 Eric S. Huseby1*
Eric S. Huseby1* Daisuke Kamimura2
Daisuke Kamimura2 Yasunobu Arima2
Yasunobu Arima2 Caitlin S. Parello1Katsuhiro Sasaki1†
Caitlin S. Parello1Katsuhiro Sasaki1† Masaaki Murakami2*
Masaaki Murakami2*- 1Department of Pathology, University of Massachusetts Medical School, Worcester, MA, USA
- 2Division of Molecular Neuroimmunology, Institute for Genetic Medicine and Graduate School of Medicine, Hokkaido University, Sapporo, Japan
Multiple Sclerosis (MS) is an inflammatory disease of the Central Nervous System (CNS) that causes the demyelination of nerve cells and destroys oligodendrocytes, neurons and axons. Historically, MS has been thought of as a T cell-mediated autoimmune disease of CNS white matter. However, recent studies have identified gray matter lesions in MS patients, suggesting that CNS antigens other than myelin proteins may be involved during the MS disease process. We have recently found that T cells targeting astrocyte-specific antigens can drive unique aspects of inflammatory CNS autoimmunity, including the targeting of gray matter and white matter of the brain and inducing heterogeneous clinical disease courses. In addition to being a target of T cells, astrocytes play a critical role in propagating the inflammatory response within the CNS induced NF-κB signaling. Here, we will discuss the pathophysiology of CNS inflammation mediated by T cell—glial cell interactions and its contributions to CNS autoimmunity.
Myelin-Specific T Cell Responses in MS and EAE
Multiple Sclerosis (MS), an inflammatory T cell-mediated autoimmune disease, is the most common neurological disease of young adults. MS causes the demyelination of nerve cells and destroys oligodendrocytes, neurons and axons (Frohman et al., 2006; Lassmann et al., 2007), with highly variable clinical manifestations. Such clinical manifestations of MS often include hyperreflexia, ataxia, spasticity and visual defects (Noseworthy et al., 2000; Keegan and Noseworthy, 2002; Hafler et al., 2005; Frohman et al., 2006; McFarland and Martin, 2007), and in some cases there are sensory defects and partial or complete paralysis. In the majority of patients, disease manifests as relapsing-remitting cycles of impairment, usually converting over time to a chronic progressive stage; 10–15% of patients present with disease that is progressive from onset (Sospedra and Martin, 2005; Frohman et al., 2006; McFarland and Martin, 2007; Steinman, 2009).
MS is thought to be primarily a CD4 T cell-mediated disease. Susceptibility to MS is genetically linked to major histocompatibility complex (MHC) genes and genes associated with T cell activation and homeostasis; however, the strongest genetic linkage occurs with certain alleles of MHC class II, which suggests a direct relationship between autoreactive CD4+ T cells and MS disease development in humans (Hillert and Olerup, 1993; Fogdell-Hahn et al., 2000; Sospedra and Martin, 2005). CD4+ T cells, in particular those that secrete IL-17, are considered to play an important role in the induction of central nervous system (CNS) autoimmunity (Korn et al., 2009). The identification of genes involved in CD4 T-cell differentiation and activation through genome wide association studies (GWAS) have further supported a role for CD4 T cells in the pathogenesis of MS (Patsopoulos et al., 2011).
The ability of myelin-reactive CD4 T cells to cause experimental autoimmune encephalomyelitis (EAE) further supports the hypothesis that myelin-reactive CD4 T cells have a central role in MS disease pathogenesis (Kuchroo et al., 2002; Sospedra and Martin, 2005; Ercolini and Miller, 2006; Hafler et al., 2007; Goverman, 2009; Steinman, 2009). MS-like clinical symptoms can be induced in animals by immunization with CNS proteins, as well as peptides derived from these CNS proteins, including myelin basic protein (MBP), proteolipid protein (PLP) and myelin oligodendrocyte glycoprotein (MOG; Ben-Nun et al., 2014). In addition, the adoptive transfer of activated CNS protein-specific CD4 T cells into naïve mice can induce paralytic diseases, allowing for in vivo study of the migratory behavior of pathogenic T cells (Jäger et al., 2009; Arima et al., 2012; Odoardi et al., 2012). However, it is unlikely that CD4 T cells are the sole mediators of disease pathogenicity, as treatments specifically targeting these cells limit neither the rate of disease relapses nor the formation of new lesions. In contrast, therapies that deplete or inhibit CNS infiltration of all lymphocyte subsets have been more successful (Lindsey et al., 1994; van Oosten et al., 1996; Rice et al., 2005).
Accumulating evidence strongly suggests that CD8 T cells also contribute to MS disease. Studies have shown that CD8 T cells are found in MS plaques—these cells are often oligoclonal, accumulate over time and can outnumber CD4 T cells regardless of the stage of activity or disease (Booss et al., 1983; Traugott et al., 1983; Hauser et al., 1986; Babbe et al., 2000; Lucchinetti et al., 2000; Frohman et al., 2006; Lassmann et al., 2007; Huseby et al., 2012). Though the antigen specificity of CNS infiltrating CD8 T cells remains unclear, a role for CD8 T cells in MS is further supported by the finding that particular MHC class I alleles can contribute to disease susceptibility (Cree et al., 2010; Healy et al., 2010).
Both a pathogenic or protective role for CNS-infiltrating CD8 T cells has been proposed. Myelin-specific CD8 T cells that are capable of killing neuronal cells in vitro have been isolated from MS patients (Tsuchida et al., 1994; Dressel et al., 1997; Medana et al., 2001; Crawford et al., 2004; Zang et al., 2004), which supports the hypothesis that CD8 T cells play a pathogenic role in the MS disease process. Further in support of this hypothesis, CD8 T cells specific for myelin proteins, including MBP, MOG, and PLP, have been shown to be pathogenic in several animal models of CNS disease (Huseby et al., 2001a; Sun et al., 2001; Ford and Evavold, 2005; Friese et al., 2008; Anderson et al., 2012). The clinical symptoms induced by such CNS-reactive CD8 T cells can be diverse. For example, mice carrying activated MBP-specific CD8 T cells succumb to a non-paralytic, acute demyelinating CNS autoimmunity that is clinically and histologically different than those of classic CD4-EAE. These atypical-EAE disease pathologies have similarities to MS patients with upper motor neuron disease (Huseby et al., 2001a). In contrast, experiments with MOG- and PLP-specific CD8 T cells resulted in CNS disease symptoms similar to classical EAE (Sun et al., 2001; Ford and Evavold, 2005; Friese et al., 2008; Anderson et al., 2012). These data suggest that myelin-specific CD8 T cells may contribute to some of the disease heterogeneity observed in MS patients.
Conversely, other studies have suggested that CD8 T cells may be suppressive during the MS disease process. CD8 T cell clones that can lyse myelin-specific CD4 T cells have been detected in MS patients (Chou et al., 1992; Zhang et al., 1993; Correale et al., 2000), and longitudinal magnetic resonance imaging (MRI) analysis has shown a negative correlation between the percentage of Tc2 cytokine-producing CD8 T cells in the periphery of MS patients and the development of lesions (Killestein et al., 2003). Moreover, protective MHC class I alleles have been identified through GWA studies, suggesting a relationship between autoreactive regulatory CD8+ T cells and MS disease development (International Multiple Sclerosis Genetics Consortium et al., 2011). In animal models, early studies found that polyclonal CD8 T cells can limit disease severity and relapses of CD4 T cell-mediated EAE (Jiang et al., 1992; Koh et al., 1992). The ability of CD8 T cells to regulate CNS autoimmune disease may occur by CD8 T cells targeting activated CD4 T cells through the recognition of peptide displayed on MHC class I and Ib molecules, as well as by secreting IL-10 and other anti-inflammatory soluble mediators (Jiang and Chess, 2006; Goverman, 2009; Kim and Cantor, 2011; Ortega et al., 2013). Thus, different subsets of CD8 T cells, like their CD4 counterparts, likely play pathogenic and immuno-regulatory roles in MS (Huseby et al., 2012).
Gray Matter Lesions in MS and EAE
MS has traditionally been thought of as a disease that targets myelin proteins within the white matter of the CNS. Recent findings indicate, however, that this may not always be the case. Using advanced MRI techniques, multiple investigators have identified gray matter lesions in MS patients that appear at the earliest stages of disease and accumulate over time (Lucchinetti et al., 2000, 2011; Peterson et al., 2001; Bo et al., 2003; Frohman et al., 2006; Calabrese et al., 2007; Lassmann et al., 2007; Fisher et al., 2008; Ontaneda et al., 2012). The presence of T cells within gray matter lesions of MS patients suggests that T cells reactive to antigens other than myelin proteins may contribute to MS disease progression. One potential cellular target of gray matter disease is astrocytes, which reside within the white and gray matter of the CNS. Astrocytes normally express low levels of MHC, however levels increase during inflammation (Wong et al., 1984; Ransohoff and Estes, 1991; De Keyser et al., 2010).
Autoreactive T cells must avoid negative selection within the thymus and be exported to the peripheral T cell repertoire in order to contribute to the CNS autoimmune disease process. Though myelin proteins, the prototypical targets of encephalogenic CD4 T cells, are primarily expressed behind the blood-brain barrier, some myelin peptide epitopes are expressed and presented in the thymus. Developing T cells that are reactive to these ligands can be subject to thymic deletion or be skewed towards low avidity or suppressive responses. These findings have lead to a differential avidity model for the development of encephalogenic T cells: strong avidity T cells targeting myelin epitopes that are presented in the thymus undergo negative selection whereas weak avidity T cells that target these same epitopes or strong avidity T cells that target myelin epitopes that are only expressed within the CNS are exported into the mature T cell repertoire and can induce autoimmunity (Liu et al., 1995; Harrington et al., 1998; Targoni and Lehmann, 1998; Huseby et al., 1999, 2001b; Klein et al., 2000; Kuchroo et al., 2002). The expectation is that T cells which target astrocytes or other CNS cell types will follow similar rules for development as those identified for T cells that target myelin.
Two proteins predominately expressed in astrocytes, Glial fibrillary acidic protein (GFAP) and S100β, have been studied as targets for autoreactive T cells. GFAP, an intermediate filament protein, is an archetypal astrocyte-specific antigen that is expressed throughout the gray matter and white matter of the brain and spinal cord (Middeldorp and Hol, 2011). GFAP is also expressed in some peripheral tissues including the thymus, intestine and pancreas, though expression levels are lower in these tissue types (Zelenika et al., 1995). In MS lesions, the expression level of GFAP increases and peptides derived from GFAP are presented by MHC class I and class II molecules (Nait-Oumesmar et al., 2007; Fissolo et al., 2009; Linker et al., 2009). S100β, a calcium binding protein, is also expressed within astroglia present within the gray and white matter of the CNS (Zimmer et al., 1995). Although both proteins are also expressed outside of the CNS, including at a low level within the thymus, T cell responses to these proteins indicate that immune tolerance towards these antigens is incomplete.
The adoptive transfer of CD4+ T cells reactive to GFAP or to S100β into rodents induces a strong inflammatory response within the spinal cord and throughout the entire CNS, including the cerebral cortex and the retina of the eye, with particularly severe inflammation observed in the gray matter (Kojima et al., 1994, 1997). These experiments demonstrate that T cell responses to non-myelin antigens are capable of being pathogenic in models of CNS autoimmunity. Compellingly, CD4+ S100β-specific T cells have been isolated from MS patients, as well as from healthy controls, indicating astrocyte-specific T cells are present in the mature T cell repertoire and may contribute to the disease process (Schmidt et al., 1997).
GFAP-Specific CD8 T Cells can Induce Relapsing/Remitting CNS Autoimmunity
The observation that CD8 T cells are present within gray matter lesions of MS patients (Peterson et al., 2001; Bo et al., 2003; Calabrese et al., 2007; Lassmann et al., 2007; Fisher et al., 2008; Lucchinetti et al., 2011; Ontaneda et al., 2012) inspired us to study astrocyte-specific CD8 T cells. We chose the astrocyte protein GFAP as the target antigen because GFAP expression and GFAP-peptide presentation by MHC class I and II molecules are increased within MS lesions (Nait-Oumesmar et al., 2007; Fissolo et al., 2009; Linker et al., 2009). Furthermore, although GFAP-specific T cells isolated from MS patients have not been studied, GFAP-specific CD8 T cells have been isolated from patients with type 1 diabetes, indicating that human T cells with this reactivity pattern populate the peripheral T cell repertoire (Standifer et al., 2006). CD8 T cells that target astrocytes and neurons have also been suggested in Rasmussen encephalitis (Schwab et al., 2009).
We have recently found that C57BL/6 mice carry CD8 T cells reactive to GFAP264–272 presented by H2-Db. We constructed TCR Tg mice expressing the GFAP-specific CD8 T cell clone, BG1 (BG1 mice), to follow the fate of naïve GFAP-specific T cells. To determine if BG1 mice maintain quiescence to GFAP over their lifetime, a cohort of WT, Rag1−/− and Gfap−/− BG1 mice were analyzed for clinical signs of CNS disease as they aged. We observed that BG1 mice do not maintain ignorance of GFAP: ~50% of WT BG1 mice and 100% of Rag−/− BG1 mice succumb to spontaneous clinical signs of CNS autoimmunity by 6 months of age. The majority of diseased BG1 mice develop balancing defects, lethargy, uneven gait and ataxia—such symptoms are referred to as atypical disease (Sasaki et al., 2014)—whereas some diseased mice also succumb to mild ascending flaccid paralysis—such symptoms are referred to as classical EAE (Stromnes and Goverman, 2006). The atypical disease symptoms that develop in BG1 mice reflect the locations within the CNS that is targeted; BG1 mice develop lesions showing prominent glial responses within the cerebellum, mid-brain and spinal cord early in a spontaneous disease course that includes both white matter and gray matter (Figure 1).
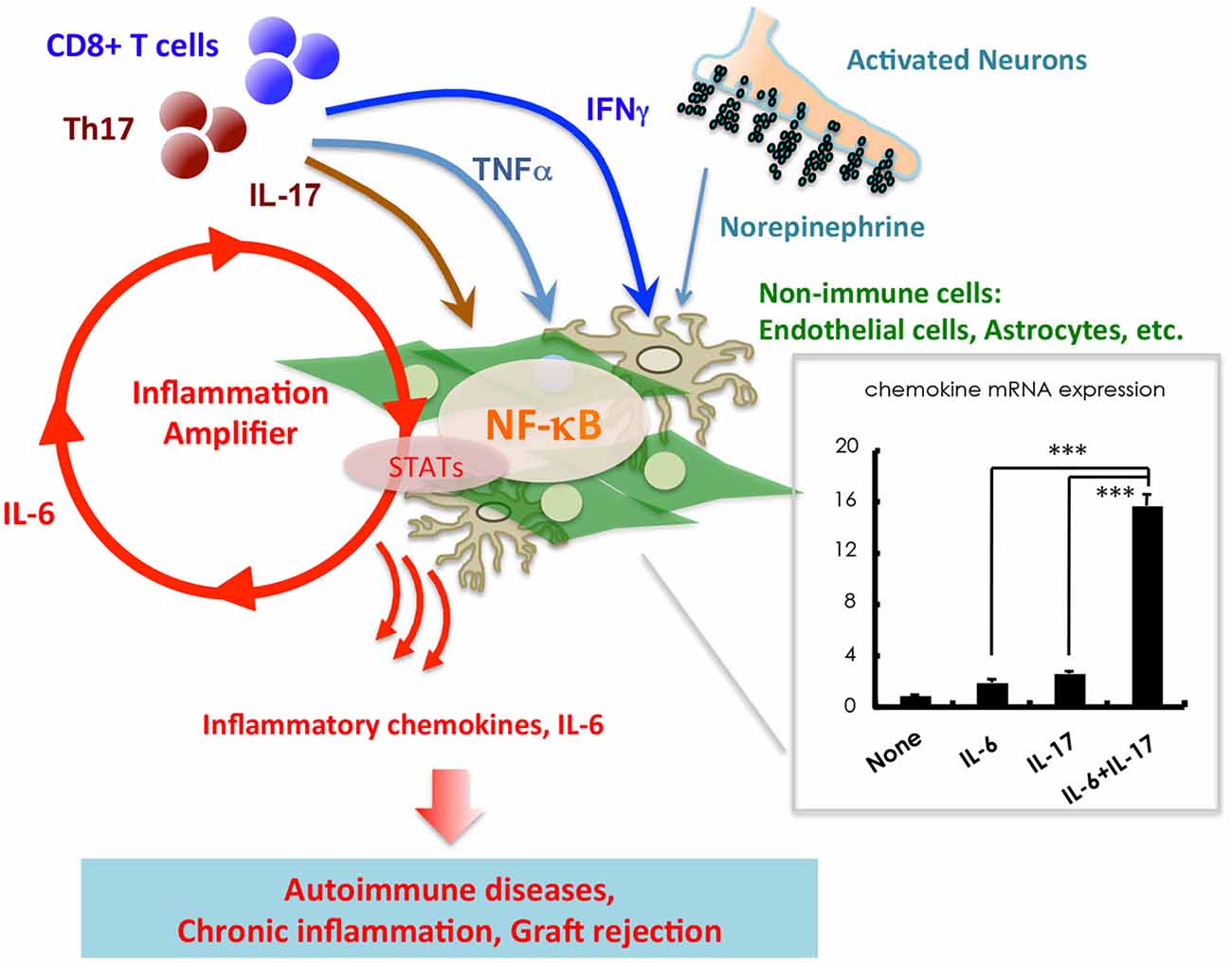
Figure 1. Stimulation of non-immune cells including endothelial cells and astrocytes with IL-17, TNFα, IFNγ, and IL-6 from T cells induces a synergistic effect on the production of inflammatory chemokines such as CCL20 and IL-6. An imaginary figure is shown. The synergistic effect requires the simultaneous activation of two transcription factors, NF-κB and STATs, in non-immune cells. Various soluble factors, including neurotransmitters from activated neurons, augment the inflammation amplifier by activating or sustaining the activation of NF-κB and STATs. Mean ± SD are shown. ***p < 0.001.
The BG1 CD8 effector T cell populations that target the CNS during spontaneous CNS disease phenotypically resemble anti-viral tissue-resident memory (TRM) cells that populate peripheral tissues following viral challenges (Schenkel and Masopust, 2014). Functionally, only low frequencies of CD8 T cells within the CNS are capable of producing IFNγ, IL-17 or granzyme B (GZB), indicating that many of the BG1 CD8 T cells present within the brain are not classic effector CD8 T cells. These data suggest that BG1 CD8 T cells that spontaneously enter into the brain interact with astrocytes to induce their differentiation into auto-reactive TRM, without gaining inflammatory cytokine expression or cytotoxic effector functions. Nevertheless, these auto-reactive TRM CD8 T cells can induce severe inflammation, glial responses and clinical disease symptoms. In contrast to CNS disease induced by auto-reactive TRM CD8 T cells, disease induced by classic IFNγ-producing pro-inflammatory CD8 T cells demonstrates severe ataxia and lethargy within 7 days, a disease pattern highly similar to those induced by in vitro or Vac-activated MBP-specific CD8 T cells (Huseby et al., 2001a; Sasaki et al., 2014). These differences in CNS disease pathologies suggest that different auto-reactive CD8 T cell lineages induce distinct CNS disease phenotypes, thereby contributing to MS disease heterogeneity. This hypothesis is consistent with studies of encephalogenic CD4 T cells. Through the observation of CD4 T cells responding to different neuroantigens and different priming protocols, it has been demonstrated that the effector lineage and activation status of CD4 T cells within the CNS influence the location of lesions within the CNS, the severity of the acute disease as well as the overall clinical outcome (Kawakami et al., 2004; Jäger et al., 2009; Pierson et al., 2012).
In both WT and Rag1−/− BG1 mice, spontaneous clinical symptoms begin as episodic bouts of functional impairment, with many mice displaying severe CNS dysfunction and then remitting to unobservable clinical symptoms. Rag1−/− BG1 mice, however, develop more severe bouts of disease, and have more relapses than WT BG1 mice, with the majority progressing to a chronic disease stage. The observed differences in the frequency and severity of spontaneous disease between WT BG1 and Rag1−/− BG1 mice suggests that GFAP-specific CD8 T cells are subject to extrinsic sources of immune regulation. To genetically map the lymphocytes that regulate GFAP-specific CD8 T cells, IAbβ−/− (MHC II-deficient) and μMT−/− (B cell-deficient) BG1 mice were generated. Spontaneous CNS disease in IAbβ−/− BG1 mice was similar in frequency and severity to WT BG1 mice. In contrast, μMT−/− BG1 mice were found to be highly susceptible to spontaneous CNS disease, with ~80% of μMT−/− BG1 mice developing chronic clinical disease, a fundamentally distinct disease course as compared to the relapsing-remitting disease most often observed in WT BG1 and Rag−/− BG1 mice (Sasaki et al., 2014). Thus, GFAP-specific CD8 T cell-mediated spontaneous relapsing-remitting and chronic disease is associated with the infiltration of tissue resident memory-like CD8 T cells into the CNS parenchyma and is regulated by polyclonal B cells. How B cells regulate CD8 T cell CNS autoimmunity, inflammation and disease remission is currently unknown.
Does the Inflammation Amplifier Regulate Relapsing/Remitting CNS Disease?
In addition to immune cells, we have demonstrated that non-immune cells, including vascular endothelial cells and glial cells, play critical roles in the induction of chronic inflammatory diseases such as EAE. Glial cells of the CNS can secrete large quantities of chemokines, growth factors and IL-6 in response to inflammatory stimuli, all of which can activate the NF-κB and STAT signaling pathways (Ogura et al., 2008; Atsumi et al., 2014). This induction of inflammation, mediated by IL-17, TNFα, IFNγ, IL-6 or various neurotransmitters, is synergistically enhanced when both the NF-κB and STAT signaling pathways are induced in glial cells. We termed this synergistic effect the inflammation amplifier (Atsumi et al., 2014). Importantly, clinical symptoms of EAE, and several additional chronic inflammatory diseases, are significantly improved in mice unable to activate the inflammation amplifier (Ogura et al., 2008; Arima et al., 2012; Lee et al., 2012; Murakami et al., 2013; Harada et al., 2015). These findings indicate that the inflammation amplifier has a central role in chronic inflammatory diseases.
The inflammation amplifier is regulated by the production of several neurotransmitters, including norepinephrine and ATP. These findings led us to hypothesize that the inflammation amplifier may link the onset and severity of CNS diseases to mental and physical stress. Indeed, regional neural activity created by gravity of the Earth on the soleus muscles enhances chemokine expressions within the CNS, resulting in inflammation occurring around the dorsal vessels of the fifth lumbar cord, during early stages of EAE. CNS inflammation and the upregulation of chemokine expression can similarly be induced artificially using electric stimulation of peripheral muscles, formally demonstrating that neuronal activity can regulate the inflammation amplifier in vivo (Arima et al., 2012). These phenomena have been termed the “gateway reflex” as these neural stimulations can create “gateways” for immune cells to enter into the CNS (Kamimura et al., 2013; Sabharwal et al., 2014).
Future Studies
The inflammation amplifier can be turned on or off in response to acute inflammation, as well as to mental and physical stress. Thus, the temporal regulation of NF-κB and STAT signaling pathways in glial cells may regulate episodic cycles of relapsing/remitting clinical disease in MS patients. Mechanistically, one way this may occur is by recruiting or limiting immune cell migration through the “gateway” present within the spinal cord and potentially through other sites within the brain. Clarifying these mechanisms, and identifying how different immune cell lineages and subsets respond to and regulate the inflammation amplifier, will provide insights into the pathogenesis of relapsing/remitting CNS diseases, and identify drug-targetable molecular pathways that can be exploited to minimize MS disease relapses.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Anderson, A. C., Chandwaskar, R., Lee, D. H., Sullivan, J. M., Solomon, A., Rodriguez-Manzanet, R., et al. (2012). A transgenic model of central nervous system autoimmunity mediated by CD4+ and CD8+ T and B cells. J. Immunol. 188, 2084–2092. doi: 10.4049/jimmunol.1102186
Arima, Y., Harada, M., Kamimura, D., Park, J. H., Kawano, F., Yull, F. E., et al. (2012). Regional neural activation defines a gateway for autoreactive T cells to cross the blood-brain barrier. Cell 148, 447–457. doi: 10.1016/j.cell.2012.01.022
Atsumi, T., Singh, R., Sabharwal, L., Bando, H., Meng, J., Arima, Y., et al. (2014). Inflammation amplifier, a new paradigm in cancer biology. Cancer Res. 74, 8–14. doi: 10.1158/0008-5472.CAN-13-2322
Babbe, H., Roers, A., Waisman, A., Lassmann, H., Goebels, N., Hohlfeld, R., et al. (2000). Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J. Exp. Med. 192, 393–404. doi: 10.1084/jem.192.3.393
Ben-Nun, A., Kaushansky, N., Kawakami, N., Krishnamoorthy, G., Berer, K., Liblau, R., et al. (2014). From classic to spontaneous and humanized models of multiple sclerosis: impact on understanding pathogenesis and drug development. J. Autoimmun. 54, 33–50. doi: 10.1016/j.jaut.2014.06.004
Bo, L., Vedeler, C. A., Nyland, H. I., Trapp, B. D., and Mørk, S. J. (2003). Subpial demyelination in the cerebral cortex of multiple sclerosis patients. J. Neuropathol. Exp. Neurol. 62, 723–732.
Booss, J., Esiri, M. M., Tourtellotte, W. W., and Mason, D. Y. (1983). Immunohistological analysis of T lymphocyte subsets in the central nervous system in chronic progressive multiple sclerosis. J. Neurol. Sci. 62, 219–232. doi: 10.1016/0022-510x(83)90201-0
Calabrese, M., De Stefano, N., Atzori, M., Bernardi, V., Mattisi, I., Barachino, L., et al. (2007). Detection of cortical inflammatory lesions by double inversion recovery magnetic resonance imaging in patients with multiple sclerosis. Arch. Neurol. 64, 1416–1422. doi: 10.1001/archneur.64.10.1416
Chou, Y. K., Henderikx, P., Jones, R. E., Kotzin, B., Hashim, G. A., Offner, H., et al. (1992). Human CD8+ T cell clone regulates autologous CD4+ myelin basic protein specific T cells. Autoimmunity 14, 111–119. doi: 10.3109/08916939209083129
Correale, J., Lund, B., McMillan, M., Ko, D. Y., McCarthy, K., and Weiner, L. P. (2000). T cell vaccination in secondary progressive multiple sclerosis. J. Neuroimmunol. 107, 130–139. doi: 10.1016/s0165-5728(00)00235-6
Crawford, M. P., Yan, S. X., Ortega, S. B., Mehta, R. S., Hewitt, R. E., Price, D. A., et al. (2004). High prevalence of autoreactive, neuroantigen-specific CD8+ T cells in multiple sclerosis revealed by novel flow cytometric assay. Blood 103, 4222–4231. doi: 10.1182/blood-2003-11-4025
Cree, B. A., Rioux, J. D., McCauley, J. L., Gourraud, P. A., Goyette, P., McElroy, J., et al. (2010). A major histocompatibility Class I locus contributes to multiple sclerosis susceptibility independently from HLA-DRB1*15:01. PLoS One 5:e11296. doi: 10.1371/journal.pone.0011296
De Keyser, J., Laureys, G., Demol, F., Wilczak, N., Mostert, J., and Clinckers, R. (2010). Astrocytes as potential targets to suppress inflammatory demyelinating lesions in multiple sclerosis. Neurochem. Int. 57, 446–450. doi: 10.1016/j.neuint.2010.02.012
Dressel, A., Chin, J. L., Sette, A., Gausling, R., Höllsberg, P., and Hafler, D. A. (1997). Autoantigen recognition by human CD8 T cell clones: enhanced agonist response induced by altered peptide ligands. J. Immunol. 159, 4943–4951.
Ercolini, A. M., and Miller, S. D. (2006). Mechanisms of immunopathology in murine models of central nervous system demyelinating disease. J. Immunol. 176, 3293–3298. doi: 10.4049/jimmunol.176.6.3293
Fisher, E., Lee, J. C., Nakamura, K., and Rudick, R. A. (2008). Gray matter atrophy in multiple sclerosis: a longitudinal study. Ann. Neurol. 64, 255–265. doi: 10.1002/ana.21436
Fissolo, N., Haag, S., de Graaf, K. L., Drews, O., Stevanovic, S., Rammensee, H. G., et al. (2009). Naturally presented peptides on major histocompatibility complex I and II molecules eluted from central nervous system of multiple sclerosis patients. Mol. Cell. Proteomics 8, 2090–2101. doi: 10.1074/mcp.M900001-MCP200
Fogdell-Hahn, A., Ligers, A., Grønning, M., Hillert, J., and Olerup, O. (2000). Multiple sclerosis: a modifying influence of HLA class I genes in an HLA class II associated autoimmune disease. Tissue Antigens 55, 140–148. doi: 10.1034/j.1399-0039.2000.550205.x
Ford, M. L., and Evavold, B. D. (2005). Specificity, magnitude and kinetics of MOG-specific CD8+ T cell responses during experimental autoimmune encephalomyelitis. Eur. J. Immunol. 35, 76–85. doi: 10.1002/eji.200425660
Friese, M. A., Jakobsen, K. B., Friis, L., Etzensperger, R., Craner, M. J., McMahon, R. M., et al. (2008). Opposing effects of HLA class I molecules in tuning autoreactive CD8(+) T cells in multiple sclerosis. Nat. Med. 14, 1227–1235. doi: 10.1038/nm.1881
Frohman, E. M., Racke, M. K., and Raine, C. S. (2006). Multiple sclerosis—the plaque and its pathogenesis. N. Engl. J. Med. 354, 942–955. doi: 10.1056/nejmra052130
Goverman, J. (2009). Autoimmune T cell responses in the central nervous system. Nat. Rev. Immunol. 9, 393–407. doi: 10.1038/nri2550
Hafler, D. A., Compston, A., Sawcer, S., Lander, E. S., Daly, M. J., De Jager, P. L., et al. (2007). Risk alleles for multiple sclerosis identified by a genomewide study. N. Engl. J. Med. 357, 851–862. doi: 10.1056/nejmoa073493
Hafler, D. A., Slavik, J. M., Anderson, D. E., O’Connor, K. C., De Jager, P., and Baecher-Allan, C. (2005). Multiple sclerosis. Immunol. Rev. 204, 208–231. doi: 10.1111/j.0105-2896.2005.00240.x
Harada, M., Kamimura, D., Arima, Y., Kohsaka, H., Nakatsuji, Y., Nishida, M., et al. (2015). Temporal expression of growth factors triggered by epiregulin regulates inflammation development. J. Immunol. 194, 1039–1046. doi: 10.4049/jimmunol.1400562
Harrington, C. J., Paez, A., Hunkapiller, T., Mannikko, V., Brabb, T., Ahearn, M., et al. (1998). Differential tolerance is induced in T cells recognizing distinct epitopes of myelin basic protein. Immunity 8, 571–580. doi: 10.1016/s1074-7613(00)80562-2
Hauser, S. L., Bhan, A. K., Gilles, F., Kemp, M., Kerr, C., and Weiner, H. L. (1986). Immunohistochemical analysis of the cellular infiltrate in multiple sclerosis lesions. Ann. Neurol. 19, 578–587. doi: 10.1002/ana.410190610
Healy, B. C., Liguori, M., Tran, D., Chitnis, T., Glanz, B., Wolfish, C., et al. (2010). HLA B*44: protective effects in MS susceptibility and MRI outcome measures. Neurology 75, 634–640. doi: 10.1212/WNL.0b013e3181ed9c9c
Hillert, J., and Olerup, O. (1993). HLA and MS. Neurology 43, 2426–2427. doi: 10.1212/wnl.43.11.2426-a
Huseby, E. S., Huseby, P. G., Shah, S., Smith, R., and Stadinski, B. D. (2012). Pathogenic CD8 T cells in multiple sclerosis and its experimental models. Front. Immunol. 3:64. doi: 10.3389/fimmu.2012.00064
Huseby, E. S., Liggitt, D., Brabb, T., Schnabel, B., Ohlén, C., and Goverman, J. (2001a). A pathogenic role for myelin-specific CD8(+) T cells in a model for multiple sclerosis. J. Exp. Med. 194, 669–676. doi: 10.1084/jem.194.5.669
Huseby, E. S., Sather, B., Huseby, P. G., and Goverman, J. (2001b). Age-dependent T cell tolerance and autoimmunity to myelin basic protein. Immunity 14, 471–481. doi: 10.1016/s1074-7613(01)00127-3
Huseby, E. S., Ohlén, C., and Goverman, J. (1999). Cutting edge: myelin basic protein-specific cytotoxic T cell tolerance is maintained in vivo by a single dominant epitope in H-2k mice. J. Immunol. 163, 1115–1118.
International Multiple Sclerosis Genetics Consortium, Wellcome Trust Case Control Consortium, Sawcer, S., Hellenthal, G., Pirinen, M., Spencer, C. C., et al. (2011). Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 476, 214–219. doi: 10.1038/nature10251
Jäger, A., Dardalhon, V., Sobel, R. A., Bettelli, E., and Kuchroo, V. K. (2009). Th1, Th17 and Th9 effector cells induce experimental autoimmune encephalomyelitis with different pathological phenotypes. J. Immunol. 183, 7169–7177. doi: 10.4049/jimmunol.0901906
Jiang, H., and Chess, L. (2006). Regulation of immune responses by T cells. N. Engl. J. Med. 354, 1166–1176. doi: 10.1056/nejmra055446
Jiang, H., Zhang, S. I., and Pernis, B. (1992). Role of CD8+ T cells in murine experimental allergic encephalomyelitis. Science 256, 1213–1215. doi: 10.1126/science.256.5060.1213
Kamimura, D., Yamada, M., Harada, M., Sabharwal, L., Meng, J., Bando, H., et al. (2013). The gateway theory: bridging neural and immune interactions in the CNS. Front. Neurosci. 7:204. doi: 10.3389/fnins.2013.00204
Kawakami, N., Lassmann, S., Li, Z., Odoardi, F., Ritter, T., Ziemssen, T., et al. (2004). The activation status of neuroantigen-specific T cells in the target organ determines the clinical outcome of autoimmune encephalomyelitis. J. Exp. Med. 199, 185–197. doi: 10.1084/jem.20031064
Keegan, B. M., and Noseworthy, J. H. (2002). Multiple sclerosis. Annu. Rev. Med. 53, 285–302. doi: 10.1146/annurev.med.53.082901.103909
Killestein, J., Eikelenboom, M. J., Izeboud, T., Kalkers, N. F., Adèr, H. J., Barkhof, F., et al. (2003). Cytokine producing CD8+ T cells are correlated to MRI features of tissue destruction in MS. J. Neuroimmunol. 142, 141–148. doi: 10.1016/s0165-5728(03)00265-0
Kim, H. J., and Cantor, H. (2011). Regulation of self-tolerance by Qa-1-restricted CD8(+) regulatory T cells. Semin. Immunol. 23, 446–452. doi: 10.1016/j.smim.2011.06.001
Klein, L., Klugmann, M., Nave, K. A., Tuohy, V. K., and Kyewski, B. (2000). Shaping of the autoreactive T-cell repertoire by a splice variant of self protein expressed in thymic epithelial cells. Nat. Med. 6, 56–61. doi: 10.1038/71540
Koh, D. R., Fung-Leung, W. P., Ho, A., Gray, D., Acha-Orbea, H., and Mak, T. W. (1992). Less mortality but more relapses in experimental allergic encephalomyelitis in CD8−/− mice. Science 256, 1210–1213. doi: 10.1126/science.256.5060.1210
Kojima, K., Berger, T., Lassmann, H., Hinze-Selch, D., Zhang, Y., Gehrmann, J., et al. (1994). Experimental autoimmune panencephalitis and uveoretinitis transferred to the Lewis rat by T lymphocytes specific for the S100 beta molecule, a calcium binding protein of astroglia. J. Exp. Med. 180, 817–829. doi: 10.1084/jem.180.3.817
Kojima, K., Wekerle, H., Lassmann, H., Berger, T., and Linington, C. (1997). Induction of experimental autoimmune encephalomyelitis by CD4+ T cells specific for an astrocyte protein, S100 beta. J. Neural Transm. Suppl. 49, 43–51. doi: 10.1007/978-3-7091-6844-8_5
Korn, T., Bettelli, E., Oukka, M., and Kuchroo, V. K. (2009). IL-17 and Th17 Cells. Annu. Rev. Immunol. 27, 485–517. doi: 10.1146/annurev.immunol.021908.132710
Kuchroo, V. K., Anderson, A. C., Waldner, H., Munder, M., Bettelli, E., and Nicholson, L. B. (2002). T cell response in experimental autoimmune encephalomyelitis (EAE): role of self and cross-reactive antigens in shaping, tuning and regulating the autopathogenic T cell repertoire. Annu. Rev. Immunol. 20, 101–123. doi: 10.1146/annurev.immunol.20.081701.141316
Lassmann, H., Brück, W., and Lucchinetti, C. F. (2007). The immunopathology of multiple sclerosis: an overview. Brain Pathol. 17, 210–218. doi: 10.1111/j.1750-3639.2007.00064.x
Lee, J., Nakagiri, T., Oto, T., Harada, M., Morii, E., Shintani, Y., et al. (2012). IL-6 amplifier, NF-κB-triggered positive feedback for IL-6 signaling, in grafts is involved in allogeneic rejection responses. J. Immunol. 189, 1928–1936. doi: 10.4049/jimmunol.1103613
Lindsey, J. W., Hodgkinson, S., Mehta, R., Mitchell, D., Enzmann, D., and Steinman, L. (1994). Repeated treatment with chimeric anti-CD4 antibody in multiple sclerosis. Ann. Neurol. 36, 183–189. doi: 10.1002/ana.410360210
Linker, R. A., Brechlin, P., Jesse, S., Steinacker, P., Lee, D. H., Asif, A. R., et al. (2009). Proteome profiling in murine models of multiple sclerosis: identification of stage specific markers and culprits for tissue damage. PLoS One 4:e7624. doi: 10.1371/journal.pone.0007624
Liu, G. Y., Fairchild, P. J., Smith, R. M., Prowle, J. R., Kioussis, D., and Wraith, D. C. (1995). Low avidity recognition of self-antigen by T cells permits escape from central tolerance. Immunity 3, 407–415. doi: 10.1016/1074-7613(95)90170-1
Lucchinetti, C., Brüuck, W., Parisi, J., Scheithauer, B., Rodriguez, M., and Lassmann, H. (2000). Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann. Neurol. 47, 707–717. doi: 10.1002/1531-8249(200006)47:6<707::aid-ana3>3.0.co;2-q
Lucchinetti, C. F., Popescu, B. F., Bunyan, R. F., Moll, N. M., Roemer, S. F., Lassmann, H., et al. (2011). Inflammatory cortical demyelination in early multiple sclerosis. N. Engl. J. Med. 365, 2188–2197. doi: 10.1056/NEJMoa1100648
McFarland, H. F., and Martin, R. (2007). Multiple sclerosis: a complicated picture of autoimmunity. Nat. Immunol. 8, 913–919. doi: 10.1038/ni1507
Medana, I., Martinic, M. A., Wekerle, H., and Neumann, H. (2001). Transection of major histocompatibility complex class I-induced neurites by cytotoxic T lymphocytes. Am. J. Pathol. 159, 809–815. doi: 10.1016/s0002-9440(10)61755-5
Middeldorp, J., and Hol, E. M. (2011). GFAP in health and disease. Prog. Neurobiol. 93, 421–443. doi: 10.1016/j.pneurobio.2011.01.005
Murakami, M., Harada, M., Kamimura, D., Ogura, H., Okuyama, Y., Kumai, N., et al. (2013). Disease-association analysis of an inflammation-related feedback loop. Cell Rep. 3, 946–959. doi: 10.1016/j.celrep.2013.01.028
Nait-Oumesmar, B., Picard-Riera, N., Kerninon, C., Decker, L., Seilhean, D., Höglinger, G. U., et al. (2007). Activation of the subventricular zone in multiple sclerosis: evidence for early glial progenitors. Proc. Natl. Acad. Sci. U S A 104, 4694–4699. doi: 10.1073/pnas.0606835104
Noseworthy, J. H., Lucchinetti, C., Rodriguez, M., and Weinshenker, B. G. (2000). Multiple sclerosis. N. Engl. J. Med. 343, 938–952. doi: 10.1056/NEJM200009283431307
Odoardi, F., Sie, C., Streyl, K., Ulaganathan, V. K., Schläger, C., Lodygin, D., et al. (2012). T cells become licensed in the lung to enter the central nervous system. Nature 488, 675–679. doi: 10.1038/nature11337
Ogura, H., Murakami, M., Okuyama, Y., Tsuruoka, M., Kitabayashi, C., Kanamoto, M., et al. (2008). Interleukin-17 promotes autoimmunity by triggering a positive-feedback loop via interleukin-6 induction. Immunity 29, 628–636. doi: 10.1016/j.immuni.2008.07.018
Ontaneda, D., Hyland, M., and Cohen, J. A. (2012). Multiple sclerosis: new insights in pathogenesis and novel therapeutics. Annu. Rev. Med. 63, 389–404. doi: 10.1146/annurev-med-042910-135833
Ortega, S. B., Kashi, V. P., Tyler, A. F., Cunnusamy, K., Mendoza, J. P., and Karandikar, N. J. (2013). The disease-ameliorating function of autoregulatory CD8 T cells is mediated by targeting of encephalitogenic CD4 T cells in experimental autoimmune encephalomyelitis. J. Immunol. 191, 117–126. doi: 10.4049/jimmunol.1300452
Patsopoulos, N. A., Esposito, F., Reischl, J., Lehr, S., Bauer, D., Heubach, J., et al. (2011). Genome-wide meta-analysis identifies novel multiple sclerosis susceptibility loci. Ann. Neurol. 70, 897–912. doi: 10.1002/ana.22609
Peterson, J. W., Bö, L., Mörk, S., Chang, A., and Trapp, B. D. (2001). Transected neurites, apoptotic neurons and reduced inflammation in cortical multiple sclerosis lesions. Ann. Neurol. 50, 389–400. doi: 10.1002/ana.1123
Pierson, E., Simmons, S. B., Castelli, L., and Goverman, J. M. (2012). Mechanisms regulating regional localization of inflammation during CNS autoimmunity. Immunol. Rev. 248, 205–215. doi: 10.1111/j.1600-065x.2012.01126.x
Ransohoff, R. M., and Estes, M. L. (1991). Astrocyte expression of major histocompatibility complex gene products in multiple sclerosis brain tissue obtained by stereotactic biopsy. Arch. Neurol. 48, 1244–1246. doi: 10.1001/archneur.1991.00530240048017
Rice, G. P., Hartung, H. P., and Calabresi, P. A. (2005). Anti-alpha4 integrin therapy for multiple sclerosis: mechanisms and rationale. Neurology 64, 1336–1342. doi: 10.1212/01.wnl.0000158329.30470.d0
Sabharwal, L., Kamimura, D., Meng, J., Bando, H., Ogura, H., Nakayama, C., et al. (2014). The gateway reflex, which is mediated by the inflammation amplifier, directs pathogenic immune cells into the CNS. J. Biochem. 156, 299–304. doi: 10.1093/jb/mvu057
Sasaki, K., Bean, A., Shah, S., Schutten, E., Huseby, P. G., Peters, B., et al. (2014). Relapsing-remitting central nervous system autoimmunity mediated by GFAP-specific CD8 T cells. J. Immunol. 192, 3029–3042. doi: 10.4049/jimmunol.1302911
Schenkel, J. M., and Masopust, D. (2014). Tissue-resident memory T cells. Immunity 41, 886–897. doi: 10.1016/j.immuni.2014.12.007
Schmidt, S., Linington, C., Zipp, F., Sotgiu, S., de Waal Malefyt, R., Wekerle, H., et al. (1997). Multiple sclerosis: comparison of the human T-cell response to S100 beta and myelin basic protein reveals parallels to rat experimental autoimmune panencephalitis. Brain 120(Pt. 8), 1437–1445. doi: 10.1093/brain/120.8.1437
Schwab, N., Bien, C. G., Waschbisch, A., Becker, A., Vince, G. H., Dornmair, K., et al. (2009). CD8+ T-cell clones dominate brain infiltrates in Rasmussen encephalitis and persist in the periphery. Brain 132, 1236–1246. doi: 10.1093/brain/awp003
Sospedra, M., and Martin, R. (2005). Immunology of multiple sclerosis. Annu. Rev. Immunol. 23, 683–747. doi: 10.1146/annurev.immunol.23.021704.115707
Standifer, N. E., Ouyang, Q., Panagiotopoulos, C., Verchere, C. B., Tan, R., Greenbaum, C. J., et al. (2006). Identification of Novel HLA-A*0201-restricted epitopes in recent-onset type 1 diabetic subjects and antibody-positive relatives. Diabetes 55, 3061–3067. doi: 10.3410/f.1047982.498017
Steinman, L. (2009). A molecular trio in relapse and remission in multiple sclerosis. Nat. Rev. Immunol. 9, 440–447. doi: 10.1038/nri2548
Stromnes, I. M., and Goverman, J. M. (2006). Active induction of experimental allergic encephalomyelitis. Nat. Protoc. 1, 1810–1819. doi: 10.1038/nprot.2006.285
Sun, D., Whitaker, J. N., Huang, Z., Liu, D., Coleclough, C., Wekerle, H., et al. (2001). Myelin antigen-specific CD8+ T cells are encephalitogenic and produce severe disease in C57BL/6 mice. J. Immunol. 166, 7579–7587. doi: 10.4049/jimmunol.166.12.7579
Targoni, O. S., and Lehmann, P. V. (1998). Endogenous myelin basic protein inactivates the high avidity T cell repertoire. J. Exp. Med. 187, 2055–2063. doi: 10.1084/jem.187.12.2055
Traugott, U., Reinherz, E. L., and Raine, C. S. (1983). Multiple sclerosis. Distribution of T cells, T cell subsets and Ia-positive macrophages in lesions of different ages. J. Neuroimmunol. 4, 201–221. doi: 10.1016/0165-5728(83)90036-x
Tsuchida, T., Parker, K. C., Turner, R. V., McFarland, H. F., Coligan, J. E., and Biddison, W. E. (1994). Autoreactive CD8+ T-cell responses to human myelin protein-derived peptides. Proc. Natl. Acad. Sci. U S A 91, 10859–10863. doi: 10.1073/pnas.91.23.10859
van Oosten, B. W., Barkhof, F., Truyen, L., Boringa, J. B., Bertelsmann, F. W., von Blomberg, B. M., et al. (1996). Increased MRI activity and immune activation in two multiple sclerosis patients treated with the monoclonal anti-tumor necrosis factor antibody cA2. Neurology 47, 1531–1534. doi: 10.1212/wnl.47.6.1531
Wong, G. H., Bartlett, P. F., Clark-Lewis, I., Battye, F., and Schrader, J. W. (1984). Inducible expression of H-2 and Ia antigens on brain cells. Nature 310, 688–691. doi: 10.1016/s0163-4453(85)91430-6
Zang, Y. C., Li, S., Rivera, V. M., Hong, J., Robinson, R. R., Breitbach, W. T., et al. (2004). Increased CD8+ cytotoxic T cell responses to myelin basic protein in multiple sclerosis. J. Immunol. 172, 5120–5127. doi: 10.4049/jimmunol.172.8.5120
Zelenika, D., Grima, B., Brenner, M., and Pessac, B. (1995). A novel glial fibrillary acidic protein mRNA lacking exon 1. Brain Res. Mol. Brain Res. 30, 251–258. doi: 10.1016/0169-328x(95)00010-p
Zhang, J., Medaer, R., Stinissen, P., Hafler, D., and Raus, J. (1993). MHC-restricted depletion of human myelin basic protein-reactive T cells by T cell vaccination. Science 261, 1451–1454. doi: 10.1126/science.7690157
Keywords: T cell, autoimmunity, glial fibrillary acidic protein, multiple sclerosis, astrocytes, experimental autoimmune encephalomyelitis, cerebellum
Citation: Huseby ES, Kamimura D, Arima Y, Parello CS, Sasaki K and Murakami M (2015) Role of T cell—glial cell interactions in creating and amplifying central nervous system inflammation and multiple sclerosis disease symptoms. Front. Cell. Neurosci. 9:295. doi: 10.3389/fncel.2015.00295
Received: 01 June 2015; Accepted: 17 July 2015;
Published: 05 August 2015.
Edited by:
Carlos Barcia, Universitat Autònoma de Barcelona, SpainReviewed by:
Stefania Ceruti, Università degli Studi di Milano, ItalyRobert Weissert, University of Regensburg, Germany
Copyright © 2015 Huseby, Kamimura, Arima, Parello, Sasaki and Murakami. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eric S. Huseby, Department of Pathology, University of Massachusetts Medical School, Sherman Center, 368 Plantation Street, Worcester, MA 01655, USA,ZXJpYy5odXNlYnlAdW1hc3NtZWQuZWR1;
Masaaki Murakami, Division of Molecular Neuroimmunology, Institute for Genetic Medicine and Graduate School of Medicine, Hokkaido University, N15 W7, Kitaku, Sapporo 060-0815, Japan,bXVyYWthbWlAaWdtLmhva3VkYWkuYWMuanA=
†Present address: Katsuhiro Sasaki, Department of Molecular and Cellular Physiology, Graduate School of Medicine, Kyoto University, Kyoto, Japan