 Mauricio A. Retamal1*
Mauricio A. Retamal1* Edison P. Reyes1,2
Edison P. Reyes1,2 Isaac E. García3
Isaac E. García3 Bernardo Pinto3
Bernardo Pinto3 Agustín D. Martínez3
Agustín D. Martínez3 Carlos González3
Carlos González3- 1Centro de Fisiología Celular e Integrativa, Facultad de Medicina, Clínica Alemana Universidad del Desarrollo, Santiago, Chile
- 2Centro de Investigación Biomédica, Universidad Autónoma de Chile, Santiago, Chile
- 3Centro Interdisciplinario de Neurociencia de Valparaíso, Instituto de Neurociencia, Facultad de Ciencias, Universidad de Valparaíso, Valparaíso, Chile
Hemichannels (HCs) and gap junction channels (GJCs) formed by protein subunits called connexins (Cxs) are major pathways for intercellular communication. While HCs connect the intracellular compartment with the extracellular milieu, GJCs allow the interchange of molecules between cytoplasm of two contacting cells. Under physiological conditions, HCs are mostly closed, but they can open under certain stimuli allowing the release of autocrine and paracrine molecules. Moreover, some pathological conditions, like ischemia or other inflammation conditions, significantly increase HCs activity. In addition, some mutations in Cx genes associated with human diseases, such as deafness or cataracts, lead to the formation of more active HCs or “leaky HCs.” In this article we will revise cellular and molecular mechanisms underlying the appearance of leaky HCs, and the consequences of their expression in different cellular systems and animal models, in seeking a common pattern or pathological mechanism of disease.
Introduction
Connexins (Cxs) are a family of transmembrane (TM) proteins formed by 21 members (Eiberger et al., 2001; Söhl and Willecke, 2004) named according to their predicted molecular weight (i.e., Cx43 has ∼43 kDa). Cxs are expressed in almost every cell type in the human body (Bruzzone et al., 1996). However, there are some differences. Thus, for example, there are Cxs widely expressed such as Cx43, which is found in the brain, kidneys, heart and reproductive organs, among others (Beyer et al., 1987, 1989; Sáez et al., 2003), or restricted to myelin-forming glial cells, as in the case of Cx29 (Söhl et al., 2001). Cxs form two types of channels; hemichannels (HCs) and gap junction channels (GJCs). HCs are formed by the oligomerization of six Cxs monomers and travel in vesicles to the plasma membrane (Vinken et al., 2006). The Cx topology in cell membrane is depicted in Figure 1 and includes four TM segments (TM1-4), which are connected through two extracellular loops (EL1-EL2) and one intracellular loop (IL); and the N-terminal (NT) and C-terminal (CT) segments oriented to the cytosol (Kumar and Gilula, 1996). HCs can form GJC in the appositional membranes of contacting cells or stay as “free” HCs anywhere on the plasma membrane (Figure 2). Free HCs are mostly closed under physiological conditions (Contreras et al., 2003), that is because they have low open probability (OP) due to one or more of the following mechanisms: (i) a blockage by extracellular Ca2+ and Mg2+ in the mM range, (ii) a negative membrane potential that closes most Cx HCs and (iii) posttranslational modification (i.e., phosphorylation) of some Cxs (Contreras et al., 2003; Gómez-Hernández et al., 2003; Johnstone et al., 2012). However, HCs can open under physiological conditions allowing communication between extracellular and intracellular space (Sáez et al., 2010). On the other hand, GJCs are formed in the appositional membrane by the serial docking of two complementary HCs, each one in the respective neighboring cell membrane (Figure 2). GJCs allow the intercellular exchange of ions and molecules such as glucose and amino acids between contacting cells (Payton et al., 1969; Goldberg et al., 2004; Ek-Vitorin and Burt, 2013). Because of these properties, Cx based channels have been associated with different cellular processes such as cellular communication and tissue coordination (Sáez et al., 2010).
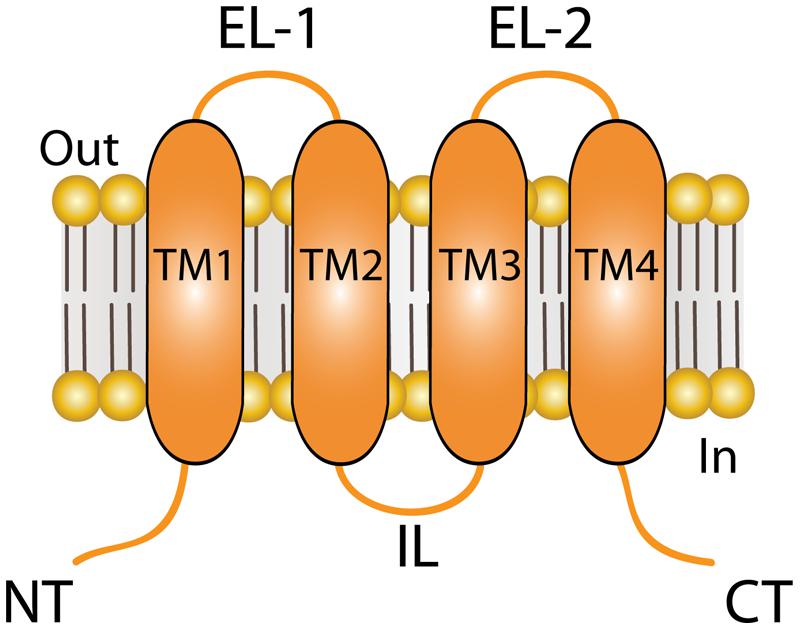
FIGURE 1. Topology of connexin (Cx) at the plasma membrane. Cartoon depicting the plasma membrane topology shared by all Cx isoforms, which includes four transmembrane (TM) segments that are connected by two extracellular loops (ELs) and one intracellular loop (IL). The amino terminal (NT) and carboxi terminal (CT) segments of each hemichannel face the cytoplasm. The length of the NT and CT segments is not intended to represent any particular Cx isoform.
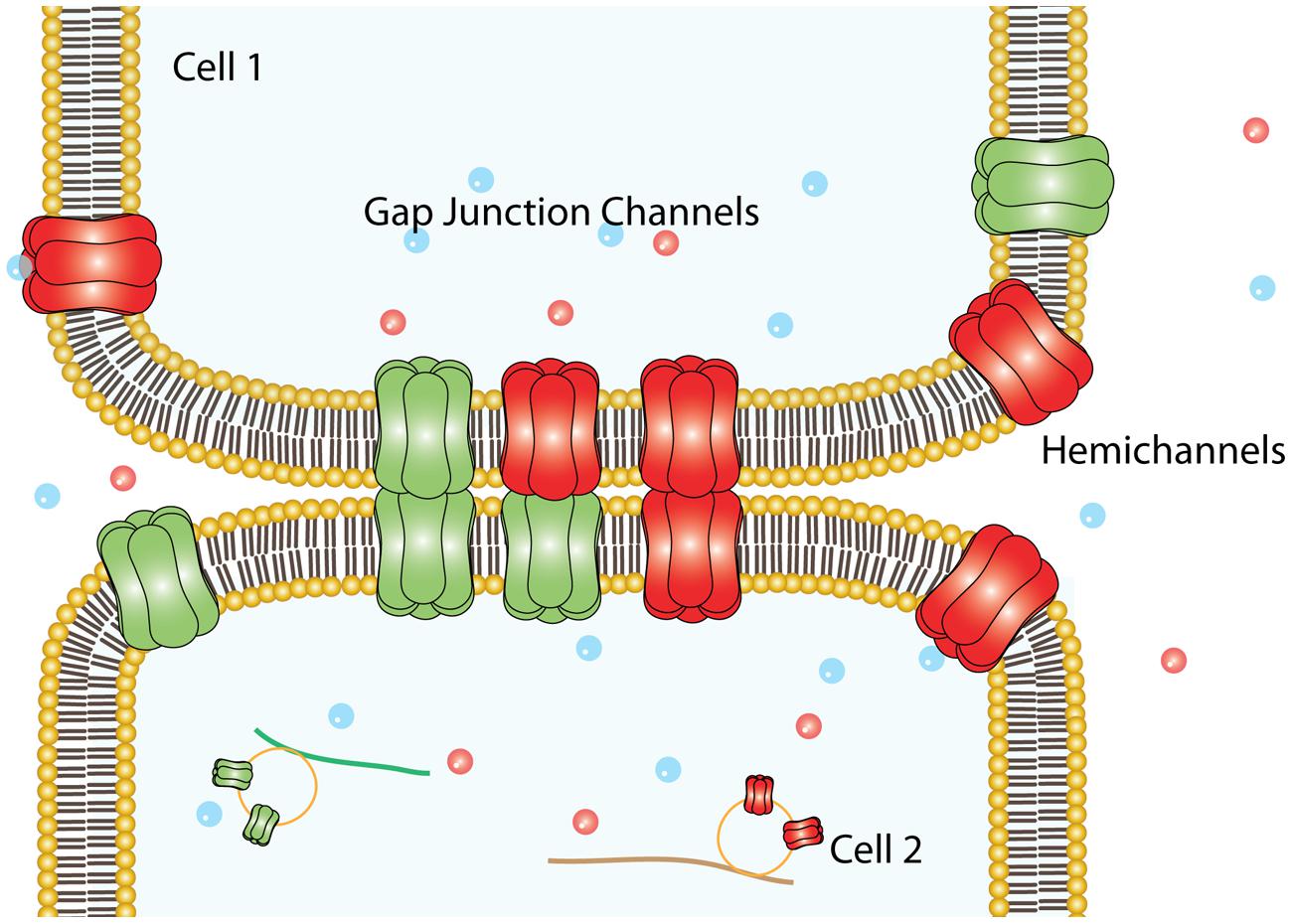
FIGURE 2. Plasma membrane arrangements of Cxs. Six Cxs oligomerize to form a HC that traveled to the non-appositional plasma membrane to form free HCs, which provide an auto/paracrine communication pathway between the cell and the extracellular milieu. Alternatively, can dock others HCs provided by an adjacent cell (appositional plasma membrane) to form intercellular aqueous pore named gap junction channels.
Role of HCs in Physiological Conditions
HCs have an estimated pore diameter ranging from 12 to 15 Å in its narrowest part (Oh et al., 1997; Gong and Nicholson, 2001; Rackauskas et al., 2010). The crystal structure of Cx26 channels shows that the NT is inside the pore, a factor that restricts the pore diameter (Maeda et al., 2009). However, recent refinements of this structure using molecular dynamic methods suggest that the pore diameter could be a little smaller (Kwon et al., 2011). Much experimental evidence shows that opening of HCs activates pathways linked to the release or uptake of paracrine and autocrine molecules such as: ATP (Anselmi et al., 2008 (Cx26); Svenningsen et al., 2013 (Cx30); Nualart-Marti et al., 2013 (Cx32); Schock et al., 2008 (Cx36); Stout et al., 2002 (Cx43)), glutamate (Takeuchi et al., 2006 (Cx32); Ye et al., 2003 (Cx43)), PGE2 (Cherian et al., 2005 (Cx43)), NAD+ (Bruzzone et al., 2001 (Cx43)) and glutathione (Rana and Dringen, 2007 (Cx43)). HCs may also mediate uptake of glucose as well as extracellular ions. (Retamal et al., 2007 (Cx43); Schalper et al., 2010 (Cx43); Sánchez et al., 2010 (Cx26); Fiori et al., 2012 (Cx26)). Research about HC permeability has been focused mostly on homomeric HCs made by Cx26, Cx32 and Cx43. However, most cell types express more than one Cx isoform, opening the possibility for the formation of heteromeric channels that would present new permeability properties (Beyer et al., 2001; Martinez et al., 2002). For example, it is known that heteromeric HCs formed by Cx26/32 (1:1 ratio) exhibits decreased permeability to (1,4,5)-IP3 compared to the respective homomeric types formed by Cx26 or Cx32 (Ayad et al., 2006). Additionally, information about the in vivo release of molecules through HCs is currently very limited. However, data available suggest that HCs are somehow involved in different physiological processes, such as the control of monocyte adhesion in mice (Wong et al., 2006), neurotransmitter release from astrocytes in the basolateral amygdala (Stehberg et al., 2012), Ca2+ signaling in adult ventricular myocytes (Li et al., 2012), sensory neuron activity (Retamal et al., 2014b), and bone cell physiology and pathology (Plotkin, 2014). Moreover, HCs may also participate in the ATP release from astrocytes to regulate basal glutamatergic synaptic transmission (Chever et al., 2014), in the control of colonic transit (McClain et al., 2014), in wound healing (Takada et al., 2014), in renal function (Sipos et al., 2009), ion flux in lens cells (Beyer and Berthoud, 2014; Mandal et al., 2015) and in the visual processing of the retina (Kamermans et al., 2001). The signaling and molecular mechanisms that control the opening of HCs under physiological conditions are poorly understood. But, at least for Cx43 HCs, one possible mechanism involves interactions between the CT and some regions of the IL (Ponsaerts et al., 2010, 2012).
The presence and functional state of HCs in the plasma membrane have been determined through several techniques, including, dye uptake of fluorescent molecules, release of substances such as ATP, electrophysiology, biotinylation, immunolocalization (Schalper et al., 2008; Wang et al., 2013a; García et al., 2015). Because the existence of other non-selective channels with big pores, like Pannexin channels, which share several characteristics with Cx HCs, there are some criterions that need to be considered in order to demonstrate unique functional properties of Cx HCs (Sáez and Leybaert, 2014). Among these criterions are: (i) cell expression of at least one Cx isoform at the plasma membrane, (ii) the ability of the cells to incorporate/release fluorescent dyes, and /or (iii) to show currents with conductance and/or properties associated to Cx HCs, (iv) the abolishment of Cx-HCs function using classical pharmacology (La3+, mefloquine, carbenoxolone) or Cx mimetic peptides (Gap 26, 27); and finally (v) to demonstrate that blocking Cx HCs exerts physiological responses.
Leaky HCs
Paul et al. (1991) provided the first evidence linking HCs with cell death, they observed that overexpression of Cx46 in Xenopus laevis oocytes, induces depolarization and lysis of oocytes 24 h after mRNA injection. Interestingly, cell death did not occur when other Cxs, like Cx32 or Cx43 were overexpressed. Later on, it was demonstrated that human Cx43 does not form functional HCs in Xenopus oocytes (Hoang et al., 2010), suggesting that formation of functional HCs depends on both Cx isoform and cell type. On the other hand, several works have shown that many pathological conditions produce uncontrolled and massive HC opening (from now called leaky HCs), which may adversely affect cellular homeostasis and induce cell death (Sáez et al., 2010). For example, it was reported that infection of the gastrointestinal tract increases Cx43 HC activity in colonocytes, which was correlated with an increase of water mobilization and appearance of diarrhea, which can be reverted by down-regulating gut expression level of Cx43 (Guttman et al., 2010). In addition, natural occurring Cx mutations have been associated with different human genetic diseases such as cataract, skin diseases, deafness, X-linked Charcot–Marie–Tooth disease, and oculodentodigital dysplasia (ODDD; Table 1). Some Cx mutations in these diseases produce leaky HCs when expressed in heterologous expression systems. Here, we will review the current knowledge about these leaky HCs and their possible pathologic mechanisms of disease.
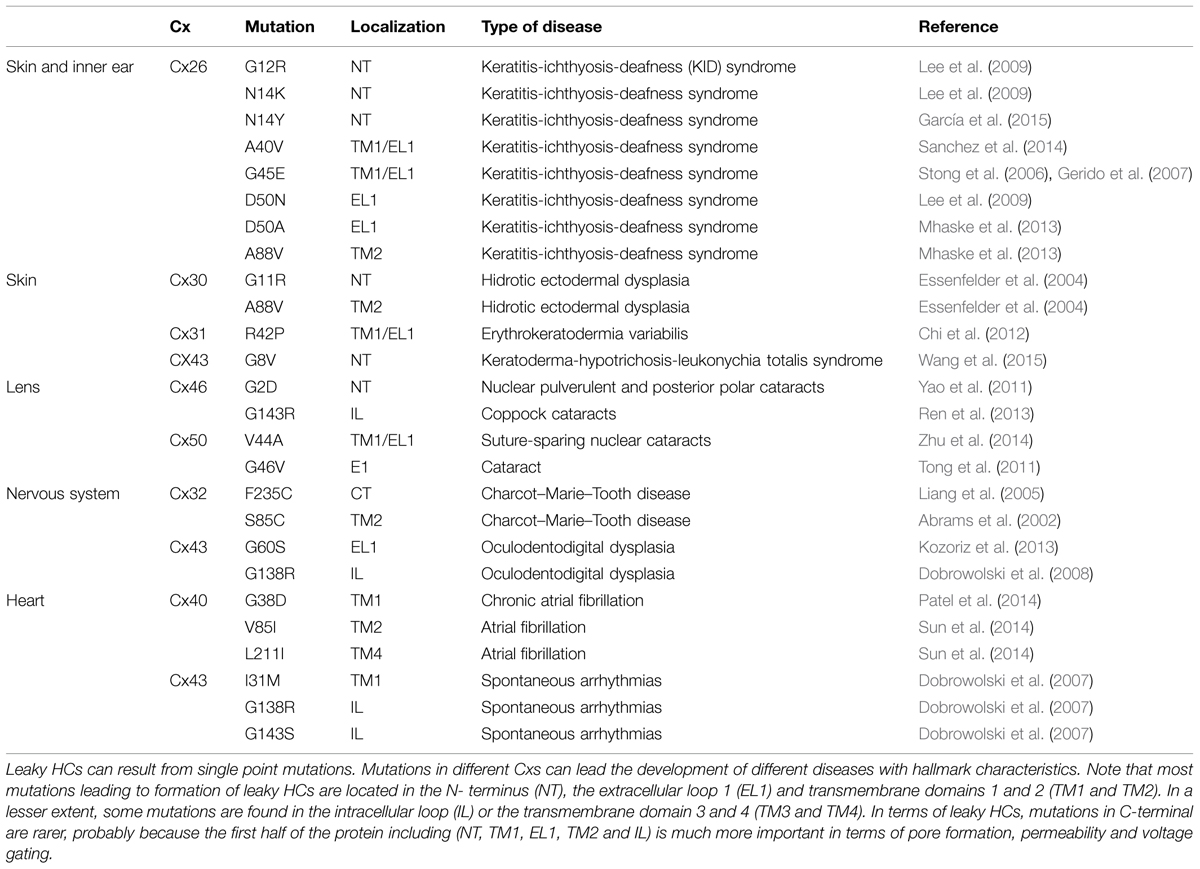
TABLE 1. Connexin (Cx) mutations associated to leaky HCs.
Cataract
Lens cells express Cx43, Cx46, and Cx50 (Beyer and Berthoud, 2014). Until now, Cx43 mutations have not been associated with cataract formation; however, the lack or malfunctioning of Cxs 46 and 50 has been associated with the development of different types of cataracts (Beyer and Berthoud, 2014). Accordingly, the expression of Cx50G46V mutant in Xenopus oocytes induces large HC currents at physiological extracellular Ca2+ concentration (Tong et al., 2011). In addition, this mutant promotes cell death when expressed in HeLa cells, which was prevented by the increase of the extracellular Ca2+ concentration (Minogue et al., 2009). Similarly, Cx50V44A mutant also induces HeLa cell death, which was reduced by HC blockers (Zhu et al., 2014). The aforementioned data suggest that some Cx50 mutations located closely to the TM1 and EL1 border result in formation of leaky HCs, which cause cataracts when expressed in humans. Additionally, different Cx46 mutations have been associated with leaky HCs. For example, mutant Cx46G143R (located in the IL) leads to the appearance of Coppock cataracts (Ren et al., 2013), and mutant Cx46G2D (located the NT) has been linked to formation of nuclear pulverulent and posterior polar cataracts (Yao et al., 2011). Lower plasma membrane expression of these mutants was enough to promote cell death when expressed in HeLa cells (Ren et al., 2013). This suggests that mutation Cx46G143R induces an important increase in the HC activity, possibly by modifying the interaction between the CT and IL, which is associated with HC opening (Ren et al., 2013). A possible explanation for the pathological mechanism of leaky Cx46 HCs is that the opening of these channels produces an excessive flow of Ca2+ through the plasma membrane (Ebihara et al., 2014; Mandal et al., 2015), which should perturb the normal ionic balance of lens cells (Figure 3).
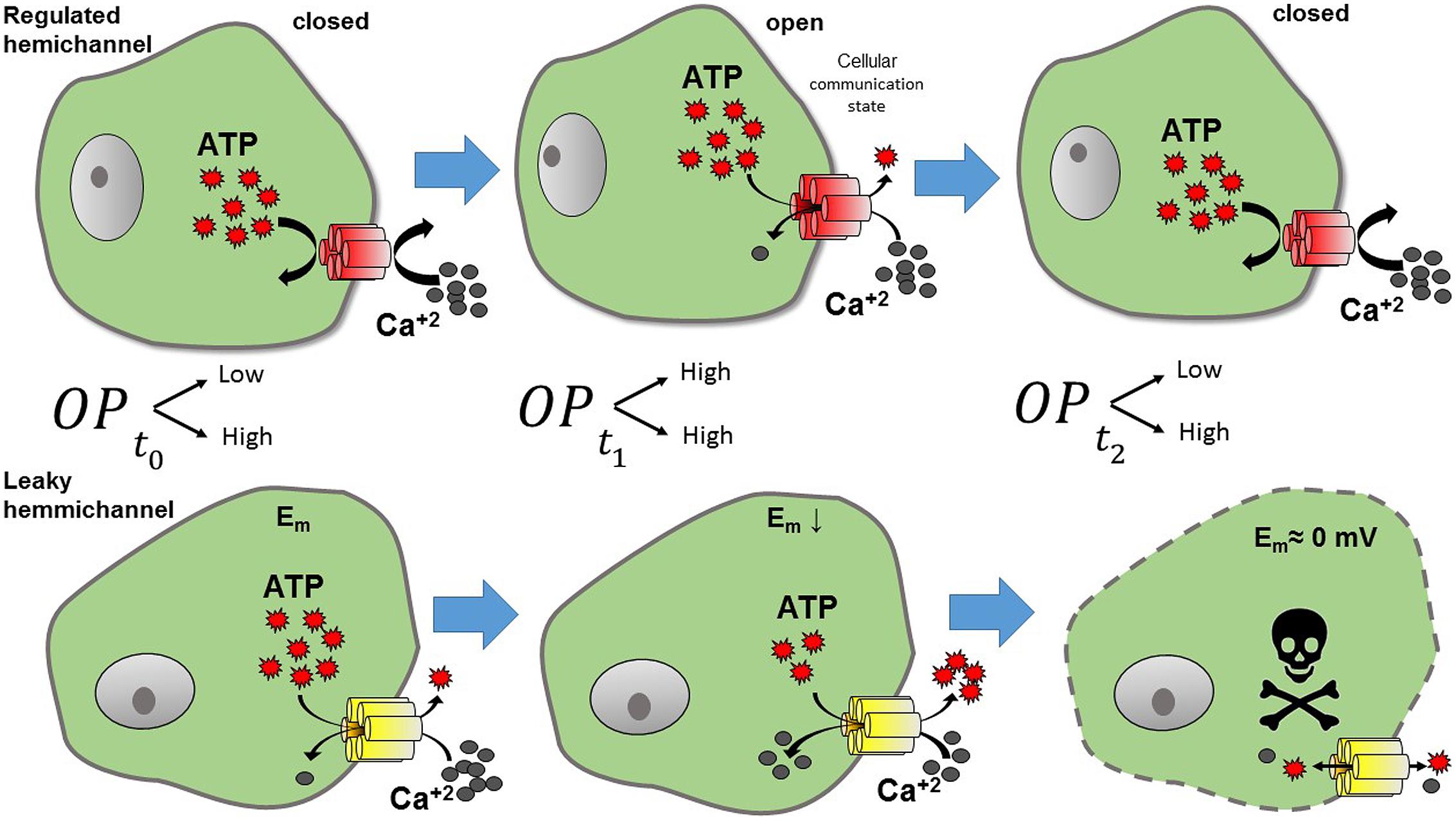
FIGURE 3. Representation of the effects of leaky HC. Under normal conditions (upper panel) HCs present a low open probability (OP). Thus, when HCs are normally closed (t0, low OP), no exchange with the extracellular milieu is observed. However, when HCs open (t1, higher OP), molecules such as ATP and Ca2+ can flow through them. Calcium may activate intracellular pathways, and ATP released from the cell, can act as a paracrine -or autocrine- signal, hence, the cell is at a communicating state. In contrast leaky HCs (lower panel) maintain a high OP, producing a continuous flow out and into the cell. Leaky HCs exchange continuously, resulting in the reduction of cell membrane potential and later cell death (t2).
Skin Diseases and Deafness
Several Cx types such as Cx26, Cx30, Cx30.3, Cx31.1, Cx37, and Cx43 are differentially expressed in the skin (Scott et al., 2012). On the other hand, while in the inner ear the sensory hair cells do not express Cxs, several Cxs (Cxs 26, 29, 30, 31, 43) are expressed in supporting epithelial cells of the organ of Corti, stria vascularis and in the interstitial cellular network that compose the wall of the scala media (Martínez et al., 2009). However, until now, only mutations in Cx26 gene are associated to syndromic (deafness plus skin disease) and non-syndromic deafness (Hoang Dinh et al., 2009; Martínez et al., 2009). Currently it is known that several missense point mutation in Cx26 – G12R, N14K, N14Y, A40V, G45E, D50N, D50A and A88V do form leaky HCs and induce both skin and hearing disorders, which together are known as keratitis-ichthyosis-deafness (KID) syndrome (Stong et al., 2006; Gerido et al., 2007; Lee et al., 2009; García et al., 2013; Mhaske et al., 2013; Meigh et al., 2014; Sanchez et al., 2014). Interestingly, García et al. (2015) showed that the mutant Cx26S17F presents decreased HC activity when expressed alone in Xenopus oocytes, but when is co-expressed with Cx43 [which does not form functional HCs in Xenopus oocytes (Hansen et al., 2014)], a large HC current is then evident (García et al., 2015). Because of these leaky HCs, HeLa cells expressing Cx26S17F and Cx43 showed almost twice the basal intracellular Ca2+ concentration (García et al., 2015). These results could explain the resulting KID syndrome of the mutant S17F, since in the human skin Cx26 and Cx43 are co-expressed in keratinocytes of the stratum basal (Wang et al., 2009). In addition, certain mutations located in the EL1 also produce leaky HCs, such as D50N, that change the Ca2+ control over HC activity through the modification of a salt bridge between D50 and K61, which is important for HC closure induced by extracellular Ca2+ (Lopez et al., 2013; Sanchez et al., 2013). Consistently, a similar mutation (Cx26D50A) also induces leaky HC and produce KID syndrome (Mhaske et al., 2013). On the other hand, mutant Cx26A40V, located in the TM1/EL1 border, increases HC activity by decreasing sensitivity to pH and divalent cations (Sanchez et al., 2014). However, other mutation in the same residue (A40G) produced non-functional HCs (Jara et al., 2012) and severe deafness without skin disease, suggesting that this residue is positioned in a pivotal region for HC gating control. Another mutant associated with KID syndrome, Cx26G45E (Griffith et al., 2006) also induces large HC currents or dye uptake that leads to cell death when expressed in Xenopus oocytes or HEK293 cells (Stong et al., 2006; Gerido et al., 2007). The phenotype induced by the Cx26G45E mutant could be the result of a lack of Ca2+ control (Zhang and Hao, 2013), reflected by enhanced Ca2+ permeability in cells expressing this mutant (Sanchez et al., 2014). In a mouse model, Cx26G45E induces a reduction of cell viability, hyperkeratosis, scaling, skinfolds, and hair loss (Mese et al., 2011). These data suggest that leaky Cx26 HCs can induce skin problems and deafness, probably due to misregulation of intracellular Ca2+ homeostasis and ATP release (García et al., 2015) (Figure 3); both conditions could affect many cellular processes, such as abnormal keratinocyte proliferation and cell death. Although, most mutations in Cx26 associated with deafness produce lack of function GJCs, only syndromic mutations produce gain of HC activity (Martínez et al., 2009; García et al., 2013). However, since syndromic mutations also affect GJCs, the pathogenic mechanism of disease may involve deregulation of both types of channels, which is a condition that would worsen the cellular homeostasis (García et al., 2015).
Mutations in Cx30 have been also linked to skin disease. Thus, mutants of Cx30- G11R (NT) and A88V (TM2) has been associated with Clouston syndrome (abnormal formation of ectodermal structures) and when they are expressed in HeLa cells induce a massive release of ATP into the extracellular medium (Essenfelder et al., 2004) and subsequent cell death (Berger et al., 2014), suggesting uncontrolled HC activity. On the other hand, the mutant Cx31R42P (which produces erythrokeratodermia variabilis) promotes an enhancement in HC activity and cell death when expressed in HeLa cells (Chi et al., 2012). It was postulated that the previous condition was mediated by an important production of free radicals as consequence of ER stress (Chi et al., 2012). Recently, a mutation in Cx43 (Cx43G8V) has been associated to keratoderma-hypotrichosis-leukonychia totalis syndrome (KHLS), a condition characterized by hyperkeratosis and alopecia, the expression of this mutant induced gain in HC activity and Ca2+ influx into the cells (Wang et al., 2015).
X-linked Charcot–Marie–Tooth Disease
This neuropathy is a hereditary disease caused by different mutations in Cx32 gene (Bergoffen et al., 1993). There are several Cx32 mutations that induce Charcot–Marie–Tooth disease (Liang et al., 2005). Patients with this disease present neurodegeneration due to altered myelin production by Schwann cells (Bergoffen et al., 1993). When the mutation Cx32F235C (CT) is expressed in Xenopus oocytes, it induces cell death after 72 h, which was associated with changes in its voltage sensitivity (Liang et al., 2005). In addition, the other pathological mutant Cx32S85C induces a decrease in the number of HCs at the plasma membrane (measured as biotinylated protein). However, when this mutant was expressed in oocytes, larger HC currents were observed under depolarization (Abrams et al., 2002). Thus, several mutations induce this disease, but to date only two of them have been reported to form leaky HCs.
Oculodentodigital dysplasia
It is a dominant negative inherited disorder caused by mutations in Cx43 encoding gene. ODDD’s patients exhibit abnormalities in fingers, toes, eyes, face and teeth. Mice expressing human mutations Cx43- G138R or G60S, mimic the phenotype observed in humans (Dobrowolski et al., 2008; Kozoriz et al., 2013). In the case of mice +/G60S, the area of cell death induced by brain ischemia was bigger compared to control mice; accordingly, astrocytes cultured from +/G60S mice, show enhanced HC activity (Kozoriz et al., 2013). In the other hand, mutant Cx43G138R lacks one of the typical phosphorylated forms of Cx43 (P2), and cells extracted from the +/G138R mice present increased ATP release (Dobrowolski et al., 2008). The previous results were consistent with the hypothesis that the phosphorylation state of the Cx43 CT regulates Cx43 HC activity.
Heart Disease
Heart cells express Cx40, Cx43, and Cx45. However, their respective expression is restricted to few types of cells in the heart (Bai, 2014). For example, Cx40 is expressed only in the atria and ventricular conduction system, while Cx43 is mostly expressed in cardiomyocytes (Bai, 2014). Several Cx40 mutations have been associated with atrial-fibrillation problems, but only mutants Cx40- G38D, V85I and L211I enhance HC activity (Patel et al., 2014; Sun et al., 2014). In the case of G38D, it was found that HCs formed by this mutant present a gain of activity when N2A cells were subjected to hyperpolarization and depolarization (Patel et al., 2014). Cx43- I31M, G143S and G138R mutants (which also induce ODDD), present spontaneous arrhythmias, which were associated with both, a decrease of GJC coupling and an increase of ATP release from cardiomyocytes (Dobrowolski et al., 2007). A few years ago, it was demonstrated that down-regulation of Cx43 in cardiac fibroblasts reduce the amount of ATP released (Lu et al., 2012). The ATP released activates the pro-fibrotic response to heart insults via activation of P2Y receptors (Lu et al., 2012). Thus, increased Cx43 HC activity after -for example- myocardial infarction (John et al., 1999; Johansen et al., 2011) will lead to cardiomyocyte malfunction due to a massive entry of Ca2+ and Na+ (Li et al., 2001). In addition, it will also contribute to cardiac fibrosis (Lu et al., 2012) increasing heart failure.
Central Nervous System Neurodegenerative Diseases
Under physiological conditions HCs participate in important functions of the nervous system (NS), as for example, in synaptic modulation (Stehberg et al., 2012; Chever et al., 2014). Moreover, it has been shown that some pathological conditions increase HC activity, in particular the activity of astrocyte HCs formed by Cx43, which have been correlated with neuronal malfunctioning and death (Orellana et al., 2012). When an ischemic episode occurs, astrocytes open their Cx43 HCs (Contreras et al., 2002; Retamal et al., 2006), probably due to dephosphorylation and S-nitrosylation of Cx43 (Retamal et al., 2006). The previous conditions induce a massive opening of astrocyte Cx43 HCs allowing the release of high amounts of ATP and glutamate from astrocytes (Orellana et al., 2011a; Li et al., 2015). This increment in extracellular ATP and glutamate concentration could induce the opening of neuronal Pannexin1 channels, perturbing neuron homeostasis causing cell death (Orellana et al., 2011a). Consistently, administration of Cx43 mimetic peptides, to block HCs, improved brain recovery after ischemia in fetal sheep (Davidson et al., 2012) and neonatal rats (Li et al., 2015).
Hyperactive HCs may also be involved in other brain diseases. Lysosomal storage diseases (LSDs) encompass a large group of inherited metabolic disorders characterized by the accumulation of storage material within lysosomes and HCs seems to have a relevant role in the progression of these diseases (Bosch and Kielian, 2014). In this line, an enhanced Cx43 HC activity was observed in astrocytes from a mouse model of LSD (CLN3Δex7/8; Finn et al., 2011; Burkovetskaya et al., 2014) which could importantly contribute to neuronal deterioration as mentioned above. On the other hand, opening of HCs could also contribute to brain deterioration in Alzheimer’s disease. Orellana et al. (2011b) reported that Aβ peptide induces massive HC opening in astrocytes, microglia, and neurons, either in culture and in hippocampal slices (Orellana et al., 2011b). This increase of HC activity is correlated with augmented release of neuroactive molecules, such as glutamate and ATP, with induction of cellular death (Orellana et al., 2011b; Bosch and Kielian, 2014). Accordingly, blockage of HCs improved memory impairment in a mouse model of Alzheimer’s disease (Takeuchi et al., 2011). Other neurodegenerative diseases in which HC have been involved are: HIV encephalitis (Eugenin and Berman, 2013; Orellana et al., 2014), amyotrophic lateral sclerosis (Boillee et al., 2006; Yamanaka et al., 2008; Takeuchi et al., 2011), Parkinson’s disease (Rufer et al., 1996; Kawasaki et al., 2009), Rasmussen encephalitis (Cepeda et al., 2015) and epilepsy (Mylvaganam et al., 2014). A common milestone of these diseases is the inflammation condition, where cytokines and reactive oxygen species (ROS) can activate HCs in glial cells (astrocytes and microglia; Retamal et al., 2007) increasing the extracellular concentration of compounds, like ATP and glutamate, that could indirectly open Pannexin1 channels leading to neuronal death (Orellana et al., 2012; Bosch and Kielian, 2014; Takeuchi and Suzumura, 2014).
Future Directions
When opened in a controlled fashion, Cx HCs allow physiological paracrine and autocrine communication between neighboring cells. However, under certain pathological conditions, these HCs open more frequently, inducing ionic imbalance and cell lysis. In particular, specific missense mutations in Cx genes associated with human genetic disease produce leaky HCs, a condition that perturbs ionic cell homeostasis, increases ATP release and Ca2+ influx, which in the extreme condition leads to cell death. Probably, the major problem in the study of Cx- based channels is the lack of specific pharmacological tools able to block or open these channels. Thus, for example, one of the most used HC blockers is La3+ (usually used at 200 μM), but this lanthanide also blocks TRP channels (Zhao et al., 2015), cGMP-activated currents (Wang et al., 2013b) and Ca2+ channels (Nelson et al., 1984). Fortunately, in the last years new tools have been developed for the study of Cx- HCs. These are based on small peptides that mimic some regions of a given Cx (Iyyathurai et al., 2013). Through the use of these mimetic peptides it has been possible to study in vitro/in vivo the role of HCs in a much more specific way. Because of their specificity and high affinity, they could be used for the treatment of diseases associated with leaky HCs. In this line of thought, mimetic peptides Gap26 or Gap27 have been observed to block cardiomyocyte death induced by ischemic-like conditions in vitro (Shintani-Ishida et al., 2007) as well as in vivo (Hawat et al., 2012). In the NS, Gap26 and Gap27 peptides blocked Cx43 HC opening induced by inflammatory conditions (Retamal et al., 2007; Froger et al., 2010), while Gap27 reduced spontaneous epileptiform activity in organotypic hippocampal slice cultures and cell death associated with this activity (Samoilova et al., 2008; Yoon et al., 2010). On the other hand, mimetic peptide Gap26 inhibits the spread of damage from the trauma zone to the penumbra in an in vitro model (Rovegno et al., 2015). Similar results have been observed in vivo in a model of spinal cord injury (Huang et al., 2008; O’Carroll et al., 2008) and post-ischemic brain injury (Davidson et al., 2012). Moreover, inhibition of Cx43 HCs with mimetic peptides in the spinal cord, significantly reduced the sensitization to neuropathic pain (Chen et al., 2014), which suggests that opening of HCs could result in an excessive release of neuroactive molecules in chronic pain. Accordingly, exposure of sensory ganglia to mimetic peptides, to block Cx43 HCs of satellite glial cells, reduced sensory neuron activity (Retamal et al., 2014a,b). Therefore, mimetic peptides could be used as the starting point to develop new and more specific pharmacologic agents to inhibit HCs to treat human diseases associated to leaky HCs.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Ms. Carolina Larrain for her corrections and comments performed on our manuscript. Also, we want to thank the financial support from the following sources: Fondecyt #1120214 and Anillo #ACT 1104 (to MR), Fondecyt #1130855 (to ADM), Fondecyt 1120802 (to CG) and Fondecyt 3150634 (to IG). The Centro Interdisciplinario de Neurociencias de Valparaíso is a Chilean Millennium Institute (P09-022-F).
References
Abrams, C. K., Bennett, M. V., Verselis, V. K., and Bargiello, T. A. (2002). Voltage opens unopposed gap junction hemichannels formed by a connexin 32 mutant associated with X-linked Charcot-Marie-Tooth disease. Proc. Natl. Acad. Sci. U.S.A. 99, 3980–3984. doi: 10.1073/pnas.261713499
Anselmi, F., Hernandez, V. H., Crispino, G., Seydel, A., Ortolano, S., Roper, S. D., et al. (2008). ATP release through connexin hemichannels and gap junction transfer of second messengers propagate Ca2+ signals across the inner ear. Proc. Natl. Acad. Sci. U.S.A. 105, 18770–18775. doi: 10.1073/pnas.0800793105
Ayad, W. A., Locke, D., Koreen, I. V., and Harris, A. L. (2006). Heteromeric, but not homomeric, connexin channels are selectively permeable to inositol phosphates. J. Biol. Chem. 281, 16727–16739. doi: 10.1074/jbc.M600136200
Bai, D. (2014). Atrial fibrillation-linked GJA5/connexin40 mutants impaired gap junctions via different mechanisms. FEBS Lett. 588, 1238–1243. doi: 10.1016/j.febslet.2014.02.064
Berger, A. C., Kelly, J. J., Lajoie, P., Shao, Q., and Laird, D. W. (2014). Mutations in Cx30 that are linked to skin disease and non-syndromic hearing loss exhibit several distinct cellular pathologies. J. Cell Sci. 127(Pt 8), 1751–1764. doi: 10.1242/jcs.138230
Bergoffen, J., Scherer, S. S., Wang, S., Scott, M. O., Bone, L. J., Paul, D. L., et al. (1993). Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science 262, 2039–2042. doi: 10.1126/science.8266101
Beyer, E. C., and Berthoud, V. M. (2014). Connexin hemichannels in the lens. Front. Physiol. 5:20. doi: 10.3389/fphys.2014.00020
Beyer, E. C., Gemel, J., Martínez, A., Berthoud, V. M., Valiunas, V., Moreno, A. P., et al. (2001). Heteromeric mixing of connexins: compatibility of partners and functional consequences. Cell Commun. Adhes. 8, 199–204. doi: 10.3109/15419060109080723
Beyer, E. C., Kistler, J., Paul, D. L., and Goodenough, D. A. (1989). Antisera directed against connexin43 peptides react with a 43-kD protein localized to gap junctions in myocardium and other tissues. J. Cell Biol. 108, 595–605. doi: 10.1083/jcb.108.2.595
Beyer, E. C., Paul, D. L., and Goodenough, D. A. (1987). Connexin43: a protein from rat heart homologous to a gap junction protein from liver. J. Cell Biol. 105, 2621–2629. doi: 10.1083/jcb.105.6.2621
Boillee, S., Yamanaka, K., Lobsiger, C. S., Copeland, N. G., Jenkins, N. A., Kassiotis, G., et al. (2006). Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312, 1389–1392. doi: 10.1126/science.1123511
Bosch, M., and Kielian, T. (2014). Hemichannels in neurodegenerative diseases: is there a link to pathology? Front. Cell. Neurosci. 8:242. doi: 10.3389/fncel.2014.00242
Bruzzone, S., Guida, L., Zocchi, E., Franco, L., and De Flora, A. (2001). Connexin 43 hemi channels mediate Ca2+-regulated transmembrane NAD+ fluxes in intact cells. FASEB J. 15, 10–12.
Bruzzone, R., White, T. W., and Paul, D. L. (1996). Connections with connexins: the molecular basis of direct intercellular signaling. Eur. J. Biochem. 238, 1–27. doi: 10.1111/j.1432-1033.1996.0001q.x
Burkovetskaya, M., Karpuk, N., Xiong, J., Bosch, M., Boska, M. D., Takeuchi, H., et al. (2014). Evidence for Aberrant astrocyte hemichannel activity in juvenile neuronal ceroid lipofuscinosis (JNCL). PLoS ONE 9:e95023. doi: 10.1371/journal.pone.0095023
Cepeda, C., Chang, J. W., Owens, G. C., Huynh, M. N., Chen, J. Y., Tran, C., et al. (2015). In Rasmussen encephalitis, hemichannels associated with microglial activation are linked to cortical pyramidal neuron coupling: a possible mechanism for cellular hyperexcitability. CNS Neurosci. Ther. 21, 152–163. doi: 10.1111/cns.12352
Chen, G., Park, C. K., Xie, R. G., Berta, T., Nedergaard, M., and Ji, R. R. (2014). Connexin-43 induces chemokine release from spinal cord astrocytes to maintain late-phase neuropathic pain in mice. Brain 137(Pt 8), 2193–2209. doi: 10.1093/brain/awu140
Cherian, P. P., Siller-Jackson, A. J., Gu, S., Wang, X., Bonewald, L. F., Sprague, E., et al. (2005). Mechanical strain opens connexin 43 hemichannels in osteocytes: a novel mechanism for the release of prostaglandin. Mol. Biol. Cell 16, 3100–3106. doi: 10.1091/mbc.E04-10-0912
Chever, O., Lee, C. Y., and Rouach, N. (2014). Astroglial connexin43 hemichannels tune basal excitatory synaptic transmission. J. Neurosci. 34, 11228–11232. doi: 10.1523/JNEUROSCI.0015-14.2014
Chi, J., Li, L., Liu, M., Tan, J., Tang, C., Pan, Q., et al. (2012). Pathogenic connexin-31 forms constitutively active hemichannels to promote necrotic cell death. PLoS ONE 7:e32531. doi: 10.1371/journal.pone.0032531
Contreras, J. E., Sáez, J. C., Bukauskas, F. F., and Bennett, M. V. (2003). Gating and regulation of connexin 43 (Cx43) hemichannels. Proc. Natl. Acad. Sci. U.S.A. 100, 11388–11393. doi: 10.1073/pnas.1434298100
Contreras, J. E., Sánchez, H. A., Eugenin, E. A., Speidel, D., Theis, M., Willecke, K., et al. (2002). Metabolic inhibition induces opening of unapposed connexin 43 gap junction hemichannels and reduces gap junctional communication in cortical astrocytes in culture. Proc. Natl. Acad. Sci. U.S.A. 99, 495–500. doi: 10.1073/pnas.012589799
Davidson, J. O., Green, C. R., Nicholson, L. F., O’Carroll, S. J., Fraser, M., Bennet, L., et al. (2012). Connexin hemichannel blockade improves outcomes in a model of fetal ischemia. Ann. Neurol. 71, 121–132. doi: 10.1002/ana.22654
Dobrowolski, R., Sasse, P., Schrickel, J. W., Watkins, M., Kim, J. S., Rackauskas, M., et al. (2008). The conditional connexin43G138R mouse mutant represents a new model of hereditary oculodentodigital dysplasia in humans. Hum. Mol. Genet. 17, 539–554. doi: 10.1093/hmg/ddm329
Dobrowolski, R., Sommershof, A., and Willecke, K. (2007). Some oculodentodigital dysplasia-associated Cx43 mutations cause increased hemichannel activity in addition to deficient gap junction channels. J. Membr. Biol. 219, 9–17. doi: 10.1007/s00232-007-9055-7
Ebihara, L., Korzyukov, Y., Kothari, S., and Tong, J. J. (2014). Cx46 hemichannels contribute to the sodium leak conductance in lens fiber cells. Am. J. Physiol. Cell Physiol. 306, C506–C513. doi: 10.1152/ajpcell.00353.2013
Eiberger, J., Degen, J., Romualdi, A., Deutsch, U., Willecke, K., and Söhl, G. (2001). Connexin genes in the mouse and human genome. Cell Commun. Adhes. 8, 163–165. doi: 10.3109/15419060109080717
Ek-Vitorin, J. F., and Burt, J. M. (2013). Structural basis for the selective permeability of channels made of communicating junctionproteins. Biochim. Biophys. Acta 1828, 51–68. doi: 10.1016/j.bbamem.2012.02.003
Essenfelder, G. M., Bruzzone, R., Lamartine, J., Charollais, A., Blanchet-Bardon, C., Barbe, M. T., et al. (2004). Connexin30 mutations responsible for hidrotic ectodermal dysplasia cause abnormal hemichannel activity. Hum. Mol. Genet. 13, 1703–1714. doi: 10.1093/hmg/ddh191
Eugenin, E. A., and Berman, J. W. (2013). Cytochrome c dysregulation induced by HIV infection of astrocytes results in bystander apoptosis of uninfected astrocytes by an IP3 and calcium-dependent mechanism. J. Neurochem. 127, 644–651. doi: 10.1111/jnc.12443
Finn, R., Kovacs, A. D., and Pearce, D. A. (2011). Altered sensitivity of cerebellar granule cells to glutamate receptor overactivation in the Cln3(Deltaex7/8)- knock-in mouse model of juvenile neuronal ceroid lipofuscinosis. Neurochem. Int. 58, 648–655. doi: 10.1016/j.neuint.2011.02.003
Fiori, M. C., Figueroa, V., Zoghbi, M. E., Saéz, J. C., Reuss, L., and Altenberg, G. A. (2012). Permeation of calcium through purified connexin 26 hemichannels. J. Biol. Chem. 287, 40826–40834. doi: 10.1074/jbc.M112.383281
Froger, N., Orellana, J. A., Calvo, C. F., Amigou, E., Kozoriz, M. G., Naus, C. C., et al. (2010). Inhibition of cytokine-induced connexin43 hemichannel activity in astrocytes is neuroprotective. Mol. Cell. Neurosci. 45, 37–46. doi: 10.1016/j.mcn.2010.05.007
García, I. E., Maripillán, J., Jara, O., Ceriani, R., Palacios-Muñoz, A., Ramachandran, J., et al. (2015). Keratitis-ichthyosis-deafness syndrome-associated Cx26 mutants produce nonfunctional gap junctions but hyperactive hemichannels when co-expressed with wild type Cx43. J. Invest. Dermatol. 135, 1338–1347. doi: 10.1038/jid.2015.20
García, I. E., Retamal, M. A., Jara, O., González, C., and Martínez, A. D. (2013). Is the gain of hemichannel activity a common feature shared by Cx26 syndromic deafness mutants? Biophysical. J. 104, 492a–493a. doi: 10.1016/j.bpj.2012.11.2715
Gerido, D. A., DeRosa, A. M., Richard, G., and White, T. W. (2007). Aberrant hemichannel properties of Cx26 mutations causing skin disease and deafness. Am. J. Physiol. Cell Physiol. 293, C337–C345. doi: 10.1152/ajpcell.00626.2006
Goldberg, G. S., Valiunas, V., and Brink, P. R. (2004). Selective permeability of gap junction channels. Biochim. Biophys. Acta 1662, 96–101. doi: 10.1016/j.bbamem.2003.11.022
Gómez-Hernández, J. M., de Miguel, M., Larrosa, B., González, D., and Barrio, L. C. (2003). Molecular basis of calcium regulation in connexin-32 hemichannels. Proc. Natl. Acad. Sci. U.S.A. 100, 16030–16035. doi: 10.1073/pnas.2530348100
Gong, X. Q., and Nicholson, B. J. (2001). Size selectivity between gap junction channels composed of different connexins. Cell Commun. Adhes. 8, 187–192. doi: 10.3109/15419060109080721
Griffith, A. J., Yang, Y., Pryor, S. P., Park, H. J., Jabs, E. W., Nadol, J. B. Jr., et al. (2006). Cochleosaccular dysplasia associated with a connexin 26 mutation in keratitis-ichthyosis-deafness syndrome. Laryngoscope 116, 1404–1408. doi: 10.1097/01.mlg.0000224549.75161.ca
Guttman, J. A., Lin, A. E., Li, Y., Bechberger, J., Naus, C. C., Vogl, A. W., et al. (2010). Gap junction hemichannels contribute to the generation of diarrhoea during infectious entericdisease. Gut 59, 218–226. doi: 10.1136/gut.2008.170464
Hansen, D. B., Braunstein, T. H., Nielsen, M. S., and MacAulay, N. (2014). Distinct permeation profiles of the connexin 30 and 43 hemichannels. FEBS Lett. 588, 1446–1457. doi: 10.1016/j.febslet.2014.01.036
Hawat, G., Hélie, P., and Baroudi, G. (2012). Single intravenous low-dose injections of connexin 43 mimetic peptides protect ischemic heart in vivo against myocardial infarction. J. Mol. Cell Cardiol. 53, 559–566. doi: 10.1016/j.yjmcc.2012.07.008
Hoang Dinh, E., Ahmad, S., Chang, Q., Tang, W., Stong, B., and Lin, X. (2009). Diverse deafness mechanisms of connexin mutations revealed by studies using in vitro approaches and mouse models. Brain Res. 1277, 52–69. doi: 10.1016/j.brainres.2009.02.008
Hoang, Q. V., Qian, H., and Ripps, H. (2010). Functional analysis of hemichannels and gap-junctional channels formed by connexins 43 and 46. Mol. Vis. 16, 1343–1352.
Huang, C., Han, X., Li, X., Lam, E., Peng, W., Lou, N., et al. (2008). Critical role of connexin 43 in secondary expansion of traumatic spinal cord injury. J. Neurosci. 32, 3333–3338. doi: 10.1523/JNEUROSCI.1216-11.2012
Iyyathurai, J., D’hondt, C., Wang, N., De Bock, M., Himpens, B., Retamal, M. A., et al. (2013). Peptides and peptide-derived molecules targeting the intracellular domains of Cx43: gap junctions versus hemichannels. Neuropharmacology 75, 491–505. doi: 10.1016/j.neuropharm.2013.04.050
Jara, O., Acuña, R., García, I. E., Maripillán, J., Figueroa, V., Sáez, J. C., et al. (2012). Critical role of the first transmembrane domain of Cx26 in regulating oligomerization and function. Mol. Biol. Cell 23, 3299–3311. doi: 10.1091/mbc.E11-12-1058
Johansen, D., Cruciani, V., Sundset, R., Ytrehus, K., and Mikalsen, S. O. (2011). Ischemia induces closure of gap junctional channels and opening of hemichannels in heart-derived cells and tissue. Cell Physiol. Biochem. 28, 103–114. doi: 10.1159/000331719
John, S. A., Kondo, R., Wang, S. Y., Goldhaber, J. I., and Weiss, J. N. (1999). Connexin-43 hemichannels opened by metabolic inhibition. J. Biol. Chem. 274, 236–240. doi: 10.1074/jbc.274.1.236
Johnstone, S. R., Billaud, M., Lohman, A. W., Taddeo, E. P., and Isakson, B. E. (2012). Posttranslational modifications in connexins and pannexins. J. Membr. Biol. 245, 319–332. doi: 10.1007/s00232-012-9453-3
Kamermans, M., Fahrenfort, I., Schultz, K., Janssen-Bienhold, U., Sjoerdsma, T., and Weiler, R. (2001). Hemichannel-mediated inhibition in the outer retina. Science 292, 1178–1180. doi: 10.1126/science.1060101
Kawasaki, A., Hayashi, T., Nakachi, K., Trosko, J. E., Sugihara, K., Kotake, Y., et al. (2009). Modulation of connexin 43 in rotenone-induced model of Parkinson’s disease. Neuroscience 160, 61–68. doi: 10.1016/j.neuroscience.2009.01.080
Kozoriz, M. G., Lai, S., Vega, J. L., Sáez, J. C., Sin, W. C., Bechberger, J. F., et al. (2013). Cerebral ischemic injury is enhanced in a model of oculodentodigital dysplasia. Neuropharmacology 75, 549–556. doi: 10.1016/j.neuropharm.2013.05.003
Kumar, N. M., and Gilula, N. B. (1996). The gap junction communication channel. Cell 84, 381–388. doi: 10.1016/S0092-8674(00)81282-9
Kwon, T., Harris, A. L., Rossi, A., and Bargiello, T. A. (2011). Molecular dynamics simulations of the Cx26 hemichannel: evaluation of structural models with Brownian dynamics. J. Gen. Physiol. 138, 475–493. doi: 10.1085/jgp.201110679
Lee, J. R., Derosa, A. M., and White, T. W. (2009). Connexin mutations causing skin disease and deafness increase hemichannel activity and cell death when expresses in Xenopus oocytes. J. Invest. Dermatol. 129, 870–878. doi: 10.1038/jid.2008.335
Li, C., Meng, Q., Yu, X., Jing, X., Xu, P., and Luo, D. (2012). Regulatory effect of connexin 43 on basal Ca2+ signaling in rat ventricular myocytes. PLoS ONE 7:e36165. doi: 10.1371/journal.pone.0036165
Li, F., Sugishita, K., Su, Z., Ueda, I., and Barry, W. H. (2001). Activation of connexin-43 hemichannels can elevate [Ca(2+)]i and [Na(+)]i in rabbit ventricular myocytes during metabolic inhibition. J. Mol. Cell Cardiol. 33, 2145–2155. doi: 10.1006/jmcc.2001.1477
Li, X., Zhao, H., Tan, X., Kostrzewa, R. M., Du, G., Chen, Y., et al. (2015). Inhibition of connexin43 improves functional recovery after ischemic brain injury in neonatal rats. Glia doi: 10.1002/glia.22826 [Epub ahead of print].
Liang, G. S., de Miguel, M., Gómez-Hernández, J. M., Glass, J. D., Scherer, S. S., Mintz, M., et al. (2005). Severe neuropathy with leaky connexin32 hemichannels. Ann. Neurol. 57, 749–754. doi: 10.1002/ana.20459
Lopez, W., Gonzalez, J., Liu, Y., Harris, A. L., and Contreras, J. E. (2013). Insights on the mechanisms of Ca(2+) regulation of connexin26 hemichannels revealed by human pathogenic mutations (D50N/Y). J. Gen. Physiol. 142, 23–35. doi: 10.1085/jgp.201210893
Lu, D., Soleymani, S., Madakshire, R., and Insel, P. A. (2012). ATP released from cardiac fibroblasts via connexin hemichannels activates profibrotic P2Y2 receptors. FASEB J. 26, 2580–2591. doi: 10.1096/fj.12-204677
Maeda, S., Nakagawa, S., Suga, M., Yamashita, E., Oshima, A., Fujiyoshi, Y., et al. (2009). Structure of the connexin 26 gap junction channel at 3.5 A resolution. Nature 458, 597–602. doi: 10.1038/nature07869
Mandal, A., Shahidullah, M., and Delamere, N. A. (2015). Calcium entry via connexin hemichannels in lens epithelium. Exp. Eye Res. 132C, 52–58. doi: 10.1016/j.exer.2015.01.012
Martínez, A. D., Acuña, R., Figueroa, V., Maripillan, J., and Nicholson, B. (2009). Gap-junction channels dysfunction in deafness and hearing loss. Antioxid. Redox. Signal. 11, 309–322. doi: 10.1089/ars.2008.2138
Martinez, A. D., Hayrapetyan, V., Moreno, A. P., and Beyer, E. C. (2002). Connexin43 and connexin45 form heteromeric gap junction channels in which individual components determine permeability and regulation. Circ. Res. 90, 1100–1107. doi: 10.1161/01.RES.0000019580.64013.31
McClain, J. L., Grubišić, V., Fried, D., Gomez-Suarez, R. A., Leinninger, G. M., Sévigny, J., et al. (2014). Ca2+ responses in enteric glia are mediated by connexin-43 hemichannels and modulate colonic transit in mice. Gastroenterology 146, 497–507.e1. doi: 10.1053/j.gastro.2013.10.061
Meigh, L., Hussain, N., Mulkey, D. K., and Dale, N. (2014). Connexin26 hemichannels with a mutation that causes KID syndrome in humans lacksensitivity to CO2. Elife 3:e04249. doi: 10.7554/eLife.04249
Mese, G., Sellitto, C., Li, L., Wang, H. Z., Valiunas, V., Richard, G., et al. (2011). The Cx26-G45E mutation displays increased hemichannel activity in a mouse model of the lethal form of keratitis-ichthyosis-deafness syndrome. Mol. Biol. Cell 22, 4776–4786. doi: 10.1091/mbc.E11-09-0778
Mhaske, P. V., Levit, N. A., Li, L., Wang, H. Z., Lee, J. R., Shuja, Z., et al. (2013). The human Cx26-D50A and Cx26-A88V mutations causing keratitis-ichthyosis-deafness syndrome display increased hemichannel activity. Am. J. Physiol. Cell Physiol. 304, C1150–C1158. doi: 10.1152/ajpcell.00374.2012
Minogue, P. J., Tong, J. J., Arora, A., Russell-Eggitt, I., Hunt, D. M., Moore, A. T., et al. (2009). A mutant connexin50 with enhanced hemichannel function leads to cell death. Invest. Ophthalmol. Vis. Sci. 50, 5837–5845. doi: 10.1167/iovs.09-3759
Mylvaganam, S., Ramani, M., Krawczyk, M., and Carlen, P. L. (2014). Roles of gap junctions, connexins, and pannexins in epilepsy. Front. Physiol. 5:172. doi: 10.3389/fphys.2014.00172
Nelson, M. T., French, R. J., and Krueger, B. K. (1984). Voltage-dependent calcium channels from brain incorporated into planar lipid bilayers. Nature 308, 77–80. doi: 10.1038/308077a0
Nualart-Marti, A., del Molino, E. M., Grandes, X., Bahima, L., Martin-Satué, M., Puchal, R., et al. (2013). Role of connexin 32 hemichannels in the release of ATP from peripheral nerves. Glia 61, 1976–1989. doi: 10.1002/glia.22568
O’Carroll, S. J., Alkadhi, M., Nicholson, L. F., and Green, C. R. (2008). Connexin 43 mimetic peptides reduce swelling, astrogliosis, and neuronal cell death after spinal cord injury. Cell Commun. Adhes. 15, 27–42. doi: 10.1080/15419060802014164
Oh, S., Ri, Y., Bennett, M. V., Trexler, E. B., Verselis, V. K., and Bargiello, T. A. (1997). Changes in permeability caused by connexin 32 mutations underlie X-linked Charcot-Marie-Tooth disease. Neuron 19, 927–938. doi: 10.1016/S0896-6273(00)80973-3
Orellana, J. A., Froger, N., Ezan, P., Jiang, J. X., Bennett, M. V., Naus, C. C., et al. (2011a). ATP and glutamate released via astroglial connexin 43 hemichannels mediate neuronal death through activation of pannexin 1 hemichannels. J. Neurochem. 118, 826–840. doi: 10.1111/j.1471-4159.2011.07210
Orellana, J. A., Shoji, K. F., Abudara, V., Ezan, P., Amigou, E., Saez, P. J., et al. (2011b). Amyloid β-induced death in neurons involves glial and neuronal hemichannels. J. Neurosci. 31, 4962–4977. doi: 10.1523/JNEUROSCI.6417-10.2011
Orellana, J. A., Saez, J. C., Bennett, M. V., Berman, J. W., Morgello, S., and Eugenin, E. A. (2014). HIV increases the release of dickkopf-1 protein from human astrocytes by a Cx43 hemichannel-dependent mechanism. J. Neurochem. 128, 752–763. doi: 10.1111/jnc.12492
Orellana, J. A., von Bernhardi, R., Giaume, C., and Sáez, J. C. (2012). Glial hemichannels and their involvement in aging and neurodegenerative diseases. Rev. Neurosci. 23, 163–177. doi: 10.1515/revneuro-2011-0065
Patel, D., Gemel, J., Xu, Q., Simon, A. R., Lin, X., Matiukas, A., et al. (2014). Atrial fibrillation-associated connexin40 mutants make hemichannels and synergistically form gap junction channels with novel properties. FEBS Lett. 588, 1458–1464. doi: 10.1016/j.febslet.2014.01.010
Paul, D. L., Ebihara, L., Takemoto, L. J., Swenson, K. I., and Goodenough, D. A. (1991). Connexin46, a novel lens gap junction protein, induces voltage-gated currents in nonjunctional plasma membrane of Xenopus oocytes. J. Cell Biol. 115, 1077–1089. doi: 10.1083/jcb.115.4.1077
Payton, B. W., Bennett, M. V., and Pappas, G. D. (1969). Permeability and structure of junctional membranes at an electrotonic synapse. Science 166, 1641–1643. doi: 10.1126/science.166.3913.1641
Plotkin, L. I. (2014). Connexin 43 hemichannels and intracellular signaling in bone cells. Front. Physiol. 5:131. doi: 10.3389/fphys.2014.00131
Ponsaerts, R., De Vuyst, E., Retamal, M., D’hondt, C., Vermeire, D., Wang, N., et al. (2010). Intramolecular loop/tail interactions are essential for connexin 43-hemichannel activity. FASEB J. 24, 4378–4395. doi: 10.1096/fj.09-153007
Ponsaerts, R., Wang, N., Himpens, B., Leybaert, L., and Bultynck, G. (2012). The contractile system as a negative regulator of the connexin43 hemichannel. Biol. Cell 104, 367–377. doi: 10.1111/boc.201100079
Rackauskas, M., Neverauskas, V., and Skeberdis, V. A. (2010). Diversity and properties of connexin gap junction channels. Medicina (Kaunas) 46, 1–12.
Rana, S., and Dringen, R. (2007). Gap junction hemichannel-mediated release of glutathione from cultured rat astrocytes. Neurosci. Lett. 415, 45–48. doi: 10.1016/j.neulet.2006.12.043
Ren, Q., Riquelme, M. A., Xu, J., Yan, X., Nicholson, B. J., Gu, S., et al. (2013). Cataract-causing mutation of human connexin 46 impairs gap junction, but increases hemichannel function and cell death. PLoS ONE 8:e74732. doi: 10.1371/journal.pone.0074732
Retamal, M. A., Alcayaga, J., Verdugo, C. A., Bultynck, G., Leybaert, L., Sáez, P. J., et al. (2014a). Opening of pannexin- and connexin-based channels increases the excitability of nodose ganglion sensory neurons. Front. Cell Neurosci. 8:158. doi: 10.3389/fncel.2014.00158
Retamal, M. A., Reyes, E. P., and Alcayaga, J. (2014b). Petrosal ganglion: a more complex role than originally imagined. Front. Physiol. 5:474. doi: 10.3389/fphys.2014.00474
Retamal, M. A., Cortés, C. J., Reuss, L., Bennett, M. V., and Sáez, J. C. (2006). S-nitrosylation and permeation through connexin 43 hemichannels in astrocytes: induction by oxidant stress and reversal by reducing agents. Proc. Natl. Acad. Sci. U.S.A. 103, 4475–4480. doi: 10.1073/pnas.0511118103
Retamal, M. A., Froger, N., Palacios-Prado, N., Ezan, P., Sáez, P. J., and Sáez, J. C., et al. (2007). Cx43 hemichannels and gap junction channels in astrocytes are regulated oppositely by proinflammatory cytokines released from activated microglia. J. Neurosci. 27, 13781–13792. doi: 10.1523/JNEUROSCI.2042-07.2007
Rovegno, M., Soto, P. A., Sáez, P. J., Naus, C. C., Sáez, J. C., and von Bernhardi, R. (2015). Connexin43 hemichannels mediate secondary cellular damage spread from the trauma zone to distal zones in astrocyte monolayers. Glia 63, 1185–1199. doi: 10.1002/glia.22808
Rufer, M., Wirth, S. B., Hofer, A., Dermietzel, R., Pastor, A., Kettenmann, H., et al. (1996). Regulation of connexin-43, GFAP, and FGF-2 is not accompanied by changes in astroglial coupling in MPTP-lesioned, FGF-2-treated parkinsonian mice. J. Neurosci. Res. 46, 606–617. doi: 10.1002/(SICI)1097-4547(19961201)46:5<606::AID-JNR9>3.0.CO;2-N
Sáez, J. C., Berthoud, V. M., Branes, M. C., Martinez, A. D., and Beyer, E. C. (2003). Plasma membrane channels formed by connexins: their regulation and functions. Physiol. Rev. 83, 1359–1400. doi: 10.1152/physrev.00007.2003
Sáez, J. C., and Leybaert, L. (2014). Hunting for connexin hemichannels. FEBS Lett. 588, 1205–1211. doi: 10.1016/j.febslet.2014.03.004
Sáez, J. C., Schalper, K. A., Retamal, M. A., Orellana, J. A., Shoji, K. F., and Bennett, M. V. (2010). Cell membrane permeabilization via connexin hemichannels in living and dying cells. Exp. Cell Res. 316, 2377–2389. doi: 10.1016/j.yexcr.2010.05.026
Samoilova, M., Wentlandt, K., Adamchik, Y., Velumian, A. A., and Carlen, P. L. (2008). Connexin 43 mimetic peptides inhibit spontaneous epileptiform activity in organotypic hippocampal slice cultures. Exp. Neurol. 210, 762–775. doi: 10.1016/j.expneurol.2008.01.005
Sanchez, H. A., Bienkowski, R., Slavi, N., Srinivas, M., and Verselis, V. K. (2014). Altered inhibition of Cx26 hemichannels by pH and Zn2+ in the A40V mutation associated with keratitis-ichthyosis-deafness syndrome. J. Biol. Chem. 289, 21519–21532. doi: 10.1074/jbc.M114.578757
Sánchez, H. A., Mese, G., Srinivas, M., White, T. W., and Verselis, V. K. (2010). Differentially altered Ca2+ regulation and Ca2+ permeability in Cx26 hemichannels formed by the A40V and G45E mutations that cause keratitis ichthyosis deafness syndrome. J. Gen. Physiol. 136, 47–62. doi: 10.1085/jgp.201010433
Sanchez, H. A., Villone, K., Srinivas, M., and Verselis, V. K. (2013). The D50N mutation and syndromic deafness: altered Cx26 hemichannel properties caused by effects on the pore and intersubunit interactions. J. Gen. Physiol. 142, 3–22. doi: 10.1085/jgp.201310962
Schalper, K. A., Palacios-Prado, N., Orellana, J. A., and Sáez, J. C. (2008). Currently used methods for identification and characterization of hemichannels. Cell Commun. Adhes. 15, 207–218. doi: 10.1080/15419060802014198
Schalper, K. A., Sánchez, H. A., Lee, S. C., Altenberg, G. A., Nathanson, M. H., and Sáez, J. C. (2010). Connexin 43 hemichannels mediate the Ca2+ influx induced by extracellular alkalinization. Am. J. Physiol. Cell Physiol. 299, C1504–C1515. doi: 10.1152/ajpcell.00015.2010
Schock, S. C., Leblanc, D., and Hakim, A. M., and Thompson, C. S. (2008). ATP release by way of connexin 36 hemichannels mediates ischemic tolerance in vitro. Biochem. Biophys. Res. Commun. 368, 138–144. doi: 10.1016/j.bbrc.2008.01.054
Scott, C. A., Tattersall, D., O’Toole, E. A., and Kelsell, D. P. (2012). Connexins in epidermal homeostasis and skin disease. Biochim. Biophys. Acta 1818, 1952–1961. doi: 10.1016/j.bbamem.2011.09.004
Shintani-Ishida, K., Uemura, K., and Yoshida, K. (2007). Hemichannels in cardiomyocytes open transiently during ischemia and contribute to reperfusion injury following brief ischemia. Am. J. Physiol. Heart Circ. Physiol. 293, H1714–H1720. doi: 10.1152/ajpheart.00022.2007
Sipos, A., Vargas, S. L., Toma, I., Hanner, F., Willecke, K., and Peti-Peterdi, J. (2009). Connexin 30 deficiency impairs renal tubular ATP release and pressure natriuresis. J. Am. Soc. Nephrol. 20, 1724–1732. doi: 10.1681/ASN.2008101099
Söhl, G., Eiberger, J., Jung, Y. T., Kozak, C. A., and Willecke, K. (2001). The mouse gap junction gene connexin29 is highly expressed in sciatic nerve and regulated during brain development. Biol. Chem. 382, 973–978. doi: 10.1515/BC.2001.122
Söhl, G., and Willecke, K. (2004). Gap junctions and the connexin protein family. Cardiovasc. Res. 62, 228–232. doi: 10.1016/j.cardiores.2003.11.013
Stehberg, J., Moraga-Amaro, R., Salazar, C., Becerra, A., Echeverría, C., Orellana, J. A., et al. (2012). Release of gliotransmitters through astroglial connexin 43 hemichannels is necessary for fear memory consolidation in the basolateral amygdala. FASEB J. 26, 3649–3657. doi: 10.1096/fj.11-198416
Stong, B. C., Chang, Q., Ahmad, S., and Lin, X. (2006). A novel mechanism for connexin 26 mutation linked deafness: cell death caused by leaky gap junction hemichannels. Laryngoscope 116, 2205–2210. doi: 10.1097/01.mlg.0000241944.77192.d2
Stout, C. E., Costantin, J. L., Naus, C. C., and Charles, A. C. (2002). Intercellular calcium signaling in astrocytes via ATP release through connexin hemichannels. J. Biol. Chem. 277, 10482–10488. doi: 10.1074/jbc.M109902200
Sun, Y., Hills, M. D., Ye, W. G., Tong, X., and Bai. D. (2014). Atrial fibrillation-linked germline GJA5/connexin40 mutants showed an increased hemichannel function. PLoS ONE 9:e95125. doi: 10.1371/journal.pone.0095125
Svenningsen, P., Burford, J. L., and Peti-Peterdi, J. (2013). ATP releasing connexin 30 hemichannels mediate flow-induced calcium signaling in the collecting duct. Front. Physiol. 4:292. doi: 10.3389/fphys.2013.00292
Takada, H., Furuya, K., and Sokabe, M. (2014). Mechanosensitive ATP release from hemichannels and Ca2+ influx through TRPC6 acceleratewound closure in keratinocytes. J. Cell Sci. 127(Pt 19), 4159–4171. doi: 10.1242/jcs.147314
Takeuchi, H., Jin, S., Wang, J., Zhang, G., Kawanokuchi, J., Kuno, R., et al. (2006). Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J. Biol. Chem. 281, 21362–21368. doi: 10.1074/jbc.M600504200
Takeuchi, H., Mizoguchi, H., Doi, Y., Jin, S., Noda, M., Liang, J., et al. (2011). Blockade of gap junction hemichannel suppresses disease progression in mouse models of amyotrophic lateral sclerosis and Alzheimer’s disease. PLoS ONE 6:e21108. doi: 10.1371/journal.pone.0021108
Takeuchi, H., and Suzumura, A. (2014). Gap junctions and hemichannels composed of connexins: potential therapeutic targets for neurodegenerative diseases. Front. Cell Neurosci. 8:189. doi: 10.3389/fncel.2014.00189
Tong, J. J., Minogue, P. J., Guo, W., Chen, T. L., Beyer, E. C., Berthoud, V. M., et al. (2011). Different consequences of cataract-associated mutations at adjacent positions in the first extracellular boundary of connexin50. Am. J. Physiol. Cell Physiol. 300, C1055–C1064. doi: 10.1152/ajpcell.00384.2010
Vinken, M., Vanhaecke, T., Papeleu, P., Snykers, S., Henkens, T., and Rogiers, V. (2006). Connexins and their channels in cell growth and cell death. Cell. Signal. 18, 592–600. doi: 10.1016/j.cellsig.2005.08.012
Wang, H., Cao, X., Lin, Z., Lee, M., Jia, X., Ren, Y., et al. (2015). Exome sequencing reveals mutation in GJA1 as a cause of keratoderma-hypotrichosis-leukonychia totalis syndrome. Hum. Mol. Genet. 24, 243–250. doi: 10.1093/hmg/ddu442
Wang, N., De Bock, M., Decrock, E., Bol, M., Gadicherla, A., Vinken, M., et al. (2013a). Paracrine signaling through plasma membrane hemichannels. Biochim. Biophys. Acta 1828, 35–50. doi: 10.1016/j.bbamem.2012.07.002
Wang, Y. F., Munemasa, S., Nishimura, N., Ren, H. M., Robert, N., Han, M., et al. (2013b). Identification of cyclic GMP-activated nonselective Ca2+-permeable cation channels and associated CNGC5 and CNGC6 genes in Arabidopsis guard cells. Plant Physiol. 163, 578–590. doi: 10.1104/pp.113.225045
Wang, X., Ramirez, A., and Budunova, I. (2009). Overexpression of connexin26 in the basal keratinocytes reduces sensitivity to tumor promoter TPA. Exp. Dermatol. 19, 633–640. doi: 10.1111/j.1600-0625.2009.01013.x
Wong, C. W., Christen, T., Roth, I., Chadjichristos, C. E., Derouette, J. P., Foglia, B. F., et al. (2006). Connexin37 protects against atherosclerosis by regulating monocyte adhesion. Nat. Med. 12, 950–954. doi: 10.1038/nm1441
Yamanaka, K., Chun, S. J., Boillee, S., Fujimori-Tonou, N., Yamashita, H., Gutmann, D. H., et al. (2008). Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci. 11, 251–253. doi: 10.1038/nn2047
Yao, K., Wang, W., Zhu, Y., Jin, C., Shentu, X., Jiang, J., et al. (2011). A novel GJA3 mutation associated with congenital nuclear pulverulent and posterior polar cataract in a Chinese family. Hum. Mutat. 32, 1367–1370. doi: 10.1002/humu.21552
Ye, Z. C., Wyeth, M. S., Baltan-Tekkok, S., and Ransom, B. R. (2003). Functional hemichannels in astrocytes: a novel mechanism of glutamate release. J. Neurosci. 23, 3588–3596.
Yoon, J. J., Green, C. R., O’Carroll, S. J., and Nicholson, L. F. (2010). Dose-dependent protective effect of connexin43 mimetic peptide against neurodegeneration in an ex vivo model of epileptiform lesion. Epilepsy Res. 92, 153–162. doi: 10.1016/j.eplepsyres.2010.08.014
Zhang, Y., and Hao, H. (2013). Conserved glycine at position 45 of major cochlear connexins constitutes a vital component of the Ca2+ sensor for gating of gap junction hemichannels. Biochem. Biophys. Res. Commun. 436, 424–429. doi: 10.1016/j.bbrc.2013.05.118
Zhao, P. Y., Gan, G., Peng, S., Wang, S. B., Chen, B., Adelman, R. A., et al. (2015). TRP channels localize to subdomains of the apical plasma membrane in human fetal retinal pigment epithelium. Invest. Ophthalmol. Vis. Sci. 56, 1916–1923. doi: 10.1167/iovs.14-15738
Keywords: connexins, leaky hemichannels, mutations, gap junction channels, cell death, disease
Citation: Retamal MA, Reyes EP, García IE, Pinto B, Martínez AD and González C (2015) Diseases associated with leaky hemichannels. Front. Cell. Neurosci. 9:267. doi: 10.3389/fncel.2015.00267
Received: 06 May 2015; Accepted: 29 June 2015;
Published: 27 July 2015.
Edited by:
Maria Cristina D’Adamo, University of Perugia, ItalyReviewed by:
Elena Ambrosini, Istituto Superiore di Sanità, ItalyMichael V. L. Bennett, Albert Einstein College of Medicine, USA
Copyright © 2015 Retamal, Reyes, García, Pinto, Martínez and González. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mauricio A. Retamal, Centro de Fisiología Celular e Integrativa, Facultad de Medicina, Clínica Alemana Universidad del Desarrollo, Avenida Las Condes #12438, Santiago, Chile,bXJldGFtYWxAdWRkLmNs