 Kazuhiko Yamamuro
Kazuhiko Yamamuro Sohei Kimoto1
Sohei Kimoto1 Kenneth M. Rosen
Kenneth M. Rosen Manabu Makinodan
Manabu Makinodan- 1Department of Psychiatry, Faculty of Medicine, Nara Medical University, Kashihara, Japan
- 2BioAxone BioSciences Inc., Cambridge, MA, USA
While neurons have long been considered the major player in multiple brain functions such as perception, emotion, and memory, glial cells have been relegated to a far lesser position, acting as merely a “glue” to support neurons. Multiple lines of recent evidence, however, have revealed that glial cells such as oligodendrocytes, astrocytes, and microglia, substantially impact on neuronal function and activities and are significantly involved in the underlying pathobiology of psychiatric disorders. Indeed, a growing body of evidence indicates that glial cells interact extensively with neurons both chemically (e.g., through neurotransmitters, neurotrophic factors, and cytokines) and physically (e.g., through gap junctions), supporting a role for these cells as likely significant modifiers not only of neural function in brain development but also disease pathobiology. Since questions have lingered as to whether glial dysfunction plays a primary role in the biology of neuropsychiatric disorders or a role related solely to their support of neuronal physiology in these diseases, informative and predictive animal models have been developed over the last decade. In this article, we review recent findings uncovered using glia-specific genetically modified mice with which we can evaluate both the causation of glia dysfunction and its potential role in neuropsychiatric disorders such as autism and schizophrenia.
Introduction
Glial cells are the non-excitable supporting cells of the central nervous system (CNS) and classified mainly as oligodendrocytes, astrocytes, and microglia. These cells are typically smaller in size, but can be far more numerous than neurons in certain brain regions such as the cerebral cortex. Overall, the ratio between neurons, and glial cells in the human CNS is approximately 4:1 (Azevedo et al., 2009), with oligodendrocytes being the most abundant type of glial cells (75.6%), followed by astrocytes (17.3%) and microglia (6.5%) in human male brains (Pelvig et al., 2008). Glial cells clearly provide “support" for both cells such as neurons and structures such as blood vessels, but also can function to increase action potential conduction velocity via saltatory conduction from one node of Ranvier to the next in myelinated axons, and also the response to damage in the CNS via gliosis, a non-specific reactive change in glial cells associated with their proliferation or hypertrophy. Convergent lines of evidence from multiple studies in neuroimaging, postmortem brains, and genome-wide association studies (GWAS) have revealed a wide range of white matter abnormalities in schizophrenia (Dwork et al., 2007; Bernstein et al., 2015). Indeed, the implication of oligodendrocytes and myelin in schizophrenia has come from analyses of postmortem brains using microarray gene expression (Iwamoto et al., 2005; Katsel et al., 2005), protein expression (Dracheva et al., 2006), electron microscopic studies (Uranova et al., 2011), and neuroimaging (Kubicki et al., 2007).
Astrocytes are regarded as neuronal partners since they hold concerted cross-talk with neighboring neurons, which is crucial for normal brain function. Astrocytes are able to sense neurogenesis, development, and maturation of brain circuits and neuronal activity leading to both homeostatic changes and increased cellular crosstalk (Wang and Bordey, 2008; Parpura et al., 2012). The homeostatic responses of astrocytes includes increases in metabolic activity, the synthesis of a neuronal preferred energy substrate lactate, clearance of neurotransmitters and buffering of extracellular K+ ions to name but a few (Kimelberg, 2007; Wang and Bordey, 2008; Parpura and Verkhratsky, 2012). The existence of bidirectional communication between astrocytes and neurons during synaptic communication and function has been conceptualized as the “tripartite synapse” with its associated alterations in neuron–glia cross talk (Newman, 2003; Perea et al., 2009).
Oligodendrocytes, the myelin forming cells of the CNS, have a small round cell body and about 4–6 branching processes, which can myelinate up to 60 axons depending on the diameter (Miller, 2002). By ensheathing axons, mature oligodendrocytes provide critical insulation to facilitate axonal conduction by increasing the resistance and reducing the effective capacitance of the axonal membrane, resulting in faster conduction speed in myelinated axons compared to unmyelinated axons of the same diameter. In addition, recent studies have provided unique roles of oligodendrocytes, indicating that myelination of the axons cannot only influence neuronal properties in ways not previously considered, but also may be a key source of trophic and metabolic support for maintaining axonal integrity (Nave, 2010).
Microglia function as not only members of the innate immune system but also participate in synaptic modulation and maturation, learning, and memory processes. Importantly, they extend a broad network of ramified processes in the CNS parenchyma. After an injury to the brain, microglia rapidly extend highly active exploratory processes into the sites of injury without any corresponding cell body movement, potentially establishing a barrier between healthy and injured tissues in which microglia actively and constantly interact with neurons and astrocytes and survey the local environment (Davalos et al., 2005; Nimmerjahn et al., 2005). Moreover, microglia can directly regulate both synaptic function and synaptic maintenance in the absence of injury or neuroinflammation (Bessis et al., 2007; Wake et al., 2009).
The focus of this review is to examine what is known regarding the relationship between altered glial cell function and the pathobiology of psychiatric disorders, and whether glial dysfunction can play a causative role. First, we examine the monogenic disorder Rett syndrome (RTT) and examine the mouse model harboring a mutant methyl-CpG binding protein 2 (MeCP2) gene where neuronal and glial biology have been extensively investigated (Table 1). Subsequently, we examine several other mouse models for schizophrenia (Table 2) and how glial cell function or dysfunction contributes both to the phenotype and pathophysiology.

TABLE 1. Summary of mutant MeCP2 mouse models.
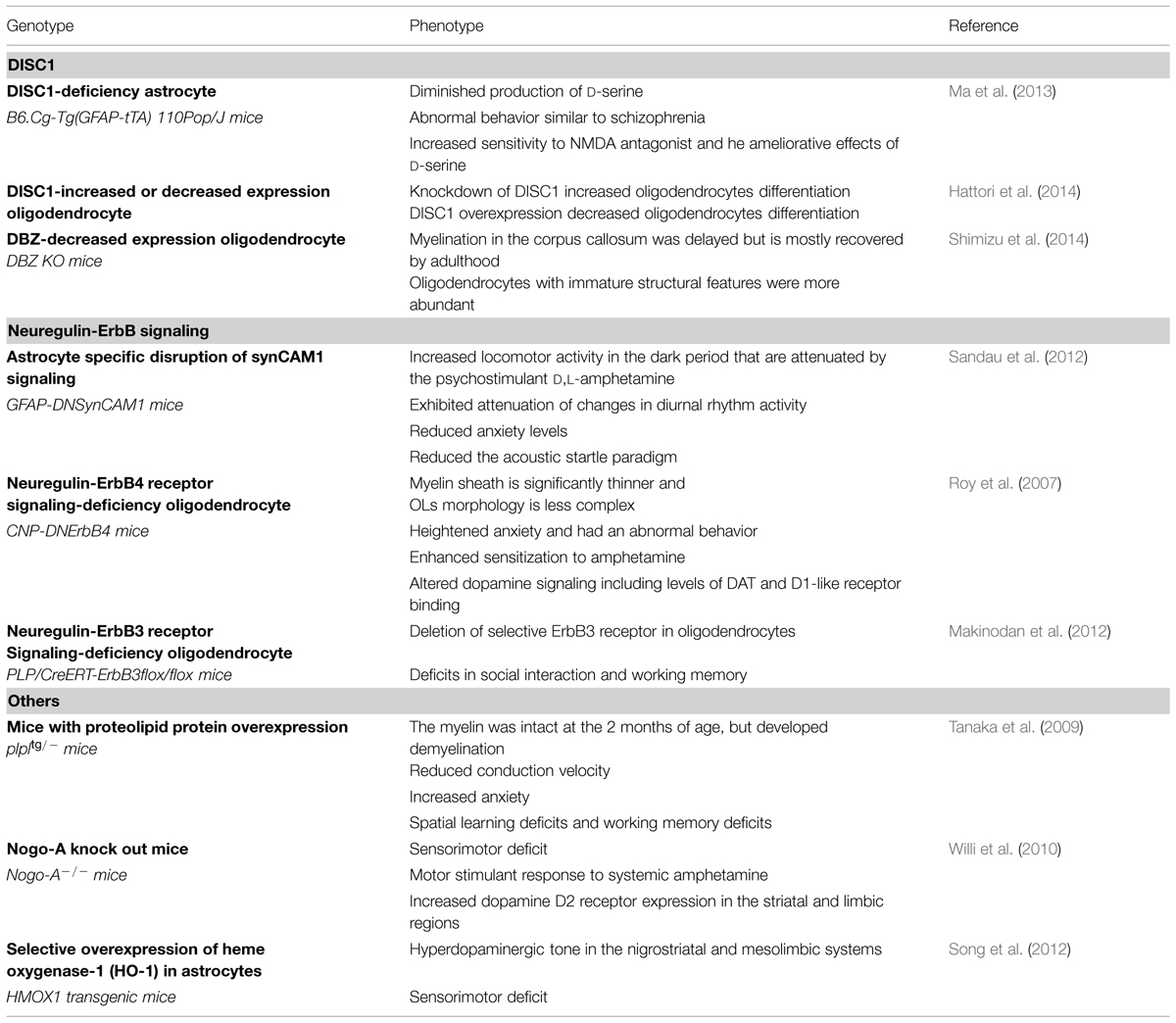
TABLE 2. Summary of mutant DISC1, neuregulin-ErbB signaling mouse models, and others.
MeCP2
Rett syndrome is currently considered a severe neurodevelopmental disorder caused by sporadic mutations in the X-chromosome-linked gene MeCP2 (Amir et al., 1999). Females born with RTT develop normally for 6–18 months and then begin to regress, losing speech, motor skills, and purposeful hand motions and suffering other severe problems including mental retardation, epileptic seizures, and overall retarded growth (Hagberg, 2002). In fact, RTT brain shares certain features with regressive type autism including small neuronal size, as well as reduced dendritic branching and spines in selected regions (Zoghbi, 2003; Armstrong, 2005).
MeCP2 Knockout Mice
In fact, male MeCP2 null mice show severe neurological symptoms at approximately 6 weeks of age, while heterozygous female mice also develop behavioral symptoms after several months (Guy et al., 2001). Loss of MeCP2 function in RTT mice leads to abnormalities in dendritic arborization (Armstrong, 2005), basal synaptic transmission (Moretti et al., 2006), excitatory synaptic plasticity (Asaka et al., 2006; Moretti et al., 2006; Chao et al., 2007) and reduced spontaneous cortical activity (Dani et al., 2005). Studies utilizing a mouse line with a conditional MeCP2 gene knockout specific to neural stem/progenitor cells, (nestin-Cre/MeCP2-/y) identified phenotypes that resemble some of the symptoms of RTT-like phenotypes (Chen et al., 2001; Guy et al., 2001). However, mice carrying a conditional knockout of MeCP2 in post-mitotic neurons driven by the calcium/calmodulin-dependent protein kinase 2 (Camk2)-cre transgene (CamkII-Cre/MeCP2-/y; Chen et al., 2001) exhibit milder and delayed RTT-like phenotypes compared with the nestin-cre driven transgenic mice (Chen et al., 2001). While the neurological symptoms of MeCP2 knockout mice are reversed by restoring MeCP2 expression (Guy et al., 2007), normal MeCP2 expression in neuronal cells is unable to prevent the phenotypes of the MeCP2 null mice (Alvarez-Saavedra et al., 2007), which implicates the specific loss of glial MeCP2 expression in the pathobiology of RTT. Thus, while MeCP2 is widely expressed throughout various cell types in the normal brain including neurons, and all types of glial cell such as astrocytes, oligodendrocytes, and microglia (Ballas et al., 2009), it appears plausible that while neuronal dysfunction was formerly viewed as a significant contributor to RTT causation, glial dysfunction actually may play a greater role in the development of RTT.
MeCP2-Deficiency in Astrocytes
With the loss of MeCP2 expression in astrocytes, there are significant abnormalities in the expression of brain-derived neurotrophic factor (BDNF); an established target of MeCP2 binding (Chang et al., 2006). Interestingly, astrocytes are known to be involved in the initiation and regulation of nervous system immune responses through the release of proinflammatory cytokines (Farina et al., 2007), and it is noteworthy that the expression of interleukin (IL)-1β and IL-6 in response to administration of lipopolysaccharide is reduced in this model compared to that of controls. In addition, p38 mitogen activated protein kinases (MARK) pathways are hyper-activated in this model irrespective of exposure to lipopolysaccharide.
A prominent neuropathological feature associated with brains of RTT is small neuronal size and reduction in dendritic branching and spine density (Armstrong, 2005). Since neurons with more extensive contact with astrocytes promote more extensive dendritic growth (van den Pol and Spencer, 2000), co-culture experiments using intact neurons and astrocytes with MeCP2 deficiency were performed. In this study, neurons cultured in the presence of the MeCP2 deficient astrocytes displayed a much less developed dendritic arborization than did neurons cultured with wild type astrocytes (Ballas et al., 2009). Furthermore, the engineered re-expression of MeCP2 in astrocytes in vivo mouse model led to both significantly improved locomotion and anxiety levels as well as respiratory state (Maezawa et al., 2009; Lioy et al., 2011).
MeCP2 Deficiency in Oligodendrocytes
When mice were engineered that lacked MeCP2 expression in oligodendrocytes, they showed a normal lifespan and the symptoms associated with the RTT-like phenotype commenced at ~10 weeks of age and were milder than those of MeCP2 null mice where the symptoms typically began at 4–5 weeks of age (Chen et al., 2001). With the observational phenotypic scoring system (score = 0–10) considering five typical RTT phenotypic traits such as mobility, gait, hindlimb clasping, tremors, and general conditions (Guy et al., 2007; Nguyen et al., 2012), while MeCP2 null mice reached a score of 6–10 between 9 and 15 weeks of age (Chen et al., 2001), oligodendrocyte MeCP2 knockout mice reached a score of 2 at 20 weeks of age. Additionally, these mice are more active and develop severe hindlimb clasping phenotypes. Restoration of MeCP2 expression solely in cells of the oligodendrocyte lineage in MeCP2 global null mice partially reverses the RTT-like phenotypes associated with loss of MeCP2 such as diminished life span, locomotor deficits, and hindlimb clasping both in male and female, and fully restored normal body weight. However, while MeCP2 expression in the oligodendrocyte lineage cells partially rescues the aberrant expression of MBP protein, it does not affect the expression of either 2′,3′-cyclic-nucleotide-3′-phosphodiesterase (CNPase), myelin oligodendrocyte glycoprotein (MOG), or myelin proteolipid protein (PLP) (Nguyen et al., 2012).
MeCP2-Deficiency in Microglia
Activated microglia release a large amount of glutamate and this microglial-associated neurotoxicity is mediated primarily by NMDA receptor signaling (Takeuchi et al., 2005). In addition to glutamate, activated microglia release pro-inflammatory cytokines such as IL-1β, IL-6, interferon (IFN)-γ, and tumor necrosis factor (TNF-α), which also promote neuronal damage (Sawada et al., 1989; Mizuno et al., 1994, 2003; Suzumura et al., 1996). TNF-α secreted from activated microglia is a major neurotoxic cytokine that induces neurodegeneration through silencing of cell survival signals and caspase-dependent cascades including promotion of signaling through Fas ligand (Greig et al., 2004; Block and Hong, 2005). Neurons treated with conditioned media from MeCP2 null microglia display damaged dendrites and the concentration of glutamate in the media is five time higher than that in control media. The blocking of both microglial glutamate synthesis by a glutaminase inhibitor and microglial glutamate release by a gap junction connexin32 (Cx32) hemichannel blocker abolishes the neurotoxic activity as well as the blocking of a glutamate receptor antagonist. These reports indicate that aberrant glutaminase activity or Cx32 expression in microglia is responsible for the increased production and release of glutamate, potentially implicating these modulators in the pathobiology of microglia-induced RTT-like symptoms (Maezawa and Jin, 2010).
Introduction of Wild Type Microglia into the MeCP2 Null Mouse
Allogeneic transplantation of wild type bone marrow into irradiation-conditioned MeCP2 null mice led to the engraftment of wild type microglia into the MeCP2 null brain parenchyma. The lifespan of these mice was significantly extended as compared with MeCP2 null mice that received either an autologous bone marrow transplant or untreated MeCP2 null mice. Similarly, body and brain weights of MeCP2-null mice recipients of wild type bone marrow recovered approximately to the level seen in wild type mice. And while the overall appearance, tremor, and gait of MeCP2 null mice recipients of wild type bone marrow were improved, the hindlimb clasping phenotype was not changed. Additionally, these mice exhibited significantly reduced numbers of apneic episodes and greatly reduced respiratory irregularities. Interestingly, these benefits resulting from engraftment with wild-type microglia were diminished when phagocytic activity was inhibited pharmacologically by using annexin V which results in substantial blocking of phagocytic activities (Lu et al., 2011; Derecki et al., 2012).
Thus, the focus of RTT studies using animal models has begun to swing from only studying potential neuronal dysfunction to include interrogation of a role for glial dysfunction and the results of the above studies lends credence to the hypothesis that glial dysfunction may play a primary rather than a secondary role in the causation of RTT-like symptoms. Similarly, studies of DISC1 knockout mice and neuregulin 1 (NRG1) knockout mice as models for schizophrenia initially focused on the role these proteins play in neurons, but more recently, their activities in glial cells have begun to be considered.
Disrupted in Schizophrenia 1 (DISC1)
Schizophrenia, which affects ~1% of the worldwide population, is also known as a neurodevelopmental disorder (Weinberger, 1987). The clinical features of schizophrenia cluster in three domains, characterized by positive symptoms (e.g., delusions, hallucinations, thought disorder), negative symptoms (e.g., social withdrawal, blunted affect, reduced motivation) and cognitive symptoms (e.g., attention and working memory deficits). Although schizophrenia is a complex disorder with polygenic and environmental antecedents, multiple lines of evidence have proposed some putative susceptibility genes for schizophrenia (Harrison and Weinberger, 2005; Schizophrenia Working Group of the Psychiatric Genomics, 2014). For example, two overlapping and opposite strand genes on chromosome 1, DISC1 and DISC2, are specifically disrupted by a t(1;11; q42.1; q14.3) balanced translocation, in a large Scottish pedigree, resulting in a cohort with several major mental illnesses such as schizophrenia, bipolar affective disorder, and recurrent major depression (St Clair et al., 1990; Millar et al., 2000, 2001; Blackwood et al., 2001; Muir et al., 2008). DISC1 is expressed in neurons within various brain areas including the olfactory bulb, cortex, hippocampus, hypothalamus, cerebellum, and brain stem, especially during development (Schurov et al., 2004). DISC1 has been shown to be involved in several neurodevelopmental processes including progenitor cell proliferation (Mao et al., 2009), radial migration (Tomita et al., 2011), dendritic arborization (Kamiya et al., 2006; Duan et al., 2007), and synapse formation (Camargo et al., 2007; Duan et al., 2007).
DISC1 Knockout Mice
Since expression of dominant negative proteins frequently has been used successfully in animal models to achieve partial loss of function for relevant proteins (Oike et al., 1999), transgenic mice harboring a C-terminally truncated, dominant negative DISC1 (DN-DISC1), expressed under the control of a promoter for αCaMKII, were generated (Hikida et al., 2007). This model displays several abnormalities including hyperactivity, disturbance in sensorimotor gating, the dynamic modulation of reward value by effortful action, progressive ratio performance, social behavior, and an anhedonia/depression-like deficit (Hikida et al., 2007; Pletnikov et al., 2008; Johnson et al., 2013). In distinction from DN-DISC1 mice, N-ethyl-N-nitrosourea (ENU) was used to induce mutations in exon 2 of mouse Disc1 gene, resulting in the occurrence of missense mutations such as Q31L (glutamine to leucine) or L100P (leucine to proline), causing an increase in depression-like behaviors in Q31L mice and schizophrenia-like behaviors including impaired prepulse inhibition (PPI) and latent inhibition in L100P mice (Clapcote et al., 2007; Shoji et al., 2012). Furthermore, the phenotypes of Q31L mutant mice were partly improved with administration of an antidepressant. Thus, DISC1 dysfunction, likely can exert its influence on neuropsychiatric disorders from its role in both neurons and glia.
An Increase or Decrease of DISC1 Expression in Oligodendrocytes
Expression of ΔhDISC1 could exert a significant influence on oligodendrocyte proliferation, differentiation, and function (Katsel et al., 2011). In fact, DISC1 is expressed in oligodendrocytes in the corpus callosum (Seshadri et al., 2010). In rat oligodendrocyte precursor cell cultures (Hattori et al., 2014), DISC1 expression decreases in the course of oligodendrocyte differentiation. Furthermore, the expression of CNPase and myelin basic protein (MBP) known to markers of myelin maturation were decreased following full length DISC1 overexpression. In contrast, the knockdown of endogenous DISC1 using RNA interference increased the expression of CNPase as well as the number of mature oligodendrocytes (Hattori et al., 2014). SRY box containing (Sox) family member, Sox10, the homeobox containing (Hox) transcription factor Nkx2.2, the basic helix-loop-helix (bHLH) family members Olig1 and Olig2 and the inhibitor of DNA binding (Id) family of proteins Id2 and Id4 have all been shown to be involved in the control of oligodendrocyte differentiation (Nicolay et al., 2007; Emery, 2010). Against this backdrop, it is perhaps not surprising that knockdown of DISC1 increased the expression of Sox10 and/or Nkx2.2, and DISC1 overexpression led to the reduced expression of these transcription factors. These findings are strongly supportive of a role for DISC1 in negatively regulating oligodendrocyte differentiation by acting upstream of Sox10 and/or Nkx2.2 to regulate their transcription (Hattori et al., 2014).
A Decrease of DBZ Expression in Oligodendrocytes
DISC1 binding zinc finger protein (DBZ), also known as ZNF365 or Su48, is a CNS specific member of the DISC1 interactome and is a novel DISC1 binding protein with a predicted C2H2-type zinc-finger motif and coil domains (Hattori et al., 2007). DBZ regulates neurite outgrowth via the DISC1-DBZ interaction in primary neurons and cultured PC12 cells in vitro (Hattori et al., 2007). In DBZ knockout mice, oligodendrocytes displaying an immature structural morphology are more abundant than in control mice and the timing of myelination in the corpus callosum is delayed (Koyama et al., 2013). Although this model implicates DBZ function in oligodendrocyte development and myelination, further studies using more specific models such as oligodendrocyte-specific DBZ knockout mice are needed to elucidate the precise function of DBZ (Shimizu et al., 2014).
DISC1-Deficiency in Astrocytes
Since the administration of the N-methyl-D-aspartic acid (NMDA) receptor antagonists phencyclidine and MK-801 induce behaviors that closely resemble those observed in schizophrenic patients, dysfunction of the NMDA receptor is regarded as a particularly strong candidate for being a component of the mechanism of schizophrenia (Javitt and Zukin, 1991; Goff and Coyle, 2001). The stereoisomer D-serine binds to the “glycine site” on the NR1 subunit and it is crucial for the activation of this receptor. D-serine acting as co-agonist at the NMDA receptor is involved in synaptic plasticity (Fossat et al., 2012; Rosenberg et al., 2013), and D-amino acid oxidase (DAAO) degrades the D-serine, modulating D-serine levels and thence NMDA receptor function (Duplantier et al., 2009; Strick et al., 2011). The biosynthesis of D-serine was clarified by the purification and molecular cloning of serine racemase (SR), which transforms L-serine to D-serine, and, interestingly, DISC1 binds to and stabilizes SR (Wolosker et al., 1999a,b; De Miranda et al., 2000). Furthermore, D-serine and SR have been predominantly localized to astrocytes ensheathing synapses, especially in brain regions with enriched NMDA receptors, suggesting that D-serine could be acting as a glial transmitter (Puyal et al., 2006; Williams et al., 2006). In this model of selective and inducible expression of mutant DISC1 in astrocytes, the expression of mutant DISC1 downregulates the level of endogenous DISC1 expression in astrocytes. The disruption of DISC1 binding to SR leads to increased ubiquitination and degradation of SR in astrocytes. The decrease of SR in astrocytes results in diminished production of D-serine in astrocytes. This mouse model displays abnormal behaviors like schizophrenia including sensitivity to an NMDA antagonist; MK-801, in an open field test and pre-pulse inhibition of acoustic startle test, and responds to the ameliorative effects of D-serine (Ma et al., 2013).
Neuregulin-ErbB Signaling
Neuregulins comprise a large family of widely expressed, alternatively spliced epidermal growth factor (EGF)-like domain-containing proteins that have been strongly implicated in neural development (Corfas et al., 2004; Mei and Xiong, 2008). NRG proteins act by binding to and activating members of the ErbB receptor tyrosine kinase family. After the initial discovery of what came to be known as NRG1, five additional NRG1 homologs (NRG2, NRG3, NRG4, NRG5, and NRG6) have been identified. NRG1 proteins bind only to either ErbB3 or ErbB4 causing a conformational change that promotes receptor dimerization and auophosphorylation, and the subsequent activation of downstream signaling pathways. However, NRG1 does not bind to the ErbB2 receptor, which functions as a co-receptor that heterodimerizes with ErbB3 or ErbB4. Additionally, ErbB4 is known to be able to function as a homodimer. In adult brains, ErbB receptors are widely and differentially expressed. In general, ErbB2 is expressed in most cells, ErbB3 is mainly found in glial populations, and ErbB4 is enriched in neurons. NRG/ErbB signaling has been widely implicated in psychiatric disorders including schizophrenia, bipolar disorder, or depression (Corfas et al., 2004; Mei and Nave, 2014).
Astrocyte Specific Disruption of synCAM1 Signaling
Synaptic cell adhesion molecule 1 (SynCAM1) is a member of the immunoglobulin (Ig) superfamily, a large group of proteins involved in cell surface recognition (Williams, 1992; Rougon and Hobert, 2003). SynCAM1 plays an important role in CNS developmental processes such as synaptic assembly (Biederer et al., 2002), enhancement of excitatory synaptic transmission (Sara et al., 2005; Fogel et al., 2007), functional presynaptic differentiation (Sara et al., 2005) and the regulation of synapse number and plasticity (Robbins et al., 2010). SynCAM1 is produced in astrocytes and plays a major role in facilitating astrocyte-to-astrocyte and astrocyte-to-neuron adhesive communication (Sandau et al., 2011). SynCAM1 is co-expressed with ErbB4 in astrocytes (Sandau et al., 2011) and is functionally related to ErbB4 receptors (Carpenter, 2003). Mice carrying a dominant-negative form of SynCAM1, specifically targeted to astrocytes, exhibited an attenuation of changes in diurnal rhythm activity. In addition, the locomotor activity in a dark field is increased and it is attenuated with the psychostimulant D, L-amphetamine, and anxiety is reduced in a zero maze test and an acoustic startle paradigm in these mice. These findings imply that these mice could be utilized as a model of neurodevelopmental disorder (Sandau et al., 2012).
Neuregulin-ErbB4 Receptors Signaling-Deficiency in Oligodendrocytes
Neuregulin 1-ErbB receptor signaling appears to play a critical role in the ontogeny of psychiatric disorders and this hypothesis has been supported by the identification of altered expression levels and/or function of NRG1, ErbB3, and ErbB4 in patients with schizophrenia (Corfas et al., 2004; Silberberg et al., 2006). Moreover, mice with reduced levels of NRG1 or ErbB4 have exhibited behavioral alterations akin to those found in schizophrenia (Gerlai et al., 2000; Golub et al., 2004; Rimer et al., 2005).
Transgenic mice expressing a dominant-negative ErbB4 receptor in oligodendrocytes exhibit thinner myelin and less complex oligodendrocyte morphology and show schizophrenia-like behaviors including high anxiety and an enhanced sensitization to amphetamine. This abnormal response to amphetamine might be due to altered dopamine signaling as aberrant expressions of dopamine transporter (DAT) and dopamine1-like receptor in the cortex, nucleus accumbens, and striatum are evident (Roy et al., 2007).
Neuregulin-ErbB3 Receptor Signaling-Deficiency in Oligodendrocytes
Neuregulin 1-ErbB signaling plays, at least in part, a critical role in oligodendrocyte development and CNS myelination (Michailov et al., 2004; Taveggia et al., 2005; Chen et al., 2006). In human prefrontal cortex, microarray analyses revealed that the level of ErbB3 was significantly reduced in schizophrenia subjects relative to a normal cohort (Hakak et al., 2001). This decrement was reproduced by another study (Tkachev et al., 2003). Consistent with the results of human studies, mice with selective ErbB3 receptor deletion in oligodendrocytes demonstrate deficits in social interaction and working memory (Makinodan et al., 2012), suggesting that NRG1-ErbB3 signaling in oligodendrocyte might contribute to the pathogenesis of schizophrenia (Makinodan et al., 2012).
Other Studies
Mice with Proteolipid Protein Overexpression
Proteolipid protein 1, a major protein in CNS myelin (Inoue et al., 1996; Mimault et al., 1999), is regarded as an “adhesive strut” that binds adjacent lamellae of the compacted myelin membrane (Boison et al., 1995).
Transgenic mice harboring extra copies of the myelin PLP1 gene have demonstrated that at the 2 months of age, the myelin was intact with a normally appearing ion channel distribution (Inoue et al., 1996), whereas the conduction velocity in all axonal tracts tested in the CNS was markedly reduced at this age (Tanaka et al., 2009). This observation was supported by subsequent analysis that these mice showed altered neuron–glia interaction with subtle changes in axonal diameters and paranodal structures, leading to schizophrenia-like behaviors including increased anxiety-related behaviors, reduced PPI, spatial learning deficits, and working memory deficits.
Nogo-A Knockout Mice
Nogo signaling plays a crucial role in restricting axonal regeneration and compensatory fiber growth in the injured adult mammalian CNS (Schwab, 2004; Yiu and He, 2006). The membrane protein Nogo-A, which is predominantly expressed in oligodendrocytes in the adult brain and in neurons mainly during development, is well-known for its role as one of several currently known inhibitors of neurite outgrowth (Huber et al., 2002; Wang et al., 2002). Postmortem and genetic studies have implicated Nogo-A and its choromosomal location in schizophrenia and bipolar disorder (Coon et al., 1998; Novak et al., 2002).
These mice showed sensorimotor deficits, disrupted latent inhibition, and perseverative behaviors. Furthermore, they displayed an enhanced response to systemic amphetamine in an open field test. These behavioral phenotypes might be due to altered monoaminergic transmitter levels in the striatal and limbic regions and/or increased dopamine D2 receptor expression in the identical brain regions. In contrast, adult mice acutely treated with anti-Nogo-A antibodies did not exhibit abnormal behaviors, but showed increased dopamine D2 receptor expression (Willi et al., 2010).
Selective Overexpression of Heme Oxygenase-1 (HO-1) in Astrocytes
The heme oxygenases (HOs), which are responsible for the degradation of heme to biliverdin/bilirubin, free iron and carbon monoxide (CO), has been strongly implicated in mammalian CNS aging and diseases (Schipper et al., 2009). Mammalian cells express two isoforms such as an inducible isoform, HO-1, and a constitutively active form, HO-2. Specifically, HO-1, encoded by the HMOX1 gene, is a 32-kDa stress protein, and the induction of the glial HMOX1 gene may lead to pathological brain iron deposition, intracellular oxidative damage, and bioenergetic failure in Alzheimer’s disease and other human CNS disorders such as Parkinson’s disease and schizophrenia (Schipper et al., 2009; Brown, 2011). This mice model displayed sensorimotor deficits, increased spontaneous horizontal movements, and stereotypy. Hyperdopaminergic signaling was identified in the striatum and substantia nigra and the associated neurochemical alterations may contribute to these behaviors (Song et al., 2012).
Conclusion
In conclusion, studies leveraging different animal models for the enhanced biochemical and physiological understanding of mental disorders have moved from strictly targeting the biology of neurons to also include an examination of the glia, especially since glial cells such as oligodendrocytes, astrocytes, and microglia have emerged as critically important modifiers of both CNS development and function. Postmortem brain analyses have clearly indicated that glial cell abnormalities are present in the brains of patients with schizophrenia. However, it remains uncertain whether the dysregulation and symptoms seen are a primary result of alterations in glial cell biology or the deficits in glial function occur as a side product of neuronal dysfunction. Nonetheless, as we have described, continued use of cell-type specific conditional knockout mice will allow us to better dissect how glial cells are implicated in nervous system dysfunction and perhaps illuminate their role in the pathobiology of psychiatric diseases such as schizophrenia.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
This work was supported by the Naito Foundation.
References
Alvarez-Saavedra, M., Saez, M. A., Kang, D., Zoghbi, H. Y., and Young, J. I. (2007). Cell-specific expression of wild-type MeCP2 in mouse models of Rett syndrome yields insight about pathogenesis. Hum. Mol. Genet. 16, 2315–2325. doi: 10.1093/hmg/ddm185
Amir, R. E., Van den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188. doi: 10.1038/13810
Armstrong, D. D. (2005). Neuropathology of Rett syndrome. J. Child Neurol. 20, 747–753. doi: 10.1177/08830738050200082401
Asaka, Y., Jugloff, D. G., Zhang, L., Eubanks, J. H., and Fitzsimonds, R. M. (2006). Hippocampal synaptic plasticity is impaired in the Mecp2-null mouse model of Rett syndrome. Neurobiol. Dis. 21, 217–227. doi: 10.1016/j.nbd.2005.07.005
Azevedo, F. A., Carvalho, L. R., Grinberg, L. T., Farfel, J. M., Ferretti, R. E., Leite, R. E., et al. (2009). Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J. Comp. Neurol. 513, 532–541. doi: 10.1002/cne.21974
Ballas, N., Lioy, D. T., Grunseich, C., and Mandel, G. (2009). Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat. Neurosci. 12, 311–317. doi: 10.1038/nn.2275
Bernstein, H. G., Steiner, J., Guest, P. C., Dobrowolny, H., and Bogerts, B. (2015). Glial cells as key players in schizophrenia pathology: recent insights and concepts of therapy. Schizophr. Res. 161, 4–18. doi: 10.1016/j.schres.2014.03.035
Bessis, A., Bechade, C., Bernard, D., and Roumier, A. (2007). Microglial control of neuronal death and synaptic properties. Glia 55, 233–238. doi: 10.1002/glia.20459
Biederer, T., Sara, Y., Mozhayeva, M., Atasoy, D., Liu, X., Kavalali, E. T., et al. (2002). SynCAM, a synaptic adhesion molecule that drives synapse assembly. Science 297, 1525–1531. doi: 10.1126/science.1072356
Blackwood, D. H., Fordyce, A., Walker, M. T., St Clair, D. M., Porteous, D. J., and Muir, W. J. (2001). Schizophrenia and affective disorders–cosegregation with a translocation at chromosome 1q42 that directly disrupts brain-expressed genes: clinical and P300 findings in a family. Am. J. Hum. Genet. 69, 428–433. doi: 10.1086/321969
Block, M. L., and Hong, J. S. (2005). Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog. Neurobiol. 76, 77–98. doi: 10.1016/j.pneurobio.2005.06.004
Boison, D., Bussow, H., D’Urso, D., Muller, H. W., and Stoffel, W. (1995). Adhesive properties of proteolipid protein are responsible for the compaction of CNS myelin sheaths. J. Neurosci. 15, 5502–5513.
Brown, A. S. (2011). The environment and susceptibility to schizophrenia. Prog. Neurobiol. 93, 23–58. doi: 10.1016/j.pneurobio.2010.09.003
Camargo, L. M., Collura, V., Rain, J. C., Mizuguchi, K., Hermjakob, H., Kerrien, S., et al. (2007). Disrupted in Schizophrenia 1 Interactome: evidence for the close connectivity of risk genes and a potential synaptic basis for schizophrenia. Mol. Psychiatry 12, 74–86. doi: 10.1038/sj.mp.4001880
Carpenter, G. (2003). ErbB-4: mechanism of action and biology. Exp. Cell Res. 284, 66–77. doi: 10.1016/S0014-4827(02)00100-3
Chang, Q., Khare, G., Dani, V., Nelson, S., and Jaenisch, R. (2006). The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron 49, 341–348. doi: 10.1016/j.neuron.2005.12.027
Chao, H. T., Zoghbi, H. Y., and Rosenmund, C. (2007). MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron 56, 58–65. doi: 10.1016/j.neuron.2007.08.018
Chen, R. Z., Akbarian, S., Tudor, M., and Jaenisch, R. (2001). Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat. Genet. 27, 327–331. doi: 10.1038/85906
Chen, S., Velardez, M. O., Warot, X., Yu, Z. X., Miller, S. J., Cros, D., et al. (2006). Neuregulin 1-erbB signaling is necessary for normal myelination and sensory function. J. Neurosci. 26, 3079–3086. doi: 10.1523/JNEUROSCI.3785-05.2006
Clapcote, S. J., Lipina, T. V., Millar, J. K., Mackie, S., Christie, S., Ogawa, F., et al. (2007). Behavioral phenotypes of Disc1 missense mutations in mice. Neuron 54, 387–402. doi: 10.1016/j.neuron.2007.04.015
Coon, H., Myles-Worsley, M., Tiobech, J., Hoff, M., Rosenthal, J., Bennett, P., et al. (1998). Evidence for a chromosome 2p13-14 schizophrenia susceptibility locus in families from Palau, Micronesia. Mol. Psychiatry 3, 521–527. doi: 10.1038/sj.mp.4000453
Corfas, G., Roy, K., and Buxbaum, J. D. (2004). Neuregulin 1-erbB signaling and the molecular/cellular basis of schizophrenia. Nat. Neurosci. 7, 575–580. doi: 10.1038/nn1258
Dani, V. S., Chang, Q., Maffei, A., Turrigiano, G. G., Jaenisch, R., and Nelson, S. B. (2005). Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. U.S.A. 102, 12560–12565. doi: 10.1073/pnas.0506071102
Davalos, D., Grutzendler, J., Yang, G., Kim, J. V., Zuo, Y., Jung, S., et al. (2005). ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 8, 752–758. doi: 10.1038/nn1472
De Miranda, J., Santoro, A., Engelender, S., and Wolosker, H. (2000). Human serine racemase: moleular cloning, genomic organization and functional analysis. Gene 256, 183–188. doi: 10.1016/S0378-1119(00)00356-5
Derecki, N. C., Cronk, J. C., Lu, Z., Xu, E., Abbott, S. B., Guyenet, P. G., et al. (2012). Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature 484, 105–109. doi: 10.1038/nature10907
Dracheva, S., Davis, K. L., Chin, B., Woo, D. A., Schmeidler, J., and Haroutunian, V. (2006). Myelin-associated mRNA and protein expression deficits in the anterior cingulate cortex and hippocampus in elderly schizophrenia patients. Neurobiol. Dis. 21, 531–540. doi: 10.1016/j.nbd.2005.08.012
Duan, X., Chang, J. H., Ge, S., Faulkner, R. L., Kim, J. Y., Kitabatake, Y., et al. (2007). Disrupted-In-Schizophrenia 1 regulates integration of newly generated neurons in the adult brain. Cell 130, 1146–1158. doi: 10.1016/j.cell.2007.07.010
Duplantier, A. J., Becker, S. L., Bohanon, M. J., Borzilleri, K. A., Chrunyk, B. A., Downs, J. T., et al. (2009). Discovery, SAR, and pharmacokinetics of a novel 3-hydroxyquinolin-2(1H)-one series of potent D-amino acid oxidase (DAAO) inhibitors. J. Med. Chem. 52, 3576–3585. doi: 10.1021/jm900128w
Dwork, A. J., Mancevski, B., and Rosoklija, G. (2007). White matter and cognitive function in schizophrenia. Int. J. Neuropsychopharmacol. 10, 513–536. doi: 10.1017/S1461145707007638
Emery, B. (2010). Regulation of oligodendrocyte differentiation and myelination. Science 330, 779–782. doi: 10.1126/science.1190927
Farina, C., Aloisi, F., and Meinl, E. (2007). Astrocytes are active players in cerebral innate immunity. Trends Immunol. 28, 138–145. doi: 10.1016/j.it.2007.01.005
Fogel, A. I., Akins, M. R., Krupp, A. J., Stagi, M., Stein, V., and Biederer, T. (2007). SynCAMs organize synapses through heterophilic adhesion. J. Neurosci. 27, 12516–12530. doi: 10.1523/JNEUROSCI.2739-07.2007
Fossat, P., Turpin, F. R., Sacchi, S., Dulong, J., Shi, T., Rivet, J. M., et al. (2012). Glial D-serine gates NMDA receptors at excitatory synapses in prefrontal cortex. Cereb. Cortex 22, 595–606. doi: 10.1093/cercor/bhr130
Gerlai, R., Pisacane, P., and Erickson, S. (2000). Heregulin, but not ErbB2 or ErbB3, heterozygous mutant mice exhibit hyperactivity in multiple behavioral tasks. Behav. Brain Res. 109, 219–227. doi: 10.1016/S0166-4328(99)00175-8
Goff, D. C., and Coyle, J. T. (2001). The emerging role of glutamate in the pathophysiology and treatment of schizophrenia. Am. J. Psychiatry 158, 1367–1377. doi: 10.1176/appi.ajp.158.9.1367
Golub, M. S., Germann, S. L., and Lloyd, K. C. (2004). Behavioral characteristics of a nervous system-specific erbB4 knock-out mouse. Behav. Brain Res. 153, 159–170. doi: 10.1016/j.bbr.2003.11.010
Greig, N. H., Mattson, M. P., Perry, T., Chan, S. L., Giordano, T., Sambamurti, K., et al. (2004). New therapeutic strategies and drug candidates for neurodegenerative diseases: p53 and TNF-alpha inhibitors, and GLP-1 receptor agonists. Ann. N. Y. Acad. Sci. 1035, 290–315. doi: 10.1196/annals.1332.018
Guy, J., Gan, J., Selfridge, J., Cobb, S., and Bird, A. (2007). Reversal of neurological defects in a mouse model of Rett syndrome. Science 315, 1143–1147. doi: 10.1126/science.1138389
Guy, J., Hendrich, B., Holmes, M., Martin, J. E., and Bird, A. (2001). A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 27, 322–326. doi: 10.1038/85899
Hagberg, B. (2002). Clinical manifestations and stages of Rett syndrome. Ment. Retard. Dev. Disabil. Res. Rev. 8, 61–65. doi: 10.1002/mrdd.10020
Hakak, Y., Walker, J. R., Li, C., Wong, W. H., Davis, K. L., Buxbaum, J. D., et al. (2001). Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 98, 4746–4751. doi: 10.1073/pnas.081071198
Harrison, P. J., and Weinberger, D. R. (2005). Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol. Psychiatry 10, 40–68. doi: 10.1038/sj.mp.4001558
Hattori, T., Baba, K., Matsuzaki, S., Honda, A., Miyoshi, K., Inoue, K., et al. (2007). A novel DISC1-interacting partner DISC1-Binding Zinc-finger protein: implication in the modulation of DISC1-dependent neurite outgrowth. Mol. Psychiatry 12, 398–407. doi: 10.1038/sj.mp.4001945
Hattori, T., Shimizu, S., Koyama, Y., Emoto, H., Matsumoto, Y., Kumamoto, N., et al. (2014). DISC1 (disrupted-in-schizophrenia-1) regulates differentiation of oligodendrocytes. PLoS ONE 9:e88506. doi: 10.1371/journal.pone.0088506
Hikida, T., Jaaro-Peled, H., Seshadri, S., Oishi, K., Hookway, C., Kong, S., et al. (2007). Dominant-negative DISC1 transgenic mice display schizophrenia-associated phenotypes detected by measures translatable to humans. Proc. Natl. Acad. Sci. U.S.A. 104, 14501–14506. doi: 10.1073/pnas.0704774104
Huber, A. B., Weinmann, O., Brosamle, C., Oertle, T., and Schwab, M. E. (2002). Patterns of Nogo mRNA and protein expression in the developing and adult rat and after CNS lesions. J. Neurosci. 22, 3553–3567.
Inoue, K., Osaka, H., Sugiyama, N., Kawanishi, C., Onishi, H., Nezu, A., et al. (1996). A duplicated PLP gene causing Pelizaeus-Merzbacher disease detected by comparative multiplex PCR. Am. J. Hum. Genet. 59, 32–39.
Iwamoto, K., Bundo, M., and Kato, T. (2005). Altered expression of mitochondria-related genes in postmortem brains of patients with bipolar disorder or schizophrenia, as revealed by large-scale DNA microarray analysis. Hum. Mol. Genet. 14, 241–253. doi: 10.1093/hmg/ddi022
Javitt, D. C., and Zukin, S. R. (1991). Recent advances in the phencyclidine model of schizophrenia. Am. J. Psychiatry 148, 1301–1308. doi: 10.1176/ajp.148.10.1301
Johnson, A. W., Jaaro-Peled, H., Shahani, N., Sedlak, T. W., Zoubovsky, S., Burruss, D., et al. (2013). Cognitive and motivational deficits together with prefrontal oxidative stress in a mouse model for neuropsychiatric illness. Proc. Natl. Acad. Sci. U.S.A. 110, 12462–12467. doi: 10.1073/pnas.1307925110
Kamiya, A., Tomoda, T., Chang, J., Takaki, M., Zhan, C., Morita, M., et al. (2006). DISC1-NDEL1/NUDEL protein interaction, an essential component for neurite outgrowth, is modulated by genetic variations of DISC1. Hum. Mol. Genet. 15, 3313–3323. doi: 10.1093/hmg/ddl407
Katsel, P., Davis, K. L., Gorman, J. M., and Haroutunian, V. (2005). Variations in differential gene expression patterns across multiple brain regions in schizophrenia. Schizophr. Res. 77, 241–252. doi: 10.1016/j.schres.2005.03.020
Katsel, P., Tan, W., Abazyan, B., Davis, K. L., Ross, C., Pletnikov, M. V., et al. (2011). Expression of mutant human DISC1 in mice supports abnormalities in differentiation of oligodendrocytes. Schizophr. Res. 130, 238–249. doi: 10.1016/j.schres.2011.04.021
Kimelberg, H. K. (2007). Supportive or information-processing functions of the mature protoplasmic astrocyte in the mammalian CNS? A critical appraisal. Neuron Glia Biol. 3, 181–189. doi: 10.1017/S1740925X08000094
Koyama, Y., Hattori, T., Shimizu, S., Taniguchi, M., Yamada, K., Takamura, H., et al. (2013). DBZ (DISC1-binding zinc finger protein)-deficient mice display abnormalities in basket cells in the somatosensory cortices. J. Chem. Neuroanat. 53, 1–10. doi: 10.1016/j.jchemneu.2013.07.002
Kubicki, M., McCarley, R., Westin, C. F., Park, H. J., Maier, S., Kikinis, R., et al. (2007). A review of diffusion tensor imaging studies in schizophrenia. J. Psychiatr. Res. 41, 15–30. doi: 10.1016/j.jpsychires.2005.05.005
Lioy, D. T., Garg, S. K., Monaghan, C. E., Raber, J., Foust, K. D., Kaspar, B. K., et al. (2011). A role for glia in the progression of Rett’s syndrome. Nature 475, 497–500. doi: 10.1038/nature10214
Lu, Z., Elliott, M. R., Chen, Y., Walsh, J. T., Klibanov, A. L., Ravichandran, K. S., et al. (2011). Phagocytic activity of neuronal progenitors regulates adult neurogenesis. Nat. Cell Biol. 13, 1076–1083. doi: 10.1038/ncb2299
Ma, T. M., Abazyan, S., Abazyan, B., Nomura, J., Yang, C., Seshadri, S., et al. (2013). Pathogenic disruption of DISC1-serine racemase binding elicits schizophrenia-like behavior via D-serine depletion. Mol. Psychiatry 18, 557–567. doi: 10.1038/mp.2012.97
Maezawa, I., and Jin, L. W. (2010). Rett syndrome microglia damage dendrites and synapses by the elevated release of glutamate. J. Neurosci. 30, 5346–5356. doi: 10.1523/JNEUROSCI.5966-09.2010
Maezawa, I., Swanberg, S., Harvey, D., LaSalle, J. M., and Jin, L. W. (2009). Rett syndrome astrocytes are abnormal and spread MeCP2 deficiency through gap junctions. J. Neurosci. 29, 5051–5061. doi: 10.1523/JNEUROSCI.0324-09.2009
Makinodan, M., Rosen, K. M., Ito, S., and Corfas, G. (2012). A critical period for social experience-dependent oligodendrocyte maturation and myelination. Science 337, 1357–1360. doi: 10.1126/science.1220845
Mao, Y., Ge, X., Frank, C. L., Madison, J. M., Koehler, A. N., Doud, M. K., et al. (2009). Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling. Cell 136, 1017–1031. doi: 10.1016/j.cell.2008.12.044
Mei, L., and Nave, K. A. (2014). Neuregulin-ERBB signaling in the nervous system and neuropsychiatric diseases. Neuron 83, 27–49. doi: 10.1016/j.neuron.2014.06.007
Mei, L., and Xiong, W. C. (2008). Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nat. Rev. Neurosci. 9, 437–452. doi: 10.1038/nrn2392
Michailov, G. V., Sereda, M. W., Brinkmann, B. G., Fischer, T. M., Haug, B., Birchmeier, C., et al. (2004). Axonal neuregulin-1 regulates myelin sheath thickness. Science 304, 700–703. doi: 10.1126/science.1095862
Millar, J. K., Christie, S., Anderson, S., Lawson, D., Hsiao-Wei Loh, D., Devon, R. S., et al. (2001). Genomic structure and localisation within a linkage hotspot of Disrupted In Schizophrenia 1, a gene disrupted by a translocation segregating with schizophrenia. Mol. Psychiatry 6, 173–178. doi: 10.1038/sj.mp.4000784
Millar, J. K., Wilson-Annan, J. C., Anderson, S., Christie, S., Taylor, M. S., Semple, C. A., et al. (2000). Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum. Mol. Genet. 9, 1415–1423. doi: 10.1093/hmg/9.9.1415
Miller, R. H. (2002). Regulation of oligodendrocyte development in the vertebrate CNS. Prog. Neurobiol. 67, 451–467. doi: 10.1016/S0301-0082(02)00058-8
Mimault, C., Giraud, G., Courtois, V., Cailloux, F., Boire, J. Y., Dastugue, B., et al. (1999). Proteolipoprotein gene analysis in 82 patients with sporadic Pelizaeus-Merzbacher disease: duplications, the major cause of the disease, originate more frequently in male germ cells, but point mutations do not. The Clinical European Network on Brain Dysmyelinating Disease. Am. J. Hum. Genet. 65, 360–369. doi: 10.1086/302483
Mizuno, T., Kawanokuchi, J., Numata, K., and Suzumura, A. (2003). Production and neuroprotective functions of fractalkine in the central nervous system. Brain Res. 979, 65–70. doi: 10.1016/S0006-8993(03)02867-1
Mizuno, T., Sawada, M., Suzumura, A., and Marunouchi, T. (1994). Expression of cytokines during glial differentiation. Brain Res. 656, 141–146. doi: 10.1016/0006-8993(94)91375-7
Moretti, P., Levenson, J. M., Battaglia, F., Atkinson, R., Teague, R., Antalffy, B., et al. (2006). Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J. Neurosci. 26, 319–327. doi: 10.1523/JNEUROSCI.2623-05.2006
Muir, W. J., Pickard, B. S., and Blackwood, D. H. (2008). Disrupted-in-Schizophrenia-1. Curr. Psychiatry Rep. 10, 140–147. doi: 10.1007/s11920-008-0025-2
Nave, K. A. (2010). Myelination and the trophic support of long axons. Nat. Rev. Neurosci. 11, 275–283. doi: 10.1038/nrn2797
Newman, E. A. (2003). New roles for astrocytes: regulation of synaptic transmission. Trends Neurosci. 26, 536–542. doi: 10.1016/S0166-2236(03)00237-6
Nguyen, M. V., Du, F., Felice, C. A., Shan, X., Nigam, A., Mandel, G., et al. (2012). MeCP2 is critical for maintaining mature neuronal networks and global brain anatomy during late stages of postnatal brain development and in the mature adult brain. J. Neurosci. 32, 10021–10034. doi: 10.1523/JNEUROSCI.1316-12.2012
Nguyen, M. V., Felice, C. A., Du, F., Covey, M. V., Robinson, J. K., Mandel, G., et al. (2013). Oligodendrocyte lineage cells contribute unique features to Rett syndrome neuropathology. J. Neurosci. 33, 18764–18774. doi: 10.1523/JNEUROSCI.2657-13.2013
Nicolay, D. J., Doucette, J. R., and Nazarali, A. J. (2007). Transcriptional control of oligodendrogenesis. Glia 55, 1287–1299. doi: 10.1002/glia.20540
Nimmerjahn, A., Kirchhoff, F., and Helmchen, F. (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318. doi: 10.1126/science.1110647
Novak, G., Kim, D., Seeman, P., and Tallerico, T. (2002). Schizophrenia and Nogo: elevated mRNA in cortex, and high prevalence of a homozygous CAA insert. Brain Res. Mol. Brain Res. 107, 183–189. doi: 10.1016/S0169-328X(02)00492-8
Oike, Y., Hata, A., Mamiya, T., Kaname, T., Noda, Y., Suzuki, M., et al. (1999). Truncated CBP protein leads to classical Rubinstein-Taybi syndrome phenotypes in mice: implications for a dominant-negative mechanism. Hum. Mol. Genet. 8, 387–396. doi: 10.1093/hmg/8.3.387
Parpura, V., Heneka, M. T., Montana, V., Oliet, S. H., Schousboe, A., Haydon, P. G., et al. (2012). Glial cells in (patho)physiology. J. Neurochem. 121, 4–27. doi: 10.1111/j.1471-4159.2012.07664.x
Parpura, V., and Verkhratsky, A. (2012). Homeostatic function of astrocytes: Ca(2+) and Na(+) signalling. Transl. Neurosci. 3, 334–344. doi: 10.2478/s13380-012-0040-y
Pelvig, D. P., Pakkenberg, H., Stark, A. K., and Pakkenberg, B. (2008). Neocortical glial cell numbers in human brains. Neurobiol. Aging 29, 1754–1762. doi: 10.1016/j.neurobiolaging.2007.04.013
Perea, G., Navarrete, M., and Araque, A. (2009). Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci. 32, 421–431. doi: 10.1016/j.tins.2009.05.001
Pletnikov, M. V., Ayhan, Y., Nikolskaia, O., Xu, Y., Ovanesov, M. V., Huang, H., et al. (2008). Inducible expression of mutant human DISC1 in mice is associated with brain and behavioral abnormalities reminiscent of schizophrenia. Mol. Psychiatry 13, 173–186, 115. doi: 10.1038/sj.mp.4002079
Puyal, J., Martineau, M., Mothet, J. P., Nicolas, M. T., and Raymond, J. (2006). Changes in D-serine levels and localization during postnatal development of the rat vestibular nuclei. J. Comp. Neurol. 497, 610–621. doi: 10.1002/cne.21016
Rimer, M., Barrett, D. W., Maldonado, M. A., Vock, V. M., and Gonzalez-Lima, F. (2005). Neuregulin-1 immunoglobulin-like domain mutant mice: clozapine sensitivity and impaired latent inhibition. Neuroreport 16, 271–275. doi: 10.1097/00001756-200502280-00014
Robbins, E. M., Krupp, A. J., Perez de Arce, K., Ghosh, A. K., Fogel, A. I., Boucard, A., et al. (2010). SynCAM 1 adhesion dynamically regulates synapse number and impacts plasticity and learning. Neuron 68, 894–906. doi: 10.1016/j.neuron.2010.11.003
Rosenberg, D., Artoul, S., Segal, A. C., Kolodney, G., Radzishevsky, I., Dikopoltsev, E., et al. (2013). Neuronal D-serine and glycine release via the Asc-1 transporter regulates NMDA receptor-dependent synaptic activity. J. Neurosci. 33, 3533–3544. doi: 10.1523/JNEUROSCI.3836-12.2013
Rougon, G., and Hobert, O. (2003). New insights into the diversity and function of neuronal immunoglobulin superfamily molecules. Annu. Rev. Neurosci. 26, 207–238. doi: 10.1146/annurev.neuro.26.041002.131014
Roy, K., Murtie, J. C., El-Khodor, B. F., Edgar, N., Sardi, S. P., Hooks, B. M., et al. (2007). Loss of erbB signaling in oligodendrocytes alters myelin and dopaminergic function, a potential mechanism for neuropsychiatric disorders. Proc. Natl. Acad. Sci. U.S.A. 104, 8131–8136. doi: 10.1073/pnas.0702157104
Sandau, U. S., Alderman, Z., Corfas, G., Ojeda, S. R., and Raber, J. (2012). Astrocyte-specific disruption of SynCAM1 signaling results in ADHD-like behavioral manifestations. PLoS ONE 7:e36424. doi: 10.1371/journal.pone.0036424
Sandau, U. S., Mungenast, A. E., McCarthy, J., Biederer, T., Corfas, G., and Ojeda, S. R. (2011). The synaptic cell adhesion molecule, SynCAM1, mediates astrocyte-to-astrocyte and astrocyte-to-GnRH neuron adhesiveness in the mouse hypothalamus. Endocrinology 152, 2353–2363. doi: 10.1210/en.2010-1434
Sara, Y., Biederer, T., Atasoy, D., Chubykin, A., Mozhayeva, M. G., Sudhof, T. C., et al. (2005). Selective capability of SynCAM and neuroligin for functional synapse assembly. J. Neurosci. 25, 260–270. doi: 10.1523/JNEUROSCI.3165-04.2005
Sawada, M., Kondo, N., Suzumura, A., and Marunouchi, T. (1989). Production of tumor necrosis factor-alpha by microglia and astrocytes in culture. Brain Res. 491, 394–397. doi: 10.1016/0006-8993(89)90078-4
Schipper, H. M., Song, W., Zukor, H., Hascalovici, J. R., and Zeligman, D. (2009). Heme oxygenase-1 and neurodegeneration: expanding frontiers of engagement. J. Neurochem. 110, 469–485. doi: 10.1111/j.1471-4159.2009.06160.x
Schizophrenia Working Group of the Psychiatric Genomics, C. (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427. doi: 10.1038/nature13595
Schurov, I. L., Handford, E. J., Brandon, N. J., and Whiting, P. J. (2004). Expression of disrupted in schizophrenia 1 (DISC1) protein in the adult and developing mouse brain indicates its role in neurodevelopment. Mol. Psychiatry 9, 1100–1110. doi: 10.1038/sj.mp.4001574
Schwab, M. E. (2004). Nogo and axon regeneration. Curr. Opin. Neurobiol. 14, 118–124. doi: 10.1016/j.conb.2004.01.004
Seshadri, S., Kamiya, A., Yokota, Y., Prikulis, I., Kano, S., Hayashi-Takagi, A., et al. (2010). Disrupted-in-Schizophrenia-1 expression is regulated by beta-site amyloid precursor protein cleaving enzyme-1-neuregulin cascade. Proc. Natl. Acad. Sci. U.S.A. 107, 5622–5627. doi: 10.1073/pnas.0909284107
Shimizu, S., Koyama, Y., Hattori, T., Tachibana, T., Yoshimi, T., Emoto, H., et al. (2014). DBZ, a CNS-specific DISC1 binding protein, positively regulates oligodendrocyte differentiation. Glia 62, 709–724. doi: 10.1002/glia.22636
Shoji, H., Toyama, K., Takamiya, Y., Wakana, S., Gondo, Y., and Miyakawa, T. (2012). Comprehensive behavioral analysis of ENU-induced Disc1-Q31L and -L100P mutant mice. BMC Res. Notes 5:108. doi: 10.1186/1756-0500-5-108
Silberberg, G., Darvasi, A., Pinkas-Kramarski, R., and Navon, R. (2006). The involvement of ErbB4 with schizophrenia: association and expression studies. Am. J. Med. Genet. B Neuropsychiatr. Genet. 141b, 142–148. doi: 10.1002/ajmg.b.30275
Song, W., Zukor, H., Lin, S. H., Hascalovici, J., Liberman, A., Tavitian, A., et al. (2012). Schizophrenia-like features in transgenic mice overexpressing human HO-1 in the astrocytic compartment. J. Neurosci. 32, 10841–10853. doi: 10.1523/JNEUROSCI.6469-11.2012
St Clair, D., Blackwood, D., Muir, W., Carothers, A., Walker, M., Spowart, G., et al. (1990). Association within a family of a balanced autosomal translocation with major mental illness. Lancet 336, 13–16. doi: 10.1016/0140-6736(90)91520-K
Strick, C. A., Li, C., Scott, L., Harvey, B., Hajos, M., Steyn, S. J., et al. (2011). Modulation of NMDA receptor function by inhibition of D-amino acid oxidase in rodent brain. Neuropharmacology 61, 1001–1015. doi: 10.1016/j.neuropharm.2011.06.029
Suzumura, A., Sawada, M., and Marunouchi, T. (1996). Selective induction of interleukin-6 in mouse microglia by granulocyte-macrophage colony-stimulating factor. Brain Res. 713, 192–198. doi: 10.1016/0006-8993(95)01535-3
Takeuchi, H., Mizuno, T., Zhang, G., Wang, J., Kawanokuchi, J., Kuno, R., et al. (2005). Neuritic beading induced by activated microglia is an early feature of neuronal dysfunction toward neuronal death by inhibition of mitochondrial respiration and axonal transport. J. Biol. Chem. 280, 10444–10454. doi: 10.1074/jbc.M413863200
Tanaka, H., Ma, J., Tanaka, K. F., Takao, K., Komada, M., Tanda, K., et al. (2009). Mice with altered myelin proteolipid protein gene expression display cognitive deficits accompanied by abnormal neuron-glia interactions and decreased conduction velocities. J. Neurosci. 29, 8363–8371. doi: 10.1523/JNEUROSCI.3216-08.2009
Taveggia, C., Zanazzi, G., Petrylak, A., Yano, H., Rosenbluth, J., Einheber, S., et al. (2005). Neuregulin-1 type III determines the ensheathment fate of axons. Neuron 47, 681–694. doi: 10.1016/j.neuron.2005.08.017
Tkachev, D., Mimmack, M. L., Ryan, M. M., Wayland, M., Freeman, T., Jones, P. B., et al. (2003). Oligodendrocyte dysfunction in schizophrenia and bipolar disorder. Lancet 362, 798–805. doi: 10.1016/S0140-6736(03)14289-4
Tomita, K., Kubo, K., Ishii, K., and Nakajima, K. (2011). Disrupted-in-Schizophrenia-1 (Disc1) is necessary for migration of the pyramidal neurons during mouse hippocampal development. Hum. Mol. Genet. 20, 2834–2845. doi: 10.1093/hmg/ddr194
Uranova, N. A., Vikhreva, O. V., Rachmanova, V. I., and Orlovskaya, D. D. (2011). Ultrastructural alterations of myelinated fibers and oligodendrocytes in the prefrontal cortex in schizophrenia: a postmortem morphometric study. Schizophr. Res. Treatment 2011:325789. doi: 10.1155/2011/325789
van den Pol, A. N., and Spencer, D. D. (2000). Differential neurite growth on astrocyte substrates: interspecies facilitation in green fluorescent protein-transfected rat and human neurons. Neuroscience 95, 603–616. doi: 10.1016/S0306-4522(99)00430-3
Wake, H., Moorhouse, A. J., Jinno, S., Kohsaka, S., and Nabekura, J. (2009). Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J. Neurosci. 29, 3974–3980. doi: 10.1523/JNEUROSCI.4363-08.2009
Wang, D. D., and Bordey, A. (2008). The astrocyte odyssey. Prog. Neurobiol. 86, 342–367. doi: 10.1016/j.pneurobio.2008.09.015
Wang, X., Chun, S. J., Treloar, H., Vartanian, T., Greer, C. A., and Strittmatter, S. M. (2002). Localization of Nogo-A and Nogo-66 receptor proteins at sites of axon-myelin and synaptic contact. J. Neurosci. 22,5505–5515.
Weinberger, D. R. (1987). Implications of normal brain development for the pathogenesis of schizophrenia. Arch. Gen. Psychiatry 44, 660–669. doi: 10.1001/archpsyc.1987.01800190080012
Willi, R., Weinmann, O., Winter, C., Klein, J., Sohr, R., Schnell, L., et al. (2010). Constitutive genetic deletion of the growth regulator Nogo-A induces schizophrenia-related endophenotypes. J. Neurosci. 30, 556–567. doi: 10.1523/JNEUROSCI.4393-09.2010
Williams, A. F. (1992). The immunoglobulin superfamily in cell surface recognition. C. R. Acad. Sci. III 314, 27–29.
Williams, S. M., Diaz, C. M., Macnab, L. T., Sullivan, R. K., and Pow, D. V. (2006). Immunocytochemical analysis of D-serine distribution in the mammalian brain reveals novel anatomical compartmentalizations in glia and neurons. Glia 53, 401–411. doi: 10.1002/glia.20300
Wolosker, H., Blackshaw, S., and Snyder, S. H. (1999a). Serine racemase: a glial enzyme synthesizing D-serine to regulate glutamate-N-methyl-D-aspartate neurotransmission. Proc. Natl. Acad. Sci. U.S.A. 96, 13409–13414. doi: 10.1073/pnas.96.23.13409
Wolosker, H., Sheth, K. N., Takahashi, M., Mothet, J. P., Brady, R. O. Jr., et al. (1999b). Purification of serine racemase: biosynthesis of the neuromodulator D-serine. Proc. Natl. Acad. Sci. U.S.A. 96, 721–725. doi: 10.1073/pnas.96.2.721
Yiu, G., and He, Z. (2006). Glial inhibition of CNS axon regeneration. Nat. Rev. Neurosci. 7, 617–627. doi: 10.1038/nrn1956
Keywords: glia, schizophrenia, autism, mouse models, astrocytes, oligodenrocytes, microglia, MeCP2
Citation: Yamamuro K, Kimoto S, Rosen KM, Kishimoto T and Makinodan M (2015) Potential primary roles of glial cells in the mechanisms of psychiatric disorders. Front. Cell. Neurosci. 9:154. doi: 10.3389/fncel.2015.00154
Received: 07 March 2015; Accepted: 06 April 2015;
Published online: 15 May 2015.
Edited by:
Tycho M. Hoogland, Netherlands Institute for Neuroscience, NetherlandsReviewed by:
Dmitry Lim, Università del Piemonte Orientale Amedeo Avogadro, ItalyZhihong Chen, Cleveland Clinic, USA
Copyright © 2015 Yamamuro, Kimoto, Rosen, Kishimoto and Makinodan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manabu Makinodan, Department of Psychiatry, Faculty of Medicine, Nara Medical University, 840 Shijo-cho, Kashihara, Nara 634-8522, Japan,bW1tQG5hcmFtZWQtdS5hYy5qcA==