 John A. Findlay
John A. Findlay David L. Hamilton
David L. Hamilton Michael L. J. Ashford
Michael L. J. Ashford- Division of Cardiovascular and Diabetes Medicine, School of Medicine, Ninewells Hospital and Medical School, University of Dundee, Dundee, UK
Glucose hypometabolism and impaired mitochondrial function in neurons have been suggested to play early and perhaps causative roles in Alzheimer's disease (AD) pathogenesis. Activity of the aspartic acid protease, beta-site amyloid precursor protein (APP) cleaving enzyme 1 (BACE1), responsible for beta amyloid peptide generation, has recently been demonstrated to modify glucose metabolism. We therefore examined, using a human neuroblastoma (SH-SY5Y) cell line, whether increased BACE1 activity is responsible for a reduction in cellular glucose metabolism. Overexpression of active BACE1, but not a protease-dead mutant BACE1, protein in SH-SY5Y cells reduced glucose oxidation and the basal oxygen consumption rate, which was associated with a compensatory increase in glycolysis. Increased BACE1 activity had no effect on the mitochondrial electron transfer process but was found to diminish substrate delivery to the mitochondria by inhibition of key mitochondrial decarboxylation reaction enzymes. This BACE1 activity-dependent deficit in glucose oxidation was alleviated by the presence of beta hydroxybutyrate or α-lipoic acid. Consequently our data indicate that raised cellular BACE1 activity drives reduced glucose oxidation in a human neuronal cell line through impairments in the activity of specific tricarboxylic acid cycle enzymes. Because this bioenergetic deficit is recoverable by neutraceutical compounds we suggest that such agents, perhaps in conjunction with BACE1 inhibitors, may be an effective therapeutic strategy in the early-stage management or treatment of AD.
Introduction
Alzheimer's disease (AD) is an age-related neurodegenerative disease, with the vast majority of cases described as sporadic and not currently linked with a specific gene defect. There are approximately 44 million people living with AD in the world, and this number is predicted to increase dramatically over the next three decades, severely impacting on healthcare systems and the socio-economic environment. AD encompasses large-scale neuronal loss and progressive memory and other cognitive decline culminating in severely impaired daily living. Indeed, there is a strong correlation between synapse loss and cognitive deterioration (DeKosky et al., 1996; Coleman and Yao, 2003). The neuronal cell death follows a characteristic spatial and temporal pattern; with signs of the disease initially presenting in layer II neurons of the entorhinal cortex (EC) and spreading through the hippocampus (Gómez-Isla et al., 1996). Selective atrophy and regional hypometabolism have also been shown to occur in the EC through the use of functional imaging (Masdeu et al., 2005). Allied to this, loss of cholinergic neurons and reduced cholinergic system enzyme activities in the basal forebrain have been proposed to drive early cognitive decline (DeKosky et al., 1992; Perry et al., 1992; Geula et al., 2008). Presently, we have a limited understanding of the molecular basis for these early events in sporadic AD etiology. Recent proteomic analyses have however highlighted a number of cellular processes, which become compromised. These include the pathways of metabolism and energy production, oxidative stress production and defense culminating in DNA and protein modifications impacting on cytoskeletal functioning (Sowell et al., 2009; Korolainen et al., 2010). All of these changes culminate in alterations in synaptic communication and precipitate memory loss (Forero et al., 2006). It has been hypothesized that a major driver of these changes is the aberrant folding and phosphorylation of the microtubule associated protein tau and aggregation of β-amyloid (Aβ) peptides (Kawaja, 2005). While the progression of AD is closely associated with dysfunctional tau protein and cholinergic deficits, it is generally observed by biomarker and other analyses that changes in the hallmark amyloid plaque pathology precedes these other pathogenic drivers (Shankar et al., 2009; Jack et al., 2013a,b; Knopman et al., 2013; Martiskainen et al., 2015). There has also been increasing interest in the role of brain mitochondrial dysfunction and reduced metabolic activity (Yao et al., 2009; Swerdlow et al., 2014; Wilkins et al., 2014).
It has long been recognized that glucose is the predominant substrate utilized by the adult brain under physiological conditions (Bouzier-Sore et al., 2006). Indeed, while only constituting 2% to body weight, the brain consumes around 20 and 25% of the total body oxygen consumption and glucose respectively, (McKenna et al., 2006). Furthermore, the respiratory quotient of the brain is almost exactly 1, indicating near universal carbohydrate metabolism (Sokoloff, 1999). Although glucose is the dominant substrate for brain metabolic activity, alternative substrates, such as glycogen and amino acids can play a role in central metabolism. However, due to limited supply and storage capacity, this is thought to be relatively minor under physiological conditions (Henderson et al., 2009). Glucose metabolism in the brain is tightly coupled to the generation of intracellular adenosine triphosphate (ATP), largely to support neurotransmitter production and release. Consequently, this reliance on glucose coupled with a high-energy consumption makes the brain, and synaptic transmission in particular, vulnerable to events, which lead to diminished metabolism.
Brain hypometabolism is a universal change observed during Alzheimer's disease (AD) progression and recently, it has been proposed to have a major causative role in AD pathogenesis (Blass, 2001; Costantini et al., 2008). Through the use of neuroimaging techniques, impaired brain glucose metabolism has been demonstrated to occur: before atrophy in autosomal AD cases, during the progression toward non-familial AD and in individuals at high risk of AD (i.e., APOE ε4 carriers) prior to symptom manifestation (Small et al., 1995; Johnson et al., 2001; Reiman et al., 2004; Mosconi et al., 2009a,b). This reduced brain glucose metabolism has been postulated to result from impaired mitochondrial functioning (Lustbader et al., 2004; Beal, 2005; Bubber et al., 2005), with similar changes observed in a variety of transgenic mouse models of AD (von Kienlin et al., 2005; Yao et al., 2009, 2011; Nilsen et al., 2014). Recent evidence suggests a putative link between neuronal glucose metabolism and the presence of Aβ peptides, considered by many to be the primary pathogenic driver of AD. Aβ peptides derive from increased cleavage of amyloid precursor protein (APP), which is ubiquitously expressed in tissues, with highest levels in the brain. APP is cleaved by two competing enzyme processes; an alpha secretase pathway, resulting in the soluble cleaved product sAPPα and no Aβ production and a beta secretase pathway, which releases sAPPβ and following additional cleavage of the remaining protein by γ-secretase, releases Aβ. Aβ accumulates in the mitochondria of AD patients and AD mouse models, prior to the appearance of amyloid deposits (Hirai et al., 2001; Devi et al., 2006; Manczak et al., 2006). Indeed, exogenous Aβ has been reported to decrease mitochondrial respiratory chain function and the activity of various mitochondrial dehydrogenase enzymes (Shearman et al., 1994; Casley et al., 2002a; Pereira et al., 2004; Caspersen et al., 2005), indicative of an Aβ-mediated bioenergetic deficit in cells.
Additionally, it has been shown that the hypometabolic state associated with AD may be driven, at least in part, by a region-specific shift in neuronal metabolism toward aerobic glycolysis (reduced oxidative metabolism in the presence of adequate oxygen supply; Vaishnavi et al., 2010). This switch in glucose metabolism was shown to closely correlate with Aβ deposition and later vulnerability to cell death during progression toward AD (Vlassenko et al., 2010). These findings clearly implicate the modulation of APP processing as a driver of the altered metabolic state observed in early, pre-AD states. A key enzyme driving excess Aβ production, as observed in AD, is the aspartyl protease, β-site APP cleaving enzyme 1 (BACE1). BACE1 was initially characterized as the enzyme controlling the rate-limiting step in Aβ generation (Hussain et al., 1999; Sinha et al., 1999; Vassar et al., 1999). More recently however, it has also been proposed to play a role in glucose metabolism with whole body knock out of BACE1 resulting in improved insulin sensitivity and glucose homeostasis (Meakin et al., 2012). Furthermore, genetic and pharmacological manipulation of APP processing can directly alter glucose uptake and metabolism in the C2C12 skeletal muscle cell line (Hamilton et al., 2014). Importantly, BACE1 is a stress-sensitive protease, with oxidative, hypoxic, inflammatory, and metabolic stress (all associated with AD initiation and/or progression) demonstrated to increase BACE1 levels and activity, causing APP processing to shift from the physiologically predominant alpha-secretase, to the beta-secretase, pathway, and increasing Aβ levels. Indeed, recent work has demonstrated a role for oxidative, lipid and metabolic stressors in regulating BACE1 gene and protein expression as well as activity (Puglielli et al., 2003; Velliquette et al., 2005; Zhang et al., 2007; O'Connor et al., 2008; Guglielmotto et al., 2009). Furthermore, alterations in its expression result from changes in micro RNA (miR) regulation of the 5′ untranslated region (UTR) of BACE1 have been demonstrated in sporadic AD cases (Nelson and Wang, 2010; Fang et al., 2012; Lei et al., 2015). Consequently, chronic stress events and altered translational regulation may in turn culminate in the consistent reports that BACE1 mRNA, protein and activity are elevated in AD brains (Fukumoto et al., 2002; Holsinger et al., 2002; Tyler et al., 2002; Yang et al., 2003; Li et al., 2004; Harada et al., 2006; Zhao et al., 2007).
Given that Aβ directly impairs mitochondrial enzyme function and that AD is associated with impaired glucose metabolism we hypothesized that manipulating APP processing through BACE1 overexpression in SH-SY5Y neuroblastoma cells would phenocopy the defects in glucose metabolism at the cellular level, allowing us to explore in more detail this initial and potentially causative change in AD progression.
Materials and Methods
Cell Culture
SH-SY5Y cells were cultured under aseptic conditions and maintained in a humidified atmosphere of 95% air and 5% CO2. Cells were maintained in Dulbecco's Modified Eagle's Medium (DMEM) F-12 media (Gibco Life Technologies, Paisley, UK) supplemented with 10% fetal bovine serum (Sera Laboratories, West Sussex, UK), 4 mM L-glutamine (Gibco) and 2% Penicillin streptomycin (100 units/ml; Gibco). SH-SY5Y cells were transfected with 12 μg DNA of pcDNA3.1 containing empty vector (SH-SY5YEV), full-length human BACE1 (SH-SY5YB1), or BACE1 active site mutant (SH-SY5YmB1) using Lipofectamine 2000 (Invitrogen Life Technologies, Paisley, UK). Stable cells were selected and lines maintained using 1 and 0.5 mg/ml G418 sulfate respectively, (Sigma-Aldrich, Gillingham, UK) as selection antibiotic. A minimum of two independently generated stable cell lines, with concurrently produced EV controls, were used for these studies. Prior to some glucose oxidation assays, cells were treated overnight, as indicated, with the pyruvate dehydrogenase inhibitor, dichloroacetate (DCA; Sigma-Aldrich) or growth media supplemented with α-lipoic acid (Sigma-Aldrich) for 48 h.
Cloning
Full length myc-his tagged human BACE1 in pcDNA3.1 was obtained from GlaxoSmithKline (GSK; Harlow, UK) and mBACE1 (a kind gift from Professor Wolfe (Brigham and Women's Hospital, Boston)) was sub-cloned into pcDNA3.1.
Immunoblotting and Gene Expression
Protein isolation and immunoblotting procedures were as described previously (Mirshamsi et al., 2004). For relative quantification of APP cleavage by alpha-secretase (sAPPα) vs. beta-secretase (sAPPβ) pathways, cells were incubated for between 20 and 24 h in Optimem (Gibco), and media concentrated (using 30 kDa Amicon Ultra 15 ml filters; Merck Millipore, Livingston, UK) by centrifugation (4000 × g) and subjected to SDS-PAGE with amounts presented relative to total protein. Protein antibodies used were: anti-Actin (Sigma-Aldrich; 1:5000), anti-APP (Ab54, GSK; 1:4000), anti-sAPPα (Cambridge Bioscience, Cambridge, UK; 1:1000), anti-sAPPβ (GSK; 1:1000), anti-BACE1 (Sigma-Aldrich; 1:1000), anti-BAD (New England Biolabs, Hitchin, UK; 1:1000), with anti-total PDH (1 mg/ml) and anti-pPDHe1α (1 mg/ml) from Drug Discovery Unit, University of Dundee.
Glucose Oxidation Assay
SH-SY5YEV, SH-SY5YB1, or SH-SY5YmB1 cells were plated into 6-well cell culture plates and any pre-treatments carried out as described above and in the Results Section. To begin the assay, cells were washed twice with Hepes-buffered saline (HBS (in mM); 140 NaCl, 20 Hepes, 5 KCl, 2.5 MgSO4, and 1 CaCl2, pH 7.4) and incubated in HBS containing 2.5 mM glucose and 74 kBq/ml D-[U-14C]glucose (PerkinElmer) along with any relevant inhibitors or treatments indicated in the Results Section for 3 h at 37°C. Media were transferred and 14CO2 liberated by acidification with 60% perchloric acid, trapped by Whatman (GF/B) filter paper discs pre-soaked with 1 M KOH and radioactivity quantified by liquid-scintillation counting. Cells from the assay were washed twice with ice-cold 0.9% NaCl and lysed with 1 ml of 50 mM NaOH, and the radioactivity contained within the lysate quantified by liquid-scintillation counting, which served as a measure of the 14C incorporation into the cell during the assay period. Total protein content was determined in the lysate via the Bradford method and used to normalize glucose incorporation and oxidation rates for each sample.
Cellular Respiration
The Seahorse Extracellular Flux Analyser utilizes solid sensors that simultaneously monitor the oxygenation and pH of the media. The rate of oxygen consumption (OCR) and extracellular acidification rates (ECAR) can therefore be assessed in near real time allowing for high resolution changes in a range of metabolic parameters. SH-SY5YEV, SH-SY5YB1, or SH-SY5YmB1 cell monolayers were seeded into XF 24-well culture microplates (Seahorse Bioscience, Copenhagen, Denmark) the day prior to treatment or assay as indicated in the Results Section. Optimal cell number was determined following a cell titration assay taking into account oxygen consumption rate (OCR) and extracellular acidification rate (ECAR), oxygen tension values and the appearance of the cell monolayers and was determined to be 40,000 cells. On the day of the assay, cells were placed in unbuffered DMEM with relevant inhibitors/treatments as indicated and placed in a non-CO2 incubator for 1 h (to de-gas solutions) prior to assay initiation. Standard 3 min mix, 2 min wait, and 3 min measure cycles were used; with five baseline measurements taken before, and a subsequent three measurements acquired following, drug additions.
Enzyme Activity Assays
Activity assays for pyruvate dehydrogenase (PDH; Abcam, Cambridge, UK), α-ketoglutarate dehydrogenase (α-KGDH; Antibodies Online, Aachen, Germany), isocitrate dehydrogenase (IDH; Sigma-Aldrich), and fumarase (Abcam) were performed according to the manufacturer's instructions.
Results
Overexpression of BACE1 Increases Amyloidogenic APP Processing and Suppresses Glucose Oxidation
Control, empty vector-treated (SH-SY5YEV) and BACE1 overexpressing (SH-SY5YB1) cells displayed equivalent APP protein levels, showing the three predominant APP transcript protein isoforms present in neurons (Huang and Jiang, 2011; Figures 1A,B). As expected, SH-SY5YB1 cells exhibited significantly higher BACE1 protein levels (Figures 1A,C), which resulted in altered APP cleavage; promoting a substantial shift from non-amyloidogenic to amyloidogenic processing (as denoted by increased sAPPβ and decreased sAPPα levels (Figures 1A,D,E). Overexpression of BACE1 resulted in a marked reduction in the rate of 14C-glucose oxidation, compared to SH-SY5YEV cells (Figure 1F), in agreement with recent findings in C2C12 skeletal muscle cells (Hamilton et al., 2014). The decrease in glucose oxidation is not due to impaired glucose incorporation into the cell (Figure 1G), and suggests that increased BACE1 protein levels and activity cause a fundamental change in the ability of SH-SY5Y cells to oxidize glucose (Figure 1H).
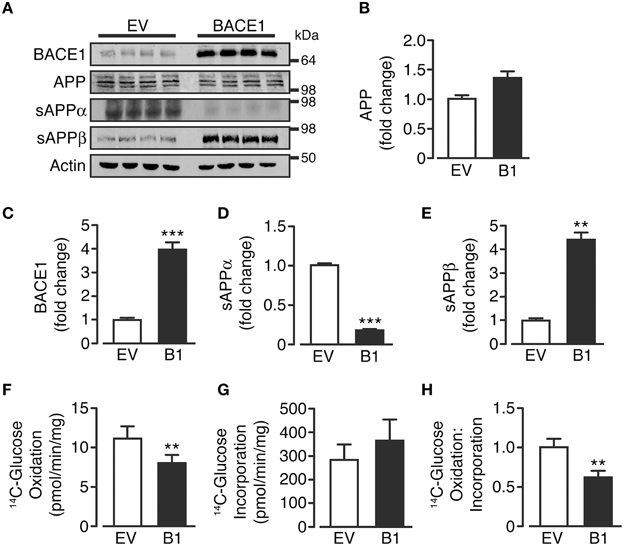
Figure 1. BACE1 overexpression alters APP processing and glucose oxidation in SH-SY5Y cells. (A) Representative immunoblots showing stable overexpression of BACE1 and resultant changes in cellular APP and sAPPα and sAPPβ shed into the culture media, compared to empty vector (EV) treated controls. (B–E) SH-SY5Y BACE1 overexpression did not change APP protein levels (B; n = 4), increased total BACE1 protein levels (C; n = 7) and induced a shift in APP processing, with a reduction in sAPPα (D; n = 4) and an increase in sAPPβ (E; n = 4). (F–H) SH-SY5Y BACE1 overexpression reduced 14C-glucose oxidation rate (F; n = 7), with no reduction in 14C-glucose incorporation rate (G; n = 9), giving an overall reduction in the ratio of 14C-glucose oxidation to incorporation (H; n = 7). Values are means ± SEM. **p < 0.01; ***p < 0.001.
In an attempt to discern whether the changes in glucose oxidation rate were dependent upon the secretase activity of BACE1, a concomitant group of cells that overexpress a mutant form of BACE1, devoid of secretase activity (SH-SY5YmB1; Mowrer and Wolfe, 2008) were examined. BACE1 protein levels in SH-SY5YmB1 cells were raised to a similar extent to that observed for SH-SY5YB1 cells, with no alteration in APP protein levels (Figures 2A–C). Interestingly, the overexpression of the secretase-dead BACE1 mutant exerted a dominant negative effect over APP processing with reduced sAPPβ release, compared with SH-SY5YB1 and SH-SY5YEV cells, into the culture media (Figures 2A,D). The reduced β-secretase cleavage of APP in the presence of mBACE1 was also mirrored in its effect on glucose oxidation rate, with no reduction in glucose oxidation concomitant with comparable glucose incorporation between cell types (Figures 2E,F), indicating that increased BACE1 activity is required to depress glucose oxidation in SH-SY5Y cells (Figure 2G).
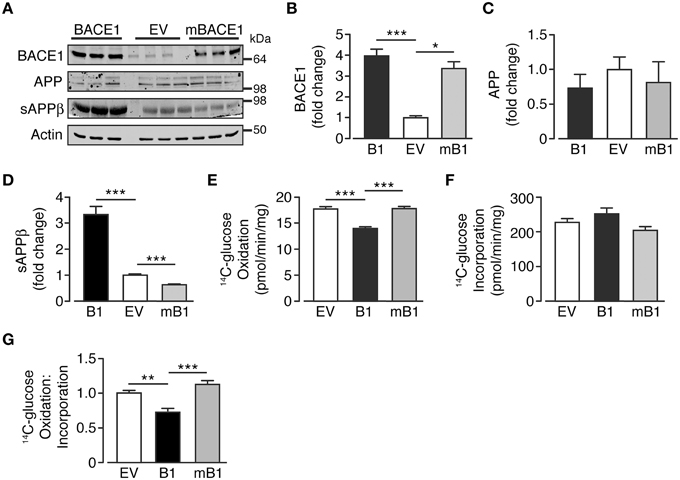
Figure 2. Glucose oxidation in SH-SY5Y cells in not impaired following overexpression of secretase-dead BACE1 protein. (A) Representative immunoblots showing stable overexpression of wild type BACE1 and a secretase-dead BACE1 mutant (mBACE1) and resultant changes in cellular APP and sAPPβ shed into the culture media. (B–D) SH-SY5Y mBACE1 overexpression increased total BACE1 to levels equivalent to BACE1 overexpressed cells and ~3-4X greater than empty vector (EV) controls (B; n = 5), with no effect on APP levels (C; n = 3), but in contrast to BACE1 overexpression, mBACE1 reduced sAPPβ to levels below that of the EV controls (D; n = 6–10). (E–G) SH-SY5Y mBACE1 overexpression had no effect on 14C-glucose oxidation compared to EV controls, in contrast to the reduction in 14C-glucose oxidation observed in BACE1 overexpressing cells (E; n = 5), with mBACE1 having no effect on 14C-glucose incorporation (F; n = 5), resulting in the ratio of 14C-glucose oxidation to incorporation being unchanged in mBACE1 cells (G; n = 5). Values are means ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
Chronic Elevation of BACE1 Stimulates Aerobic Glycolysis in SH-SY5Y Cells
The cellular pathways that facilitate glucose metabolism are glycolysis (and the pentose phosphate pathway), which occurs in the cytoplasm and the TCA cycle and oxidative phosphorylation, which are present in the matrix and inner mitochondrial membrane, respectively. Actively respiring mitochondria consume oxygen and therefore oxygen consumption rate (OCR) can be taken as a measure of substrate flux through the oxidative phosphorylation pathway while extracellular acidification rate (ECAR) reflects the release of lactate (lactic acid) converted from pyruvate following glycolysis. SH-SY5YB1 cells exhibit decreased OCR concurrent with increased ECAR in comparison to SH-SY5YEV cells (Figures 3A,B). Taken together these changes reflect a robust shift between these metabolic processes as shown by the change in the ratio of oxidative phosphorylation to glycolytic metabolism in cells with increased BACE1 activity (Figure 3C).
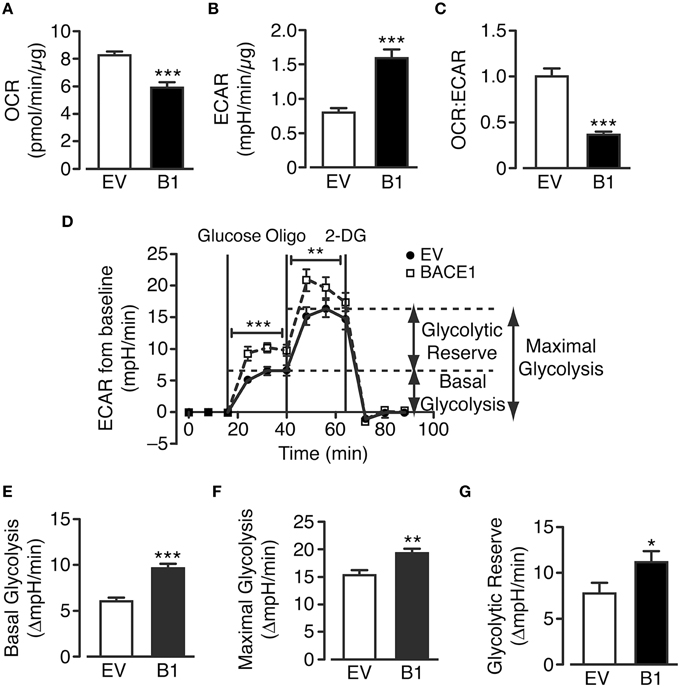
Figure 3. BACE1 overexpression alters glucose metabolism in SH-SY5Y cells. (A–C) BACE1 overexpression reduces oxygen consumption rate (OCR) (A) and increases the extracellular acidification rate (ECAR) (B) resulting in a reduced OCR:ECAR ratio (C), compared to EV cells (n = 7–12). (D) Glycolysis stress test temporal profile for EV and BACE1 SH-SY5Y cells showing the effects on ECAR of the sequential addition of 2.5 mM glucose, 1 μM oligomycin (Oligo) and 25 mM 2-deoxyglucose (2DG). (E–G) BACE1 overexpression increased basal glycolysis (E), maximal glycolysis (F) and glycolytic reserve (G) compared to EV-treated cells (n = 18–21). Values are means ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
To further investigate these changes, a glycolysis stress test (which assesses basal glycolysis, glycolytic capacity and glycolytic reserve) was performed (Figure 3D). SH-SY5YEV and SH-SY5YB1 cells were incubated in media containing zero glucose, 2.5 mM pyruvate and 4 mM L-glutamine for 100 min, following which stimulation of basal glycolysis was achieved by the injection of 2.5 mM glucose into the assay media. The increase in basal ECAR was significantly higher in SH-SY5YB1 cells, compared to control cells, indicating that the raised BACE1 activity augmented aerobic glycolysis (Figure 3E). Inhibition of F1F0 ATP synthase by oligomycin leaves cells wholly reliant on glycolysis for ATP generation (termed maximal glycolysis). This rate was also significantly increased in SH-SY5YB1, compared to control, cells following BACE1 overexpression (Figure 3F). Finally, the cellular glycolytic reserve is given by the difference between basal and maximal glycolysis, with a significantly higher glycolytic reserve observed in the SH-SY5YB1 cells (Figure 3G). Collectively, these data indicate that raised BACE1 activity depresses glucose oxidation in SH-SY5Y cells and, as a result, there is a compensatory increase in glucose metabolism through aerobic glycolysis.
Mitochondrial Efficiency is Unaltered by Raised BACE1 Activity in SH-SY5Y Cells
The marked reduction in oxidative phosphorylation, concurrent with increased glycolytic metabolism in SH-SY5YB1 cells, indicated that increased BACE1 activity reduced substrate delivery to the mitochondria and/or impaired mitochondrial function. To test mitochondrial efficiency in these cells, a modified Mitochondrial Stress Test (Seahorse Bioscience) protocol was utilized. As previously reported, a limited maximal respiration rate is achievable in undifferentiated SH-SY5Y cells (Xun et al., 2012) in the absence of pyruvate in the assay media. Consequently, we therefore performed a split assay to investigate the effects of mitochondrial metabolism perturbation: the first part measuring the proportion of respiration dedicated to ATP generation (ATP synthase inhibition with oligomycin), the maintenance of mitochondrial leak (a combination of rotenone (complex I inhibition) and antimycin A (complex III inhibition) to minimize mitochondrial respiration) and the relative contribution of non-mitochondrial OCR (Figure 4A). However, increased BACE1 activity in SH-SY5Y cells had no effect on any of these oxidative parameters (Figures 4B–D). The second part encompasses a test of the cellular reserve capacity via the induction of maximal respiration through addition of the proton ionophore FCCP. This drug collapses the mitochondrial membrane potential, leading to uncoupled (no ATP generation) substrate flux through the mitochondria, promoting an increase in glycolysis in an attempt to maintain intracellular ATP levels. Raised BACE1 activity also had no effect on SH-SY5Y mitochondrial reserve capacity (Figures 4E,F). The results from these assays show that chronic elevation of BACE1 protein expression and activity does not impair mitochondrial electron transfer function. Consequently, this result strongly indicated that the BACE1 activity-driven decrease in glucose oxidation was the result of diminished substrate delivery to the mitochondria.
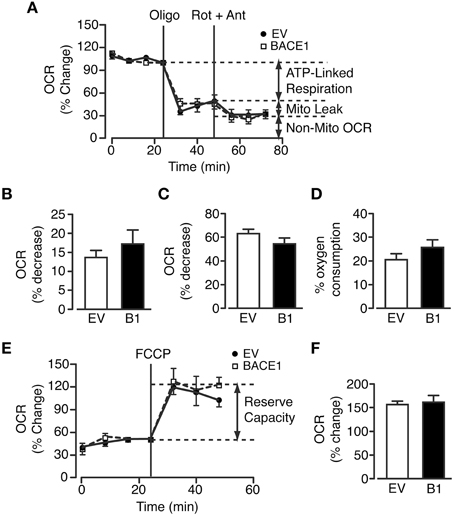
Figure 4. BACE1 overexpression has no effect on ATP-linked, maximal or leak mitochondrial oxygen consumption. (A) Modified mitochondrial stress temporal profile for EV and BACE1 SH-SY5Y cells showing the effects on the percentage change in normalized OCR of the sequential addition of 1 μM oligomycin (oligo) and 2 μM rotenone and antimycin (Rot + Ant). (B–D) BACE1 overexpression had no effect on ATP-linked OCR (B), on the OCR required to maintain the mitochondrial leak (C) or the non-mitochondrial OCR (D) compared to EV cells (n = 8). (E) Temporal profile showing the normalized baseline OCR for EV and BACE1 cells and the maximal OCR (reserve capacity) attained following addition of 0.2 μM carboyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP). (F) BACE1 overexpression has no effect on the percentage OCR increase following FCCP addition compared to EV cells (n = 9–10). Values are means ± SEM.
Raised BACE1 Activity Lesions Key Metabolic Pathways in Oxidative Glucose Metabolism
There are three key control points involved in the regulation of neuronal metabolism, these are the generation of glucose-6-phosphate, pyruvate and acetyl CoA. In neurons, pyruvate is predominantly produced directly from metabolism of glucose or indirectly, through the provision of extracellular lactate via the astrocyte-neuron lactate shuttle (Magistretti and Pellerin, 1999; Itoh et al., 2003). Therefore, in an attempt to better understand the changes in cellular metabolism induced by increased BACE1 activity, lactate utilization by SH-SY5Y cells was assessed. SH-SY5YB1 cells displayed significantly reduced lactate consumption as assessed by the ability of lactate to repress 14C-labeled glucose oxidation (Figure 5A). This deficit could also be observed as a reduction in OCR when cells were provided physiological concentrations (0.5 or 2 mM; Phypers and Pierce, 2006) of lactate as sole substrate, although the reduction in lactate metabolism was overcome by the presentation of a higher (4 mM) lactate concentration (Figures 5A,B). As the oxidation of both glucose and lactate was diminished, we hypothesized that raised BACE1 activity resulted in impairment of the pyruvate dehydrogenase complex activity (PDH). To address this we used an indirect PDH activity assay and demonstrated that SH-SY5YB1 cells displayed a marked reduction of PDH activity (Figure 5C). Consistent with this outcome, SH-SY5YB1cells displayed reduced OCR when provided with pyruvate (2.5 mM) as the sole substrate (Figure 5D).
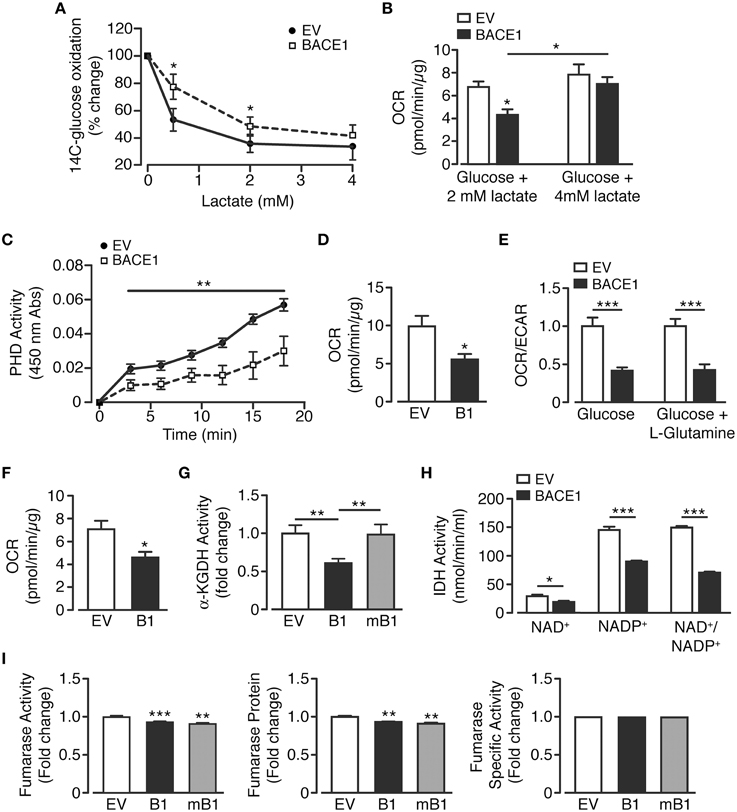
Figure 5. BACE1 overexpression induces specific mitochondrial TCA cycle enzyme lesions. (A,B) SH-SY5Y cells overexpressing BACE1 exhibit reduced lactate usage at levels ≤ 2mM compared to EV cells as demonstrated by (A) reduced repression of 14C-glucose oxidation (n = 6) and (B) direct OCR when lactate provided as sole substrate (n = 6–11). (C) Pyruvate dehydrogenase activity (PDH) is reduced in BACE1 overexpressing, compared to EV, cells (n = 5). (D) OCR when EV and BACE1 cells provided 2.5 mM pyruvate as sole substrate (n = 5). (E) OCR of EV and BACE1 SH-SY5Y cells when provided with 2.5 mM glucose alone or 2.5 mM glucose + 4 mM glutamine (n = 4–10). (F) direct OCR when 4 mM glutamine provided as sole substrate (n = 8). (G) BACE1, but not mBACE1, overexpression, reduced the enzyme activity of α-KGDH, compared to EV cells (n = 6). (H) IDH activity (NAD+ and NADP+ isoforms) is reduced by BACE1 overexpression compared to EV cells (n = 4). (I) BACE1 or mBACE1 overexpression has no effect on fumarase enzyme activity, protein levels or specific activity compared to EV cells (n = 7). Values are means ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
In response to cellular injury or stress, compensatory metabolic strategies are employed in an attempt to bypass reduced glucose utilization associated with decreased PDH activity and obviate impaired mitochondrial TCA function. For example, glutamine metabolism is diverted from an oxidative to reductive route (Wise et al., 2011; Mullen et al., 2012), causing increased conversion to glutamate and, through glutamate dehydrogenase, raised levels of α-ketoglutarate. This additional source of α-ketoglutarate should supplement ATP generation through the TCA cycle as it bypasses the block at PDH. Consequently, we examined whether such an alternative route for TCA substrate replenishment was capable of recovering oxidative metabolism. Thus, OCR and ECAR were measured when SH-SY5YEV and SH-SY5YB1 cells were provided glucose (2.5 mM) alone or glucose (2.5 mM) + glutamine (4 mM). However, the presence of glutamine did not affect the attenuation of OCR or enhancement of ECAR in SH-SY5YB1 cells (Figure 5E). Moreover, OCR was reduced in SH-SY5YB1 cells when glutamine was provided as the exclusive substrate (Figure 5F). These data indicate that either glutamine does not play a significant role in SH-SY5Y basal metabolism and/or that raised BACE1 activity blunts additional TCA enzymes, such as α-ketoglutarate dehydrogenase (α-KGDH), which converts α-ketoglutarate to succinyl-CoA. Indeed, SH-SY5YB1 cells showed a large, BACE1 secretase activity-dependent, reduction in α-KGDH activity, which is a rate controlling step of the TCA cycle (Figure 5G).
Because, PDH and α-KGDH catalyze decarboxylation reactions involved in mitochondrial bioenergetics, we assayed the activity of the third enzyme utilizing this process, isocitrate dehydrogenase (IDH). Three isoforms of IDH are present in cells, IDH3 in the mitochondrial matrix, which reduces NAD+ to NADH, and IDH1 (cytoplasmic and peroxisomal) and IDH2 (mitochondrial) reducing NADP+ to NADPH. Consequently, specific isoform function was determined by differential supplementation (NAD+ or NADP+) of the reaction mixes. In agreement with the results for the decarboxylation enzymes above, raised BACE1 activity resulted in depression of NAD+- and NADP+-dependent IHD activity (Figure 5H). However, impairment of TCA cycle enzyme activities by increased BACE1 activity was not universal. For example, although fumarase activity was reduced slightly in SH-SY5YB1 cells, this was matched in SH-SY5YmB1 cells, with the protein levels of fumarase also decreased in both these cell lines, resulting in unaltered specific activity of fumarase by raised BACE1 in SH-SY5Y cells (Figure 5I). Thus, manipulation of APP cleavage toward amyloidogenic processing by increased BACE1 activity in SH-SY5Y cells reduces the overall catabolic capacity of the TCA cycle through specific enzyme lesions rather than a wholesale down-regulation.
Rescue of Glucose Oxidation in SH-SY5YB1 Cells
As glutamine supplementation was unable to recover BACE1-mediated inhibition of glucose oxidation, we focused on PDH as a potential target for pharmacological or alternative nutrient interventions. A key regulator of PDH activity is by phosphorylation of its E1α subunit (Patel et al., 2014), which is predominantly controlled by the inhibitory effect of pyruvate dehydrogenase kinases (PDKs) vs. activation through de-phosphorylation by pyruvate dehydrogenase phosphatase (PDP). Dichloroacetate (DCA) is a PDK1 inhibitor, which enhances oxidative phosphorylation in the brain (Itoh et al., 2003) and has been promoted clinically for its anti-neoplastic effects (Kankotia and Stacpoole, 2014). Consistent with PDH inhibition, SH-SY5YB1 cells exhibited increased phosphorylation of the e1α subunit at serine 293 compared to SH-SY5YEV cells (Figure 6A). Treatment with DCA (10 and 100 μM) resulted in decreased levels of Ser293 e1α phosphorylation of both SH-SY5Y cell types, although Ser293 e1α phosphorylation remained significantly higher in SH-SY5YB1 cells (Figures 6A,B). In agreement with the reduction in e1α phosphorylation, incubation of SH-SY5YEV cells with DCA (100 μM) increased the glucose oxidation rate (Figure 6C). However, although glucose oxidation of SH-SY5YB1 cells was also enhanced by DCA treatment, this remained significantly lower than that of the SH-SY5YEV cells, mirroring e1α phosphorylation status.
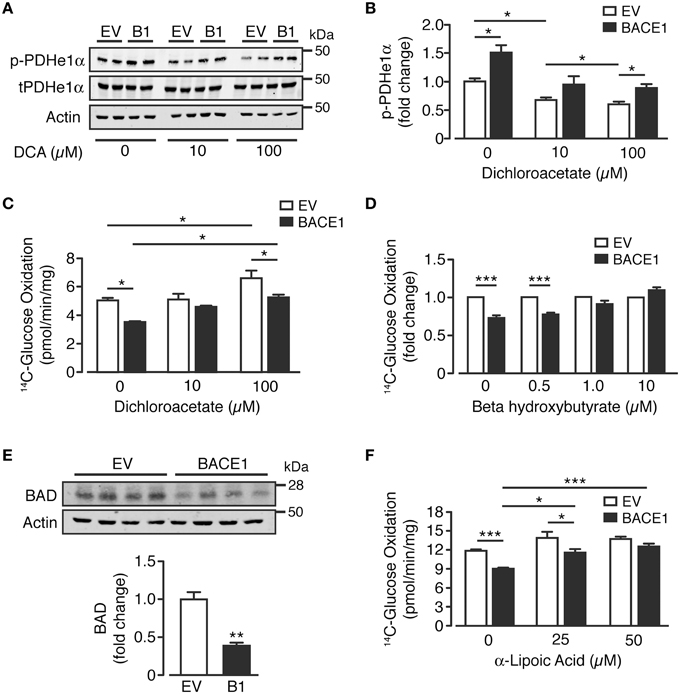
Figure 6. Reversal of BACE1 mediated impaired glucose oxidation in SH-SY5Y cells. (A,B) Representative immunoblots (A) of phosphorylated PDHe1α (p-PDHe1α) subunit, total PDHe1α and actin loading control in control (EV) and BACE1 overexpressing cells in the absence and presence of 10 μM or 100 μM dichloroacetate (DCA), with quantification of the immunoblot data shown in (B) (n = 4). (C) DCA partially reverses BACE1 mediated impairment of glucose oxidation, but also increases OCR of EV cells (n = 5). (D) reduced OCR associated with BACE1 cells is recovered to EV control levels by addition of 1 μM and 10 μM beta hydroxybutyrate (n = 4–6). (E) Representative immunoblots of BCL-2-associated agonist of cell death (BAD) and actin loading control with quantification of the immunoblot data (n = 4). (F) BACE1-mediated reduction in OCR is recovered to EV control levels by addition of 50 μM α-lipoic acid (n = 6). Values are means ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
An alternative means of bypassing PDH is through the application of ketones, such as beta-hydroxybutyrate (BHB), which is metabolized to acetyl-CoA and enters the TCA cycle at oxaloacetate. The presence of BHB (0.5–10 μM) recovered the relative deficit in glucose oxidation of SH-SY5YB1 cells, compared to SH-SY5YEV cells, in a concentration-dependent manner (Figure 6D). Previous studies (Danial, 2008; Giménez-Cassina et al., 2012) have indicated that diminished mitochondrial consumption of glucose in neurons and the ability to switch substrate preference to ketones is dependent on the presence and activity of BAD (BCL-2-associated agonist of cell death), a member of the BCL-2 gene family member of apoptotic control proteins, with increased mitochondrial usage of BHB in Bad−∕− cortical neurons. In agreement with this model, we find that the increased ability of SH-SY5YB1 cells to utilize BHB over SH-SY5YEV cells and enhance glucose oxidation, is associated with a large reduction of BAD protein expression (Figure 6E). Finally we also investigated the ability of the naturally occurring enzyme co-factor, α-lipoic acid to modify glucose oxidation of SH-SY5YB1 cells as previous studies have shown this compound to up-regulate mitochondrial bioenergetics and promote glucose uptake (Packer and Cadenas, 2011; Sancheti et al., 2013). Indeed PDH and α-KGDH protein complexes require α-lipoic acid as a co-factor for their acyl transferase activity. Supplementation of the growth media with α-lipoic acid (25 or 50 μM for 48 h) resulted in a robust concentration-dependent attenuation of the impaired glucose oxidation rate displayed by SH-SY5YB1, compared to SH-SY5YEV cells (Figure 6F). Taken together, these results demonstrate the applicability of alternative nutrient (BHB) or co-factor (α–lipoic acid) supplementation to alleviate or by-pass the impaired glucose metabolism elicited by raised BACE1 activity in SH-SY5Y cells.
Discussion
In this study we demonstrate that raised BACE1 activity by overexpression in SH-SY5Y cells, and subsequent modification of APP metabolism, induces a shift from the physiologically predominant alpha-secretase cleavage pathway to the amyloidogenic beta-secretase cleavage pathway, which results in decreased glucose metabolism. This bioenergetic impairment was dependent upon the secretase activity of BACE1 and produced a fundamental shift in the cellular metabolic profile, characterized by reduced glucose oxidation in association with a compensatory increase in glycolysis, in an attempt to maintain ATP production.
Decreased brain glucose metabolism is an invariant pathophysiological event occurring in AD progression, and it has been hypothesized that this may occur years, and even decades prior to symptom presentation (Mosconi et al., 2009a,b; Bateman et al., 2012; Jack et al., 2013c). Impaired glucose metabolism has also been noted in central tissues taken from AD animal models (von Kienlin et al., 2005; Sancheti et al., 2013; Nilsen et al., 2014). Furthermore, despite the observation that reduced glucose metabolism is predictive of later cognitive decline and AD symptom presentation, relatively little is known about the cellular mechanisms underlying these changes. Interestingly, the metabolic shift we have observed in favor of aerobic glycolysis under conditions of increased BACE1 activity mirrors early and predictive changes occurring in the brains of people who later develop AD. The brain areas that display this profile overlap with regions showing the greatest prevalence of Aβ pathology, future susceptibility to cell death and are predictive of cognitive decline (Vaishnavi et al., 2010; Vlassenko et al., 2010).
The mechanisms underlying glucose hypometabolism in AD are not well-understood. However, mitochondrial dysfunction has been widely reported in clinical and experimental AD studies (Lustbader et al., 2004; Beal, 2005; Bubber et al., 2005) and Aβ has been reported to accumulate in the mitochondria of AD patients and transgenic AD mouse models prior to amyloid deposition (Hirai et al., 2001; Devi et al., 2006; Manczak et al., 2006; Du et al., 2008). Although there are numerous reports of impaired electron transfer in mitochondria, for example via diminished activity in complexes I to V in AD subjects (Parker et al., 1994; Maurer et al., 2000; Kim et al., 2001; Bosetti et al., 2002), and diminished complex III and IV activity in a transgenic AD mouse model (Mucke et al., 2000) we found no effect of raised BACE1 activity on mitochondrial electron transfer function in SH-SY5Y cells. It may be that additional injurious processes, such as increased oxidative and/or inflammatory stress are required to act concurrently with raised BACE1 activity and increased levels of Aβ to elicit this outcome.
Thus the driving force for decreased glucose oxidation in SH-SY5YB1 cells appears to be through reduced substrate delivery to mitochondria by a BACE1 activity-dependent impairment of specific TCA cycle enzymes; PDH, αKGDH and IDH each of which utilize decarboxylation reactions. Previous studies on post-mortem AD brains indicate reduced levels and/or activity of these key enzymes: PDH, αKGDH and IDH (Sorbi et al., 1983; Butterworth and Besnard, 1990; Mastrogiacoma et al., 1996; Gibson et al., 2000; Ko et al., 2001; Bubber et al., 2005), with PDH and αKGDH exhibiting the largest decreases in activity. Furthermore, Aβ peptides have been demonstrated to inhibit PDH and αKGDH directly (Shearman et al., 1994; Casley et al., 2002a,b). In contrast we find no change in the activity of the TCA enzyme fumarase, the activity of which is also unaltered in AD brains (Bubber et al., 2005). Our results add to these findings by implicating raised BACE1 activity and manipulation of APP processing as central to these enzyme deficits and metabolic adaptations at the cellular level, indicating a key role for BACE1 and its up-regulation in response to oxidative and inflammatory stress, which are associated with the very early stages of AD (Nunomura et al., 2001; Zhang et al., 2007; Guglielmotto et al., 2009). Indeed, these results show that raised BACE1 activity effectively phenocopies some of the earliest changes in glucose metabolism seen in the brain during the progression toward AD.
In situations of diminished glucose utilization, there are a number of strategies that may be engaged in an attempt to bypass this block in metabolism. For example, in brain ischemic-reperfusion injury there is also decline in glucose oxidation and PDH activity (Bogaert et al., 1994; Zaidan et al., 1998). Application of glutamine has been demonstrated to offer some protection against ischemic-reperfusion injury in peripheral tissues by providing an alternative energy source for the cell to maintain ATP content (Arsenian, 1998; Wischmeyer et al., 2003). In addition to providing a source of carbon for neurotransmitter production, glutamine can also feed into the TCA cycle and be metabolized to increase levels of α-ketoglutarate and succinate. However, our data demonstrate that glutamine supplementation is unable to restore oxidative glucose metabolism in cells overexpressing BACE1. Furthermore, SH-SY5YB1 cells exhibited diminished oxidation of glutamine when applied as sole substrate, in line with the reduced activity of α-KGDH observed in these cells.
The inhibition of PDH activity in SH-SY5YB1 cells was associated with increased phosphorylation of the e1α subunit at Ser293, which is the main inhibitory site for PDH activity (Jha et al., 2012). Although, PDK1 inhibition with dichloroacetate reduced e1α phosphorylation levels, this was also observed in SH-SY5YEV cells, in conjunction with a maintained relative lower glucose oxidation in SH-SY5YB1 cells. This outcome may be owing to the presence of BACE1 activity-sensitive DCA-resistant PDK isoenzyme, or PDP, or an alternative kinase that phosphorylates e1α at Ser293 (e.g. GSK3). In contrast, application of the ketone, BHB, to SH-SY5YB1 cells demonstrates complete recovery of glucose oxidation, relative to SH-SY5YEV cells, presumably by directly increasing the availability of acetyl CoA to the TCA cycle, thus circumventing PDH. The enhanced ability to utilize ketone bodies to aid glucose oxidation in SH-SY5YB1 cells may result from their reduced BAD levels, compared to SH-SY5YEV cells. This finding agrees with the work of Nikita Danial's group showing that neurons derived from BAD knock out animals display an augmented bioenergetic profile in the presence of ketone bodies (Giménez-Cassina et al., 2012).
α-lipoic acid is synthesized in mitochondria and is a necessary cofactor for PDH and α-KGDH activity. Our finding that exogenously applied α-lipoic acid also increases glucose oxidation in SH-SY5YB1, but not SH-SY5YEV cells suggests that raised BACE1 activity either reduces mitochondrial α-lipoic acid levels or lessens the ability of α-lipoic acid to enhance PDH and α-KGDH activity. The latter mechanism may be favored as there is evidence that excess α-lipoic acid provides protection to neuronal cells from the deleterious effects of exogenously applied Aβ peptides on PDH activity (Bielarczyk et al., 2006) and Aβ-induced toxicity (Zhang et al., 2001). Regarding the potential to reverse the metabolic deficits associated with age-dependent cognitive decline and early-stage or mild AD, the data presented herein demonstrate the capacity of α-lipoic acid and BHB to attenuate impaired glucose oxidation at a cellular level in face of chronically raised BACE1 activity and increased levels of Aβ peptides. BHB and α-lipoic acid have been demonstrated to exhibit some positive effects on cognitive decline in elderly dementia and mild AD patients (Hager et al., 2001; Reger et al., 2004; Costantini et al., 2008; Henderson et al., 2009; Henderson and Poirier, 2011).
Taken together, our findings are supportive for the rationale of targeting these TCA enzyme deficits in early cognitive impairment and AD. However, so far clinical trials based on raising circulating levels of ketones or giving α-lipoic acid as a dietary supplement have provided mixed outcomes and further trials examining α-lipoic acid are currently in progress. Perhaps a future strategy for AD management and treatment may be through the combination of this neutraceutical approach with new therapeutics, such as BACE1 inhibitors, currently in clinical trials (Vassar et al., 2014).
Author Contributions
JF and DH performed experiments and analyzed data. JF, DH, and MA contributed to the conception and design of experiments, interpretation of data and drafting and revising the manuscript. MA supervised the study and JF and MA wrote the manuscript. All authors approved the final version. MA is the guarantor of this work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Dundee Cell Products and Professor Wolfe for help with the cloning and provision of the mBACE1 construct, respectively. This work was supported by the Cruden Foundation Ltd and by grants from Alzheimer's Research UK (ART-PhD2010-12 and ARUK-EG2012A-2) and Diabetes UK (0003681).
References
Arsenian, M. (1998). Potential cardiovascular applications of glutamate, aspartate, and other amino acids. Clin. Cardiol. 21, 620–624. doi: 10.1002/clc.4960210904
Bateman, R. J., Xiong, C., Benzinger, T. L., Fagan, A. M., Goate, A., Fox, N. C., et al. (2012). Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N. Engl. J. Med. 367, 795–804. doi: 10.1056/NEJMoa1202753
Beal, M. F. (2005). Oxidative damage as an early marker of Alzheimer's disease and mild cognitive impairment. Neurobiol. Aging 26, 585–586. doi: 10.1016/j.neurobiolaging.2004.09.022
Bielarczyk, H., Gul, S., Ronowska, A., Bizon-Zygmanska, D., Pawelczyk, T., and Szutowicz, A. (2006). RS-alpha-lipoic acid protects cholinergic cells against sodium nitroprusside and amyloid-beta neurotoxicity through restoration of acetyl-CoA level. J. Neurochem. 98, 1242–1251. doi: 10.1111/j.1471-4159.2006.03966.x
Blass, J. P. (2001). Brain metabolism and brain disease: is metabolic deficiency the proximate cause of Alzheimer dementia? J. Neurosci. Res. 66, 851–856. doi: 10.1002/jnr.10087
Bogaert, Y. E., Rosenthal, R. E., and Fiskum, G. (1994). Postischemic inhibition of cerebral cortex pyruvate dehydrogenase. Free Radic. Biol. Med. 16, 811–820 doi: 10.1016/0891-5849(94)90197-X
Bosetti, F., Brizzi, F., Barogi, S., Mancuso, M., Siciliano, G., Tendi, E. A., et al. (2002). Cytochrome c oxidase and mitochondrial F1F0-ATPase (ATP synthase) activities in platelets and brain from patients with Alzheimer's disease. Neurobiol. Aging 23, 371–376. doi: 10.1016/S0197-4580(01)00314-1
Bouzier-Sore, A. K., Voisin, P., Bouchaud, V., Bezancon, E., Franconi, J. M., and Pellerin, L. (2006). Competition between glucose and lactate as oxidative energy substrates in both neurons and astrocytes: a comparative NMR study. Eur. J. Neurosci. 24, 1687–1694. doi: 10.1111/j.1460-9568.2006.05056.x
Bubber, P., Haroutunian, V., Fisch, G., Blass, J. P., and Gibson, G. E. (2005). Mitochondrial abnormalities in Alzheimer brain: mechanistic implications. Ann. Neurol. 57, 695–703. doi: 10.1002/ana.20474
Butterworth, R. F., and Besnard, A. M. (1990). Thiamine-dependent enzyme changes in temporal cortex of patients with Alzheimer's disease. Metab. Brain Dis. 5, 179–184. doi: 10.1007/BF00997071
Casley, C. S., Canevari, L., Land, J. M., Clark, J. B., and Sharpe, M. A. (2002a). Beta-amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J. Neurochem. 80, 91–100. doi: 10.1046/j.0022-3042.2001.00681.x
Casley, C. S., Land, J. M., Sharpe, M. A., Clark, J. B., Duchen, M. R., and Canevari, L. (2002b). Beta-amyloid fragment 25-35 causes mitochondrial dysfunction in primary cortical neurons. Neurobiol. Dis. 10, 258–267. doi: 10.1006/nbdi.2002.0516
Caspersen, C., Wang, N., Yao, J., Sosunov, A., Chen, X., Lustbader, J. W., et al. (2005). Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer's disease. FASEB J. 19, 2040–2041. doi: 10.1096/fj.05-3735fje
Coleman, P. D., and Yao, P. J. (2003). Synaptic slaughter in Alzheimer's disease. Neurobiol. Aging 24, 1023–1027. doi: 10.1016/j.neurobiolaging.2003.09.001
Costantini, L. C., Barr, L. J., Vogel, J. L., and Henderson, S. T. (2008). Hypometabolism as a therapeutic target in Alzheimer's disease. BMC Neurosci. 9(Suppl. 2):S16. doi: 10.1186/1471-2202-9-s2-s16
Danial, N. N. (2008). BAD: undertaker by night, candyman by day. Oncogene 27(Suppl 1), S53–S70. doi: 10.1038/onc.2009.44
DeKosky, S. T., Harbaugh, R. E., Schmitt, F. A., Bakay, R. A., Chui, H. C., Knopman, D. S., et al. (1992). Cortical biopsy in Alzheimer's disease: diagnostic accuracy and neurochemical, neuropathological, and cognitive correlations. Intraventricular Bethanecol Study Group. Ann. Neurol. 32, 625–632. doi: 10.1002/ana.410320505
DeKosky, S. T., Scheff, S. W., and Styren, S. D. (1996). Structural correlates of cognition in dementia: quantification and assessment of synapse change. Neurodegeneration 5, 417–421. doi: 10.1006/neur.1996.0056
Devi, L., Prabhu, B. M., Galati, D. F., Avadhani, N. G., and Anandatheerthavarada, H. K. (2006). Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer's disease brain is associated with mitochondrial dysfunction. J. Neurosci. 26, 9057–9068. doi: 10.1523/JNEUROSCI.1469-06.2006
Du, H., Guo, L., Fang, F., Chen, D., Sosunov, A. A., McKhann, G. M., et al. (2008). Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat. Med. 14, 1097–1105. doi: 10.1038/nm.1868
Fang, M., Wang, J., Zhang, X., Geng, Y., Hu, Z., Rudd, J. A., et al. (2012). The miR-124 regulates the expression of BACE1/beta-secretase correlated with cell death in Alzheimer's disease. Toxicol. Lett. 209, 94–105. doi: 10.1016/j.toxlet.2011.11.032
Forero, D. A., Casadesus, G., Perry, G., and Arboleda, H. (2006). Synaptic dysfunction and oxidative stress in Alzheimer's disease: emerging mechanisms. J. Cell. Mol. Med. 10, 796–805. doi: 10.1111/j.1582-4934.2006.tb00439.x
Fukumoto, H., Cheung, B. S., Hyman, B. T., and Irizarry, M. C. (2002). Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch. Neurol. 59, 1381–1389. doi: 10.1001/archneur.59.9.1381
Geula, C., Nagykery, N., Nicholas, A., and Wu, C. K. (2008). Cholinergic neuronal and axonal abnormalities are present early in aging and in Alzheimer disease. J. Neuropathol. Exp. Neurol. 67, 309–318. doi: 10.1097/NEN.0b013e31816a1df3
Gibson, G. E., Haroutunian, V., Zhang, H., Park, L. C., Shi, Q., Lesser, M., et al. (2000). Mitochondrial damage in Alzheimer's disease varies with apolipoprotein E genotype. Ann. Neurol. 48, 297–303. doi: 10.1002/1531-8249(200009)48:3<297::AID-ANA3>3.0.CO;2-Z
Giménez-Cassina, A., Martínez-Francois, J. R., Fisher, J. K., Szlyk, B., Polak, K., Wiwczar, J., et al. (2012). BAD-dependent regulation of fuel metabolism and K(ATP) channel activity confers resistance to epileptic seizures. Neuron 74, 719–730. doi: 10.1016/j.neuron.2012.03.032
Gómez-Isla, T., Price, J. L., McKeel, D. W. Jr., Morris, J. C., Growdon, J. H., and Hyman, B. T. (1996). Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer's disease. J. Neurosci. 16, 4491–4500.
Guglielmotto, M., Aragno, M., Autelli, R., Giliberto, L., Novo, E., Colombatto, S., et al. (2009). The up-regulation of BACE1 mediated by hypoxia and ischemic injury: role of oxidative stress and HIF1alpha. J. Neurochem. 108, 1045–1056. doi: 10.1111/j.1471-4159.2008.05858.x
Hager, K., Marahrens, A., Kenklies, M., Riederer, P., and Munch, G. (2001). Alpha-lipoic acid as a new treatment option for Alzheimer [corrected] type dementia. Arch. Gerontol. Geriatr. 32, 275–282. doi: 10.1016/S0167-4943(01)00104-2
Hamilton, D. L., Findlay, J. A., Montagut, G., Meakin, P. J., Bestow, D., Jalicy, S. M., et al. (2014). Altered amyloid precursor protein processing regulates glucose uptake and oxidation in cultured rodent myotubes. Diabetologia 57, 1684–1692. doi: 10.1007/s00125-014-3269-x
Harada, H., Tamaoka, A., Ishii, K., Shoji, S., Kametaka, S., Kametani, F., et al. (2006). Beta-site APP cleaving enzyme 1 (BACE1) is increased in remaining neurons in Alzheimer's disease brains. Neurosci. Res. 54, 24–29. doi: 10.1016/j.neures.2005.10.001
Henderson, S. T., and Poirier, J. (2011). Pharmacogenetic analysis of the effects of polymorphisms in APOE, IDE and IL1B on a ketone body based therapeutic on cognition in mild to moderate Alzheimer's disease; a randomized, double-blind, placebo-controlled study. BMC Med. Genet. 12:137. doi: 10.1186/1471-2350-12-137
Henderson, S. T., Vogel, J. L., Barr, L. J., Garvin, F., Jones, J. J., and Costantini, L. C. (2009). Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer's disease: a randomized, double-blind, placebo-controlled, multicenter trial. Nutr. Metab. 6:31. doi: 10.1186/1743-7075-6-31
Hirai, K., Aliev, G., Nunomura, A., Fujioka, H., Russell, R. L., Atwood, C. S., et al. (2001). Mitochondrial abnormalities in Alzheimer's disease. J. Neurosci. 21, 3017–3023. doi: 10.1017/S1431927611001875
Holsinger, R. M., McLean, C. A., Beyreuther, K., Masters, C. L., and Evin, G. (2002). Increased expression of the amyloid precursor beta-secretase in Alzheimer's disease. Ann. Neurol. 51, 783–786. doi: 10.1002/ana.10208
Huang, H. C., and Jiang, Z. F. (2011). Amyloid-beta protein precursor family members: a review from homology to biological function. J. Alzheimers Dis. 26, 607–626. doi: 10.3233/JAD−2011−110335
Hussain, I., Powell, D., Howlett, D. R., Tew, D. G., Meek, T. D., Chapman, C., et al. (1999). Identification of a novel aspartic protease (Asp 2) as beta-secretase. Mol. Cell. Neurosci. 14, 419–427. doi: 10.1006/mcne.1999.0811
Itoh, Y., Esaki, T., Shimoji, K., Cook, M., Law, M. J., Kaufman, E., et al. (2003). Dichloroacetate effects on glucose and lactate oxidation by neurons and astroglia in vitro and on glucose utilization by brain in vivo. Proc. Natl. Acad. Sci. U.S.A. 100, 4879–4884. doi: 10.1073/pnas.0831078100
Jack, C. R. Jr., Knopman, D. S., Jagust, W. J., Petersen, R. C., Weiner, M. W., Aisen, P. S., et al. (2013a). Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 12, 207–216. doi: 10.1016/S1474-4422(12)70291-0
Jack, C. R. Jr., Wiste, H. J., Weigand, S. D., Knopman, D. S., Lowe, V., Vemuri, P., et al. (2013b). Amyloid-first and neurodegeneration-first profiles characterize incident amyloid PET positivity. Neurology 81, 1732–1740. doi: 10.1212/01.wnl.0000435556.21319.e4
Jack, C. R. Jr., Barrio, J. R., and Kepe, V. (2013c). Cerebral amyloid PET imaging in Alzheimer's disease. Acta Neuropathol. 126, 643–657. doi: 10.1007/s00401-013-1185-7
Jha, M. K., Jeon, S., and Suk, K. (2012). Pyruvate Dehydrogenase Kinases in the Nervous System: their Principal Functions in Neuronal-glial Metabolic Interaction and Neuro-metabolic Disorders. Curr. Neuropharmacol. 10, 393–403. doi: 10.2174/157015912804499528
Johnson, K. A., Lopera, F., Jones, K., Becker, A., Sperling, R., Hilson, J., et al. (2001). Presenilin-1-associated abnormalities in regional cerebral perfusion. Neurology 56, 1545–1551. doi: 10.1212/WNL.56.11.1545
Kankotia, S., and Stacpoole, P. W. (2014). Dichloroacetate and cancer: new home for an orphan drug? Biochim. Biophys. Acta 1846, 617–629. doi: 10.1016/j.bbcan.2014.08.005
Kawaja, M. D. (2005). A proteomic approach to assess intraneuronal inclusions associated with neurodegenerative disorders. Curr. Opin. Mol. Ther. 7, 565–568.
Kim, S. H., Vlkolinsky, R., Cairns, N., Fountoulakis, M., and Lubec, G. (2001). The reduction of NADH ubiquinone oxidoreductase 24- and 75-kDa subunits in brains of patients with Down syndrome and Alzheimer's disease. Life Sci. 68, 2741–2750. doi: 10.1016/S0024-3205(01)01074-8
Knopman, D. S., Jack, C. R. Jr., Wiste, H. J., Weigand, S. D., Vemuri, P., Lowe, V. J., et al. (2013). Selective worsening of brain injury biomarker abnormalities in cognitively normal elderly persons with beta-amyloidosis. JAMA Neurol. 70, 1030–1038. doi: 10.1001/jamaneurol.2013.182
Ko, L. W., Sheu, K. F., Thaler, H. T., Markesbery, W. R., and Blass, J. P. (2001). Selective loss of KGDHC-enriched neurons in Alzheimer temporal cortex: does mitochondrial variation contribute to selective vulnerability? J. Mol. Neurosci. 17, 361–369. doi: 10.1385/JMN:17:3:361
Korolainen, M. A., Nyman, T. A., Aittokallio, T., and Pirttilä, T. (2010). An update on clinical proteomics in Alzheimer's research. J. Neurochem. 112, 1386–1414. doi: 10.1111/j.1471-4159.2009.06558.x
Lei, X., Lei, L., Zhang, Z., Zhang, Z., and Cheng, Y. (2015). Downregulated miR-29c correlates with increased BACE1 expression in sporadic Alzheimer's disease. Int. J. Clin. Exp. Pathol. 8, 1565–1574.
Li, R., Lindholm, K., Yang, L. B., Yue, X., Citron, M., Yan, R., et al. (2004). Amyloid beta peptide load is correlated with increased beta-secretase activity in sporadic Alzheimer's disease patients. Proc. Natl. Acad. Sci. U.S.A. 101, 3632–3637. doi: 10.1073/pnas.0205689101
Lustbader, J. W., Cirilli, M., Lin, C., Xu, H. W., Takuma, K., Wang, N., et al. (2004). ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science 304, 448–452. doi: 10.1126/science.1091230
Magistretti, P. J., and Pellerin, L. (1999). Cellular mechanisms of brain energy metabolism and their relevance to functional brain imaging. Philos. Trans. R. Soc. Lond. B Biol. Sci. 354, 1155–1163. doi: 10.1098/rstb.1999.0471
Manczak, M., Anekonda, T. S., Henson, E., Park, B. S., Quinn, J., and Reddy, P. H. (2006). Mitochondria are a direct site of A beta accumulation in Alzheimer's disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 15, 1437–1449. doi: 10.1093/hmg/ddl066
Martiskainen, H., Helisalmi, S., Viswanathan, J., Kurki, M., Hall, A., Herukka, S. K., et al. (2015). Effects of Alzheimer's disease-associated risk loci on cerebrospinal fluid biomarkers and disease progression: a polygenic risk score approach. J. Alzheimers. Dis. 43, 565–573. doi: 10.3233/JAD−140777
Masdeu, J. C., Zubieta, J. L., and Arbizu, J. (2005). Neuroimaging as a marker of the onset and progression of Alzheimer's disease. J. Neurol. Sci. 236, 55–64. doi: 10.1016/j.jns.2005.05.001
Mastrogiacoma, F., Lindsay, J. G., Bettendorff, L., Rice, J., and Kish, S. J. (1996). Brain protein and alpha-ketoglutarate dehydrogenase complex activity in Alzheimer's disease. Ann. Neurol. 39, 592–598. doi: 10.1002/ana.410390508
Maurer, I., Zierz, S., and Möller, H. J. (2000). A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol. Aging 21, 455–462 doi: 10.1016/S0197-4580(00)00112-3
McKenna, M. C., Waagepetersen, H. S., Schousboe, A., and Sonnewald, U. (2006). Neuronal and astrocytic shuttle mechanisms for cytosolic-mitochondrial transfer of reducing equivalents: current evidence and pharmacological tools. Biochem. Pharmacol. 71, 399–407. doi: 10.1016/j.bcp.2005.10.011
Meakin, P. J., Harper, A. J., Hamilton, D. L., Gallagher, J., McNeilly, A. D., Burgess, L. A., et al. (2012). Reduction in BACE1 decreases body weight, protects against diet-induced obesity and enhances insulin sensitivity in mice. Biochem. J. 441, 285–296. doi: 10.1042/BJ20110512
Mirshamsi, S., Laidlaw, H. A., Ning, K., Anderson, E., Burgess, L. A., Gray, A., et al. (2004). Leptin and insulin stimulation of signalling pathways in arcuate nucleus neurones: PI3K dependent actin reorganization and KATP channel activation. BMC Neurosci. 5:54. doi: 10.1186/1471-2202-5-54
Mosconi, L., Mistur, R., Switalski, R., Brys, M., Glodzik, L., Rich, K., et al. (2009a). Declining brain glucose metabolism in normal individuals with a maternal history of Alzheimer disease. Neurology 72, 513–520. doi: 10.1212/01.wnl.0000333247.51383.43
Mosconi, L., Mistur, R., Switalski, R., Tsui, W. H., Glodzik, L., Li, Y., et al. (2009b). FDG-PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer's disease. Eur. J. Nucl. Med. Mol. Imaging 36, 811–822. doi: 10.1007/s00259-008-1039-z
Mowrer, K. R., and Wolfe, M. S. (2008). Promotion of BACE1 mRNA alternative splicing reduces amyloid beta-peptide production. J. Biol. Chem. 283, 18694–18701. doi: 10.1074/jbc.M801322200
Mucke, L., Masliah, E., Yu, G. Q., Mallory, M., Rockenstein, E. M., Tatsuno, G., et al. (2000). High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J. Neurosci. 20, 4050–4058.
Mullen, A. R., Wheaton, W. W., Jin, E. S., Chen, P. H., Sullivan, L. B., Cheng, T., et al. (2012). Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 481, 385–388.
Nelson, P. T., and Wang, W. X. (2010). MiR-107 is reduced in Alzheimer's disease brain neocortex: validation study. J. Alzheimers Dis. 21, 75–79. doi: 10.3233/JAD−2010−091603
Nilsen, L. H., Witter, M. P., and Sonnewald, U. (2014). Neuronal and astrocytic metabolism in a transgenic rat model of Alzheimer's disease. J. Cereb. Blood Flow Metab. 34, 906–914. doi: 10.1038/jcbfm.2014.37
Nunomura, A., Perry, G., Aliev, G., Hirai, K., Takeda, A., Balraj, E. K., et al. (2001). Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 60, 759–767.
O'Connor, T., Sadleir, K. R., Maus, E., Velliquette, R. A., Zhao, J., Cole, S. L., et al. (2008). Phosphorylation of the translation initiation factor eIF2alpha increases BACE1 levels and promotes amyloidogenesis. Neuron 60, 988–1009. doi: 10.1016/j.neuron.2008.10.047
Packer, L., and Cadenas, E. (2011). Lipoic acid: energy metabolism and redox regulation of transcription and cell signaling. J. Clin. Biochem. Nutr. 48, 26–32. doi: 10.3164/jcbn.11-005FR
Parker, W. D. Jr., Parks, J., Filley, C. M., and Kleinschmidt-DeMasters, B. K. (1994). Electron transport chain defects in Alzheimer's disease brain. Neurology 44, 1090–1096 doi: 10.1212/wnl.44.6.1090
Patel, M. S., Nemeria, N. S., Furey, W., and Jordan, F. (2014). The pyruvate dehydrogenase complexes: structure-based function and regulation. J. Biol. Chem. 289, 16615–16623. doi: 10.1074/jbc.R114.563148
Pereira, C., Ferreiro, E., Cardoso, S. M., and de Oliveira, C. R. (2004). Cell degeneration induced by amyloid-beta peptides: implications for Alzheimer's disease. J. Mol. Neurosci. 23, 97–104. doi: 10.1385/JMN:23:1-2:097
Perry, E. K., Johnson, M., Kerwin, J. M., Piggott, M. A., Court, J. A., Shaw, P. J., et al. (1992). Convergent cholinergic activities in aging and Alzheimer's disease. Neurobiol. Aging 13, 393–400. doi: 10.1016/0197-4580(92)90113-C
Phypers, B., and Pierce, J. T. (2006). Lactate physiology in health and disease. Contin. Educ. Anaesth. Crit. Care Pain 6, 128–132. doi: 10.1093/bjaceaccp/mkl018
Puglielli, L., Ellis, B. C., Saunders, A. J., and Kovacs, D. M. (2003). Ceramide stabilizes beta-site amyloid precursor protein-cleaving enzyme 1 and promotes amyloid beta-peptide biogenesis. J. Biol. Chem. 278, 19777–19783. doi: 10.1074/jbc.M300466200
Reger, M. A., Henderson, S. T., Hale, C., Cholerton, B., Baker, L. D., Watson, G. S., et al. (2004). Effects of beta-hydroxybutyrate on cognition in memory-impaired adults. Neurobiol. Aging 25, 311–314. doi: 10.1016/S0197-4580(03)00087-3
Reiman, E. M., Chen, K., Alexander, G. E., Caselli, R. J., Bandy, D., Osborne, D., et al. (2004). Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer's dementia. Proc. Natl. Acad. Sci. U.S.A. 101, 284–289. doi: 10.1073/pnas.2635903100
Sancheti, H., Akopian, G., Yin, F., Brinton, R. D., Walsh, J. P., and Cadenas, E. (2013). Age-dependent modulation of synaptic plasticity and insulin mimetic effect of lipoic acid on a mouse model of Alzheimer's disease. PLoS ONE 8:e69830. doi: 10.1371/journal.pone.0069830
Shankar, G. M., Leissring, M. A., Adame, A., Sun, X., Spooner, E., Masliah, E., et al. (2009). Biochemical and immunohistochemical analysis of an Alzheimer's disease mouse model reveals the presence of multiple cerebral Abeta assembly forms throughout life. Neurobiol. Dis. 36, 293–302. doi: 10.1016/j.nbd.2009.07.021
Shearman, M. S., Ragan, C. I., and Iversen, L. L. (1994). Inhibition of PC12 cell redox activity is a specific, early indicator of the mechanism of beta-amyloid-mediated cell death. Proc. Natl. Acad. Sci. U.S.A. 91, 1470–1474. doi: 10.1073/pnas.91.4.1470
Sinha, S., Anderson, J. P., Barbour, R., Basi, G. S., Caccavello, R., Davis, D., et al. (1999). Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature 402, 537–540. doi: 10.1038/990114
Small, G. W., Mazziotta, J. C., Collins, M. T., Baxter, L. R., Phelps, M. E., Mandelkern, M. A., et al. (1995). Apolipoprotein E type 4 allele and cerebral glucose metabolism in relatives at risk for familial Alzheimer disease. JAMA 273, 942–947. doi: 10.1001/jama.1995.03520360056039
Sokoloff, L. (1999). Energetics of functional activation in neural tissues. Neurochem. Res. 24, 321–329. doi: 10.1023/A:1022534709672
Sorbi, S., Bird, E. D., and Blass, J. P. (1983). Decreased pyruvate dehydrogenase complex activity in Huntington and Alzheimer brain. Ann. Neurol. 13, 72–78. doi: 10.1002/ana.410130116
Sowell, R. A., Owen, J. B., and Butterfield, D. A. (2009). Proteomics in animal models of Alzheimer's and Parkinson's diseases. Ageing Res. Rev. 8, 1–17. doi: 10.1016/j.arr.2008.07.003
Swerdlow, R. H., Burns, J. M., and Khan, S. M. (2014). The Alzheimer's disease mitochondrial cascade hypothesis: progress and perspectives. Biochim. Biophys. Acta 1842, 1219–1231. doi: 10.1016/j.bbadis.2013.09.010
Tyler, S. J., Dawbarn, D., Wilcock, G. K., and Allen, S. J. (2002). alpha- and beta-secretase: profound changes in Alzheimer's disease. Biochem. Biophys. Res. Commun. 299, 373–376. doi: 10.1016/S0006-291X(02)02635-9
Vaishnavi, S. N., Vlassenko, A. G., Rundle, M. M., Snyder, A. Z., Mintun, M. A., and Raichle, M. E. (2010). Regional aerobic glycolysis in the human brain. Proc. Natl. Acad. Sci. U.S.A. 107, 17757–17762. doi: 10.1073/pnas.1010459107
Vassar, R., Bennett, B. D., Babu-Khan, S., Kahn, S., Mendiaz, E. A., Denis, P., et al. (1999). Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741. doi: 10.1126/science.286.5440.735
Vassar, R., Kuhn, P. H., Haass, C., Kennedy, M. E., Rajendran, L., Wong, P. C., et al. (2014). Function, therapeutic potential and cell biology of BACE proteases: current status and future prospects. J. Neurochem. 130, 4–28. doi: 10.1111/jnc.12715
Velliquette, R. A., O'Connor, T., and Vassar, R. (2005). Energy inhibition elevates beta-secretase levels and activity and is potentially amyloidogenic in APP transgenic mice: possible early events in Alzheimer's disease pathogenesis. J. Neurosci. 25, 10874–10883. doi: 10.1523/JNEUROSCI.2350-05.2005
Vlassenko, A. G., Vaishnavi, S. N., Couture, L., Sacco, D., Shannon, B. J., Mach, R. H., et al. (2010). Spatial correlation between brain aerobic glycolysis and amyloid-beta (Abeta) deposition. Proc. Natl. Acad. Sci. U.S.A. 107, 17763–17767. doi: 10.1073/pnas.1010461107
von Kienlin, M., Künnecke, B., Metzger, F., Steiner, G., Richards, J. G., Ozmen, L., et al. (2005). Altered metabolic profile in the frontal cortex of PS2APP transgenic mice, monitored throughout their life span. Neurobiol. Dis. 18, 32–39. doi: 10.1016/j.nbd.2004.09.005
Wilkins, H. M., Harris, J. L., Carl, S. M., Lu, J., Eva Selfridge, J., Roy, N., et al. (2014). Oxaloacetate activates brain mitochondrial biogenesis, enhances the insulin pathway, reduces inflammation and stimulates neurogenesis. Hum. Mol. Genet. 23, 6528–6541. doi: 10.1093/hmg/ddu371
Wischmeyer, P. E., Jayakar, D., Williams, U., Singleton, K. D., Riehm, J., Bacha, E. A., et al. (2003). Single dose of glutamine enhances myocardial tissue metabolism, glutathione content, and improves myocardial function after ischemia-reperfusion injury. JPEN J. Parenter. Enteral Nutr. 27, 396–403. doi: 10.1177/0148607103027006396
Wise, D. R., Ward, P. S., Shay, J. E., Cross, J. R., Gruber, J. J., Sachdeva, U. M., et al. (2011). Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. U.S.A. 108, 19611–19616. doi: 10.1073/pnas.1117773108
Xun, Z., Lee, D. Y., Lim, J., Canaria, C. A., Barnebey, A., Yanonne, S. M., et al. (2012). Retinoic acid-induced differentiation increases the rate of oxygen consumption and enhances the spare respiratory capacity of mitochondria in SH-SY5Y cells. Mech. Ageing Dev. 133, 176–185. doi: 10.1016/j.mad.2012.01.008
Yang, L. B., Lindholm, K., Yan, R., Citron, M., Xia, W., Yang, X. L., et al. (2003). Elevated beta-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat. Med. 9, 3–4. doi: 10.1038/nm0103-3
Yao, J., Irwin, R. W., Zhao, L., Nilsen, J., Hamilton, R. T., and Brinton, R. D. (2009). Mitochondrial bioenergetic deficit precedes Alzheimer's pathology in female mouse model of Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 106, 14670–14675. doi: 10.1073/pnas.0903563106
Yao, J., Chen, S., Mao, Z., Cadenas, E., and Brinton, R. D. (2011). 2-Deoxy-D-glucose treatment induces ketogenesis, sustains mitochondrial function, and reduces pathology in female mouse model of Alzheimer's disease. PLoS ONE 6:e21788. doi: 10.1371/journal.pone.0021788
Zaidan, E., Sheu, K. F., and Sims, N. R. (1998). The pyruvate dehydrogenase complex is partially inactivated during early recirculation following short-term forebrain ischemia in rats. J. Neurochem. 70, 233–241. doi: 10.1046/j.1471-4159.1998.70010233.x
Zhang, L., Xing, G. Q., Barker, J. L., Chang, Y., Maric, D., Ma, W., et al. (2001). Alpha-lipoic acid protects rat cortical neurons against cell death induced by amyloid and hydrogen peroxide through the Akt signalling pathway. Neurosci. Lett. 312, 125–128. doi: 10.1016/S0304-3940(01)02205-4
Zhang, X., Zhou, K., Wang, R., Cui, J., Lipton, S. A., Liao, F. F., et al. (2007). Hypoxia-inducible factor 1alpha (HIF-1alpha)-mediated hypoxia increases BACE1 expression and beta-amyloid generation. J. Biol. Chem. 282, 10873–10880. doi: 10.1074/jbc.M608856200
Zhao, J., Fu, Y., Yasvoina, M., Shao, P., Hitt, B., O'Connor, T., et al. (2007). Beta-site amyloid precursor protein cleaving enzyme 1 levels become elevated in neurons around amyloid plaques: implications for Alzheimer's disease pathogenesis. J. Neurosci. 27, 3639–3649. doi: 10.1523/JNEUROSCI.4396-06.2007
Keywords: glucose metabolism, BACE1, amyloid, pyruvate dehydrogenase, mitochondria, alpha lipoic acid
Citation: Findlay JA, Hamilton DL and Ashford MLJ (2015) BACE1 activity impairs neuronal glucose oxidation: rescue by beta-hydroxybutyrate and lipoic acid. Front. Cell. Neurosci. 9:382. doi: 10.3389/fncel.2015.00382
Received: 14 July 2015; Accepted: 14 September 2015;
Published: 01 October 2015.
Edited by:
Tommaso Pizzorusso, University of Florence and CNR Institute of Neuroscience, ItalyReviewed by:
Hermona Soreq, The Hebrew University of Jerusalem, IsraelMario Costa, Italian National Research Council, Italy
Copyright © 2015 Findlay, Hamilton and Ashford. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael L. J. Ashford, Division of Cardiovascular and Diabetes Medicine, School of Medicine, Ninewells Hospital and Medical School, University of Dundee, Dundee DD1 9SY, UK,bS5sLmouYXNoZm9yZEBkdW5kZWUuYWMudWs=
†Present Address: John A. Findlay, Faculty of Natural and Environmental Sciences Life Sciences, Centre for Biological Sciences, University of Southampton, Southampton, England