 Juhyun Song
Juhyun Song Jong Eun Lee
Jong Eun Lee- 1Department of Anatomy, Yonsei University College of Medicine, Seoul, South Korea
- 2Brain Korea 21 Plus Project for Medical Sciences, Brain Research Institute, Yonsei University College of Medicine, Seoul, South Korea
Hyperglycemia results in oxidative stress and leads to neuronal apoptosis in the brain. Diabetes studies show that microglia participate in the progression of neuropathogenesis through their involvement in inflammation in vivo and in vitro. In high-glucose-induced inflammation, apoptosis signal regulating kinase 1 (ASK1) triggers the release of apoptosis cytokines and apoptotic gene expression. MicroRNA-Let7A (miR-Let7A) is reported to be a regulator of inflammation. In the present study, we investigated whether miR-Let7A regulates the function of microglia by controlling ASK1 in response to high-glucose-induced oxidative stress. We performed reverse transcription (RT) polymerase chain reaction, Taqman assay, real-time polymerase chain reaction, and immunocytochemistry to confirm the alteration of microglia function. Our results show that miR-Let7A is associated with the activation of ASK1 and the expression of anti-inflammatory cytokine (interleukin (IL)-10) and Mycs (c-Myc and N-Myc). Thus, the relationship between Let-7A and ASK1 could be a novel target for enhancing the beneficial function of microglia in central nervous system (CNS) disorders.
Introduction
Microglia are major glial cells with an important role in defending the brain against invading microorganisms, as they are the resident immunocompetent and phagocytic cells in the central nervous system (CNS) (Kreutzberg, 1996). However, the improper activation of microglia is related to neuronal damage in CNS diseases via the overproduction of free radicals and the secretion of various cytokines (Dringen, 2005; Krady et al., 2005; Frank-Cannon et al., 2009). Microglia activation is linked to changes in both their morphology, including cell hypertrophy (Tsuda et al., 2008), and their function (Wodarski et al., 2009; Xu et al., 2009). Recently, microglia have been found to exhibit two broad phenotypes: M1 and M2 types (Colton, 2009) Activated M1-type microglia release various neuroinflammatory mediators such as reactive oxygen species (ROS) (Quan et al., 2007) and pro-inflammatory cytokines (Min et al., 2003; Zhang et al., 2013), including interleukin (IL)-1β, IL-6, and tumor necrosis factor (TNF)-α, in the CNS (Hanisch, 2002). By contrast, activated M2-type microglia appear to suppress inflammation by producing anti-inflammatory cytokines, including IL-10 (Yang et al., 2009), tumor growth factor (TGF)-β (Mantovani et al., 2004; Martinez et al., 2008), and IL-4 (Stein et al., 1992). Hyperglycemia impairs neuronal function and leads to oxidative stress (Catanzaro et al., 2013). Cerebral cortex neurons become damaged by high-glucose-induced oxidative stress according to an experimental diabetes study (Arnal et al., 2010). The state of high-glucose induced by hyperglycemia leads to severe neuronal apoptosis in the brain (Knudsen et al., 1989; Sredy et al., 1991; Li and Sima, 2004; Sima et al., 2004) and impaired cognition (Li et al., 2002, 2003). Several studies report that activated microglia are associated with neurodegeneration (Klein et al., 2004) and structural changes, including hippocampal injury and cerebral atrophy, in animal models of diabetes (Musen et al., 2006; Kodl et al., 2008) as well as the progression of brain atrophy and cognitive decline in elderly diabetes patients (van Elderen et al., 2010). Furthermore, a recent in vitro study demonstrates that hyperglycemia increases microglial vulnerability to lipopolysaccharide (LPS)-induced inflammation (J. Y. Wang et al., 2001) and induces the secretion of pro-inflammatory cytokines and ROS and the activation of apoptotic signaling pathways, such as the nuclear factor kappa B (NF-κB) signaling pathway (Quan et al., 2007). Some research demonstrates that the inhibition of microglia activation attenuates diabetes-induced inflammatory cytokine production and reduces apoptosis (Krady et al., 2005). How microglia activation is modulated in high-glucose stress is a crucial problem to solve for hyperglycemia-induced neuroinflammatory diseases. Hyperglycemia-induced oxidative stress accelerates cell senescence and activates apoptosis signal regulating kinase 1 (ASK1) (Yokoi et al., 2006), which is a member of the mitogen-activated protein (MAP) kinase kinase kinase group associated with apoptosis signaling (Ichijo et al., 1997; Tobiume et al., 2001). ASK1 can cause apoptosis through non-transcription- or transcription-dependent mechanisms (Tobiume et al., 2001). MicroRNAs (miRNAs), which are endogenous 22-nucleotide RNAs that bind to the 3′-untranslated region of a messenger RNA (mRNA) target, regulate oxidative and inflammatory state (Marin et al., 2013) by affecting gene expression (Martinez and Tuschl, 2004; Du and Zamore, 2005; Schamberger et al., 2012). Several microRNAs including miR-155 (Arango et al., 2015; Cheng et al., 2015; Tan et al., 2015), miR-146 (Schulte et al., 2013; Echavarria et al., 2015), and miR-21(Quinn and O'Neill, 2011) reported the relationship with inflammation in various condition. miR-21, miR-126, and miR-210 are downregulated under in vitro diabetic conditions and in Type 1 diabetic patients (Osipova et al., 2014). In addition, miR-146a (Baldeon et al., 2014; Wang et al., 2014), miR-155(Lin et al., 2014), miR-133 (Xiao et al., 2007), miR-1, and miR-206 (Shan et al., 2010) regulate diverse mechanisms in high-glucose stress. Among miRNAs, miR-Let7 in particular is known to regulate glucose metabolism (Zhu et al., 2011) and is linked to inflammatory pathways (Iliopoulos et al., 2009). Here, we investigated the relationship between miR-Let7A and ASK1 activation in microglia function against high-glucose stress. Our results increase our understanding of the mechanisms underlying microglia function in hyperglycemia-induced oxidative stress states.
Materials and Methods
Microglia Culture and D-Glucose Treatment
Murine BV2 microglial cells were obtained from Prof. Eun-hye Joe (Ajou University School of Medicine, Chronic Inflammatory Disease Research Center) and cultured in Dulbecco's Modified Eagle's Medium (Gibco, NY, USA) supplemented with 10% fetal bovine serum (Gibco, NY, USA) and 100 μg/ml penicillin-streptomycin (Gibco, NY, USA) at 37°C in a humidified atmosphere containing 5% CO2. Also we treated D-glucose 25 mM, 60 mM and 90 mM, and 120 mM (Sigma, St. Louis, MO, USA) in microglia cells, considering other in vitro studies in high glucose concentrations (25 mM -120 mM) (Quan et al., 2007, 2011; Dey et al., 2015; Zhang et al., 2015).
miRNA Transfection
BV2 microglia were treated with miR-Let7A mimic or anti-miR-Let7A. Let-7A mimic (Let-7A precursor molecules are considered to be processed into mature miRNA by mimicking the Let-7A natural shearing process) and anti-Let-7A (Let7A inhibitor) were purchased from Ambion (Austin, TX, USA). For the transfection of RNA duplexes, a 20 nM final concentration of Let-7A miRNA mimic or Let-7A inhibitor was mixed with lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) in Opti-MEM medium and incubated at room temperature for 10 min. The mixture was added to cells in 6-well plates. After cells reached 60% confluence, they were harvested for total protein or RNA extraction. ASK1 inhibitor (NQDI-1) was purchased from Tocris Bioscience (Bristol, UK). BV2 microglial cells were pretreated with ASK1 inhibitor (600 nM) to inhibit ASK1 activation 2 h before high-glucose injury.
Cell Viability Assay
BV2 microglia (2 × 105 cells/ml) were seeded in 96-well plates to examine the effects of all experimental treatments. Cells were then rinsed with phosphate-buffered saline (PBS), and culture medium was replaced with serum-free medium. Next, 100 μl 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma, St. Louis, MO, USA) solution (5 mg/ml in PBS) was added per well. After 1 h of incubation, the medium was removed, and dimethyl sulfoxide was added to solubilize the purple formazan product of the MTT reaction. The supernatant from each well was measured using an ELISA plate reader at a wavelength of 570 nm with background subtraction at 650 nm. All experiments were repeated at least six times. Cell viability was not considered 100% for any treatment group. Cell viability was expressed as a percentage relative to the control group value.
Reverse Transcription Polymerase Chain Reaction (RT-PCR)
To examine the expression of IL-10, c-Myc, and N-Myc in BV2 microglia, RT-PCR was performed using each primer. Briefly, samples were lysed with Trizol reagent (Invitrogen, Carlsbad, CA, USA), and total RNA was extracted according to the manufacturer's protocol. cDNA synthesis from mRNA and sample normalization were performed. PCR was performed using the following thermal cycling conditions: 95°C for 10 min; 35 cycles of denaturing at 95°C for 15 s, annealing at 70°C for 30 s, elongation at 72°C for 30 s; final extension at 72°C for 10 min; and holding at 4°C. PCR was performed using the following primers (5′–3′); IL-10 (F): CCAAGCCTTATCGGAAATGA, (R) TTTTCACAGGGGAGAAATCG; c-Myc (F): TCAAGAGGCGAACACACAAC, (R): GGCCTTTTCATTGTTTTCCA; N-Myc (F): GTCACCACATTCACCATCACTGT, (R): AGCGTGTTCAATTTTCTTTAGCA; GAPDH (F): GGCATGGACTGTGGTCATGAG, (R): TGCACCACCAACTGCTTAGC. PCR products were electrophoresed in 1.5% agarose gels and stained with ethidium bromide.
Quantitative Real-time PCR
To examine the amount of TNF-α, IL-6, IL-10, and ASK1 mRNA in BV2 cells under high-glucose conditions, quantitative real-time PCR was performed using each primer. Total cellular RNA was extracted from BV2 microglia using Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. RNA was mixed with One Step SYBR® Prime Script TM RT-PCR Kit II (Takara, Otsu, Shiga, Japan) and specific primers in a total reaction volume of 20 μl. PCR was performed using the following primers (5′–3′); TNF-α (F): CAAGGGACAAGGCTGCCCCG, (R): GCAGGGGCTCTTGACGGCAG; IL-6 (F): AACGATGATGCACTTGCAGA, (R): CTCTGAAGGACTCTGGCTTTG; IL-10 (F): CCAAGCCTTATCGGAAATGA, (R): TTTTCACAGGGGAGAAATCG; ASK1 (F): AGGACGGAGACTGTGAGGGT, (R): GTCCTGCATAGACGATCCCAT; GAPDH (F): GGCATGGACTGTGGTCATGAG, (R): TGCACCACCACTGCTTAGC. Amplification cycles were carried out at 42°C for 5 min, 95°C for 10 s, 95°C for 5 s, 60°C for 34 s, and 65°C for 15 s. Quantitative SYBR Green real-time PCR was performed with an ABI prism 7500 Real-Time PCR System (Life Technologies Corporation, CA, USA) and analyzed with comparative Ct quantification (Popivanova et al., 2008). GAPDH was amplified as an internal control. The Ct value for GAPDH was subtracted from the Ct values for each gene (ΔCt). The ΔCt values of treated cells were compared with those of untreated cells.
Taqman Assay
Total RNA was extracted from cells using Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. For quantitative analysis of miR-Let7A, reverse transcription (RT) was performed using Taqman Micro RNA RT kit (Takara, Otsu, Shiga, Japan) according to the manufacturer's instructions with 10 ng total RNA. PCR reactions were then performed according to manufacturer's instructions to quantify the expression level of miR-Let7A using Taqman Universal PCR Master Mix, No Amp Erase UNG (Applied Biosystems, Foster City, CA, USA), and Taqman microRNA assay (Takara, Otsu, Shiga, Japan) for the miR-Let7A of interest. The PCR amplification was performed in ABI 7500 Real Time PCR (Life Technologies Corporation, CA, USA) at 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 60 s. The PCR incubation profile was extended to 40 cycles for miR-Let7A. The PCR primers for Let-7A were (F): 5′-GCGCCTGAGGTAGTAGGTTG-3′ and (R): 5′-CAGTGCAGGGTCCGAGGT-3′. The PCR primers for U6 were (F): 5′-CTCGCTTCGGCAGCACATATACT-3′ and (R): 5′-ACGCTTCACGAATTTGCGTGTC-3′, respectively. The data were uniformly normalized to the internal control U6, and relative expression levels were evaluated using the 2−ΔΔCt method. All experiments were run in triplicate (Zhang et al., 2014).
Western Blot Analysis
After drug treatment and exposure to high glucose stress, BV2 cells were washed twice with ice-cold PBS, scraped, and collected. BV2 cell pellets were lysed with ice-cold RIPA buffer (Sigma-Aldrich, St. Louis, MO, USA). The lysates were centrifuged at 13,200 rpm for 1 h at 4°C to produce whole-cell extracts. Protein content was quantified using the BCA protein assay kit (Pierce, IL, USA). Protein (30 μg) was separated on a 10% SDS-polyacrylamide gel and transferred onto a polyvinylidene difluoride membrane. After blocking with 5% bovine serum albumin in TBS/Tween (20 nM Tris (pH 7.2), 150 mM NaCl, 0.1% Tween 20) for 1 h at room temperature, immunoblots were incubated overnight at 4°C with primary antibodies that specifically detect phosphorylation-ASK1 (p-ASK1) (1:1000; Cell signaling biotechnology, Danvers, MA, USA), ASK1 (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA, USA), or β-actin (1:2000; Cell Signaling, Danvers, MA, USA). Blots were then incubated with horseradish peroxidase-linked anti-mouse or -rabbit IgG antibodies (Abcam, Cambridge, MA, USA) for 1 h at room temperature. Protein bands were detected and analyzed using enhanced chemiluminescence (ECL; Pierce, IL, USA)(Song et al., 2014).
Immunocytochemistry
BV2 microglia were washed three times with PBS for immunostaining and blocked for 30 min. BV2 microglia were then incubated with primary antibodies overnight at 4°C. The following primary antibodies were used: anti-goat CD68 (1:200, Millipore, Billerica, MA, USA) and anti-rabbit CD206 (1:200, Millipore, Billerica, MA, USA). After incubating BV2 microglia with the primary antibodies, cells were washed with PBS three times for 3 min each. Next, samples were incubated with fluorescein isothiocyanate (FITC)-conjugated donkey anti-goat (1:200, Jackson Immunoresearch, West Grove, PA, USA) or rhodamine-conjugated goat anti-rabbit (1:200, Jackson Immunoresearch, West Grove, PA, USA) for 2 h at room temperature. BV2 microglia were washed with PBS three times for 3 min each and then counterstained with 1 μg/ml 4′, 6-diamidino-2-phenylindole (DAPI) (1:100, Invitrogen, Carlsbad, CA, USA) for 10 min at room temperature. Fixed samples were imaged using a Zeiss LSM 700 confocal microscope (Carl Zeiss, Thornwood, NY, USA).
Statistical Analysis
Statistical analyses were carried out using SPSS 18.0 software (IBM Corp., Armonk, NY, USA). All data are expressed as mean ± standard error of the mean (S.E.M.). Significant group differences were determined by one-way analysis of variance followed by Bonferroni post-hoc multiple comparisons tests. Each experiment consisted of three replicates per condition. Differences were considered statistically significant at *p < 0.05 and **p < 0.001.
Results
Inhibition of ASK1 Activation Enhances Cell Viability against High-glucose Stress
To investigate the viability of BV2 microglia in high-glucose conditions, we conducted an MTT assay with different concentration of glucose (Figure 1A). Cell viability was reduced by glucose treatment (25, 60, 90, or 120 mM). After pre-treatment with ASK1 inhibitor (600 nM, TOCRIS, Bristol, UK), the viability of high-glucose-exposed microglia significantly increased by 20% compared with that of the high-glucose treatment alone group (Figure 1A). Because cell viability showed the largest change by ASK1 inactivation in the 120 mM glucose condition, we used this concentration in subsequent experiments. To determine the whether ASK1 directly participates in the alteration of cell viability against high-glucose stress, we conducted quantitative real time-PCR using ASK1-specific primer (Figures 1B,C). ASK1 expression was increased in the 25 mM glucose condition and was markedly reduced after ASK1 inhibitor pre-treatment (Figure 1B). ASK1 expression was increased in the 120 mM glucose condition and was significantly reduced after ASK1 inhibitor pre-treatment (Figure 1C).
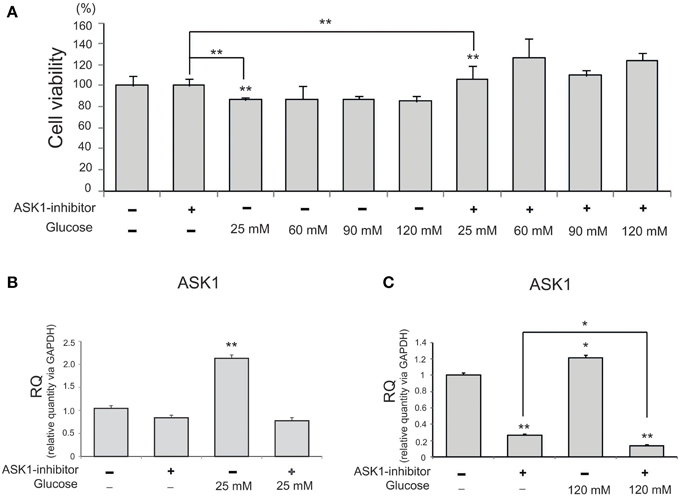
Figure 1. The measurement of cell viability and the expression of ASK1 in microglia under high glucose condition. (A) MTT assay. The graph showed that BV2 microglia in the high glucose treatment groups (25, 60, 90, 120 mM) exhibited decrease of cell viability compared to the normal control group. BV2 microglia in all ASK1 inhibitor pre-treatment groups exhibited increase of cell viability after high glucose injury compared to the normal control group. Especially, the microglia in the glucose 120 mM with ASK1 inhibitor conditions showed markedly the increase of cell viability comparison with ASK1 inhibitor treatment group. Each experiment included the six repeats per condition. (B) The measurement of relative quantitative mRNA level. The graph showed the mRNA level of ASK1 in glucose condition. In high glucose 25 mM treatment group, the level of ASK1 mRNA increased evidently compared to the no treatment group. ASK1 inhibitor pre-treatment attenuated the expression of ASK1 in microglia in spite of 25 mM high glucose condition. GAPDH was used as a control. Each experiment included the three repeats per condition. Each experiment included the three repeats per condition. Data were expressed as mean ± S.E.M, and were analyzed statistically using One-Way ANOVA, followed by Bonferroni's post hoc. **p < 0.001. (C) The measurement of relative quantitative mRNA level. The graph showed the mRNA level of ASK1 in glucose condition. In high glucose 120 mM treatment group, the level of ASK1 mRNA increased evidently compared to the no treatment group. ASK1 inhibitor pre-treatment attenuated the expression of ASK1 in microglia in spite of 120 mM high glucose condition. GAPDH was used as a control. Each experiment included the three repeats per condition. Each experiment included the three repeats per condition. Data were expressed as mean ± S.E.M, and were analyzed statistically using One-Way ANOVA, followed by Bonferroni's post hoc. *p < 0.05, **p < 0.001. RQ, relative quantitative value of mRNA via GAPDH.
Measurement of M1- and M2-type Microglia after High-glucose Injury
To observe the function of microglia, we investigated the expression of CD68 (Figure 2A) and CD206 (Figure 2B) in 25 mM and 120 mM high glucose concentration using immunocytochemical analysis. The expression of CD68 was considerably increased in the high-glucose treatment group (120 mM) and was attenuated by the inhibition of ASK1 activation (Figure 2A). The expression of CD206 was considerably reduced in the high-glucose treatment group (120 mM) and was slightly increased by the inhibition of ASK1 (Figure 2B).
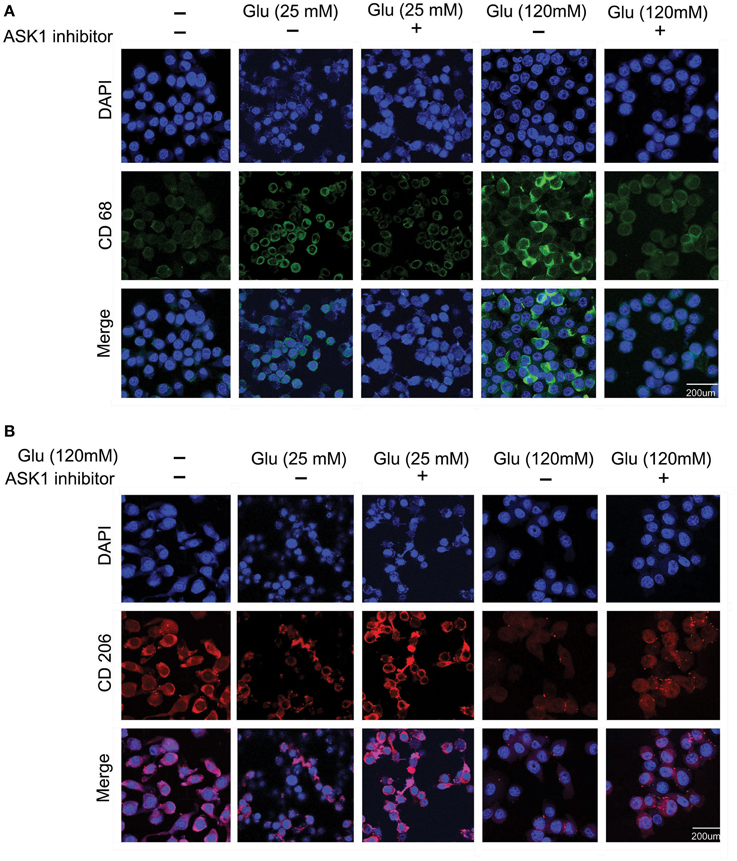
Figure 2. The immunocytochemical images to determine the microglia phenotype. (A) The expression of CD 68 (considered as the marker of M1 type microglia) was evaluated by immunocytochemistry. This image showed that high glucose 120 mM treated microglia expressed more the CD 68 protein in microglia than the untreated microglia. Also, the ASK1 inhibitor pre-treated microglia showed the attenuation of CD 68 expression compared with the normal microglia. (B) The expression of CD 206 (considered as the marker of M2 type microglia) was examined by immunocytochemistry. This image showed that high glucose 120 mM treated microglia expressed lesser the CD 206 protein in microglia than the untreated microglia. In addition, the ASK1 inhibitor pre-treated microglia showed the increase of CD 206 expression compared with the normal microglia. Scale bar: 200 μm, 4′, 6-diamidino-2-phenylindole (DAPI): blue, CD 68: green, CD 206: red. Glu (25 mM): glucose 25 mM treatment, Glu (120 mM): glucose 120 mM treatment.
Measurement of Cytokine Levels in Microglia after High-glucose Injury
To examine the mRNA level of cytokines in microglia following the inhibition of ASK1 activation in high-glucose injury (25 mM and 120 mM), we conducted quantitative real-time PCR (Figures 3A–D) and RT-PCR (Figure 3E). The mRNA level of pro-inflammatory cytokine TNF-α in microglia was increased in the high-glucose treatment group (Figures 3A,C) and was dramatically reduced by ASK1 inhibition (Figures 3A,C). The mRNA level of pro-inflammatory cytokine IL-6 in microglia was significantly increased in the high-glucose treatment group (Figures 3B,D) and was decreased by ASK1 inhibition (Figure 3B). The mRNA level of anti-inflammatory cytokine IL-10 in microglia was significantly decreased in the high-glucose treatment group (Figure 3E) and tended to increase after ASK1 inhibition (Figure 3E). Taken together, these results indicate that the inhibition of ASK1 activation regulates the expression of cytokines.
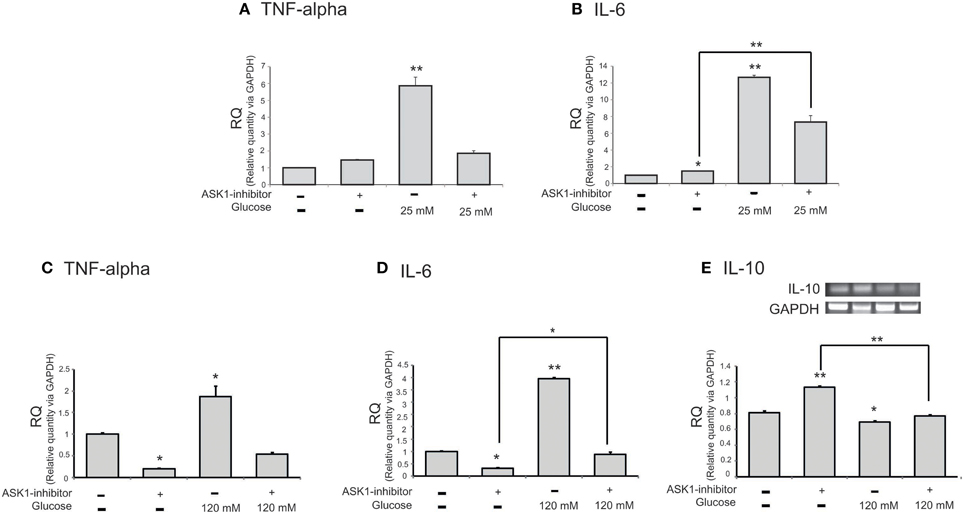
Figure 3. The mRNA level of cytokines on microglia in high glucose stress. The mRNA levels of TNF-alpha, IL-6, IL-10 were measured using quantitative real time–PCR (A–D) and reverse transcription-PCR (E). (A) The mRNA level of TNF-alpha was increased in the high glucose treatment group (25 mM) and reduced in ASK1 inhibitor pre-treated high glucose group. (B) The mRNA amount of IL-6 was increased in the high glucose treatment group (25 mM) and significantly decreased in ASK1 inhibitor pre-treated high glucose group. (C) The mRNA level of TNF-alpha was increased in the high glucose treatment group (120 mM) and reduced in ASK1 inhibitor pre-treated high glucose group. (D) The mRNA amount of IL-6 was increased in the high glucose treatment group (120 mM) and significantly decreased in ASK1 inhibitor pre-treated high glucose group (120 mM). (E) The mRNA level of IL-10 exhibited decrease of expression in the high glucose treatment group (120 mM) whereas it significantly increased in ASK1 inhibitor pre-treated high glucose group (120 mM). GAPDH was used as a control. Each experiment included the 3 repeats per condition. Data were expressed as mean ± S.E.M, and were analyzed statistically using One-Way ANOVA, followed by Bonferroni's post hoc. *p < 0.05, **p < 0.001. RQ, relative quantitative value of mRNA via GAPDH.
Measurement of miR-Let7A Level in BV2 Microglia after High-glucose Injury
To examine levels of miR-Let7A in microglia following the inhibition of ASK1 activation in high-glucose injury (25 mM and 120 mM), we conducted a Taqman assay (Figure 4). The level of miR-Let7A in microglia was significantly increased in the high-glucose treatment group [25 mM (Figure 4A) and 120 mM (Figure 4B)] and was reduced by ASK1 inhibition (Figure 4). The level of miR-Let7A in microglia in the high-glucose treatment group was almost the same as that in microglia with overexpression of Let7A plus ASK1 inactivation in the high-glucose condition (Figure 4).
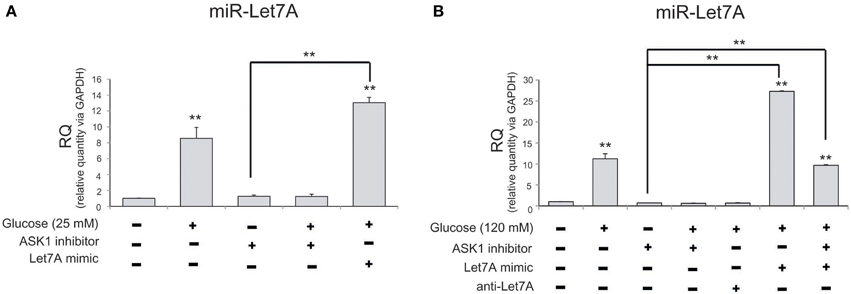
Figure 4. The measurement of miR-Let7A on microglia in high glucose stress. (A) In high glucose 25 mM treatment group, miR-Le7A level was considerably increased in comparison with the no treatment group. Under high glucose stress, ASK1 inhibitor inhibited the reduced level of miR-Let7A compared with the high glucose treatment group. The co-treatment with glucose 25 mM and miR-Let7A mimic showed higher level of miR-Le7A than high glucose treatment group. U6 was used as a control. Each experiment included the three repeats per condition. Data were expressed as mean ± S.E.M, and were analyzed statistically using One-Way ANOVA, followed by Bonferroni's post hoc. **p < 0.001. (B) In high glucose 120 mM treatment group, miR-Le7A level was considerably increased compared to the no treatment group. Under high glucose stress, ASK1 inhibitor inhibited the reduced level of miR-Let7A compared with the high glucose treatment group. The co-treatment with ASK1 inhibitor and miR-Let7A mimic showed lesser level of miR-Le7A than high glucose treatment group. U6 was used as a control. Each experiment included the 3 repeats per condition. Data were expressed as mean ± S.E.M, and were analyzed statistically using One-Way ANOVA, followed by Bonferroni's post hoc. **p < 0.001. RQ, relative quantitative value of mRNA via U6; ASK1 inhibitor, ASK1 inhibitor (600 nM) pre-treatment and high glucose injury group/The inhibition of ASK1 activation group; Let7A mimic, Let7A over expression group in high glucose condition; anti-Let7A, Let7A inhibition in high glucose condition; Glucose (25 mM), glucose 25 mM treatment; Glucose (120 mM), glucose 120 mM treatment.
Relationship between ASK1 and miR-Let7A in High-glucose Injury
To confirm a relationship between miR-Let7A and ASK1 activation in microglia in high-glucose injury, we conducted quantitative real-time PCR (Figure 5A) and RT PCR (Figures 5B,C) and western blot analysis (Figures 5D–F). The mRNA level of ASK1 in microglia was significantly increased in the high-glucose treatment group and was reduced in the miR-Let7A mimic transfection group (Figure 5A). Also, the mRNA level of ASK1 was reduced by Let7A mimic transfection under glucose 120 mM treatment condition (Figure 5A). The mRNA level of ASK1 in BV2 cells was reduced by Let7A mimic treatment (Figure 5B), and ASK1 inhibitor treatment (Figure 5C) in comparison with the only glucose 120 mM treatment group (Figures 5B,C). The activation of ASK1 [the phosphorylation of ASK1(p-ASK1)/ASK1 protein level] was increased in glucose 120 mM treatment group, while the increased phosphorylation of ASK1 in glucose 120 mM treatment was attenuated by the inhibition of ASK1 (Figure 5E). The activation of ASK1 was reduced in Let7A treatment group (Figure 5F).
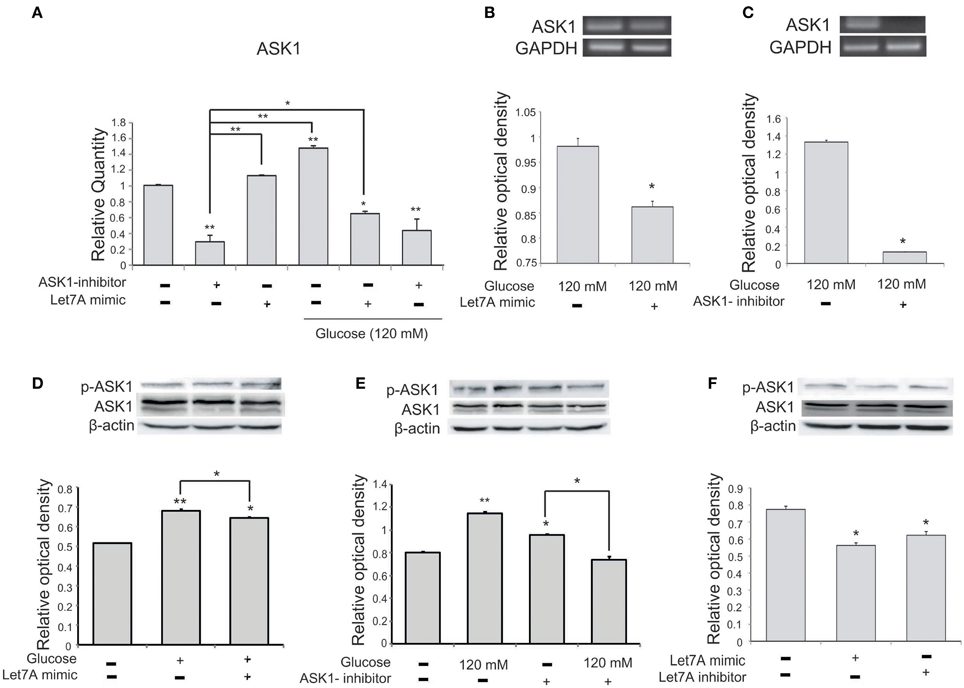
Figure 5. The measurement of ASK1 on microglia in high glucose stress. (A) The mRNA level of ASK1 was evidently increased by glucose treatment. Under high glucose stress, ASK1 inhibitor and the miR-Let7A overexpression group inhibited the decreased level of miR-Let7A compared to the normal control group. GAPDH was used as a control. Each experiment included the three repeats per condition. Data were expressed as mean ± S.E.M, and were analyzed statistically using One-Way ANOVA, followed by Bonferroni's post hoc. *p < 0.05, **p < 0.001, (B) The overexpression of Let7A was significantly decreased the mRNA of ASK1 in glucose 120 mM treatment condition in comparison with the only glucose treatment group. GAPDH was used as a control. Each experiment included the three repeats per condition. Data were expressed as mean ± S.E.M, and were analyzed statistically using One-Way ANOVA, followed by Bonferroni's post hoc. *p < 0.05. (C) The inhibition of ASK1 was significantly attenuated the mRNA of ASK1 in glucose 120 mM treatment condition in comparison with the only glucose treatment group. GAPDH was used as a control. Each experiment included the three repeats per condition. Data were expressed as mean ± S.E.M, and were analyzed statistically using One-Way ANOVA, followed by Bonferroni's post hoc. *p < 0.05. (D) The phosphorylation of ASK1/ASK1 level was increased by glucose 120 mM treatment compared to the normal control group. Also the activation of ASK1 was reduced in the glucose with Let7A overexpression in comparison with the only glucose treatment group. ß-actin was used as a control. Each experiment included the three repeats per condition. Data were expressed as mean ± S.E.M, and were analyzed statistically using One-Way ANOVA, followed by Bonferroni's post hoc. *p < 0.05, **p < 0.001. (E) The activation of ASK1 was reduced by ASK1 inhibitor treatment under glucose 120 mM treatment in comparison with the ASK1 inhibitor treatment group. β-actin was used as a control. Each experiment included the three repeats per condition. Data were expressed as mean ± S.E.M, and were analyzed statistically using One-Way ANOVA, followed by Bonferroni's post hoc. *p < 0.05, **p < 0.001. (F) The activation of ASK1 was reduced by Let7A mimic treatment compared with the no treatment group. β-actin was used as a control. Each experiment included the three repeats per condition. Data were expressed as mean ± S.E.M, and were analyzed statistically using One-Way ANOVA, followed by Bonferroni's post hoc. *p < 0.05. ASK1 inhibitor, ASK1 inhibitor (600 nM) pre-treatment and high glucose injury group/The inhibition of ASK1 activation group; Let7A mimic, Let7A over expression group in high glucose condition; anti-Let7A, Let7A inhibition in high glucose condition; p-ASK1: the phosphorylation of ASK1.
Effect of ASK1 Inhibition and miR-Let7A Overexpression on Cytokine Expression in High-glucose Injury
To examine changes in cytokine mRNA levels in microglia after ASK1 inactivation and miR-Let7A overexpression under high-glucose injury, we conducted quantitative real-time PCR. The mRNA level of pro-inflammatory cytokine IL-6 in microglia was significantly increased in the high-glucose treatment group (Figure 6A) and was also increased in the Let7A mimic transfection group. In addition, the expression of IL-6 was decreased in the ASK1 inactivation group and in the ASK1 inhibition plus Let7A overexpression group after high-glucose injury (Figure 6A). The mRNA level of anti-inflammatory cytokine IL-10 in microglia was reduced in the high-glucose treatment group (Figure 6B) and was increased in the miR-Let7A overexpression group. In addition, the expression of IL-10 was increased in the ASK1 inactivation group in the high-glucose condition compared with that in the high-glucose treatment alone group. In the inhibition of ASK1 plus Let7A overexpression in high-glucose injury group, the expression of IL-10 was slightly increased compared with that in the Let7A mimic transfection group (Figure 6B).
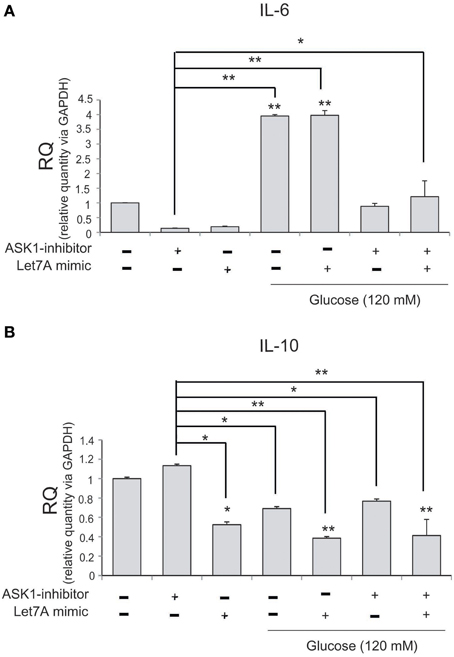
Figure 6. The mRNA level of cytokines on microglia in high glucose stress. The mRNA levels of IL-6, IL-10 in ASK1 inhibition and miR-Let7A overexpression were measured using quantitative real time–PCR. (A) The mRNA level of IL-6 was increased in the high glucose treatment group and reduced in ASK1 inhibitor pre-treatment group. In high glucose condition, IL-6 mRNA level was attenuated as the inhibition of ASK1 activation. (B) The mRNA level of IL-10 was reduced in the high glucose treatment group and increased in ASK1 inhibitor pre-treatment group. In high glucose condition, IL-10 mRNA level was attenuated through the overexpression of miR-Let7A whereas the inhibition of ASK1 activation was promoted the expression of IL-10. GAPDH was used as a control. Each experiment included the three repeats per condition. Data were expressed as mean ± S.E.M, and were analyzed statistically using One-Way ANOVA, followed by Bonferroni's post hoc. *p < 0.05, **p < 0.001. RQ, relative quantitative value of mRNA.
Alteration of N-Myc and c-Myc Expression by ASK1 Inhibition and Let7A Overexpression in High-glucose Injury
To examine the regulation of N-Myc and c-Myc by ASK1 inhibition and miR-Le7A overexpression, we analyzed the mRNA level of these factors using RT-PCR (Figure 7). The mRNA level of N-Myc was increased in the high-glucose condition (Figure 7A). The inhibition of ASK1 activation markedly decreased N-Myc expression, whereas miR-Let7A overexpression did not result in large changes in N-Myc mRNA levels in the high-glucose condition (Figure 7A). Moreover, the inhibition of ASK1 in the Let7A mimic transfection condition decreased N-Myc mRNA level to almost the same value as that for ASK1 inhibition under the high-glucose condition (Figure 7A). Based on our findings, we assume that miR-Let7A links ASK1 to N-Myc in the high-glucose condition. In addition, the mRNA level of c-Myc was significantly increased in the high-glucose condition (Figure 7B). The inhibition of ASK1 activation decreased c-Myc expression, and miR-Let7A overexpression led to a slight decrease of c-Myc mRNA level under the high-glucose condition compared with that for the high-glucose treatment alone group (Figure 7B). Moreover, the inhibition of ASK1 plus Let7A overexpression increased c-Myc mRNA level in the high-glucose condition (Figure 7B).
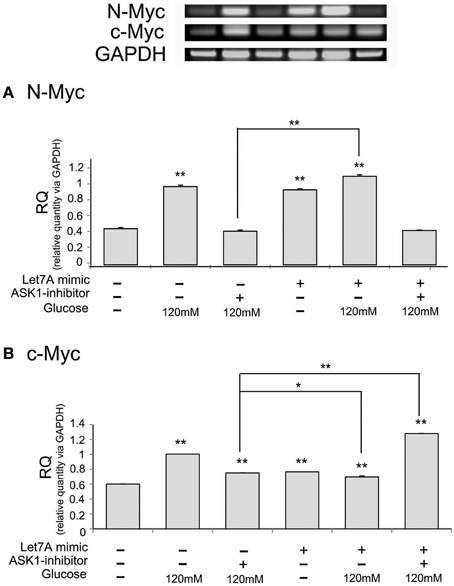
Figure 7. The mRNA level of N-Myc and c-Myc on microglia in high glucose stress. The mRNA levels of N-Myc and c-Myc were measured using reverse transcription—PCR (A,B). (A) The mRNA level of N-Myc evidently exhibited increase of expression in the high glucose treatment group whereas it was decreased in ASK1 inhibitor pre-treated group. In ASK1 inhibition with Let7A overexpression group, microglia expressed lower N-Myc mRNA level in high glucose condition than only high glucose treatment group. (B) The mRNA level of c-Myc showed increase of expression in the high glucose treatment group whereas it significantly was attenuated in ASK1 inhibitor pre-treated high glucose group. In ASK1 inhibition with Let7A overexpression group, microglia was increased c-Myc mRNA level in high glucose condition than only high glucose treatment group. GAPDH was used as a control. Each experiment included the four repeats per condition. Data were expressed as mean ± S.E.M, and were analyzed statistically using One-Way ANOVA, followed by Bonferroni's post hoc. *p < 0.05, **p < 0.001. RQ, relative quantitative value of mRNA. ASK1 inhibitor, ASK1 inhibitor (600 nM) pre-treatment and high glucose injury group/The inhibition of ASK1 activation group; Let7A mimic. Let7A over expression group in high glucose condition.
Discussion
In the CNS, elevated glucose levels lead to severe neuronal death and impaired cognition (Duarte et al., 2008; McCrimmon et al., 2012). In hyperglycemic conditions, ASK1 is activated, involving apoptotic gene expression (F. Wang et al., 2015). miRNAs are small, 19− to 23−nucleotide RNA molecules that play a role in regulating protein expression in cells (Cai et al., 2010; Feng et al., 2010; Lu et al., 2010). miRNAs have been implicated in various cellular processes such as proliferation and apoptosis (Bartel, 2004; Bentwich et al., 2005). In particular, previous studies suggest that miR-Let7A is associated with apoptotic signaling in high-glucose conditions (Urbich et al., 2008; Iliopoulos et al., 2009; Zhu et al., 2011; Perez et al., 2013). Our results confirm the involvement of ASK1 in microglia cell viability and activation in high-glucose conditions, consistent with findings that ASK1 is associated with high-glucose-induced endoplasmic reticulum stress and apoptosis (Jiang et al., 2015; Wang et al., 2015). In addition, our results regarding the M1-type marker CD 68 (Lemstra et al., 2007; Wojtera et al., 2012) and M2-type marker CD 206 (Stein et al., 1992; Mulder et al., 2014) indicate that the inhibition of ASK1 may induce activation of M2-type microglia and affect signaling that suppresses the inflammation response, suggesting that M2-type macrophages protect cells from inflammation (Varin and Gordon, 2009; Colton and Wilcock, 2010; Cherry et al., 2014). In the present study, a high-glucose state led to increases in the levels of pro-inflammatory cytokines such as TNF-α and IL-6 (Varnum and Ikezu, 2012) and decreases in the levels of anti-inflammatory cytokine such as IL-10 (Ledeboer et al., 2000). Furthermore, we found that inhibition of ASK1 attenuated the release of pro-inflammatory cytokines and promoted the release of anti-inflammatory cytokine such as IL-10 in high-glucose stress. Moreover, more miR-Let7A expression occurred in the ASK1 activation condition than in the normal condition. Based on our results, we assume that miR-Let7A may be linked to the activation of ASK1 and may be involved in the expression of cytokines in hyperglycemia. In the present study, the transfection of miR-Let7A and anti-miR-Let7A led to ASK1 activation and altered the expression of cytokines. In particular, miR-Let7A tended to reduce pro-inflammatory cytokines more than anti-inflammatory cytokines. miR-Let7A regulated the expression of cytokines more in the ASK1 inhibitor co-treatment group than the miR-Let7A mimic transfection only group. Several studies demonstrate that anti-inflammatory cytokines are regulated through inhibition of ASK1 activation (Liu et al., 2012), and ASK1 is involved in the production of inflammatory cytokines in macrophages (Matsuzawa et al., 2005; Osaka et al., 2007; Yuk et al., 2009). Considering previous evidence, our results indicate that Let7A miRNA may strongly regulate the expression of inflammatory cytokines by controlling ASK1 activation. Specifically, our results show that miR-Let7A may affect the expression of anti-inflammatory cytokines including IL-10 by regulating ASK1 activation under glucose-induced stress. IL-10 is an anti-inflammatory cytokine that inhibits the production of pro-inflammatory cytokines including TNF-α, IL-1, and IL-6 (Joyce et al., 1994; Joyce and Steer, 1996; Sawada et al., 1999) in response to oxidative stress (Ledeboer et al., 2000; Molina-Holgado et al., 2001; Szczepanik et al., 2001), and its expression is also correlated with protection against neuronal damage (de Waal Malefyt et al., 1991; Fiorentino et al., 1991; Froen et al., 2002). Also, gene delivery of either IL-10 in the rat hippocampus alleviates deficits in learning and memory function (Lynch et al., 2004; Nolan et al., 2005). Based on our results, the promotion of IL-10 by miR-Let7A transfection may suppress the inflammatory response and protect neurons via ASK1 activation in hyperglycemic conditions. The Myc protein, as an unstable nuclear phosphorylated protein, is involved in many cellular mechanisms (Hann and Eisenman, 1984; Ramsay et al., 1986). c-Myc induces apoptosis signaling with mitogenic signals (Prendergast, 1999). c-Myc protein is phosphorylated at both Ser-62 and Ser-71 by JNK (Noguchi et al., 1999) and is linked to ASK1 signaling (Noguchi et al., 2001). c-Myc triggers the activation of ASK1 and the mitochondrial apoptosis pathway (Evan and Littlewood, 1998; Noguchi et al., 1999; Desbiens et al., 2003) and induces cell proliferation (Sears et al., 1999). Furthermore, c-Myc is a crucial factor in both ribosome biogenesis and p53 stabilization (Menne et al., 2007; Dai and Lu, 2008; Kim et al., 2011). A recent study demonstrates that the inhibition of p38 and ASK1 reduces N-myc transcript levels (Guldal et al., 2012). N-myc triggers the apoptosis of neuroblastoma in hypoxia stress (Rossler et al., 2001). In hepatocytes, N-myc also leads to apoptosis following serum deprivation (Ueda and Ganem, 1996). N-myc was originally identified by oncogene expression profiling of human neuroblastoma cells (Schwab et al., 1983). The elevated expression of N-myc increases the susceptibility of neuroblastoma cells to diverse types of cell death signaling (Schweigerer et al., 1990; Malynn et al., 2000; Rossler et al., 2001). In addition, N-myc plays a crucial role in microglia function by regulating inflammatory mediators (Jung et al., 2005). Several researchers have discovered that the N-myc downstream-regulated gene 2 (Ndrg2) plays a key role in reactive astrogliosis involving IL-6/STAT3 signaling (Takarada-Iemata et al., 2014), and N-myc controls cytokine-induced regulator IRE1 protein and c-Jun N-terminal kinase in pancreatic beta cells (Brozzi et al., 2014). Considering our results, we speculate that miR-Let7A regulates the expression of c-Myc and N-myc by involving ASK1 activation in high-glucose conditions and that miR-Let7A may also be associated with the cytokine secretion pathway by regulating c-Myc and N-Myc protein. In conclusion, we suggest that miR-Let7A may affect microglia by involving ASK1 activation in hyperglycemia. The results of the present study increase our understanding of the relationship between miR-Let7A and ASK1 and the function of microglia in hyperglycemia-induced oxidative stress.
Author Contributions
JS performed the experiences and drafted the manuscript. JL discussed the findings and helped draft the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science, and Technology (NRF-2014R1A2A2A01006556).
References
Arango, D., Diosa-Toro, M., Rojas-Hernandez, L. S., Cooperstone, J. L., Schwartz, S. J., Mo, X., et al. (2015). Dietary apigenin reduces LPS-induced expression of miR-155 restoring immune balance during inflammation. Mol. Nutr. Food Res. 59, 763–772. doi: 10.1002/mnfr.201400705
Arnal, E., Miranda, M., Barcia, J., Bosch-Morell, F., and Romero, F. J. (2010). Lutein and docosahexaenoic acid prevent cortex lipid peroxidation in streptozotocin-induced diabetic rat cerebral cortex. Neuroscience 166, 271–278. doi: 10.1016/j.neuroscience.2009.12.028
Baldeon, R. L., Weigelt, K., de Wit, H., Ozcan, B., van Oudenaren, A., Sempertegui, F., et al. (2014). Decreased serum level of miR-146a as sign of chronic inflammation in type 2 diabetic patients. PLoS ONE 9:e115209. doi: 10.1371/journal.pone.0115209
Bartel, D. P. (2004). MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297. doi: 10.1016/S0092-8674(04)00045-5
Bentwich, I., Avniel, A., Karov, Y., Aharonov, R., Gilad, S., Barad, O., et al. (2005). Identification of hundreds of conserved and nonconserved human microRNAs. Nat. Genet. 37, 766–770. doi: 10.1038/ng1590
Brozzi, F., Gerlo, S., Grieco, F. A., Nardelli, T. R., Lievens, S., Gysemans, C., et al. (2014). A combined “omics” approach identifies N-Myc interactor as a novel cytokine-induced regulator of IRE1 protein and c-Jun N-terminal kinase in pancreatic beta cells. J. Biol. Chem. 289, 20677–20693. doi: 10.1074/jbc.M114.568808
Cai, B., Pan, Z., and Lu, Y. (2010). The roles of microRNAs in heart diseases: a novel important regulator. Curr. Med. Chem. 17, 407–411. doi: 10.2174/092986710790226129
Catanzaro, O., Capponi, J. A., Michieli, J., Labal, E., Di Martino, I., and Sirois, P. (2013). Bradykinin B(1) antagonism inhibits oxidative stress and restores Na+K+ ATPase activity in diabetic rat peripheral nervous system. Peptides 44, 100–104. doi: 10.1016/j.peptides.2013.01.019
Cheng, Y. Q., Ren, J. P., Zhao, J., Wang, J. M., Zhou, Y., Li, G. Y., et al. (2015). miR-155 regulates IFN-gamma production in NK cells via Tim-3 signaling in chronic HCV infection. Immunology. doi: 10.1111/imm.12463. [Epub ahead of print].
Cherry, J. D., Olschowka, J. A., and O'Banion, M. K. (2014). Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J. Neuroinflammation 11:98. doi: 10.1186/1742-2094-11-98
Colton, C., and Wilcock, D. M. (2010). Assessing activation states in microglia. CNS Neurol. Disord. Drug Targets 9, 174–191. doi: 10.2174/187152710791012053
Colton, C. A. (2009). Heterogeneity of microglial activation in the innate immune response in the brain. J. Neuroimmune Pharmacol. 4, 399–418. doi: 10.1007/s11481-009-9164-4
Dai, M. S., and Lu, H. (2008). Crosstalk between c-Myc and ribosome in ribosomal biogenesis and cancer. J. Cell. Biochem. 105, 670–677. doi: 10.1002/jcb.21895
Desbiens, K. M., Deschesnes, R. G., Labrie, M. M., Desfosses, Y., Lambert, H., Landry, J., et al. (2003). c-Myc potentiates the mitochondrial pathway of apoptosis by acting upstream of apoptosis signal-regulating kinase 1 (Ask1) in the p38 signalling cascade. Biochem. J. 372, 631–641. doi: 10.1042/BJ20021565
de Waal Malefyt, R., Abrams, J., Bennett, B., Figdor, C. G., and de Vries, J. E. (1991). Interleukin 10(IL-10) inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL-10 produced by monocytes. J. Exp. Med. 174, 1209–1220. doi: 10.1084/jem.174.5.1209
Dey, N., Bera, A., Das, F., Ghosh-Choudhury, N., Kasinath, B. S., and Choudhury, G. G. (2015). High glucose enhances microRNA-26a to activate mTORC1 for mesangial cell hypertrophy and matrix protein expression. Cell. Signal. 27, 1276–1285. doi: 10.1016/j.cellsig.2015.03.007
Dringen, R. (2005). Oxidative and antioxidative potential of brain microglial cells. Antioxid. Redox Signal. 7, 1223–1233. doi: 10.1089/ars.2005.7.1223
Du, T., and Zamore, P. D. (2005). microPrimer: the biogenesis and function of microRNA. Development 132, 4645–4652. doi: 10.1242/dev.02070
Duarte, A. I., Santos, P., Oliveira, C. R., Santos, M. S., and Rego, A. C. (2008). Insulin neuroprotection against oxidative stress is mediated by Akt and GSK-3beta signaling pathways and changes in protein expression. Biochim. Biophys. Acta 1783, 994–1002. doi: 10.1016/j.bbamcr.2008.02.016
Echavarria, R., Mayaki, D., Neel, J. C., Harel, S., Sanchez, V., and Hussain, S. N. (2015). Angiopoietin-1 inhibits Toll-like receptor 4 signaling in cultured endothelial cells: role of miR-146b-5p. Cardiovasc. Res. doi: 10.1093/cvr/cvv120. [Epub ahead of print].
Evan, G., and Littlewood, T. (1998). A matter of life and cell death. Science 281, 1317–1322. doi: 10.1126/science.281.5381.1317
Feng, B., Chen, S., George, B., Feng, Q., and Chakrabarti, S. (2010). miR133a regulates cardiomyocyte hypertrophy in diabetes. Diabetes Metab. Res. Rev. 26, 40–49. doi: 10.1002/dmrr.1054
Fiorentino, D. F., Zlotnik, A., Mosmann, T. R., Howard, M., and O'Garra, A. (1991). IL-10 inhibits cytokine production by activated macrophages. J. Immunol. 147, 3815–3822.
Frank-Cannon, T. C., Alto, L. T., McAlpine, F. E., and Tansey, M. G. (2009). Does neuroinflammation fan the flame in neurodegenerative diseases? Mol. Neurodegener. 4:47. doi: 10.1186/1750-1326-4-47
Froen, J. F., Munkeby, B. H., Stray-Pedersen, B., and Saugstad, O. D. (2002). Interleukin-10 reverses acute detrimental effects of endotoxin-induced inflammation on perinatal cerebral hypoxia-ischemia. Brain Res. 942, 87–94. doi: 10.1016/S0006-8993(02)02700-2
Guldal, C. G., Ahmad, A., Korshunov, A., Squatrito, M., Awan, A., Mainwaring, L. A., et al. (2012). An essential role for p38 MAPK in cerebellar granule neuron precursor proliferation. Acta Neuropathol. doi: 10.1007/s00401-012-0946-z. [Epub ahead of print].
Hanisch, U. K. (2002). Microglia as a source and target of cytokines. Glia 40, 140–155. doi: 10.1002/glia.10161
Hann, S. R., and Eisenman, R. N. (1984). Proteins encoded by the human c-myc oncogene: differential expression in neoplastic cells. Mol. Cell. Biol. 4, 2486–2497.
Ichijo, H., Nishida, E., Irie, K., ten Dijke, P., Saitoh, M., Moriguchi, T., et al. (1997). Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 275, 90–94. doi: 10.1126/science.275.5296.90
Iliopoulos, D., Hirsch, H. A., and Struhl, K. (2009). An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell 139, 693–706. doi: 10.1016/j.cell.2009.10.014
Jiang, Q., Yuan, Y., Zhou, J., Wu, Y., Zhou, Q., Gui, S., et al. (2015). Apoptotic events induced by high glucose in human hepatoma HepG2 cells involve endoplasmic reticulum stress and MAPK's activation. Mol. Cell. Biochem. 399, 113–122. doi: 10.1007/s11010-014-2238-5
Joyce, D. A., Gibbons, D. P., Green, P., Steer, J. H., Feldmann, M., and Brennan, F. M. (1994). Two inhibitors of pro-inflammatory cytokine release, interleukin-10 and interleukin-4, have contrasting effects on release of soluble p75 tumor necrosis factor receptor by cultured monocytes. Eur. J. Immunol. 24, 2699–2705. doi: 10.1002/eji.1830241119
Joyce, D. A., and Steer, J. H. (1996). IL-4, IL-10 and IFN-gamma have distinct, but interacting, effects on differentiation-induced changes in TNF-alpha and TNF receptor release by cultured human monocytes. Cytokine 8, 49–57. doi: 10.1006/cyto.1996.0007
Jung, D. Y., Lee, H., and Suk, K. (2005). Pro-apoptotic activity of N-myc in activation-induced cell death of microglia. J. Neurochem. 94, 249–256. doi: 10.1111/j.1471-4159.2005.03186.x
Kim, H. D., Kim, T. S., and Kim, J. (2011). Aberrant ribosome biogenesis activates c-Myc and ASK1 pathways resulting in p53-dependent G1 arrest. Oncogene 30, 3317–3327. doi: 10.1038/onc.2011.47
Klein, J. P., Hains, B. C., Craner, M. J., Black, J. A., and Waxman, S. G. (2004). Apoptosis of vasopressinergic hypothalamic neurons in chronic diabetes mellitus. Neurobiol. Dis. 15, 221–228. doi: 10.1016/j.nbd.2003.10.009
Knudsen, G. M., Jakobsen, J., Barry, D. I., Compton, A. M., and Tomlinson, D. R. (1989). Myo-inositol normalizes decreased sodium permeability of the blood-brain barrier in streptozotocin diabetes. Neuroscience 29, 773–777. doi: 10.1016/0306-4522(89)90148-6
Kodl, C. T., Franc, D. T., Rao, J. P., Anderson, F. S., Thomas, W., Mueller, B. A., et al. (2008). Diffusion tensor imaging identifies deficits in white matter microstructure in subjects with type 1 diabetes that correlate with reduced neurocognitive function. Diabetes 57, 3083–3089. doi: 10.2337/db08-0724
Krady, J. K., Basu, A., Allen, C. M., Xu, Y., LaNoue, K. F., Gardner, T. W., et al. (2005). Minocycline reduces proinflammatory cytokine expression, microglial activation, and caspase-3 activation in a rodent model of diabetic retinopathy. Diabetes 54, 1559–1565. doi: 10.2337/diabetes.54.5.1559
Kreutzberg, G. W. (1996). Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 19, 312–318. doi: 10.1016/0166-2236(96)10049-7
Ledeboer, A., Breve, J. J., Poole, S., Tilders, F. J., and Van Dam, A. M. (2000). Interleukin-10, interleukin-4, and transforming growth factor-beta differentially regulate lipopolysaccharide-induced production of pro-inflammatory cytokines and nitric oxide in co-cultures of rat astroglial and microglial cells. Glia 30, 134–142. doi: 10.1002/(SICI)1098-1136(200004)30:2<134::AID-GLIA3>3.0.CO;2-3
Lemstra, A. W., Groen in't Woud, J. C., Hoozemans, J. J., van Haastert, E. S., Rozemuller, A. J., Eikelenboom, P., et al. (2007). Microglia activation in sepsis: a case-control study. J. Neuroinflammation 4:4. doi: 10.1186/1742-2094-4-4
Li, Z. G., and Sima, A. A. (2004). C-peptide and central nervous system complications in diabetes. Exp. Diabesity Res. 5, 79–90. doi: 10.1080/15438600490424550
Li, Z. G., Zhang, W., Grunberger, G., and Sima, A. A. (2002). Hippocampal neuronal apoptosis in type 1 diabetes. Brain Res. 946, 221–231. doi: 10.1016/S0006-8993(02)02887-1
Li, Z. G., Zhang, W., and Sima, A. A. (2003). C-peptide enhances insulin-mediated cell growth and protection against high glucose-induced apoptosis in SH-SY5Y cells. Diabetes Metab. Res. Rev. 19, 375–385. doi: 10.1002/dmrr.389
Lin, X., You, Y., Wang, J., Qin, Y., Huang, P., and Yang, F. (2014). MicroRNA-155 deficiency promotes nephrin acetylation and attenuates renal damage in hyperglycemia-induced nephropathy. Inflammation 38, 546–554. doi: 10.1007/s10753-014-9961-7
Liu, H., Xu, R., Feng, L., Guo, W., Cao, N., Qian, C., et al. (2012). A novel chromone derivative with anti-inflammatory property via inhibition of ROS-dependent activation of TRAF6-ASK1-p38 pathway. PLoS ONE 7:e37168. doi: 10.1371/journal.pone.0037168
Lu, H., Buchan, R. J., and Cook, S. A. (2010). MicroRNA-223 regulates Glut4 expression and cardiomyocyte glucose metabolism. Cardiovasc. Res. 86, 410–420. doi: 10.1093/cvr/cvq010
Lynch, A. M., Walsh, C., Delaney, A., Nolan, Y., Campbell, V. A., and Lynch, M. A. (2004). Lipopolysaccharide-induced increase in signalling in hippocampus is abrogated by IL-10–a role for IL-1 beta? J. Neurochem. 88, 635–646. doi: 10.1046/j.1471-4159.2003.02157.x
Malynn, B. A., de Alboran, I. M., O'Hagan, R. C., Bronson, R., Davidson, L., DePinho, R. A., et al. (2000). N-myc can functionally replace c-myc in murine development, cellular growth, and differentiation. Genes Dev. 14, 1390–1399.
Mantovani, A., Sica, A., Sozzani, S., Allavena, P., Vecchi, A., and Locati, M. (2004). The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 25, 677–686. doi: 10.1016/j.it.2004.09.015
Marin, T., Gongol, B., Chen, Z., Woo, B., Subramaniam, S., Chien, S., et al. (2013). Mechanosensitive microRNAs-role in endothelial responses to shear stress and redox state. Free Radic. Biol. Med. 64, 61–68. doi: 10.1016/j.freeradbiomed.2013.05.034
Martinez, F. O., Sica, A., Mantovani, A., and Locati, M. (2008). Macrophage activation and polarization. Front. Biosci. 13, 453–461. doi: 10.2741/2692
Martinez, J., and Tuschl, T. (2004). RISC is a 5′ phosphomonoester-producing RNA endonuclease. Genes Dev. 18, 975–980. doi: 10.1101/gad.1187904
Matsuzawa, A., Saegusa, K., Noguchi, T., Sadamitsu, C., Nishitoh, H., Nagai, S., et al. (2005). ROS-dependent activation of the TRAF6-ASK1-p38 pathway is selectively required for TLR4-mediated innate immunity. Nat. Immunol. 6, 587–592. doi: 10.1038/ni1200
McCrimmon, R. J., Ryan, C. M., and Frier, B. M. (2012). Diabetes and cognitive dysfunction. Lancet 379, 2291–2299. doi: 10.1016/S0140-6736(12)60360-2
Menne, T. F., Goyenechea, B., Sanchez-Puig, N., Wong, C. C., Tonkin, L. M., Ancliff, P. J., et al. (2007). The Shwachman-Bodian-Diamond syndrome protein mediates translational activation of ribosomes in yeast. Nat. Genet. 39, 486–495. doi: 10.1038/ng1994
Min, K. J., Jou, I., and Joe, E. (2003). Plasminogen-induced IL-1beta and TNF-alpha production in microglia is regulated by reactive oxygen species. Biochem. Biophys. Res. Commun. 312, 969–974. doi: 10.1016/j.bbrc.2003.11.010
Molina-Holgado, F., Grencis, R., and Rothwell, N. J. (2001). Actions of exogenous and endogenous IL-10 on glial responses to bacterial LPS/cytokines. Glia 33, 97–106. doi: 10.1002/1098-1136(200102)33:2<97::AID-GLIA1009>3.0.CO;2-N
Mulder, R., Banete, A., and Basta, S. (2014). Spleen-derived macrophages are readily polarized into classically activated (M1) or alternatively activated (M2) states. Immunobiology 219, 737–745. doi: 10.1016/j.imbio.2014.05.005
Musen, G., Lyoo, I. K., Sparks, C. R., Weinger, K., Hwang, J., Ryan, C. M., et al. (2006). Effects of type 1 diabetes on gray matter density as measured by voxel-based morphometry. Diabetes 55, 326–333. doi: 10.2337/diabetes.55.02.06.db05-0520
Noguchi, K., Kitanaka, C., Yamana, H., Kokubu, A., Mochizuki, T., and Kuchino, Y. (1999). Regulation of c-Myc through phosphorylation at Ser-62 and Ser-71 by c-Jun N-terminal kinase. J. Biol. Chem. 274, 32580–32587. doi: 10.1074/jbc.274.46.32580
Noguchi, K., Kokubu, A., Kitanaka, C., Ichijo, H., and Kuchino, Y. (2001). ASK1-signaling promotes c-Myc protein stability during apoptosis. Biochem. Biophys. Res. Commun. 281, 1313–1320. doi: 10.1006/bbrc.2001.4498
Nolan, Y., Maher, F. O., Martin, D. S., Clarke, R. M., Brady, M. T., Bolton, A. E., et al. (2005). Role of interleukin-4 in regulation of age-related inflammatory changes in the hippocampus. J. Biol. Chem. 280, 9354–9362. doi: 10.1074/jbc.M412170200
Osaka, N., Takahashi, T., Murakami, S., Matsuzawa, A., Noguchi, T., Fujiwara, T., et al. (2007). ASK1-dependent recruitment and activation of macrophages induce hair growth in skin wounds. J. Cell Biol. 176, 903–909. doi: 10.1083/jcb.200611015
Osipova, J., Fischer, D. C., Dangwal, S., Volkmann, I., Widera, C., Schwarz, K., et al. (2014). Diabetes-associated microRNAs in pediatric patients with type 1 diabetes mellitus: a cross-sectional cohort study. J. Clin. Endocrinol. Metab. 99, E1661–E1665. doi: 10.1210/jc.2013-3868
Perez, L. M., Bernal, A., San Martin, N., Lorenzo, M., Fernandez-Veledo, S., and Galvez, B. G. (2013). Metabolic rescue of obese adipose-derived stem cells by Lin28/Let7 pathway. Diabetes 62, 2368–2379. doi: 10.2337/db12-1220
Popivanova, B. K., Kitamura, K., Wu, Y., Kondo, T., Kagaya, T., Kaneko, S., et al. (2008). Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J. Clin. Invest. 118, 560–570.
Prendergast, G. C. (1999). Mechanisms of apoptosis by c-Myc. Oncogene 18, 2967–2987. doi: 10.1038/sj.onc.1202727
Quan, Y., Du, J., and Wang, X. (2007). High glucose stimulates GRO secretion from rat microglia via ROS, PKC, and NF-kappaB pathways. J. Neurosci. Res. 85, 3150–3159. doi: 10.1002/jnr.21421
Quan, Y., Jiang, C. T., Xue, B., Zhu, S. G., and Wang, X. (2011). High glucose stimulates TNFalpha and MCP-1 expression in rat microglia via ROS and NF-kappaB pathways. Acta Pharmacol. Sin. 32, 188–193. doi: 10.1038/aps.2010.174
Quinn, S. R., and O'Neill, L. A. (2011). A trio of microRNAs that control Toll-like receptor signalling. Int. Immunol. 23, 421–425. doi: 10.1093/intimm/dxr034
Ramsay, G., Stanton, L., Schwab, M., and Bishop, J. M. (1986). Human proto-oncogene N-myc encodes nuclear proteins that bind DNA. Mol. Cell. Biol. 6, 4450–4457.
Rossler, J., Schwab, M., Havers, W., and Schweigerer, L. (2001). Hypoxia promotes apoptosis of human neuroblastoma cell lines with enhanced N-myc expression. Biochem. Biophys. Res. Commun. 281, 272–276. doi: 10.1006/bbrc.2001.4342
Sawada, M., Suzumura, A., Hosoya, H., Marunouchi, T., and Nagatsu, T. (1999). Interleukin-10 inhibits both production of cytokines and expression of cytokine receptors in microglia. J. Neurochem. 72, 1466–1471. doi: 10.1046/j.1471-4159.1999.721466.x
Schamberger, A., Sarkadi, B., and Orban, T. I. (2012). Human mirtrons can express functional microRNAs simultaneously from both arms in a flanking exon-independent manner. RNA Biol. 9, 1177–1185. doi: 10.4161/rna.21359
Schulte, L. N., Westermann, A. J., and Vogel, J. (2013). Differential activation and functional specialization of miR-146 and miR-155 in innate immune sensing. Nucleic Acids Res. 41, 542–553. doi: 10.1093/nar/gks1030
Schwab, M., Alitalo, K., Klempnauer, K. H., Varmus, H. E., Bishop, J. M., Gilbert, F., et al. (1983). Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature 305, 245–248. doi: 10.1038/305245a0
Schweigerer, L., Scheurich, P., and Fotsis, T. (1990). Enhanced MYCN oncogene expression in human neuroblastoma cells results in increased susceptibility to growth inhibition by TNF alpha. Biochem. Biophys. Res. Commun. 170, 1301–1307. doi: 10.1016/0006-291X(90)90535-U
Sears, R., Leone, G., DeGregori, J., and Nevins, J. R. (1999). Ras enhances Myc protein stability. Mol. Cell 3, 169–179. doi: 10.1016/S1097-2765(00)80308-1
Shan, Z. X., Lin, Q. X., Deng, C. Y., Zhu, J. N., Mai, L. P., Liu, J. L., et al. (2010). miR-1/miR-206 regulate Hsp60 expression contributing to glucose-mediated apoptosis in cardiomyocytes. FEBS Lett. 584, 3592–3600. doi: 10.1016/j.febslet.2010.07.027
Sima, A. A., Kamiya, H., and Li, Z. G. (2004). Insulin, C-peptide, hyperglycemia, and central nervous system complications in diabetes. Eur. J. Pharmacol. 490, 187–197. doi: 10.1016/j.ejphar.2004.02.056
Song, J., Cheon, S. Y., Jung, W., Lee, W. T., and Lee, J. E. (2014). Resveratrol induces the expression of interleukin-10 and brain-derived neurotrophic factor in BV2 microglia under hypoxia. Int. J. Mol. Sci. 15, 15512–15529. doi: 10.3390/ijms150915512
Sredy, J., Sawicki, D. R., and Notvest, R. R. (1991). Polyol pathway activity in nervous tissues of diabetic and galactose-fed rats: effect of dietary galactose withdrawal or tolrestat intervention therapy. J. Diabet. Complications 5, 42–47. doi: 10.1016/0891-6632(91)90010-M
Stein, M., Keshav, S., Harris, N., and Gordon, S. (1992). Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J. Exp. Med. 176, 287–292. doi: 10.1084/jem.176.1.287
Szczepanik, A. M., Funes, S., Petko, W., and Ringheim, G. E. (2001). IL-4, IL-10 and IL-13 modulate A beta(1–42)-induced cytokine and chemokine production in primary murine microglia and a human monocyte cell line. J. Neuroimmunol. 113, 49–62. doi: 10.1016/S0165-5728(00)00404-5
Takarada-Iemata, M., Kezuka, D., Takeichi, T., Ikawa, M., Hattori, T., Kitao, Y., et al. (2014). Deletion of N-myc downstream-regulated gene 2 attenuates reactive astrogliosis and inflammatory response in a mouse model of cortical stab injury. J. Neurochem. 130, 374–387. doi: 10.1111/jnc.12729
Tan, Y., Yang, J., Xiang, K., Tan, Q., and Guo, Q. (2015). Suppression of MicroRNA-155 Attenuates neuropathic pain by regulating SOCS1 signalling pathway. Neurochem. Res. 40, 550–560. doi: 10.1007/s11064-014-1500-2
Tobiume, K., Matsuzawa, A., Takahashi, T., Nishitoh, H., Morita, K., Takeda, K., et al. (2001). ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep. 2, 222–228. doi: 10.1093/embo-reports/kve046
Tsuda, M., Ueno, H., Kataoka, A., Tozaki-Saitoh, H., and Inoue, K. (2008). Activation of dorsal horn microglia contributes to diabetes-induced tactile allodynia via extracellular signal-regulated protein kinase signaling. Glia 56, 378–386. doi: 10.1002/glia.20623
Ueda, K., and Ganem, D. (1996). Apoptosis is induced by N-myc expression in hepatocytes, a frequent event in hepadnavirus oncogenesis, and is blocked by insulin-like growth factor II. J. Virol. 70, 1375–1383.
Urbich, C., Kuehbacher, A., and Dimmeler, S. (2008). Role of microRNAs in vascular diseases, inflammation, and angiogenesis. Cardiovasc. Res. 79, 581–588. doi: 10.1093/cvr/cvn156
van Elderen, S. G., de Roos, A., de Craen, A. J., Westendorp, R. G., Blauw, G. J., Jukema, J. W., et al. (2010). Progression of brain atrophy and cognitive decline in diabetes mellitus: a 3-year follow-up. Neurology 75, 997–1002. doi: 10.1212/WNL.0b013e3181f25f06
Varin, A., and Gordon, S. (2009). Alternative activation of macrophages: immune function and cellular biology. Immunobiology 214, 630–641. doi: 10.1016/j.imbio.2008.11.009
Varnum, M. M., and Ikezu, T. (2012). The classification of microglial activation phenotypes on neurodegeneration and regeneration in Alzheimer's disease brain. Arch. Immunol. Ther. Exp. (Warsz). 60, 251–266. doi: 10.1007/s00005-012-0181-2
Wang, F., Wu, Y., Gu, H., Reece, E. A., Fang, S., Gabbay-Benziv, R., et al. (2015). Ask1 gene deletion blocks maternal diabetes-induced endoplasmic reticulum stress in the developing embryo by disrupting the unfolded protein response signalosome. Diabetes 64, 973–988. doi: 10.2337/db14-0409
Wang, H. J., Huang, Y. L., Shih, Y. Y., Wu, H. Y., Peng, C. T., and Lo, W. Y. (2014). MicroRNA-146a decreases high glucose/thrombin-induced endothelial inflammation by inhibiting NAPDH oxidase 4 expression. Mediators Inflamm. 2014:379537. doi: 10.1155/2014/379537
Wang, J. Y., Yang, J. M., Wang, J. Y., Tao, P. L., and Yang, S. N. (2001). Synergistic apoptosis induced by bacterial endotoxin lipopolysaccharide and high glucose in rat microglia. Neurosci. Lett. 304, 177–180. doi: 10.1016/S0304-3940(01)01780-3
Wodarski, R., Clark, A. K., Grist, J., Marchand, F., and Malcangio, M. (2009). Gabapentin reverses microglial activation in the spinal cord of streptozotocin-induced diabetic rats. Eur. J. Pain 13, 807–811. doi: 10.1016/j.ejpain.2008.09.010
Wojtera, M., Sobow, T., Kloszewska, I., Liberski, P. P., Brown, D. R., and Sikorska, B. (2012). Expression of immunohistochemical markers on microglia in Creutzfeldt-Jakob disease and Alzheimer's disease: morphometric study and review of the literature. Folia Neuropathol. 50, 74–84.
Xiao, J., Luo, X., Lin, H., Zhang, Y., Lu, Y., Wang, N., et al. (2007). MicroRNA miR-133 represses HERG K+ channel expression contributing to QT prolongation in diabetic hearts. J. Biol. Chem. 282, 12363–12367. doi: 10.1074/jbc.C700015200
Xu, H., Chen, M., and Forrester, J. V. (2009). Para-inflammation in the aging retina. Prog. Retin. Eye Res. 28, 348–368. doi: 10.1016/j.preteyeres.2009.06.001
Yang, J., Jiang, Z., Fitzgerald, D. C., Ma, C., Yu, S., Li, H., et al. (2009). Adult neural stem cells expressing IL-10 confer potent immunomodulation and remyelination in experimental autoimmune encephalitis. J. Clin. Invest. 119, 3678–3691. doi: 10.1172/JCI37914
Yokoi, T., Fukuo, K., Yasuda, O., Hotta, M., Miyazaki, J., Takemura, Y., et al. (2006). Apoptosis signal-regulating kinase 1 mediates cellular senescence induced by high glucose in endothelial cells. Diabetes 55, 1660–1665. doi: 10.2337/db05-1607
Yuk, J. M., Shin, D. M., Yang, C. S., Kim, K. H., An, S. J., Rho, J., et al. (2009). Role of apoptosis-regulating signal kinase 1 in innate immune responses by Mycobacterium bovis bacillus Calmette-Guerin. Immunol. Cell Biol. 87, 100–107. doi: 10.1038/icb.2008.74
Zhang, X., Dong, H., Zhang, S., Lu, S., Sun, J., and Qian, Y. (2015). Enhancement of LPS-induced microglial inflammation response via TLR4 under high glucose conditions. Cel. Physiol. Biochem. 35, 1571–1581. doi: 10.1159/000373972
Zhang, Y. P., Eber, A., Yuan, Y., Yang, Z., Rodriguez, Y., Levitt, R. C., et al. (2013). Prophylactic and antinociceptive effects of coenzyme Q10 on diabetic neuropathic pain in a mouse model of type 1 diabetes. Anesthesiology 118, 945–954. doi: 10.1097/ALN.0b013e3182829b7b
Zhang, Z., Huang, L., Yu, Z., Chen, X., Yang, D., Zhan, P., et al. (2014). Let-7a functions as a tumor suppressor in Ewing's sarcoma cell lines partly by targeting cyclin-dependent kinase 6. DNA Cell Biol. 33, 136–147. doi: 10.1089/dna.2013.2179
Keywords: Microglia BV2, microRNA-Let7A (miR-Let7A), apoptosis signal regulating kinase 1 (ASK1), cytokine, N-Myc, c-Myc
Citation: Song J and Lee JE (2015) ASK1 modulates the expression of microRNA Let7A in microglia under high glucose in vitro condition. Front. Cell. Neurosci. 9:198. doi: 10.3389/fncel.2015.00198
Received: 03 February 2015; Accepted: 07 May 2015;
Published: 20 May 2015.
Edited by:
Fabio Blandini, National Institute of Neurology C. Mondino Foundation, ItalyReviewed by:
Philippe Georgel, Strasbourg University, FrancePeixin Yang, University of Maryland School of Medicine, USA
Copyright © 2015 Song and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jong Eun Lee, Yonsei Brain Korea 21 Project for Medical Science Yonsei University, Department of Anatomy, Yonsei University College of Medicine, 50 Yonsei-ro, Seodaemun-gu, Seoul, 120-752, South Korea,amVsZWVAeXVocy5hYw==