 Katherine Conant
Katherine Conant Megan Allen
Megan Allen Seung T. Lim
Seung T. Lim- Department of Neuroscience and Interdisciplinary Program in Neuroscience, Georgetown University Medical Center, Washington, DC, USA
Spatially localized proteolysis represents an elegant means by which neuronal activity dependent changes in synaptic structure, and thus experience dependent learning and memory, can be achieved. In vitro and in vivo studies suggest that matrix metalloproteinase and adamalysin activity is concentrated at the cell surface, and emerging evidence suggests that increased peri-synaptic expression, release and/or activation of these proteinases occurs with enhanced excitatory neurotransmission. Synaptically expressed cell adhesion molecules (CAMs) could therefore represent important targets for neuronal activity-dependent proteolysis. Several CAM subtypes are expressed at the synapse, and their cleavage can influence the efficacy of synaptic transmission through a variety of non-mutually exclusive mechanisms. In the following review, we discuss mechanisms that regulate neuronal activity-dependent synaptic CAM shedding, including those that may be calcium dependent. We also highlight CAM targets of activity-dependent proteolysis including neuroligin and intercellular adhesion molecule-5 (ICAM-5). We include discussion focused on potential consequences of synaptic CAM shedding, with an emphasis on interactions between soluble CAM cleavage products and specific pre- and post-synaptic receptors.
A Brief Overview of the Players: Excitatory Synapses, Metalloproteinases, and Cell Adhesion Molecules
Excitatory Synapses in the Central Nervous System (CNS)
Changes in the number, structure, and/or function excitatory glutamatergic synapses are critical to experience dependent plasticity (Moser et al., 1994; Kopec et al., 2006). In a simplified view, the majority of these synapses are dipartite structures consisting of pre-synaptic axon terminals from which transmitter is released and post-synaptic neurotransmitter receptor-bearing dendritic spines. The latter are small protrusions of varied size and shape that emerge from the dendritic shaft of glutamate-responsive neurons (Alvarez and Sabatini, 2007). Spines with relatively large diameter heads or a mushroom-like morphology tend to be comparatively more stable and to have an increased α-amino-3-hydroxy-5methyl-4-isoxazoleproprionic acid (AMPA) receptor (GluA) number (Matsuzaki et al., 2004; Kopec et al., 2006; Bourne and Harris, 2007; Matsuzaki, 2007; Kasai et al., 2010). Importantly, neuronal activity dependent spine head enlargement, with a concomitant increase in synaptic incorporation of GluAs, is thought to underlie lasting enhancement of synaptic transmission or long-term potentiation (LTP). In a more complex view, glutamatergic synapses can be appreciated as multipartite sites in which glial cell processes approximate pre- and post-synaptic contact sites. Glial cells, or their soluble products, may thus modulate the structural and functional dynamics of neurotransmission (Dityatev and Rusakov, 2011).
Metalloproteinases in the CNS
Metzincin proteases are zinc-dependent endopeptidases that include cell-secreted matrix metalloproteinases (MMPs) and transmembrane spanning adamalysins [a disintegrin and metalloproteinase (ADAMs)]. These proteases are increasingly appreciated as important effectors of brain function [reviewed in Rivera et al. (2010), Huntley (2012), Sonderegger and Matsumoto-Miyai (2014)]. Though a variety of MMPs and ADAMs are expressed in man, including more than 23 MMPs identified to date (Page-McCaw et al., 2007), it should be noted that a select subset is likely relevant to physiological and pathological CNS plasticity. Family members with well-described expression in neurons, astrocytes, or microglia include MMP-1, MMP-2, MMP-3, MMP-7, MMP-9, MMP-12, MMP-13, MMP-14, and a disintegrin and metalloproteinase-10 (ADAM-10; Yong et al., 2001; Van Hove et al., 2012a).
The cell regulates overall activity of specific MMP and ADAM family members at several levels including that of gene expression. At the transcriptional level, activator protein 1 (AP-1) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) increase the expression of varied family members such as MMP-9 (Ganguly et al., 2013; see also Table 1 of Berry et al., 2013 for more on transcription factors and select MMPs). In contrast the transcription factor Yin Yang 1 (YY1) directs histone modification to strongly repress transcription of MMP-9 (Rylski et al., 2008). While less is known about the regulation of ADAM family member expression, we do know that the promoter for ADAM10 contains several transcription factor binding-sites including a retinoic acid-responsive element where retinoic acid receptors and retinoic X receptors can bind and thereby activate gene expression (Prinzen et al., 2005; Tippmann et al., 2009). Retinoic acid receptors are present in synaptoneurosomes and dendrites and contribute to select forms of synaptic plasticity (Aoto et al., 2008; Groth and Tsien, 2008).
Post-translational regulation of MMPs and ADAMs is also important with respect to enzymatic activity. Since MMPs typically act on extracellular substrates, release mechanisms represent a potentially important point of control. In a study that activated fibrosarcoma cells with phorbol myristate acetate (PMA), it was shown that subsequent release of MMP-2 and -9 is soluble NSF attachment protein receptor (SNARE) dependent (Kean et al., 2009). The SNARE protein family is critical for calcium-dependent vesicular fusion and release from neurons (Gerber and Sudhof, 2002; Sudhof, 2013). Since neuronal activity can increase intracellular calcium through mechanisms including activation of voltage gated calcium channels, it is tempting to speculate that calcium-dependent MMP release could be facilitated with the same. Of interest, MMP-2 and MMP-9 containing vesicles are observed in the somatodendritic compartment and found in dendritic spines (Sbai et al., 2008; Wilczynski et al., 2008). Moreover, stimuli that may increase intra-neuronal calcium and can induce LTP, can also evoke local MMP-9 release (Wang et al., 2008).
With respect to transmembrane spanning ADAMs, localization is also regulated. For example, ADAM10 and ADAM17 are mainly associated with the endoplasmic reticulum (ER) and Golgi apparatus, with little protein present at the plasma membrane (Schlondorff et al., 2000; Gutwein et al., 2003). ADAM10 contains an ER retention signal at its C-terminus, suggesting that unidentified proteins are required for the ER exit and transport of this protease to the plasma membrane (Marcello et al., 2010, 2012). The binding of tetraspanins to ADAM10 (Xu et al., 2009; Prox et al., 2012) can promote ADAM10 exit from the ER. Synapse-associated protein-97 (SAP-97), a cargo protein involved in protein trafficking at excitatory synapses, can bind to proline-rich sequences in the cytosolic domain of ADAM10 (Marcello et al., 2012). N-methyl-D-aspartate receptor (GluN) activation has been shown to affect phosphorylation of SAP97, as well as the transport of ADAM10 from Golgi outposts to synaptic membranes (Saraceno et al., 2014).
The activation of appropriately localized proteases represents an additional point of control. In the case of MMPs, this is typically achieved following release from the cell through cleavage of the pro-domain by other metalloproteinases or plasmin (Nagase et al., 1990). Cleavage of the pro-domain disrupts a critical Cys-Zn2+ interaction that otherwise blocks substrate processing (Van Wart and Birkedal-Hansen, 1990). Non-proteolytic activation of MMPs, however, also occurs. For example, nitration or oxidation may alter tertiary structure to activate pro-forms (Gu et al., 2002). Though less well-studied, MMPs may also be active within the cell (Wang et al., 2002).
Finally, MMP activity can be quenched by processes including low density lipoprotein receptor dependent internalization (Hahn-Dantona et al., 2001), and non-covalent interactions with endogenous tissue inhibitors of metalloproteinases (TIMPs; Gardner and Ghorpade, 2003; Visse and Nagase, 2003; Brew and Nagase, 2010).
In terms of brain plasticity related mechanisms that impact MMP expression, release and/or activity, several stimuli or stressors have been studied (see Table 1 for a partial summary). These include seizure activity (Zhang et al., 1998; Szklarczyk et al., 2002), cytokines (Khuth et al., 2001; Ogier et al., 2005; Ben-Hur et al., 2006), neurotrophins (Kuzniewska et al., 2013), chronic stress (van der Kooij et al., 2014), spatial learning (Wright et al., 2003; Meighan et al., 2006), head trauma (Phillips and Reeves, 2001; Kim et al., 2005), cocaine (Brown et al., 2008; Smith et al., 2014), methamphetamine (Liu et al., 2008), modafinil (He et al., 2011), ischemia (Planas et al., 2001; Rivera et al., 2002), and viral infection of the central nervous system (CNS; Conant et al., 1999; Johnston et al., 2002; Patrick et al., 2002; Zhang et al., 2003). Upregulation of MMP-9 mRNA and enzymatic activity has also been documented in response to neuronal depolarization by KCl (Von Gertten et al., 2003) and kainate (Szklarczyk et al., 2002; Konopacki et al., 2007; Wilczynski et al., 2008; Rylski et al., 2009). MMP-9 levels are increased with LTP (Nagy et al., 2006), and MMP-9 mRNA can be transported to dendrites to undergo local translation and protein release following glutamate stimulation (Dziembowska et al., 2012). Brain-derived neurotrophin factor (BDNF) also upregulates MMP-9 at the mRNA, protein, and enzymatic activity level in dendrites. This process requires engagement of TrkB receptors with subsequent activation of extracellular signal-regulated kinase 1/2 (ERK1/2) and binding of c-Fos to the proximal MMP-9 promoter region (Kuzniewska et al., 2013). In contrast, MMP gene expression can be suppressed by factors including TGF-β, the anesthetic propofol (Zhang et al., 2013), retinoic acid (Osteen et al., 1996; Li et al., 2011; Ye et al., 2011), or sleep deprivation (Taishi et al., 2001).
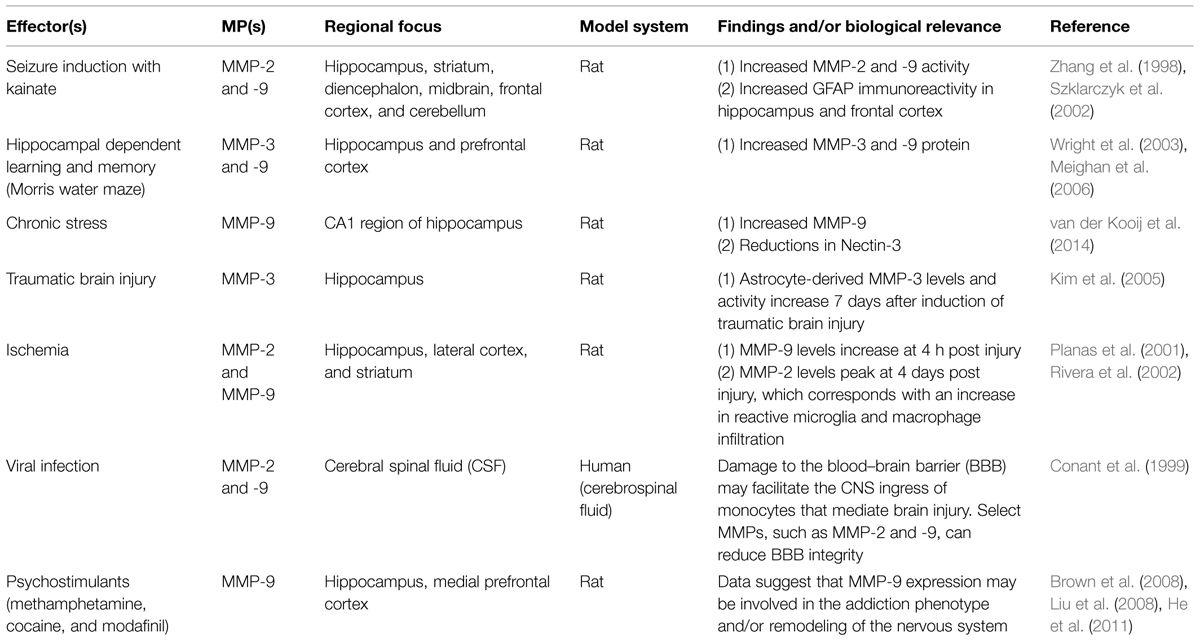
TABLE 1. Stimuli and stressors linked to altered MMP levels in the CNS.
While a wide variety of studies have examined MMP and ADAM expression in brain or brain-derived cell cultures as a function of specific stimuli, CNS changes have also been examined in a limited number of mutant mouse models. A summary of results from studies using this approach is shown in Table 2.
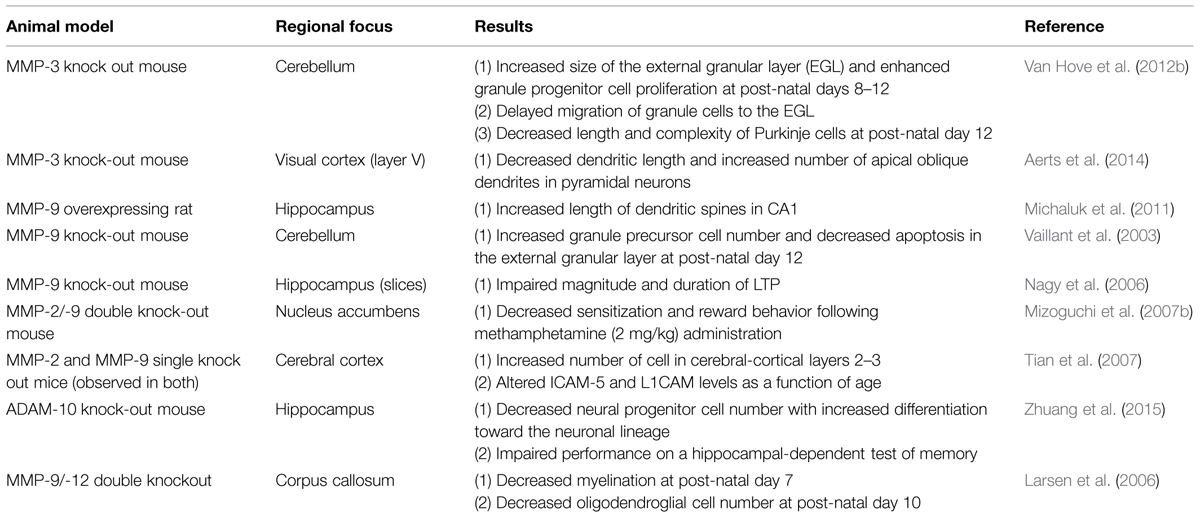
TABLE 2. Altered neuronal migration and/or plasticity in mutant mouse models.
CAMs in the CNS
Cell adhesion molecules represent transmembrane adhesion molecules expressed at cell contact sites including the synapse. These molecules typically belong to one of several superfamilies which include cadherins, neurexins/neuroligins, and Ig-domain containing members [reviewed in Benson and Huntley (2012)]. CAMs can mediate stable cell–cell junctions and select family members also play a role in the initiation of synapse formation. Cell surface levels of these molecules, as well as adhesive contact strength, are modified by processes including clathrin-dependent endocytosis (Kamiguchi and Lemmon, 2000). In addition, varied transmembrane CAMs have important intracellular interactions. For example, while integrin cytoplasmic tails do not possess endogenous kinase activity, they interact with critical effectors of intracellular protein phosphorylation cascades (Clark and Brugge, 1995). Finally, through cis-interactions, transmembrane CAMs may influence the localization of synaptic proteins. As a potential example, N-cadherin, GluN1 and L1 are found together in large multiprotein complexes (Husi et al., 2000) suggesting that GluN may be part of a membrane adhesion complex (Sheng and Lee, 2000).
Accumulating evidence demonstrates that disrupted CAM expression can influence experience dependent plasticity. For example, ablation of N-cadherin from excitatory forebrain synapses of post-natal mice is associated with an alteration in the composition of glutamatergic synapses, so that levels of the GluA1 subunit and PSD95 are diminished (Nikitczuk et al., 2014). Earlier work by the same group has shown that a conditional N-cadherin knockout causes a reduction in the maintenance, but not induction, of LTP (Bozdagi et al., 2010). These studies are of particular relevance in that conditional ablation of N-cadherin addresses potential confounds that might be associated with knockout effects on early brain development.
Neuron-specific deletion of dystroglycan, a transmembrane protein that links extracellular matrix and the cytoskeleton, also reduces LTP. Specifically, neuron-specific deletion of this protein is associated with a blunting of high frequency stimulation (HFS) induced LTP at CA3–CA1 synapses (Satz et al., 2010). Dystroglycan is expressed by varied cell types including glia, and glial expression of the molecule is involved in forebrain development (Satz et al., 2010).
A number of studies have also investigated LTP in mice that lack specific Ig-domain CAM family members [comprehensively reviewed in Dityatev et al. (2008)]. Neural cell adhesion molecule (NCAM) is a homophilic binding protein that is expressed on the surface of neurons and glia and has been implicated in neurite outgrowth and synaptic plasticity. Indeed, NCAM-deficient mice show impaired LTP in area CA3 (Cremer et al., 1998), and impaired LTP in NCAM knockouts can be rescued by increasing GluN dependent glutamate transmission (Kochlamazashvili et al., 2012). In related work, Cremer et al. (1998) studied mice with a targeted deletion of a polysialyltransferase that attaches polysialic acid (PSA) to NCAM, and that is expressed predominantly in post-natal life (Eckhardt et al., 2000). These animals were shown to have lower post-natal levels of PSA in the brain as well as impaired LTP in CA1 that is evident by 4 weeks of age. Mice that are deficient in ICAM-5, an additional Ig-domain family member expressed on excitatory neurons of the telencephalon (Oka et al., 1990; Benson et al., 1998), also show changes in glutamatergic synapses. These animals show an increase in the dendritic spine/filopodia ratio at P7, suggesting that full length ICAM-5 may delay spine maturation (Matsuno et al., 2006). Though full length ICAM-5 is gradually excluded from spines during their developmental maturation, it remains in approximately 60 percent of spines in adult hippocampal neurons (Sakurai et al., 1998; Matsuno et al., 2006). An antibody directed against ICAM-5, which would presumably disrupt adhesive interactions important to filopodial maintenance, inhibits LTP in rat hippocampus (Sakurai et al., 1998). In mouse hippocampus, however, LTP is relatively increased in an ICAM-5 null animal (Nakamura et al., 2001). Though confounds include antibody specificity, as well as developmental and compensatory effects in the knockout, results are of interest with respect to ICAM-5 as a potential modulator of glutamatergic function.
Specific neuroligin family members have also been investigated with respect to glutamatergic transmission. These are cell adhesion proteins on the post-synaptic membrane that mediate the formation and maintenance of synapses between neurons. Neuroligins act as ligands for β-Neurexins, which are located on the presynaptic membrane. Of particular interest is a study of an autism-associated point mutation in the neuroligin tail that was evaluated following generation of a knock-in mouse (Etherton et al., 2011). Whole-cell voltage-clamp recordings in hippocampal CA1 pyramidal neurons from the knock-in showed a decrease in mini excitatory post-synaptic current (mEPSC) frequency but not amplitude. Changes in GluA receptor subunit composition or presynaptic release possibility were excluded by additional studies, and it was suggested that the neuroligin-3 cytoplasmic tail modulates recruitment of GluAs to post-synaptic sites of excitatory synapses (Etherton et al., 2011).
In addition to in vitro and animal model based studies, human genetic studies are consistent with an important role for CAMs in neuroplasticity. Mutations in contactin-associated protein 2, which may promote neuronal circuit assembly during development (Anderson et al., 2012), predispose to autism. Moreover, polymorphisms in CAMs including cadherin 13 (Johnson et al., 2008; Uhl et al., 2014) are associated with addiction risk.
Synaptic CAMs: Perfectly Poised Substrates for Neuronal Activity Dependent Cleavage
Though a role for CAMs in processes such as LTP could be in whole or large part secondary to the function of full-length molecules, it should also be considered that synaptically localized CAMs represent especially attractive targets for neuronal activity dependent proteolysis. CAM cleavage could disrupt stable interactions with exogenous CAM ligands and/or cause additional effects, including generation of bioactive or dominant negative receptor fragments. Varied CAMs are expressed at synaptic contacts including N-cadherin, L1-CAM, ICAM-5, DSCAM, syndecan 2, syncam 2, and neuroligin (Benson et al., 1998; Peixoto et al., 2012; Sonderegger and Matsumoto-Miyai, 2014), and activity-dependent, membrane-proximal cleavage of these molecules is supported by in vitro studies that have demonstrated juxtamembrane shedding for specific family members (Peixoto et al., 2012; Sonderegger and Matsumoto-Miyai, 2014). Data from analysis of cerebrospinal spinal fluid samples also supports shedding of CAMs (Strekalova et al., 2006). In this case, shed CAMs likely access the interstitial space from where they in turn gain access to CSF. In one study, an increase in levels of soluble ICAM-5 ectodomain was detected in the CSF of patients with epilepsy and/or infection (Lindsberg et al., 2002; Tian et al., 2008). In related work, N terminal sequencing of NCAM fragments from the CSF of patients with schizophrenia was performed and a disease-associated increase in levels of ectodomain fragments observed (Vawter et al., 2001).
Regulated cleavage of synaptic CAMs can influence glutamatergic transmission through several non-mutually exclusive mechanisms including reduced synaptic stability, conversion of N-terminal CAM ectodomains into soluble effectors of plasticity, and increased generation of intracellular domains (ICDs) that influence transcription. With respect to ICD generation, it should be noted that ectodomain shedding of CAMs is frequently followed by intramembranous gamma secretase cleavage to generate specific C terminal fragments [reviewed in Jordan and Kreutz (2009)].
Activity Dependent Cleavage of Neuroligin and N-Cadherin
Though it has been suggested that ectodomain shedding is highly regulated with only 2% of cell surface proteins released by this process (Hayashida et al., 2010), emerging evidence suggests that neuronal activity dependent CAM shedding represents an important mechanism by which synaptic structure and function are modulated. For example, neuronal activity dependent cleavage of neuroligin-1 is triggered by GluN activation and dependent on MMP or ADAM activity (Peixoto et al., 2012; Suzuki et al., 2012). It occurs in a membrane proximal location and results in destabilization of neuroligin-1’s presynaptic partner, neurexin-1β. Destabilization of neurexin is in turn thought to reduce the probability of presynatic neurotransmitter release (Peixoto et al., 2012).
GluN agonists, as well as ADAM and MMP family members that are regulated in a neuronal activity dependent manner, have also been linked to N-cadherin shedding (Reiss et al., 2005; Uemura et al., 2007; Williams et al., 2010; Paudel et al., 2013; Porlan et al., 2014). One of many potential sequelae of this event is the associated generation of a C terminal fragment which is quickly processed by gamma secretase to generate a smaller intracellular fragment that destabilizes a protein critical for CREB dependent transcription (Marambaud et al., 2003).
Activity Dependent Cleavage of Ig-domain CAMs
In work related to a potential role of ICAM-5 shedding in developmental plasticity, it has been shown that long term treatment (16 h) of DIV 14 hippocampal neurons with 5 μM NMDA or AMPA stimulated an MMP-dependent increase in supernatant levels of shed ICAM-5. ICAM-5 is expressed on dendritic elements of excitatory/spiny neurons in the telenchephalon (Benson et al., 1998). Since full length ICAM-5 may be a negative regulator of filopodia-to-spine transition (Matsuno et al., 2006), these findings are consistent with the possibility that ICAM-5 shedding contributes to developmental spine maturation.
Studies related to the possibility that ICAM-5 cleavage may occur in a relatively rapid manner to influence activity dependent glutamatergic transmission in the adult CNS have also been performed. NMDA stimulation of cultured hippocampal neurons and high frequency tetanic stimulation of hippocampal slices have both been linked to relatively rapid MMP-dependent ICAM-5 shedding (Conant et al., 2010b). In cultured cells, appreciable release of soluble ICAM-5 into culture supernatants can be detected within 5 min of NMDA exposure (Conant et al., 2010b).
Additional studies have examined neuronal activity dependent cleavage of nectin-1, an Ig-like adhesion molecule expressed at puncta adherentia junctions in the CA3 pyramidal region of adult mouse hippocampus (Lim et al., 2012). Of interest, in vitro over-expression of cleavage resistant mutants of nectin 1 is associated with an increase in the density of dendritic spines (Lim et al., 2012). One possibility is that cleavage resistant mutants might lead to an increase in the stability of spines.
Elegant work on a related adhesion molecule, demonstrated enhanced MMP-9 dependent cleavage of nectin-3 in perisynaptic CA1 in the setting of chronic stress (van der Kooij et al., 2014). Intriguingly, inhibition of MMP-9 activity or GluN activation led to a reduction chronic stress related behavioral alterations.
IgLON family members, abundant GPI anchored transmembrane proteins, are also processed in a metalloproteinase dependent manner. The IgLON family is a subgroup of the immunoglobulin superfamily cell adhesion molecules (CAMs) and composed of limbic system-associated protein (LAMP), opioid binding cell adhesion molecule (OBCAM), neurotrimin (NTM) and Kilon. Long term treatment of hippocampal neurons with a broad spectrum MMP inhibitor and subsequent pull down of surface proteins demonstrated that inhibitor-treated neurons show increased levels of specific IgLON family members including NTM (Sanz et al., 2014).
Glutamate and MMP dependent shedding of synaptic cell adhesion molecule 2 (SynCAM-2) has also been described (Bajor et al., 2012), which is of interest given the role of this molecule in synapse organization and function (Biederer et al., 2002; Fogel et al., 2007).
Emerging and Future Studies of Neuronal Activity Dependent CAM Cleavage
Matrix metalloproteinases and ADAMs can also act on a variety of less traditional CAMs including nerve-glia antigen 2 (NG2), β-dystroglycan, and amyloid precursor protein (APP) and netrin-G ligand-3 (Ahmad et al., 2006; Michaluk et al., 2007; Lee et al., 2014; Sakry et al., 2014). There is evidence that these molecules are shed in a neuronal activity-dependent manner, and that they play a role in developmental and/or adult plasticity. For example, recent work suggests that glutamatergic transmission is altered in NG2 knockout animals (Sakry et al., 2014). Future studies will be necessary to further explore mechanisms by which shedding of these proteins can influence plasticity.
Future studies will also be necessary address issues related to shedding of dimers versus monomers, as well as issues of whether single nucleotide polymorphisms (SNPs) influence shedding. Results from recent work suggest that ADAM dependent shedding of neuregulin-1 requires prior dimerization (Hartmann et al., 2015), and analysis of soluble ICAM-1 in pleural fluid suggests that this molecule may also be shed as a dimer (Melis et al., 2003).
Future studies could additionally explore the question of whether post-translational modifications such as glycosylation can influence the cleavage and/or bioactivity of protease-generated CAM fragments. And finally, unbiased proteomics could be utilized to examine interactions between shed CAMs and other proteins in the background of select physiological and pathological processes.
Metalloproteinases and Synaptic Transmission
Despite their ability to stimulate effects that could both enhance or depress neurotransmission, the majority of studies support a view in which non-pathological neuronal activity stimulates an MMP dependent enhancement of long term memory and its correlates. For example, several groups have demonstrated that MMP inhibitors reduce LTP stimulated by HFS and/or theta burst stimulation (TBS; Nagy et al., 2006; Meighan et al., 2007; Conant et al., 2010b). Inhibition of MMP activity also reduces chemical LTP (cLTP) associated increases in the firing rate and bursting of dissociated cultures of primary hippocampal neurons (Niedringhaus et al., 2012).
Consistent with their effects on hippocampal LTP, varied biochemical and behavioral studies support a role for MMPs in hippocampal dependent learning and/or memory. For example, knockout of MMP-9 impairs contextual fear conditioning (Nagy et al., 2007). Interestingly, in wild-type animals contextual fear conditioning increases hippocampal MMP-9 protein levels as well as MMP-9 dependent cleavage of dystroglycan (Ganguly et al., 2013). These data suggest that MMP-9 plays a role in hippocampal memory association and/or retention. It has also been shown that hippocampal MMP-3 and -9 mRNA levels are increased with Morris water maze (MWM) training, as are levels of active MMP-3 and -9 protein (Meighan et al., 2006). Moreover, treatment with the non-competitive GluN antagonist, MK801, reduces training-associated increases in specific MMP levels, as well as post-training performance assessed by latency to reach platform. Intra-hippocampal or intra-cerebral ventricular injection of a broad-spectrum chemical MMP inhibitor, as compared to artificial CSF control injection, can also reduce time spent in the target quadrant during the MWM probe trial (Meighan et al., 2006). Of interest with respect to anesthetic-modulation of learning and memory, it has been shown that while MWM training can induce a gradual increase in pro- and active-MMP-9, propofol can reduce this increase and also disrupt spatial memory retention 24 h after training (Zhang et al., 2013). In contrast, the wake promoting agent modafinil increases MMP-9 expression in dorsal hippocampal CA3 in a model of REM sleep deprivation (He et al., 2011). In this same model, modafinil increases synapsin 1 expression in an MMP-9 dependent manner. In addiction-related plasticity work, it has been shown that context dependent learning of nicotine induced conditioned place preference (CPP) is associated with an increase in hippocampal MMP-2, -3, and -9 expression, and that exposure to a chemical MMP inhibitor during nicotine induced CPP training can block CPP acquisition (Natarajan et al., 2013). In addition, methamphetamine-induced behavioral sensitization is reduced in mice lacking MMP-2 or MMP-9 (Mizoguchi et al., 2007b).
Matrix metalloproteinase activity can also contribute to enhanced glutamatergic transmission in regions including striatum and amygdala. For example, a chemical MMP inhibitor can disrupt reconsolidation of a fear memory associated with a conditioned stimulus that is independent of contextual cues (Brown et al., 2009). In studies with MMP-9 null mice, Kaczmarek and colleagues have shown that MMP activity in the central amygdala is required for appetitive but not aversive learning (Knapska et al., 2013). In recent work related to cocaine and MMP levels in nucleus accumbens core, an increase in gelatinase activity as detected by in situ zymography was detected along neuronal soma and dendrites (Smith et al., 2014). AMPA/NMDA ratios were also increased in medium spiny neurons in cocaine extinguished rats and further increased by cue-induced reinstatement, in an MMP dependent manner. MSNs also showed MMP dependent changes in MSN spine head diameter and/or number in cocaine extinguished and reinstated animals (Smith et al., 2014).
Mechanisms by which MMPs Influence Neurotransmission; A Focus on CAM Cleavage as a Means to Generate Integrin-Binding Ligands
In terms of the mechanisms by which MMPs modulate actin and spine dynamics to enhance glutamatergic transmission, it should be noted that despite their potential to act on varied substrates such as proneurotrophins (Lee et al., 2001), evidence suggests that their ability to enhance LTP is β1 integrin dependent (Nagy et al., 2006; Meighan et al., 2007). Since changes in the number and size of dendritic spines are thought to underlie LTP, Huntley and colleagues (Wang et al., 2008) also monitored spine size and EPSPs simultaneously in hippocampal neurons with combined 2-photon time-lapse imaging and whole-cell recordings. These investigators observed that persistent spine enlargement and synaptic potentiation required both MMP activity and β1 integrins (Wang et al., 2008). In related studies, we have previously shown that a β1 integrin blocking antibody prevents cLTP associated increases in the overall firing rate of hippocampal-derived neurons (Niedringhaus et al., 2012).
Integrin signaling, and signaling through β1 containing integrins in particular, has been well-associated with changes in dendritic and spine actin dynamics (Huang et al., 2006; Shi and Ethell, 2006). The majority of hippocampal integrin heterodimers contain a β1 subunit (Pinkstaff et al., 1998; Chan et al., 2006; Huntley, 2012). In addition, β1 integrins are expressed on dendrites (Mortillo et al., 2012) and implicated in the regulation of post-natal dendritic arbor and synapse density (Warren et al., 2012). Antagonism of β1 integrin signaling by function blocking antibodies or GRGDS peptide administration is associated with a decay of LTP, and hippocampal infusion of function blocking antibodies to β1 blocks formation of long-term object location memory (Babayan et al., 2012). Integrin like kinase is also involved in the induction and maintenance of cocaine sensitization, and its silencing prevents sensitization-associated serine-845 phosphorylation of GluA1 (Chen et al., 2010). Though a role for integrin signaling has been implicated in potentiated synaptic activity (Bernard-Trifilo et al., 2005; Nagy et al., 2006; Meighan et al., 2007), the identity of physiologically relevant ligands and important mechanisms by which these ligands are generated has yet to be fully explored.
Recent work is consistent with the possibility that MMP-dependent CAM shedding represents a potential mechanism by which excitatory transmission generates integrin-binding ligands. As opposed to larger ECM components, CAM ectodomains may be relatively soluble. Indeed, previous studies have shown that cleavage of relatively large molecules does not necessarily disrupt three dimensional integrity of the same (Huganir and Racker, 1980). Numerous studies, including those that have demonstrated the integrity of CAM N-terminal fragments in spinal fluid and tissue, support the concept of ectodomain solubility and stability (Lindsberg et al., 2002).
Cell adhesion molecule ectodomains are known to possess integrin-binding motifs and indeed stable CAM/integrin interactions have been described (Conant et al., 2010a; Kelly et al., 2013; Ning et al., 2013). CAM ectodomains can also stimulate integrin dependent signaling. For example, shedding of the L1 adhesion molecule has been shown to stimulate integrin dependent cell migration (Mechtersheimer et al., 2001). In addition, we have observed that soluble ICAM-5 can stimulate a β1 dependent increase in action potential frequency in cultured hippocampal neurons (Niedringhaus et al., 2012). We also observe co-immunoprecipiation of both full length and shed N terminal ICAM-5 with β1 in hippocampal lystates from methamphetamine challenged mice, suggesting that the shed ectodomain may interact with β1 in vivo (Conant et al., 2010a). NCAM and NG2 can also interact with integrins, and though not yet tested for effects on integrin-dependent neurotransmission, dorsal hippocampal injection of PSA-NCAM has been shown to partially restore impaired contextual memory in NCAM deficient mice (Senkov et al., 2006). Of interest, mice that overexpress the NCAM ectodomain show memory impairments that are similar to those observed in the knockout (Pillai-Nair et al., 2005). This is consistent with a dominant negative effect as well as the possibility that dysregulated ectodomain shedding is deleterious, and it underlines our need to better explore the bioactivity of CAM ectodomains in both physiological and pathological conditions. A schematic of CAM ectodomain shedding from glia and/or neurons, with subsequent integrin binding, is shown in Figure 1.
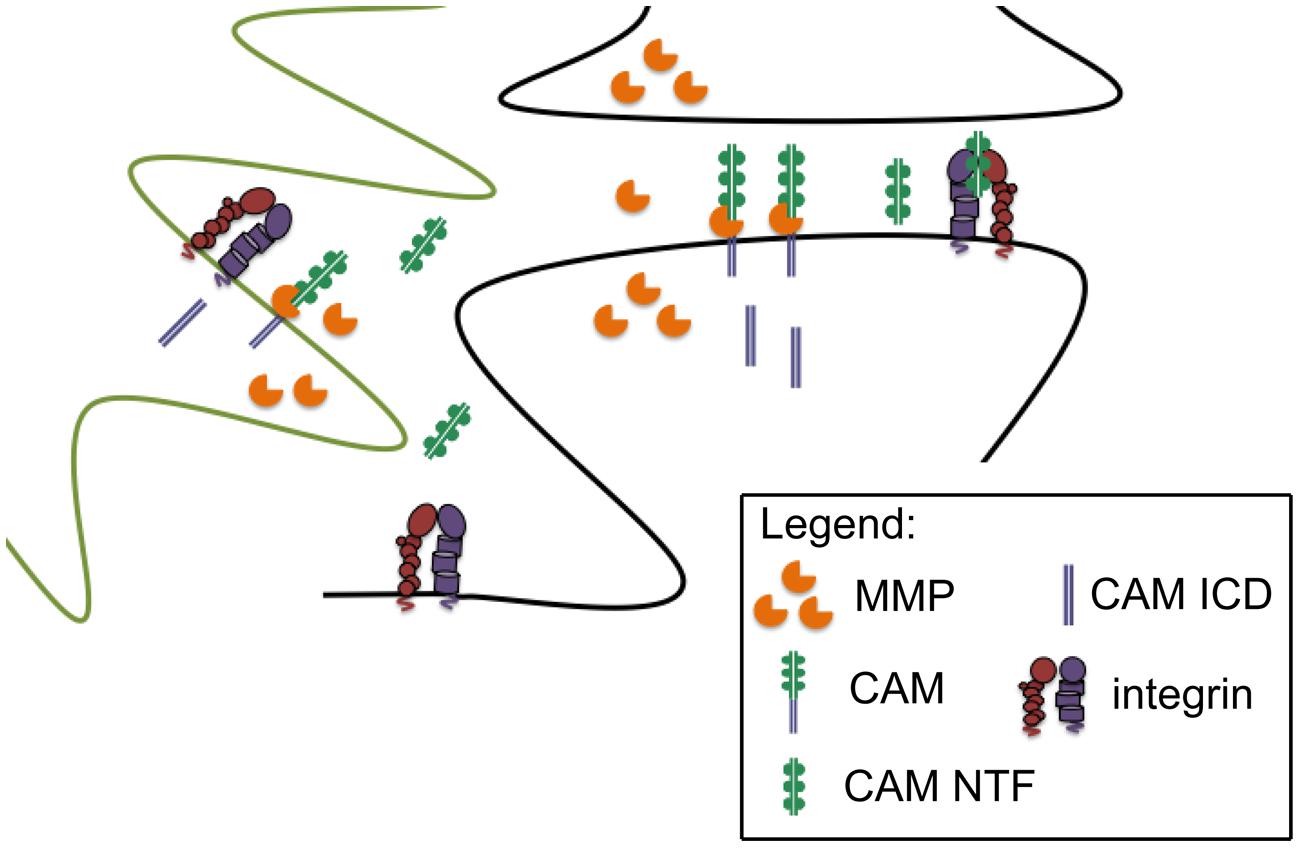
FIGURE 1. Synaptically localized CAM cleavage. Pre and post synaptic components (black outline) and glial components (green outline) of the synapse are shown. MMPs can be released from neurons and/or glia to cleave peri-synaptic CAMs, thus generating CAM N-terminal fragments with the potential to stimulate intregrin dependent signaling. Though not the focus of this review, cleavage generated CAM intracellular domains (ICDs) may also influence synaptic transmission.
In terms of the functional consequences of MMPs and/or CAM ectodomains at single synapses to in turn affect LTP, several non-mutually exclusive possibilities exist as shown in Figure 2. In the first, supported by high resolution imaging (Wang et al., 2008), MMP activity causes a change in actin dynamics with a subsequent widening of spines. This would likely bring an increase in functional GluA receptors to the spine head and increase amplitude of mEPSCs. A second possibility is that MMP generated integrin binding ligands could stimulate the growth of new spines. Integrin signaling has been linked to the same (Shi and Ethell, 2006), and though we did not observe a significant increase in spine number in ICAM-5 ectodomain stimulated DIV 14 rat hippocampal neurons at 1 or 24 h (Lonskaya et al., 2013), it would be premature to rule out the possibility that this measure could be increased at other time points or following exposure to additional CAM ectodomains. The potential for integrin binding ligands to cause an unsilencing of post-synaptic components that were previously silent due to deficient synaptic levels of GluA receptors should be considered as a third possibility. Integrin signaling can activate protein kinases that would in turn phosphorylate specific GluA subunits to enhance their synaptic entry (Lim et al., 2008; Chen et al., 2010). Consistent with this possibility, in previous work we have observed both an ICAM-5 ectodomain stimulated increase in the phosphorylation and membrane localization of GluA1, and an increase in the frequency of mEPSCs (Lonskaya et al., 2013). A fourth possibility is that MMP-dependent signaling stimulates in increase in spine head protrusions to affect glutamatergic transmission. While LTP is generally thought to represent an experience dependent increase in dendritic number and/or size (Kopec et al., 2006), increased complexity of spines might also occur. It has been shown that MMP-9 can stimulate an increase in spine head protrusions (Szepesi et al., 2013). This finding is of significance in that these protrusions may be PSD-95 and GluA positive (Richards et al., 2005; Szepesi et al., 2013), and they may be functionally active in terms of mediating glutamateric neurotransmission (Richards et al., 2005).
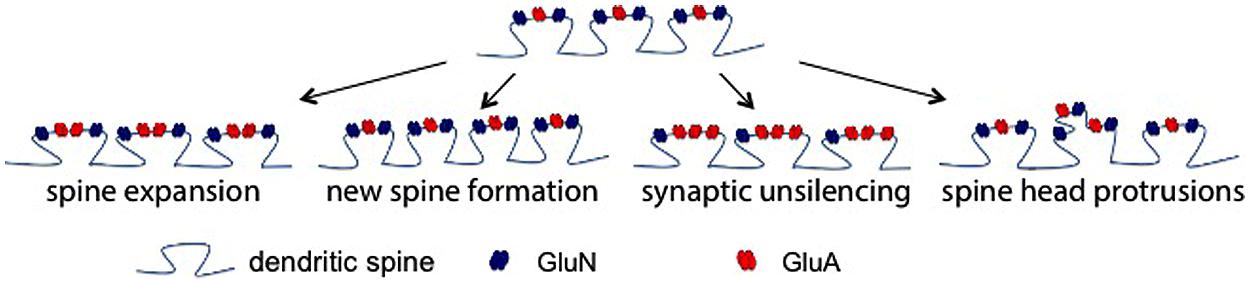
FIGURE 2. Post-synaptic mechanisms by which MMP activity could enhance glutamatergic transmission. There are several non-mutually exclusive possibilities by which MMP activity could influence excitatory neurotransmission. Shown (top) is a dendritic segment with representative spines. As the schematic suggests with arrows to show potential changes, existing spine expansion, new spine formation, synaptic unsilencing, and formation of spine head protrusions represent post synaptic changes that might contribute to MMP and/or integrin dependent changes in the post synaptic element.
Future studies related to CAM-integrin interactions will be necessary to examine a variety of additional questions including that of which integrin binding ligands are generated with learning and memory in vivo. Further study of whether ectodomain shedding plays and important role in select MMP dependent endpoints including changes in spine size or number (Shi and Ethell, 2006; Wang et al., 2008), the development of spine head protrusions (Szepesi et al., 2013), and developmental changes in neurite outgrowth and dendritic arbor (Van Hove et al., 2012a) may also be warranted. MMPs have also been linked to changes in neuronal excitability (Wojtowicz and Mozrzymas, 2014), and since integrin signaling can also influence ion channel function (Wildering et al., 2002), this could represent a parallel topic for future exploration.
An additional avenue for exploration includes the question of whether the downstream effects of CAM shedding can synergize with events that follow from MMP-dependent processing of additional synaptic substrates. As an example, we consider protease activated receptor-1 (PAR-1). A select subset of MMPs target PAR-1, a G protein coupled receptor that is activated by cleavage in N-terminal domain and consequent exposition of a tethered peptide ligand (Vergnolle et al., 2001; Soh et al., 2010). The receptor is expressed on select neuronal subpopulations and has been detected in synaptoneurosomes (Han et al., 2011; Maggio et al., 2013a,b). While activation of neuronal PARs has the potential to enhance GluN subunit phosphorylation and GluN function (Gingrich et al., 2000), whether PAR activation enhances neuronal integrin signaling is unknown. In non-neural cells, however, it has been shown that PAR-1 activation can enhance integrin affinity for ligands (Shattil et al., 2010).
CAM Shedding: From Physiology to Pathology
While regulated physiological release of MMPs can contribute to adaptive plasticity, it is important to note that dysregulated release has the potential to disrupt the same (Wojtowicz and Mozrzymas, 2014). Consistent with this, MMP inhibitors have been shown to ameliorate neuronal injury in a number of disease models. In many of these studies, however, reduced MMP-dependent damage to blood brain barrier integrity is likely to account for much of the observed neuroprotection (Asahi et al., 2001). There is, however, an increasing appreciation synaptic proteolysis in particular as significant a contributor to neurological disease. To follow, we will briefly discuss two disorders in which altered synaptic proteolysis may be critical to disease expression. The first is fragile X syndrome (FXS) and the second is psychostimulant addiction.
Fragile X Syndrome
Fragile X syndrome is a leading genetic cause of intellectual disability and autism (Santoro et al., 2012). At present, no cure is available. Symptoms include developmental delay and increased susceptibility to seizures, while pathological findings include relatively dense and immature dendritic spines (Galvez and Greenough, 2005; McKinney et al., 2005; Pan et al., 2010; Santoro et al., 2012). Expansion of the trinucleotide CGG repeat in excess of 200 repeats located in the 5′ untranslated region of the X chromosome-linked FMR1 gene cause FXS (Verkerk et al., 1991; Sutcliffe et al., 1992). This leads to transcriptional silencing and a consequent lack of functional protein product, fragile X mental retardation protein (FMRP). FMRP regulates expression of a subset of dendritically localized mRNAs, and thus levels of dendritically localized proteins may be altered in FXS (Darnell et al., 2012; Santoro et al., 2012). FMRP generally acts to inhibit the translation of target genes, but in select cases it may have actions that lead instead to enhanced translation (Darnell et al., 2012; Santoro et al., 2012). For example, FMRP is thought to enhance translation of superoxide dismutase (Bechara et al., 2009), and thus antioxidant enzyme activity might be reduced in FXS. This has implications for glial activation, which has been observed in association with the disease (Rossignol and Frye, 2012). Moreover, neuronal and/or glial expression of MMPs may be elevated in response to increased glial activation and/or oxidant stress (Gu et al., 2002).
Published studies have linked MMP activity to FXS. Levels of MMP-9 are increased in affected humans and in a murine model (Sidhu et al., 2014). Increased dendritic translation of MMP-9 has also been observed (Janusz et al., 2013). Recent work suggests that expression of additional MMPs may be increased as well. For example, transcripts for MMP-2, -3, -7, -9, and -24 are increased in heavy polysomes from Fmr1 mutant mice (Gkogkas et al., 2014). In terms of functional consequences, dendritic spine abnormalities in a mouse model of FXS can be reduced by minocycline, an inhibitor of MMP activity that can access the brain (Bilousova et al., 2009). Spine abnormalities are also reduced in the background of MMP-9 deficiency (Sidhu et al., 2014). Moreover, exogenous MMP administration to cultured neurons has been associated with relevant changes in spine morphology (Bilousova et al., 2006). Interestingly, MMP knockout can also reduce neuronal circuit defects in a drosophila model of disease (Siller and Broadie, 2011).
The mechanism(s) by which excess MMP activity stimulates the FXS phenotype have yet to be determined. Several possibilities have been suggested, including increased signaling by MMP-dependent activation of pro-neurotrophins and/or generation of integrin-binding laminin fragments (Sidhu et al., 2014). It is tempting to speculate, however, that excess generation of integrin-binding CAM fragments might play a role. Future studies to address this question may therefore be warranted.
Addiction
Matrix metalloproteinase-dependent changes in synaptic structure and function are also thought to contribute to the maladaptive learning and memory associated with addiction to stimulants including methamphetamine. Methamphetamine is a widely abused illicit drug that has high addictive potential. A variety of studies suggest that methamphetamine is linked to metabolic changes in the brain as well as to synaptic injury (Pu et al., 1996; Volkow et al., 2001; Chang et al., 2009). Evidence for increased MMP expression, release, and/or activation in the setting of methamphetamine exposure comes from several groups. This stimulant can increase release of MMP-1 from cultured neural cells (Conant et al., 2004). This observation is consistent with results from rodent studies in which methamphetamine stimulates increased binding of AP-1, a transcription factor critical to the expression of MMPs including MMP-1 (Akiyama et al., 1996). Additional studies have demonstrated that 5 days of exposure to the drug (2 mg/kg/day) is associated with increased MMP-2 and -9 protein in the frontal cortex and nucleus accumbens of rats (Mizoguchi et al., 2007a). Moreover, an acute high dose of MA (40 mg/kg) is followed by increased mRNA expression of MMP-9 in murine CNS (Liu et al., 2008). Similarly, cocaine, which is similar to methamphetamine in its potential to increase catecholamine levels, has been shown to increase MMP-9 activity in the medial prefrontal cortex at 1, 3, and 24 h post-administration (Brown et al., 2008).
Methamphetamine has the potential to increase MMP expression through several non-mutually exclusive mechanisms including increased catecholamine dependent signaling, activation of glutamate receptor signaling, and increased oxidant stress. For example, methamphetamine associated increases dopamine can act on D1 type dopamine receptors to enhance substrate proteolysis (Iwakura et al., 2011). Since both D1 and D2 type are linked to βγ subunits that can activate PKC and release of intracellular calcium, activation of either receptor type might stimulate calcium dependent MMP release and/or PKC dependent activation of a transmembrane MMP. Another possibility is that MA increases levels of glutamate, as has been shown by Yamamoto and colleagues (Mark et al., 2004), and that glutamate signaling can in turn stimulate increased MMP expression and/or activity. Relatively high concentrations of MA also stimulate an increase in signaling by reactive oxygen intermediates (Lee et al., 2002), which can enhance both the expression and the activation of select MMPs (Gu et al., 2002).
Increased MMP activity may also contribute to synaptic and behavioral changes observed with stimulant exposure. It has been shown that methamphetamine-induced behavioral sensitization is reduced in mice lacking MMP-2 or MMP-9 (Mizoguchi et al., 2007b). Protease activity has also been shown to contribute to cocaine associated CPP (Brown et al., 2007). In recent work focused on structural and function changes at the levels of the synapse in the setting of cocaine exposure paradigms, an increase in the AMPA/NMDA current ratio was increased in extinguished rats, and further increased 15 min following cue-induced reinstatement (Smith et al., 2014). An increase in the AMPA/NMDA ratio after extinction was restored to control by a selective MMP-2 inhibitor, while the altered reinstatement ratio was restored by either and MMP-2 or -9 inhibitor (Smith et al., 2014). Complementary measures of spine density and spine size supported a view in which MMP-2 could increase the density and head size of spines in extinguished animals and that MMP-9 activity contributed to an increase in spine size with reinstatement.
Overall, studies related to the role of MMPs in addiction are exciting and should stimulate further work to address underlying mechanistic components.
Summary
Matrix metalloproteinases were named for their ability to cleave extracellular matrix proteins such as laminin and collagen. While matrix remodeling effects may be essential during development and wound healing, proteolysis of cell surface receptors including CAMs could represent a critical means by which MMPs can fine tune synaptic structure and function in a more stable or relatively mature CNS. This possibility is supported by work showing that neuronal activity stimulates proteolysis of synaptically localized CAMs, and that CAM shedding can influence varied measures of synaptic transmission. Future studies will be necessary to examine CAM cleavage as affected by variables including cell type, brain region, stimulus type/duration, and developmental stage. Future studies will also be necessary to determine which CAM cleavage products are most likely to influence MMP dependent plasticity in vivo, and to determine the extent to which CAM shedding combines with additional MMP-stimulated events to influence experience dependent plasticity.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
We would like to acknowledge funding from the National Institutes of Health (R01NS083410 and T32NS041218).
References
Aerts, J., Nys, J., Moons, L., Hu, T. T., and Arckens, L. (2014). Altered neuronal architecture and plasticity in the visual cortex of adult MMP-3-deficient mice. Brain Struct. Funct. doi: 10.1007/s00429-014-0819-4 [Epub ahead of print].
Ahmad, M., Takino, T., Miyamori, H., Yoshizaki, T., Furukawa, M., and Sato, H. (2006). Cleavage of amyloid-beta precursor protein (APP) by membrane-type matrix metalloproteinases. J. Biochem. 139, 517–526. doi: 10.1093/jb/mvj054
Akiyama, K., Ishihara, T., and Kashihara, K. (1996). Effect of acute and chronic administration of methamphetamine on activator protein-1 binding activities in the rat brain regions. Ann. N. Y. Acad. Sci. 801, 13–28. doi: 10.1111/j.1749-6632.1996.tb17428.x
Alvarez, V. A., and Sabatini, B. L. (2007). Anatomical and physiological plasticity of dendritic spines. Annu. Rev. Neurosci. 30, 79–97. doi: 10.1146/annurev.neuro.30.051606.094222
Anderson, G. R., Galfin, T., Xu, W., Aoto, J., Malenka, R. C., and Sudhof, T. C. (2012). Candidate autism gene screen identifies critical role for cell-adhesion molecule CASPR2 in dendritic arborization and spine development. Proc. Natl. Acad. Sci. U.S.A. 109, 18120–18125. doi: 10.1073/pnas.1216398109
Aoto, J., Nam, C. I., Poon, M. M., Ting, P., and Chen, L. (2008). Synaptic signaling by all-trans retinoic acid in homeostatic synaptic plasticity. Neuron 60, 308–320. doi: 10.1016/j.neuron.2008.08.012
Asahi, M., Wang, X., Mori, T., Sumii, T., Jung, J. C., Moskowitz, M. A., et al. (2001). Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J. Neurosci. 21, 7724–7732.
Babayan, A. H., Kramar, E. A., Barrett, R. M., Jafari, M., Haettig, J., Chen, L. Y., et al. (2012). Integrin dynamics produce a delayed stage of long-term potentiation and memory consolidation. J. Neurosci. 32, 12854–12861. doi: 10.1523/JNEUROSCI.2024-12.2012
Bajor, M., Michaluk, P., Gulyassy, P., Kekesi, A. K., Juhasz, G., and Kaczmarek, L. (2012). Synaptic cell adhesion molecule-2 and collapsin response mediator protein-2 are novel members of the matrix metalloproteinase-9 degradome. J. Neurochem. 122, 775–788. doi: 10.1111/j.1471-4159.2012.07829.x
Bechara, E. G., Didiot, M. C., Melko, M., Davidovic, L., Bensaid, M., Martin, P., et al. (2009). A novel function for fragile X mental retardation protein in translational activation. PLoS Biol. 7:e16. doi: 10.1371/journal.pbio.1000016
Ben-Hur, T., Ben-Yosef, Y., Mizrachi-Kol, R., Ben-Menachem, O., and Miller, A. (2006). Cytokine-mediated modulation of MMPs and TIMPs in multipotential neural precursor cells. J. Neuroimmunol. 175, 12–18. doi: 10.1016/j.jneuroim.2006.02.002
Benson, D. L., and Huntley, G. W. (2012). Synapse adhesion: a dynamic equilibrium conferring stability and flexibility. Curr. Opin. Neurobiol. 22, 397–404. doi: 10.1016/j.conb.2011.09.011
Benson, D. L., Yoshihara, Y., and Mori, K. (1998). Polarized distribution and cell type-specific localization of telencephalin, an intercellular adhesion molecule. J. Neurosci. Res. 52, 43–53. doi: 10.1002/(SICI)1097-4547(19980401)52:1<43::AID-JNR5>3.0.CO;2-K
Bernard-Trifilo, J. A., Kramar, E. A., Torp, R., Lin, C. Y., Pineda, E. A., Lynch, G., et al. (2005). Integrin signaling cascades are operational in adult hippocampal synapses and modulate NMDA receptor physiology. J. Neurochem. 93, 834–849. doi: 10.1111/j.1471-4159.2005.03062.x
Berry, E., Bosonea, A. M., Wang, X., and Fernandez-Patron, C. (2013). Insights into the activity, differential expression, mutual regulation, and functions of matrix metalloproteinases and a disintegrin and metalloproteinases in hypertension and cardiac disease. J. Vasc. Res. 50, 52–68. doi: 10.1159/000345240
Biederer, T., Sara, Y., Mozhayeva, M., Atasoy, D., Liu, X., Kavalali, E. T., et al. (2002). SynCAM, a synaptic adhesion molecule that drives synapse assembly. Science 297, 1525–1531. doi: 10.1126/science.1072356
Bilousova, T. V., Dansie, L., Ngo, M., Aye, J., Charles, J. R., Ethell, D. W., et al. (2009). Minocycline promotes dendritic spine maturation and improves behavioural performance in the fragile X mouse model. J. Med. Genet. 46, 94–102. doi: 10.1136/jmg.2008.061796
Bilousova, T. V., Rusakov, D. A., Ethell, D. W., and Ethell, I. M. (2006). Matrix metalloproteinase-7 disrupts dendritic spines in hippocampal neurons through NMDA receptor activation. J. Neurochem. 97, 44–56. doi: 10.1111/j.1471-4159.2006.03701.x
Bourne, J., and Harris, K. M. (2007). Do thin spines learn to be mushroom spines that remember? Curr. Opin. Neurobiol. 17, 381–386. doi: 10.1016/j.conb.2007.04.009
Bozdagi, O., Wang, X. B., Nikitczuk, J. S., Anderson, T. R., Bloss, E. B., Radice, G. L., et al. (2010). Persistence of coordinated long-term potentiation and dendritic spine enlargement at mature hippocampal CA1 synapses requires N-cadherin. J. Neurosci. 30, 9984–9989. doi: 10.1523/JNEUROSCI.1223-10.2010
Brew, K., and Nagase, H. (2010). The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim. Biophys. Acta 1803, 55–71. doi: 10.1016/j.bbamcr.2010.01.003
Brown, T. E., Forquer, M. R., Cocking, D. L., Jansen, H. T., Harding, J. W., and Sorg, B. A. (2007). Role of matrix metalloproteinases in the acquisition and reconsolidation of cocaine-induced conditioned place preference. Learn. Mem. 14, 214–223. doi: 10.1101/lm.476207
Brown, T. E., Forquer, M. R., Harding, J. W., Wright, J. W., and Sorg, B. A. (2008). Increase in matrix metalloproteinase-9 levels in the rat medial prefrontal cortex after cocaine reinstatement of conditioned place preference. Synapse 62, 886–889. doi: 10.1002/syn.20562
Brown, T. E., Wilson, A. R., Cocking, D. L., and Sorg, B. A. (2009). Inhibition of matrix metalloproteinase activity disrupts reconsolidation but not consolidation of a fear memory. Neurobiol. Learn. Mem. 91, 66–72. doi: 10.1016/j.nlm.2008.09.003
Chan, C. S., Weeber, E. J., Zong, L., Fuchs, E., Sweatt, J. D., and Davis, R. L. (2006). Beta 1-integrins are required for hippocampal AMPA receptor-dependent synaptic transmission, synaptic plasticity, and working memory. J. Neurosci. 26, 223–232. doi: 10.1523/JNEUROSCI.4110-05.2006
Chang, L., Cloak, C., Jiang, C. S., Farnham, S., Tokeshi, B., Buchthal, S., et al. (2009). Altered neurometabolites and motor integration in children exposed to methamphetamine in utero. Neuroimage 48, 391–397. doi: 10.1016/j.neuroimage.2009.06.062
Chen, Q., Zhu, X., Zhang, Y., Wetsel, W. C., Lee, T. H., and Zhang, X. (2010). Integrin-linked kinase is involved in cocaine sensitization by regulating PSD-95 and synapsin I expression and GluR1 Ser845 phosphorylation. J. Mol. Neurosci. 40, 284–294. doi: 10.1007/s12031-009-9218-3
Clark, E. A., and Brugge, J. S. (1995). Integrins and signal transduction pathways: the road taken. Science 268, 233–239. doi: 10.1126/science.7716514
Conant, K., Lonskaya, I., Szklarczyk, A., Krall, C., Steiner, J., Maguire-Zeiss, K., et al. (2010a). Methamphetamine-associated cleavage of the synaptic adhesion molecule intercellular adhesion molecule-5. J. Neurochem. 118, 521–532. doi: 10.1111/j.1471-4159.2010.07153.x
Conant, K., Wang, Y., Szklarczyk, A., Dudak, A., Mattson, M. P., and Lim, S. T. (2010b). Matrix metalloproteinase-dependent shedding of intercellular adhesion molecule-5 occurs with long-term potentiation. Neuroscience 166, 508–521. doi: 10.1016/j.neuroscience.2009.12.061
Conant, K., Mcarthur, J. C., Griffin, D. E., Sjulson, L., Wahl, L. M., and Irani, D. N. (1999). Cerebrospinal fluid levels of MMP-2, 7, and 9 are elevated in association with human immunodeficiency virus dementia. Ann. Neurol. 46, 391–398. doi: 10.1002/1531-8249(199909)46:3<391::AID-ANA15>3.0.CO;2-0
Conant, K., St Hillaire, C., Anderson, C., Galey, D., Wang, J., and Nath, A. (2004). Human immunodeficiency virus type 1 Tat and methamphetamine affect the release and activation of matrix-degrading proteinases. J. Neurovirol. 10, 21–28. doi: 10.1080/13550280490261699
Cremer, H., Chazal, G., Carleton, A., Goridis, C., Vincent, J. D., and Lledo, P. M. (1998). Long-term but not short-term plasticity at mossy fiber synapses is impaired in neural cell adhesion molecule-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 95, 13242–13247. doi: 10.1073/pnas.95.22.13242
Darnell, J. C., Van Driesche, S. J., Zhang, C., Hung, K. Y., Mele, A., Fraser, C. E., et al. (2012). FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261. doi: 10.1016/j.cell.2011.06.013
Dityatev, A., Bukalo, O., and Schachner, M. (2008). Modulation of synaptic transmission and plasticity by cell adhesion and repulsion molecules. Neuron Glia Biol. 4, 197–209. doi: 10.1017/S1740925X09990111
Dityatev, A., and Rusakov, D. A. (2011). Molecular signals of plasticity at the tetrapartite synapse. Curr. Opin. Neurobiol. 21, 353–359. doi: 10.1016/j.conb.2010.12.006
Dziembowska, M., Milek, J., Janusz, A., Rejmak, E., Romanowska, E., Gorkiewicz, T., et al. (2012). Activity-dependent local translation of matrix metalloproteinase-9. J. Neurosci. 32, 14538–14547. doi: 10.1523/JNEUROSCI.6028-11.2012
Eckhardt, M., Bukalo, O., Chazal, G., Wang, L., Goridis, C., Schachner, M., et al. (2000). Mice deficient in the polysialyltransferase ST8SiaIV/PST-1 allow discrimination of the roles of neural cell adhesion molecule protein and polysialic acid in neural development and synaptic plasticity. J. Neurosci. 20, 5234–5244.
Etherton, M. R., Tabuchi, K., Sharma, M., Ko, J., and Sudhof, T. C. (2011). An autism-associated point mutation in the neuroligin cytoplasmic tail selectively impairs AMPA receptor-mediated synaptic transmission in hippocampus. EMBO J. 30, 2908–2919. doi: 10.1038/emboj.2011.182
Fogel, A. I., Akins, M. R., Krupp, A. J., Stagi, M., Stein, V., and Biederer, T. (2007). SynCAMs organize synapses through heterophilic adhesion. J. Neurosci. 27, 12516–12530. doi: 10.1523/JNEUROSCI.2739-07.2007
Galvez, R., and Greenough, W. T. (2005). Sequence of abnormal dendritic spine development in primary somatosensory cortex of a mouse model of the fragile X mental retardation syndrome. Am. J. Med. Genet. A 135, 155–160. doi: 10.1002/ajmg.a.30709
Ganguly, K., Rejmak, E., Mikosz, M., Nikolaev, E., Knapska, E., and Kaczmarek, L. (2013). Matrix metalloproteinase (MMP) 9 transcription in mouse brain induced by fear learning. J. Biol. Chem. 288, 20978–20991. doi: 10.1074/jbc.M113.457903
Gardner, J., and Ghorpade, A. (2003). Tissue inhibitor of metalloproteinase (TIMP)-1: the TIMPed balance of matrix metalloproteinases in the central nervous system. J. Neurosci. Res. 74, 801–806. doi: 10.1002/jnr.10835
Gerber, S. H., and Sudhof, T. C. (2002). Molecular determinants of regulated exocytosis. Diabetes 51(Suppl. 1), S3–S11. doi: 10.2337/diabetes.51.2007.S3
Gingrich, M. B., Junge, C. E., Lyuboslavsky, P., and Traynelis, S. F. (2000). Potentiation of NMDA receptor function by the serine protease thrombin. J. Neurosci. 20, 4582–4595.
Gkogkas, C. G., Khoutorsky, A., Cao, R., Jafarnejad, S. M., Prager-Khoutorsky, M., Giannakas, N., et al. (2014). Pharmacogenetic inhibition of eIF4E-dependent Mmp9 mRNA translation reverses fragile X syndrome-like phenotypes. Cell Rep. 9, 1742–1755. doi: 10.1016/j.celrep.2014.10.064
Groth, R. D., and Tsien, R. W. (2008). A role for retinoic acid in homeostatic plasticity. Neuron 60, 192–194. doi: 10.1016/j.neuron.2008.10.003
Gu, Z., Kaul, M., Yan, B., Kridel, S. J., Cui, J., Strongin, A., et al. (2002). S-nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science 297, 1186–1190. doi: 10.1126/science.1073634
Gutwein, P., Mechtersheimer, S., Riedle, S., Stoeck, A., Gast, D., Joumaa, S., et al. (2003). ADAM10-mediated cleavage of L1 adhesion molecule at the cell surface and in released membrane vesicles. FASEB J. 17, 292–294.
Hahn-Dantona, E., Ruiz, J. F., Bornstein, P., and Strickland, D. K. (2001). The low density lipoprotein receptor-related protein modulates levels of matrix metalloproteinase 9 (MMP-9) by mediating its cellular catabolism. J. Biol. Chem. 276, 15498–15503. doi: 10.1074/jbc.M100121200
Han, K. S., Mannaioni, G., Hamill, C. E., Lee, J., Junge, C. E., Lee, C. J., et al. (2011). Activation of protease activated receptor 1 increases the excitability of the dentate granule neurons of hippocampus. Mol. Brain 4, 32. doi: 10.1186/1756-6606-4-32
Hartmann, M., Parra, L. M., Ruschel, A., Lindner, C., Morrison, H., Herrlich, A., et al. (2015). Inside-out regulation of ectodomain cleavage of cluster-of-differentiation-44 [CD44] and of neuregulin-1 requires substrate dimerization. J. Biol. Chem. 290, 17041–17054. doi: 10.1074/jbc.M114.610204
Hayashida, K., Bartlett, A. H., Chen, Y., and Park, P. W. (2010). Molecular and cellular mechanisms of ectodomain shedding. Anat. Rec. (Hoboken) 293, 925–937. doi: 10.1002/ar.20757
He, B., Peng, H., Zhao, Y., Zhou, H., and Zhao, Z. (2011). Modafinil treatment prevents REM sleep deprivation-induced brain function impairment by increasing MMP-9 expression. Brain Res. 1426, 38–42. doi: 10.1016/j.brainres.2011.09.002
Huang, Z., Shimazu, K., Woo, N. H., Zang, K., Muller, U., Lu, B., et al. (2006). Distinct roles of the beta 1-class integrins at the developing and the mature hippocampal excitatory synapse. J. Neurosci. 26, 11208–11219. doi: 10.1523/JNEUROSCI.3526-06.2006
Huganir, R. L., and Racker, E. (1980). Endogenous and exogenous proteolysis of the acetylcholine receptor from Torpedo californica. J. Supramol. Struct. 14, 13–19. doi: 10.1002/jss.400140103
Huntley, G. W. (2012). Synaptic circuit remodelling by matrix metalloproteinases in health and disease. Nat. Rev. Neurosci. 13, 743–757. doi: 10.1038/nrn3320
Husi, H., Ward, M. A., Choudhary, J. S., Blackstock, W. P., and Grant, S. G. (2000). Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nat. Neurosci. 3, 661–669. doi: 10.1038/76615
Iwakura, Y., Wang, R., Abe, Y., Piao, Y. S., Shishido, Y., Higashiyama, S., et al. (2011). Dopamine-dependent ectodomain shedding and release of epidermal growth factor in developing striatum: target-derived neurotrophic signaling (Part 2). J. Neurochem. 118, 57–68. doi: 10.1111/j.1471-4159.2011.07295.x
Janusz, A., Milek, J., Perycz, M., Pacini, L., Bagni, C., Kaczmarek, L., et al. (2013). The Fragile X mental retardation protein regulates matrix metalloproteinase 9 mRNA at synapses. J. Neurosci. 33, 18234–18241. doi: 10.1523/JNEUROSCI.2207-13.2013
Johnson, C., Drgon, T., Liu, Q. R., Zhang, P. W., Walther, D., Li, C. Y., et al. (2008). Genome wide association for substance dependence: convergent results from epidemiologic and research volunteer samples. BMC Med. Genet. 9:113. doi: 10.1186/1471-2350-9-113
Johnston, J. B., Silva, C., and Power, C. (2002). Envelope gene-mediated neurovirulence in feline immunodeficiency virus infection: induction of matrix metalloproteinases and neuronal injury. J. Virol. 76, 2622–2633. doi: 10.1128/JVI.76.6.2622-2633.2002
Jordan, B. A., and Kreutz, M. R. (2009). Nucleocytoplasmic protein shuttling: the direct route in synapse-to-nucleus signaling. Trends Neurosci. 32, 392–401. doi: 10.1016/j.tins.2009.04.001
Kamiguchi, H., and Lemmon, V. (2000). Recycling of the cell adhesion molecule L1 in axonal growth cones. J. Neurosci. 20, 3676–3686.
Kasai, H., Fukuda, M., Watanabe, S., Hayashi-Takagi, A., and Noguchi, J. (2010). Structural dynamics of dendritic spines in memory and cognition. Trends Neurosci. 33, 121–129. doi: 10.1016/j.tins.2010.01.001
Kean, M. J., Williams, K. C., Skalski, M., Myers, D., Burtnik, A., Foster, D., et al. (2009). VAMP3, syntaxin-13 and SNAP23 are involved in secretion of matrix metalloproteinases, degradation of the extracellular matrix and cell invasion. J. Cell Sci. 122, 4089–4098. doi: 10.1242/jcs.052761
Kelly, E. A., Tremblay, M. E., Gahmberg, C. G., Tian, L., and Majewska, A. K. (2013). Interactions between intercellular adhesion molecule-5 positive elements and their surroundings in the rodent visual cortex. Commun. Integr. Biol. 6, e27315. doi: 10.4161/cib.27315
Khuth, S. T., Akaoka, H., Pagenstecher, A., Verlaeten, O., Belin, M. F., Giraudon, P., et al. (2001). Morbillivirus infection of the mouse central nervous system induces region-specific upregulation of MMPs and TIMPs correlated to inflammatory cytokine expression. J. Virol. 75, 8268–8282. doi: 10.1128/JVI.75.17.8268-8282.2001
Kim, H. J., Fillmore, H. L., Reeves, T. M., and Phillips, L. L. (2005). Elevation of hippocampal MMP-3 expression and activity during trauma-induced synaptogenesis. Exp. Neurol. 192, 60–72. doi: 10.1016/j.expneurol.2004.10.014
Knapska, E., Lioudyno, V., Kiryk, A., Mikosz, M., Gorkiewicz, T., Michaluk, P., et al. (2013). Reward learning requires activity of matrix metalloproteinase-9 in the central amygdala. J. Neurosci. 33, 14591–14600. doi: 10.1523/JNEUROSCI.5239-12.2013
Kochlamazashvili, G., Bukalo, O., Senkov, O., Salmen, B., Gerardy-Schahn, R., Engel, A. K., et al. (2012). Restoration of synaptic plasticity and learning in young and aged NCAM-deficient mice by enhancing neurotransmission mediated by GluN2A-containing NMDA receptors. J. Neurosci. 32, 2263–2275. doi: 10.1523/JNEUROSCI.5103-11.2012
Konopacki, F. A., Rylski, M., Wilczek, E., Amborska, R., Detka, D., Kaczmarek, L., et al. (2007). Synaptic localization of seizure-induced matrix metalloproteinase-9 mRNA. Neuroscience 150, 31–39. doi: 10.1016/j.neuroscience.2007.08.026
Kopec, C. D., Li, B., Wei, W., Boehm, J., and Malinow, R. (2006). Glutamate receptor exocytosis and spine enlargement during chemically induced long-term potentiation. J. Neurosci. 26, 2000–2009. doi: 10.1523/JNEUROSCI.3918-05.2006
Kuzniewska, B., Rejmak, E., Malik, A. R., Jaworski, J., Kaczmarek, L., and Kalita, K. (2013). Brain-derived neurotrophic factor induces matrix metalloproteinase 9 expression in neurons via the serum response factor/c-Fos pathway. Mol. Cell Biol. 33, 2149–2162. doi: 10.1128/MCB.00008-13
Larsen, P. H., Dasilva, A. G., Conant, K., and Yong, V. W. (2006). Myelin formation during development of the CNS is delayed in matrix metalloproteinase-9 and -12 null mice. J. Neurosci. 26, 2207–2214. doi: 10.1523/JNEUROSCI.1880-05.2006
Lee, H., Lee, E. J., Song, Y. S., and Kim, E. (2014). Long-term depression-inducing stimuli promote cleavage of the synaptic adhesion molecule NGL-3 through NMDA receptors, matrix metalloproteinases and presenilin/gamma-secretase. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369, 20130158. doi: 10.1098/rstb.2013.0158
Lee, R., Kermani, P., Teng, K. K., and Hempstead, B. L. (2001). Regulation of cell survival by secreted proneurotrophins. Science 294, 1945–1948. doi: 10.1126/science.1065057
Lee, Y. W., Son, K. W., Flora, G., Hennig, B., Nath, A., and Toborek, M. (2002). Methamphetamine activates DNA binding of specific redox-responsive transcription factors in mouse brain. J. Neurosci. Res. 70, 82–89. doi: 10.1002/jnr.10370
Li, W., Chang, L., Rong, Z., and Liu, W. (2011). Retinoic acid diminished the expression of MMP-2 in hyperoxia-exposed premature rat lung fibroblasts through regulating mitogen-activated protein kinases. J. Huazhong. Univ. Sci. Technolog. Med. Sci. 31, 251–257. doi: 10.1007/s11596-011-0262-1
Lim, C. J., Kain, K. H., Tkachenko, E., Goldfinger, L. E., Gutierrez, E., Allen, M. D., et al. (2008). Integrin-mediated protein kinase A activation at the leading edge of migrating cells. Mol. Biol. Cell. 19, 4930–4941. doi: 10.1091/mbc.E08-06-0564
Lim, S. T., Chang, A., Giuliano, R. E., and Federoff, H. J. (2012). Ectodomain shedding of nectin-1 regulates the maintenance of dendritic spine density. J. Neurochem. 120, 741–751. doi: 10.1111/j.1471-4159.2011.07592.x
Lindsberg, P. J., Launes, J., Tian, L., Valimaa, H., Subramanian, V., Siren, J., et al. (2002). Release of soluble ICAM-5, a neuronal adhesion molecule, in acute encephalitis. Neurology 58, 446–451. doi: 10.1212/WNL.58.3.446
Liu, Y., Brown, S., Shaikh, J., Fishback, J. A., and Matsumoto, R. R. (2008). Relationship between methamphetamine exposure and matrix metalloproteinase 9 expression. Neuroreport 19, 1407–1409. doi: 10.1097/WNR.0b013e32830dd606
Lonskaya, I., Partridge, J., Lalchandani, R. R., Chung, A., Lee, T., Vicini, S., et al. (2013). Soluble ICAM-5, a product of activity dependent proteolysis, increases mEPSC frequency and dendritic expression of GluA1. PLoS ONE 8:e69136. doi: 10.1371/journal.pone.0069136
Maggio, N., Cavaliere, C., Papa, M., Blatt, I., Chapman, J., and Segal, M. (2013a). Thrombin regulation of synaptic transmission: implications for seizure onset. Neurobiol. Dis. 50, 171–178. doi: 10.1016/j.nbd.2012.10.017
Maggio, N., Itsekson, Z., Dominissini, D., Blatt, I., Amariglio, N., Rechavi, G., et al. (2013b). Thrombin regulation of synaptic plasticity: implications for physiology and pathology. Exp. Neurol. 247, 595–604. doi: 10.1016/j.expneurol.2013.02.011
Marambaud, P., Wen, P. H., Dutt, A., Shioi, J., Takashima, A., Siman, R., et al. (2003). A CBP binding transcriptional repressor produced by the PS1/epsilon-cleavage of N-cadherin is inhibited by PS1 FAD mutations. Cell 114, 635–645. doi: 10.1016/j.cell.2003.08.008
Marcello, E., Epis, R., Saraceno, C., Gardoni, F., Borroni, B., Cattabeni, F., et al. (2012). SAP97-mediated local trafficking is altered in Alzheimer disease patients’ hippocampus. Neurobiol. Aging 33, 422.e1–10. doi: 10.1016/j.neurobiolaging.2010.09.015
Marcello, E., Gardoni, F., Di Luca, M., and Perez-Otano, I. (2010). An arginine stretch limits ADAM10 exit from the endoplasmic reticulum. J. Biol. Chem. 285, 10376–10384. doi: 10.1074/jbc.M109.055947
Mark, K. A., Soghomonian, J. J., and Yamamoto, B. K. (2004). High-dose methamphetamine acutely activates the striatonigral pathway to increase striatal glutamate and mediate long-term dopamine toxicity. J. Neurosci. 24, 11449–11456. doi: 10.1523/JNEUROSCI.3597-04.2004
Matsuno, H., Okabe, S., Mishina, M., Yanagida, T., Mori, K., and Yoshihara, Y. (2006). Telencephalin slows spine maturation. J. Neurosci. 26, 1776–1786. doi: 10.1523/JNEUROSCI.2651-05.2006
Matsuzaki, M. (2007). Factors critical for the plasticity of dendritic spines and memory storage. Neurosci. Res. 57, 1–9. doi: 10.1016/j.neures.2006.09.017
Matsuzaki, M., Honkura, N., Ellis-Davies, G. C., and Kasai, H. (2004). Structural basis of long-term potentiation in single dendritic spines. Nature 429, 761–766. doi: 10.1038/nature02617
McKinney, B. C., Grossman, A. W., Elisseou, N. M., and Greenough, W. T. (2005). Dendritic spine abnormalities in the occipital cortex of C57BL/6 Fmr1 knockout mice. Am. J. Med. Genet. B Neuropsychiatr. Genet. 136B, 98–102. doi: 10.1002/ajmg.b.30183
Mechtersheimer, S., Gutwein, P., Agmon-Levin, N., Stoeck, A., Oleszewski, M., Riedle, S., et al. (2001). Ectodomain shedding of L1 adhesion molecule promotes cell migration by autocrine binding to integrins. J. Cell Biol. 155, 661–673. doi: 10.1083/jcb.200101099
Meighan, P. C., Meighan, S. E., Davis, C. J., Wright, J. W., and Harding, J. W. (2007). Effects of matrix metalloproteinase inhibition on short- and long-term plasticity of schaffer collateral/CA1 synapses. J. Neurochem. 102, 2085–2096. doi: 10.1111/j.1471-4159.2007.04682.x
Meighan, S. E., Meighan, P. C., Choudhury, P., Davis, C. J., Olson, M. L., Zornes, P. A., et al. (2006). Effects of extracellular matrix-degrading proteases matrix metalloproteinases 3 and 9 on spatial learning and synaptic plasticity. J. Neurochem. 96, 1227–1241. doi: 10.1111/j.1471-4159.2005.03565.x
Melis, M., Pace, E., Siena, L., Spatafora, M., Tipa, A., Profita, M., et al. (2003). Biologically active intercellular adhesion molecule-1 is shed as dimers by a regulated mechanism in the inflamed pleural space. Am. J. Respir. Crit. Care Med. 167, 1131–1138. doi: 10.1164/rccm.200207-654OC
Michaluk, P., Kolodziej, L., Mioduszewska, B., Wilczynski, G. M., Dzwonek, J., Jaworski, J., et al. (2007). Beta-dystroglycan as a target for MMP-9, in response to enhanced neuronal activity. J. Biol. Chem. 282, 16036–16041. doi: 10.1074/jbc.M700641200
Michaluk, P., Wawrzyniak, M., Alot, P., Szczot, M., Wyrembek, P., Mercik, K., et al. (2011). Influence of matrix metalloproteinase MMP-9 on dendritic spine morphology. J. Cell Sci. 124, 3369–3380. doi: 10.1242/jcs.090852
Mizoguchi, H., Yamada, K., Mouri, A., Niwa, M., Mizuno, T., Noda, Y., et al. (2007a). Role of matrix metalloproteinase and tissue inhibitor of MMP in methamphetamine-induced behavioral sensitization and reward: implications for dopamine receptor down-regulation and dopamine release. J. Neurochem. 102, 1548–1560. doi: 10.1111/j.1471-4159.2007.04623.x
Mizoguchi, H., Yamada, K., Niwa, M., Mouri, A., Mizuno, T., Noda, Y., et al. (2007b). Reduction of methamphetamine-induced sensitization and reward in matrix metalloproteinase-2 and -9-deficient mice. J. Neurochem. 100, 1579–1588.
Mortillo, S., Elste, A., Ge, Y., Patil, S. B., Hsiao, K., Huntley, G. W., et al. (2012). Compensatory redistribution of neuroligins and N-cadherin following deletion of synaptic beta1-integrin. J. Comp. Neurol. 520, 2041–2052. doi: 10.1002/cne.23027
Moser, M. B., Trommald, M., and Andersen, P. (1994). An increase in dendritic spine density on hippocampal CA1 pyramidal cells following spatial learning in adult rats suggests the formation of new synapses. Proc. Natl. Acad. Sci. U.S.A. 91, 12673–12675. doi: 10.1073/pnas.91.26.12673
Nagase, H., Enghild, J. J., Suzuki, K., and Salvesen, G. (1990). Stepwise activation mechanisms of the precursor of matrix metalloproteinase 3 (stromelysin) by proteinases and (4-aminophenyl)mercuric acetate. Biochemistry 29, 5783–5789. doi: 10.1021/bi00476a020
Nagy, V., Bozdagi, O., and Huntley, G. W. (2007). The extracellular protease matrix metalloproteinase-9 is activated by inhibitory avoidance learning and required for long-term memory. Learn. Mem. 14, 655–664. doi: 10.1101/lm.678307
Nagy, V., Bozdagi, O., Matynia, A., Balcerzyk, M., Okulski, P., Dzwonek, J., et al. (2006). Matrix metalloproteinase-9 is required for hippocampal late-phase long-term potentiation and memory. J. Neurosci. 26, 1923–1934. doi: 10.1523/JNEUROSCI.4359-05.2006
Nakamura, K., Manabe, T., Watanabe, M., Mamiya, T., Ichikawa, R., Kiyama, Y., et al. (2001). Enhancement of hippocampal LTP, reference memory and sensorimotor gating in mutant mice lacking a telencephalon-specific cell adhesion molecule. Eur. J. Neurosci. 13, 179–189. doi: 10.1046/j.0953-816X.2000.01366.x
Natarajan, R., Harding, J. W., and Wright, J. W. (2013). A role for matrix metalloproteinases in nicotine-induced conditioned place preference and relapse in adolescent female rats. J. Exp. Neurosci. 7, 1–14. doi: 10.4137/JEN.S11381
Niedringhaus, M., Chen, X., Dzakpasu, R., and Conant, K. (2012). MMPs and soluble ICAM-5 increase neuronal excitability within in vitro networks of hippocampal neurons. PLoS ONE 7:e42631. doi: 10.1371/journal.pone.0042631
Nikitczuk, J. S., Patil, S. B., Matikainen-Ankney, B. A., Scarpa, J., Shapiro, M. L., Benson, D. L., et al. (2014). N-cadherin regulates molecular organization of excitatory and inhibitory synaptic circuits in adult hippocampus in vivo. Hippocampus 24, 943–962. doi: 10.1002/hipo.22282
Ning, L., Tian, L., Smirnov, S., Vihinen, H., Llano, O., Vick, K., et al. (2013). Interactions between ICAM-5 and beta1 integrins regulate neuronal synapse formation. J. Cell Sci. 126, 77–89. doi: 10.1242/jcs.106674
Ogier, C., Creidy, R., Boucraut, J., Soloway, P. D., Khrestchatisky, M., and Rivera, S. (2005). Astrocyte reactivity to Fas activation is attenuated in TIMP-1 deficient mice, an in vitro study. BMC Neurosci. 6:68. doi: 10.1186/1471-2202-6-68
Oka, S., Mori, K., and Watanabe, Y. (1990). Mammalian telencephalic neurons express a segment-specific membrane glycoprotein, telencephalin. Neuroscience 35, 93–103. doi: 10.1016/0306-4522(90)90124-M
Osteen, K. G., Bruner, K. L., and Sharpe-Timms, K. L. (1996). Steroid and growth factor regulation of matrix metalloproteinase expression and endometriosis. Semin. Reprod. Endocrinol. 14, 247–255. doi: 10.1055/s-2007-1016334
Page-McCaw, A., Ewald, A. J., and Werb, Z. (2007). Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 8, 221–233. doi: 10.1038/nrm2125
Pan, F., Aldridge, G. M., Greenough, W. T., and Gan, W. B. (2010). Dendritic spine instability and insensitivity to modulation by sensory experience in a mouse model of fragile X syndrome. Proc. Natl. Acad. Sci. U.S.A. 107, 17768–17773. doi: 10.1073/pnas.1012496107
Patrick, M. K., Johnston, J. B., and Power, C. (2002). Lentiviral neuropathogenesis: comparative neuroinvasion, neurotropism, neurovirulence, and host neurosusceptibility. J. Virol. 76, 7923–7931. doi: 10.1128/JVI.76.16.7923-7931.2002
Paudel, S., Kim, Y. H., Huh, M. I., Kim, S. J., Chang, Y., Park, Y. J., et al. (2013). ADAM10 mediates N-cadherin ectodomain shedding during retinal ganglion cell differentiation in primary cultured retinal cells from the developing chick retina. J. Cell. Biochem. 114, 942–954. doi: 10.1002/jcb.24435
Peixoto, R. T., Kunz, P. A., Kwon, H., Mabb, A. M., Sabatini, B. L., Philpot, B. D., et al. (2012). Transsynaptic signaling by activity-dependent cleavage of neuroligin-1. Neuron 76, 396–409. doi: 10.1016/j.neuron.2012.07.006
Phillips, L. L., and Reeves, T. M. (2001). Interactive pathology following traumatic brain injury modifies hippocampal plasticity. Restor. Neurol. Neurosci. 19, 213–235.
Pillai-Nair, N., Panicker, A. K., Rodriguiz, R. M., Gilmore, K. L., Demyanenko, G. P., Huang, J. Z., et al. (2005). Neural cell adhesion molecule-secreting transgenic mice display abnormalities in GABAergic interneurons and alterations in behavior. J. Neurosci. 25, 4659–4671. doi: 10.1523/JNEUROSCI.0565-05.2005
Pinkstaff, J. K., Lynch, G., and Gall, C. M. (1998). Localization and seizure-regulation of integrin beta 1 mRNA in adult rat brain. Brain Res. Mol. Brain Res. 55, 265–276. doi: 10.1016/S0169-328X(98)00007-2
Planas, A. M., Sole, S., and Justicia, C. (2001). Expression and activation of matrix metalloproteinase-2 and -9 in rat brain after transient focal cerebral ischemia. Neurobiol. Dis. 8, 834–846. doi: 10.1006/nbdi.2001.0435
Porlan, E., Marti-Prado, B., Morante-Redolat, J. M., Consiglio, A., Delgado, A. C., Kypta, R., et al. (2014). MT5-MMP regulates adult neural stem cell functional quiescence through the cleavage of N-cadherin. Nat. Cell Biol. 16, 629–638. doi: 10.1038/ncb2993
Prinzen, C., Muller, U., Endres, K., Fahrenholz, F., and Postina, R. (2005). Genomic structure and functional characterization of the human ADAM10 promoter. FASEB J. 19, 1522–1524. doi: 10.1096/fj.04-3619fje
Prox, J., Rittger, A., and Saftig, P. (2012). Physiological functions of the amyloid precursor protein secretases ADAM10, BACE1, and presenilin. Exp. Brain Res. 217, 331–341. doi: 10.1007/s00221-011-2952-0
Pu, C., Broening, H. W., and Vorhees, C. V. (1996). Effect of methamphetamine on glutamate-positive neurons in the adult and developing rat somatosensory cortex. Synapse 23, 328–334. doi: 10.1002/(SICI)1098-2396(199608)23:4<328::AID-SYN11>3.0.CO;2-T
Reiss, K., Maretzky, T., Ludwig, A., Tousseyn, T., De Strooper, B., Hartmann, D., et al. (2005). ADAM10 cleavage of N-cadherin and regulation of cell-cell adhesion and beta-catenin nuclear signalling. EMBO J. 24, 742–752. doi: 10.1038/sj.emboj.7600671
Richards, D. A., Mateos, J. M., Hugel, S., De Paola, V., Caroni, P., Gahwiler, B. H., et al. (2005). Glutamate induces the rapid formation of spine head protrusions in hippocampal slice cultures. Proc. Natl. Acad. Sci. U.S.A. 102, 6166–6171. doi: 10.1073/pnas.0501881102
Rivera, S., Khrestchatisky, M., Kaczmarek, L., Rosenberg, G. A., and Jaworski, D. M. (2010). Metzincin proteases and their inhibitors: foes or friends in nervous system physiology? J. Neurosci. 30, 15337–15357. doi: 10.1523/JNEUROSCI.3467-10.2010
Rivera, S., Ogier, C., Jourquin, J., Timsit, S., Szklarczyk, A. W., Miller, K., et al. (2002). Gelatinase B and TIMP-1 are regulated in a cell- and time-dependent manner in association with neuronal death and glial reactivity after global forebrain ischemia. Eur. J. Neurosci. 15, 19–32. doi: 10.1046/j.0953-816x.2001.01838.x
Rossignol, D. A., and Frye, R. E. (2012). A review of research trends in physiological abnormalities in autism spectrum disorders: immune dysregulation, inflammation, oxidative stress, mitochondrial dysfunction and environmental toxicant exposures. Mol. Psychiatry 17, 389–401. doi: 10.1038/mp.2011.165
Rylski, M., Amborska, R., Zybura, K., Michaluk, P., Bielinska, B., Konopacki, F. A., et al. (2009). JunB is a repressor of MMP-9 transcription in depolarized rat brain neurons. Mol. Cell. Neurosci. 40, 98–110. doi: 10.1016/j.mcn.2008.09.005
Rylski, M., Amborska, R., Zybura, K., Mioduszewska, B., Michaluk, P., Jaworski, J., et al. (2008). Yin Yang 1 is a critical repressor of matrix metalloproteinase-9 expression in brain neurons. J. Biol. Chem. 283, 35140–35153. doi: 10.1074/jbc.M804540200
Sakry, D., Neitz, A., Singh, J., Frischknecht, R., Marongiu, D., Biname, F., et al. (2014). Oligodendrocyte precursor cells modulate the neuronal network by activity-dependent ectodomain cleavage of glial NG2. PLoS Biol. 12:e1001993. doi: 10.1371/journal.pbio.1001993
Sakurai, E., Hashikawa, T., Yoshihara, Y., Kaneko, S., Satoh, M., and Mori, K. (1998). Involvement of dendritic adhesion molecule telencephalin in hippocampal long-term potentiation. Neuroreport 9, 881–886. doi: 10.1097/00001756-199803300-00022
Santoro, M. R., Bray, S. M., and Warren, S. T. (2012). Molecular mechanisms of fragile X syndrome: a twenty-year perspective. Annu. Rev. Pathol. 7, 219–245. doi: 10.1146/annurev-pathol-011811-132457
Sanz, R., Ferraro, G. B., and Fournier, A. E. (2014). IgLON cell adhesion molecules are shed from the cell surface of cortical neurons to promote neuronal growth. J. Biol. Chem. 290, 4330–4342. doi: 10.1074/jbc.M114.628438
Saraceno, C., Marcello, E., Di Marino, D., Borroni, B., Claeysen, S., Perroy, J., et al. (2014). SAP97-mediated ADAM10 trafficking from Golgi outposts depends on PKC phosphorylation. Cell Death Dis. 5:e1547. doi: 10.1038/cddis.2014.492
Satz, J. S., Ostendorf, A. P., Hou, S., Turner, A., Kusano, H., Lee, J. C., et al. (2010). Distinct functions of glial and neuronal dystroglycan in the developing and adult mouse brain. J. Neurosci. 30, 14560–14572. doi: 10.1523/JNEUROSCI.3247-10.2010
Sbai, O., Ferhat, L., Bernard, A., Gueye, Y., Ould-Yahoui, A., Thiolloy, S., et al. (2008). Vesicular trafficking and secretion of matrix metalloproteinases-2, -9 and tissue inhibitor of metalloproteinases-1 in neuronal cells. Mol. Cell Neurosci. 39, 549–568. doi: 10.1016/j.mcn.2008.08.004
Schlondorff, J., Becherer, J. D., and Blobel, C. P. (2000). Intracellular maturation and localization of the tumour necrosis factor alpha convertase (TACE). Biochem. J. 347(Pt 1), 131–138. doi: 10.1042/0264-6021:3470131
Senkov, O., Sun, M., Weinhold, B., Gerardy-Schahn, R., Schachner, M., and Dityatev, A. (2006). Polysialylated neural cell adhesion molecule is involved in induction of long-term potentiation and memory acquisition and consolidation in a fear-conditioning paradigm. J. Neurosci. 26, 10888–10898. doi: 10.1523/JNEUROSCI.0878-06.2006
Shattil, S. J., Kim, C., and Ginsberg, M. H. (2010). The final steps of integrin activation: the end game. Nat. Rev. Mol. Cell Biol. 11, 288–300. doi: 10.1038/nrm2871
Sheng, M., and Lee, S. H. (2000). Growth of the NMDA receptor industrial complex. Nat. Neurosci. 3, 633–635. doi: 10.1038/76576
Shi, Y., and Ethell, I. M. (2006). Integrins control dendritic spine plasticity in hippocampal neurons through NMDA receptor and Ca2+/calmodulin-dependent protein kinase II-mediated actin reorganization. J. Neurosci. 26, 1813–1822. doi: 10.1523/JNEUROSCI.4091-05.2006
Sidhu, H., Dansie, L. E., Hickmott, P. W., Ethell, D. W., and Ethell, I. M. (2014). Genetic removal of matrix metalloproteinase 9 rescues the symptoms of fragile X syndrome in a mouse model. J. Neurosci. 34, 9867–9879. doi: 10.1523/JNEUROSCI.1162-14.2014
Siller, S. S., and Broadie, K. (2011). Neural circuit architecture defects in a Drosophila model of Fragile X syndrome are alleviated by minocycline treatment and genetic removal of matrix metalloproteinase. Dis. Model. Mech. 4, 673–685. doi: 10.1242/dmm.008045
Smith, A. C., Kupchik, Y. M., Scofield, M. D., Gipson, C. D., Wiggins, A., Thomas, C. A., et al. (2014). Synaptic plasticity mediating cocaine relapse requires matrix metalloproteinases. Nat. Neurosci. 17, 1655–1657. doi: 10.1038/nn.3846
Soh, U. J., Dores, M. R., Chen, B., and Trejo, J. (2010). Signal transduction by protease-activated receptors. Br. J. Pharmacol. 160, 191–203. doi: 10.1111/j.1476-5381.2010.00705.x
Sonderegger, P., and Matsumoto-Miyai, K. (2014). Activity-controlled proteolytic cleavage at the synapse. Trends Neurosci. 37, 413–423. doi: 10.1016/j.tins.2014.05.007
Strekalova, H., Buhmann, C., Kleene, R., Eggers, C., Saffell, J., Hemperly, J., et al. (2006). Elevated levels of neural recognition molecule L1 in the cerebrospinal fluid of patients with Alzheimer disease and other dementia syndromes. Neurobiol. Aging 27, 1–9. doi: 10.1016/j.neurobiolaging.2004.11.013
Sudhof, T. C. (2013). Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron 80, 675–690. doi: 10.1016/j.neuron.2013.10.022
Sutcliffe, J. S., Nelson, D. L., Zhang, F., Pieretti, M., Caskey, C. T., Saxe, D., et al. (1992). DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum. Mol. Genet. 1, 397–400. doi: 10.1093/hmg/1.6.397
Suzuki, K., Hayashi, Y., Nakahara, S., Kumazaki, H., Prox, J., Horiuchi, K., et al. (2012). Activity-dependent proteolytic cleavage of neuroligin-1. Neuron 76, 410–422. doi: 10.1016/j.neuron.2012.10.003
Szepesi, Z., Bijata, M., Ruszczycki, B., Kaczmarek, L., and Wlodarczyk, J. (2013). Matrix metalloproteinases regulate the formation of dendritic spine head protrusions during chemically induced long-term potentiation. PLoS ONE 8:e63314. doi: 10.1371/journal.pone.0063314
Szklarczyk, A., Lapinska, J., Rylski, M., Mckay, R. D., and Kaczmarek, L. (2002). Matrix metalloproteinase-9 undergoes expression and activation during dendritic remodeling in adult hippocampus. J. Neurosci. 22, 920–930.
Taishi, P., Sanchez, C., Wang, Y., Fang, J., Harding, J. W., and Krueger, J. M. (2001). Conditions that affect sleep alter the expression of molecules associated with synaptic plasticity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 281,R839–R845.
Tian, L., Lappalainen, J., Autero, M., Hanninen, S., Rauvala, H., and Gahmberg, C. G. (2008). Shedded neuronal ICAM-5 suppresses T-cell activation. Blood 111, 3615–3625. doi: 10.1182/blood-2007-09-111179
Tian, L., Stefanidakis, M., Ning, L., Van Lint, P., Nyman-Huttunen, H., Libert, C., et al. (2007). Activation of NMDA receptors promotes dendritic spine development through MMP-mediated ICAM-5 cleavage. J. Cell Biol. 178, 687–700. doi: 10.1083/jcb.200612097
Tippmann, F., Hundt, J., Schneider, A., Endres, K., and Fahrenholz, F. (2009). Up-regulation of the alpha-secretase ADAM10 by retinoic acid receptors and acitretin. FASEB J. 23, 1643–1654. doi: 10.1096/fj.08-121392
Uemura, K., Kuzuya, A., Aoyagi, N., Ando, K., Shimozono, Y., Ninomiya, H., et al. (2007). Amyloid beta inhibits ectodomain shedding of N-cadherin via down-regulation of cell-surface NMDA receptor. Neuroscience 145, 5–10. doi: 10.1016/j.neuroscience.2006.12.022
Uhl, G. R., Drgonova, J., and Hall, F. S. (2014). Curious cases: altered dose-response relationships in addiction genetics. Pharmacol. Ther. 141, 335–346. doi: 10.1016/j.pharmthera.2013.10.013
Vaillant, C., Meissirel, C., Mutin, M., Belin, M. F., Lund, L. R., and Thomasset, N. (2003). MMP-9 deficiency affects axonal outgrowth, migration, and apoptosis in the developing cerebellum. Mol. Cell. Neurosci. 24, 395–408. doi: 10.1016/S1044-7431(03)00196-9
van der Kooij, M. A., Fantin, M., Rejmak, E., Grosse, J., Zanoletti, O., Fournier, C., et al. (2014). Role for MMP-9 in stress-induced downregulation of nectin-3 in hippocampal CA1 and associated behavioural alterations. Nat. Commun. 5, 4995. doi: 10.1038/ncomms5995
Van Hove, I., Lemmens, K., Van De Velde, S., Verslegers, M., and Moons, L. (2012a). Matrix metalloproteinase-3 in the central nervous system: a look on the bright side. J. Neurochem. 123, 203–216. doi: 10.1111/j.1471-4159.2012.07900.x
Van Hove, I., Verslegers, M., Buyens, T., Delorme, N., Lemmens, K., Stroobants, S., et al. (2012b). An aberrant cerebellar development in mice lacking matrix metalloproteinase-3. Mol. Neurobiol. 45, 17–29. doi: 10.1007/s12035-011-8215-z
Van Wart, H. E., and Birkedal-Hansen, H. (1990). The cysteine switch: a principle of regulation of metalloproteinase activity with potential applicability to the entire matrix metalloproteinase gene family. Proc. Natl. Acad. Sci. U.S.A. 87, 5578–5582. doi: 10.1073/pnas.87.14.5578
Vawter, M. P., Usen, N., Thatcher, L., Ladenheim, B., Zhang, P., Vanderputten, D. M., et al. (2001). Characterization of human cleaved N-CAM and association with schizophrenia. Exp. Neurol. 172, 29–46. doi: 10.1006/exnr.2001.7790
Vergnolle, N., Wallace, J. L., Bunnett, N. W., and Hollenberg, M. D. (2001). Protease-activated receptors in inflammation, neuronal signaling and pain. Trends Pharmacol Sci 22, 146–152. doi: 10.1016/S0165-6147(00)01634-5
Verkerk, A. J., Pieretti, M., Sutcliffe, J. S., Fu, Y. H., Kuhl, D. P., Pizzuti, A., et al. (1991). Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65, 905–914. doi: 10.1016/0092-8674(91)90397-H
Visse, R., and Nagase, H. (2003). Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ. Res. 92, 827–839. doi: 10.1161/01.RES.0000070112.80711.3D
Volkow, N. D., Chang, L., Wang, G. J., Fowler, J. S., Franceschi, D., Sedler, M., et al. (2001). Loss of dopamine transporters in methamphetamine abusers recovers with protracted abstinence. J. Neurosci. 21, 9414–9418.
Von Gertten, C., Holmin, S., Mathiesen, T., and Nordqvist, A. C. (2003). Increases in matrix metalloproteinase-9 and tissue inhibitor of matrix metalloproteinase-1 mRNA after cerebral contusion and depolarisation. J. Neurosci. Res. 73, 803–810. doi: 10.1002/jnr.10729
Wang, W., Schulze, C. J., Suarez-Pinzon, W. L., Dyck, J. R., Sawicki, G., and Schulz, R. (2002). Intracellular action of matrix metalloproteinase-2 accounts for acute myocardial ischemia and reperfusion injury. Circulation 106, 1543–1549. doi: 10.1161/01.CIR.0000028818.33488.7B
Wang, X. B., Bozdagi, O., Nikitczuk, J. S., Zhai, Z. W., Zhou, Q., and Huntley, G. W. (2008). Extracellular proteolysis by matrix metalloproteinase-9 drives dendritic spine enlargement and long-term potentiation coordinately. Proc. Natl. Acad. Sci. U.S.A. 105, 19520–19525. doi: 10.1073/pnas.0807248105
Warren, M. S., Bradley, W. D., Gourley, S. L., Lin, Y. C., Simpson, M. A., Reichardt, L. F., et al. (2012). Integrin beta1 signals through Arg to regulate postnatal dendritic arborization, synapse density, and behavior. J. Neurosci. 32, 2824–2834. doi: 10.1523/JNEUROSCI.3942-11.2012
Wilczynski, G. M., Konopacki, F. A., Wilczek, E., Lasiecka, Z., Gorlewicz, A., Michaluk, P., et al. (2008). Important role of matrix metalloproteinase 9 in epileptogenesis. J. Cell Biol. 180, 1021–1035. doi: 10.1083/jcb.200708213
Wildering, W. C., Hermann, P. M., and Bulloch, A. G. (2002). Rapid neuromodulatory actions of integrin ligands. J. Neurosci. 22, 2419–2426.
Williams, H., Johnson, J. L., Jackson, C. L., White, S. J., and George, S. J. (2010). MMP-7 mediates cleavage of N-cadherin and promotes smooth muscle cell apoptosis. Cardiovasc. Res. 87, 137–146. doi: 10.1093/cvr/cvq042
Wojtowicz, T., and Mozrzymas, J. W. (2014). Matrix metalloprotease activity shapes the magnitude of EPSPs and spike plasticity within the hippocampal CA3 network. Hippocampus 24, 135–153. doi: 10.1002/hipo.22205
Wright, J. W., Masino, A. J., Reichert, J. R., Turner, G. D., Meighan, S. E., Meighan, P. C., et al. (2003). Ethanol-induced impairment of spatial memory and brain matrix metalloproteinases. Brain Res. 963, 252–261. doi: 10.1016/S0006-8993(02)04036-2
Xu, D., Sharma, C., and Hemler, M. E. (2009). Tetraspanin12 regulates ADAM10-dependent cleavage of amyloid precursor protein. FASEB J. 23, 3674–3681. doi: 10.1096/fj.09-133462
Ye, H., Cai, P. C., Zhou, Q., and Ma, W. L. (2011). Transforming growth factor-beta1 suppresses the up-regulation of matrix metalloproteinase-2 by lung fibroblasts in response to tumor necrosis factor-alpha. Wound Repair Regen. 19, 392–399. doi: 10.1111/j.1524-475X.2011.00680.x
Yong, V. W., Power, C., Forsyth, P., and Edwards, D. R. (2001). Metalloproteinases in biology and pathology of the nervous system. Nat. Rev. Neurosci. 2, 502–511. doi: 10.1038/35081571
Zhang, J. W., Deb, S., and Gottschall, P. E. (1998). Regional and differential expression of gelatinases in rat brain after systemic kainic acid or bicuculline administration. Eur. J. Neurosci. 10, 3358–3368. doi: 10.1046/j.1460-9568.1998.00347.x
Zhang, J., Zhang, X., and Jiang, W. (2013). Propofol impairs spatial memory consolidation and prevents learning-induced increase in hippocampal matrix metalloproteinase-9 levels in rat. Neuroreport 24, 831–836. doi: 10.1097/WNR.0b013e328364fe69
Zhang, K., Mcquibban, G. A., Silva, C., Butler, G. S., Johnston, J. B., Holden, J., et al. (2003). HIV-induced metalloproteinase processing of the chemokine stromal cell derived factor-1 causes neurodegeneration. Nat. Neurosci. 6, 1064–1071. doi: 10.1038/nn1127
Keywords: metalloproteases, MMP, adhesion, CAM, glutamate, dendritic spine
Citation: Conant K, Allen M and Lim ST (2015) Activity dependent CAM cleavage and neurotransmission. Front. Cell. Neurosci. 9:305. doi: 10.3389/fncel.2015.00305
Received: 31 March 2015; Accepted: 27 July 2015;
Published: 11 August 2015.
Edited by:
Jerzy W. Mozrzymas, Wroclaw Medical University, PolandReviewed by:
Daniela Tropea, Trinity College Dublin, IrelandAnnalisa Scimemi, The State University of New York at Albany, USA
Copyright © 2015 Conant, Allen and Lim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katherine Conant, Department of Neuroscience and Interdisciplinary Program in Neuroscience, Georgetown University Medical Center, 3970 Reservoir Road, Washington, DC 20007, USA,a2VjODRAZ2VvcmdldG93bi5lZHU=