Following an i.p. injection of CNO, most of the physiological phenotypes were observed in 5 min and peak in 30 ~ 45 min (Agulhon et al., 2013). Increased temporal and spatial resolution can be obtained by either microinjecting CNO into the region of interest or uncaging CNO with laser pulse activation; the latter approach has temporal and spatial resolution similar to optogenetic activation; caged CNO has recently been prepared (Brian Roth, personal communication). Overall, DREADD technology enables activation of the entire fabric of endogenous signaling cascades in specific cell types that are generally stimulated by GPCRs; this is a striking advantage over most other methods used to activate glial signaling in situ or in vivo.
Selective Astrocyte Gene Rescue in Mice with Global Gene Deletion
The optogenetic and pharmacogenetic approaches for activating astrocyte in vivo share a common pitfall of potential over-activation. Recently, a conditional endogenous gene repair approach was used to isolate the role of astrocyte-specific endogenous signaling in vivo in a mouse model of Rett's syndrome (RTT).
RTT is an X-chromosome-linked autism spectrum disorder due to the loss of function of the transcriptional regulator methyl-CpG-binding protein 2 (MeCP2) in the brain (Amir et al., 1999; Guy et al., 2007). Because MeCP2 is expressed in all CNS cell types (Ballas et al., 2009), a conditional knock-in mice, MeCP2loxp mice, was developed to study cell-specific disease mechanisms. In this model, the endogenous Mecp2 gene is silenced by insertion of a loxP-Stop cassette, but can be activated when combined with Cre- or Cre-ER system (Guy et al., 2007). When MeCP2loxp mice were crossed to hGFAP-CreERT2 mice (Hirrlinger et al., 2006), the expression of MeCP2 was selectively restored in GFAP+ astrocytes when mice were treated with tamoxifen (Lioy et al., 2011). The specific re-expression of MeCP2 in astrocytes significantly improved RTT phenotype possibly by restoring normal dendritic morphology and levels of the excitatory glutamate transporter VGLUT1 (Lioy et al., 2011). This model illustrates the potential of using conditional astrocyte-specific rescue model to isolate the function of astrocyte signaling in vivo and in disease.
Spatial Control of Astrocytic Signaling via Viral Delivery—Advantages and Disadvantages
At this time, there are no astrocyte transcriptional units that can be used to target specific populations of astrocytes in mature brain. Consequently, a large number of investigators have used viral vectors to perturb signaling in subpopulations of astrocytes using both adeno-associated viruses (AAV) and lentiviral vectors (Figueiredo et al., 2014). Several AAV serotypes show tropism toward astrocytes, including AAV 2/5 and AAV 8 (Koerber et al., 2009; Aschauer et al., 2013; Petrosyan et al., 2014). In combination with an astrocytic selective promoter (Lee et al., 2008; Pfrieger and Slezak, 2012), these AAVs are expected to express the gene of interest selectively in astrocytes in specific brain regions.
The disadvantages of using viral vectors to express genes in astrocytes should not be overlooked. The most obvious concerns are tissue damage and reactive gliosis induced by viral injection. Reactive astrocytes display more robust, frequent and widely-spread intracellular Ca2+ activity, and intercellular coupling and Ca2+ waves are exaggerated among reactive astrocytes (Agulhon et al., 2012). A recent study showed that AAV 2/5 vector can be used to induce astrogliosis and disrupt the glutamate-glutamine cycle in astrocytes, which led to glutamate-reversible hyperactivity of nearby neurons (Ortinski et al., 2010). The primary methods for assessing whether or not astrocytes are pathologically transformed by viral infection are through morphological studies and GFAP expression levels (Xie et al., 2010; Shigetomi et al., 2013a). However, it is likely that more subtle undetected changes in astrocytes occur that could influence their functional interactions with surrounding cells.
Another challenge of using viral injection to express constructs in astrocytes is cell-specificity. AAVs show tropism to all cell types in the CNS (Aschauer et al., 2013; Gholizadeh et al., 2013; Petrosyan et al., 2014; Yang et al., 2014). Even with an astrocytic-specific promoter, it is important to carefully verify astrocyte specific transduction using the most sensitive methods available. A low level of GPCR expression in non-astrocyte cell types could lead to significant downstream signaling and confound the interpretation of findings.
One final consideration to keep in mind is that it is currently impossible to transduce functionally distinct populations of astrocytes in a manner analogous to transducing a subpopulation of functionally distinct neurons; this will only be solved as new subpopulation specific astrocyte transcriptional units are identified.
Current Knock-Out Models for Selective Inactivation of Astrocytic Signaling
Specific GPCR Knockout Models
To date, most investigators have used optogenetic, pharmacogenetic or pharmacological tools to determine the consequences of activating astrocyte signaling cascades (Agulhon et al., 2008). None of these approaches recapitulate the complex regulation of signaling cascades occurring in vivo. Findings from these studies (see review by Agulhon et al., 2008) provide insight into the potential outcome(s) of astrocyte signaling. However, one has to keep in mind that these highly artificial types of stimulation may lead to outcomes that rarely, or perhaps never, occur in physiology. To determine the functional significance of astrocytic signaling, a more powerful approach is to demonstrate that the loss-of-function of a particular pathway affects physiological processes such as synaptic transmission or behavior. Here we review current knockout (KO) and conditional KO (cKO) models for deleting specific astrocytic signaling pathways (Table 2).
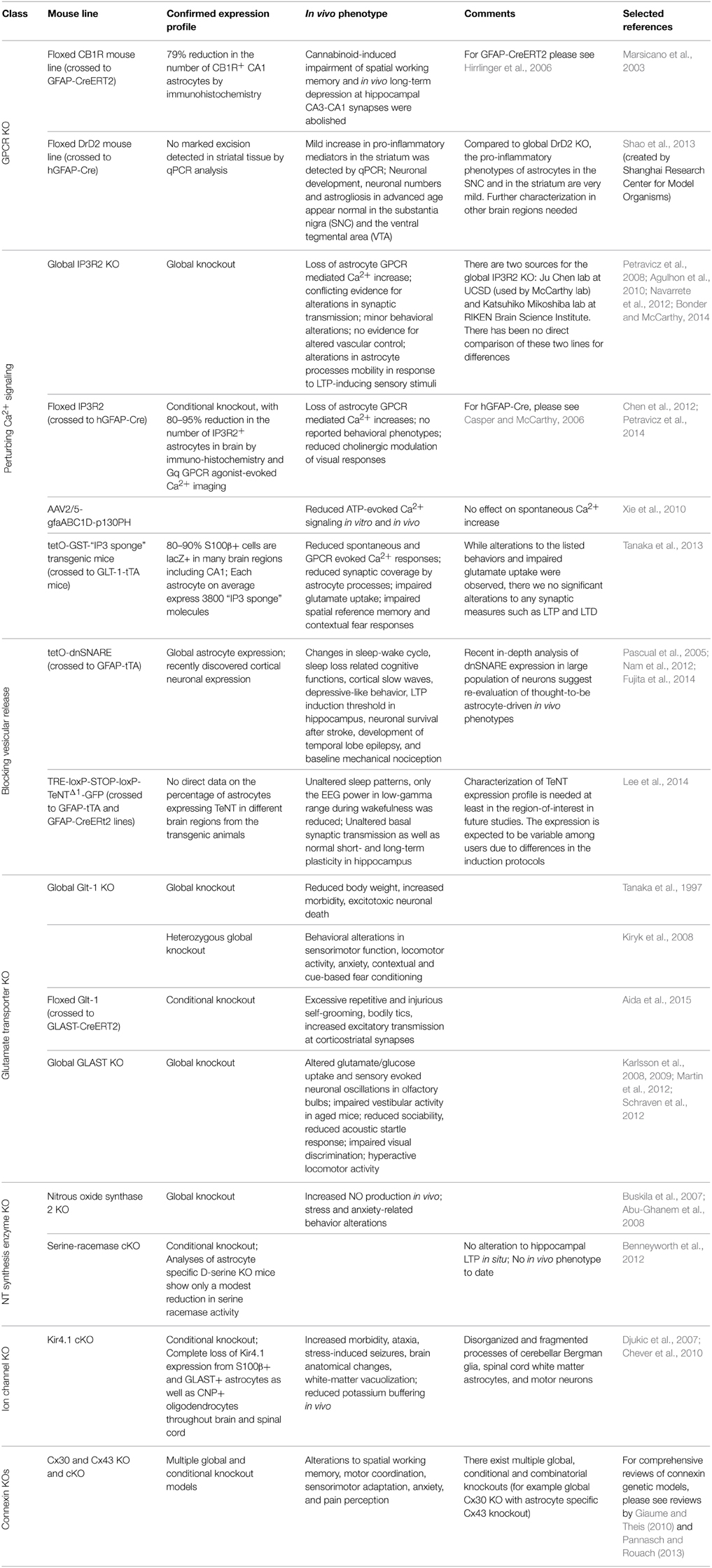
Table 2. Current molecular tools for perturbing astrocyte signaling in vivo.
GFAP-CB1R-KO
One of the first GPCRs targeted for a cKO in astrocytes was the cannabinoid type-1 receptor (CB1R), a GPCR predominately coupled to Gi signaling (Han et al., 2012). GFAP-CB1R cKO mouse line was accomplished by crossing floxed CB1R mouse line with an inducible Cre system driven by the GFAP promoter (GFAP-CreERT2) (Hirrlinger et al., 2006). Following tamoxifen administration, CB1R expression was reduced in GFAP+ astrocytes in the brain, providing the first model to study the contribution of astrocytes in cannabinoid induced working memory impairment in vivo. Han et al. found that conditional knockout of astrocytic CB1R abolished cannabinoid-induced impairment of spatial working memory and in vivo long-term depression at hippocampal CA3-CA1 synapses. In contrast, a cKO of CB1R in glutamatergic or GABAergic neurons did not rescue cannabinoid induced deficit (Han et al., 2012). This study revealed significant impact of a single astrocyte GPCR signaling system on synaptic modulation and behavior, as well as presented an elegant in vivo system for temporal control of astrocytic signaling.
GFAP-DRD2-cKO
In 2013, an astrocyte dopamine D2 receptor (DRD2) conditional knockout mouse was generated to study the role of astrocytic GPCR signaling in aging-related neuroinflammation (Shao et al., 2013). DRD2 couples to Gi (Missale et al., 1998) and is expressed in astrocytes in vivo (Khan et al., 2001). During aging, DRD2 expression is downregulated in the brain (Kaasinen et al., 2000), suggesting potential involvement of DRD2 in aging related neuroinflammation. In this study DRD2-deficient astrocytes were found to produce more proinflammatory mediators compared to wild-type astrocytes. Further, this effect appears to be mediated through a decrease in αB-crystallin (CRYAB) signaling, a small heat-shock protein known to negatively regulate pro-inflammatory mediator production and to display neuro-protective effect (Ousman et al., 2007). Interestingly, DRD2-deficient astrocytes also show robust GFAP upregulation, and a reactive morphology in the substantia nigra and the striatum of aged mice (Shao et al., 2013), suggesting the possible link between astrocytic GPCR signaling and age-related impairments in cognitive and motor function.
Models for Removing or Partially Removing Astrocytic Ca2+ Fluxes In Vivo
IP3R2 Germline and Conditional IP3R2 Knockout
Astrocytic Gq-GPCR/PLC/IP3 signaling is the most intensely studied pathway in the proposed modulation of neuronal activity and cerebral blood flow by astrocytes and has been the subject of numerous reviews (Haydon and Carmignoto, 2006; Agulhon et al., 2008, 2012; Fiacco et al., 2009; Halassa and Haydon, 2010; Hamilton and Attwell, 2010). The activation of this pathway results in the release of Ca2+ from IP3 receptor (IP3R) regulated intracellular stores in the endoplasmic reticulum. Astrocytes express only one of three subtypes of this receptor, IP3R type 2 (IP3R2) (Sharp et al., 1999; Holtzclaw et al., 2002; Foskett et al., 2007; Hertle and Yeckel, 2007). The first functional report to confirm this was published by Petravicz et al. (2008), in which the germline IP3R2 knockout mouse (generated by the Ju Chen lab; Li et al., 2005) was found to lack somatic increases in intracellular Ca2+ upon activation of multiple subtypes of Gq-GPCRs known to exist on astrocytes. Further experiments using this model have confirmed that astrocytes lack Gq-GCPR elicited, IP3R-dependent Ca2+ signals in their processes and soma (Agulhon et al., 2010; Di Castro et al., 2011; Panatier et al., 2011; Takata et al., 2011; Navarrete et al., 2012; Tamamushi et al., 2012; Nizar et al., 2013). The IP3R2 KO mouse is fertile, displays no overt alterations in brain development, and displays no obvious behavioral alterations (in contrast to the IP3R1 KO, the primary neuronal IP3R Matsumoto and Nagata, 1999). Due to these features, this mouse model has become one the most utilized mouse models in astrocyte research. Use of this model has led to novel findings (Panatier et al., 2011; Navarrete et al., 2012; Haustein et al., 2014; Perez-Alvarez et al., 2014), several of which have been contradictory with previously held theories concerning astrocyte-neuron communication and vascular control (Fiacco et al., 2007; Petravicz et al., 2008; Agulhon et al., 2010; Nizar et al., 2013; Takata et al., 2013; Bonder and McCarthy, 2014). Recently, evidence of residual Ca2+ signaling of a non-IP3R origin has been published using genetically encoded Ca2+ indicators (GECIs) in the IP3R2 KO model (Haustein et al., 2014; Kanemaru et al., 2014), further illustrating the model's usefulness for discovery of novel signaling events in astrocytes; importantly, there is no evidence suggesting that this residual Ca2+ signaling is regulated by neuronal activity.
Due to its restricted expression pattern in the CNS, a germline knockout of IP3R2 provides a clean and reliable model to block the release of intracellular Ca2+ in astrocytes elicited by Gq-GPCR activity in vivo. However, this model suffers in that it lacks tissue specificity, as do all germline knockout models. IP3R2 is expressed in multiple tissues outside the CNS including the heart (Li et al., 2005), pancreas (Orabi et al., 2012), lungs, liver, and kidneys (Fujino et al., 1995); this leads to potential confounding issues in the use of this model in vivo when assessing the role of astrocyte GPCR-dependent Ca2+ fluxes in behavior. Additionally, concerns over compensation due to the role of intracellular Ca2+ signaling during development have been raised regarding this model; however to date no evidence for altered development leading to compensation has been reported.
In an attempt to address some of these issues, a conditional IP3R2 knockout mouse model was developed by our laboratory. This model was generated by crossing the original floxed IP3R2 mouse developed by Dr. Ju Chen at UCSF (Li et al., 2005) to a GFAP-Cre recombinase mouse (Stehlik et al., 2006) to restrict the deletion of IP3R2 to GFAP+ cells in the CNS. This model recombines the floxed IP3R2 allele at a high rate (>80–85% GFAP+ cells lack IP3R2), significantly reducing the number of astrocytes responding to Gq-GPCR activation or neuronal activity in multiple brain regions (Petravicz et al., 2008; Chen et al., 2012). The use of the GFAP-Cre system spatially and temporally restricts the IP3R2 deletion to GFAP+ glia, thereby making it more appropriate for in vivo analysis such as behavioral characterization and reduces potential developmental compensation. Our lab recently published a behavioral analysis of the IP3R2 cKO mice and found no significant alteration to behavior (Petravicz et al., 2014). Most importantly, no alterations to learning and memory as assessed by the Morris Water Maze test were observed in these mice, despite previous literature proposing an important role for astrocyte IP3R-mediated Ca2+ signaling in hippocampal LTP. It is unlikely that developmental compensation occurs such that alternative ions substitute for Ca2+ in physiological processes or that global rewiring of neuronal circuits occurs to compensate for the loss of astrocyte Ca2+ fluxes. Nevertheless, to completely rule out developmental compensation, it will be necessary to prepare inducible IP3R2 cKO mice using mice expressing floxed IP3R2 and an astrocyte specific inducible Cre system.
IP3 Sponges
Activation of Phospholipase C beta (PLCβ) and sequential release of IP3 are the key steps in Gq-GPCR mediated intracellular Ca2+ elevations. Traditional approaches to abolish Gq-GPCR mediated Ca2+ elevation in astrocytes include chelating intracellular Ca2+ with BAPTA, or preventing IP3-mediated release of Ca2+ from ER using IP3R2 KO mice. Recently, the Pleckstrin Homology domain of PLC-like protein (p130PH), which binds cytosolic IP3 molecules, was used to suppress astrocytic Ca2+ signaling in vitro and ATP-induced astrocytic Ca2+ responses in vivo (Xie et al., 2010). In this study, p130PH was selectively expressed in cortical astrocytes in vivo using rAAV2/5 vector in combination with a specific astrocyte promoter, gfaABC(1)D (Lee et al., 2008; Xie et al., 2010). p130PH transduced astrocytes in the somatosensory cortex exhibited reduced amplitude and frequency of Ca2+ activity in response to direct ATP application on cortex compared to non-transduced astrocytes, whereas the characteristics of spontaneous Ca2+ activity in p130PH-transduced astrocytes remained unchanged (Xie et al., 2010). Therefore, p130PH serves as a more selective tool to suppress Gq-GPCR induced Ca2+ elevations without chelating Ca2+ activity in astrocytes completely. This system serves as a nice addition to IP3R2KO and IP3R2 cKO mouse lines to distinguish astrocyte functions regulated by ER released Ca2+ vs. channel mediated Ca2+ in vivo.
The expression of a fragment of the IP3 binding domain of IP3R1, an “IP3 sponge” (Iwasaki et al., 2002) can also be used to suppress IP3 induced Ca2+ release in astrocytes in vivo (Tanaka et al., 2013). These investigators found that suppression of astrocyte Ca2+ responses affected several behavioral responses, but no underlying evidence for alterations to synaptic transmission was found. Surprisingly, astrocytes expressing the “IP3 sponge” exhibited process retraction surrounding synapses, which was attributed to underlie the behavioral phenotypes. Recently, evidence that astrocyte processes retract in response to LTP-inducing stimuli in vivo was reported, and that this feature was lacking in IP3R2 KO mice (Perez-Alvarez et al., 2014). These findings appear to contradict those reported by Tanaka et al. (2013). Further comparison between these two methods of blocking astrocyte Ca2+ increases will be required to clarify this contradiction.
Blocking Vesicular Release from Astrocytes with dnSNARE or Tetanus Toxin
Ca2+ dependent release of neurotransmitters from astrocytes, termed “gliotransmission,” is one of the most important concepts presented in glial biology over the past several decades (Araque et al., 2014). While several mechanisms have been suggested to underlie gliotransmission, most studies support a process dependent on a vesicular release system (i.e., a SNARE dependent process) similar to that found in neurons (Zorec et al., 2012; Sahlender et al., 2014). To test the significance of SNARE-mediated gliotransmission to synaptic function, a mouse line that expresses a dominant negative form of SNARE (dnSNARE) in astrocytes was developed in our laboratory in 2005 (Pascual et al., 2005). This line was prepared by coinjecting three independent constructs (tetO-lacZ, tetO-dnSNARE, and tetO-eGFP) into fertilized zygotes. In dnSNARE transgenic mice, the expression of the cytosolic portion of the SNARE domain of synaptobrevin 2, lacZ, and eGFP are controlled by tetracycline regulatory system. When crossed with GFAP-tTA mice, the expression of dnSNARE, lacZ, and eGFP are independently controlled by doxycycline. In the absence of doxycycline, dnSNARE is expressed and interferes with SNARE-dependent vesicular release. A limitation of this model is that since dnSNARE was not directly tagged, there is no way in situ to verify that it is not expressed in cells other than astrocytes. Nevertheless, the dnSNARE mice have been used in a large number of studies to demonstrate a role for gliotransmission in synaptic transmission, synaptic plasticity, as well as behavior. However, a recent paper performed an in-depth analysis of dnSNARE mice and found that that the expression of dnSNARE was also expressed by a large population of neurons (Fujita et al., 2014). Given the critical role of the SNARE complex in neurotransmitter release, these findings bring into question the validity of this model and the findings obtained using this system.
Studies using dnSNARE mice suggest that SNARE-mediated astrocytic release of ATP and subsequent adenosine receptor activation regulates neuronal excitability and synaptic plasticity in many brain regions as well as modulate certain behaviors including sleep (Nam et al., 2012). The dnSNARE mice exhibit a weak sleep phenotype under basal conditions, as well as an attenuated “rebound” response to sleep deprivation (Halassa et al., 2009). Cortical slow oscillations, a rhythm characterizing non-rapid eye movement (non-REM) sleep was also found impaired in dnSNARE mice (Fellin et al., 2009). Further, the hippocampal dependent memory deficits produced by sleep deprivation were rescued in dnSNARE mice (Florian et al., 2011). These studies suggest that astrocytes release ATP in vesicular manner and that this plays an important role in sleep patterns and cortical oscillations.
Tenanus toxins (TeNTs) are known to interfere with synaptic vesicular release as well as other processes dependent on vesicular protein trafficking (Galli et al., 1994). Recently, a transgenic model system was developed using TeNT to block vesicular release in astrocytes in vivo (Lee et al., 2014). This transgenic model took advantage of both the tetracycline inducible regulatory system and the Cre-dependent inducible system to block vesicular release from astrocytes. A transgenic line was prepared that contained the tetracycline response element (TRE) followed by a floxed stop cassette and a cassette that when expressed led to the expression of eGFP tagged TeNT (TRE-loxP-STOP-loxP-TeNTΔ1-GFP). In this system, the expression of TeNT required Cre expression to remove the floxed stop cassette and tetracycline transactivator (tTA) to activate the TRE. TRE-loxP-STOP-loxP-TeNTΔ1-GFP mice were crossed with GFAP-tTA and GFAP-CreERt2 lines to create a triple transgenic mouse line. In the triple transgenic mice TeNT can be expressed in astrocytes following tamoxifen treatment; the expression of TeNT is suppressed in the presence of doxycycline. In this model, astrocyte expression of TeNT requires that two different transgenes driven by the GFAP promoter be expressed in the same astrocyte markedly increasing the probability of astrocyte specific expression. Further, as TeNT was directly tagged with eGFP, it is possible to identify all cells expressing TeNT. In contrast to the impaired sleep pattern in dnSNARE mice, astrocytic TeNT-expressing mice showed unaltered sleep patterns compared to triple transgenic mice without tamoxifen or double transgenic mice with tamoxifin (Lee et al., 2014). Studies using this mouse line indicate that basal synaptic transmission as well as normal short- and long-term plasticity in hippocampus in situ is not altered by the expression of TeNT (Lee et al., 2014); these findings bring into question the concept that astrocytes release gliotransmitters that modulate synaptic transmission and plasticity via a vesicular dependent process. Interestingly, the EEG power in low-gamma range during wakefulness was reduced, whereas the EEG power during but not non-REM sleep remain unchanged (Lee et al., 2014). These observations in sleep regulation from astrocytic TeNT-expressing mice do not match with those from dnSNARE mice.
Memory deficit was also detected in the mice expressing TeNT in astrocytes using novel object recognition test (Lee et al., 2014). Fast local field potential oscillations in the gamma frequency are closely correlated with many cognitive functions, including learning, memory storage and retrieval and attention (Basar-Eroglu et al., 1996). Astrocytic TeNT-expressing mice did not show deficit in other behavior tests that involves simpler form of memory processing or are less dependent on cortical processing. The reduction of gamma oscillation power and significant deficit in novel object recognition suggest that the fast neural circuit oscillations are regulated by astrocytes (Lee et al., 2014).
Mouse Models for Assessing Astrocyte Neurotransmitter Regulation
Glutamate Transporter Knockout Mouse Models
Astrocytes are responsible for 80–90% of glutamate reuptake in the brain (Tzingounis and Wadiche, 2007), and two of the five glutamate transporters (GluTs) are primarily expressed in astrocytes: Glt-1/EAAT2 and GLAST/EAAT1 (Danbolt, 2001; Huang and Bergles, 2004). There are currently germline knockout mouse models for both Glt-1 (Tanaka et al., 1997) and GLAST (Harada et al., 1998); however their use in vivo has been limited. The Glt-1 germline knockout mouse model suffers from reduced body weight, increased morbidity, and progressive neuronal death due to excitotoxocity (Tanaka et al., 1997). This limits the ability to conduct in vivo experiments, which typically require older mice, to examine the role of astrocytic glutamate reuptake via Glt-1. Recently, the heterozygous Glt-1 (Glt-1 Het) knockout model has become an attractive model for studying the role of Glt-1 in vivo. The Glt-1 Het model does not suffer from the more obvious adverse effects of the full Glt-1 knockout, and displays several interesting behavioral phenotypes and has allowed for the study of Glt-1 in several brain pathologies (Kiryk et al., 2008). The GLAST knockout mice are viable, enabling in vivo studies. GLAST is primarily expressed in the cerebellum and olfactory bulb (Regan et al., 2007), and this mouse model has led to interesting findings concerning the physiological (Martin et al., 2012; Schraven et al., 2012) and pathological (Karlsson et al., 2008, 2009) functions of GLAST. Recently, a Glt-1 floxed mouse model has been developed (Aida et al., 2015). Induced knockout of Glt-1 in adult animals resulted in development of repetitive behaviors and alterations to excitatory transmission due to reduced glutamate uptake. The floxed Glt-1 model when combined with inducible Cre systems will open up new avenues of research into the role of Glt-1 in vivo that were not possible due to the lethality of the germline Glt-1 KO.
Nitrous Oxide Synthase 2 Knockout
In the central nervous system nitric oxide serves a number of roles, and has been shown to act at glutamatergic synapses to enhance glutamate release (Garthwaite, 2008). While neurons in many brain regions are known to produce and release NO via nitric oxide synthase (nNOS), this does not account fully for the activity of NO in several brain regions where excitatory neurons lack nNOS expression. Astrocytes are known to express all three isoforms of NOS, and are the sole expressers of an inducible form of NOS (iNOS or NOS2) that is activated in response to physiological stress in a Ca2+ dependent manner (Murphy, 2000; Buskila et al., 2005; Amitai, 2010). Astrocyte-derived NO has been shown to enhance LTP of presynaptic afferents in the spinal cord, as well as enhance synaptic transmission in the neocortex in acute slice preparations (Ikeda and Murase, 2004; Buskila and Amitai, 2010). In vivo evidence for astrocytic-derived NO being a modulator of neuronal transmission has primarily come from the use of a NOS2 knockout mouse, in which it was discovered that deletion of the calmodulin-binding domain of NOS2 led to a net increase in overall NO concentrations in the brains of mutant mice. Interesting, the increase in NO originated from astrocytes through an alternate mechanism without alterations in the relative levels of NOS isoforms (Buskila et al., 2007). Further, these mice display stress and anxiety-related alterations to behavior suggesting a role for astrocyte-derived NO in the modulation of neural circuits (Abu-Ghanem et al., 2008). Currently, the role of astrocyte-derived NO in modulation of neuronal circuit activity remains an understudied area of glial research.
Astrocytic Serine-Racemase Conditional Knockout
D-serine has long been considered one of the three primary gliotransmitters along with ATP/adenosine and glutamate, with putative astrocyte derived D-serine reported to modulate neuronal NMDA receptors (Panatier et al., 2006; Billard, 2008; Oliet and Mothet, 2009; Martineau, 2013; Shigetomi et al., 2013b; Sild and Van Horn, 2013). The enzyme responsible for D-serine production (serine racemase, SR) was initially found to be primarily expressed by astrocytes with some modest expression in neurons (Schell et al., 1995; Mothet et al., 2005). However, more recent studies have called this expression pattern into question (Miya et al., 2008; Ding et al., 2011; Ehmsen et al., 2013). The most recent study of SR localization in mice and human brains finds that nearly all immunostaining for SR is found in neurons and not astrocytes (Balu et al., 2014). Recently, cell type specific knockouts of SR were generated to examine the relative contributions of astrocytes and neurons in the forebrain of mice (Benneyworth et al., 2012). The astrocyte specific knockout of SR led to a modest (~15%) reduction in SR expression, while the neuronal knockout in forebrain neurons reduced SR by much larger amounts (~65%). Further, the neuronal specific SR knockout displayed alterations to LTP at hippocampal synapses that were not found in the astrocyte SR knockout. These findings raise new questions in how astrocytes may be regulating D-serine availability in the brain, which the astrocyte specific SR knockout will be crucial to resolving.
Knockout Models for Astrocyte Membrane Channels
Kir4.1
One of the major roles in the CNS for astrocytes is the buffering of potassium ions in response to neuronal activity. Astrocytes express a variety of potassium channels, but among them Kir4.1 plays a predominant role in their K+ buffering capacity (Takumi et al., 1995; Higashi et al., 2001; Djukic et al., 2007). A germline full Kir4.1 knockout model has provided insight into the role of potassium buffering in response to hyperammonemic conditions (Stephan et al., 2012) and the channel's role in regulating astrocyte membrane potential during development (Seifert et al., 2009). An astrocyte conditional knockout model for Kir4.1 was generated by the McCarthy lab to provide a cleaner animal model alternative to the germline KO (Djukic et al., 2007). The conditional knockout astrocytes display reduced glutamate clearance and decreased resting membrane voltage, while neuronal plasticity was enhanced implicating Kir4.1 as an important mediator of extracellular potassium regulation. These finding have been confirmed in an in vivo study utilizing the Kir4.1 cKO, with the cKO mice found to have reduced capacity to regulate extracellular potassium levels compared to controls (Chever et al., 2010). Usage of this model in awake mice to examine the effect of altered potassium homeostasis may provide unique insights into how astrocytes regulate neuronal networks.
Connexins
Astrocytes in the brain exist not only as single units occupying a discrete domain, but also as a network of cells connected by connexin (Cx) gap junctions. Astrocytes express two major connexin proteins (Cx43 and Cx30) with germline and conditional knockout mouse models existing (Dermietzel et al., 2000; Teubner et al., 2003; Wiencken-Barger et al., 2007). These mouse models display a variety of alterations to behavior, synaptic transmission, metabolic support, and ion homeostasis (Giaume and Theis, 2010; Pannasch and Rouach, 2013). However, there are a large number of open questions concerning the role of gap junction communication and astrocyte networks in vivo. Currently, the only in vivo experiments in these models have involved behavioral studies, indicating a significant impact on neuronal circuit function (for an excellent review please see Pannasch and Rouach, 2013). Further exploration of neuronal activity in vivo utilizing these models, as well as generation of inducible knockout systems, represents novel avenues of research for understanding astrocyte network function.
Current Limitations in Genetically Targeting Astrocytes
At this time, there is not a single gene delivery system that can be used to exclusively express transgenes or recombine naïve genes in astrocytes. The common astrocyte marker proteins (GFAP, S100b, glutamine synthetase, aquaporin 4, connexin43, GLAST, GLT1, ALDH1L1) are either expressed in alternate subsets of mature CNS cells (Dunham et al., 1992; Zhuo et al., 2001; Su et al., 2004; Hachem et al., 2005; Regan et al., 2007; Donato et al., 2013), in progenitor CNS cells that give rise to multiple CNS cell types (Hartfuss et al., 2001; Casper and McCarthy, 2006), or are expressed outside the CNS (Jessen et al., 1990; Rinholm et al., 2007; Darlot et al., 2008; Meabon et al., 2012; Rutkovskiy et al., 2012; Jesus et al., 2014; Kato et al., 2014). The GFAP transcriptional regulatory unit (TRU) is probably the best characterized and most frequently used TRU for regulating gene expression in astrocytes and serves as a good example to illustrate the difficulties in targeting genes to astrocytes. The GFAP TRU is active in progenitor cells that give rise to astrocytes, neurons, and oligodendrocytes (Casper and McCarthy, 2006). Even in the mature CNS, certain populations of neurons express GFAP (Zhuo et al., 2001; Su et al., 2004; Regan et al., 2007). Further, in the periphery, non-myelinating peripheral glia (Jessen et al., 1990) as well as certain populations of non-neural cells (e.g., stellate cells in the liver) are GFAP+ (Lim et al., 2008) and will express transgenes driven by the GFAP TRU. To avoid transgene expression in progenitor cells, many laboratories have developed inducible gene regulatory systems (Casper and McCarthy, 2006; Hirrlinger et al., 2006; Mori et al., 2006). This approach circumvents gene expression in progenitors during development but does not affect the expression of transgenes in peripheral glia, adult stem cells or small populations of neurons that normally express GFAP. How much of a problem this presents depends on the question being asked. When studying a transgene in the mature CNS that can be assumed not to affect developmental processes nor lead to a peripheral phenotype it is reasonable to use many of the available astrocyte TRU to drive transgenes to astrocytes. For example, the GFAP TRU can be used to express eGFP, GCaMP, or DREADD receptors that must be activated by an exogenous ligand without preventative concern about developmental expression. However, when using the GFAP TRU to drive bioactive molecules such as DREADD receptors or inducible Cre recombinase, it is important to remain cognizant that in addition to astrocytes, peripheral GFAP+ cells, adult stem cells, and certain neurons will also be affected. Alternatively, when expressing molecules that innately affect biological processes (e.g., a dominant negative mutation, constitutively active signaling molecule or Cre recombinase), it is very important to consider the consequences of expression during development.
The above discussion assumes that the astrocyte TRU is acting with the fidelity of the endogenous TRU. Unfortunately, this is often not the case and is largely dependent on the genomic construct used to prepare the TRU. Most typically, investigators use a fragment of the TRU to drive transgene expression. As transgenes generally integrate somewhat randomly at active sites in the genome, the activity of surrounding genomic regulatory units can markedly affect transgene expression levels as well as the cells the transgene is expressed. This problem is markedly reduced using a BAC approach where very large genomic segments containing the TRU and inserted transgene are used to prepare transgenic lines. One final difficulty is that there are no astrocyte TRU systems that can be used to target subpopulations of astrocytes. Currently, the only way to genetically-manipulate subpopulations of astrocytes is to transduce these cells using viral vectors. Unfortunately, this generally restricts the size of the TRU used to target astrocytes and requires viral injection that may lead to subtle or striking changes in astrocyte function.
In summary, genetic manipulation of astrocytes is a very powerful tool for assessing the role of these cells in physiology, disease, and behavior. However, just as one has to verify the specificity of pharmacological reagents, great care must be used to insure the specificity of genetic manipulations.
Emerging Technologies for Manipulating Astrocytic Signaling In Vivo
Temporal Control of GPCR Signaling Using Optogenetic GPCRs
Optogenetically activated GPCR signaling is a reasonable alternative to ChR2 stimulation in astrocytes. There are two systems based on adrenergic receptors: Opto-alpha-1 (Gq linked) and opto-beta-1 (Gs linked) (Airan et al., 2009). Activation of these systems use similar experimental methods as optogenetics, but have the advantage of activating endogenous signaling cascades that exist in astrocytes. To date, these systems have not been tested for astrocytes in vivo, only in astroglia in culture (Figueiredo et al., 2014). While the Gq-linked opto-alpha-1 predictably elicited Ca2+ increases via release from intracellular stores, the Gs-linked opto-beta-1 was found to also trigger Ca2+ increases in a cyclic AMP-dependent manner. In culture systems, it has been shown that the Gβγ subunit of Gs-coupled GPCRs is capable of directly gating IP3Rs (Zeng et al., 2003). Recently, experiments in HEK293 cells discovered that IP3R2 complexes with Gαs and type 6 adenyl cyclase (AC6), and facilitates crosstalk between the two signaling pathways (Tovey et al., 2008, 2010). Further study into the mechanism behind increases in Ca2+ in astrocytes may lead to the identification of novel pathways regulating Ca2+ signaling.
Recently, the concept of using opsin based-pigments to develop optogenetic tools for modulating GPCR signaling was suggested (Koyanagi and Terakita, 2014). One of the candidates is melanopsin (OPN4), a Gq-coupled opsin that is originally found in a subtype of retinal ganglion cells (Hatori and Panda, 2010; Sexton et al., 2012). Several groups have used ectopic expression of OPN4 to control intracellular Ca2+ dynamics in neurons (Koizumi et al., 2013). In 2013, Karunarathne and colleagues used non-rhodopsin opsins to activate native Gq, Gi/o, and Gs signaling in localized regions of a single cell, and were able to gain spatial-temporal control over immune cell migration (Karunarathne et al., 2013b) as well as neurite initiation and extension (Karunarathne et al., 2013a). These studies suggest high potential of opsins as optogenetic GPCRs for in vivo astrocyte research.
Studying G-Protein Independent Signaling Using Biased DREADDs
One important aspect of GPCR signaling in astrocytes that has rarely been explored is the role of G-protein independent signaling. Endogenous GPCR activation not only initiates signaling via heterotrimeric G proteins, but also recruits proteins of the arrestin family, which act as scaffolding proteins and promote G protein-independent signaling (Pierce et al., 2002; Rajagopal et al., 2010; Shukla et al., 2011). Research has shown that arrestin 3 (β-arrestin 2) is expressed in astrocytes ex vivo (Bruchas et al., 2006; McLennan et al., 2008), and it is involved in kappa opioid receptor (KOR)-induced proliferation (McLennan et al., 2008; Miyatake et al., 2009), reduction of chemical-induced apoptosis (Zhu and Reiser, 2014), CXCR7 mediated inflammatory response (Odemis et al., 2012; Lipfert et al., 2013) and beta 2-adrenergic receptor (β2AR)-mediated glycogenolysis (Dong et al., 2001; Du et al., 2010) in astrocytes. As a scaffolding protein, β-arrestins also mediate internalization and ubiquitylation for many ion channels and transporters expressed in astrocytes (Shukla et al., 2011). As the list of signaling pathways that β-arrestins can regulate in astrocytes grows, it is important to dissect the relative contribution of G-protein independent signaling pathway to known functions of astrocytic GPCR signaling.
Recently, a modified Gq-DREADD that has strong biases toward arrestin-signaling was developed (Nakajima and Wess, 2012). This receptor was generated by introducing a point mutation within the highly conserved DRY motif [Rq(R165L)] of Gq-DREADD, which results in lack of ability to activate heterotrimetic G proteins. Therefore, CNO-induced Rq(R165L) activation has no effect on the levels of conventional second messages, but can promote CNO-dependent and arrestin-dependent signaling in biological systems (Nakajima and Wess, 2012). This novel GPCR represents an excellent tool to study the relative contribution of G protein-dependent and independent pathways in the known function of astrocytes, as well as reveal the physiological roles of astrocytic-arrestin signaling in vivo.
Conditional Gαq/Gα11 KO
GPCR KO mouse lines for all the known Gα subunits have been developed to analyze the physiological function of GPCR signaling in vivo (Offermanns, 1999), many of which show deficiencies in CNS-related physiology (Offermanns, 2001). Although these KO mouse lines are not astrocyte specific, inducible and conditional Gαq/Gα11 KO mice are available (Wettschureck et al., 2001) and can be combined with astrocytic specific Cre mouse line to selectively knock out Gq-GPCR signaling in astrocytes.
The conditional Gαq/Gα11 KO system was developed to study the role of Gαq/Gα11 signaling in specific tissues without developmental problems exhibited in the constitutive KO mouse line for both genes. This mouse line was developed using the Cre/LoxP system where the Gαq gene (gnaq) is conditionally inactivated in Gα11 KO (gna11–/–) mice, which do not have obvious behavior defect (Stanislaus et al., 1998). Therefore, mice homozygous for gnaqflox gene appear normal until Cre recombinase is introduced. The conditional Gαq/Gα11 KO mouse was first used with MLC2a-Cre to obtain cardiomyocyte-specific Gαq/Gα11 deficiency, resulting in a nearly complete recombination of gnaqflox in cardiomyocytes (Wettschureck et al., 2001).
In 2006, the Gαq/Gα11 cKO mice was crossed to mice that express Cre under the control of the promoter of the Ca2+/calmodulin-dependent protein kinase IIα gene (Camkcre4 mice) to generate forebrain specific and neuronal Gαq/Gα11 double KO mice (Wettschureck et al., 2004, 2006; Broicher et al., 2008), resulting in impaired endocannabinoid levels, increased seizure susceptibility (Wettschureck et al., 2006), and lack of maternal behavior in females (Wettschureck et al., 2004). The Gαq/Gα11 cKO line was also used to disrupt glial Gq/11 signaling in combination with Nestin-Cre mouse (Wettschureck et al., 2005), resulting in loss of Gq/11 in the neural stem cells that gives rise to both neurons and astrocytes. Although the Gq/11 signaling deficiency did not cause gross morphological changes in the developing nervous system, pups with insufficient Gq/11 signaling suffers from hypothalamic growth hormone deficiency and somatotroph hypoplasia, dwarfism, and anorexia (Wettschureck et al., 2005). Given the availability of astrocytic-specific Cre mice, it will be possible to isolate the contribution of astrocytic Gq/11 signaling in vivo.
Summary and Future Directions
There is no doubt that genetic tools will play an important role in understanding the role of astrocytes in physiology, behavior, and neurological disorders. Studies cited above provide strong evidence that astrocytes are doing much more than simply insulating synapses and providing nutrients to neurons. It is not surprising that findings using genetic tools may conflict with previous findings using pharmacological methods to perturb astrocyte function. It is also not surprising that different genetic approaches (e.g., dnSNARE and tetanus toxin expression) may yield different results. These conflicts require additional studies using multiple tools (pharmacological and genetic) to refine our understanding of astrocyte function. Currently, there are several important limitations in the genetic tools available for perturbing astrocyte function in vivo. First, we are extremely limited in the transcriptional units (promoters) available for expressing molecules in astrocytes. Currently, all available transcriptional units drive transgene expression in cells other than astrocytes. Further, during development, most astrocyte promoters drive gene expression in progenitor cells that give rise to neurons, oligodendrocytes, and astrocytes (Casper and McCarthy, 2006). Consequently, it is necessary to use inducible systems linked to astrocyte promoters to avoid transgene expression in progenitors during development. Second, when expressing transgenes that drive function, it is important to remember that the transgene may be overexpressed and targeted to cellular compartments not normally found. While this can be partially overcome using an inducible gene regulatory system, it is likely that this will remain a caveat until we know a great deal more about the cell machinery in astrocytes that target molecules to specific cellular compartments. Third, we currently lack promoters that can be used to drive transgene expression in subtypes of astrocytes. Subtype specific astrocyte promoters will enable important advances with respect to the heterogeneity of astrocytes as well as their different functional roles.
In summary, while genetic manipulation of astrocytes is in its early stages of development, genetic models have already provided important insight into the role of astrocytes in physiology, behavior, and neurological diseases. It seems very likely that future advances in this field will depend largely on genetic approaches currently available as well as those under development.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. T. Kendall Harden for his generous help with the preparation of Figure 1. The work in McCarthy laboratory is supported by NINDS RO1-NS20212-26 and RO1-MH099564.
References
Abu-Ghanem, Y., Cohen, H., Buskila, Y., Grauer, E., and Amitai, Y. (2008). Enhanced stress reactivity in nitric oxide synthase type 2 mutant mice: findings in support of astrocytic nitrosative modulation of behavior. Neuroscience 156, 257–265. doi: 10.1016/j.neuroscience.2008.07.043
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ackman, J. B., Ramos, R. L., Sarkisian, M. R., and Loturco, J. J. (2007). Citron kinase is required for postnatal neurogenesis in the hippocampus. Dev. Neurosci. 29, 113–123. doi: 10.1159/000096216
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Agulhon, C., Boyt, K. M., Xie, A. X., Friocourt, F., Roth, B. L., and McCarthy, K. D. (2013). Modulation of the autonomic nervous system and behaviour by acute glial cell Gq protein-coupled receptor activation in vivo. J. Physiol. 591, 5599–5609. doi: 10.1113/jphysiol.2013.261289
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Agulhon, C., Fiacco, T. A., and McCarthy, K. D. (2010). Hippocampal short- and long-term plasticity are not modulated by astrocyte Ca2+ signaling. Science 327, 1250–1254. doi: 10.1126/science.1184821
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Agulhon, C., Petravicz, J., McMullen, A. B., Sweger, E. J., Minton, S. K., Taves, S. R., et al. (2008). What is the role of astrocyte calcium in neurophysiology? Neuron 59, 932–946. doi: 10.1016/j.neuron.2008.09.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Agulhon, C., Sun, M. Y., Murphy, T., Myers, T., Lauderdale, K., and Fiacco, T. A. (2012). Calcium signaling and gliotransmission in normal vs. reactive astrocytes. Front. Pharmacol. 3:139. doi: 10.3389/fphar.2012.00139
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Aida, T., Yoshida, J., Nomura, M., Tanimura, A., Iino, Y., Soma, M., et al. (2015). Astroglial glutamate transporter deficiency increases synaptic excitability and leads to pathological repetitive behaviors in mice. Neuropsychopharmacology. doi: 10.1038/npp.2015.26. [Epub ahead of print].
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Airan, R. D., Thompson, K. R., Fenno, L. E., Bernstein, H., and Deisseroth, K. (2009). Temporally precise in vivo control of intracellular signalling. Nature 458, 1025–1029. doi: 10.1038/nature07926
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Alexander, G. M., Rogan, S. C., Abbas, A. I., Armbruster, B. N., Pei, Y., Allen, J. A., et al. (2009). Remote control of neuronal activity in transgenic mice expressing evolved G protein-coupled receptors. Neuron 63, 27–39. doi: 10.1016/j.neuron.2009.06.014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Amir, R. E., Van Den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188. doi: 10.1038/13810
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Amitai, Y. (2010). Physiologic role for “inducible” nitric oxide synthase: a new form of astrocytic-neuronal interface. Glia 58, 1775–1781. doi: 10.1002/glia.21057
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Araque, A., Carmignoto, G., Haydon, P. G., Oliet, S. H. R., Robitaille, R., and Volterra, A. (2014). Gliotransmitters travel in time and space. Neuron 81, 728–739. doi: 10.1016/j.neuron.2014.02.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Armbruster, B. N., Li, X., Pausch, M. H., Herlitze, S., and Roth, B. L. (2007). Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc. Natl. Acad. Sci. U.S.A. 104, 5163–5168. doi: 10.1073/pnas.0700293104
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Aschauer, D. F., Kreuz, S., and Rumpel, S. (2013). Analysis of transduction efficiency, tropism and axonal transport of AAV serotypes 1, 2, 5, 6, 8 and 9 in the mouse brain. PLoS ONE 8:e76310. doi: 10.1371/journal.pone.0076310
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Atkinson, P. J., Dellovade, T., Albers, D., Von Schack, D., Saraf, K., Needle, E., et al. (2009). Sonic Hedgehog signaling in astrocytes is dependent on p38 mitogen-activated protein kinase and G-protein receptor kinase 2. J. Neurochem. 108, 1539–1549. doi: 10.1111/j.1471-4159.2009.05900.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ballas, N., Lioy, D. T., Grunseich, C., and Mandel, G. (2009). Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat. Neurosci. 12, 311–317. doi: 10.1038/nn.2275
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Balu, D. T., Takagi, S., Puhl, M. D., Benneyworth, M. A., and Coyle, J. T. (2014). D-serine and serine racemase are localized to neurons in the adult mouse and human forebrain. Cell. Mol. Neurobiol. 34, 419–435. doi: 10.1007/s10571-014-0027-z
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Basar-Eroglu, C., Struber, D., Schurmann, M., Stadler, M., and Basar, E. (1996). Gamma-band responses in the brain: a short review of psychophysiological correlates and functional significance. Int. J. Psychophysiol. 24, 101–112. doi: 10.1016/S0167-8760(96)00051-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bender, D., Holschbach, M., and Stocklin, G. (1994). Synthesis of n.c.a. carbon-11 labelled clozapine and its major metabolite clozapine-N-oxide and comparison of their biodistribution in mice. Nucl. Med. Biol. 21, 921–925. doi: 10.1016/0969-8051(94)90080-9
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Benesova, J., Hock, M., Butenko, O., Prajerova, I., Anderova, M., and Chvatal, A. (2009). Quantification of astrocyte volume changes during ischemia in situ reveals two populations of astrocytes in the cortex of GFAP/EGFP mice. J. Neurosci. Res. 87, 96–111. doi: 10.1002/jnr.21828
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Benneyworth, M. A., Li, Y., Basu, A. C., Bolshakov, V. Y., and Coyle, J. T. (2012). Cell selective conditional null mutations of serine racemase demonstrate a predominate localization in cortical glutamatergic neurons. Cell. Mol. Neurobiol. 32, 613–624. doi: 10.1007/s10571-012-9808-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Beppu, K., Sasaki, T., Tanaka, K. F., Yamanaka, A., Fukazawa, Y., Shigemoto, R., et al. (2014). Optogenetic countering of glial acidosis suppresses glial glutamate release and ischemic brain damage. Neuron 81, 314–320. doi: 10.1016/j.neuron.2013.11.011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Billard, J. M. (2008). D-serine signalling as a prominent determinant of neuronal-glial dialogue in the healthy and diseased brain. J. Cell. Mol. Med. 12, 1872–1884. doi: 10.1111/j.1582-4934.2008.00315.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bonder, D. E., and McCarthy, K. D. (2014). Astrocytic Gq-GPCR-Linked IP3R-Dependent Ca2+ signaling does not mediate neurovascular coupling in mouse visual cortex in vivo. J. Neurosci. 34, 13139–13150. doi: 10.1523/JNEUROSCI.2591-14.2014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Broicher, T., Wettschureck, N., Munsch, T., Coulon, P., Meuth, S. G., Kanyshkova, T., et al. (2008). Muscarinic ACh receptor-mediated control of thalamic activity via G(q)/G (11)-family G-proteins. Pflugers Arch. 456, 1049–1060. doi: 10.1007/s00424-008-0473-x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bruchas, M. R., Macey, T. A., Lowe, J. D., and Chavkin, C. (2006). Kappa opioid receptor activation of p38 MAPK is GRK3- and arrestin-dependent in neurons and astrocytes. J. Biol. Chem. 281, 18081–18089. doi: 10.1074/jbc.M513640200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bull, C., Freitas, K. C., Zou, S., Poland, R. S., Syed, W. A., Urban, D. J., et al. (2014). Rat nucleus accumbens core astrocytes modulate reward and the motivation to self-administer ethanol after abstinence. Neuropsychopharmacology 39, 2835–2845. doi: 10.1038/npp.2014.135
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Burkhardt, U., Wojcik, B., Zimmermann, M., and Klein, J. (2014). Phospholipase D is a target for inhibition of astroglial proliferation by ethanol. Neuropharmacology 79, 1–9. doi: 10.1016/j.neuropharm.2013.11.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bushong, E. A., Martone, M. E., Jones, Y. Z., and Ellisman, M. H. (2002). Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J. Neurosci. 22, 183–192.
Buskila, Y., Abu-Ghanem, Y., Levi, Y., Moran, A., Grauer, E., and Amitai, Y. (2007). Enhanced astrocytic nitric oxide production and neuronal modifications in the neocortex of a NOS2 mutant mouse. PLoS ONE 2:e843. doi: 10.1371/journal.pone.0000843
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Buskila, Y., and Amitai, Y. (2010). Astrocytic iNOS-dependent enhancement of synaptic release in mouse neocortex. J. Neurophysiol. 103, 1322–1328. doi: 10.1152/jn.00676.2009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Buskila, Y., Farkash, S., Hershfinkel, M., and Amitai, Y. (2005). Rapid and reactive nitric oxide production by astrocytes in mouse neocortical slices. Glia 52, 169–176. doi: 10.1002/glia.20217
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cao, X., Li, L. P., Wang, Q., Wu, Q., Hu, H. H., Zhang, M., et al. (2013). Astrocyte-derived ATP modulates depressive-like behaviors. Nat. Med. 19, 773–777. doi: 10.1038/nm.3162
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Casper, K. B., and McCarthy, K. D. (2006). GFAP-positive progenitor cells produce neurons and oligodendrocytes throughout the CNS. Mol. Cell. Neurosci. 31, 676–684. doi: 10.1016/j.mcn.2005.12.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen, N., Sugihara, H., Sharma, J., Perea, G., Petravicz, J., Le, C., et al. (2012). Nucleus basalis-enabled stimulus-specific plasticity in the visual cortex is mediated by astrocytes. Proc. Natl. Acad. Sci. U.S.A. 109, E2832–E2841. doi: 10.1073/pnas.1206557109
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chever, O., Djukic, B., McCarthy, K. D., and Amzica, F. (2010). Implication of Kir4.1 channel in excess potassium clearance: an in vivo study on anesthetized glial-conditional Kir4.1 knock-out mice. J. Neurosci. 30, 15769–15777. doi: 10.1523/JNEUROSCI.2078-10.2010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Danbolt, N. C. (2001). Glutamate uptake. Prog. Neurobiol. 65, 1–105. doi: 10.1016/S0301-0082(00)00067-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Danussi, C., Akavia, U. D., Niola, F., Jovic, A., Lasorella, A., Pe'er, D., et al. (2013). RHPN2 drives mesenchymal transformation in malignant glioma by triggering RhoA activation. Cancer Res. 73, 5140–5150. doi: 10.1158/0008-5472.CAN-13-1168-T
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Darlot, F., Artuso, A., Lautredou-Audouy, N., and Casellas, D. (2008). Topology of Schwann cells and sympathetic innervation along preglomerular vessels: a confocal microscopic study in protein S100B/EGFP transgenic mice. Am. J. Physiol. Renal Physiol. 295, F1142–F1148. doi: 10.1152/ajprenal.00599.2007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dermietzel, R., Gao, Y., Scemes, E., Vieira, D., Urban, M., Kremer, M., et al. (2000). Connexin43 null mice reveal that astrocytes express multiple connexins. Brain Res. Brain Res. Rev. 32, 45–56. doi: 10.1016/S0165-0173(99)00067-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Devaraju, P., Sun, M. Y., Myers, T. L., Lauderdale, K., and Fiacco, T. A. (2013). Astrocytic group I mGluR-dependent potentiation of astrocytic glutamate and potassium uptake. J. Neurophysiol. 109, 2404–2414. doi: 10.1152/jn.00517.2012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Di Castro, M. A., Chuquet, J., Liaudet, N., Bhaukaurally, K., Santello, M., Bouvier, D., et al. (2011). Local Ca2+ detection and modulation of synaptic release by astrocytes. Nat. Neurosci. 14, 1276–1284. doi: 10.1038/nn.2929
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ding, X., Ma, N., Nagahama, M., Yamada, K., and Semba, R. (2011). Localization of D-serine and serine racemase in neurons and neuroglias in mouse brain. Neurol. Sci. 32, 263–267. doi: 10.1007/s10072-010-0422-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
DiNuzzo, M., Mangia, S., Maraviglia, B., and Giove, F. (2013). Regulatory mechanisms for glycogenolysis and K+ uptake in brain astrocytes. Neurochem. Int. 63, 458–464. doi: 10.1016/j.neuint.2013.08.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Djukic, B., Casper, K. B., Philpot, B. D., Chin, L. S., and McCarthy, K. D. (2007). Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J. Neurosci. 27, 11354–11365. doi: 10.1523/JNEUROSCI.0723-07.2007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Donato, R., Cannon, B. R., Sorci, G., Riuzzi, F., Hsu, K., Weber, D. J., et al. (2013). Functions of S100 proteins. Curr. Mol. Med. 13, 24–57. doi: 10.2174/156652413804486214
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dong, S., Rogan, S. C., and Roth, B. L. (2010). Directed molecular evolution of DREADDs: a generic approach to creating next-generation RASSLs. Nat. Protoc. 5, 561–573. doi: 10.1038/nprot.2009.239
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dong, X., Han, S., Zylka, M. J., Simon, M. I., and Anderson, D. J. (2001). A diverse family of GPCRs expressed in specific subsets of nociceptive sensory neurons. Cell 106, 619–632. doi: 10.1016/S0092-8674(01)00483-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Du, T., Li, B., Li, H., Li, M., Hertz, L., and Peng, L. (2010). Signaling pathways of isoproterenol-induced ERK1/2 phosphorylation in primary cultures of astrocytes are concentration-dependent. J. Neurochem. 115, 1007–1023. doi: 10.1111/j.1471-4159.2010.06995.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Duffy, H. S., Ashton, A. W., O'Donnell, P., Coombs, W., Taffet, S. M., Delmar, M., et al. (2004). Regulation of connexin43 protein complexes by intracellular acidification. Circ. Res. 94, 215–222. doi: 10.1161/01.RES.0000113924.06926.11
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dunham, B., Liu, S., Taffet, S., Trabka-Janik, E., Delmar, M., Petryshyn, R., et al. (1992). Immunolocalization and expression of functional and nonfunctional cell-to-cell channels from wild-type and mutant rat heart connexin43 cDNA. Circ. Res. 70, 1233–1243. doi: 10.1161/01.RES.70.6.1233
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ehmsen, J. T., Ma, T. M., Sason, H., Rosenberg, D., Ogo, T., Furuya, S., et al. (2013). D-serine in glia and neurons derives from 3-phosphoglycerate dehydrogenase. J. Neurosci. 33, 12464–12469. doi: 10.1523/JNEUROSCI.4914-12.2013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fellin, T., Halassa, M. M., Terunuma, M., Succol, F., Takano, H., Frank, M., et al. (2009). Endogenous nonneuronal modulators of synaptic transmission control cortical slow oscillations in vivo. Proc. Natl. Acad. Sci. U.S.A. 106, 15037–15042. doi: 10.1073/pnas.0906419106
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fiacco, T. A., Agulhon, C., and McCarthy, K. D. (2009). Sorting out astrocyte physiology from pharmacology. Annu. Rev. Pharmacol. Toxicol. 49, 151–174. doi: 10.1146/annurev.pharmtox.011008.145602
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fiacco, T. A., Agulhon, C., Taves, S. R., Petravicz, J., Casper, K. B., Dong, X., et al. (2007). Selective stimulation of astrocyte calcium in situ does not affect neuronal excitatory synaptic activity. Neuron 54, 611–626. doi: 10.1016/j.neuron.2007.04.032
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Figueiredo, M., Lane, S., Stout, R. F. Jr. Liu, B., Parpura, V., Teschemacher, A. G., et al. (2014). Comparative analysis of optogenetic actuators in cultured astrocytes. Cell Calcium 56, 208–214. doi: 10.1016/j.ceca.2014.07.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Florian, C., Vecsey, C. G., Halassa, M. M., Haydon, P. G., and Abel, T. (2011). Astrocyte-derived adenosine and A1 receptor activity contribute to sleep loss-induced deficits in hippocampal synaptic plasticity and memory in mice. J. Neurosci. 31, 6956–6962. doi: 10.1523/JNEUROSCI.5761-10.2011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Foskett, J. K., White, C., Cheung, K. H., and Mak, D. O. (2007). Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 87, 593–658. doi: 10.1152/physrev.00035.2006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fujino, I., Yamada, N., Miyawaki, A., Hasegawa, M., Furuichi, T., and Mikoshiba, K. (1995). Differential expression of type 2 and type 3 inositol 1,4,5-trisphosphate receptor mRNAs in various mouse tissues: in situ hybridization study. Cell Tissue Res. 280, 201–210. doi: 10.1007/s004410050345
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fujita, T., Chen, M. J., Li, B., Smith, N. A., Peng, W., Sun, W., et al. (2014). Neuronal transgene expression in dominant-negative SNARE mice. J. Neurosci. 34, 16594–16604. doi: 10.1523/JNEUROSCI.2585-14.2014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Galli, T., Chilcote, T., Mundigl, O., Binz, T., Niemann, H., and De Camilli, P. (1994). Tetanus toxin-mediated cleavage of cellubrevin impairs exocytosis of transferrin receptor-containing vesicles in CHO cells. J. Cell Biol. 125, 1015–1024. doi: 10.1083/jcb.125.5.1015
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Garthwaite, J. (2008). Concepts of neural nitric oxide-mediated transmission. Eur. J. Neurosci. 27, 2783–2802. doi: 10.1111/j.1460-9568.2008.06285.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gholizadeh, S., Tharmalingam, S., MacAldaz, M. E., and Hampson, D. R. (2013). Transduction of the central nervous system after intracerebroventricular injection of adeno-associated viral vectors in neonatal and juvenile mice. Hum. Gene Ther. Methods 24, 205–213. doi: 10.1089/hgtb.2013.076
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Giaume, C., and Theis, M. (2010). Pharmacological and genetic approaches to study connexin-mediated channels in glial cells of the central nervous system. Brain Res. Rev. 63, 160–176. doi: 10.1016/j.brainresrev.2009.11.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gourine, A. V., Kasymov, V., Marina, N., Tang, F., Figueiredo, M. F., Lane, S., et al. (2010). Astrocytes control breathing through pH-dependent release of ATP. Science 329, 571–575. doi: 10.1126/science.1190721
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Guettier, J. M., Gautam, D., Scarselli, M., Ruiz De Azua, I., Li, J. H., Rosemond, E., et al. (2009). A chemical-genetic approach to study G protein regulation of beta cell function in vivo. Proc. Natl. Acad. Sci. U.S.A. 106, 19197–19202. doi: 10.1073/pnas.0906593106
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Guy, J., Gan, J., Selfridge, J., Cobb, S., and Bird, A. (2007). Reversal of neurological defects in a mouse model of Rett syndrome. Science 315, 1143–1147. doi: 10.1126/science.1138389
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ha, J. S., Dho, S. H., Youm, T. H., Kwon, K. S., and Park, S. S. (2014). Astrocytic phospholipase A2 contributes to neuronal glutamate toxicity. Brain Res. 1590, 97–106. doi: 10.1016/j.brainres.2014.10.015
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hachem, S., Aguirre, A., Vives, V., Marks, A., Gallo, V., and Legraverend, C. (2005). Spatial and temporal expression of S100B in cells of oligodendrocyte lineage. Glia 51, 81–97. doi: 10.1002/glia.20184
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Halassa, M. M., Florian, C., Fellin, T., Munoz, J. R., Lee, S. Y., Abel, T., et al. (2009). Astrocytic modulation of sleep homeostasis and cognitive consequences of sleep loss. Neuron 61, 213–219. doi: 10.1016/j.neuron.2008.11.024
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Halassa, M. M., and Haydon, P. G. (2010). Integrated brain circuits: astrocytic networks modulate neuronal activity and behavior. Annu. Rev. Physiol. 72, 335–355. doi: 10.1146/annurev-physiol-021909-135843
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hamilton, N. B., and Attwell, D. (2010). Do astrocytes really exocytose neurotransmitters? Nat. Rev. Neurosci. 11, 227–238. doi: 10.1038/nrn2803
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Han, J., Kesner, P., Metna-Laurent, M., Duan, T., Xu, L., Georges, F., et al. (2012). Acute cannabinoids impair working memory through astroglial CB1 receptor modulation of hippocampal LTD. Cell 148, 1039–1050. doi: 10.1016/j.cell.2012.01.037
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Harada, T., Harada, C., Watanabe, M., Inoue, Y., Sakagawa, T., Nakayama, N., et al. (1998). Functions of the two glutamate transporters GLAST and GLT-1 in the retina. Proc. Natl. Acad. Sci. U.S.A. 95, 4663–4666. doi: 10.1073/pnas.95.8.4663
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hartfuss, E., Galli, R., Heins, N., and Gotz, M. (2001). Characterization of CNS precursor subtypes and radial glia. Dev. Biol. 229, 15–30. doi: 10.1006/dbio.2000.9962
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hatori, M., and Panda, S. (2010). The emerging roles of melanopsin in behavioral adaptation to light. Trends Mol. Med. 16, 435–446. doi: 10.1016/j.molmed.2010.07.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Haustein, M. D., Kracun, S., Lu, X. H., Shih, T., Jackson-Weaver, O., Tong, X., et al. (2014). Conditions and constraints for astrocyte calcium signaling in the hippocampal mossy fiber pathway. Neuron 82, 413–429. doi: 10.1016/j.neuron.2014.02.041
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Haydon, P. G., and Carmignoto, G. (2006). Astrocyte control of synaptic transmission and neurovascular coupling. Physiol. Rev. 86, 1009–1031. doi: 10.1152/physrev.00049.2005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hertle, D. N., and Yeckel, M. F. (2007). Distribution of inositol-1,4,5-trisphosphate receptor isotypes and ryanodine receptor isotypes during maturation of the rat hippocampus. Neuroscience 150, 625–638. doi: 10.1016/j.neuroscience.2007.09.058
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Higashi, K., Fujita, A., Inanobe, A., Tanemoto, M., Doi, K., Kubo, T., et al. (2001). An inwardly rectifying K(+) channel, Kir4.1, expressed in astrocytes surrounds synapses and blood vessels in brain. Am. J. Physiol. Cell Physiol. 281, C922–C931.
Hirrlinger, P. G., Scheller, A., Braun, C., Hirrlinger, J., and Kirchhoff, F. (2006). Temporal control of gene recombination in astrocytes by transgenic expression of the tamoxifen-inducible DNA recombinase variant CreERT2. Glia 54, 11–20. doi: 10.1002/glia.20342
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Holtje, M., Hoffmann, A., Hofmann, F., Mucke, C., Grosse, G., Van Rooijen, N., et al. (2005). Role of Rho GTPase in astrocyte morphology and migratory response during in vitro wound healing. J. Neurochem. 95, 1237–1248. doi: 10.1111/j.1471-4159.2005.03443.x
Holtzclaw, L. A., Pandhit, S., Bare, D. J., Mignery, G. A., and Russell, J. T. (2002). Astrocytes in adult rat brain express type 2 inositol 1,4,5-trisphosphate receptors. Glia 39, 69–84. doi: 10.1002/glia.10085
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Huang, Y. H., and Bergles, D. E. (2004). Glutamate transporters bring competition to the synapse. Curr. Opin. Neurobiol. 14, 346–352. doi: 10.1016/j.conb.2004.05.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ikeda, H., and Murase, K. (2004). Glial nitric oxide-mediated long-term presynaptic facilitation revealed by optical imaging in rat spinal dorsal horn. J. Neurosci. 24, 9888–9896. doi: 10.1523/JNEUROSCI.2608-04.2004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Iwasaki, H., Chiba, K., Uchiyama, T., Yoshikawa, F., Suzuki, F., Ikeda, M., et al. (2002). Molecular characterization of the starfish inositol 1,4,5-trisphosphate receptor and its role during oocyte maturation and fertilization. J. Biol. Chem. 277, 2763–2772. doi: 10.1074/jbc.M108839200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jain, S., Ruiz De Azua, I., Lu, H., White, M. F., Guettier, J. M., and Wess, J. (2013). Chronic activation of a designer G(q)-coupled receptor improves beta cell function. J. Clin. Invest. 123, 1750–1762. doi: 10.1172/JCI66432
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jessen, K. R., Morgan, L., Stewart, H. J., and Mirsky, R. (1990). Three markers of adult non-myelin-forming Schwann cells, 217c(Ran-1), A5E3 and GFAP: development and regulation by neuron-Schwann cell interactions. Development 109, 91–103.
Jesus, T. T., Bernardino, R. L., Martins, A. D., Sa, R., Sousa, M., Alves, M. G., et al. (2014). Aquaporin-4 as a molecular partner of cystic fibrosis transmembrane conductance regulator in rat Sertoli cells. Biochem. Biophys. Res. Commun. 446, 1017–1021. doi: 10.1016/j.bbrc.2014.03.046
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jo, A., Park, H., Lee, S. H., Ahn, S. H., Kim, H. J., Park, E. M., et al. (2014). SHP-2 binds to caveolin-1 and regulates Src activity via competitive inhibition of CSK in response to H2O2 in astrocytes. PLoS ONE 9:e91582. doi: 10.1371/journal.pone.0091582
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kaasinen, V., Vilkman, H., Hietala, J., Nagren, K., Helenius, H., Olsson, H., et al. (2000). Age-related dopamine D2/D3 receptor loss in extrastriatal regions of the human brain. Neurobiol. Aging 21, 683–688. doi: 10.1016/S0197-4580(00)00149-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kalman, M., and Szabo, A. (2001). Immunohistochemical investigation of actin-anchoring proteins vinculin, talin and paxillin in rat brain following lesion: a moderate reaction, confined to the astroglia of brain tracts. Exp. Brain Res. 139, 426–434. doi: 10.1007/s002210100789
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kanemaru, K., Sekiya, H., Xu, M., Satoh, K., Kitajima, N., Yoshida, K., et al. (2014). In vivo visualization of subtle, transient, and local activity of astrocytes using an ultrasensitive Ca indicator. Cell Rep. 8, 311–318. doi: 10.1016/j.celrep.2014.05.056
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Karlsson, R. M., Tanaka, K., Heilig, M., and Holmes, A. (2008). Loss of glial glutamate and aspartate transporter (excitatory amino acid transporter 1) causes locomotor hyperactivity and exaggerated responses to psychotomimetics: rescue by haloperidol and metabotropic glutamate 2/3 agonist. Biol. Psychiatry 64, 810–814. doi: 10.1016/j.biopsych.2008.05.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Karlsson, R. M., Tanaka, K., Saksida, L. M., Bussey, T. J., Heilig, M., and Holmes, A. (2009). Assessment of glutamate transporter GLAST (EAAT1)-deficient mice for phenotypes relevant to the negative and executive/cognitive symptoms of schizophrenia. Neuropsychopharmacology 34, 1578–1589. doi: 10.1038/npp.2008.215
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Karunarathne, W. K., Giri, L., Kalyanaraman, V., and Gautam, N. (2013a). Optically triggering spatiotemporally confined GPCR activity in a cell and programming neurite initiation and extension. Proc. Natl. Acad. Sci. U.S.A. 110, E1565–E1574. doi: 10.1073/pnas.1220697110
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Karunarathne, W. K., Giri, L., Patel, A. K., Venkatesh, K. V., and Gautam, N. (2013b). Optical control demonstrates switch-like PIP3 dynamics underlying the initiation of immune cell migration. Proc. Natl. Acad. Sci. U.S.A. 110, E1575–E1583. doi: 10.1073/pnas.1220755110
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kato, J., Takai, Y., Hayashi, M. K., Kato, Y., Tanaka, M., Sohma, Y., et al. (2014). Expression and localization of aquaporin-4 in sensory ganglia. Biochem. Biophys. Res. Commun. 451, 562–567. doi: 10.1016/j.bbrc.2014.08.026
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kefas, B., Floyd, D. H., Comeau, L., Frisbee, A., Dominguez, C., Dipierro, C. G., et al. (2013). A miR-297/hypoxia/DGK-alpha axis regulating glioblastoma survival. Neuro-oncology 15, 1652–1663. doi: 10.1093/neuonc/not118
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Khan, Z. U., Koulen, P., Rubinstein, M., Grandy, D. K., and Goldman-Rakic, P. S. (2001). An astroglia-linked dopamine D2-receptor action in prefrontal cortex. Proc. Natl. Acad. Sci. U.S.A. 98, 1964–1969. doi: 10.1073/pnas.98.4.1964
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kiryk, A., Aida, T., Tanaka, K., Banerjee, P., Wilczynski, G. M., Meyza, K., et al. (2008). Behavioral characterization of GLT1 (+/-) mice as a model of mild glutamatergic hyperfunction. Neurotox. Res. 13, 19–30. doi: 10.1007/BF03033364
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kleinlogel, S., Feldbauer, K., Dempski, R. E., Fotis, H., Wood, P. G., Bamann, C., et al. (2011). Ultra light-sensitive and fast neuronal activation with the Ca(2)+-permeable channelrhodopsin CatCh. Nat. Neurosci. 14, 513–518. doi: 10.1038/nn.2776
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Koerber, J. T., Klimczak, R., Jang, J. H., Dalkara, D., Flannery, J. G., and Schaffer, D. V. (2009). Molecular evolution of adeno-associated virus for enhanced glial gene delivery. Mol. Ther. 17, 2088–2095. doi: 10.1038/mt.2009.184
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Koizumi, A., Tanaka, K. F., and Yamanaka, A. (2013). The manipulation of neural and cellular activities by ectopic expression of melanopsin. Neurosci. Res. 75, 3–5. doi: 10.1016/j.neures.2012.07.010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kong, M., Munoz, N., Valdivia, A., Alvarez, A., Herrera-Molina, R., Cardenas, A., et al. (2013). Thy-1-mediated cell-cell contact induces astrocyte migration through the engagement of alphaVbeta3 integrin and syndecan-4. Biochim. Biophys. Acta 1833, 1409–1420. doi: 10.1016/j.bbamcr.2013.02.013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Koyanagi, M., and Terakita, A. (2014). Diversity of animal opsin-based pigments and their optogenetic potential. Biochim. Biophys. Acta 1837, 710–716. doi: 10.1016/j.bbabio.2013.09.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lau, C. L., O'Shea, R. D., Broberg, B. V., Bischof, L., and Beart, P. M. (2011). The Rho kinase inhibitor Fasudil up-regulates astrocytic glutamate transport subsequent to actin remodelling in murine cultured astrocytes. Br. J. Pharmacol. 163, 533–545. doi: 10.1111/j.1476-5381.2011.01259.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lee, H. S., Ghetti, A., Pinto-Duarte, A., Wang, X., Dziewczapolski, G., Galimi, F., et al. (2014). Astrocytes contribute to gamma oscillations and recognition memory. Proc. Natl. Acad. Sci. U.S.A. 111, E3343–E3352. doi: 10.1073/pnas.1410893111
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lee, S. M., Cho, Y. S., Kim, T. H., Jin, M. U., Ahn, D. K., Noguchi, K., et al. (2012). An ultrastructural evidence for the expression of transient receptor potential ankyrin 1 (TRPA1) in astrocytes in the rat trigeminal caudal nucleus. J. Chem. Neuroanat. 45, 45–49. doi: 10.1016/j.jchemneu.2012.07.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lee, Y., Messing, A., Su, M., and Brenner, M. (2008). GFAP promoter elements required for region-specific and astrocyte-specific expression. Glia 56, 481–493. doi: 10.1002/glia.20622
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, D., Herault, K., Isacoff, E. Y., Oheim, M., and Ropert, N. (2012). Optogenetic activation of LiGluR-expressing astrocytes evokes anion channel-mediated glutamate release. J. Physiol. 590, 855–873. doi: 10.1113/jphysiol.2011.219345
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, X., Zima, A. V., Sheikh, F., Blatter, L. A., and Chen, J. (2005). Endothelin-1-induced arrhythmogenic Ca2+ signaling is abolished in atrial myocytes of inositol-1,4,5-trisphosphate(IP3)-receptor type 2-deficient mice. Circ. Res. 96, 1274–1281. doi: 10.1161/01.RES.0000172556.05576.4c
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lim, M. C., Maubach, G., and Zhuo, L. (2008). Glial fibrillary acidic protein splice variants in hepatic stellate cells–expression and regulation. Mol. Cells 25, 376–384.
Lin, J. Y. (2012). Optogenetic excitation of neurons with channelrhodopsins: light instrumentation, expression systems, and channelrhodopsin variants. Prog. Brain Res. 196, 29–47. doi: 10.1016/B978-0-444-59426-6.00002-1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lioy, D. T., Garg, S. K., Monaghan, C. E., Raber, J., Foust, K. D., Kaspar, B. K., et al. (2011). A role for glia in the progression of Rett's syndrome. Nature 475, 497–500. doi: 10.1038/nature10214
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lipfert, J., Odemis, V., and Engele, J. (2013). Grk2 is an essential regulator of CXCR7 signalling in astrocytes. Cell. Mol. Neurobiol. 33, 111–118. doi: 10.1007/s10571-012-9876-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Marsicano, G., Goodenough, S., Monory, K., Hermann, H., Eder, M., Cannich, A., et al. (2003). CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science 302, 84–88. doi: 10.1126/science.1088208
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Martin, C., Houitte, D., Guillermier, M., Petit, F., Bonvento, G., and Gurden, H. (2012). Alteration of sensory-evoked metabolic and oscillatory activities in the olfactory bulb of GLAST-deficient mice. Front. Neural Circuits 6:1. doi: 10.3389/fncir.2012.00001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Martineau, M. (2013). Gliotransmission: focus on exocytotic release of L-glutamate and D-serine from astrocytes. Biochem. Soc. Trans. 41, 1557–1561. doi: 10.1042/BST20130195
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Masaki, H., Wakayama, Y., Hara, H., Jimi, T., Unaki, A., Iijima, S., et al. (2010). Immunocytochemical studies of aquaporin 4, Kir4.1, and alpha1-syntrophin in the astrocyte endfeet of mouse brain capillaries. Acta Histochem. Cytochem. 43, 99–105. doi: 10.1267/ahc.10016
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Matsumoto, M., and Nagata, E. (1999). Type 1 inositol 1,4,5-trisphosphate receptor knock-out mice: their phenotypes and their meaning in neuroscience and clinical practice. J. Mol. Med. 77, 406–411. doi: 10.1007/s001090050370
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
McLennan, G. P., Kiss, A., Miyatake, M., Belcheva, M. M., Chambers, K. T., Pozek, J. J., et al. (2008). Kappa opioids promote the proliferation of astrocytes via Gbetagamma and beta-arrestin 2-dependent MAPK-mediated pathways. J. Neurochem. 107, 1753–1765. doi: 10.1111/j.1471-4159.2008.05745.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Meabon, J. S., Lee, A., Meeker, K. D., Bekris, L. M., Fujimura, R. K., Yu, C. E., et al. (2012). Differential expression of the glutamate transporter GLT-1 in pancreas. J. Histochem. Cytochem. 60, 139–151. doi: 10.1369/0022155411430095
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Miller, R. L., and Loewy, A. D. (2013). ENaC gamma-expressing astrocytes in the circumventricular organs, white matter, and ventral medullary surface: sites for Na+ regulation by glial cells. J. Chem. Neuroanat. 53, 72–80. doi: 10.1016/j.jchemneu.2013.10.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Missale, C., Nash, S. R., Robinson, S. W., Jaber, M., and Caron, M. G. (1998). Dopamine receptors: from structure to function. Physiol. Rev. 78, 189–225.
Miya, K., Inoue, R., Takata, Y., Abe, M., Natsume, R., Sakimura, K., et al. (2008). Serine racemase is predominantly localized in neurons in mouse brain. J. Comp. Neurol. 510, 641–654. doi: 10.1002/cne.21822
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Miyatake, M., Rubinstein, T. J., McLennan, G. P., Belcheva, M. M., and Coscia, C. J. (2009). Inhibition of EGF-induced ERK/MAP kinase-mediated astrocyte proliferation by mu opioids: integration of G protein and beta-arrestin 2-dependent pathways. J. Neurochem. 110, 662–674. doi: 10.1111/j.1471-4159.2009.06156.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Molotkov, D., Zobova, S., Arcas, J. M., and Khiroug, L. (2013). Calcium-induced outgrowth of astrocytic peripheral processes requires actin binding by Profilin-1. Cell Calcium 53, 338–348. doi: 10.1016/j.ceca.2013.03.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mori, T., Tanaka, K., Buffo, A., Wurst, W., Kuhn, R., and Gotz, M. (2006). Inducible gene deletion in astroglia and radial glia-a valuable tool for functional and lineage analysis. Glia 54, 21–34. doi: 10.1002/glia.20350
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mothet, J. P., Pollegioni, L., Ouanounou, G., Martineau, M., Fossier, P., and Baux, G. (2005). Glutamate receptor activation triggers a calcium-dependent and SNARE protein-dependent release of the gliotransmitter D-serine. Proc. Natl. Acad. Sci. U.S.A. 102, 5606–5611. doi: 10.1073/pnas.0408483102
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Murk, K., Blanco Suarez, E. M., Cockbill, L. M., Banks, P., and Hanley, J. G. (2013). The antagonistic modulation of Arp2/3 activity by N-WASP, WAVE2 and PICK1 defines dynamic changes in astrocyte morphology. J. Cell Sci. 126, 3873–3883. doi: 10.1242/jcs.125146
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Murphy, S. (2000). Production of nitric oxide by glial cells: regulation and potential roles in the CNS. Glia 29, 1–13. doi: 10.1002/(SICI)1098-1136(20000101)29:13.0.CO;2-N
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nagel, G., Brauner, M., Liewald, J. F., Adeishvili, N., Bamberg, E., and Gottschalk, A. (2005). Light activation of channelrhodopsin-2 in excitable cells of Caenorhabditis elegans triggers rapid behavioral responses. Curr. Biol. 15, 2279–2284. doi: 10.1016/j.cub.2005.11.032
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nagel, G., Szellas, T., Huhn, W., Kateriya, S., Adeishvili, N., Berthold, P., et al. (2003). Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc. Natl. Acad. Sci. U.S.A. 100, 13940–13945. doi: 10.1073/pnas.1936192100
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nakajima, K., and Wess, J. (2012). Design and functional characterization of a novel, arrestin-biased designer G protein-coupled receptor. Mol. Pharmacol. 82, 575–582. doi: 10.1124/mol.112.080358
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nam, H. W., McIver, S. R., Hinton, D. J., Thakkar, M. M., Sari, Y., Parkinson, F. E., et al. (2012). Adenosine and glutamate signaling in neuron-glial interactions: implications in alcoholism and sleep disorders. Alcohol. Clin. Exp. Res. 36, 1117–1125. doi: 10.1111/j.1530-0277.2011.01722.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Navarrete, M., Perea, G., Fernandez De Sevilla, D., Gomez-Gonzalo, M., Nunez, A., Martin, E. D., et al. (2012). Astrocytes mediate in vivo cholinergic-induced synaptic plasticity. PLoS Biol. 10:e1001259. doi: 10.1371/journal.pbio.1001259
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nichols, C. D., and Roth, B. L. (2009). Engineered G-protein coupled receptors are powerful tools to investigate biological processes and behaviors. Front. Mol. Neurosci. 2:16. doi: 10.3389/neuro.02.016.2009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nijboer, C. H., Heijnen, C. J., Degos, V., Willemen, H. L., Gressens, P., and Kavelaars, A. (2013). Astrocyte GRK2 as a novel regulator of glutamate transport and brain damage. Neurobiol. Dis. 54, 206–215. doi: 10.1016/j.nbd.2012.12.013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nizar, K., Uhlirova, H., Tian, P., Saisan, P. A., Cheng, Q., Reznichenko, L., et al. (2013). In vivo stimulus-induced vasodilation occurs without IP3 receptor activation and may precede astrocytic calcium increase. J. Neurosci. 33, 8411–8422. doi: 10.1523/JNEUROSCI.3285-12.2013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Odemis, V., Lipfert, J., Kraft, R., Hajek, P., Abraham, G., Hattermann, K., et al. (2012). The presumed atypical chemokine receptor CXCR7 signals through G(i/o) proteins in primary rodent astrocytes and human glioma cells. Glia 60, 372–381. doi: 10.1002/glia.22271
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Offermanns, S. (1999). New insights into the in vivo function of heterotrimeric G-proteins through gene deletion studies. Naunyn Schmiedebergs. Arch. Pharmacol. 360, 5–13. doi: 10.1007/s002109900030
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Offermanns, S. (2001). In vivo functions of heterotrimeric G-proteins: studies in Galpha-deficient mice. Oncogene 20, 1635–1642. doi: 10.1038/sj.onc.1204189
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Oliet, S. H., and Mothet, J. P. (2009). Regulation of N-methyl-D-aspartate receptors by astrocytic D-serine. Neuroscience 158, 275–283. doi: 10.1016/j.neuroscience.2008.01.071
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ono, K., Suzuki, H., Higa, M., Tabata, K., and Sawada, M. (2014). Glutamate release from astrocyte cell-line GL261 via alterations in the intracellular ion environment. J. Neural Transm. 121, 245–257. doi: 10.1007/s00702-013-1096-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Orabi, A. I., Luo, Y., Ahmad, M. U., Shah, A. U., Mannan, Z., Wang, D., et al. (2012). IP3 receptor type 2 deficiency is associated with a secretory defect in the pancreatic acinar cell and an accumulation of zymogen granules. PLoS ONE 7:e48465. doi: 10.1371/journal.pone.0048465
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ortinski, P. I., Dong, J., Mungenast, A., Yue, C., Takano, H., Watson, D. J., et al. (2010). Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nat. Neurosci. 13, 584–591. doi: 10.1038/nn.2535
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ousman, S. S., Tomooka, B. H., Van Noort, J. M., Wawrousek, E. F., O'Connor, K. C., Hafler, D. A., et al. (2007). Protective and therapeutic role for alphaB-crystallin in autoimmune demyelination. Nature 448, 474–479. doi: 10.1038/nature05935
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Panatier, A., Theodosis, D. T., Mothet, J. P., Touquet, B., Pollegioni, L., Poulain, D. A., et al. (2006). Glia-derived D-serine controls NMDA receptor activity and synaptic memory. Cell 125, 775–784. doi: 10.1016/j.cell.2006.02.051
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Panatier, A., Vallee, J., Haber, M., Murai, K. K., Lacaille, J. C., and Robitaille, R. (2011). Astrocytes are endogenous regulators of basal transmission at central synapses. Cell 146, 785–798. doi: 10.1016/j.cell.2011.07.022
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pannasch, U., and Rouach, N. (2013). Emerging role for astroglial networks in information processing: from synapse to behavior. Trends Neurosci. 36, 405–417. doi: 10.1016/j.tins.2013.04.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pascual, O., Casper, K. B., Kubera, C., Zhang, J., Revilla-Sanchez, R., Sul, J. Y., et al. (2005). Astrocytic purinergic signaling coordinates synaptic networks. Science 310, 113–116. doi: 10.1126/science.1116916
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pascual-Lucas, M., Fernandez-Lizarbe, S., Montesinos, J., and Guerri, C. (2014). LPS or ethanol triggers clathrin- and rafts/caveolae-dependent endocytosis of TLR4 in cortical astrocytes. J. Neurochem. 129, 448–462. doi: 10.1111/jnc.12639
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Perea, G., Yang, A., Boyden, E. S., and Sur, M. (2014). Optogenetic astrocyte activation modulates response selectivity of visual cortex neurons in vivo. Nat. Commun. 5, 3262. doi: 10.1038/ncomms4262
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Perez-Alvarez, A., Navarrete, M., Covelo, A., Martin, E. D., and Araque, A. (2014). Structural and functional plasticity of astrocyte processes and dendritic spine interactions. J. Neurosci. 34, 12738–12744. doi: 10.1523/JNEUROSCI.2401-14.2014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Persson, A., Lindwall, C., Curtis, M. A., and Kuhn, H. G. (2010). Expression of ezrin radixin moesin proteins in the adult subventricular zone and the rostral migratory stream. Neuroscience 167, 312–322. doi: 10.1016/j.neuroscience.2010.01.035
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Petravicz, J., Boyt, K. M., and McCarthy, K. D. (2014). Astrocyte IP3R2-dependent Ca(2+) signaling is not a major modulator of neuronal pathways governing behavior. Front. Behav. Neurosci. 8:384. doi: 10.3389/fnbeh.2014.00384
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Petravicz, J., Fiacco, T. A., and McCarthy, K. D. (2008). Loss of IP3 receptor-dependent Ca2+ increases in hippocampal astrocytes does not affect baseline CA1 pyramidal neuron synaptic activity. J. Neurosci. 28, 4967–4973. doi: 10.1523/JNEUROSCI.5572-07.2008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Petrosyan, H. A., Alessi, V., Singh, V., Hunanyan, A. S., Levine, J. M., and Arvanian, V. L. (2014). Transduction efficiency of neurons and glial cells by AAV-1, -5, -9, -rh10 and -hu11 serotypes in rat spinal cord following contusion injury. Gene Ther. 21, 991–1000. doi: 10.1038/gt.2014.74
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pfrieger, F. W., and Slezak, M. (2012). Genetic approaches to study glial cells in the rodent brain. Glia 60, 681–701. doi: 10.1002/glia.22283
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pierce, K. L., Premont, R. T., and Lefkowitz, R. J. (2002). Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 3, 639–650. doi: 10.1038/nrm908
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Porter, J. T., and McCarthy, K. D. (1997). Astrocytic neurotransmitter receptors in situ and in vivo. Prog. Neurobiol. 51, 439–455. doi: 10.1016/S0301-0082(96)00068-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rajagopal, S., Rajagopal, K., and Lefkowitz, R. J. (2010). Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat. Rev. Drug Discov. 9, 373–386. doi: 10.1038/nrd3024
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Regan, M. R., Huang, Y. H., Kim, Y. S., Dykes-Hoberg, M. I., Jin, L., Watkins, A. M., et al. (2007). Variations in promoter activity reveal a differential expression and physiology of glutamate transporters by glia in the developing and mature CNS. J. Neurosci. 27, 6607–6619. doi: 10.1523/JNEUROSCI.0790-07.2007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rinholm, J. E., Slettalokken, G., Marcaggi, P., Skare, O., Storm-Mathisen, J., and Bergersen, L. H. (2007). Subcellular localization of the glutamate transporters GLAST and GLT at the neuromuscular junction in rodents. Neuroscience 145, 579–591. doi: 10.1016/j.neuroscience.2006.12.041
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rogan, S. C., and Roth, B. L. (2011). Remote control of neuronal signaling. Pharmacol. Rev. 63, 291–315. doi: 10.1124/pr.110.003020
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rossello, X. S., Igbavboa, U., Weisman, G. A., Sun, G. Y., and Wood, W. G. (2012). AP-2beta regulates amyloid beta-protein stimulation of apolipoprotein E transcription in astrocytes. Brain Res. 1444, 87–95. doi: 10.1016/j.brainres.2012.01.017
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rutkovskiy, A., Stenslokken, K. O., Mariero, L. H., Skrbic, B., Amiry-Moghaddam, M., Hillestad, V., et al. (2012). Aquaporin-4 in the heart: expression, regulation and functional role in ischemia. Basic Res. Cardiol. 107, 280. doi: 10.1007/s00395-012-0280-6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sahlender, D. A., Savtchouk, I., and Volterra, A. (2014). What do we know about gliotransmitter release from astrocytes? Philos. Trans. R. Soc. Lond. B. Biol. Sci. 369, 20130592. doi: 10.1098/rstb.2013.0592
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sanchez, J. F., Sniderhan, L. F., Williamson, A. L., Fan, S., Chakraborty-Sett, S., and Maggirwar, S. B. (2003). Glycogen synthase kinase 3beta-mediated apoptosis of primary cortical astrocytes involves inhibition of nuclear factor kappaB signaling. Mol. Cell. Biol. 23, 4649–4662. doi: 10.1128/MCB.23.13.4649-4662.2003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sanchez-Fernandez, G., Cabezudo, S., Garcia-Hoz, C., Beninca, C., Aragay, A. M., Mayor, F. Jr., et al. (2014). Galphaq signalling: the new and the old. Cell. Signal. 26, 833–848. doi: 10.1016/j.cellsig.2014.01.010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Santama, N., Er, C. P., Ong, L. L., and Yu, H. (2004). Distribution and functions of kinectin isoforms. J. Cell Sci. 117, 4537–4549. doi: 10.1242/jcs.01326
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schell, M. J., Molliver, M. E., and Snyder, S. H. (1995). D-serine, an endogenous synaptic modulator: localization to astrocytes and glutamate-stimulated release. Proc. Natl. Acad. Sci. U.S.A. 92, 3948–3952. doi: 10.1073/pnas.92.9.3948
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schoenenberger, P., Scharer, Y. P., and Oertner, T. G. (2011). Channelrhodopsin as a tool to investigate synaptic transmission and plasticity. Exp. Physiol. 96, 34–39. doi: 10.1113/expphysiol.2009.051219
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schraven, S. P., Franz, C., Ruttiger, L., Lowenheim, H., Lysakowski, A., Stoffel, W., et al. (2012). Altered phenotype of the vestibular organ in GLAST-1 null mice. J. Assoc. Res. Otolaryngol. 13, 323–333. doi: 10.1007/s10162-011-0311-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Seifert, G., Huttmann, K., Binder, D. K., Hartmann, C., Wyczynski, A., Neusch, C., et al. (2009). Analysis of astroglial K+ channel expression in the developing hippocampus reveals a predominant role of the Kir4.1 subunit. J. Neurosci. 29, 7474–7488. doi: 10.1523/JNEUROSCI.3790-08.2009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Servitja, J. M., Masgrau, R., Pardo, R., Sarri, E., Von Eichel-Streiber, C., Gutkind, J. S., et al. (2003). Metabotropic glutamate receptors activate phospholipase D in astrocytes through a protein kinase C-dependent and Rho-independent pathway. Neuropharmacology 44, 171–180. doi: 10.1016/S0028-3908(02)00361-1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sexton, T., Buhr, E., and Van Gelder, R. N. (2012). Melanopsin and mechanisms of non-visual ocular photoreception. J. Biol. Chem. 287, 1649–1656. doi: 10.1074/jbc.R111.301226
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shao, W., Zhang, S. Z., Tang, M., Zhang, X. H., Zhou, Z., Yin, Y. Q., et al. (2013). Suppression of neuroinflammation by astrocytic dopamine D2 receptors via alphaB-crystallin. Nature 494, 90–94. doi: 10.1038/nature11748
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sharp, A. H., Nucifora, F. C. Jr. Blondel, O., Sheppard, C. A., Zhang, C., Snyder, S. H., et al. (1999). Differential cellular expression of isoforms of inositol 1,4,5-triphosphate receptors in neurons and glia in brain. J. Comp. Neurol. 406, 207–220.
Shigetomi, E., Bushong, E. A., Haustein, M. D., Tong, X., Jackson-Weaver, O., Kracun, S., et al. (2013a). Imaging calcium microdomains within entire astrocyte territories and endfeet with GCaMPs expressed using adeno-associated viruses. J. Gen. Physiol. 141, 633–647. doi: 10.1085/jgp.201210949
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shigetomi, E., Jackson-Weaver, O., Huckstepp, R. T., O'Dell, T. J., and Khakh, B. S. (2013b). TRPA1 channels are regulators of astrocyte basal calcium levels and long-term potentiation via constitutive D-serine release. J. Neurosci. 33, 10143–10153. doi: 10.1523/JNEUROSCI.5779-12.2013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shinohara, R., Thumkeo, D., Kamijo, H., Kaneko, N., Sawamoto, K., Watanabe, K., et al. (2012). A role for mDia, a Rho-regulated actin nucleator, in tangential migration of interneuron precursors. Nat. Neurosci. 15, 373–380, S371–S372. doi: 10.1038/nn.3020
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shukla, A. K., Xiao, K., and Lefkowitz, R. J. (2011). Emerging paradigms of beta-arrestin-dependent seven transmembrane receptor signaling. Trends Biochem. Sci. 36, 457–469. doi: 10.1016/j.tibs.2011.06.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sild, M., and Van Horn, M. R. (2013). Astrocytes use a novel transporter to fill gliotransmitter vesicles with D-serine: evidence for vesicular synergy. J. Neurosci. 33, 10193–10194. doi: 10.1523/JNEUROSCI.1665-13.2013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sorensen, S. D., and Conn, P. J. (2003). G protein-coupled receptor kinases regulate metabotropic glutamate receptor 5 function and expression. Neuropharmacology 44, 699–706. doi: 10.1016/S0028-3908(03)00053-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stanislaus, D., Janovick, J. A., Ji, T., Wilkie, T. M., Offermanns, S., and Conn, P. M. (1998). Gonadotropin and gonadal steroid release in response to a gonadotropin-releasing hormone agonist in Gqalpha and G11alpha knockout mice. Endocrinology 139, 2710–2717. doi: 10.1210/endo.139.6.5942
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stehlik, I., Caspersen, J. P., and Barrett, S. C. (2006). Spatial ecology of mating success in a sexually polymorphic plant. Proc. Biol. Sci. 273, 387–394. doi: 10.1098/rspb.2005.3317
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stephan, J., Haack, N., Kafitz, K. W., Durry, S., Koch, D., Hochstrate, P., et al. (2012). Kir4.1 channels mediate a depolarization of hippocampal astrocytes under hyperammonemic conditions in situ. Glia 60, 965–978. doi: 10.1002/glia.22328
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Su, M., Hu, H., Lee, Y., D'Azzo, A., Messing, A., and Brenner, M. (2004). Expression specificity of GFAP transgenes. Neurochem. Res. 29, 2075–2093. doi: 10.1007/s11064-004-6881-1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Takata, N., Mishima, T., Hisatsune, C., Nagai, T., Ebisui, E., Mikoshiba, K., et al. (2011). Astrocyte calcium signaling transforms cholinergic modulation to cortical plasticity in vivo. J. Neurosci. 31, 18155–18165. doi: 10.1523/JNEUROSCI.5289-11.2011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Takata, N., Nagai, T., Ozawa, K., Oe, Y., Mikoshiba, K., and Hirase, H. (2013). Cerebral blood flow modulation by Basal forebrain or whisker stimulation can occur independently of large cytosolic Ca2+ signaling in astrocytes. PLoS ONE 8:e66525. doi: 10.1371/journal.pone.0066525
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Takumi, T., Ishii, T., Horio, Y., Morishige, K., Takahashi, N., Yamada, M., et al. (1995). A novel ATP-dependent inward rectifier potassium channel expressed predominantly in glial cells. J. Biol. Chem. 270, 16339–16346. doi: 10.1074/jbc.270.27.16339
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tamamushi, S., Nakamura, T., Inoue, T., Ebisui, E., Sugiura, K., Bannai, H., et al. (2012). Type 2 inositol 1,4,5-trisphosphate receptor is predominantly involved in agonist-induced Ca(2+) signaling in Bergmann glia. Neurosci. Res. 74, 32–41. doi: 10.1016/j.neures.2012.06.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tanaka, K. F., Ahmari, S. E., Leonardo, E. D., Richardson-Jones, J. W., Budreck, E. C., Scheiffele, P., et al. (2010). Flexible Accelerated STOP Tetracycline operator-knockin (FAST): a versatile and efficient new gene modulating system. Biol. Psychiatry 67, 770–773. doi: 10.1016/j.biopsych.2009.12.020
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tanaka, K. F., Matsui, K., Sasaki, T., Sano, H., Sugio, S., Fan, K., et al. (2012). Expanding the repertoire of optogenetically targeted cells with an enhanced gene expression system. Cell Rep. 2, 397–406. doi: 10.1016/j.celrep.2012.06.011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tanaka, K., Watase, K., Manabe, T., Yamada, K., Watanabe, M., Takahashi, K., et al. (1997). Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 276, 1699–1702. doi: 10.1126/science.276.5319.1699
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tanaka, M., Shih, P. Y., Gomi, H., Yoshida, T., Nakai, J., Ando, R., et al. (2013). Astrocytic Ca2+ signals are required for the functional integrity of tripartite synapses. Mol. Brain 6:6. doi: 10.1186/1756-6606-6-6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Teubner, B., Michel, V., Pesch, J., Lautermann, J., Cohen-Salmon, M., Sohl, G., et al. (2003). Connexin30 (Gjb6)-deficiency causes severe hearing impairment and lack of endocochlear potential. Hum. Mol. Genet. 12, 13–21. doi: 10.1093/hmg/ddg001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tovey, S. C., Dedos, S. G., Rahman, T., Taylor, E. J., Pantazaka, E., and Taylor, C. W. (2010). Regulation of inositol 1,4,5-trisphosphate receptors by cAMP independent of cAMP-dependent protein kinase. J. Biol. Chem. 285, 12979–12989. doi: 10.1074/jbc.M109.096016
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tovey, S. C., Dedos, S. G., Taylor, E. J., Church, J. E., and Taylor, C. W. (2008). Selective coupling of type 6 adenylyl cyclase with type 2 IP3 receptors mediates direct sensitization of IP3 receptors by cAMP. J. Cell Biol. 183, 297–311. doi: 10.1083/jcb.200803172
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tsunematsu, T., Tabuchi, S., Tanaka, K. F., Boyden, E. S., Tominaga, M., and Yamanaka, A. (2013). Long-lasting silencing of orexin/hypocretin neurons using archaerhodopsin induces slow-wave sleep in mice. Behav. Brain Res. 255, 64–74. doi: 10.1016/j.bbr.2013.05.021
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tzingounis, A. V., and Wadiche, J. I. (2007). Glutamate transporters: confining runaway excitation by shaping synaptic transmission. Nat. Rev. Neurosci. 8, 935–947. doi: 10.1038/nrn2274
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Unichenko, P., Myakhar, O., and Kirischuk, S. (2012). Intracellular Na+ concentration influences short-term plasticity of glutamate transporter-mediated currents in neocortical astrocytes. Glia 60, 605–614. doi: 10.1002/glia.22294
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vedrenne, C., and Hauri, H. P. (2006). Morphogenesis of the endoplasmic reticulum: beyond active membrane expansion. Traffic 7, 639–646. doi: 10.1111/j.1600-0854.2006.00419.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Verkhratsky, A., Noda, M., Parpura, V., and Kirischuk, S. (2013). Sodium fluxes and astroglial function. Adv. Exp. Med. Biol. 961, 295–305. doi: 10.1007/978-1-4614-4756-6_25
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vitkovic, L., Aloyo, V. J., Maeda, S., Benzil, D. L., Bressler, J. P., and Hilt, D. C. (2005). Identification of myristoylated alanine-rich C kinase substrate (MARCKS) in astrocytes. Front. Biosci. 10:160–165. doi: 10.2741/1517
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, F., Smith, N. A., Xu, Q., Fujita, T., Baba, A., Matsuda, T., et al. (2012). Astrocytes modulate neural network activity by Ca(2)+-dependent uptake of extracellular K+. Sci. Signal. 5, ra26. doi: 10.1126/scisignal.2002334
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, F., Smith, N. A., Xu, Q., Goldman, S., Peng, W., Huang, J. H., et al. (2013). Photolysis of caged Ca2+ but not receptor-mediated Ca2+ signaling triggers astrocytic glutamate release. J. Neurosci. 33, 17404–17412. doi: 10.1523/JNEUROSCI.2178-13.2013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, H., Ubl, J. J., Stricker, R., and Reiser, G. (2002). Thrombin (PAR-1)-induced proliferation in astrocytes via MAPK involves multiple signaling pathways. Am. J. Physiol. Cell Physiol. 283, C1351–C1364. doi: 10.1152/ajpcell.00001.2002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wess, J., Nakajima, K., and Jain, S. (2013). Novel designer receptors to probe GPCR signaling and physiology. Trends Pharmacol. Sci. 34, 385–392. doi: 10.1016/j.tips.2013.04.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wettschureck, N., Moers, A., Hamalainen, T., Lemberger, T., Schutz, G., and Offermanns, S. (2004). Heterotrimeric G proteins of the Gq/11 family are crucial for the induction of maternal behavior in mice. Mol. Cell. Biol. 24, 8048–8054. doi: 10.1128/MCB.24.18.8048-8054.2004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wettschureck, N., Moers, A., Wallenwein, B., Parlow, A. F., Maser-Gluth, C., and Offermanns, S. (2005). Loss of Gq/11 family G proteins in the nervous system causes pituitary somatotroph hypoplasia and dwarfism in mice. Mol. Cell. Biol. 25, 1942–1948. doi: 10.1128/MCB.25.5.1942-1948.2005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wettschureck, N., Rutten, H., Zywietz, A., Gehring, D., Wilkie, T. M., Chen, J., et al. (2001). Absence of pressure overload induced myocardial hypertrophy after conditional inactivation of Galphaq/Galpha11 in cardiomyocytes. Nat. Med. 7, 1236–1240. doi: 10.1038/nm1101-1236
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wettschureck, N., Van Der Stelt, M., Tsubokawa, H., Krestel, H., Moers, A., Petrosino, S., et al. (2006). Forebrain-specific inactivation of Gq/G11 family G proteins results in age-dependent epilepsy and impaired endocannabinoid formation. Mol. Cell. Biol. 26, 5888–5894. doi: 10.1128/MCB.00397-06
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wiencken-Barger, A. E., Djukic, B., Casper, K. B., and McCarthy, K. D. (2007). A role for Connexin43 during neurodevelopment. Glia 55, 675–686. doi: 10.1002/glia.20484
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Woerner, B. M., Luo, J., Brown, K. R., Jackson, E., Dahiya, S. M., Mischel, P., et al. (2012). Suppression of G-protein-coupled receptor kinase 3 expression is a feature of classical GBM that is required for maximal growth. Mol. Cancer Res. 10, 156–166. doi: 10.1158/1541-7786.MCR-11-0411
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Xie, Y., Wang, T., Sun, G. Y., and Ding, S. (2010). Specific disruption of astrocytic Ca2+ signaling pathway in vivo by adeno-associated viral transduction. Neuroscience 170, 992–1003. doi: 10.1016/j.neuroscience.2010.08.034
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yamashita, A., Hamada, A., Suhara, Y., Kawabe, R., Yanase, M., Kuzumaki, N., et al. (2014). Astrocytic activation in the anterior cingulate cortex is critical for sleep disorder under neuropathic pain. Synapse 68, 235–247. doi: 10.1002/syn.21733
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yang, B., Li, S., Wang, H., Guo, Y., Gessler, D. J., Cao, C., et al. (2014). Global CNS transduction of adult mice by intravenously delivered rAAVrh.8 and rAAVrh.10 and nonhuman primates by rAAVrh.10. Mol. Ther. 22, 1299–1309. doi: 10.1038/mt.2014.68
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zeng, W., Mak, D. O., Li, Q., Shin, D. M., Foskett, J. K., and Muallem, S. (2003). A new mode of Ca2+ signaling by G protein-coupled receptors: gating of IP3 receptor Ca2+ release channels by Gbetagamma. Curr. Biol. 13, 872–876. doi: 10.1016/S0960-9822(03)00330-0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhu, Z., and Reiser, G. (2014). PAR-1 activation rescues astrocytes through the PI3K/Akt signaling pathway from chemically induced apoptosis that is exacerbated by gene silencing of beta-arrestin 1. Neurochem. Int. 67, 46–56. doi: 10.1016/j.neuint.2013.12.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhuo, L., Theis, M., Alvarez-Maya, I., Brenner, M., Willecke, K., and Messing, A. (2001). hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis 31, 85–94. doi: 10.1002/gene.10008
CrossRef Full Text | PubMed Abstract | Full Text | Google Scholar
Zorec, R., Araque, A., Carmignoto, G., Haydon, P. G., Verkhratsky, A., and Parpura, V. (2012). Astroglial excitability and gliotransmission: an appraisal of Ca2+ as a signalling route. ASN Neuro. 4, 103–119. doi: 10.1042/AN20110061
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: astrocyte, in vivo, GPCR signaling, DREADD, IP3R2 KO
Citation: Xie AX, Petravicz J and McCarthy KD (2015) Molecular approaches for manipulating astrocytic signaling in vivo. Front. Cell. Neurosci. 9:144. doi: 10.3389/fncel.2015.00144
Received: 05 February 2015; Accepted: 27 March 2015;
Published: 21 April 2015.
Edited by:
Tycho M. Hoogland, Netherlands Institute for Neuroscience, NetherlandsReviewed by:
Eliana Scemes, Albert Einstein College of Medicine, USAErlend Arnulf Nagelhus, University of Oslo, Norway
Copyright © 2015 Xie, Petravicz and McCarthy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ken D. McCarthy, Department of Pharmacology, School of Medicine, UNC-Chapel Hill, 120 Mason Farm Road, Chapel Hill, NC 27599-7365, USAa2VuX21jY2FydGh5QG1lZC51bmMuZWR1