 Wan Y. Leong
Wan Y. Leong Zhi H. Lim1
Zhi H. Lim1 Vladimir Korzh
Vladimir Korzh Thomas Pietri
Thomas Pietri Eyleen L. K. Goh
Eyleen L. K. Goh- 1Program in Neuroscience and Behavioral Disorder, Duke-NUS Graduate Medical School, Singapore, Singapore
- 2Institute of Molecular and Cell Biology, Singapore, Singapore
- 3Department of Biological Sciences, National University of Singapore, Singapore, Singapore
- 4Institut de Biologie de l’École Normale Supérieure, Institut National de la Santé et de la Recherche Médicale U1024, Centre National de la Recherche Scientifique UMR 8197, Paris, France
- 5Department of Physiology, Yong Loo Lin School of Medicine, National University of Singapore, Singapore, Singapore
- 6KK Research Centre, KK Women’s and Children’s Hospital, Singapore, Singapore
Mutations in the gene encoding the MECP2 underlies Rett syndrome, a neurodevelopmental disorder in young females. Although reduced pain sensitivity in Rett syndrome patients and in partial MeCP2 deficient mice had been reported, these previous studies focused predominantly on motor impairments. Therefore, it is still unknown how MeCP2 is involved in these sensory defects. In addition, the human disease manifestations where males with mutations in MECP2 gene normally do not survive and females show typical neurological symptoms only after 18 months of age, is profoundly different in MeCP2-deficient mouse where all animals survived, and males but not females displayed Rett syndrome phenotypes at an early age. Thus, the mecp2-deficient zebrafish serves as an additional animal model to aid in deciphering the role and mechanisms of Mecp2 in neurodevelopment. Here, we used two independent methods of silencing expression of Mecp2 in zebrafish to uncover a novel role of Mecp2 in trigeminal ganglion sensory neurons during the embryonic development. mecp2-null mutation and morpholino-mediated silencing of Mecp2 in the zebrafish embryos resulted in defects in peripheral innervation of trigeminal sensory neurons and consequently affecting the sensory function. These defects were demonstrated to be dependent on the expression of Sema5b and Robo2. The expression of both proteins together could better overcome the defects caused by Mecp2 deficiency as compared to the expression of either Sema5b or Robo2 alone. Sema5b and Robo2 were downregulated upon Mecp2 silencing or in mecp2-null embryos, and Chromatin immunoprecipitation (ChIP) assay using antibody against Mecp2 was able to pull down specific regions of both Sema5b and Robo2 promoters, showing interaction between Mecp2 and the promoters of both genes. In addition, cell-specific expression of Mecp2 can overcome the innervation and sensory response defects in Mecp2 morphants indicating that these MeCP2-mediated defects are cell-autonomous. The sensory deficits caused by Mecp2 deficiency mirror the diminished sensory response observed in Rett syndrome patients. This suggests that zebrafish could be an unconventional but useful model for this disorder manifesting defects that are not easily studied in full using rodent models.
Introduction
Mutations in the gene encoding the Methyl-CpG binding protein 2 (MECP2) underlies Rett syndrome, a neurodevelopmental disorder presented with mental retardation, autistic behavior, compromised sensory sensations and loss of previously acquired cognitive milestones, including purposeful hand use and expressive language, in young females. Other clinical features of Rett syndrome include impairment of sleep pattern, breathing disturbance when awake, peripheral vasomotor disturbance, autonomic dysfunction (cold, blue extremities), dystonia, progressive scoliosis and diminished response to pain (Hagberg et al., 1983; Amir et al., 1999; Armstrong, 2005; Downs et al., 2010; Neul et al., 2010). It has been documented that about 90% of all Rett syndrome cases resulted from mutations in the X-linked MECP2 (Amir et al., 1999; Shahbazian and Zoghbi, 2001; Armstrong, 2005; Bienvenu and Chelly, 2006).
MeCP2 is a multifunctional protein that was first identified by its ability to bind to methylated DNA (Lewis et al., 1992; Bird, 2008; Guy et al., 2011). Earlier studies on MeCP2 demonstrated its role as transcriptional repressor for a selected set of target genes (Nan et al., 1997; Chandler et al., 1999). Subsequent studies showed that MeCP2 may be involved in both transcriptional repression or activation, depending on the molecular context (Chahrour et al., 2008; Ben-Shachar et al., 2009; Guy et al., 2011). In addition, MeCP2 was shown to interact with the RNA-binding protein Y box-binding protein 1 and regulates the splicing of reporter minigenes, and is possibly responsible for the aberrant alternative splicing patterns in a mouse model of Rett syndrome (Young et al., 2005). Thus, mutations in MECP2 are expected to alter expression of its downstream target genes with the consequences of impaired neuronal development and function.
Alterations in MeCP2 expression have been detected in autism spectrum disorders as well as in non-syndromic mental retardations (Chahrour and Zoghbi, 2007). MeCP2 is ubiquitously expressed, but its critical function in the mammalian brain is suggested by the abundant expression of MeCP2 in the CNS (Skene et al., 2010). Therefore, most of the current studies on Rett syndrome and MeCP2 are focused on the development of neurons in the developing or adult CNS (Ma et al., 2015; Zhao et al., 2015). Moreover, the panembryonic Mecp2 gene knockout or the brain-specific gene knockout in mice showed similar neurological phenotypes (Chen et al., 2001; Guy et al., 2001). These studies suggested a requirement for MeCP2 in the normal development of the nervous system. The importance of MeCP2 in embryonic development as well as postnatal physiological processes underlies the majority of disease etiologies associated with Rett syndrome. Although reduced pain sensitivity in Rett syndrome patients and reduced pain recognition in partial MeCP2 deficient mice have been reported (Samaco et al., 2008; Downs et al., 2010), it is still unknown how MeCP2 is involved in these sensory responses.
Like many X-linked disorders, Rett syndrome patients displayed mosaic expression of mutant and normal MeCP2 protein in different cell types, resulting in significant variations in phenotypes and clinical severity (Shahbazian and Zoghbi, 2001; Christodoulou and Weaving, 2003; Skene et al., 2010). Moreover, not much is known with regards to the differences of the various MeCP2 protein isoforms and mutations during early development as well as in disease pathology. The MECP2 gene is also present in non-mammalian vertebrates, including the zebrafish Danio rerio (Coverdale et al., 2004) and mecp2-null zebrafish also shows altered motor behaviors (Pietri et al., 2013). The mecp2-null zebrafish model enables the screening for genes involved in early development and will aid in deciphering the role and mechanisms of Mecp2 in vertebrate neurodevelopment. Using both mecp2-null mutants and mecp2 morpholino-mediated knockdown approach, we have identified an important role for and also established underlying mechanisms of Mecp2 function in vertebrate neurodevelopment. Specifically, we found Mecp2 regulating projections of sensory neurons and sensory responses, at least in part, through directly activating transcription of specific axon guidance cues, Sema5b and Robo2.
Results
Mecp2 Plays a Role in Embryonic Development of Zebrafish
In situ-hybridization showed that mecp2 in the zebrafish is ubiquitously expressed in the central nervous system (CNS), with the highest level of expression found in the forebrain (Figure 1A). We next compared genomic organization of mammalian (human) MECP2 and zebrafish mecp2 gene (Figure 1B). Zebrafish mecp2 contains three exons while human MECP2 contains four exons. Although zebrafish mecp2 does not have exon 2 of human MECP2, it also contains two main domains critical for its functions. These are a methyl-cytosine binding domain (MBD) and a transcriptional repression domain (TRD) that shares 85.9 and 54% identity respectively with mammalian species (Coverdale et al., 2004). mecp2-null mutants (Q63∗) was a nonsense mutation generated through N-ethyl-N-nitrosourea (ENU)-mutagenesis. This Q63∗ position is located directly before the MBD domain leading to a loss of function (Pietri et al., 2013). Since there are no available antibodies against zebrafish Mecp2, we designed MO oligonucleotide corresponding to the sequence of the splice acceptor site at the intron 1-exon 2 border on zebrafish mecp2. This is so that the efficiency and specificity of splicing blocker MO can be determined by RT-PCR (Figures 1C,D). Using two sets of primers (a forward primer on exon 1 and a reverse primer on exon 2 or exon 3) (Figure 1B), a deletion of the 372 bp fragment in the exon 2 was detected in morphant pre-mRNA resulting in absence (primer set 1 – Figure 1C) or shorter PCR product (primer set 2 –Figure 1D). MO corresponding to the sequence of the ATG translation initiation site was also tested and the morphants showed similar phenotypes as those injected with splicing blocker MO (data not shown).
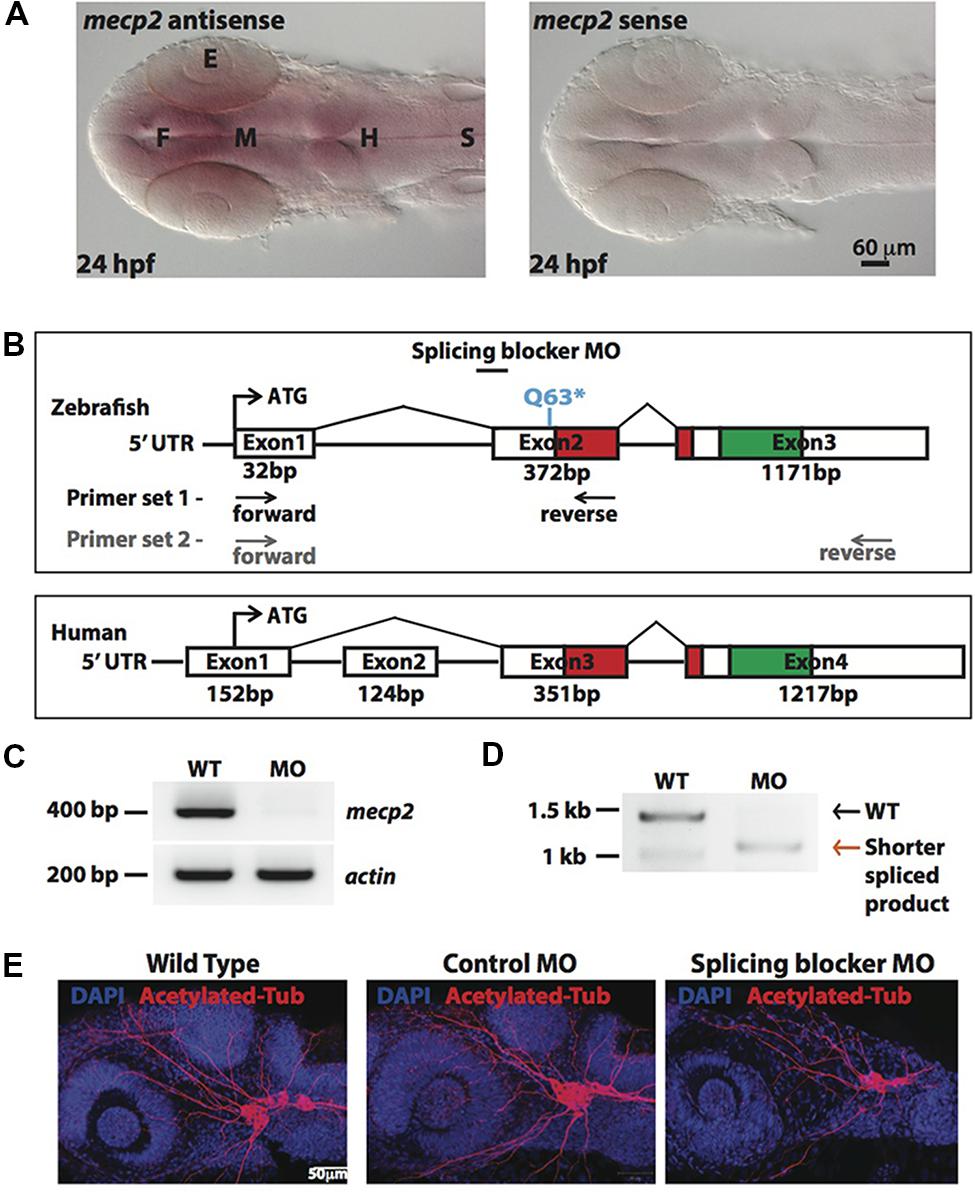
FIGURE 1. Knocking down Mecp2 in zebrafish. Mecp2 is ubiquitously expressed throughout the CNS at 24 hpf (A). The embryos are displayed as whole mounts, dorsal view (F- forebrain, M - midbrain and H-hindbrain, S - spinal cord, E- eye) Scale bar = 60 μm. Schematic diagram shows gene structure of the zebrafish mecp2 and the mammalian (human or mouse) MeCP2α isoform (B). The zebrafish mecp2 gene consists of 3 exons with the main domains highlighted in red (Methyl-CpG-binding domain) and green (transcriptional repressor domain). The splice blocking morpholino targets the intron 1/exon 2 boundary on mecp2 pre-mRNA. Arrows below Zebrafish mecp2 gene show the positions where the forward and reverse primers were targeted [primer set 1 with black arrows for experiment in (C) and primer set 2 with gray arrows for (D)]. The specificity of morpholino was determined by RT-PCR (B,C). Representative gel images showing normal splicing of mecp2 as a band of 400 bp with actin (206 bp) as the control (C). Representative gel image showing normal splicing of mecp2 in WT (1455 bp) or blocked splicing band (exon skipping) (1086 bp) in mecp2 spliced MO injected embryos (D). Representative images showing gross morphological changes in TG neurons projections induced by mecp2 MO during development of zebrafish at 24 hpf (E). Scale bar = 50 μm.
MeCP2 was previously shown to regulate neurite growth and axon targeting in the hippocampus, motor cortex and olfactory system of the mouse (Matarazzo et al., 2004; Belichenko et al., 2009; Degano et al., 2009; Palmer et al., 2012). Thus, we first examined gross axonal projections using an axon marker, acetylated tubulin (AcTub) at 24 h post fertilization (hpf). The projections of the sensory neurons of the trigeminal ganglion (TG) are among the earliest detected with anti-AcTub antibody (Metcalfe et al., 1990). Uninjected WT embryos or embryos injected with control or mecp2 MO at one-cell stage were examined at 24 hpf for gross morphologies of their projections from TG neurons (Figure 1D). Uninjected WT and control MO-injected embryos exhibited similar gross morphology of TG neurons and their projections. However, Mecp2 morphants showed significantly less peripheral projections from TG (Figure 1E).
Mecp2 Knockdown Decreases Projections from Sensory Neurons
Visibly less arborizations of peripheral projections from TG were observed in Mecp2 morphants (Figure 1E). Thus, we next quantify this defect by measuring the total projection length in each embryo (as mean projection length in μm). Knocking down of Mecp2 indeed inhibits growth and branching of the peripheral axons, resulting on average a 45% decrease in total length of peripheral axon projections as compared to controls. A potential caveat in interpreting these results is that some MOs can activate p53, thus causing non-specific neural defects (Robu et al., 2007; Eisen and Smith, 2008). We therefore routinely used p53 MO to relieve possible non-specific neural cell death caused by toxicity effects of MOs (Robu et al., 2007). We found that embryos injected with mecp2 MO together with p53 MO showed similar phenotypes in sensory neurons compared to those injected with mecp2 MO alone. To further ensure that these phenotypes are not due to off-target effects, we performed rescue experiments by co-injection with mecp2 mRNA (Figures 2A,B). We found phenotypes of Mecp2 morphants could be reversed by co-injection with mecp2 mRNA (Figures 2A,B). These results suggest that Mecp2 plays a key role in axonal development in zebrafish sensory neurons.
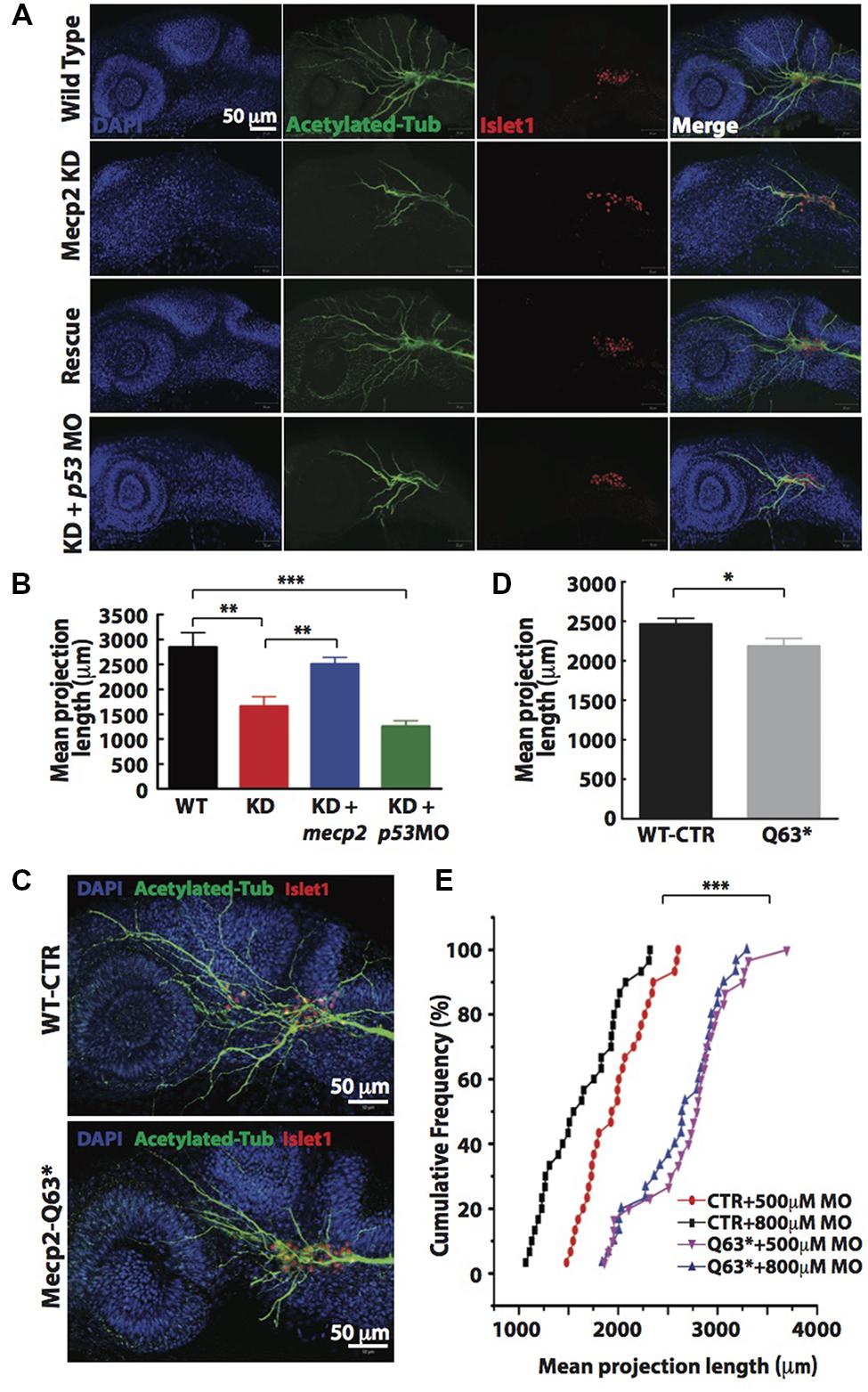
FIGURE 2. Knockdown of Mecp2 affects the axonal projection of trigeminal ganglion (TG) cells. Structure of peripheral dendritic projection and nerve pathways revealed by axon marker, acetylated tubulin (green), a general nucleus marker, DAPI (blue) and a neuronal marker, islet1 (red) at 24 hpf (A). Representative images showing peripheral axonal projections from sensory neurons of TG in wild-type controls (WT, top panel), Mecp2 morphants (second panel from top), Mecp2 morphants rescued by coinjecting mRNA of Mecp2 (third panel from top) and Mecp2 morphants coinjected with p53 MO (lower panel) (A). Quantitative analysis of total peripheral projections of trigeminal sensory neurons, with graph presenting total length of peripheral projections from trigeminal ganglia per zebrafish (B) N > 10. Representative images showing peripheral axonal projections from sensory neurons of TG in wild-type controls (WT, top panel) and mecp2Q63∗/Q63∗ mutants (Mecp2-Q63, bottom panel) (C). Graph showing quantitative analysis of peripheral projections (total length) from trigeminal ganglia per zebrafish in wild-type controls and mecp2Q63∗/Q63∗ mutants (D). Graph showing total neurite length of controls (CTR) and mecp2Q63∗/Q63∗ mutants (Q63) injected with either 500 or 800 μM of mecp2 MO (E). Scale bar = 50 μm. Each individual dot on graph (E) represents individual embryos. N = 30 in each group. (∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.0001, student t-test for (D) and one-way ANOVA with Bonferroni’s post hoc test for (B,E)].
To further verify the specificity of these morpholino-mediated phenotypes, we also examined neurite projections in mecp2-null mutants (Q63∗). These mecp2-null fish exhibit clear altered behavioral alterations such as spontaneous and sensory-evoked motor anomalies, as well as defective thigmotaxis and have a relatively shorter lifespan than WT fish (Pietri et al., 2013). These mecp2-null embryos also show defect in TG projections as compared to WT control embryos, with about 15–20% decrease in the mean dendrite length of TG neurons as compared to WT control embryos (Figures 2C,D). Since this mecp2-null mutant is generated from a mutation that putatively truncated the protein at amino acid position 63, the truncated Mecp2 protein may still have residual Mecp2 activity. Thus, we next carried out titration experiments by injecting two different doses (500 and 800 μM) of mecp2 MO in WT-CTR or Q63∗ mutant embryos (Figure 2E). This is to further rule out any off-target effects from mecp2 MO and to determine if this truncated Mecp2 protein in mecp2-null embryos is still functional in promoting TG projections. WT-CTR embryos showed more severe defects in the mean projection length upon injection with higher concentration of Mecp2 MO. However, the increasing concentrations of mecp2 MO did not cause further projection defects in Q63∗ mutants (Figure 2E), suggesting that there is no residual Mecp2 activity in mecp2-null mutant embryos, at least in these sensory neuron projections at the time point examined. These data obtained by titrating the Mecp2 MO in a null genetic background as compared to control embryos, further confirmed the specificity of our mecp2 MO.
Cell Autonomous Effect of Mecp2 on Sensory Neurons During Embryonic Development
Since a high rate of transient apoptosis during neurodevelopment is crucial for normal neurodevelopment in zebrafish (Cole and Ross, 2001), we examined the morphants using terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick-end labeling (TUNEL) labeling at 16 and 24 hpf. Knocking down Mecp2 increases the level of apoptosis at 16 and 24 hpf as compared to control (Figures 3A,B). There were an average of 7.5 and 14.0 TUNEL-positive cells per area of 12 mm × 12 mm in the head of morphants at 16 and 24 hpf respectively, as compared to the 2.0–4.0 TUNEL-positive cells in control embryos at both time points (Figures 3A,B). However, very few of the TG neurons were TUNEL-positive as shown by a low number of TUNEL-positive cells colocalized with a pan neuronal marker, HuC that labels differentiating neural cells (Figure 3A). The expression pattern of HuC in the early differentiating neurons was previously described (Park et al., 2000; Sato et al., 2006). HuC highlighted the first differentiated trigeminal neurons at 11 hpf. By 24 hpf, the TG has an average of 37 ± 3 neurons, and all these neurons express HuC. Other proteins such as Islet1, Trpa1b and P2x3b are also expressed in the TG, but are only expressed in different subpopulations of the trigeminal sensory neurons (Caron et al., 2008). Therefore, HuC was used as a marker to identify whether specific genes of interest are localized in all or only a subset of trigeminal sensory neurons (Pan et al., 2012). These studies indicated HuC as an ideal promoter to identify neurons in the TG.
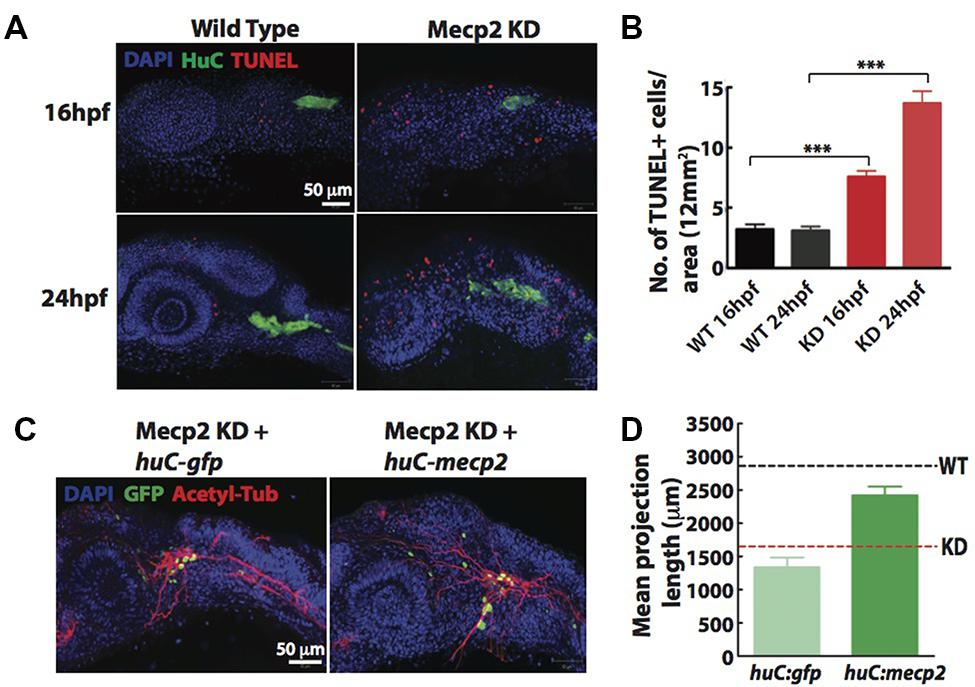
FIGURE 3. Cell autonomous effect of Mecp2 on sensory neurons during embryonic development. Embryos were injected with mecp2 MO and labeled with TUNEL. Antibody against HuC was used to label cell bodies in TG. Representative images showing TUNEL labeling in TG in wild-type controls (WT, left panel), Mecp2 morphants (right panel) (Blue – DAPI, Green – HuC and Red – TUNEL) (A). Quantitative analysis of TUNEL-labeled cells (indicative of apoptosis) at 16 and 24 hpf were presented in graphs showing total number of TUNEL positive cells per 12 mm × 12 mm in the head (B). Representative images showing peripheral axonal projections from sensory neurons of TG in Mecp2 morphants rescued by coinjection with huC-gfp control plasmids (left panel) or huC-mecp2 plasmids (right panel) (C). Quantitative analysis of total peripheral projections of trigeminal sensory neurons, with graphs presenting total length of peripheral projections from trigeminal ganglia per zebrafish (D). N > 10 in each group. Scale bar = 50 μm (∗∗∗P < 0.0001, one-way ANOVA with Bonferroni’s post hoc test).
To rule out non-cell-autonomous effect on sensory neurons from this general increase in apoptosis of the surrounding cells, we generated a construct with HuC promoter driving the expression of Mecp2 in neurons only. Since GFP intensity varies in different cells and low GFP intensity in most projections does not allow proper and unbiased tracings, we quantified total length and number of projections using acetylated tubulin as a marker (Figures 3C,D). Expression of huC-mecp2 in Mecp2 morphants was able to rescue reduction in axonal projections in TG neurons (Figures 3C,D). The rescue is not as efficient as compared to that of mecp2 mRNA, because not all TG cells express Mecp2 under the HuC promoter. Nonetheless, the rescue by huC-mecp2 is significant and huC-gfp control does not have any effect on axonal projections in Mecp2 morphants. Taken together, these results confirm that the morphant phenotype is not caused by an increase in apoptotic cells in the vicinity of TG. Our data suggest that defects in sensory neurons resulting from Mecp2 knockdown represent a cell-autonomous effect.
Mecp2 Regulates Transcriptional Activation of Axon Guidance Cues
The known function of MeCP2 mainly involves its binding to methylated-CpG islands as well as activating and suppressing transcription. We therefore sought to examine the role of Mecp2 in regulating gene expression in zebrafish. Expression profiling was undertaken using a 135K-zebrafish gene expression array (∼38,000 known transcripts), comparing the transcriptome of wild-type controls with Mecp2 morphants. A total of 1123 genes had a minimum of 2-fold change over control (p < 0.05), but only 1005 of which had a mammalian (human or mouse) ortholog (http://www.ncbi.nlm.nih.gov/geo/ – accession number GSE71173). Out of these 1005 genes, 607 were down-regulated and 398 were up-regulated (minimum twofold change; significance was established at p < 0.05) in Mecp2 morphants, and 118 genes (43 down-regulated and 75 up-regulated) are without human orthologs (Figure 4A). These results strongly suggest that knocking down Mecp2 could either activate or repress transcription. Functional analysis of these 1123 genes in silico using the program Database for Annotation, Visualization and Integrated Discovery (DAVID) yield the following. Mecp2-regulated genes in zebrafish appear to function in diverse cellular processes (Figures 4B,C). Many of the up-regulated genes had roles in proliferation, cell communication and metabolism. By contrast, many of the down-regulated genes had functions related to neuronal development, including neurogenesis, neuronal projection morphogenesis, cell migration and synaptogenesis (Figures 4B,C). Since Mecp2 knockdown resulted in projection defects in sensory neurons, we searched for genes with axon guidance functions in our microarray that can potentially be involved in Mecp2-mediated effects. sema3f, sema5b and robo2 were found in functional clusters involved in mammalian brain and nervous system development, specifically in neuronal projection morphogenesis, and they were strongly down-regulated by knocking down Mecp2.
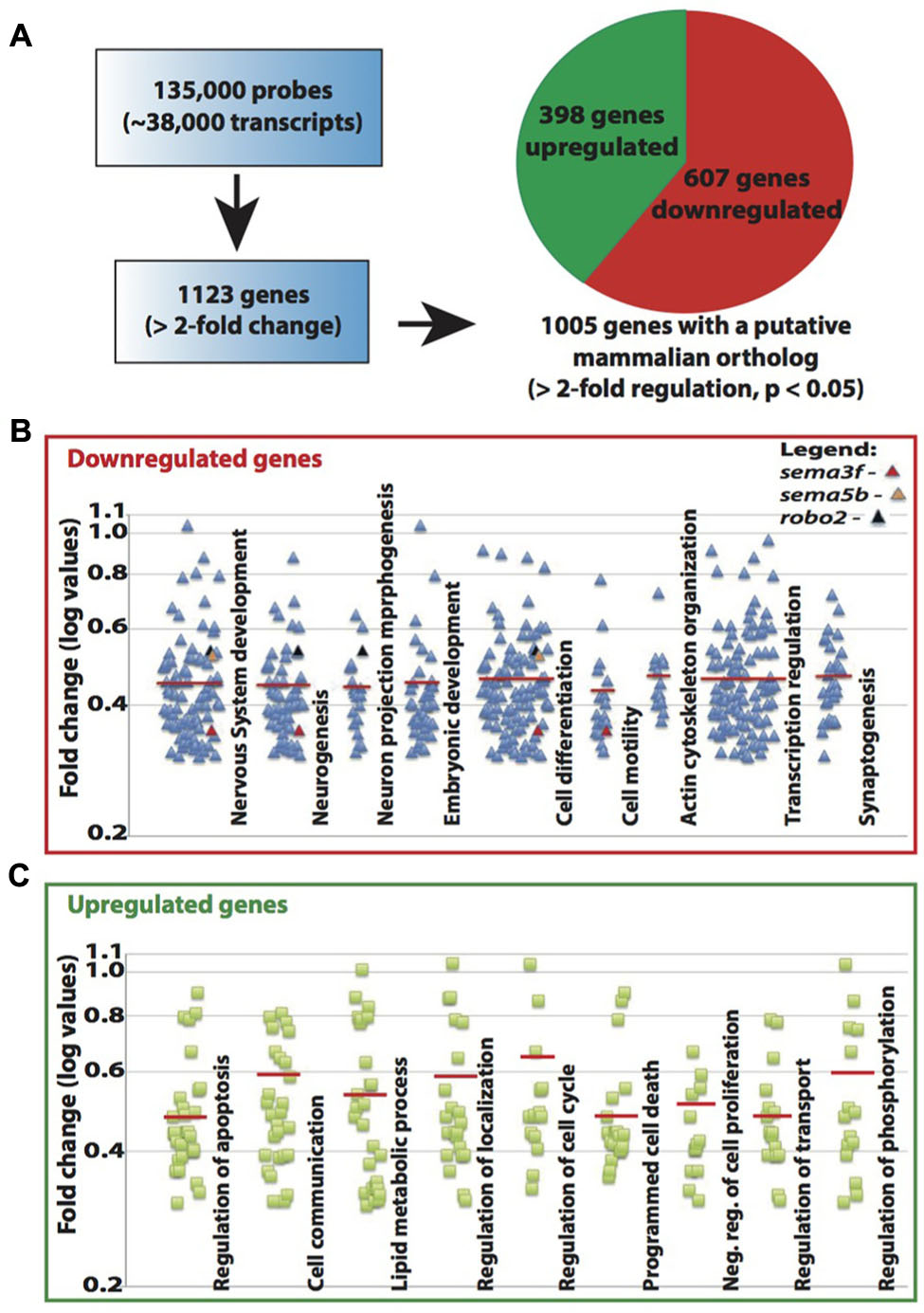
FIGURE 4. Mecp2 morphants showed decreased expression of axon guidance cues. Schematic diagram showing the number of genes that are upregulated or downregulated in Mecp2 deficient embryos (A). Out of the 1005 genes that were found to have putative mammalian orthologs, 607 were downregulated and 398 were upregulated (minimum twofold change; significance was established at p < 0.05) in Mecp2 morphants (A). Dot plots showing downregulated (B) and upregulated (C) genes. Each dot represent a single gene with at least twofold increased or decreased in expression presented as log values (base10) on y-axis. Relative fold change of downregulated genes, sema3f (0.352568), sema5b (0.523226) and robo2 (0.537693) were highlighted as colored dots (red, orange, and black respectively) in their respective functional groups.
Non-quantitative and quantitative RT-PCR validated that mRNA levels of sema3f, sema5b and robo2 were indeed reduced in Mecp2 morphants (Figures 5A,B). Our results suggest that Mecp2 plays an important role in the expression of these axon guidance cues. To determine if these genes are also downregulated in mecp2-null mutants, we examined mRNA expression of sema3f, sema5b and robo2 in control and mecp2-null embryos (Figure 5C). There are modest but significant changes in sema3f, sema5b and robo2 mRNA expression level in mecp2-null mutant as compared to WT control embryos. The modest change in the mRNA expression in mecp2-null mutants likely contributed to the subtle defects in TG projections in mecp2-null mutants. Whereas the bigger change in expression of these axon guidance cues in mecp2 MO-mediated knockdown embryos contributed to the more severe defects. Moreover, injection of mecp2 MO into mecp2-null embryos did not affect mRNA levels of sema3f, sema5b or robo2. These results confirmed that there are possibly compensatory effects in mecp2-null mutants as compared to mecp2 MO injected embryos.
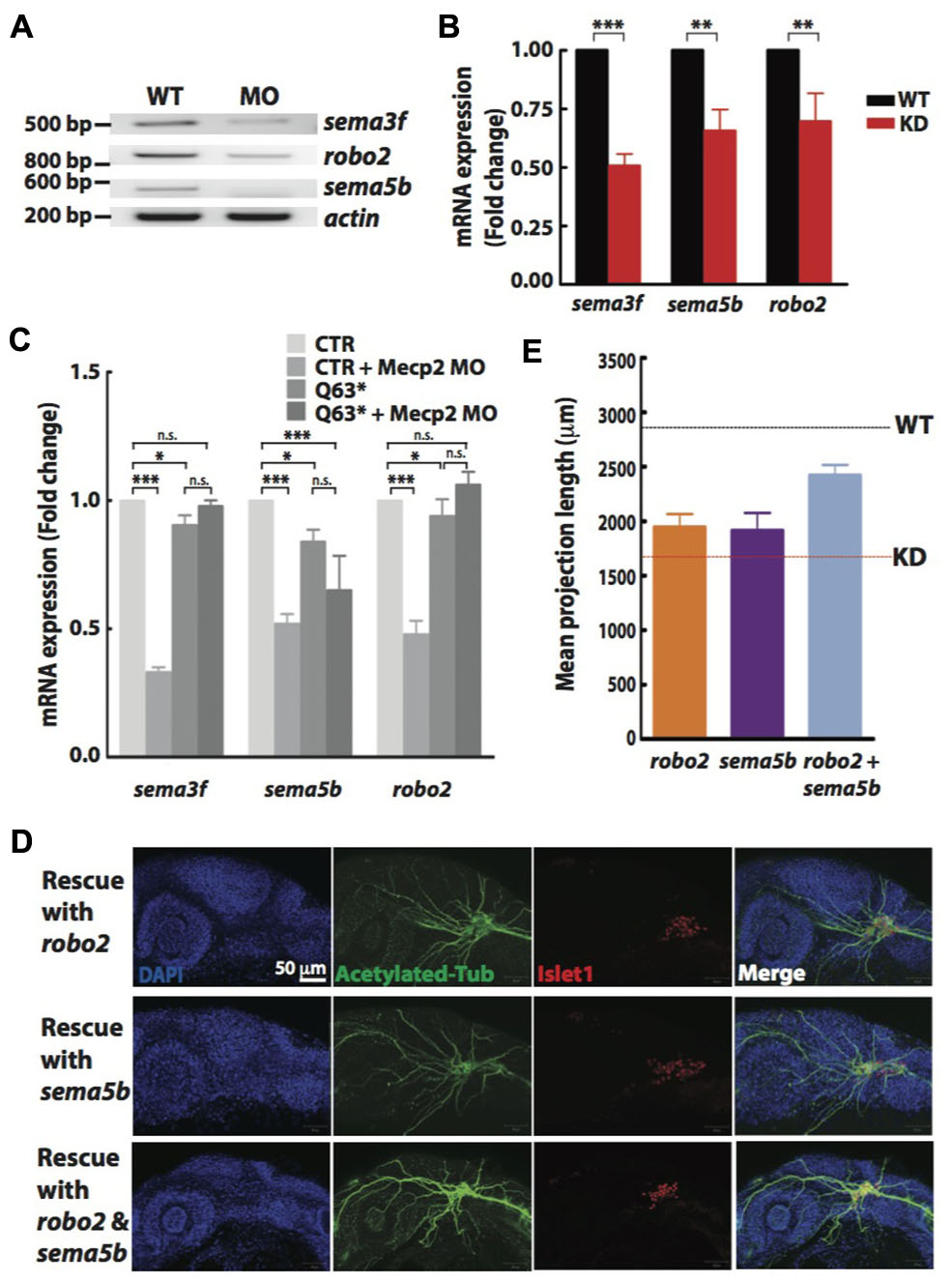
FIGURE 5. Sema5b and Robo2 rescue Mecp2-induced defects in axonal projections in TG. Representative gel images showing sema3f, sema5b and robo2 mRNA levels regulated by Mecp2 (A). Graph presenting quantitative RT-PCR of the three above mentioned genes in WT or Mecp2 MO injected embryos (B). Graph showing expression of sema3f, sema5b and robo2 mRNA in CTR and mecp2Q63∗/Q63∗ mutants using RT-PCR (C). Data are presented as fold expression of each gene normalized with housekeeping gene, beta-actin to value 1. (∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001, two-way ANOVA with Bonferroni’s post hoc test). Sema5b and Robo2 rescue axonal projection of TG cells. Representative images showing peripheral projections of trigeminal sensory neurons in embryos injected with mecp2 MO together with mRNA of Sema5b, Robo2 or both (D). Quantitative analysis of total peripheral projections from trigeminal sensory neurons, with graph presenting total length of peripheral projections from trigeminal ganglia per zebrafish (E) (Blue – DAPI, Green – acetylated tubulin, Red – islet1). N > 10 in each group, Scale bar = 50 μm.
Sema5b and Robo2 are Essential for Mecp2-Mediated Defects of Axon Projections
Although there are changes in expression levels of class 3 semaphorins and their receptors in Mecp2-null mice, neither Sema3A or 3F has effects on repulsion or neurite outgrowth response of Mecp2-null olfactory axons (Degano et al., 2009). We also did not observe significant defects in embryos injected with morpholino targeting sema3f (Data not shown). Therefore, to further establish Robo2 and Sema5b-mediated Mecp2-dependent neuronal projection morphogenesis, we asked if the defects seen in Mecp2 morphants could be rescued by Robo2 and Sema5b. We co-injected mRNAs of robo2 or sema5b or both together with mecp2 MO. Our data showed that excess Robo2 or Sema5b alone modestly rescued defects observed in Mecp2 morphants (average of 70 and 67% of control, respectively as compared to the 50% in Mecp2 KD group) (Figures 5D,E). Importantly, coexpression of both Robo2 and Sema5b in Mecp2 morphants showed more projections from TG neurons (average of 90% of control) as compared to single gene rescue (Figures 5D,E). These results suggest that Robo2 and Sema5b expression plays a role in the neurodevelopmental phenotype associated with loss of Mecp2.
Mecp2 Directly Regulates Transcription of sema5b and robo2
Next, we investigated whether Mecp2 directly regulates Robo2 and Sema5b expression by interacting with their promoters. We carried out conventional chromatin immunoprecipitation (ChIP) assay on 24 hpf wild-type embryos injected with mecp2-gfp using GFP antibody, followed by site-specific PCR analysis. Primers were designed within 2000 bp region upstream of translational start site of both sema5b and robo2 promoters. Our results showed that specific regions on the promoters of these two mentioned genes co-immunoprecipitated with Mecp2 in vivo, suggesting that their transcription is directly subjected to Mecp2 regulation (Figure 6A). Quantification of PCR products showed weak binding of Mecp2 to the promoter of sema5b at P4 (within –1824 to -1723) and relatively strong binding at P1, P2, and P3 (within -155 to -57, -792 to -681, and -1310 to -1177) regions (Figures 6A,C). On the other hand, Mecp2 binds strongly to robo2 promoter at P1 and P4 regions (within -349 to -222 and -2045 to -1916), and does not bind or only binds very weakly to P2 and P3 regions (-849 to -730 and -1374 to -1236) (Figures 6A,B). These results suggest that Mecp2 is likely to regulate transcription of Robo2 and Sema5b, through its association with the promoters of both genes.
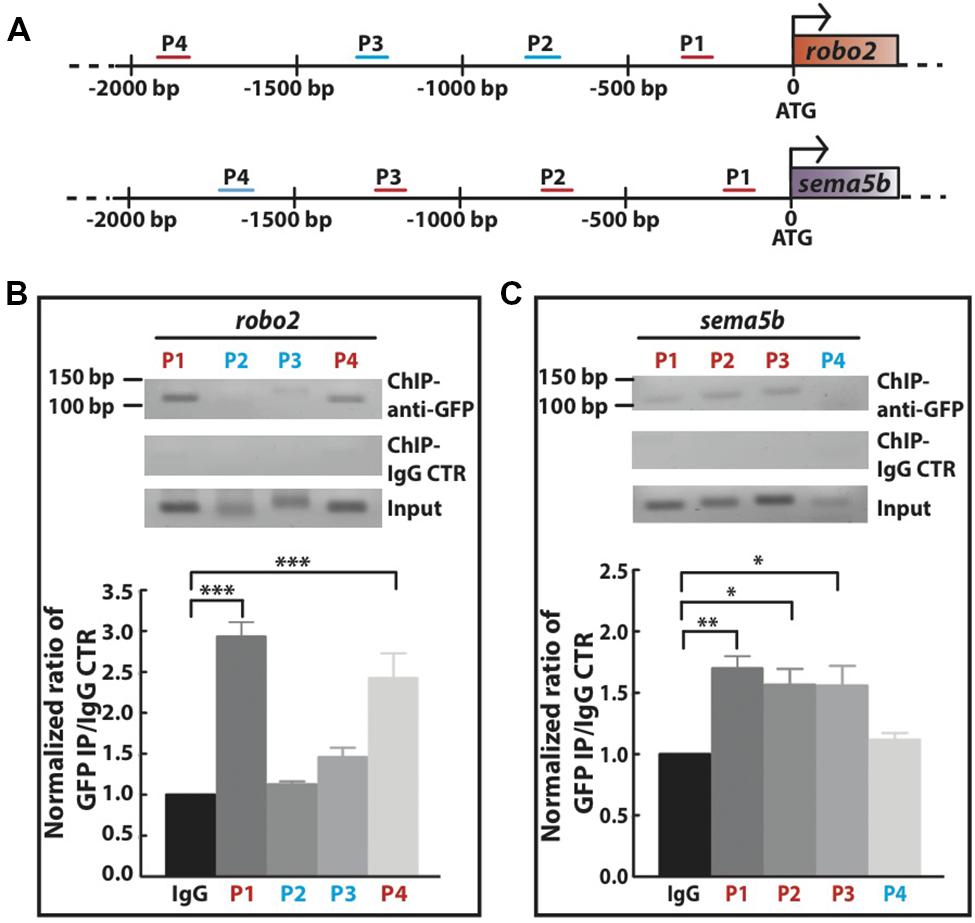
FIGURE 6. Mecp2 directly regulates transcription of robo2 and sema5b genes. Chromatin immunoprecipitation (ChIP) analysis shows the recruitment of Mecp2 to robo2 and sema5b promoters. Schematic diagram of the 5′ promoter region of robo2 and sema5b. Short horizontal lines (red and blue) labeled P1-4 indicate the relative positions of PCR products from the respective sets of primers on both promoter regions (A). Representative gel images (top panel – anti-GFP pulldown, middle panel – IgG control pulldown and bottom panel – input) and quantitative analysis of ChIP assay performed on 24 hpf wild-type embryos overexpressing Mecp2 protein (GFP-tagged Mecp2) (B,C). Anti-GFP antibody pulled down specific robo2 and sema5b promoter regions [strong binding as indicated by the PCR products (amplify by their respective primers) in red, and very weak or no binding as indicated in blue]. Graphs show fold difference ratio of ChIP with anti-GFP over ChIP with IgG control normalized to value 1 (B,C). (∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001, two-way ANOVA with Bonferroni’s post hoc test).
Mecp2-Mediated Defects of Axon Projections Leads to Diminished Sensory Response
Innervation of sensory neurons is essential for sensory functions, which can be assessed by the ability of the embryo to respond to tactile stimuli (Sagasti et al., 2005; Carmean and Ribera, 2010; Low et al., 2011). We found normal muscle striations in Mecp2 deficient embryos (Figure 7A), but these embryos are less responsive to tactile stimuli as compared to controls at 48 h after mecp2 MO injection (Figures 7B,C). WT control embryos responded to head tactile stimuli within an average of approximately 0.5 s upon tactile stimulation, whereas Mecp2 morphants took an average of 10.0 s to respond.
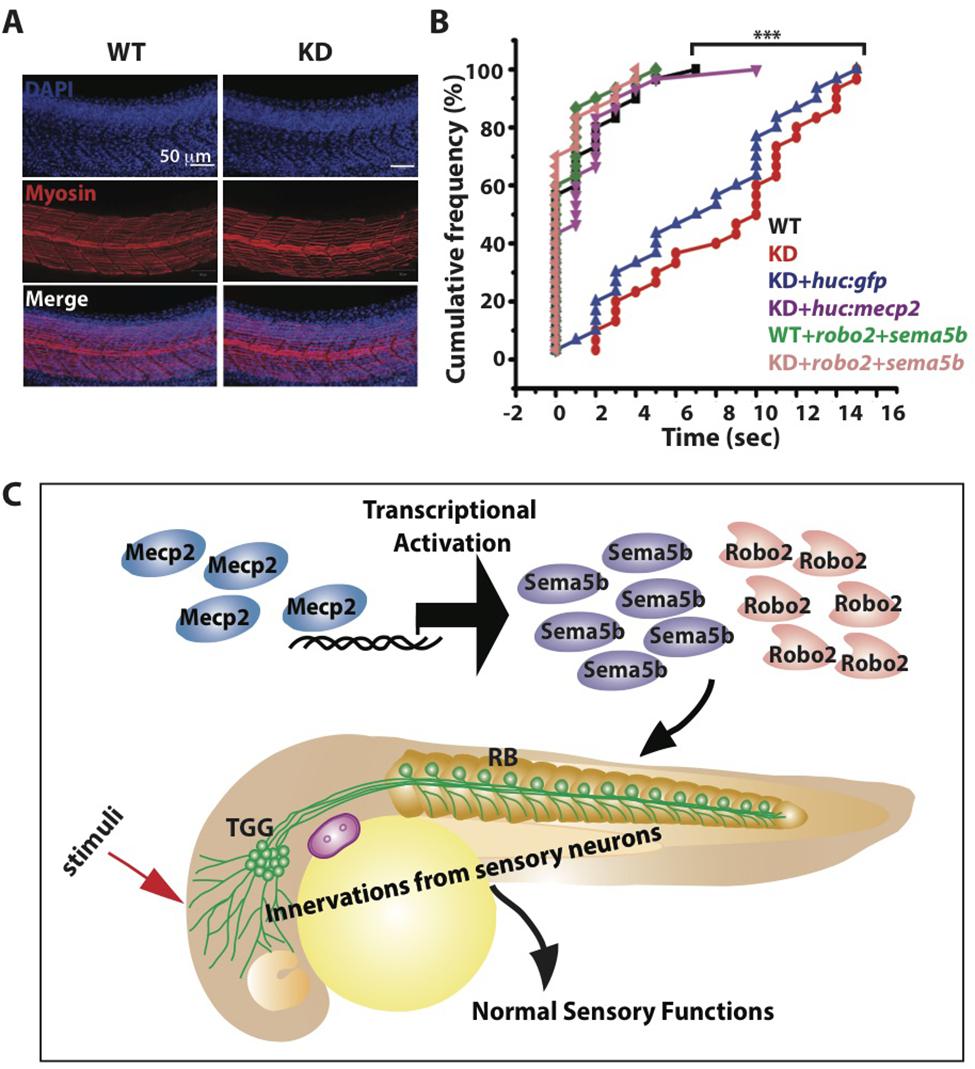
FIGURE 7. Mecp2-mediated defects of axon projections leads to diminished sensory response. Representative images showing arrays of muscle fibers in WT control and Mecp2 morphants (KD) embryos immunostained with monoclonal antibody A4.1025 (myosin) (A). In all panels, anterior is to the left. (Blue-DAPI, red-myosin) Scale bar = 50 μm. Graphs showing rates of response upon tactile stimuli (red arrow in C) from the six experimental groups of WT and KD, including WT and KD coinjected with various rescue constructs indicated on the graph, upon tactile stimuli applied to the same region specified (B,C). Proposed model shows Mecp2 plays a part in primary sensory projections through regulating transcription of Sema5b and Robo2 (C). Each individual dot (B) represents individual embryos. N > 30. (∗∗∗P < 0.0001, one-way ANOVA with Bonferroni’ post hoc test).
Importantly, HuC promoter specific expression of Mecp2 in TG neurons rescued the diminished sensory response in Mecp2 morphants, reaffirming that the sensory behavioral phenotype is a cell autonomous effect resulting from a loss of Mecp2 (Figure 7B). This diminished sensory response in Mecp2 morphants mirrors the sensory deficit phenotypes observed in Rett patients, mouse models (Devarakonda et al., 2009; Downs et al., 2010; Battaglia, 2011; Samaco et al., 2013) and zebrafish mecp2-null mutant (Pietri et al., 2013). To determine if Robo2 and Sema5b directly play a role in Mecp2–mediated sensory response, we coexpressed Robo2 and Sema5b in Mecp2 morphants (Figure 7B). This led to restoration of sensory responses in Mecp2 morphants to levels comparable to WT controls, indicating that Robo2 and Sema5b are downstream proteins of Mecp2 that are responsible for mediating MeCP2-dependent sensory responses (Figure 7C).
Taken together, our results show that Mecp2 plays a role in embryonic development of zebrafish. Mecp2 could function by regulating the expression of genes acting during formation of peripheral neurites by sensory TG neurons, which is essential for sensory response (working model illustrated in Figure 7C).
Discussion
Human males with mutations in MECP2 gene normally do not survive, whereas females showed normal postnatal development till 6–18 months of age, when typical neurological symptoms started to appear (Amir et al., 1999; Armstrong, 2005). Male rhesus and cynomolgous fetuses carrying MECP2 mutations were miscarried, while females did not show any obvious phenotypes at least for the first 4 months after birth during the study period (Liu et al., 2014). The human disease manifestations were profoundly different from those seen in mouse Rett syndrome models where all animals survived, and males but not females displayed Rett syndrome phenotypes at an early age (Guy et al., 2011). Despite showing clear behavioral alterations during early development, mecp2Q63∗ mutant fish also survived but has relatively shorter lifespan than WT fish (Pietri et al., 2013). In addition, this mecp2Q63∗ mutant exhibits motor defects compatible with motor phenotypes observed in Mecp2-null mouse models and Rett syndrome patients. This mecp2Q63∗ mutant fish also showed delayed response to tactile stimulations (Pietri et al., 2013) which is in agreement with defects exhibited by Mecp2 morphants and MeCP2 deficiency in mice. Partial MeCP2 deficiency in mice resulted in reduced pain recognition in a hot plate assay (Samaco et al., 2008). Mecp2 knockdown in zebrafish resulted in retarded responses to tactile stimuli while exhibiting normal muscle striations as compared to controls indicating that Mecp2-knockdown-induced defect in sensory neuron projections is indeed responsible for the diminished touch sensitivity observed.
The presumably null mecp2Q63∗ mutation is a nonsense mutation generated using ENU mutagenesis approach, putatively truncated the protein at position 63 and does not correspond to any known human MECP2 mutations. In mice, the two Mecp2 splice variants (Mecp2-e1 and Mecp2-e2) differing only in the first 26 amino acids, show dramatically different functions such that forced expression of MeCP2-e2 but not MeCP2-e1 promotes cell death (Dastidar et al., 2012). Although the non-lethality of this mutant fish could be that the truncated protein still had functions essential for sensory neuronal projections in zebrafish, our data from titration experiments using different concentrations of mecp2 MO rule out this possibility. Injection of mecp2 MO into WT-CTR but not these mecp2Q63∗ mutants affected innervations of TG neurons and expression of the axon guidance cues examined, confirming specificity of our mecp2 MO. Despite the absence of functional Mecp2, the expression of axon guidance cues such as Sema5b and Robo2, are only modestly but significantly decreased in these mecp2Q63∗ mutants as compared to Mecp2 morphants. Thus, explaining the milder TG projection phenotypes observed in mecp2Q63∗ mutants as compared to Mecp2 morphants and also suggesting possible compensatory effects in mecp2Q63∗ mutants. Possible compensatory mechanisms include the presence of another genetic alteration in the mecp2-null mutants that can suppress effects of the mecp2Q63∗ mutation, functional compensation through other proteins that have MBD and TRD domains or Mecp2-independent positive feedback loop for regulating Mecp2 target genes (Pietri et al., 2013). In fact, it has recently been shown that genetic compensation in zebrafish mutants but not morphants is the key reason underlying the difference in phenotypes between mutants and knockdowns (Rossi et al., 2015).
Zebrafish and human MeCP2 proteins share only about 43% amino acid sequence identity (and 46.3% identity with Xenopus), but the conserved domains are apparent (Coverdale et al., 2004) and Mecp2 may thus serve similar functions in mammals and fish. Zebrafish Mecp2 protein is more similar to the mammalian MeCP2α isoform than the MeCP2β in terms of N-terminus and genomic organization (Coverdale et al., 2004). Studies with mutant Mecp2 in Xenopus showed deficits in binding of Mecp2 to a corepressor complex (Stancheva et al., 2003). Mecp2 knockdown in zebrafish reduces metabotropic-type glutamate receptor 2 (mGluR2), which was also observed in Rett syndrome patients (Blue et al., 1999). Some of the other Mecp2-regulated genes that we found in zebrafish, such as atxn1 and pax6, were previously implicated in developmental delays in humans (Kim et al., 2011; Celestino-Soper et al., 2012). Therefore, MeCP2-regulated genes found in mammalians are likely to be involved in similar processes in zebrafish.
Functional categorization of all the Mecp2-regulated genes on our array suggest that Mecp2 is involved in transcriptional down-regulation of genes with roles in general cell maintenance, but up-regulation of genes with roles in neuronal development, specifically in neuronal differentiation, axon projections and synaptogenesis. One possible explanation for MeCP2 acting as transcription activator or repressor, is the binding of MeCP2 to either 5-hydroxymethylcytosine (5hmC) or 5-methylcytosine (5mC) as shown in a recent study using mice. MeCP2 binding to 5hmC can facilitate transcription in neural cell types, but represses transcription when bound to 5mC containing DNA (Mellén et al., 2012). However, the 5hmC and 5mC content in the same genes vary between different cell types. These variations can therefore explain, or at least in part, for the very few overlaps in MeCP2-dependent regulated genes found from different studies, especially from studies using different cells and different parts of the mammalian brains (Guy et al., 2011). For example, one of the most characterized target gene of MeCP2 – BDNF, does not consistently appear to be regulated by MeCP2 in all the arrays (Guy et al., 2011). Ratios of 5hmC/5mC in mouse Sema5b and Robo2 (the two genes that we found transcriptionally activated by Mecp2 in zebrafish) are approximately 1 or much higher than 1 (ratio of 3) in purkinje cells (PCs) and specialized Bergmann glial (BG) cell population, but is only about 0.2–0.4 in granule cells (GCs) (Mellén et al., 2012). Therefore, suggesting that MeCP2 probably activates transcription of both Sema5b and Robo2 in PC and BG but represses transcription of both genes in GC cells.
Our results show that Mecp2 knockdown causes defects in projections from sensory neurons through activating transcription of axon guidance cues represented by Sema5b and Robo2. These defects in zebrafish emulate sensory and neuromotor deficits seen in Rett syndrome patients (Samaco et al., 2008; Devarakonda et al., 2009; Downs et al., 2010; Battaglia, 2011). Although Rett syndrome patients have sensory deficits, this aspect of the disorder is still largely uncharacterized. sema3f, sema5b and robo2 are genes that have roles in axon projections and synaptogenesis and were significantly down-regulated in Mecp2 morphants (Figures 4 and 5). These known axon repelling guidance cues appear to have axon promoting functions in sensory neurons in zebrafish (at least for both Sema5b and Robo2). Previous studies in mice showed guidance cues such as Slits (ligand for Robo receptor) that typically act as axon repellents may also act to attract some axons (such as DRG, trigeminal sensory neurons and retinal axons inside the optic fiber layer) (Wang et al., 1999; Nguyen Ba-Charvet et al., 2001; Jin et al., 2003; Ma and Tessier-Lavigne, 2007). Semaphorins typically cause repulsion and collapse of axons but could on the other hand promote dendrite growth in hippocampal neurons (Ng et al., 2013). Therefore, it could be possible that semaphorins can also promote growth of some axons as observed for Slit/Robo. Semaphorins are widely expressed in the developing nervous system of mice, but there are no known roles of Sema5b specifically. Previous studies in mouse models demonstrated that semaphorins act as guidance cues for axon targeting, cell migration, cell death or synapse formation during the development of nervous system (Koncina et al., 2007). Severe abnormality in peripheral nerve projections, which includes trigeminal, facial, vagus, accessory and glossopharyngeal nerves were observed in Sema3D knockout mouse (Taniguchi et al., 1997).
The Robo2 protein is highly conserved from fly to humans, and is known to function in developing nervous systems with roles in axon guidance, synaptogenesis and cell migration (Yeo et al., 2004; Campbell et al., 2007; López-Bendito et al., 2007; Andrews et al., 2008; Pan et al., 2012). Mice deficient in Robo2 demonstrated obvious axon guidance errors, with a more prominent effect on mice deficient in both Robo1 and Robo2 (López-Bendito et al., 2007). Like Robo2, Sema5b is also a ligand-binding receptor. Besides its role as a guidance cue, little else is known about its role in neuronal development. Interestingly, Robo2 or Sema5b can both modestly rescue defective projections from TG neurons in Mecp2 morphants. Although there are no known expression studies on Sema5b in zebrafish, Robo2 was found to be expressed in a subpopulation of HuC positive cells of the TG in zebrafish embryos at 24 hpf (Pan et al., 2012). Therefore, partial rescue of the phenotype observed upon expressing Robo2 alone could be because not all TG neurons express Robo2. It is also possible that both Robo2 and Sema5b are working together and are not able to achieve significant rescue effect as an individual. Concurrent expression of both Robo2 and Sema5b can rescue this defect to approximately control levels, suggesting that Robo2 and Sema5b plays important role in mediating the effects of Mecp2 in regulating projections of sensory neurons. In contrast, although we found a decrease of Sema3f expression in Mecp2 morphants, this decrease did not translate to significant defects, at least in the gross morphology of zebrafish embryos and also in projections of sensory neurons, which is likely due to compensatory effects from other class 3 semaphorins. Similar observation on mice was reported, where abnormal levels of class 3 semaphorins and their receptors were also observed in the olfactory system in Mecp2-null mice. But neither Sema3A nor 3F elicited any difference in repulsion or neurite outgrowth response of Mecp2-null olfactory axons (Degano et al., 2009).
In summary, our data using mecp2-null mutants and mecp2 MO-mediated knockdown approach show that Mecp2 is important for peripheral innervation of sensory neurons in the zebrafish. Mecp2 knockdown in zebrafish model recapitulates sensory deficits of Rett syndrome in humans. We also found expression of well-known axon guidance cues such as Sema5b and Robo2 positively regulated by Mecp2 through its interactions with specific regions on both promoters. These guidance cues are essential for the Mecp2-dependent innervations from sensory neurons and sensory response during embryonic development.
Materials and Methods
Injection of Zebrafish
Wild-type zebrafish of the AB strain were maintained under standard conditions of fish husbandry. Freshly fertilized zebrafish eggs were injected with mRNA (100 ng/μl), plasmid DNA (40 ng/μl), mecp2 morpholino (800 μM), robo2 morpholino (200 μM) and sema5b morpholino (500 μM) at the one- to two-cell stage in a volume of approximately 1 nl. Approximately 200 embryos were injected for each morpholinos or overexpresson constructs. At least 30 embryos were analyzed for each experimental group used per experiment. For experiments using mecp2-null zebrafish embryos, mecp2 splice blocking morpholino was also injected into mecp2Q63∗/Q63∗ mutant (mecp2-null) and its wild-type control embryos (WT-CTR) (both in the Nacre background). The injected embryos were cultured at 28°C, and embryos were fixed at specific developmental stages for further analysis. Morpholinos were purchased from GeneTools. Splice-blocking morpholino (5′-CTCACCTCTGCTGACAACAAAATAA-3′) was selected for knocking down Mecp2. This splice-blocking MO that allows efficiency of MO to be determined through PCR, was used for all the Mecp2 morphants shown here. The control morpholino (5′-CCTCTTACCTCAGTTACAATTTATA-3′) and p53 morpholino (5′-GCGCCATTGCTTTGCAAGAATTG-3′) used were the scrambled sequence and a translational blocker respectively from Gene Tools. All zebrafish experiments were in compliance and approved by the Singapore National Advisory Committee on Laboratory Animal Research.
Cloning of Zebrafish mecp2, sema5b, and robo2
cDNAs were synthesized from 24 hpf wild-type embryos using cDNA synthesis kit (Invitrogen). These cDNA templates were used for PCR amplification of the full-length zebrafish mecp2, sema5b and robo2. The PCR products were cloned into TOPO vector and subsequently sub-cloned into either pCS2-GFPxlt or pSP64T expression vector for over expression studies and rescue experiment. Full-length zebrafish mecp2 was cloned into huC promoter plasmid to induce specific expression in subsets of TG neurons. GFP-expressing embryos were screened by fluorescent microscopy to determine for expression of injected constructs. Only embryos that expressed GFP were used for all downstream experiments and analysis.
Whole-Mount In Situ Hybridization
Purified plasmid was linearized by selected restriction enzymes and used as templates for in vitro transcription using T7 or SP6 RNA polymerase to generate Digoxigenin (DIG)-labeled sense and antisense probes using DIG RNA labeling kit (Roche). Whole-mount in situ hybridizations were performed following routine protocols.
Antibody Staining and Terminal Deoxynucleotidyl Transferase (TdT)-Mediated dUTP Nick-End Labeling (TUNEL) Assay
Whole-mount antibody staining on zebrafish embryos was performed according to standard protocols. The following antibodies were used: rabbit anti-GFP (1:1000; Abcam), monoclonal anti-acetylated tubulin (1:500; Sigma), monoclonal anti-islet [1:100; The Developmental Studies Hybridoma Bank (DSHB)], monoclonal antibody A4.1025 (skeletal muscle myosin) (1:20; DSHB) and mouse anti-HuC/D (1:1000; Invitrogen). For confocal microscopy, appropriate Alexa Fluor-conjugated secondary antibodies (1:500; Molecular Probes) were used for signal detection. Embryos were counterstained with 4,6-diamidino-2-phenylindole (DAPI) to visualize cell nuclei when required. For TUNEL assay, the embryos were made more permeable by treating with proteinase K and 0.1% sodium citrate prior to the standard protocol as recommended by the manufacturer (Roche).
In Vitro Transcription of Capped mRNA
Plasmids encoding mecp2, sema5b and robo2 were linearized, and capped, full-length mRNA was transcribed from this template using the mMessage mMachine Kit (Ambion). The mRNA was injected into one- to two-cell stage embryos either alone or in combination with a morpholino.
Zebrafish Gene Expression Microarray
Three independent pools of 24 hpf zebrafish Mecp2 morphant embryos were compared to three independent pools of 24 hpf control embryos. RNA extraction was done using RNeasy mini kit (QIAGEN). The triplicate embryo pools made a total of six hybridizations. cDNA synthesis, cDNA labeling, microarray hybridization and washing were performed following the manufacturer’s instructions (Roche NimbleGen). The 135K NimbleGen array contains 135,000 probes targeting 38,489 transcripts derived from the Ensembl database built on the Zv7 assembly (37,157 zebrafish genes identified). Array hybridization was done using Maui Hybridization System and was scanned using Axon GenePix 4000B Scanner. Data extraction and image processing were performed using NimbleScan software v.2.6 and the Robust Multichip Average (RMA) algorithm used to generate gene expression values. The normalized data was subsequently analyzed using ArrayStar. Threshold was set at twofold change with a 95% confidence interval by student’s t-test with Benjamini Hochberg False Discovery Rate analysis.
Quantitative Real-Time PCR
One microgram of total RNA was used as starting material. Reverse transcription was carried out with Invitrogen’s SuperScript III First-Strand Synthesis kit using oligo(dT) primers. Gene specific primers were used to amplify sema3f (fw-5′ CAACCAGTACTGCCAAGACTAC 3′; rv-5′ TCCTGGTGGTGTCTCCTATT 3′), sema5b (fw-5′ CTGCTGTCTTTCCTGGTGTATG 3′; rv-5′ GTGGTGTTGCCCTTGTAGTT 3′) and robo2 (fw-5′ GACATACCTCCATCAGGGTTTAG 3′; rv-5′ GAAACTGCAGCAGGAGAAGA 3′) by qPCR on a Biorad CFX96 RT PCT using SYBR Select master mix (Invitrogen). A sample volume of 12 μl was used for all assays. All samples and standards were run in triplicates. Three biological replicates were performed for each set of data normalized using housekeeping gene beta-actin.
Chromatin Immunoprecipitation (ChIP)
Wild-type embryos were injected at one-cell stage with GFP-tagged mecp2 RNA. They were harvested manually at 24 hpf, then dechorionated, deyolked and immediately crosslinked to DNA by direct addition of formaldehyde at a final concentration of 1% for 10 min at room temperature. Embryo tissues were lysed and sonicated (Sonics Vibra-Cell) to produce DNA fragments that are between 300 and 800 bp. After sonication, ChIP assay was performed according to Millipore protocol (Magna ChIP G Tissue Kit). Protein-DNA complexes were immunoprecipitated overnight at 4°C in the presence of specific anti-GFP antibody (Abcam) or control rabbit IgG (Santa Cruz Biotechnology). DNA was purified and used directly for PCR of sema5b and robo2 promoters. Primers were designed within 2000 bp region upstream of translational start site of sema5b promoter (Primer 1: fw-5′ CTCTGCTGATCGTAAAGGTGTAT 3′; rv-5′ GTTAAAGAGGACAGACGGAGTG 3′, Primer 2: fw-5′ TCCAGCTCAGTTCAGTTCAAG 3′; rv-5′ TTGCGATTTCCAACATATACATAGC 3′, Primer 3: fw-5′ GGGTTCAATTCCCTACAGTCTT 3′; rv-5′ GGATGGATGGATTGATGGTTAGA 3′, Primer 4: fw-5′ TGTTCCACTTCTGGTCAGTTT 3′; rv-5′ TCTGAGGAGACCCAGCTTTA 3′) or robo2 promoter (Primer 1: fw-5′ GAGGATCGTAAGGTGCTTCTG 3′; rv-5′ ACACCATCTCCTACTGTGTTTG 3′, Primer 2: fw-5′ ACACTGACATATTGGTGGTCAT 3′; rv-5′ CAGCTTTCCGTGGTGTTTATTC 3′, Primer 3: fw-5′ CTTAGAGCTGACACTGGAAGATG 3′; rv-5′ TGGGCGAACGGATAGATAGA 3′, Primer 4: fw-5′ TGCTGACACACCGCTAATC 3′; rv-5′ GAGGAATGCAAGGACCAATTTC 3).
Behavior Studies
Wild-type embryos injected with either control morpholino (800 μM), mecp2 morpholino (800 μM), or in combination with rescue constructs (∼40 ng for DNA and 100 ng for mRNA) were allowed to develop to 48 hpf under normal growth conditions before scoring was done. These embryos were dechorionated manually and subjected to tactile stimuli at the head region. Manual scoring was done by taking down the time required by the embryo to produce a response upon stimulation applied with the tip of a mounting needle. Zero seconds was recorded for embryos that gave an immediate reaction to the tactile stimuli.
Imaging, Analysis, and Figure Preparation
Stained embryos were dissected from their yolk and mounted in 70% glycerol. High-resolution images of embryos were captured using a Zeiss LSM 710 confocal microscope (Carl Zeiss Pte Ltd, Singapore). Low-resolution images were obtained using a Zeiss SteREO Discovery V8 stereoscope equipped with an Axiocam ICc3 color digital camera (Carl Zeiss Pte Ltd, Singapore). For analysis of the neurite structure of TG cells, three-dimensional (3-D) reconstructions of the processes of all labeled cells were made from Z-series stacks of confocal images. The projection images were semi-automatically traced with NIH ImageJ using the NeuronJ plugin. The total length of processes in each individual embryo were subsequently measured and analyzed. All data were scrambled and only decoded after analysis was done blinded. A total of >30 embryos from >3 individual experiments were analyzed per group. Gel quantification of DNA for ChIP PCR was done using NIH ImageJ. Statistical significance (P < 0.05) was assessed using student t-test, one-way or two-way ANOVA with Bonferroni’ post hoc test, after normalization checked using Shapiro–Wilk normality test. Figures were assembled using Adobe Illustrator CS4.
Author Contributions
WL designed and performed all experiments and analyzed all data; ZL analyzed data for microarray; VK designed some experiments and made critical edits on the manuscript; TP contributed key materials for the experiments and made critical inputs to the manuscript; EG initiated and directed the entire study, designed experiments, analyzed data and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank G. Augustine, S. Jesuthasan, S. Shenolikar, R. Krishnan, D. Purves, and B. L. Tang for comments on the manuscript and helpful discussion, K. Kwan for Robo2 cDNA, H. Okamoto for HuC:kaede construct, S. Choksi for advice on microarray analysis and Agency for Science, Technology and Research (A∗STAR), Singapore core zebrafish facility for providing embryos, J. Tay for technical support. This work was supported by Duke-NUS Graduate Signature Research Program funded by A∗STAR and the Ministry of Health, Singapore, the Academic Center of Excellence (ACE) research award from GlaxoSmithKline (GSK), Abbott Nutrition and the National Research Foundation Singapore under its Competitive Research Program (NRF 2008 NRF-CRP 002-082) to EG.
References
Amir, R. E., Van den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188. doi: 10.1038/13810
Andrews, W., Barber, M., Hernadez-Miranda, L. R., Xian, J., Rakic, S., Sundaresan, V., et al. (2008). The role of Slit-Robo signaling in the generation, migration and morphological differentiation of cortical interneurons. Dev. Biol. 313, 648–658. doi: 10.1016/j.ydbio.2007.10.052
Armstrong, D. D. (2005). Neuropathology of Rett syndrome. J. Child Neurol. 20, 747–753. doi: 10.1177/08830738050200090901
Battaglia, A. (2011). Sensory impairment in mental retardation: a potential role for NGF. Arch. Ital. Biol. 149, 193–203. doi: 10.4449/aib.v149i2.1362
Belichenko, P. V., Wright, E. E., Belichenko, N. P., Masliah, E., Li, H. H., Mobley, W. C., et al. (2009). Widespread changes in dendritic and axonal morphology in Mecp2-mutant mouse models of Rett syndrome: evidence for disruption of neuronal networks. J. Comp. Neurol. 514, 240–258. doi: 10.1002/cne.22009
Ben-Shachar, S., Chahrour, M., Thaller, C., Shaw, C. A., and Zoghbi, H. Y. (2009). Mouse models of MeCP2 disorders share gene expression changes in the cerebellum and hypothalamus. Hum. Mol. Genet. 18, 2431–2442. doi: 10.1093/hmg/ddp181
Bienvenu, T., and Chelly, J. (2006). Molecular genetics of Rett syndrome: when DNA methylation goes unrecognized. Nat. Rev. Genet. 7, 415–426. doi: 10.1038/nrg1878
Bird, A. (2008). The methyl-CpG-binding protein MeCP2 and neurological disease. Biochem. Soc. Trans. 36, 575–583. doi: 10.1042/BST0360575
Blue, M. E., Naidu, S., and Johnston, M. V. (1999). Altered development of glutamate and GABA receptors in the basal ganglia of girls with rett syndrome. Exp. Neurol. 156, 345–352. doi: 10.1006/exnr.1999.7030
Campbell, D. S., Stringham, S. A., Timm, A., Xiao, T., Law, M.-Y., Baier, H., et al. (2007). Slit1a inhibits retinal ganglion cell arborization and synaptogenesis via Robo2-dependent and -independent pathways. Neuron 55, 231–245. doi: 10.1016/j.neuron.2007.06.034
Carmean, V., and Ribera, A. B. (2010). Genetic analysis of the touch response in zebrafish (Danio rerio). Int. J. Comp. Psychol. 23, 91–102.
Caron, S. J. C., Prober, D., Choy, M., and Schier, A. F. (2008). In vivo birthdating by BAPTISM reveals that trigeminal sensory neuron diversity depends on early neurogenesis. Development 135, 3259–3269. doi: 10.1242/dev.023200
Celestino-Soper, P. B., Skinner, C., Schroer, R., Eng, P., Shenai, J., Nowaczyk, M. M., et al. (2012). Deletions in chromosome 6p22.3-p24.3, including ATXN1, are associated with developmental delay and autism spectrum disorders. Mol. Cytogenet. 5:17. doi: 10.1186/1755-8166-5-17
Chahrour, M., Jung, S. Y., Shaw, C., Zhou, X., Wong, S. T. C., Qin, J., et al. (2008). MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320, 1224–1229. doi: 10.1126/science.1153252
Chahrour, M., and Zoghbi, H. Y. (2007). The story of Rett syndrome: from clinic to neurobiology. Neuron 56, 422–437. doi: 10.1016/j.neuron.2007.10.001
Chandler, S. P., Guschin, D., Landsberger, N., and Wolffe, A. P. (1999). The methyl-CpG binding transcriptional repressor MeCP2 stably associates with nucleosomal DNA. Biochemistry 38, 7008–7018. doi: 10.1021/bi990224y
Chen, R. Z., Akbarian, S., Tudor, M., and Jaenisch, R. (2001). Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat. Genet. 27, 327–331. doi: 10.1038/85906
Christodoulou, J., and Weaving, L. S. (2003). MECP2 and beyond: phenotype-genotype correlations in Rett syndrome. J. Child Neurol. 18, 669–674. doi: 10.1177/08830738030180100901
Cole, L. K., and Ross, L. S. (2001). Apoptosis in the developing zebrafish embryo. Dev. Biol. 240, 123–142. doi: 10.1006/dbio.2001.0432
Coverdale, L. E., Martyniuk, C. J., Trudeau, V. L., and Martin, C. C. (2004). Differential expression of the methyl-cytosine binding protein 2 gene in embryonic and adult brain of zebrafish. Brain Res. Dev. Brain Res. 153, 281–287. doi: 10.1016/j.devbrainres.2004.08.009
Dastidar, S. G., Bardai, F. H., Ma, C., Price, V., Rawat, V., Verma, P., et al. (2012). Isoform-specific toxicity of Mecp2 in postmitotic neurons: suppression of neurotoxicity by FoxG1. J. Neurosci. 32, 2846–2855. doi: 10.1523/JNEUROSCI.5841-11.2012
Degano, A. L., Pasterkamp, R. J., and Ronnett, G. V. (2009). MeCP2 deficiency disrupts axonal guidance, fasciculation, and targeting by altering Semaphorin 3F function. Mol. Cell. Neurosci. 42, 243–254. doi: 10.1016/j.mcn.2009.07.009
Devarakonda, K. M., Lowthian, D., and Raghavendra, T. (2009). A case of Rett syndrome with reduced pain sensitivity. Paediatr. Anaesth. 19, 625–627. doi: 10.1111/j.1460-9592.2009.03018.x
Downs, J., Géranton, S. M., Bebbington, A., Jacoby, P., Bahi-Buisson, N., Ravine, D., et al. (2010). Linking MECP2 and pain sensitivity: the example of Rett syndrome. Am. J. Med. Genet. A 152A, 1197–1205. doi: 10.1002/ajmg.a.33314
Eisen, J. S., and Smith, J. C. (2008). Controlling morpholino experiments: don’t stop making antisense. Development 135, 1735–1743. doi: 10.1242/dev.001115
Guy, J., Cheval, H., Selfridge, J., and Bird, A. (2011). The role of MeCP2 in the brain. Annu. Rev. Cell Dev. Biol. 27, 631–652. doi: 10.1146/annurev-cellbio-092910-154121
Guy, J., Hendrich, B., Holmes, M., Martin, J. E., and Bird, A. (2001). A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 27, 322–326. doi: 10.1038/85899
Hagberg, B., Aicardi, J., Dias, K., and Ramos, O. (1983). A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett’s syndrome: report of 35 cases. Ann. Neurol. 14, 471–479. doi: 10.1002/ana.410140412
Jin, Z., Zhang, J., Klar, A., Chedotal, A., Rao, Y., Cepko, C. L., et al. (2003). Irx4-mediated regulation of Slit1 expression contributes to the definition of early axonal paths inside the retina. Development 130, 1037–1048. doi: 10.1242/dev.00326
Kim, K.-Y., Hysolli, E., and Park, I.-H. (2011). Neuronal maturation defect in induced pluripotent stem cells from patients with Rett syndrome. Proc. Natl. Acad. Sci. U.S.A. 108, 14169–14174. doi: 10.1073/pnas.1018979108
Koncina, E., Roth, L., Gonthier, B., and Bagnard, D. (2007). Role of semaphorins during axon growth and guidance. Adv. Exp. Med. Biol. 621, 50–64. doi: 10.1007/978-0-387-76715-4_4
Lewis, J. D., Meehan, R. R., Henzel, W. J., Maurer-Fogy, I., Jeppesen, P., Klein, F., et al. (1992). Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell 69, 905–914. doi: 10.1016/0092-8674(92)90610-O
Liu, H., Chen, Y., Niu, Y., Zhang, K., Kang, Y., Ge, W., et al. (2014). TALEN-mediated gene mutagenesis in rhesus and cynomolgus monkeys. Cell Stem. Cell 14, 323–328. doi: 10.1016/j.stem.2014.01.018
López-Bendito, G., Flames, N., Ma, L., Fouquet, C., Di Meglio, T., Chedotal, A., et al. (2007). Robo1 and Robo2 cooperate to control the guidance of major axonal tracts in the mammalian forebrain. J. Neurosci. 27, 3395–3407. doi: 10.1523/JNEUROSCI.4605-06.2007
Low, S. E., Amburgey, K., Horstick, E., Linsley, J., Sprague, S. M., Cui, W. W., et al. (2011). TRPM7 is required within zebrafish sensory neurons for the activation of touch-evoked escape behaviors. J. Neurosci. 31, 11633–11644. doi: 10.1523/JNEUROSCI.4950-10.2011
Ma, D., Yoon, S.-I., Yang, C.-H., Marcy, G., Zhao, N., Leong, W.-Y., et al. (2015). Rescue of methyl-CpG binding protein 2 dysfunction-induced Defects in newborn neurons by pentobarbital. Neurotherapeutics 12, 477–490. doi: 10.1007/s13311-015-0343-0
Ma, L., and Tessier-Lavigne, M. (2007). Dual branch-promoting and branch-repelling actions of Slit/Robo signaling on peripheral and central branches of developing sensory axons. J. Neurosci. 27, 6843–6851. doi: 10.1523/JNEUROSCI.1479-07.2007
Matarazzo, V., Cohen, D., Palmer, A. M., Simpson, P. J., Khokhar, B., Pan, S.-J., et al. (2004). The transcriptional repressor Mecp2 regulates terminal neuronal differentiation. Mol. Cell. Neurosci. 27, 44–58. doi: 10.1016/j.mcn.2004.05.005
Mellén, M., Ayata, P., Dewell, S., Kriaucionis, S., and Heintz, N. (2012). MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell 151, 1417–1430. doi: 10.1016/j.cell.2012.11.022
Metcalfe, W. K., Myers, P. Z., Trevarrow, B., Bass, M. B., and Kimmel, C. B. (1990). Primary neurons that express the L2/HNK-1 carbohydrate during early development in the zebrafish. Development 110, 491–504.
Nan, X., Campoy, F. J., and Bird, A. (1997). MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell 88, 471–481. doi: 10.1016/S0092-8674(00)81887-5
Neul, J. L., Kaufmann, W. E., Glaze, D. G., Christodoulou, J., Clarke, A. J., Bahi-Buisson, N., et al. (2010). Rett syndrome: revised diagnostic criteria and nomenclature. Ann. Neurol. 68, 944–950. doi: 10.1002/ana.22124
Ng, T., Ryu, J. R., Sohn, J. H., Tan, T., Song, H., Ming, G.-L., et al. (2013). Class 3 semaphorin mediates dendrite growth in adult newborn neurons through Cdk5/FAK pathway. PLoS ONE 8:e65572. doi: 10.1371/journal.pone.0065572.s003
Nguyen Ba-Charvet, K. T., Brose, K., Marillat, V., Sotelo, C., Tessier-Lavigne, M., and Chédotal, A. (2001). Sensory axon response to substrate-bound Slit2 is modulated by laminin and cyclic GMP. Mol. Cell. Neurosci. 17, 1048–1058. doi: 10.1006/mcne.2001.0994
Palmer, A. M., Degano, A. L., Park, M. J., Ramamurthy, S., and Ronnett, G. V. (2012). Normal mitral cell dendritic development in the setting of Mecp2 mutation. Neuroscience 202, 108–116. doi: 10.1016/j.neuroscience.2011.11.044
Pan, Y. A., Choy, M., Prober, D. A., and Schier, A. F. (2012). Robo2 determines subtype-specific axonal projections of trigeminal sensory neurons. Development 139, 591–600. doi: 10.1242/dev.076588
Park, H.-C., Kim, C.-H., Bae, Y.-K., Yeo, S.-Y., Kim, S.-H., Hong, S.-K., et al. (2000). Analysis of upstream elements in the HuC promoter leads to the establishment of transgenic zebrafish with fluorescent neurons. Dev. Biol. 227, 279–293. doi: 10.1006/dbio.2000.9898
Pietri, T., Roman, A.-C., Guyon, N., Romano, S. A., Washbourne, P., Moens, C. B., et al. (2013). The first mecp2-null zebrafish model shows altered motor behaviors. Front. Neural Circuits 7:118. doi: 10.3389/fncir.2013.00118
Robu, M. E., Larson, J. D., Nasevicius, A., Beiraghi, S., Brenner, C., Farber, S. A., et al. (2007). p53 activation by knockdown technologies. PLoS Genet. 3:e78. doi: 10.1371/journal.pgen.0030078
Rossi, A., Kontarakis, Z., Gerri, C., Nolte, H., Hölper, S., Krüger, M., et al. (2015). Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature 524, 230–233. doi: 10.1038/nature14580
Sagasti, A., Guido, M. R., Raible, D. W., and Schier, A. F. (2005). Repulsive interactions shape the morphologies and functional arrangement of zebrafish peripheral sensory arbors. Curr. Biol. 15, 804–814. doi: 10.1016/j.cub.2005.03.048
Samaco, R. C., Fryer, J. D., Ren, J., Fyffe, S., Chao, H.-T., Sun, Y., et al. (2008). A partial loss of function allele of methyl-CpG-binding protein 2 predicts a human neurodevelopmental syndrome. Hum. Mol. Genet. 17, 1718–1727. doi: 10.1093/hmg/ddn062
Samaco, R. C., McGraw, C. M., Ward, C. S., Sun, Y., Neul, J. L., and Zoghbi, H. Y. (2013). Female Mecp2(+/-) mice display robust behavioral deficits on two different genetic backgrounds providing a framework for pre-clinical studies. Hum. Mol. Genet. 22, 96–109. doi: 10.1093/hmg/dds406
Sato, T., Takahoko, M., and Okamoto, H. (2006). HuC:Kaede, a useful tool to label neural morphologies in networks in vivo. Genesis 44, 136–142. doi: 10.1002/gene.20196
Shahbazian, M. D., and Zoghbi, H. Y. (2001). Molecular genetics of Rett syndrome and clinical spectrum of MECP2 mutations. Curr. Opin. Neurol. 14, 171–176. doi: 10.1097/00019052-200104000-00006
Skene, P. J., Illingworth, R. S., Webb, S., Kerr, A. R. W., James, K. D., Turner, D. J., et al. (2010). Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol. Cell 37, 457–468. doi: 10.1016/j.molcel.2010.01.030
Stancheva, I., Collins, A. L., Van den Veyver, I. B., Zoghbi, H., and Meehan, R. R. (2003). A mutant form of MeCP2 protein associated with human Rett syndrome cannot be displaced from methylated DNA by notch in Xenopus embryos. Mol. Cell 12, 425–435. doi: 10.1016/S1097-2765(03)00276-4
Taniguchi, M., Yuasa, S., Fujisawa, H., Naruse, I., Saga, S., Mishina, M., et al. (1997). Disruption of semaphorin III/D gene causes severe abnormality in peripheral nerve projection. Neuron 19, 519–530. doi: 10.1016/S0896-6273(00)80368-2
Wang, K. H., Brose, K., Arnott, D., Kidd, T., Goodman, C. S., Henzel, W., et al. (1999). Biochemical purification of a mammalian slit protein as a positive regulator of sensory axon elongation and branching. Cell 96, 771–784. doi: 10.1016/S0092-8674(00)80588-7
Yeo, S.-Y., Miyashita, T., Fricke, C., Little, M. H., Yamada, T., Kuwada, J. Y., et al. (2004). Involvement of Islet-2 in the Slit signaling for axonal branching and defasciculation of the sensory neurons in embryonic zebrafish. Mech. Dev. 121, 315–324. doi: 10.1016/j.mod.2004.03.006
Young, J. I., Hong, E. P., Castle, J. C., Crespo-Barreto, J., Bowman, A. B., Rose, M. F., et al. (2005). Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc. Natl. Acad. Sci. U.S.A. 102, 17551–17558. doi: 10.1073/pnas.0507856102
Keywords: Mecp2, trigeminal ganglion, sensory functions, Rett syndrome, neurodevelopmental disorder, axon guidance cues, Robo2, Sema5b
Citation: Leong WY, Lim ZH, Korzh V, Pietri T and Goh ELK (2015) Methyl-CpG Binding Protein 2 (Mecp2) Regulates Sensory Function Through Sema5b and Robo2. Front. Cell. Neurosci. 9:481. doi: 10.3389/fncel.2015.00481
Received: 22 September 2015; Accepted: 30 November 2015;
Published: 21 December 2015.
Edited by:
Tommaso Pizzorusso, University of Florence, ItalyReviewed by:
Daniela Tropea, Trinity College Dublin, IrelandElsa Fabbretti, University of Nova Gorica, Slovenia
Copyright © 2015 Leong, Lim, Korzh, Pietri and Goh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eyleen L. K. Goh, ZXlsZWVuLmdvaEBkdWtlLW51cy5lZHUuc2c=