 Tadhg Crowley1
Tadhg Crowley1 John-Mark Fitzpatrick1
John-Mark Fitzpatrick1 Teun Kuijper1
Teun Kuijper1 John F. Cryan1,2Orna O’Toole3
John F. Cryan1,2Orna O’Toole3 Olivia F. O’Leary1,2*†
Olivia F. O’Leary1,2*† Eric J. Downer1,4*†
Eric J. Downer1,4*†
- 1Department of Anatomy and Neuroscience, University College Cork, Cork, Ireland
- 2Alimentary Pharmabiotic Centre, University College Cork, Cork, Ireland
- 3Mercy University Hospital, Cork, Ireland
- 4School of Medicine, Discipline of Physiology, Trinity Biomedical Sciences Institute, Trinity College, Dublin, Ireland
The GABAB receptor agonist, baclofen, is used to treat muscle tightness and cramping caused by spasticity in a number of disorders including multiple sclerosis (MS), but its precise mechanism of action is unknown. Neuroinflammation drives the central pathology in MS and is mediated by both immunoreactive glial cells and invading lymphocytes. Furthermore, a body of data indicates that the Toll-like receptor (TLR) family of innate immune receptors is implicated in MS progression. In the present study we investigated whether modulation of GABAB receptors using baclofen can exert anti-inflammatory effects by targeting TLR3 and(or) TLR4-induced inflammatory signaling in murine glial cells and human peripheral blood mononuclear cells (PBMCs) isolated from healthy control individuals and patients with the relapse-remitting (RR) form of MS. TLR3 and TLR4 stimulation promoted the nuclear sequestration of NF-κB and pro-inflammatory cytokine expression in murine glia, while TLR4, but not TLR3, promoted pro-inflammatory cytokine expression in PBMCs isolated from both healthy donors and RR-MS patients. Importantly, this effect was exacerbated in RR-MS patient immune cells. We present further evidence that baclofen dose-dependently attenuated TLR3- and TLR4-induced inflammatory signaling in primary glial cells. Pre-exposure of PBMCs isolated from healthy donors to baclofen attenuated TLR4-induced TNF-α expression, but did not affect TLR4-induced TNF-α expression in RR-MS patient PBMCs. Interestingly, mRNA expression of the GABAB receptor was reduced in PBMCs from RR-MS donors when compared to healthy controls, an effect that might contribute to the differential sensitivity to baclofen seen in healthy and RR-MS patient cells. Overall these findings indicate that baclofen differentially regulates TLR3 and TLR4 signaling in glia and immune cells, and offers insight on the role of baclofen in the treatment of neuroinflammatory disease states including MS.
Introduction
Autoimmunity drives the development of multiple sclerosis (MS), involving central nervous system (CNS) infiltration of immune cells, myelin degradation, reactive changes in glia and axonal loss (Compston and Coles, 2002). The innate immune system has received much interest as having a defined role in the progression and/or etiology of MS (O’Brien et al., 2008; Downer, 2011). Innate immunity is regulated by complex mechanisms involving pattern-recognition receptors (PRRs) that recognize molecular signatures of microbes. Intracellular signaling triggered by PRRs leads to transcriptional expression of inflammatory mediators that coordinate the elimination of pathogens (Akira et al., 2006). If left unchecked, or not tightly regulated, dysregulation of this system can lead to conditions such as sepsis and autoimmunity (Lehnardt, 2010).
Toll-like receptors (TLRs) belong to the family of signaling PRRs (Jeannin et al., 2008) that initiate innate immune reactions by activating transcription factors such as nuclear factor (NF)-κB, in addition to inducing the expression of interferons (IFNs) and cytokines. Thus far, 12 functional TLRs have been identified in mice, and 10 in humans (O’Neill, 2004; Kawai and Akira, 2006). TLRs are localized in endosomal compartments (including TLR3, TLR7, TLR8, TLR9), or are cell membrane-bound (as with all other TLRs; O’Neill, 2004), and couple to specific signaling cascades, inducing gene transcription and controlling immune processes, with this specificity reliant on the TLR adaptor proteins recruited (O’Neill, 2004). All TLRs (with the exception of TLR3), recruit the adaptor myeloid differentiation factor 88 (MyD88; Medzhitov et al., 1998). TLR3 (and TLR4) induces MyD88-independent signaling to couple to NF-κB via Toll-Interleukin-1 Receptor (TIR)-domain-containing adaptor-inducing IFN-β (TRIF) protein. TLRs are key players in CNS diseases, and with respect to MS, key roles of TLRs have been shown in murine models of MS (Touil et al., 2006), while the expression of TLRs characterized on immune cells and CNS glia and neurons (Nishimura and Naito, 2005).
Gamma amino butyric acid (GABA) is the major inhibitory amino acid neurotransmitter in the brain (Cobb et al., 1995). GABA exerts its effects through the ionotropic receptors, GABAA/GABAA-ρ and the metabotropic receptor GABAB (Bormann, 2000; Campagna-Slater and Weaver, 2007; Pinard et al., 2010), and GABA receptor transcripts are present in neurons, glia (Lee et al., 2011; Vélez-Fort et al., 2012), and immune cells (Bhat et al., 2010). There is a growing body of evidence pointing to the key role of GABA receptors in neuroinflammation (Ziegler et al., 1980; Tyagi et al., 2015), and recent reports has identified that modulation of the GABAergic system occurs in MS (Han et al., 2008; Carmans et al., 2013). Indeed, GABA insufficiency has been identified in MS patients (Demakova et al., 2003), while intrathecal administration of the GABAB receptor agonist, baclofen, is used as a treatment strategy to control spasticity in patients with MS (Gunnarsson and Samuelsson, 2015). Furthermore, enhancing endogenous GABA via administration of the GABA-degrading enzyme GABA-transaminase (GABA-T), has been shown to be protective in an experimental autoimmune encephalomyelitis (EAE) mouse model (Carmans et al., 2013), indicating that increasing endogenous GABA also has therapeutic potential in the murine model of MS. In support of these in vivo findings, in vitro studies have shown that baclofen reduces TLR4-induced release of pro-inflammatory cytokines from primary murine microglia (Kuhn et al., 2004), indicating that cross talk may exist between the GABAergic and TLR systems, with relevance to inflammatory signaling events.
GABAB receptors are metabotropic Gi/Go-coupled receptors (Padgett and Slesinger, 2010) which are distributed throughout the CNS and periphery (Ong and Kerr, 1990; Hyland and Cryan, 2010). GABAB receptors can function to regulate ion channels (activate K+ and inhibit Ca2+ channels) and cellular signaling (adenylate cyclase, MAPK; Kornau, 2006; Jiang et al., 2012), limit the release of neurotransmitters (GABA, glutamate; Pinard et al., 2010; Gassmann and Bettler, 2012), and dampen depolarisation induced by excitatory neurotransmitters. GABAB receptors are implicated in a variety of neurodegenerative, neuroinflammatory, and pathophysiological disorders including depression, spasticity, pain, and schizophrenia (Reyes-García et al., 2007; Bjurstöm et al., 2008; Garcia-Oscos et al., 2012). Given previous reports linking TLR’s and the GABAB receptor subtype with neuroinflammation, particularly with relevance to MS pathogenesis, we sought to explore the impact of baclofen (the GABAB receptor agonist) on TLR3 and TLR4-induced inflammatory signaling both centrally and in the periphery, using murine glial cells and human peripheral blood mononuclear cells (PBMCs) isolated from healthy individuals and newly-diagnosed patients with the relapsing-remitting (RR) form of MS patients. This study identifies baclofen as a differential regulator of TLR3 and TLR4 signaling in glia and immune cells, and offers insight on the role of baclofen in regulating the innate immune response in cellular pathology associated with MS.
Materials and Methods
Preparation of Primary Mixed Glial Cultures and Treatments
Primary mixed glia were prepared from the whole brain of 1-day-old C57/BL6 mice and plated (at 5 × 105 cells/ml) as previously described (Downer et al., 2010). All experiments were performed under a license issued by the Health Products Regulatory Authority (Ireland) and in accordance with the European Directive 86/609/EEC and the guidelines laid down by the Animal Experimentation Ethics Committee of University College Cork. After 14 days in culture, mixed glia were treated with the TLR4 ligand LPS (100 ng/ml; Sigma–Aldrich, UK), the TLR3 ligand Poly(I:C) (10 μg/ml; InvivoGen, France) or vehicle (sterile H2O) for timepoints ranging from 10 min–24 h. In a second series of experiments, mixed glia were exposed to the GABAB agonist baclofen (10, 30, and 100 μM; Sigma–Aldrich, Germany) for 30 min prior to LPS (100 ng/ml; 30 min and 24 h) or Poly(I:C) (10 μg/ml; 6 h and 24 h) exposure.
Patients and Blood Samples
Healthy donors and RR-MS patients attending outpatient clinics at the Mercy University Hospital, Cork, were recruited for this study. Written informed consent was obtained from each participant and the study received ethical approval from the Clinical Research Ethics committee of the Cork Teaching Hospitals (CREC). Patient recruitment into the study was via a Senior Consultant Neurologist and patients had to meet the revised MacDonald diagnostic criteria for clinically defined MS (Polman et al., 2011) including patient history, clinical signs and symptoms, physical examination, and adjunctive diagnostic tools including MRI. The Disability Status Scale scores were taken using the Expanded Disability Status Scale (EDSS) by a Senior Consultant Neurologist in an outpatient clinic. All confirmed MS patients had a RR form of MS as defined by the revised McDonald criteria. Disease severity was scored at time of collection using the Kurtzke’s (1983) EDSS. Patients with RR-MS were clinically stable and naïve to disease modifying therapies including IFN-β, glatiramer acetate, fingolimod, teriflunomide, dimethyl fumarate, natalizumab, and alemtuzumab. PBMCs were collected from venous blood of healthy controls participants (mean age 30.3 ± 1.4 years; n = 16) and RR-MS patients (mean age 39.3 ± 3.2 years; n = 8) by way of venipuncture as previously described (Downer et al., 2013b). Details of the patient demographics are presented in Figure 3A. Human PBMCs were prepared from heparinized venous whole blood samples (50 ml per donor) from individuals by density separation over LymphoprepTM (Axis-Shield, Norway). PBMCs were plated (1 × 106 cells/ml) on 24-well plates prior to treatment. Plasma samples were separated following centrifugation, aliquoted, and stored at -80°C for subsequent analysis via ELISA.
Cytokine Analysis in Culture Supernatants
Mixed glia (5 × 105 cells/ml) and PBMCs (1 × 106 cells/ml) were seeded overnight in 24-well plates. Cells were incubated with LPS (100 ng/ml) or Poly(I:C) (10 μg/ml) for timepoints ranging from 10 min–24 h. Mixed glia/PBMCs were also pre-exposed (30 min) to baclofen (10, 30, and 100 μM) prior to LPS (100 ng/ml; 24 h) or Poly(I:C) (10 μg/ml; 24 h) exposure. Supernatants were assayed for tumor necrosis factor-α (TNF-α) and IL-8 concentration by ELISA according to manufacturer’s instructions (Duoset, R&D Systems, Abingdon, UK).
Quantitative Real-Time PCR
RNA was extracted from PBMCs (untreated) using a NucleoSpin® RNAII isolation kit (Macherey-Nagel Inc., Geschäftsführer, Germany). The concentration of RNA was determined using a UV/V is spectrophotometer. cDNA synthesis was performed on 1 μg RNA using a High Capacity cDNA RT Kit (Applied Biosystems, Carlsbad, CA, USA) according to the manufacturer’s instructions. Equal amounts of cDNA were used for RT-PCR amplification. Real-time PCR primers were delivered as “Taqman® Gene Expression Assays” containing forward and reverse primers, and a FAM-labeled MGB Taqman probe for the target gene (Applied Biosystems). GABAB1 primers were used to assess the expression of the target gene (Taqman Gene Expression Assay no. Hs00181306_m1). cDNA (1:4 dilution) was prepared and real-time PCR performed using Applied Biosystems 7300 Real-time PCR System. cDNA was mixed with qPCRTM Mastermix Plus (Applied Biosystems) and the respective gene assay in a 25 μl volume (10 μl of diluted cDNA, 12.5 μl Taqman® Universal PCR Mastermix, 1.25 μl target primer and 1.25 μl 18S). Eukaryotic 18S rRNA was used as an endogenous control and expression was conducted using a gene expression assay containing forward and reverse primers and a VIC-labeled MGB Taqman probe (#4319413E; Applied Biosystems, USA). Samples were run in duplicate and 40 cycles were run as follows: 10 min at 95°C and for each cycle, 15 s at 95°C and 1 min at 60°C. Gene expression was calculated relative to the endogenous control and analysis was performed using the 2-ΔΔCT method. In all experiments no change in relative 18S rRNA expression between treatment groups was observed.
TNF-α and IL-8 Measurement in Plasma
Plasma samples from healthy donors and RR-MS patients were analyzed for concentrations of TNF-α and IL-8 by ELISA (Duoset, R&D Systems, UK) according to manufacturer’s instructions.
Immunocytochemistry
Mixed glia (5 × 105 cells/ml) and PBMCs (1 × 106 cells/ml) were seeded overnight on 13-mm diameter coverslips coated with poly-l-lysine (Sigma–Aldrich) and grown for 24 h. Cells were incubated with LPS (100 ng/ml) or Poly(I:C) (10 μg/ml) for timepoints ranging from 10 min–24 h. Mixed glia/PBMCs were also pre-exposed (30 min) to baclofen (10, 30 and 100 μM) prior to LPS (100 ng/ml; 30 min) or Poly(I:C) (10 μg/ml; 6 h) exposure. Cells were then fixed in ice-cold methanol for 10 min, permeabilized with 0.2% Triton X-100 (Thermo Fisher Scientific, Waltham, IL, USA) in PBS for 10 min at room temperature and blocked with 10% goat serum (Vector Laboratories, Peterborough, UK) for 2 h. Cells were treated overnight at 4°C with rabbit polyclonal NF-κB p65 antibody (1:200 in 5% goat serum; Santa Cruz Biotechnology, Santa Cruz, CA, USA). Cells were then washed and incubated with goat anti-rabbit Alexa488 secondary antibody (1:1000 in 5% goat serum; Invitrogen, Dublin, Ireland) and 4′,6-diamidino-2-phenylindole (DAPI; 1.5 μg/ml) in PBS, washed, and mounted (Vectashield; Vector Laboratories). Cells were imaged using an Olympus IX70 inverted microscope. The fluorescence intensity in the nucleus of individual cells stained for NF-κB p65 was measured using the Image J analysis software (Rasband,WJ, http://rsb.info.nih.gov/ij/). The relative fluorescence intensity was calculated as the intensity after subtraction of the background noise. For each treatment, six coverslips were stained and five fields of view were captured per coverslip. Negative control experiments were performed by replacing the primary antibody with PBS and using equal gain settings during acquisition and analysis.
Statistical Analysis
Data were analyzed using one-way or two-way analysis of variance (ANOVA) as appropriate. When analysis by ANOVA indicated significance (P < 0.05), the post hoc Student Newman–Keuls test was used. Correlation tests were performed using two-tailed Spearman (non-parametric) correlation coefficient. Data are expressed as means ± SEs of the mean (SEM).
Results
TLR4 Activation Time-Dependently Increases Nuclear NF-κB p65 Expression and TNF-α Release in Mixed Glia
TLR4 and TLR3 were initially targeted given their involvement in EAE progression (Touil et al., 2006) and evidence that their expression is dysregulated in MS lesions (Bsibsi et al., 2002). To initially characterize the impact of TLR4 stimulation on pro-inflammatory signaling in primary murine mixed glial cells, we temporally assessed the impact of LPS on the distribution of the NF-κB p65 subunit in response to LPS treatment. Firstly, mixed glia were stimulated with LPS for various timepoints ranging from 10 min–24 h, and the localization of endogenous p65 was assessed by immunofluorescence (Figures 1A,B). In vehicle-treated glial cells, p65 is predominantly cytoplasmic, as evidenced by the detection of 488-conjugated immunocomplexes outside of the DAPI-stained regions (Figure 1B). Stimulation of glia with LPS time-dependently promoted the accumulation of p65 in the nucleus, peaking at 30 min and 1 h post-LPS exposure (Figures 1A,B). We next characterized the impact of TLR4 activation on TNF-α protein production in primary mixed glia. LPS time-dependently enhanced TNF-α expression in primary murine mixed glial cells, with mean maximal stimulatory effects on protein expression observed at 6–24 h (Figure 1C), indicating that TLR4 stimulation promotes inflammatory signaling in primary mouse glial cells in our culture system.
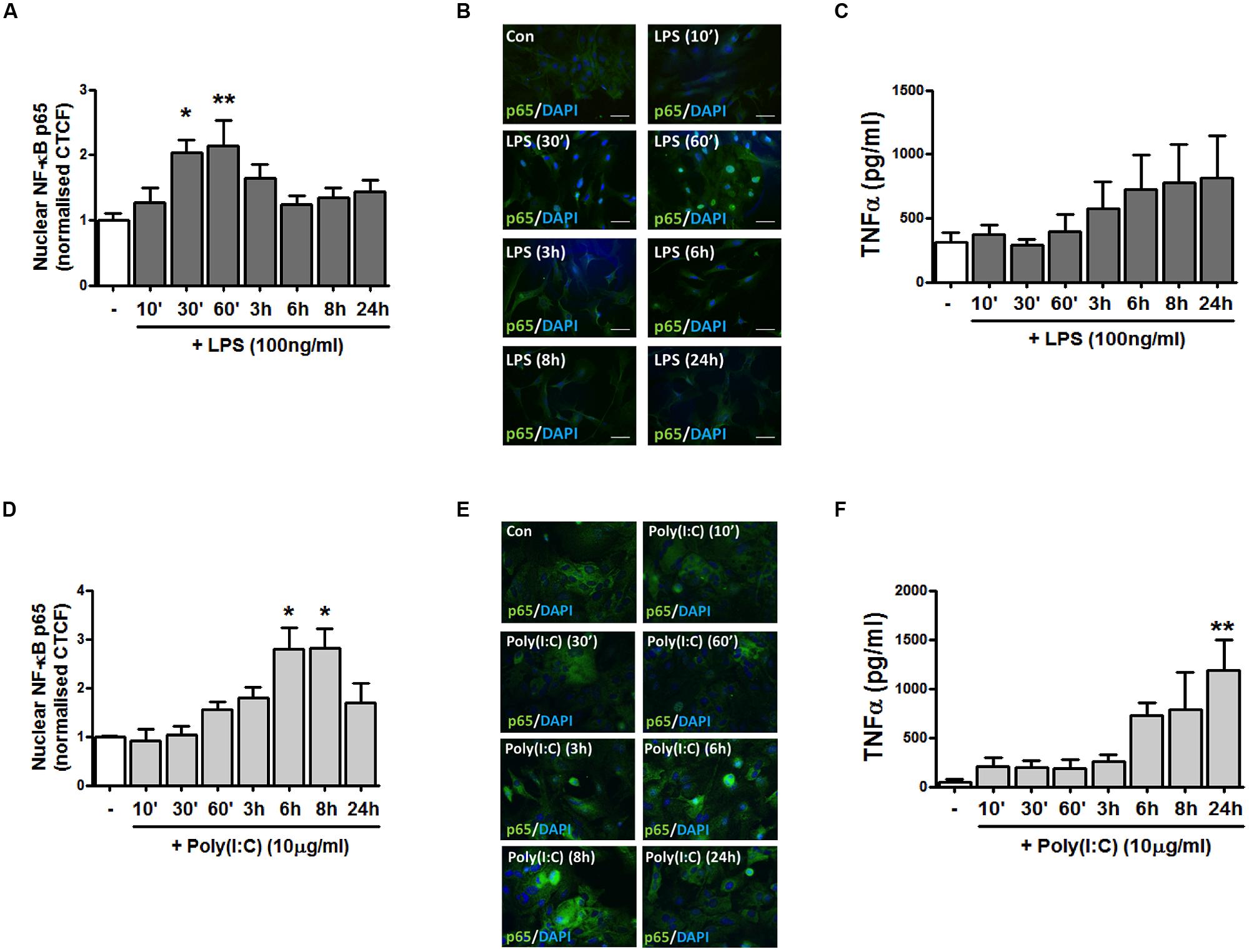
FIGURE 1. TLR4 and TLR3 stimulation promotes NF-κB p65 distribution to the nucleus and pro-inflammatory TNF-α expression in primary mixed glial cell cultures. (A,D) The relative fluorescence intensity (corrected total cell fluorescence; CTCF) of nuclear NF-κB p65 subunit was determined using Image J to assess the subcellular localization of p65 in mixed glial cultures following exposure to (A) LPS and (D) poly(I:C) for the indicated time periods. (B,E) Immunofluorescent images assessing the subcellular localization of p65 in mixed glial cultures following exposure to (B) LPS and (E) poly(I:C) for the indicated time periods. Nuclei were stained with DAPI, and images obtained using an Olympus IX70 inverted microscope equipped with the appropriate filter sets. Data are presented as the mean ± S.E.M and are representative of six animals. Scale bar = 20μm. Mixed glial cells were incubated with (C) LPS and (F) poly(I:C) (timepoints from 10 min–24 h) and supernatants were analyzed for TNF-α production using ELISA. Data are presented as the mean ± SEM of triplicate determinations and are representative of six animals. ∗p < 0.05, ∗∗p < 0.01 compared with vehicle-treated cells.
TLR3 Activation Enhances Nuclear NF-κB p65 Expression and TNF-α Release in Mixed Glia
To assess the impact of TLR3 stimulation on inflammatory signaling events in mixed glial cells, we next assessed the effect of poly(I:C) exposure on the distribution of the NF-κB p65 subunit. Primary murine glial cells were incubated with poly(I:C) for timepoints ranging from 10 min–24 h, and the localization of the p65 subunit assessed by immunofluorescence (Figures 1D,E). TLR3 stimulation time-dependently promoted the accumulation of NF-κB in the nucleus, peaking at 6 and 8 h post-poly(I:C) exposure (Figures 1D,E). This temporal profile differed in response to TLR4 stimulation, where maximal sequestration of NF-κB p65 was observed at 30 min and 1 h post-LPS treatment (Figure 1A). Poly(I:C) significantly enhanced TNF-α expression in mixed glial cells, with mean maximal stimulatory effects on protein expression observed at 24 h (Figure 1F). This indicates that TLR3 and TLR4 activation in primary glia robustly enhances pro-inflammatory signaling in primary murine glial cells.
Effect of the GABAB Receptor Agonist Baclofen on TLR4-Induced NF-κB-p65 Nuclear Expression and Pro-Inflammatory Cytokine Release in Mixed Glia
We next examined the impact of the GABAB agonist baclofen on TLR4-induced inflammatory signaling events in primary mixed glia. Glial cultures were pre-treated (30 min) with baclofen (10, 30, and 100 μM) prior to LPS exposure (30 min) and nuclear expression of NF-κB-p65 measured by fluorescence microscopy (Figures 2A,B). Stimulation of glia with LPS promoted the accumulation of p65 in the nucleus (Figure 2A). Pre-exposure to baclofen (at 30 μM) significantly attenuated the LPS-induced accumulation of p65 in the nucleus, while pre-incubation of glia with the GABAB agonist at concentrations of 10 μM (p = 0.09) and 100 μM (p = 0.85), failed to significantly impact LPS-induced nuclear p65 sequestration (Figures 2A,B). Similarly, Figure 2C demonstrates that baclofen attenuated LPS-induced TNF-α production in glia at concentrations of 10 and 30 μM, although this did not reach statistical significance. These findings suggest that the proclivity of baclofen to impact TLR4 signaling in primary mixed glia is concentration dependent.
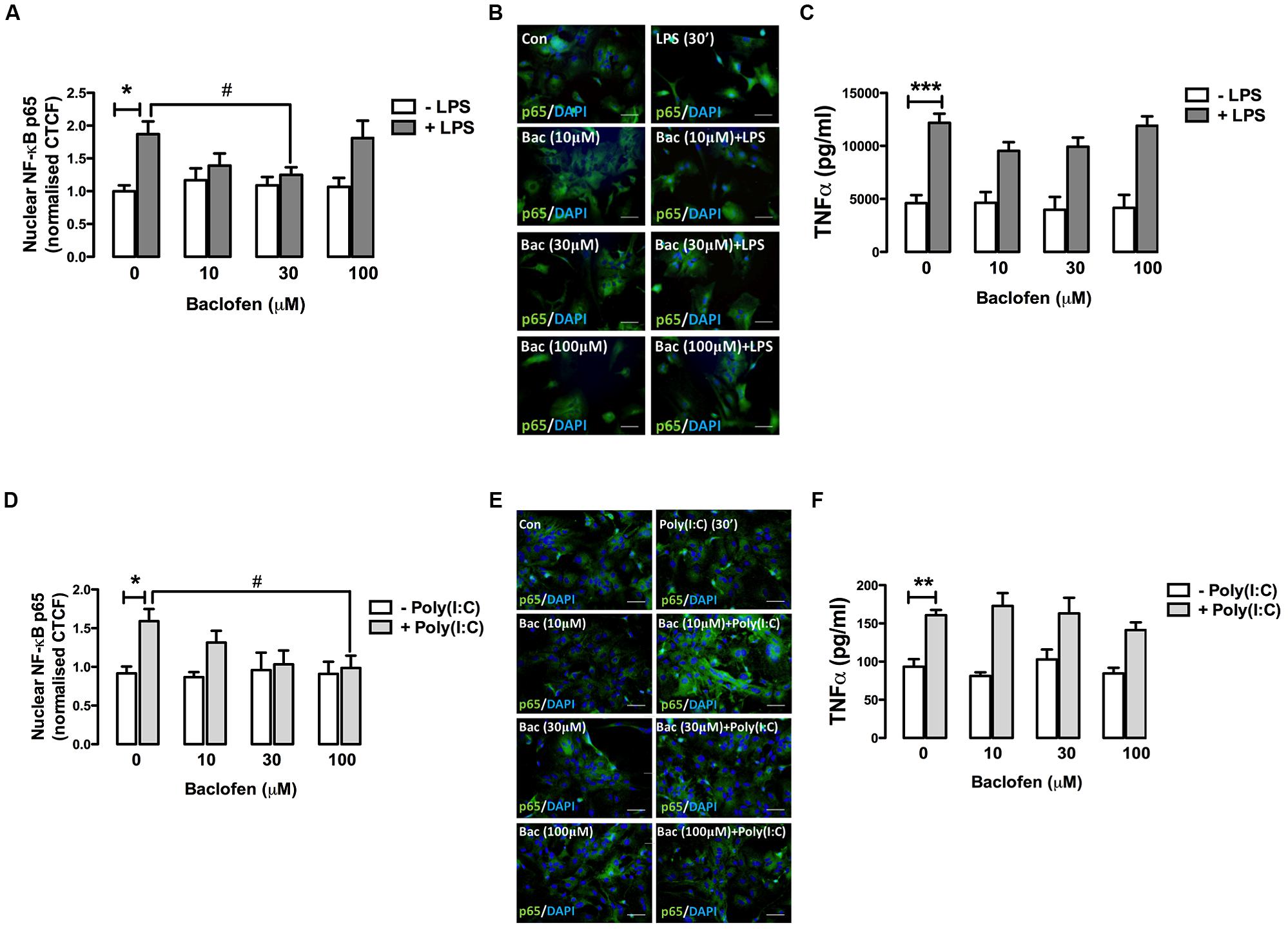
FIGURE 2. GABAB receptor activation modulates TLR4- and TLR3-induced NF-κB-p65 nuclear expression and TNF-α expression release in glia. (A,D) CTCF of nuclear NF-κB p65 subunit in mixed glial cultures following exposure to (A) LPS (30 min) and (D) poly(I:C) (6 h) in the absence and presence of baclofen (10, 30, and 100 μM). (B,E) Immunofluorescent images demonstrating the subcellular localization of p65 in glia following exposure to (B) LPS ± baclofen and (E) poly(I:C) ± baclofen. Nuclei were stained with DAPI, and images obtained using an Olympus IX70 inverted microscope equipped with the appropriate filter sets. Data are presented as the mean ± SEM and are representative of six animals. Scale bar = 20 μm. Mixed glial cells were pre-incubated with baclofen (10, 30, and 100 μM) prior to (C) LPS and (F) poly(I:C) (both at 24 h) and supernatants analyzed for TNF-α expression by ELISA. Data are presented as the mean ± SEM of triplicate determinations from six animals. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 compared with vehicle-treated cells. #p < 0.05 compared to cells treated with LPS or Poly(I:C) in the absence of baclofen.
GABAB Receptor Activation Modulates TLR3-Induced NF-κB-p65 Nuclear Expression and TNF-α Expression in Glia
Given that GABAB activation modulates TLR4-induced signaling in glia, we next assessed the impact of baclofen on TLR3-induced inflammatory signaling in glial cells. Pre-incubation of glia with baclofen dose-dependently attenuated poly(I:C)-induced nuclear sequestration (at 6 h) of NF-κB, reaching significance at 100 μM (Figures 2D,E). Baclofen reduced poly(I:C)-induced TNF-α production in glia at concentrations of 100 μM, although this did not reach statistical significance (Figure 2F). These findings suggest that GABAB activation regulates TLR3 signaling to NF-κB in glia in a concentration-dependent manner.
Demographic Data of Human Participants
A total of 24 subjects were assessed consisting of healthy control participants (n = 16) and newly diagnosed RR-MS patients (n = 8). All RR-MS patients were drug naïve at the time of inclusion. Mean disease duration in the RR-MS cohort was 1.2 ± 0.3 years and subjects had mild to moderate disability as reflected by EDSS scores of 2.6 ± 0.4 on average. The mean age of the 24 participants who enrolled in the investigation was 33.1 ± 7.8 years (range 23–40 years in control cohort; range 31–54 years in RR-MS cohort) and 46% of the sample was female. The overall demographics of the participants are reported in Figure 3A.
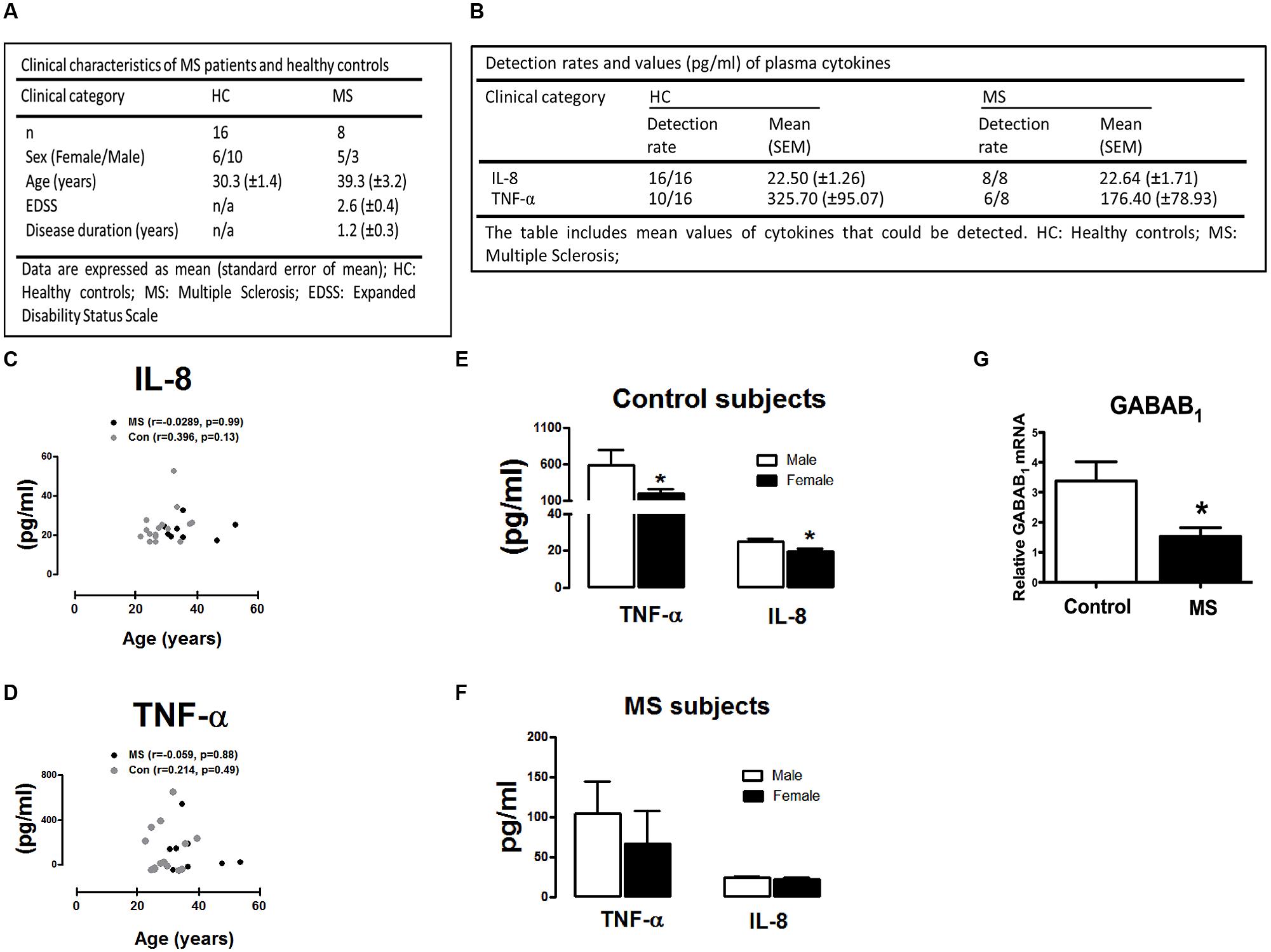
FIGURE 3. Demographic and clinical characteristics of MS patients and control subjects included in the study. (A) Demographics of healthy control participants and RR-MS patients. Expanded Disability Status Scale (EDSS). (B) Plasma levels of IL-8 and TNF-α in control (n = 16) and MS (n = 8) cohort. Detection rates and mean plasma expression values of IL-8 and TNF-α are summarized. (C,D) Correlation between IL-8/TNF-α plasma levels and age of study participants. Correlation plots demonstrating (C) IL-8 and (D) TNF-α plotted against subject age. No correlation was observed between age in control and MS groups analyzed (two-tailed Spearman non-parametric correlation coefficient, correlation r-values, and significance p-values, are indicated in the graphs). Levels of TNF-α and IL-8 in (E) healthy control participants and (F) MS patients in plasma obtained from male (n = 13) and female (n = 11) participants. (G) Peripheral blood mononuclear cells (PBMCs) isolated from healthy subjects and newly diagnosed RR-MS patients were assessed for GABAB mRNA expression, cDNA generated and assayed by quantitative real-time PCR for levels of GABAB1 mRNA. Data are presented as the mean ± SEM of triplicate determinations. ∗p < 0.05 compared with male control subjects (E). ∗p < 0.05 compared with all control subjects (G).
Cytokine and Chemokine Analysis in Human Plasma
To determine if RR-MS is associated with alterations in peripheral inflammatory signature, we initially assessed plasma levels of the inflammatory chemokine IL-8 and cytokine TNF-α in plasma from control and RR-MS groups. No significant difference in plasma IL-8 (p = 0.94) and TNF-α (p = 0.28) concentration was found between groups (Figure 3B). Detection rates and mean values of plasma TNF-α and IL-8 are summarized in Figure 3B. In addition, no clear correlation between the levels of plasma cytokines and participant age was determined in either RR-MS patient or control groups (two-tailed Spearman non-parametric correlation coefficient; Figures 3C,D), indicating that participant age had no impact on peripheral cytokine/chemokine expression. We next assessed the impact of gender on plasma cytokine and chemokine levels. Interestingly, compared to male control subjects, the expression levels of TNF-α and IL-8 were significantly reduced in female control subjects (Figure 3E). No significant difference was determined between males vs. females in the RR-MS cohort (Figure 3F). This data indicates that the level of cytokine/chemokine expression in healthy individuals is higher in males than females, but that this gender difference is not apparent in the RR-MS cohort.
GABAB Expression in Human PBMCs
To determine the expression profile of the metabotropic receptor GABAB in human PBMCs, and whether this profile was altered in disease, we assessed the expression of GABAB1 subunit in PBMCs isolated from healthy volunteers and RR-MS patients. Detectable levels of the GABAB1 subunit was determined in human PBMCs, with RR-MS associated with a reduction (twofold) in endogenous GABAB1 expression when compared to PBMCs isolated from healthy subjects (Figure 3G). This indicates that downregulation of GABAB receptors occurs in PBMCs isolated from newly diagnosed RR-MS patients, when compared to immune cells from healthy subjects.
Effect of TLR3 and TLR4 Stimulation on PBMCs Isolated from Control and RR-MS Cohorts
Given the defined role of TLR3 and TLR4 in EAE progression (Touil et al., 2006), in addition to evidence that their expression is up-regulated in MS lesions (Bsibsi et al., 2002), we next set out to assess the impact of TLR3 and TLR4 stimulation on inflammatory responses of immune cells isolated from control and RR-MS patients. Firstly, supernatants from unstimulated PBMCs isolated from RR-MS patients displayed enhanced TNF-α (Figures 4A,C) and IL-8 (Figures 4B,D) expression, when compared to PBMCs from healthy subjects, indicating the RR-MS patient PBMCs display an enhanced endogenous inflammatory signature. LPS enhanced TNF-α production in PBMCs from both groups (Figure 4A), and importantly the production of TNF-α was exacerbated in PBMCs prepared from the RR-MS group, compared with the control cohort (Figure 4A). In terms of IL-8 expression, two-way ANOVA revealed a significant influence of disease status on IL-8 levels [F(1,30) = 4.773, p = 0.0369], but no significant influence of TLR4 stimulation on overall variation [F(2,30) = 0.7174, p = 0.4962] (Figure 4B). However, post hoc analysis revealed a trend toward a statistically significant increase in IL-8 release following LPS treatment in PBMCs isolated from healthy controls (p = 0.0612), but not in RR-MS patients. Interestingly, PBMCs isolated from healthy subjects and RR-MS patients were unresponsive to poly(I⋅C) stimulation, both in terms of TNF-α and IL-8 production (Figures 4C,D), indicating that PBMCs from control individuals and RR-MS patients are refractory to TLR3 stimulation, in terms of TNF-α and IL-8 signaling.
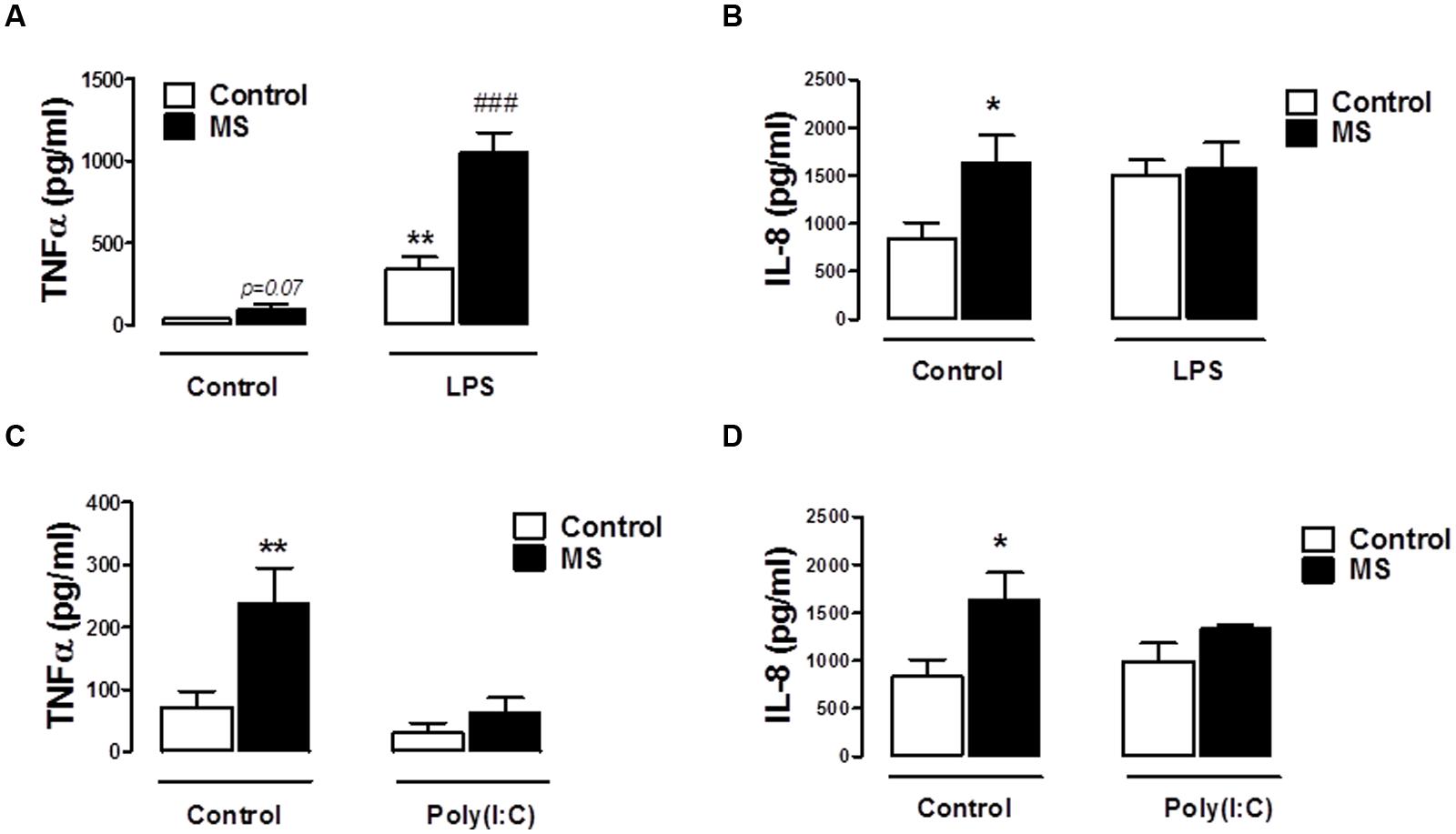
FIGURE 4. Peripheral blood mononuclear cells from RR-MS patients display enhanced inflammatory signaling. PBMCs prepared from healthy subjects and MS patients were seeded into 24-well plates and treated with vehicle, LPS (100 ng/ml) or poly(I⋅C) (10 μg/ml) for 24 h. Supernatants were analyzed for TNF-α (A,C) and IL-8 (B,D) production using ELISA. Data are presented as the mean ± SEM of triplicate determinations. ∗p < 0.05 and ∗∗p < 0.01 compared with vehicle-treated PBMCs from healthy subjects. ###p < 0.001 compared with vehicle-treated cells from MS patients.
Baclofen Attenuates TLR4-Induced TNF-α Cytokine Release in Human PBMCs Isolated from Healthy, but not RR-MS, Subjects
Given that GABAB activation modulates TLR4-induced signaling in primary mixed glia (Figure 2), and that TLR4 stimulation enhances TNF-α signaling in PBMCs from control and RR-MS patients (Figure 4), we next assessed the proclivity of baclofen to modulate TLR4-induced TNF-α release in PBMCs isolated from healthy subjects and RR-MS patients. PBMCs from healthy subjects (Figure 5A) and RR-MS patients (Figure 5B) were pre-treated (30 min) with baclofen (10, 30, and 100 μM) prior to LPS exposure (24 h) and TNF-α expression assessed. PBMCs isolated from healthy subjects were responsive to LPS with an increase in TNF-α release observed, whereas baclofen modestly attenuated this (Figure 5A). In contrast, pre-exposure (30 min) to baclofen prior to LPS exposure (24 h) failed to significantly impact TLR4-induced TNF-α expression in PBMCs from RR-MS patients at each dose tested (Figure 5B). Importantly, this indicates a differential sensitivity of cells from healthy and RR-MS subjects in terms of the ability of baclofen to modulate TLR-induced inflammatory signaling, which may have implications in terms of the design of GABAB receptor therapeutics to treat neuroinflammatory disorders including MS.
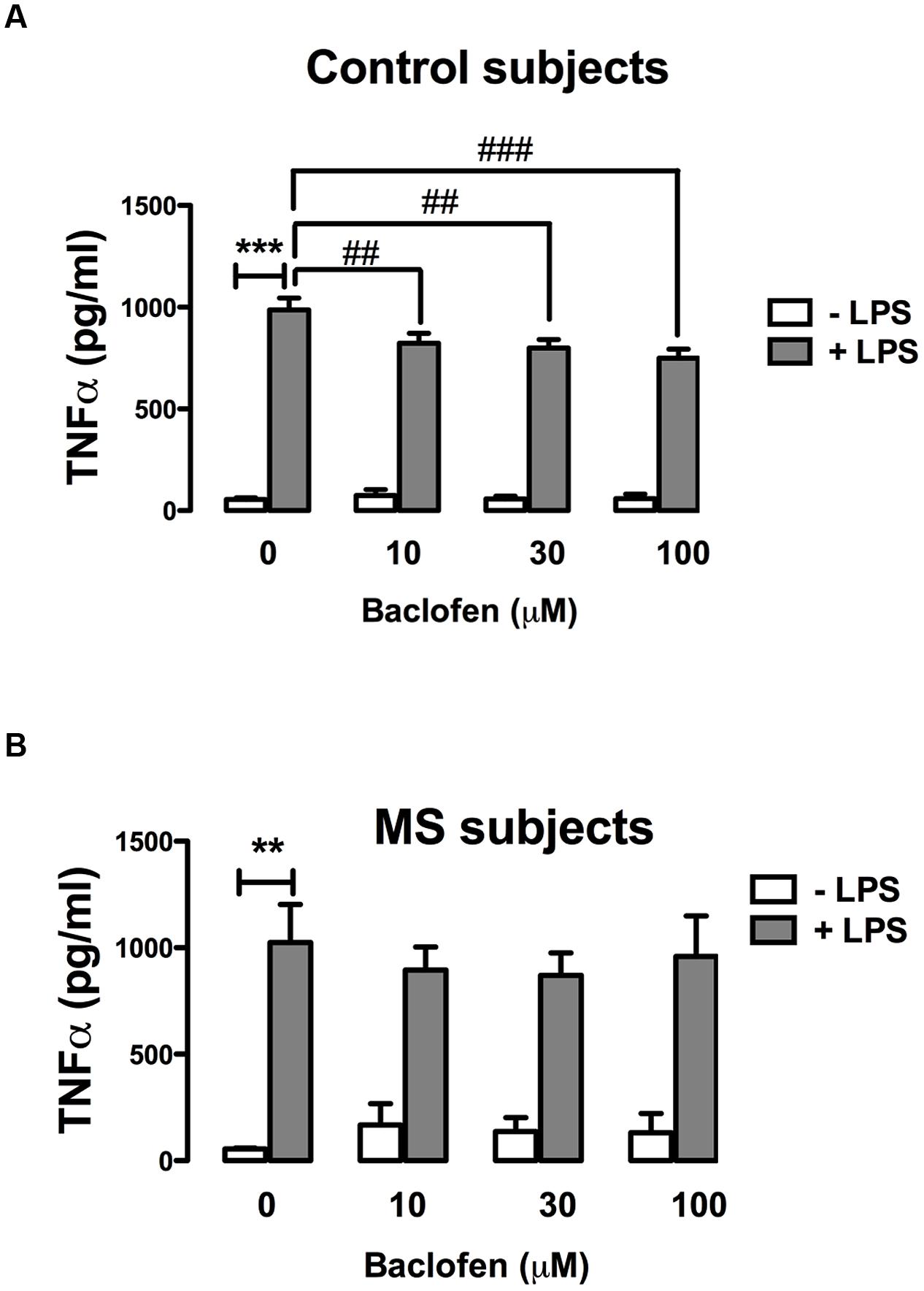
FIGURE 5. GABAB activation attenuates TLR4-induced TNF-α production in PBMCs from healthy, but not MS, subjects. PBMCs isolated from (A) healthy subjects and (B) MS patients were seeded in 24-well plates and pre-treated with baclofen (10, 30, and 100 μM) in the absence and presence of LPS (100 ng/ml) for 24 h. Supernatants were analyzed for TNF-α production using ELISA. Data are presented as the mean ± SEM of triplicate determinations. ∗∗p < 0.01 and ∗∗∗p < 0.001 compared with vehicle-treated cells. ##p < 0.01 and ###p < 0.001 compared to cells treated with LPS in the absence of baclofen.
Discussion
Understanding the mechanisms underlying neuroinflammatory processes in MS may pave the way to novel therapeutic strategies. Data herein demonstrate that the GABAB receptor agonist baclofen has the proclivity to differentially target TLR4 and TLR3 inflammatory signaling events both in CNS glial cells, and in peripheral immune cells. Baclofen reduced TLR3/4-induced nuclear NF-κB and TNF-α (albeit modestly) expression in glial cells, with distinct dose-dependency identified for these effects. GABAB receptor activation attenuated TLR4-induced TNF-α expression in PBMCs isolated from healthy individuals, while baclofen had no impact on TLR4 signaling in RR-MS patient PBMCs. Furthermore, PBMCs from RR-MS patients expressed lower levels of the GABAB receptor mRNA. Significantly, we also determined that immune cells from the RR-MS patient group displayed hypersensitivity to LPS exposure in terms of TNF-α expression.
TLR family members are expressed in cells of the CNS (Downer et al., 2013a; Nakano et al., 2015) and TLR ligands regulate CNS inflammation (Lehnardt, 2010). We initially targeted TLR3 and TLR4 signaling given the role of these receptors in neuroinflammatory events linked with MS (Bsibsi et al., 2002; Touil et al., 2006). Both NF-κB activation and pro-inflammatory cytokine expression are known downstream signaling events following ligation of TLR3 and TLR4 (Moynagh, 2005), and signaling via both cascades is perturbed following TLR3 (Cavassani et al., 2008) and TLR4 (Devaraj et al., 2011) knockout. We examined the ability of LPS and poly(I:C) to couple to TLR4 and TLR3 inflammatory signaling in primary glia, respectively. Data presented herein indicate that LPS and poly(I:C) enhanced NF-κB activation and TNF-α cytokine expression in glia, with the kinetics of this response mirroring data described elsewhere (Park et al., 2006; Horvath et al., 2008).
This study highlights the anti-inflammatory potential of baclofen in glial cells, by virtue of its inhibitory effects on the NF-κB-TNF-α pathway induced by TLR3/4 activation. This is consistent with data elsewhere indicating that baclofen reduces LPS-induced production of IL-6/IL-12 in mouse microglia (Kuhn et al., 2004), and reduces phospho-p65-NF-κB and IL-6/TNF-α expression in human astrocytes and microglial cells stimulated with IFN-γ/LPS (Lee et al., 2011). GABAB receptors are expressed on astrocytes and microglia (Charles et al., 2003), and evidence indicates that glial GABAB receptors are functional (Kuhn et al., 2004; Oka et al., 2006). Given the expression and functional activity of GABAB receptors on glia, alongside evidence that baclofen negatively regulates TLR3/4-induced inflammatory signaling, it is likely that GABAB receptors play an integral role in the course of neuroinflammation by targeting glia. The ability of baclofen to target TLR3/4-induced inflammatory signaling in glia in vitro may contribute to an anti-inflammatory efficacy in vivo, and this will be the focus of further studies.
Interestingly, different effects of GABAB receptor modulation on the immune system have been indicated, suggesting that GABA is a regulator of immune cell activity and inflammation. Indeed, GABA receptors have been identified on neutrophils, macrophages, dendritic cells, and T-cells (Rane et al., 2005; Jin et al., 2013), and cells of the immune system possess the metabolic machinery for the synthesis of GABA (Auteri et al., 2015). Given the evidence that GABA is a neuroimmune modulator, alongside our findings that baclofen modulates TLR-induced inflammatory signaling in central glial cells, we next assessed the impact of baclofen on TLR signaling in peripheral immune cells. Firstly, our findings indicate that TLR4 activation promotes TNF-α and IL-8 cytokine expression in human PBMCs, and these findings are consistent with data elsewhere (Kaplin et al., 2009; Downer et al., 2013b). Importantly, data presented herein indicate an anti-inflammatory propensity of baclofen in human immune cells, demonstrating that baclofen blunts LPS-induced TNF-α expression in peripheral immune cells from healthy subjects. This finding is in line with data demonstrating that exposure of human PBMCs to baclofen ameliorates phytohemagglutinin-induced TNF-α release (Duthey et al., 2010). A significant finding here is that this is the first direct evidence that baclofen can target TLR signaling in human PBMCs, and hence has relevance across a range of neuroimmune disorders.
Our results identify that PBMCs isolated from healthy subjects were responsive to baclofen, with a significant, albeit moderate, decrease in LPS-induced TNF-α expression observed following baclofen treatment. In contrast, baclofen failed to regulate TLR4-induced TNF-α expression in PBMCs from RR-MS subjects. This is significant as intrathecal administration of baclofen can be used to treat spasticity in MS, in addition to spinal cord injury, cerebral palsy, and acquired brain injury (Gunnarsson and Samuelsson, 2015). Furthermore, there is evidence of a loss of GABAergic neurons at lesion sites in post-mortem MS brain (Redondo et al., 2014). The differential sensitivity of cells from healthy and RR-MS subjects to baclofen indicates that MS drives a desensitizing signal in terms of GABAB receptor function. Indeed, our findings indicate that downregulation of GABAB receptors occurs in PBMCs in newly diagnosed RR-MS patients, which may govern the differential sensitivity to baclofen exposure seen in healthy and MS patient cells. In support of this, the level of GABA is reduced in the blood serum of patients with MS as compared to the controls (Demakova et al., 2003). In addition, baclofen treatment has been shown to reduce GABAB receptor density in rat spinal cord (Kroin et al., 1993), which may be postulated as a molecular mechanism for development of tolerance. It is especially interesting to note that treatment failure has been shown with baclofen in MS patients (Stroet et al., 2013), and the inability of baclofen to target TLR4-induced pro-inflammatory cytokine production might contribute to these effects.
Whether the effects of baclofen identified in the present and other studies are a direct result of its action at the GABAB receptor is not yet entirely clear, particularly given the recent discovery that baclofen, and several GABAB receptor antagonists, are allosteric modulators of CXCR4, a chemokine receptor involved in neuroimmune crosstalk (Guyon et al., 2013). Indeed, determining whether the effects of baclofen that were observed in this study are specifically due to its action at the GABAB receptor rather than at CXCR4 will be the focus of future studies. Such studies will be challenging because several GABAB receptor antagonists also act as allosteric modulators of CXCR4 (Guyon et al., 2013). Future studies aimed at identifying GABAB receptor antagonists that do not affect CXCR4 activity will be required in order to address this question directly. Nevertheless, since baclofen and some GABAB receptor antagonists exert the same effects on CXCR4 activity (Guyon et al., 2013), then it would be expected that prevention of the effects of baclofen by a GABAB receptor antagonist would be independent of CXCR4 activity (since both baclofen and the GABAB receptor antagonists behave as CXCR4 antagonists). Alternatively, experiments could examine the effects of baclofen under conditions whereby CXCR4 expression or activity is altered, however, such experiments might be complicated by the modulation of inflammatory mediators by CXCR4 itself. Despite these limitations, there is accumulating evidence that many components of the GABA neurotransmitter system, including not only the GABAB receptor but also the GABAA receptor, can have anti-inflammatory effects (Song et al., 1998; Bhat et al., 2010; Lee et al., 2011).
The rationale behind the present study was to determine if baclofen could target TLR-induced signaling in RR-MS, particularly given the growing body of data indicating that the TLR system is a key player in MS pathogenesis (O’Brien et al., 2008; Downer, 2011). However, intrathecal administration of baclofen is used to manage spasticity in patients with a range of conditions including spinal cord injury, cerebral palsy and acquired brain injury in addition to MS, and hence in future studies it would be interesting to compare the effects of baclofen on the PBMCs of patients with other neurological conditions.
The concentrations of cytokines/chemokines are altered in MS (Ubogu et al., 2006), suggesting that cytokine/chemokine signatures may indicate disease progression in patient groups (O’Connell et al., 2014). Given that evidence indicates that TNF-α (Sharief and Hentges, 1991) and IL-8 (Lund et al., 2004) are higher in serum of patients with MS, we examined the relative expression of TNF-α and IL-8 in plasma isolated from newly diagnosed RR-MS patients. Our analysis revealed no difference in plasma IL-8/TNF-α between control and RR-MS groups. Systemic inflammation in MS is strongly associated with periods of intense relapse (Confavreux et al., 2000), hence it is plausible that alterations in peripheral cytokines in MS may be phase dependent. In support of this, Mellergard et al. (2010) indicate that MS patients with EDSS measurements in the range comparable to the RR-MS cohort assessed in present study demonstrate no alteration in plasma TNF-α when compared to healthy subjects (Mellergard et al., 2010).
Neuroinflammatory changes develop with age, with defects in the immune system identified in elderly individuals (Allman and Miller, 2005). In addition, an increased plasma level of pro-inflammatory cytokines has been demonstrated in elderly, compared to young, individuals (Forsey et al., 2003). Our findings indicate no clear correlation between the levels of plasma cytokine/chemokine and participant age in either RR-MS or control groups, indicating that the age of subjects did not impact cytokine/chemokine profiles. Interestingly, compared to male control subjects, the expression levels of TNF-α and IL-8 were significantly reduced in female control subjects. This indicates that particular expression profiles of cytokines/chemokines may be gender specific, which is confirmed elsewhere (Asai et al., 2001).
Marked alterations in the expression profile of TLRs has been determined in MS lesions in human brain samples, in the brain samples from mice that have undergone EAE, and in CSF cells isolated from MS patients (Marta, 2009). With this in mind we investigated the effect of TLR4 and TLR3 stimulation on the pro-inflammatory cytokine and chemokine, TNF-α and IL-8. PBMCs without stimulation, as well as after stimulation with LPS, displayed increased production of TNF-α and IL-8 in the RR-MS sample cohort, when compared to the control group. These results suggest that immune cells from the RR-MS patient group have an enhanced endogenous inflammatory signature. In addition, RR-MS patient PBMCs displayed hypersensitivity to LPS exposure in terms of TNF-α expression. TLR4 expression is upregulated in CNS lesions in mice following EAE and in PBMCs from RR-MS and secondary progressive (SP) MS patients (Andersson et al., 2008), which in support of our study, indicates that TLR4 signaling participates in an innate immune response that may shape the inflammatory response in both forms of MS. It is intriguing that the hypersensitivity of RR-MS patient cells to LPS is only relevant in the context of TNF-α induction, since LPS shows comparable efficacy in inducing IL-8 in cells from healthy and RR-MS patients. Hence, any form of TLR4 hypersensitivity that may exist in MS patient cells appears to be restricted to the pathway leading to TNF-α expression, and further studies will probe the intracellular signaling events in patient cells.
Our studies also probed the effects of TLR3 stimulation on TNF-α/IL-8 in PBMCs. Surprisingly, PBMCs isolated from healthy donors and RR-MS patients did not respond to TLR3 stimulation by enhancing TNF-α or IL-8 expression, suggesting that the TLR3 pathway leading to cytokine and chemokine expression may be desensitized in our participant cohorts. Indeed, we have previously demonstrated that the TLR3-IFN-β pathway is desensitized in MS patient PBMCs, suggesting that MS patients may be pre-sensitized to viral infection showing some form of TLR3 tolerance. However, the non-responsiveness of healthy donor PBMCs to poly(I⋅C) is in line (Wesch et al., 2006) and in contrast (Myles et al., 2012) to findings elsewhere in studies which investigated freshly isolated PBMCs; differences in dose and timecourse for treatment regimen used with poly(I:C) in the present study may underlie the differences observed.
Although baclofen demonstrates therapeutic effects in MS, the mechanism(s) of action are poorly understood. We present evidence that the innate arm of the immune system is a target for baclofen anti-inflammatory action, and demonstrate that baclofen differentially targets TLR4 and TLR3 inflammatory signaling events both in primary murine glial cells and in PBMCs isolated from human blood samples. Baclofen can exert anti-inflammatory properties at specific doses, by down-regulating TLR-induced activation of NF-κB and induction of pro-inflammatory cytokines. Significantly, our findings also indicate that inflammatory signaling, and sensitivity to TLR4 stimulation, was enhanced in PBMCs of patients with RR-MS, highlighting that TLR4 may play a role at least in the RR form of MS pathogenesis. We targeted this particular patient cohort because they were newly-diagnosed and naïve to disease modifying therapies. However, investigating TLR functioning and its modulation by baclofen in patients with secondary chronic progressive MS would be an important future study. Overall, the present study provides novel insight into the cellular effects of targeting central and peripheral GABAB receptors in the modulation of TLR functioning.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Translational Research Access Programme (TRAP), the College of Medicine and Health (University College Cork) and the Department of Anatomy and Neuroscience, University College Cork. The authors would like to thank A-M. Cusack, P. Kirwan, and J. Kelly for assistance with phlebotomy. The authors would also like to acknowledge the Department of Anatomy and Neuroscience Imaging Centre, BioSciences Institute, University College Cork, for assistance in preparing and imaging specimens for this research.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fncel.2015.00284
References
Akira, S., Uematsu, S., and Takeuchi, O. (2006). Pathogen recognition and innate immunity. Cell 124, 783–801. doi: 10.1016/j.cell.2006.02.015
Allman, D., and Miller, J. P. (2005). The aging of early B-cell precursors. Immunol. Rev. 205, 18–29. doi: 10.1111/j.0105-2896.2005.00269.x
Andersson, A., Covacu, R., Sunnemark, D., Danilov, A. I., Dal Bianco, A., Khademi, M., et al. (2008). Pivotal advance: HMGB1 expression in active lesions of human and experimental multiple sclerosis. J. Leukoc. Biol. 84, 1248–1255. doi: 10.1189/jlb.1207844
Asai, K., Hiki, N., Mimura, Y., Ogawa, T., Unou, K., and Kaminishi, M. (2001). Gender differences in cytokine secretion by human peripheral blood mononuclear cells: role of estrogen in modulating LPS-induced cytokine secretion in an ex vivo septic model. Shock 16, 340–343. doi: 10.1097/00024382-200116050-00003
Auteri, M., Zizzo, M. G., and Serio, R. (2015). GABA and GABA receptors in the gastrointestinal tract: from motility to inflammation. Pharmacol. Res. 93, 11–21. doi: 10.1016/j.phrs.2014.12.001
Bhat, R., Axtell, R., Mitra, A., Miranda, M., Lock, C., Tsien, R. W., et al. (2010). Inhibitory role for GABA in autoimmune inflammation. Proc. Natl. Acad. Sci. U.S.A. 107, 2580–2585. doi: 10.1073/pnas.0915139107
Bjurstöm, H., Wang, J., Ericsson, I., Bengtsson, M., Liu, Y., Kumar-Mendu, S., et al. (2008). GABA, a natural immunomodulator of T lymphocytes. J. Neuroimmunol. 205, 44–50. doi: 10.1016/j.jneuroim.2008.08.017
Bormann, J. (2000). The ‘ABC’ of GABA receptors. Trends Pharmacol. Sci. 21, 16–19. doi: 10.1016/S0165-6147(99)01413-3
Bsibsi, M., Ravid, R., Gveric, D., and Van Noort, J. M. (2002). Broad expression of Toll-like receptors in the human central nervous system. J. Neuropathol. Exp. Neurol. 61, 1013–1021.
Campagna-Slater, V., and Weaver, D. F. (2007). Molecular modelling of the GABAA ion channel protein. J. Mol. Graph. Mod. 25, 721–730. doi: 10.1016/j.jmgm.2006.06.001
Carmans, S., Hendriks, J. J. A., Slaets, H., Thewissen, K., Stinissen, P., Rigo, J.-M., et al. (2013). Systemic treatment with the inhibitory neurotransmitter γ-aminobutyric acid aggravates experimental autoimmune encephalomyelitis by affecting proinflammatory immune responses. J. Neuroimmunol. 255, 45–53. doi: 10.1016/j.jneuroim.2012.11.001
Cavassani, K. A., Ishii, M., Wen, H., Schaller, M. A., Lincoln, P. M., Lukacs, N. W., et al. (2008). TLR3 is an endogenous sensor of tissue necrosis during acute inflammatory events. J. Exp. Med. 205, 2609–2621. doi: 10.1084/jem.20081370
Charles, K. J., Deuchars, J., Davies, C. H., and Pangalos, M. N. (2003). GABA B receptor subunit expression in glia. Mol. Cell. Neurosci. 24, 214–223. doi: 10.1016/S1044-7431(03)00162-3
Cobb, S. R., Buhl, E. H., Halasy, K., Paulsen, O., and Somogyi, P. (1995). Synchronization of neuronal activity in hippocampus by individual GABAergic interneurons. Nature 378, 75–78. doi: 10.1038/378075a0
Compston, A., and Coles, A. (2002). Multiple sclerosis. Lancet 359, 1221–1231. doi: 10.1016/S0140-6736(02)08220-X
Confavreux, C., Vukusic, S., Moreau, T., and Adeleine, P. (2000). Relapses and progression of disability in multiple sclerosis. N. Engl. J. Med. 343, 1430–1438. doi: 10.1056/NEJM200011163432001
Demakova, E. V., Korobov, V. P., and Lemkina, L. M. (2003). [Determination of gamma-aminobutyric acid concentration and activity of glutamate decarboxylase in blood serum of patients with multiple sclerosis]. Klin. Lab. Diagn. 4, 15–17.
Devaraj, S., Tobias, P., and Jialal, I. (2011). Knockout of toll-like receptor-4 attenuates the pro-inflammatory state of diabetes. Cytokine 55, 441–445. doi: 10.1016/j.cyto.2011.03.023
Downer, E. J. (2011). Cannabinoids and innate immunity: taking a toll on neuroinflammation. ScientificWorldJournal 11, 855–865. doi: 10.1100/tsw.2011.84
Downer, E. J., Cowley, T. R., Lyons, A., Mills, K. H., Berezin, V., Bock, E., et al. (2010). A novel anti-inflammatory role of NCAM-derived mimetic peptide, FGL. Neurobiol. Aging 31, 118–128. doi: 10.1016/j.neurobiolaging.2008.03.017
Downer, E. J., Johnston, D. G., and Lynch, M. A. (2013a). Differential role of Dok1 and Dok2 in TLR2-induced inflammatory signaling in glia. Mol. Cell. Neurosci. 56, 148–158. doi: 10.1016/j.mcn.2013.04.007
Downer, E. J., Jones, R. S., Mcdonald, C. L., Greco, E., Brennan, S., Connor, T. J., et al. (2013b). Identifying early inflammatory changes in monocyte-derived macrophages from a population with IQ-discrepant episodic memory. PLoS ONE 8:e63194. doi: 10.1371/journal.pone.0063194
Duthey, B., Hübner, A., Diehl, S., Boehncke, S., Pfeffer, J., and Boehncke, W.-H. (2010). Anti-inflammatory effects of the GABA(B) receptor agonist baclofen in allergic contact dermatitis. Exp. Dermatol. 19, 661–666. doi: 10.1111/j.1600-0625.2010.01076.x
Forsey, R. J., Thompson, J. M., Ernerudh, J., Hurst, T. L., Strindhall, J., Johansson, B., et al. (2003). Plasma cytokine profiles in elderly humans. Mech. Ageing Dev. 124, 487–493. doi: 10.1016/S0047-6374(03)00025-3
Garcia-Oscos, F., Salgado, H., Hall, S., Thomas, F., Farmer, G. E., Bermeo, J., et al. (2012). The stress-induced cytokine interleukin-6 decreases the inhibition/excitation ratio in the rat temporal cortex via trans-signaling. Biol. Psychiatry 71, 574–582. doi: 10.1016/j.biopsych.2011.11.018
Gassmann, M., and Bettler, B. (2012). Regulation of neuronal GABA(B) receptor functions by subunit composition. Nat. Rev. Neurosci. 13, 380–394. doi: 10.1038/nrn3249
Gunnarsson, S., and Samuelsson, K. (2015). Patient experiences with intrathecal baclofen as a treatment for spasticity - a pilot study. Disabil. Rehabil. 37, 834–841. doi: 10.3109/09638288.2014.943844
Guyon, A., Kussrow, A., Olmsted, I. R., Sandoz, G., Bornhop, D. J., and Nahon, J.-L. (2013). Baclofen and other GABAB receptor agents are allosteric modulators of the CXCL12 chemokine receptor CXCR4. J. Neurosci. 33, 11643–11654. doi: 10.1523/JNEUROSCI.6070-11.2013
Han, M. H., Hwang, S. I., Roy, D. B., Lundgren, D. H., Price, J. V., Ousman, S. S., et al. (2008). Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature 451, 1076–1081. doi: 10.1038/nature06559
Horvath, R. J., Nutile-Mcmenemy, N., Alkaitis, M. S., and Deleo, J. A. (2008). Differential migration, LPS-induced cytokine, chemokine, and NO expression in immortalized BV-2 and HAPI cell lines and primary microglial cultures. J. Neurochem. 107, 557–569. doi: 10.1111/j.1471-4159.2008.05633.x
Hyland, N. P., and Cryan, J. F. (2010). A gut feeling about GABA: focus on GABAB receptors. Front. Pharmacol. 1:124. doi: 10.3389/fphar.2010.00124
Jeannin, P., Jaillon, S., and Delneste, Y. (2008). Pattern recognition receptors in the immune response against dying cells. Curr. Opin. Immunol. 20, 530–537. doi: 10.1016/j.coi.2008.04.013
Jiang, X., Su, L., Zhang, Q., He, C., Zhang, Z., Yi, P., et al. (2012). GABAB receptor complex as a potential target for tumor therapy. J. Histochem. Cytochem. 60, 269–279. doi: 10.1369/0022155412438105
Jin, Z., Mendu, S. K., and Birnir, B. (2013). GABA is an effective immunomodulatory molecule. Amino Acids 45, 87–94. doi: 10.1007/s00726-011-1193-7
Kaplin, A., Carroll, K. A., Cheng, J., Allie, R., Lyketsos, C. G., Calabresi, P., et al. (2009). IL-6 release by LPS-stimulated peripheral blood mononuclear cells as a potential biomarker in Alzheimer’s disease. Int. Psychogeriatr. 21, 413–414. doi: 10.1017/S1041610208008107
Kawai, T., and Akira, S. (2006). TLR signaling. Cell Death Differ. 13, 816–825. doi: 10.1038/sj.cdd.4401850
Kornau, H. C. (2006). GABA(B) receptors and synaptic modulation. Cell Tissue Res. 326, 517–533. doi: 10.1007/s00441-006-0264-7
Kroin, J. S., Bianchi, G. D., and Penn, R. D. (1993). Intrathecal baclofen down-regulates GABAB receptors in the rat substantia gelatinosa. J. Neurosurg. 79, 544–549. doi: 10.3171/jns.1993.79.4.0544
Kuhn, S. A., Van Landeghem, F. K. H., Zacharias, R., Färber, K., Rappert, A., Pavlovic, S., et al. (2004). Microglia express GABA(B) receptors to modulate interleukin release. Mol. Cell. Neurosci. 25, 312–322. doi: 10.1016/j.mcn.2003.10.023
Kurtzke, J. F. (1983). Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology 33, 1444–1452. doi: 10.1212/WNL.33.11.1444
Lee, M., Schwab, C., and Mcgeer, P. L. (2011). Astrocytes are GABAergic cells that modulate microglial activity. Glia 59, 152–165. doi: 10.1002/glia.21087
Lehnardt, S. (2010). Innate immunity and neuroinflammation in the CNS: the role of microglia in Toll-like receptor-mediated neuronal injury. Glia 58, 253–263.
Lund, B. T., Ashikian, N., Ta, H. Q., Chakryan, Y., Manoukian, K., Groshen, S., et al. (2004). Increased CXCL8 (IL-8) expression in multiple sclerosis. J. Neuroimmunol. 155, 161–171. doi: 10.1016/j.jneuroim.2004.06.008
Marta, M. (2009). Toll-like receptors in multiple sclerosis mouse experimental models. Ann. N. Y. Acad. Sci. 1173, 458–462. doi: 10.1111/j.1749-6632.2009.04849.x
Medzhitov, R., Preston-Hurlburt, P., Kopp, E., Stadlen, A., Chen, C., Ghosh, S., et al. (1998). MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol. Cell 2, 253–258. doi: 10.1016/S1097-2765(00)80136-7
Mellergard, J., Edstrom, M., Vrethem, M., Ernerudh, J., and Dahle, C. (2010). Natalizumab treatment in multiple sclerosis: marked decline of chemokines and cytokines in cerebrospinal fluid. Mult. Scler. 16, 208–217. doi: 10.1177/1352458509355068
Moynagh, P. N. (2005). TLR signalling and activation of IRFs: revisiting old friends from the NF-kappaB pathway. Trends Immunol. 26, 469–476. doi: 10.1016/j.it.2005.06.009
Myles, A., Rahman, M. T., and Aggarwal, A. (2012). Membrane-bound toll-like receptors are overexpressed in peripheral blood and synovial fluid mononuclear cells of enthesitis-related arthritis category of juvenile idiopathic arthritis (JIA-ERA) patients and lead to secretion of inflammatory mediators. J. Clin. Immunol. 32, 488–496. doi: 10.1007/s10875-011-9640-5
Nakano, Y., Furube, E., Morita, S., Wanaka, A., Nakashima, T., and Miyata, S. (2015). Astrocytic TLR4 expression and LPS-induced nuclear translocation of STAT3 in the sensory circumventricular organs of adult mouse brain. J. Neuroimmunol. 278, 144–158. doi: 10.1016/j.jneuroim.2014.12.013
Nishimura, M., and Naito, S. (2005). Tissue-specific mRNA expression profiles of human toll-like receptors and related genes. Biol. Pharm. Bull. 28, 886–892. doi: 10.1248/bpb.28.886
O’Brien, K., Fitzgerald, D. C., Naiken, K., Alugupalli, K. R., Rostami, A. M., and Gran, B. (2008). Role of the innate immune system in autoimmune inflammatory demyelination. Curr. Med. Chem. 15, 1105–1115. doi: 10.2174/092986708784221458
O’Connell, K. E., Mok, T., Sweeney, B., Ryan, A. M., and Dev, K. K. (2014). The use of cytokine signature patterns: separating drug naive, interferon and natalizumab-treated multiple sclerosis patients. Autoimmunity 47, 505–511. doi: 10.3109/08916934.2014.930734
Oka, M., Wada, M., Wu, Q., Yamamoto, A., and Fujita, T. (2006). Functional expression of metabotropic GABAB receptors in primary cultures of astrocytes from rat cerebral cortex. Biochem. Biophys. Res. Commun. 341, 874–881. doi: 10.1016/j.bbrc.2006.01.039
O’Neill, L. A. J. (2004). TLRs: Professor Mechnikov, sit on your hat. Trends Immunol. 25, 687–693. doi: 10.1016/j.it.2004.10.005
Ong, J., and Kerr, D. I. (1990). GABA-receptors in peripheral tissues. Life Sci. 46, 1489–1501. doi: 10.1016/0024-3205(90)90421-M
Padgett, C. L., and Slesinger, P. A. (2010). GABAB receptor coupling to G-proteins and ion channels. Adv. Pharmacol. 58, 123–147. doi: 10.1016/S1054-3589(10)58006-2
Park, C., Lee, S., Cho, I. H., Lee, H. K., Kim, D., Choi, S. Y., et al. (2006). TLR3-mediated signal induces proinflammatory cytokine and chemokine gene expression in astrocytes: differential signaling mechanisms of TLR3-induced IP-10 and IL-8 gene expression. Glia 53, 248–256. doi: 10.1002/glia.20278
Pinard, A., Seddik, R., and Bettler, B. (2010). GABAB receptors: physiological functions and mechanisms of diversity. Adv. Pharmacol. 58, 231–255. doi: 10.1016/S1054-3589(10)58010-4
Polman, C. H., Reingold, S. C., Banwell, B., Clanet, M., Cohen, J. A., Filippi, M., et al. (2011). Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann. Neurol. 69, 292–302. doi: 10.1002/ana.22366
Rane, M. J., Gozal, D., Butt, W., Gozal, E., Pierce, W. M. Jr., Guo, S. Z., et al. (2005). Gamma-amino butyric acid type B receptors stimulate neutrophil chemotaxis during ischemia-reperfusion. J. Immunol. 174, 7242–7249. doi: 10.4049/jimmunol.174.11.7242
Redondo, J., Kemp, K., Hares, K., Rice, C., Scolding, N., and Wilkins, A. (2014). Purkinje cell pathology and loss in multiple sclerosis cerebellum. Brain Pathol. doi: 10.1111/bpa.12230 [Epub ahead of print].
Reyes-García, M. G., Hernández-Hernández, F., Hernández-Téllez, B., and García-Tamayo, F. (2007). GABA (A) receptor subunits RNA expression in mice peritoneal macrophages modulate their IL-6/IL-12 production. J. Neuroimmunol. 188, 64–68. doi: 10.1016/j.jneuroim.2007.05.013
Sharief, M. K., and Hentges, R. (1991). Association between tumor necrosis factor-alpha and disease progression in patients with multiple sclerosis. N. Engl. J. Med. 325, 467–472. doi: 10.1056/NEJM199108153250704
Song, D. K., Suh, H. W., Huh, S. O., Jung, J. S., Ihn, B. M., Choi, I. G., et al. (1998). Central GABAA and GABAB receptor modulation of basal and stress-induced plasma interleukin-6 levels in mice. J. Pharmacol. Exp. Ther. 287, 144–149.
Stroet, A., Trampe, N., and Chan, A. (2013). Treatment failure of intrathecal baclofen and supra-additive effect of nabiximols in multiple sclerosis-related spasticity: a case report. Ther. Adv. Neurol. Disord. 6, 199–203. doi: 10.1177/1756285613475835
Touil, T., Fitzgerald, D., Zhang, G. X., Rostami, A., and Gran, B. (2006). Cutting edge: TLR3 stimulation suppresses experimental autoimmune encephalomyelitis by inducing endogenous IFN-beta. J. Immunol. 177, 7505–7509. doi: 10.4049/jimmunol.177.11.7505
Tyagi, R. K., Bisht, R., Pant, J., Kumar, P., Majeed, A. B., and Prakash, A. (2015). Possible role of GABA-B receptor modulation in MPTP induced Parkinson’s disease in rats. Exp. Toxicol. Pathol. 67, 211–217. doi: 10.1016/j.etp.2014.12.001
Ubogu, E. E., Cossoy, M. B., and Ransohoff, R. M. (2006). The expression and function of chemokines involved in CNS inflammation. Trends Pharmacol. Sci. 27, 48–55. doi: 10.1016/j.tips.2005.11.002
Vélez-Fort, M., Audinat, E., and Angulo, M. C. (2012). Central role of GABA in neuron-glia interactions. Neuroscientist 18, 237–250. doi: 10.1177/1073858411403317
Wesch, D., Beetz, S., Oberg, H. H., Marget, M., Krengel, K., and Kabelitz, D. (2006). Direct costimulatory effect of TLR3 ligand poly(I:C) on human gamma delta T lymphocytes. J. Immunol. 176, 1348–1354. doi: 10.4049/jimmunol.176.3.1348
Keywords: glia, TLR, multiple sclerosis, GABA, baclofen, inflammation, innate immunity
Citation: Crowley T, Fitzpatrick J-M, Kuijper T, Cryan JF, O’Toole O, O’Leary OF and Downer EJ (2015) Modulation of TLR3/TLR4 inflammatory signaling by the GABAB receptor agonist baclofen in glia and immune cells: relevance to therapeutic effects in multiple sclerosis. Front. Cell. Neurosci. 9:284. doi: 10.3389/fncel.2015.00284
Received: 14 May 2015; Accepted: 10 July 2015;
Published: 28 July 2015.
Edited by:
Carlos Barcia, Universitat Autònoma de Barcelona, SpainCopyright © 2015 Crowley, Fitzpatrick, Kuijper, Cryan, O’Toole, O’Leary and Downer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eric J. Downer, School of Medicine, Discipline of Physiology, Trinity Biomedical Sciences Institute, Trinity College, Dublin 2, Ireland,ZWRvd25lckB0Y2QuaWU=; Olivia F. O’Leary, Department of Anatomy and Neuroscience, Western Gateway Building, University College Cork, Cork, Ireland,by5vbGVhcnlAdWNjLmll
†Olivia F. O’Leary and Eric J. Downer are co-last authors.