 Sónia S. Leal
Sónia S. Leal Cláudio M. Gomes
Cláudio M. Gomes- 1Faculdade de Ciências, Biosystems and Integrative Sciences Institute and Department of Chemistry and Biochemistry, Universidade de Lisboa, Campo Grande, Lisboa, Portugal
- 2Instituto Tecnologia Química e Biológica, Universidade Nova de Lisboa, Oeiras, Portugal
More than 20 distinct gene loci have so far been implicated in amyotrophic lateral sclerosis (ALS), a fatal neurodegenerative disorder characterized by progressive neurodegeneration of motor neurons (MN) and death. Most of this distinct set of ALS-related proteins undergoes toxic deposition specifically in MN for reasons which remain unclear. Here we overview a recent body of evidence indicative that mutations in ALS-related proteins can disrupt fundamental Ca2+ signalling pathways in MN, and that Ca2+ itself impacts both directly or indirectly in many ALS critical proteins and cellular processes that result in MN neurodegeneration. We argue that the inherent vulnerability of MN to dysregulation of intracellular Ca2+ is deeply associated with discriminating pathogenicity and aberrant crosstalk of most of the critical proteins involved in ALS. Overall, Ca2+ deregulation in MN is at the cornerstone of different ALS processes and is likely one of the factors contributing to the selective susceptibility of these cells to this particular neurodegenerative disease.
Sporadic and Familial ALS Aggregates Share Identical Proteins
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by the selective degeneration of motor neurons (MN) in the spinal cord, brainstem and cerebral cortex (Rowland and Shneider, 2001). Most cases of ALS are sporadic (sALS) with no known genetic linkage, while approximately 10% are associated with familial forms (fALS), presenting mutations in over 20 genes encoding for distinct proteins with varied functions (Table 1). Despite the heterogeneous genetics of fALS and unknown etiology of sALS and its clear multifactorial character, most ALS patients present similar phenotypes with formation of cytoplasmic proteinaceous aggregates in the affected MN (Al-Chalabi et al., 2012). Notably, many proteins involved in fALS forms are also found in sALS toxic aggregates (Maekawa et al., 2009; Deng et al., 2010; Forsberg et al., 2010; Blokhuis et al., 2013) and found to cross-talk and impact on each other in ALS pathology (Kanekura et al., 2004; Volkening et al., 2009; Tudor et al., 2010; Nihei et al., 2012; Pokrishevsky et al., 2012; Stoica et al., 2014; Osaka et al., 2015). This suggests that apart from mutations, additional chemical and/or biological factors influence the selective involvement of these proteins also in sALS neurodegeneration. In agreement, most of the proteins which are involved in ALS are ordinarily expressed in many distinct cell tissues other than the nervous system (e.g., SOD1, FUS, TDP-43, VAPB, matrin-3, ataxin-2, alsin) but are only found to generate toxicity among MN. The selective vulnerability of such cells suggests that environmental triggers within those neurons are mandatory for the onset of ALS.
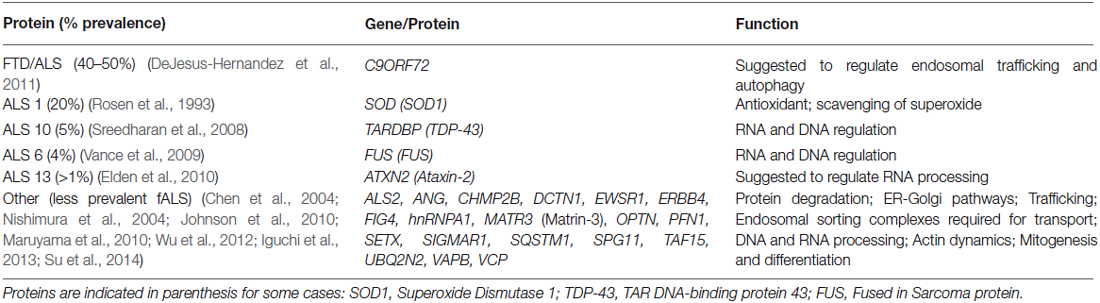
Table 1. Heterogeneity of fALS causative genes.
Calcium Dysregulation in ALS-Affected Motor Neurons—The Factual Case of ALS1
A particular feature that distinguishes ALS affected MN from other cells relates to their inherent vulnerability to Ca2+ overload; indeed, these neurons highly express Ca2+ permeable AMPA receptors (Williams et al., 1997; Shaw and Eggett, 2000; Van Den Bosch et al., 2000; Vandenberghe et al., 2000; Guatteo et al., 2007) concurrently with a low Ca2+ buffering capacity due to endogenous low expression of Ca2+ buffering proteins (CaBPs) such as parvalbumin and calbindin (Alexianu et al., 1994; Palecek et al., 1999; Jaiswal, 2013) albeit the presence of EF-hand Ca2+ binding proteins in MN (Migheli et al., 1999; Zhang et al., 2014). This combination of inherent physiological features of MN to manage Ca2+ levels, though essential for normal functioning (von Lewinski and Keller, 2005), are likely a predisposition risk for the systematic intracellular Ca2+ overload that is detected in ALS1 affected MN (Siklós et al., 1996, 1998; Kruman et al., 1999; Grosskreutz et al., 2010; Kawamata and Manfredi, 2010). Interestingly, the levels of calretinin and parvalbumin in MN axons is found further decreased in ALS patients (Hayashi et al., 2013), thus establishing an increased deficit in MN Ca2+ buffering capability under pathological conditions.
In fact, a direct outcome of the low expression of CaBPs in MN is that mitochondria are likely to assume a major role in buffering calcium in these cells. In agreement, it might not be a coincidence that in ALS1, mutated SOD1 was shown to abnormally accumulate in the mitochondrial intermembrane space (Jaarsma et al., 2001; Liu et al., 2004), affecting mitochondrial function leading to disturbance of Ca2+ homeostasis and ERMCC cycle (Jaiswal and Keller, 2009; Lautenschläger et al., 2013). Moreover, studies on cellular and animal ALS1 models have shown that Ca2+ overload is linked with SOD1 aggregation (Tateno et al., 2004; Tradewell et al., 2011). Indeed, we have recently shown that Ca2+ can bind to SOD1 immature states promoting its aggregation (Leal et al., 2013; Estácio et al., 2015) thus establishing an additional ALS1 pathological pathway for the impact of Ca2+ overload on SOD1 toxic deposition. In agreement, a decrease of intracellular Ca2+ overload in ALS1 models through AMPA channel antagonists or overexpression of calcium-buffering proteins has been shown to reduce SOD1 toxic aggregation and neurodegeneration (Beers et al., 2001; Van Damme et al., 2003; Tateno et al., 2004; Tortarolo et al., 2006; Yin et al., 2007; Parone et al., 2013).
Crossways of ALS Critical Proteins with Calcium
In spite of the scarcity of available data regarding more recently discovered fALS models linked with systematic Ca2+ deregulation, compelling evidence argues that many critical proteins involved in ALS (other than SOD1), are directly or indirectly involved with Ca2+. It is noteworthy that in such instances, ALS-associated mutations in many of these ALS critical proteins happen to potentiate Ca2+ deregulation and/or result in an increased vulnerability to the effects of Ca2+. For example, VAP-B which is involved in ALS8, is a ER-membrane MAM protein directly engaged in Ca2+ exchange between the ER and mitochondria (De Vos et al., 2012). The ALS VAPBP56S variant was shown to disrupt Ca2+ homeostasis leading to a perturbation of the anterograde mitochondrial axonal transport and affecting the Miro1/kinesin-1 interaction with tubulin (Mórotz et al., 2012). Alsin, a protein implicated in juvenile ALS2, is involved in endossome/membrane trafficking that undergoes Ca2+ dependent binding to the NCS regulating neurocalcin alpha protein (Masutani et al., 2008). This suggests that alsin membrane binding might be a Ca2+ dependent process, and therefore passible to become affected by dysregulation of Ca2+ levels. Moreover, alsin is also found to play a role in AMPAR trafficking, where ALS2 mutations lead to distinct subcellular GRIP1 localization and reduction of the calcium-impermeable GluR2 containing AMPA receptors, thus likely rendering neurons susceptible to deviant Ca2+ influxes (Lai et al., 2006). Matrin 3, a multifunctional nuclear matrix protein involved in ALS21, is suggested to be regulated through a Ca2+ dependent interaction with CaM (Valencia et al., 2007) and is therefore likely to be affected by deregulated Ca2+ levels. As only very recently matrin 3 has been implicated in ALS (Johnson et al., 2014), future studies will be needed to clarify this possibility. Ataxin-2, which is involved in ALS13 is an ubiquitous cytoplasmic protein proposed to induce defects in the ER–Golgi pathway and disrupt Ca2+ signalling (van den Heuvel et al., 2014). The possibility that ataxin-2 influences ER–Golgi function is inferred from a recent study where it was shown that intermediate-length polyQ expansions in ataxin-2 mutants enhance FUS-induced ER stress and Golgi fragmentation (Farg et al., 2013). The suggestion that the ALS13 related intermediate-length polyQ expansions in ataxin-2 can lead to Ca2+ signalling disruption derives from the causal association that polyQ-expanded forms of ataxin-2, or huntingtin and ataxin-3 in other diseases, associates with the C-terminal domain of the intracellular calcium release channel receptor—InsP3R1 and enhance InsP3R1-mediated calcium release in neurons (Tang et al., 2003; Chen et al., 2008; Liu et al., 2009). Thus, it is tempting to speculate that intermediate-length polyQ expansions in ataxin-2 may also cause it to associate with InsP3R1 and thereby modulate calcium signaling, though this was not yet shown. In ALS6, the FUS protein leads to CAMK2N2 up-regulation (Convertini et al., 2013), an inhibitor of CAMKII that regulates neuronal synaptic plasticity through phosphorylation of AMPA receptors. Given that it has been shown that abnormal CAMKII inhibition by small molecules and peptides results in dysregulation of Ca2+/glutamate signalling (Ashpole et al., 2012), it may be hypothesized that CAMKII inhibitor—CAMK2N2 up-regulation by mutant FUS will also result in Ca2+ dysregulation in ALS6. Moreover, deregulated Ca2+ levels can activate the Ca2+-dependent calpain protease that cleaves TDP-43 at the C-terminal, generating aggregation prone N-terminal segments that are found misallocated in the majority of ALS patients (even in those that do not carry TDP-43 mutations associated with ALS10) driving TDP-43 toxicity across ALS pathology (Aggad et al., 2014; Yamashita and Kwak, 2014). In addition, TDP-43 is simultaneously found to interact with other critical proteins in ALS namely matrin-3 (Johnson et al., 2014), ataxin-2 (Nihei et al., 2012), VAPB (Stoica et al., 2014), and SOD1 (Volkening et al., 2009) and is therefore rather tempting to conjecture about a tight interrelation between Ca2+ dyshomeostasis and the involvement of critical proteins in ALS.
ALS Toxic Processes and the Role of Calcium
In fact, major pathological processes in ALS involving excitotoxicity and the ER-mitochondria Ca2+ cycle are deeply connected and potentially trigger or/and are enhanced by intracellular Ca2+ deregulation: (a) Glutamatergic excitotoxicity is tough to be mediated by an excessive influx of extracellular ions, including Ca2+, resulting in elevated intracellular Ca2+ levels that can activate cytoplasmatic Ca2+-dependent apoptotic proteins (e.g., calcineurin, calpain) which promote cell death (Wang et al., 1999; Kim et al., 2002); (b) elevated intracellular levels of Ca2+ also lead to mitochondrial Ca2+ overload, that is deeply interconnected with mitochondrial dysfunction resulting in ROS production, oxidative stress and eventually to apoptosis or necrosis (Kawamata and Manfredi, 2010; Cozzolino and Carrì, 2012); and (c) depletion of Ca2+ levels in the ER which is suggested to occur via a persistent shift of Ca2+ from the ER to the mitochondria due to deregulated ER MCC leads to protein folding dysfunction and proteasome impairment, resulting in ER stress and apoptosis (Prell et al., 2013; Tadic et al., 2014). Mutations in critical proteins associated with ALS actually seem to increase the susceptibility for these toxic processes to occur. For example, misfolded and aggregated SOD1 mutants localized within the mitochondrial membrane of spinal cord MN cause dysfunction in oxidative phosphorylation and bind aberrantly to Bcl-2, generating toxicity (Jung et al., 2002; Mattiazzi et al., 2002; Liu et al., 2004; Vande Velde et al., 2008; Pedrini et al., 2010); also, the ALS linked P56S mutation in VAPB, or th A4V, G85R and G93A SOD1 mutations leads to toxic protein aggregation and ER stress (Prosser et al., 2008; Kim et al., 2010; Atkin et al., 2014).
Conclusions
Overall, we here argue that Ca2+ deregulation seems to establish a converging point for major ALS dysfunctional pathways and critical associated proteins, and can therefore be a key environmental factor to better understand ALS etiology and its pathomechanisms. However, we do not intend to ground that all proteins implicated in ALS will necessarily lead to Ca2+ deregulation; rather, we seek to discuss that processes involving Ca2+ could directly or indirectly (e.g., via Ca2+ effects on processes dependent of other divalent cations) account for their mutual involvement in ALS. Interestingly, the so-called “calcium hypothesis” establishing a close link between Ca2+ deregulation and neurodegeneration, has also been suggested to play a central role in other neurodegenerative disorders such as Alzheimer’s, Ataxia, Parkinson’s and Huntington Diseases (Bezprozvanny, 2010; Kasumu and Bezprozvanny, 2012), where Ca2+ channels and proteins involved in neuronal Ca2+ signalling systems are likely potential targets for therapeutic strategies (Zundorf and Reiser, 2011).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by Fundação para a Ciência e a Tecnologia through grant PTDC/QUI-BIQ/117789/2010 (to CMG), post-doctoral fellowship SFRH/BPD/47477/2008 (to SSL), strategic grant PEst-OE/EQB/LA0004/2011 (to ITQB-Laboratόrio Associado) and grant UID/MULTI/04046/2013 from FCT/MCTES/PIDDAC (to BioISI).
References
Aggad, D., Vérièpe, J., Tauffenberger, A., and Parker, J. A. (2014). TDP-43 toxicity proceeds via calcium dysregulation and necrosis in aging Caenorhabditis elegans motor neurons. J. Neurosci. 34, 12093–12103. doi: 10.1523/jneurosci.2495-13.2014
Al-Chalabi, A., Jones, A., Troakes, C., King, A., Al-Sarraj, S., and van den Berg, L. H. (2012). The genetics and neuropathology of amyotrophic lateral sclerosis. Acta Neuropathol. 124, 339–352. doi: 10.1007/s00401-012-1022-4
Alexianu, M. E., Ho, B. K., Mohamed, A. H., La Bella, V., Smith, R. G., and Appel, S. H. (1994). The role of calcium-binding proteins in selective motoneuron vulnerability in amyotrophic lateral sclerosis. Ann. Neurol. 36, 846–858. doi: 10.1002/ana.410360608
Ashpole, N. M., Song, W., Brustovetsky, T., Engleman, E. A., Brustovetsky, N., Cummins, T. R., et al. (2012). Calcium/calmodulin-dependent protein kinase II (CaMKII) inhibition induces neurotoxicity via dysregulation of glutamate/calcium signaling and hyperexcitability. J. Biol. Chem. 287, 8495–8506. doi: 10.1074/jbc.m111.323915
Atkin, J. D., Farg, M. A., Soo, K. Y., Walker, A. K., Halloran, M., Turner, B. J., et al. (2014). Mutant SOD1 inhibits ER-Golgi transport in amyotrophic lateral sclerosis. J. Neurochem. 129, 190–204. doi: 10.1111/jnc.12493
Beers, D. R., Ho, B. K., Siklós, L., Alexianu, M. E., Mosier, D. R., Mohamed, A. H., et al. (2001). Parvalbumin overexpression alters immune-mediated increases in intracellular calcium and delays disease onset in a transgenic model of familial amyotrophic lateral sclerosis. J. Neurochem. 79, 499–509. doi: 10.1046/j.1471-4159.2001.00582.x
Blokhuis, A. M., Groen, E. J., Koppers, M., van den Berg, L. H., and Pasterkamp, R. J. (2013). Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 125, 777–794. doi: 10.1007/s00401-013-1125-6
Chen, Y. Z., Bennett, C. L., Huynh, H. M., Blair, I. P., Puls, I., Irobi, J., et al. (2004). DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am. J. Hum. Genet. 74, 1128–1135. doi: 10.1086/421054
Chen, X., Tang, T. S., Tu, H., Nelson, O., Pook, M., Hammer, R., et al. (2008). Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 3. J. Neurosci. 28, 12713–12724. doi: 10.1523/JNEUROSCI.3909-08.2008
Convertini, P., Zhang, J., de la Grange, P., Hayward, L. J., Zhu, H., and Stamm, S. (2013). Genome wide array analysis indicates that an amyotrophic lateral sclerosis mutation of FUS causes an early increase of CAMK2N2 in vitro. Biochim. Biophys. Acta 1832, 1129–1135. doi: 10.1016/j.bbadis.2013.03.015
Cozzolino, M., and Carrì, M. T. (2012). Mitochondrial dysfunction in ALS. Prog. Neurobiol. 97, 54–66. doi: 10.1016/j.pneurobio.2011.06.003
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
Deng, H. X., Zhai, H., Bigio, E. H., Yan, J., Fecto, F., Ajroud, K., et al. (2010). FUS-immunoreactive inclusions are a common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosis. Ann. Neurol. 67, 739–748. doi: 10.1002/ana.22051
De Vos, K. J., Mórotz, G. M., Stoica, R., Tudor, E. L., Lau, K. F., Ackerley, S., et al. (2012). VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum. Mol. Genet. 21, 1299–1311. doi: 10.1093/hmg/ddr559
Elden, A. C., Kim, H. J., Hart, M. P., Chen-Plotkin, A. S., Johnson, B. S., Fang, X., et al. (2010). Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466, 1069–1075. doi: 10.1038/nature09320
Estácio, S. G., Leal, S. S., Cristóvão, J. S., Faísca, P. F., and Gomes, C. M. (2015). Calcium binding to gatekeeper residues flanking aggregation-prone segments underlies non-fibrillar amyloid traits in superoxide dismutase 1 (SOD1). Biochim. Biophys. Acta 1854, 118–126. doi: 10.1016/j.bbapap.2014.11.005
Farg, M. A., Soo, K. Y., Warraich, S. T., Sundaramoorthy, V., Blair, I. P., and Atkin, J. D. (2013). Ataxin-2 interacts with FUS and intermediate-length polyglutamine expansions enhance FUS-related pathology in amyotrophic lateral sclerosis. Hum. Mol. Genet. 22, 717–728. doi: 10.1093/hmg/dds479
Forsberg, K., Jonsson, P. A., Andersen, P. M., Bergemalm, D., Graffmo, K. S., Hultdin, M., et al. (2010). Novel antibodies reveal inclusions containing non-native SOD1 in sporadic ALS patients. PLoS One 5:e11552. doi: 10.1371/journal.pone.0011552
Grosskreutz, J., Van Den Bosch, L., and Keller, B. U. (2010). Calcium dysregulation in amyotrophic lateral sclerosis. Cell Calcium 47, 165–174. doi: 10.1016/j.ceca.2009.12.002
Guatteo, E., Carunchio, I., Pieri, M., Albo, F., Canu, N., Mercuri, N. B., et al. (2007). Altered calcium homeostasis in motor neurons following AMPA receptor but not voltage-dependent calcium channels’ activation in a genetic model of amyotrophic lateral sclerosis. Neurobiol. Dis. 28, 90–100. doi: 10.1016/j.nbd.2007.07.002
Hayashi, S., Amari, M., and Okamoto, K. (2013). Loss of calretinin- and parvalbumin-immunoreactive axons in anterolateral columns beyond the corticospinal tracts of amyotrophic lateral sclerosis spinal cords. J. Neurol. Sci. 331, 61–66. doi: 10.1016/j.jns.2013.05.008
Iguchi, Y., Katsuno, M., Ikenaka, K., Ishigaki, S., and Sobue, G. (2013). Amyotrophic lateral sclerosis: an update on recent genetic insights. J. Neurol. 260, 2917–2927. doi: 10.1007/s00415-013-7112-y
Jaarsma, D., Rognoni, F., van Duijn, W., Verspaget, H. W., Haasdijk, E. D., and Holstege, J. C. (2001). CuZn superoxide dismutase (SOD1) accumulates in vacuolated mitochondria in transgenic mice expressing amyotrophic lateral sclerosis-linked SOD1 mutations. Acta Neuropathol. 102, 293–305. doi: 10.1007/s004010100399
Jaiswal, M. K. (2013). Calcium, mitochondria and the pathogenesis of ALS: the good, the bad and the ugly. Front. Cell. Neurosci. 7:199. doi: 10.3389/fncel.2013.00199
Jaiswal, M. K., and Keller, B. U. (2009). Cu/Zn superoxide dismutase typical for familial amyotrophic lateral sclerosis increases the vulnerability of mitochondria and perturbs Ca2+ homeostasis in SOD1G93A mice. Mol. Pharmacol. 75, 478–489. doi: 10.1124/mol.108.050831
Johnson, J. O., Mandrioli, J., Benatar, M., Abramzon, Y., Van Deerlin, V. M., Trojanowski, J. Q., et al. (2010). Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68, 857–864. doi: 10.1016/j.neuron.2010.11.036
Johnson, J. O., Pioro, E. P., Boehringer, A., Chia, R., Feit, H., Renton, A. E., et al. (2014). Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat. Neurosci. 17, 664–666. doi: 10.1038/nn.3688
Jung, C., Higgins, C. M., and Xu, Z. (2002). Mitochondrial electron transport chain complex dysfunction in a transgenic mouse model for amyotrophic lateral sclerosis. J. Neurochem. 83, 535–545. doi: 10.1046/j.1471-4159.2002.01112.x
Kanekura, K., Hashimoto, Y., Niikura, T., Aiso, S., Matsuoka, M., and Nishimoto, I. (2004). Alsin, the product of ALS2 gene, suppresses SOD1 mutant neurotoxicity through RhoGEF domain by interacting with SOD1 mutants. J. Biol. Chem. 279, 19247–19256. doi: 10.1074/jbc.m313236200
Kasumu, A., and Bezprozvanny, I. (2012). Deranged calcium signaling in Purkinje cells and pathogenesis in spinocerebellar ataxia 2 (SCA2) and other ataxias. Cerebellum 11, 630–639. doi: 10.1007/s12311-010-0182-9
Kawamata, H., and Manfredi, G. (2010). Mitochondrial dysfunction and intracellular calcium dysregulation in ALS. Mech. Ageing Dev. 131, 517–526. doi: 10.1016/j.mad.2010.05.003
Kim, M. J., Jo, D. G., Hong, G. S., Kim, B. J., Lai, M., Cho, D. H., et al. (2002). Calpain-dependent cleavage of cain/cabin1 activates calcineurin to mediate calcium-triggered cell death. Proc. Natl. Acad. Sci. U S A 99, 9870–9875. doi: 10.1073/pnas.152336999
Kim, S., Leal, S. S., Ben Halevy, D., Gomes, C. M., and Lev, S. (2010). Structural requirements for VAP-B oligomerization and their implication in amyotrophic lateral sclerosis-associated VAP-B(P56S) neurotoxicity. J. Biol. Chem. 285, 13839–13849. doi: 10.1074/jbc.m109.097345
Kruman, I. I., Pedersen, W. A., Springer, J. E., and Mattson, M. P. (1999). ALS-linked Cu/Zn-SOD mutation increases vulnerability of motor neurons to excitotoxicity by a mechanism involving increased oxidative stress and perturbed calcium homeostasis. Exp. Neurol. 160, 28–39. doi: 10.1006/exnr.1999.7190
Lai, C., Xie, C., McCormack, S. G., Chiang, H. C., Michalak, M. K., Lin, X., et al. (2006). Amyotrophic lateral sclerosis 2-deficiency leads to neuronal degeneration in amyotrophic lateral sclerosis through altered AMPA receptor trafficking. J. Neurosci. 26, 11798–11806. doi: 10.1523/jneurosci.2084-06.2006
Lautenschläger, J., Prell, T., Ruhmer, J., Weidemann, L., Witte, O. W., and Grosskreutz, J. (2013). Overexpression of human mutated G93A SOD1 changes dynamics of the ER mitochondria calcium cycle specifically in mouse embryonic motor neurons. Exp. Neurol. 247, 91–100. doi: 10.1016/j.expneurol.2013.03.027
Leal, S. S., Cardoso, I., Valentine, J. S., and Gomes, C. M. (2013). Calcium ions promote superoxide dismutase 1 (SOD1) aggregation into non-fibrillar amyloid: a link to toxic effects of calcium overload in amyotrophic lateral sclerosis (ALS)? J. Biol. Chem. 288, 25219–25228. doi: 10.1074/jbc.M113.470740
Liu, J., Lillo, C., Jonsson, P. A., Vande Velde, C., Ward, C. M., Miller, T. M., et al. (2004). Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron 43, 5–17. doi: 10.1016/j.neuron.2004.06.016
Liu, J., Tang, T. S., Tu, H., Nelson, O., Herndon, E., Huynh, D. P., et al. (2009). Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 2. J. Neurosci. 29, 9148–9162. doi: 10.1523/JNEUROSCI.0660-09.2009
Maekawa, S., Leigh, P. N., King, A., Jones, E., Steele, J. C., Bodi, I., et al. (2009). TDP-43 is consistently co-localized with ubiquitinated inclusions in sporadic and Guam amyotrophic lateral sclerosis but not in familial amyotrophic lateral sclerosis with and without SOD1 mutations. Neuropathology 29, 672–683. doi: 10.1111/j.1440-1789.2009.01029.x
Maruyama, H., Morino, H., Ito, H., Izumi, Y., Kato, H., Watanabe, Y., et al. (2010). Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465, 223–226. doi: 10.1038/nature08971
Masutani, T., Taguchi, K., Kumanogoh, H., Nakamura, S., and Maekawa, S. (2008). Molecular interaction of neurocalcin alpha with alsin (ALS2). Neurosci. Lett. 438, 26–28. doi: 10.1016/j.neulet.2008.04.066
Mattiazzi, M., D’Aurelio, M., Gajewski, C. D., Martushova, K., Kiaei, M., Beal, M. F., et al. (2002). Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J. Biol. Chem. 277, 29626–29633. doi: 10.1074/jbc.m203065200
Migheli, A., Cordera, S., Bendotti, C., Atzori, C., Piva, R., and Schiffer, D. (1999). S-100beta protein is upregulated in astrocytes and motor neurons in the spinal cord of patients with amyotrophic lateral sclerosis. Neurosci. Lett. 261, 25–28. doi: 10.1016/s0304-3940(98)01001-5
Mórotz, G. M., De Vos, K. J., Vagnoni, A., Ackerley, S., Shaw, C. E., and Miller, C. C. (2012). Amyotrophic lateral sclerosis-associated mutant VAPBP56S perturbs calcium homeostasis to disrupt axonal transport of mitochondria. Hum. Mol. Genet. 21, 1979–1988. doi: 10.1093/hmg/dds011
Nihei, Y., Ito, D., and Suzuki, N. (2012). Roles of ataxin-2 in pathological cascades mediated by TAR DNA-binding protein 43 (TDP-43) and Fused in Sarcoma (FUS). J. Biol. Chem. 287, 41310–41323. doi: 10.1074/jbc.m112.398099
Nishimura, A. L., Mitne-Neto, M., Silva, H. C., Richieri-Costa, A., Middleton, S., Cascio, D., et al. (2004). A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am. J. Hum. Genet. 75, 822–831. doi: 10.1086/425287
Osaka, M., Ito, D., Yagi, T., Nihei, Y., and Suzuki, N. (2015). Evidence of a link between ubiquilin 2 and optineurin in amyotrophic lateral sclerosis. Hum. Mol. Genet. 24, 1617–1629. doi: 10.1093/hmg/ddu575
Palecek, J., Lips, M. B., and Keller, B. U. (1999). Calcium dynamics and buffering in motoneurones of the mouse spinal cord. J. Physiol. 520(Pt. 2), 485–502. doi: 10.1111/j.1469-7793.1999.00485.x
Parone, P. A., Da Cruz, S., Han, J. S., McAlonis-Downes, M., Vetto, A. P., Lee, S. K., et al. (2013). Enhancing mitochondrial calcium buffering capacity reduces aggregation of misfolded SOD1 and motor neuron cell death without extending survival in mouse models of inherited amyotrophic lateral sclerosis. J. Neurosci. 33, 4657–4671. doi: 10.1523/jneurosci.1119-12.2013
Pedrini, S., Sau, D., Guareschi, S., Bogush, M., Brown, R. H. Jr., Naniche, N., et al. (2010). ALS-linked mutant SOD1 damages mitochondria by promoting conformational changes in Bcl-2. Hum. Mol. Genet. 19, 2974–2986. doi: 10.1093/hmg/ddq202
Pokrishevsky, E., Grad, L. I., Yousefi, M., Wang, J., Mackenzie, I. R., and Cashman, N. R. (2012). Aberrant localization of FUS and TDP43 is associated with misfolding of SOD1 in amyotrophic lateral sclerosis. PLoS One 7:e35050. doi: 10.1371/journal.pone.0035050
Prell, T., Lautenschläger, J., and Grosskreutz, J. (2013). Calcium-dependent protein folding in amyotrophic lateral sclerosis. Cell Calcium 54, 132–143. doi: 10.1016/j.ceca.2013.05.007
Prosser, D. C., Tran, D., Gougeon, P. Y., Verly, C., and Ngsee, J. K. (2008). FFAT rescues VAPA-mediated inhibition of ER-to-Golgi transport and VAPB-mediated ER aggregation. J. Cell. Sci. 121, 3052–3061. doi: 10.1242/jcs.028696
Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Sapp, P., Hentati, A., et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62. doi: 10.1038/362059a0
Rowland, L. P., and Shneider, N. A. (2001). Amyotrophic lateral sclerosis. N. Engl. J. Med. 344, 1688–1700. doi: 10.1056/NEJM200105313442207
Shaw, P. J., and Eggett, C. J. (2000). Molecular factors underlying selective vulnerability of motor neurons to neurodegeneration in amyotrophic lateral sclerosis. J. Neurol. 247(Suppl. 1), I17–I27. doi: 10.1007/bf03161151
Siklós, L., Engelhardt, J. I., Alexianu, M. E., Gurney, M. E., Siddique, T., and Appel, S. H. (1998). Intracellular calcium parallels motoneuron degeneration in SOD-1 mutant mice. J. Neuropathol. Exp. Neurol. 57, 571–587. doi: 10.1097/00005072-199806000-00005
Siklós, L., Engelhardt, J., Harati, Y., Smith, R. G., Joó, F., and Appel, S. H. (1996). Ultrastructural evidence for altered calcium in motor nerve terminals in amyotropic lateral sclerosis. Ann. Neurol. 39, 203–216. doi: 10.1002/ana.410390210
Sreedharan, J., Blair, I. P., Tripathi, V. B., Hu, X., Vance, C., Rogelj, B., et al. (2008). TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672. doi: 10.1126/science.1154584
Stoica, R., De Vos, K. J., Paillusson, S., Mueller, S., Sancho, R. M., Lau, K. F., et al. (2014). ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 5:3996. doi: 10.1038/ncomms4996
Su, X. W., Broach, J. R., Connor, J. R., Gerhard, G. S., and Simmons, Z. (2014). Genetic heterogeneity of amyotrophic lateral sclerosis: implications for clinical practice and research. Muscle Nerve 49, 786–803. doi: 10.1002/mus.24198
Tadic, V., Prell, T., Lautenschlaeger, J., and Grosskreutz, J. (2014). The ER mitochondria calcium cycle and ER stress response as therapeutic targets in amyotrophic lateral sclerosis. Front. Cell. Neurosci. 8:147. doi: 10.3389/fncel.2014.00147
Tang, T. S., Tu, H., Chan, E. Y., Maximov, A., Wang, Z., Wellington, C. L., et al. (2003). Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5) triphosphate receptor type 1. Neuron 39, 227–239. doi: 10.1016/S0896-6273(03)00366-0
Tateno, M., Sadakata, H., Tanaka, M., Itohara, S., Shin, R. M., Miura, M., et al. (2004). Calcium-permeable AMPA receptors promote misfolding of mutant SOD1 protein and development of amyotrophic lateral sclerosis in a transgenic mouse model. Hum. Mol. Genet. 13, 2183–2196. doi: 10.1093/hmg/ddh246
Tortarolo, M., Grignaschi, G., Calvaresi, N., Zennaro, E., Spaltro, G., Colovic, M., et al. (2006). Glutamate AMPA receptors change in motor neurons of SOD1G93A transgenic mice and their inhibition by a noncompetitive antagonist ameliorates the progression of amytrophic lateral sclerosis-like disease. J. Neurosci. Res. 83, 134–146. doi: 10.1002/jnr.20715
Tradewell, M. L., Cooper, L. A., Minotti, S., and Durham, H. D. (2011). Calcium dysregulation, mitochondrial pathology and protein aggregation in a culture model of amyotrophic lateral sclerosis: mechanistic relationship and differential sensitivity to intervention. Neurobiol. Dis. 42, 265–275. doi: 10.1016/j.nbd.2011.01.016
Tudor, E. L., Galtrey, C. M., Perkinton, M. S., Lau, K. F., De Vos, K. J., Mitchell, J. C., et al. (2010). Amyotrophic lateral sclerosis mutant vesicle-associated membrane protein-associated protein-B transgenic mice develop TAR-DNA-binding protein-43 pathology. Neuroscience 167, 774–785. doi: 10.1016/j.neuroscience.2010.02.035
Valencia, C. A., Ju, W., and Liu, R. (2007). Matrin 3 is a Ca2+/calmodulin-binding protein cleaved by caspases. Biochem. Biophys. Res. Commun. 361, 281–286. doi: 10.1016/j.bbrc.2007.06.156
Vance, C., Rogelj, B., Hortobágyi, T., De Vos, K. J., Nishimura, A. L., Sreedharan, J., et al. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211. doi: 10.1126/science.1165942
Van Damme, P., Leyssen, M., Callewaert, G., Robberecht, W., and Van Den Bosch, L. (2003). The AMPA receptor antagonist NBQX prolongs survival in a transgenic mouse model of amyotrophic lateral sclerosis. Neurosci. Lett. 343, 81–84. doi: 10.1016/s0304-3940(03)00314-8
Vandenberghe, W., Robberecht, W., and Brorson, J. R. (2000). AMPA receptor calcium permeability, GluR2 expression and selective motoneuron vulnerability. J. Neurosci. 20, 123–132.
Van Den Bosch, L., Vandenberghe, W., Klaassen, H., Van Houtte, E., and Robberecht, W. (2000). Ca(2+)-permeable AMPA receptors and selective vulnerability of motor neurons. J. Neurol. Sci. 180, 29–34. doi: 10.1016/s0022-510x(00)00414-7
van den Heuvel, D. M., Harschnitz, O., Van Den Berg, L. H., and Pasterkamp, R. J. (2014). Taking a risk: a therapeutic focus on ataxin-2 in amyotrophic lateral sclerosis? Trends Mol. Med. 20, 25–35. doi: 10.1016/j.molmed.2013.09.001
Vande Velde, C., Miller, T. M., Cashman, N. R., and Cleveland, D. W. (2008). Selective association of misfolded ALS-linked mutant SOD1 with the cytoplasmic face of mitochondria. Proc. Natl. Acad. Sci. U S A 105, 4022–4027. doi: 10.1073/pnas.0712209105
Volkening, K., Leystra-Lantz, C., Yang, W., Jaffee, H., and Strong, M. J. (2009). Tar DNA binding protein of 43 kDa (TDP-43), 14–3-3 proteins and copper/zinc superoxide dismutase (SOD1) interact to modulate NFL mRNA stability. Implications for altered RNA processing in amyotrophic lateral sclerosis (ALS). Brain Res. 1305, 168–182. doi: 10.1016/j.brainres.2009.09.105
von Lewinski, F., and Keller, B. U. (2005). Ca2+, mitochondria and selective motoneuron vulnerability: implications for ALS. Trends Neurosci. 28, 494–500. doi: 10.1016/j.tins.2005.07.001
Wang, H. G., Pathan, N., Ethell, I. M., Krajewski, S., Yamaguchi, Y., Shibasaki, F., et al. (1999). Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science 284, 339–343. doi: 10.1126/science.284.5412.339
Williams, T. L., Day, N. C., Ince, P. G., Kamboj, R. K., and Shaw, P. J. (1997). Calcium-permeable alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors: a molecular determinant of selective vulnerability in amyotrophic lateral sclerosis. Ann. Neurol. 42, 200–207. doi: 10.1002/ana.410420211
Wu, C. H., Fallini, C., Ticozzi, N., Keagle, P. J., Sapp, P. C., Piotrowska, K., et al. (2012). Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 488, 499–503. doi: 10.1038/nature11280
Yamashita, T., and Kwak, S. (2014). The molecular link between inefficient GluA2 Q/R site-RNA editing and TDP-43 pathology in motor neurons of sporadic amyotrophic lateral sclerosis patients. Brain Res. 1584, 28–38. doi: 10.1016/j.brainres.2013.12.011
Yin, H. Z., Tang, D. T., and Weiss, J. H. (2007). Intrathecal infusion of a Ca(2+)-permeable AMPA channel blocker slows loss of both motor neurons and of the astrocyte glutamate transporter, GLT-1 in a mutant SOD1 rat model of ALS. Exp. Neurol. 207, 177–185. doi: 10.1016/j.expneurol.2007.07.011
Zhang, M. D., Tortoriello, G., Hsueh, B., Tomer, R., Ye, L., Mitsios, N., et al. (2014). Neuronal calcium-binding proteins 1/2 localize to dorsal root ganglia and excitatory spinal neurons and are regulated by nerve injury. Proc. Natl. Acad. Sci. U S A 111, E1149–E1158. doi: 10.1073/pnas.1402318111
Keywords: neurodegenerative diseases, calcium homeostasis, ALS, proteinopathies, SOD1
Citation: Leal SS and Gomes CM (2015) Calcium dysregulation links ALS defective proteins and motor neuron selective vulnerability. Front. Cell. Neurosci. 9:225. doi: 10.3389/fncel.2015.00225
Received: 21 April 2015; Accepted: 28 May 2015;
Published online: 16 June 2015.
Edited by:
Manoj Kumar Jaiswal, Center for Neuroscience and Regenerative Medicine, USAReviewed by:
Adán Dagnino-Acosta, Baylor College of Medicine, USAMarco Martina, Northwestern University, USA
Karim Mekhail, University of Toronto, Canada
Copyright © 2015 Leal and Gomes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cláudio M. Gomes, Faculdade de Ciências, Biosystems and Integrative Sciences Institute and Department of Chemistry and Biochemistry, Universidade de Lisboa, Ed C8, Campo Grande, Lisboa 1749-016, Portugal,Y21nb21lc0BmYy51bC5wdA==;
folding.fc.ul.pt