 Yixing Du1,2
Yixing Du1,2 Conrad M. Kiyoshi1
Conrad M. Kiyoshi1 Qi Wang1,3
Qi Wang1,3 Wei Wang4
Wei Wang4 Baofeng Ma1Catherine C. Alford1Shiying Zhong1Qi Wan2
Baofeng Ma1Catherine C. Alford1Shiying Zhong1Qi Wan2 Haijun Chen5
Haijun Chen5 Eric E. Lloyd6
Eric E. Lloyd6 Robert M. Bryan Jr.6
Robert M. Bryan Jr.6 Min Zhou1*
Min Zhou1*- 1Department of Neuroscience, The Ohio State University Wexner Medical Center, Columbus, OH, USA
- 2Department of Neurology, The First Affiliated Hospital of Nanjing Medical University, Nanjing, China
- 3Department of Neurology, Meitan General Hospital, Xibahe Nanli, Beijing, China
- 4Department of Physiology, Institute of Brain Research, School of Basic Medicine, Huazhong University of Science and Technology, Wuhan, China
- 5Department of Biological Science, University at Albany, State University of New York, Albany, NY, USA
- 6Department of Anesthesiology, Baylor College of Medicine, Houston, TX, USA
We have recently shown that a linear current-to-voltage (I-V) relationship of membrane conductance (passive conductance) reflects the intrinsic property of K+ channels in mature astrocytes. While passive conductance is known to underpin a highly negative and stable membrane potential (VM) essential for the basic homeostatic function of astrocytes, a complete repertoire of the involved K+ channels remains elusive. TREK-1 two-pore domain K+ channel (K2P) is highly expressed in astrocytes, and covalent association of TREK-1 with TWIK-1, another highly expressed astrocytic K2P, has been reported as a mechanism underlying the trafficking of heterodimer TWIK-1/TREK-1 channel to the membrane and contributing to astrocyte passive conductance. To decipher the individual contribution of TREK-1 and address whether the appearance of passive conductance is conditional to the co-expression of TWIK-1/TREK-1 in astrocytes, TREK-1 single and TWIK-1/TREK-1 double gene knockout mice were used in the present study. The relative quantity of mRNA encoding other astrocyte K+ channels, such as Kir4.1, Kir5.1, and TREK-2, was not altered in these gene knockout mice. Whole-cell recording from hippocampal astrocytes in situ revealed no detectable changes in astrocyte passive conductance, VM, or membrane input resistance (Rin) in either kind of gene knockout mouse. Additionally, TREK-1 proteins were mainly located in the intracellular compartments of the hippocampus. Altogether, genetic deletion of TREK-1 alone or together with TWIK-1 produced no obvious alteration in the basic electrophysiological properties of hippocampal astrocytes. Thus, future research focusing on other K+ channels may shed light on this long-standing and important question in astrocyte physiology.
Introduction
Mature hippocampal astrocytes exhibit a membrane K+ conductance characterized by a linear current-to-voltage (I-V) relationship (passive conductance), and a highly negative membrane potential (VM) (Zhou et al., 2006; Kafitz et al., 2008). Both of these electrophysiological features are fundamental for the basic homeostatic functions of astrocytes in the brain (Verkhratsky and Steinhauser, 2000; Walz, 2000; Olsen and Sontheimer, 2008; Wang and Bordey, 2008; Kimelberg, 2010).
While passive behavior and low membrane resistance (RM) represent intrinsic properties of membrane ion channels (Du et al., 2015), the molecular identity of K+ channels underlying this unique membrane conductance remains an issue to be fully resolved. Now, the inwardly rectifying K+ channel Kir4.1 and two-pore domain K+ channels (K2P) TWIK-1(K2P 1.1) and TREK-1(K2P 2.1) are considered the three major K+ channels, and the important contribution of Kir4.1 to astrocyte passive conductance has been well documented (Butt and Kalsi, 2006; Neusch et al., 2006; Djukic et al., 2007; Kucheryavykh et al., 2007; Seifert et al., 2009; Tong et al., 2014; Olsen et al., 2015).
In the hippocampus, Kir4.1 shows only moderate expression compared to other brain regions (Nwaobi et al., 2014). Consistently, our functional analysis indicated that 48% of passive conductance is mediated by Kir4.1 channels (Ma et al., 2014). To determine the contribution of other K+ channels to passive conductance, we initially considered TWIK-1 and TREK-1 as the two K2P candidates based on an astrocyte transcriptome database (Cahoy et al., 2008; Zhou et al., 2009). However, TWIK-1 gene knockout affected the passive conductance minimally because of the retention of large amounts of TWIK-1 channels in intracellular compartments and the behavior of this channel as a non-selective cation channel in the membrane (Wang et al., 2013, 2015).
To explore a possible contribution of TREK-1 to passive conductance, TREK-1 gene knockout mice were introduced in this study. TREK-1 is an outwardly rectifying leak type K+ channel that follows Goldman-Hodgkin-Katz (GHK) constant field rectification (Fink et al., 1996). Thus, a combined expression of TREK-1 and Kir4.1 may hypothetically explain the observed passive conductance in astrocytes. TREK-1 is highly dynamic and subject to regulation by various changes occurring in the interstitial environment, such as pH, temperature, osmolarity, and mechanic force (Patel et al., 1998; Maingret et al., 1999; Lesage and Lazdunski, 2000; Chemin et al., 2005; Murbartián et al., 2005). This makes TREK-1 an attractive candidate as it may confer astrocytes’ ability to interact with neurons and integrate neuron-glia function.
The covalent association of TWIK-1/TREK-1 as a dimer via a disulfide bridge has been reported to underlie the trafficking of this heterodimer channel to the membrane and contribution to astrocyte passive conductance (Hwang et al., 2014). To address this question further, TWIK-1/TREK-1 double gene knockout mice were created to determine whether absence of both K2P isoforms may affect the passive conductance.
We show that the basic electrophysiological properties of mature hippocampal astrocytes were not altered in either TREK-1 single or TWIK-1/TREK-1 double gene knockout mice, which calls attention to explore alternative K+ channels responsible for the still mysterious passive conductance in astrocytes.
Materials and Methods
Animals
All the experimental procedures were performed in accordance with a protocol approved by the Animal Care and Use Committees of The Ohio State University. TREK-1 gene (Kcnk2) knockout (TREK-1−/−) mice were generated on a mixed background of C57BL6J and SV129 strains. Kcnk2 spans 136 Kbp of the mouse genome and has eight exons. As previously reported, for the mutant Kcnk2, a 4 Kbp genomic DNA sequence, from the position 14 bp of the second exon to the position 23 bp of the third intron, was replaced by a β-galactosidase/neomycin (LacZ/Neo) selection cassette (Namiranian et al., 2010). TWIK-1 gene (Kcnk1) knockout (TWIK-1−/−) mice were created with C57BL/6J genetic background, where the exon 2 of TWIK-1 gene (Kcnk1) was genetically deleted (Nie et al., 2005). TWIK-1/TREK-1 double knockout (TWIK-1−/−/TREK-1−/−) mice were generated by crossing TWIK-1−/− and TREK-1−/− mice.
Both male and female mice at postnatal day (P) of 21–28 were used, and the animals were divided into (1) wild type (WT); (2) TREK-1−/−; (3) TWIK-1−/−; and (4) TWIK-1−/−/TREK-1−/− groups in this study.
PCR Genotyping
To confirm the gene deletion, PCR genotyping was performed using total DNA isolated from tails of mice. Two pairs of primers were designed to recognize exon 3 of Kcnk2 in WT mice and the LacZ/Neo selection cassette in TREK-1−/− mice. TREK-1 WT primers: forward, GCTGGGTGAAGTTCTTCAGC; reverse, CATTACCTGGATGAGTTCGTC. TREK-1−/− primers: forward, GCAGCGCATCGCCTTCTATC; reverse, AGGAGATGAAGACCTCTGCAAAGG. To detect the WT TWIK-1 gene (Kcnk1) and the truncated form of Kcnk1 in TWIK-1−/− mice, two pairs of primers were set to recognize exon 2 and to surround the exon 2, respectively. TWIK-1 WT primers: forward, TCCAGGGGAAGGCTACAA; reverse, GGAGACGGCAGGCAGTAA. TWIK-1−/− primers: forward, CCCTTACTCTTATCAATCGG; reverse, GTTTGCTTGTGAATGTGC.
RNA Extraction from Freshly Dissociated Single Astrocytes
Mice were anesthetized by intraperitoneal injection of 8% chloral hydrate in 0.9% NaCl saline (0.05 ml/100 g body weight). The brain was removed from the skull and placed in oxygenated (95% O2/5% CO2) Ca2+-free artificial cerebral spinal fluid (Ca2+-free aCSF; in mM):125 NaCl, 25 NaHCO3, 1.25 NaH2PO4, 3.5 KCl, 1 MgCl2, 1 Na-pyruvate, and 10 glucose (Zhou and Kimelberg, 2001; Xie et al., 2007). Coronal slices of 400 μm thickness were sectioned with a Vibratome (Pelco, 1500) and transferred to 34°C Ca2+-free aCSF supplemented with 1 μM SR101 (Invitrogen, New York, NY, USA; Nimmerjahn et al., 2004). After incubation with SR101 for 30 min, the brain slices were transferred to Ca2+-free aCSF for another 30 min at room temperature (20–22°C). Hippocampal regions were dissected out from the brain slices and digested for 25 min with 24 μ/ml papain in aCSF containing (in mM): 125 NaCl, 25 NaHCO3, 1.25 NaH2PO4, 3.5 KCl, 2 CaCl2, 1 MgCl2, and 10 glucose (osmolarity, 295 ± 5 mOsm; pH 7.3–7.4), supplemented with 0.8 mg/ml L-cysteine. After digestion, the hippocampal slices were returned to the Ca2+-free aCSF for recovery for at least 1 h. The loosened hippocampal slices were gently triturated into a cell suspension. To harvest single dissociated astrocytes, the cells were identified by their positive SR101 fluorescent staining and well-preserved soma and spatial domain (Bushong et al., 2002; Du et al., 2015). In each run, thirty astrocytes were collected for each genotype group by a glass electrode (diameter ~10 μm) attached to a micromanipulator. RNA extraction was done using RNeasy Mini Kit (Qiagen, Valencia, CA, USA) right after cell harvesting.
Real-Time Quantitative Reverse Transcription PCR (QRT-PCR) Analysis
Immediately after RNA extraction, RNA was converted into cDNA using Applied Biosystem’s High Capacity cDNA Reverse Transcription Kit (Grand Islands, NY, USA). The PCR primer pairs for identification of TWIK-1, TREK-1, TREK-2, Kir4.1, Kir5.1, KV6.3 and GAPDH gene are shown in Table 1. All primer pairs spanned at least two exons to avoid amplification of genomic DNA. Each primer pair was tested by conventional PCR before quantitative reverse transcription PCR (qRT-PCR) to ensure that the amplicon yielded an anticipated and distinct single band. SYBR® Select Master Mix (Invitrogen, New York, NY, USA) was used, and the qRT-PCR was run on a Step One Plus equipment (Life technologies, New York, NY, USA). Each assay was performed in triplicate samples from the same mouse. A minimum of three repeats were done for each genotype group. For each repetition, a negative control with no template was always present. Gapdh was used as the internal reference and routinely run in parallel with targeted genes. Data were obtained as Ct values (threshold cycle). The expression levels of target genes were expressed as 2−ΔCT, where ΔCT was referred to the Ct difference between gene of interest and Gapdh.
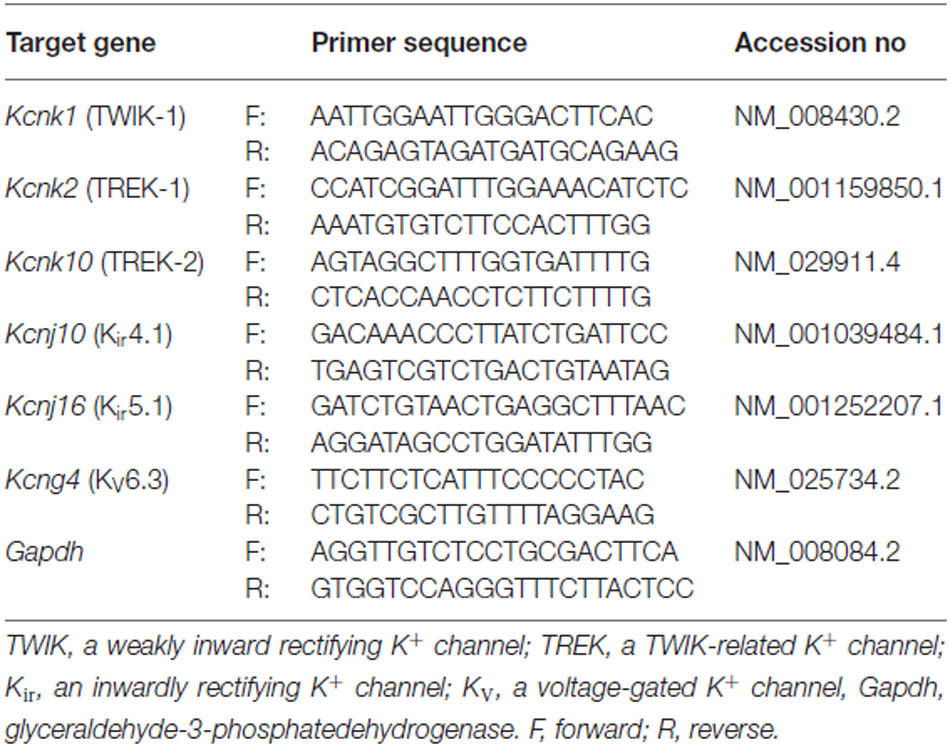
Table 1. Primers for qRT-PCR analysis.
Preparation of Acute Hippocampal Slices
Hippocampal slices were prepared as we described previously (Ma et al., 2012a). Briefly, brains were rapidly removed from skulls and placed into ice-cold oxygenated (95% O2/5% CO2) slice cutting aCSF with reduced Ca2+ and increased Mg2+ (in mM): 125 NaCl, 3.5 KCl, 25 NaHCO3, 1.25 NaH2PO4, 0.1 CaCl2, 3 MgCl2 and 10 Glucose. Coronal hippocampal slices at 250 μm thickness were cut at 4°C and transferred to the oxygenated standard aCSF, recovering at room temperature for at least 1 h before SR101 incubation and electrophysiological recording.
Electrophysiology
For in situ recording of astrocytes, individual hippocampal slices were transferred to the recording chamber mounted on an Olympus BX51WI microscope, with constant perfusion of oxygenated aCSF (osmolarity, 295 ± 5 mOsm; pH 7.3–7.4; 2.0 ml/min). Astrocytes located in the CA1 region were visualized using an infrared differential interference contrast (IR-DIC) video camera with the 40× water-immersion objective. The morphologically identified astrocytes could be further confirmed by their positive SR101 fluorescent staining. Whole-cell patch clamp recordings were performed using a MultiClamp 700A (or MultiClamp 700B) amplifier and pClamp 9.2 software (Molecular Devices, Sunnyvale, CA, USA). Borosilicate glass pipettes (Warner Instrument) were pulled from a Micropipette Puller (Model P-87, Sutter Instrument). The recording electrodes had a resistance of 3–7 MΩ when filled with the electrode solution containing (in mM): 140 K-gluconate, 13.4 Na-gluconate, 0.5 CaCl2, 1.0 MgCl2, 5 EGTA, 10 HEPES, 3 Mg-ATP, and 0.3 Na2-GTP (pH 7.23~7.25, 280 ± 5 mOsm). The electrode solution contained 3 mM low Cl− and 14 mM high Na+, similar to physiological conditions (Kelly et al., 2009; Ma et al., 2012a).
The liquid junction potential was compensated for prior to forming the cell-attached mode in all recordings. After establishing whole-cell recording, the resting VM was either read in “I = 0” mode or recorded under current clamp mode. In current clamp recording, the input resistance (Rin) was measured by the built-in “Resistance test” function (63 pA/600 ms pulse) before and after recording. We have recently shown that mature hippocampal astrocytes exhibit a very low RM of 6.4 MΩ, and a routinely achievable access resistance (Ra) reading from “Membrane test” function is ~15 MΩ for astrocytes older than P21 (Zhou et al., 2009; Ma et al., 2014). Recordings with initial Rin greater than 20 MΩ or Rin varying more than 10% during recording were discarded.
In pharmacological experiments, 100 μM BaCl2 and 400 μM quinine were dissolved directly in aCSF for bath application to block astrocytic Kir4.1 and K2Ps, respectively. The hypoosmotic bath solution, 205 ± 5 mOsm, was achieved by reducing 50 mM NaCl in normal aCSF. A temperature controller (Warner Instruments, TC-324C) was used to elevate the solution temperature in recording chamber to 32 ± 1°C. Unless noted specifically, most of the recordings were conducted at room temperature of ~22°C.
Western Blot Analysis
Fractionation of Hippocampal Proteins from Different Subcellular Regions
Mice were anesthetized as described above, and then the hippocampus was quickly removed from anesthetized mice and separated into hydrophilic (cytoplasmic) and hydrophobic (membrane) proteins by Mem-PER Eukaryotic Membrane Protein Extraction Kit (Thermo Fisher Scientific, Rockford, IL, USA). To remove chemicals that may interfere with the following BCA protein assay and SDS-polyacrylamide gel electrophoresis (SDS-PAGE) analysis, all the fractioned protein samples were cleaned by SDS-PAGE Sample Prep Kit (Thermo Fisher Scientific, Rockford, IL, USA).
SDS-PAGE and Immunoblotting
Protein concentration was determined with the BCA protein assay kit (Thermo Fisher Scientific, Rockford, IL, USA). Samples were mixed with a 5× reducing loading buffer containing 100 mM DTT (Thermo Fisher Scientific, Rockford, IL, USA) and heated at 95°C for 5 min. Equal amounts of protein (25 μg/lane) were separated on a 4–12% tris-glycine gel (Bio-Rad, Hercules, CA, USA) and subsequently transferred to a nitrocellulose membrane (Micron Separations Inc., Westborough, MA). The membranes were blocked with 5% non-fat milk/TBST (Tris-buffered saline with 0.05% Tween 20) for 1 h at room temperature. The membranes were incubated with anti-TREK-1 antibodies (1:2000, Alomone Labs, Jerusalem, Israel) at 4°C overnight. After washing, the membranes were incubated with secondary antibody (Jackson ImmunoResearch Laboratories, Maine, USA). Immunoreactivity was detected with an enhanced chemiluminescent detection (Thermo Fisher Scientific, Rockford, IL, USA). Blots were scanned and quantified by Quantity One software (Bio-Rad, Hercules, CA, USA). After detecting TREK-1 immunoreactivity, the original membranes were stripped with stripping buffer (0.4 M Glycine, 0.2% SDS and 2% Tween 20, pH2.0) and re-probed with the following primary antibodies sequentially to determine the quality of protein fractionation: anti-Na+/K+ ATPase alpha 2(+) polypeptide (ATP1α2; 1:1000, Abgent, San Diego, CA, USA) and anti-glial fibrillary acidic protein (GFAP; 1:500, DAKO, Carpinteria, CA, USA). This antibody incubation and blot quantification followed the same procedure described above.
Data Analysis
The patch clamp recording data were analyzed by Clampfit 9.2 (Molecular Devices, Sunnyvale, CA, USA) and Origin 8.0 (OriginLab, Northhampton, MA, USA). All the results are given as means ± SEM. Statistical analysis was performed using Student’s t-test of two independent samples. For multiple comparison tests, WT was used as control group; TWIK-1−/−, TREK-1−/−, and TWIK-1−/−/TREK-1−/− were compared to against it. Thus, One-Way ANOVA followed by Dunnett’s test were combined for these tests. Significance level was set at P < 0.05.
Results
TREK-1 Single and TWIK-1/TREK-1 Double Gene Knockout Mice
In TREK-1−/− mice, four transmembrane domains, including two pore-forming regions, were genetically targeted for deletion as previously reported (Figure 1A, upper; Namiranian et al., 2010). The targeted deletion in TWIK-1−/− mice included the second and third transmembrane domains (Figure 1A, lower; Nie et al., 2005; Wang et al., 2013). The targeted gene deletions were confirmed by PCR genotyping analysis. Specifically, to detect TREK-1−/−, one pair of primers was set to recognize exon 3 of WT Kcnk2, whereas another pair was set to recognize the LacZ-Neo selection cassette in the TREK-1 mutant. These resulted in the anticipated 453 bp for the WT Kcnk2 and 200 bp for the Kcnk2 mutant, respectively (Figure 1B, upper; Namiranian et al., 2010). To detect the truncated Kcnk1 in TWIK-1−/−, one pair of primers was set to recognize exon 2 in WT, and another pair was set to surround exon 2. These resulted in the anticipated bands of 588 bp and 355 bp in amplicons (Figure 1B, lower; Wang et al., 2013). TWIK-1−/−/TREK-1−/− showed an anticipated band at ~200 bp for Kcnk2 mutant as well as a 355 bp band for the truncated Kcnk1 (Figure 1B).
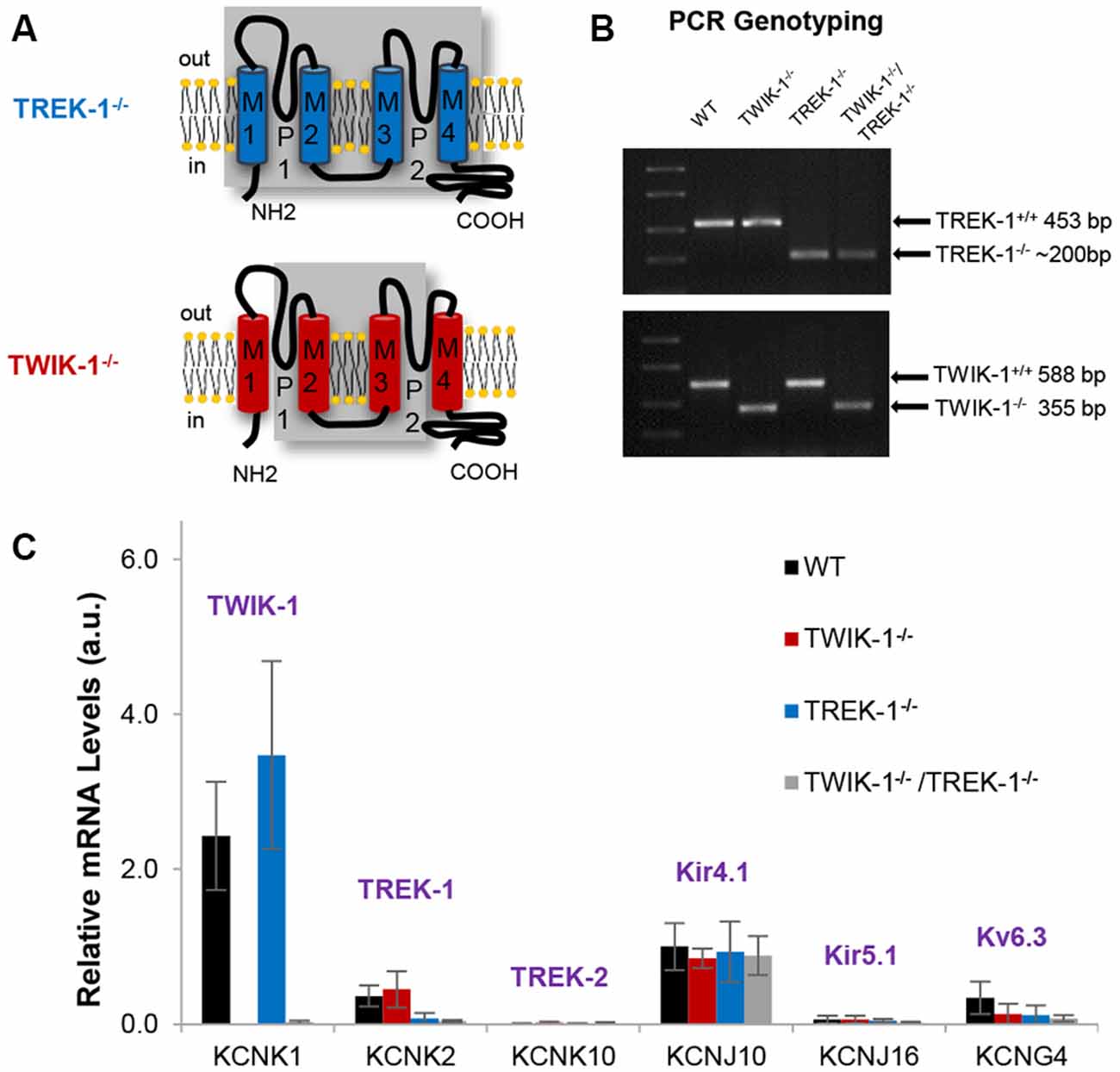
Figure 1. Expression of astrocyte K+ channels in TREK-1, TWIK-1 single, and TWIK-1/TREK-1 double gene knockout mice. (A) Schematic illustrations of genetic deletion of four transmembrane domains, including two pore-forming regions, in TREK-1 knockout (TREK-1−/−, upper), and TWIK-1 knockout (TWIK-1−/−, lower) mice. (B) PCR genotyping confirmation of successful genetic knockout of the targeted genes in TWIK-1−/−, TREK-1−/−, and TWIK-1/TREK-1 double knockout (TWIK-1−/−/TREK-1−/−) mice. (C) The relative quantity of mRNA of K+ channels in freshly dissociated mature astrocytes. The results were examined from wild type (WT), TWIK-1−/−, TREK-1−/−, and TWIK-1−/−/TREK-1−/− astrocytes using qRT-PCR.
As reported previously, the growth, fertility and gross anatomy of TREK-1−/− and TWIK-1−/− do not differ from their age- and gender-matched WT mice, and their offspring from heterozygote mating followed a Mendelian ratio (Nie et al., 2005; Namiranian et al., 2010; Wang et al., 2013). TWIK-1−/−/TREK-1−/− mice were generated by crossing two mouse lines. The resulted mice essentially exhibit similar characteristics as the WT mice, and their offspring from heterozygote mating followed a Mendelian ratio.
TREK-1 and TWIK-1 Gene Knockout does not Alter the Expression of Other Astrocyte K+ Channels at Trascript Levels
To clarify whether single TREK-1, TWIK-1, or double TWIK-1/TREK-1 gene knockout would interfere with the expression of the other astrocyte K+ channels or produce compensation for the loss of these two K+ channels, we compared the expression levels of mRNA for a group of K+ channels that were selected based on the following considerations. First, we included the three major astrocyte K+ channels known to express relative mRNA levels of TWIK-1 > Kir4.1 > TREK-1 in isolated cortical astrocytes (Cahoy et al., 2008) and freshly isolated hippocampal astrocytes (Seifert et al., 2009). Second, for other K2P members, we selected TREK-2 in the TREK subfamily, as it distributes extensively in the central nervous system (CNS) and its mRNA expression overlaps with TREK-1 expression in many brain regions (Medhurst et al., 2001; Talley et al., 2001). As TWIK-2 and TWIK-3 in the TWIK subfamily were undetectable in our previously study in both WT and TWIK-1−/− mice (Wang et al., 2013), these two K2P isoforms were not included in this analysis. Third, we included Kir5.1 and KV6.3, which showed relatively high astrocytic expression among other K+ channels in Kir and KV subfamilies, respectively (Hibino et al., 2004; Cahoy et al., 2008).
In isolated WT hippocampal astrocytes, qRT-PCR analysis revealed an order of mRNA expression levels of TWIK-1 > Kir4.1 > TREK-1 > KV6.3. TREK-2 and Kir5.1 were barely detectable. In knockout mice, qRT-PCR results confirmed the anticipated deletion of TREK-1 and TWIK-1 either alone or together (Figure 1C). Furthermore, in all tested genotypes, the expression levels and the relative abundance of other astrocyte K+ channels were not altered compared to WT. Thus, targeted TREK-1 and TWIK-1 gene deletion, either alone or together, did not result in an altered profile of K+ channel expression, nor compensatory up-regulation in any of the candidate K+ channels (Figure 1C). Nevertheless, these results could not fully rule out a potential compensation of passive conductance by other unknown K+ channels in TREK-1 and/or TWIK-1 gene knockout mice.
In summary, the anticipated single and double TWIK-1 and TREK-1 gene knockouts were successful in the mouse lines used in this study, and these gene deletions did not alter the expression pattern of other known astrocytic K+ channels that have the potential to functionally compensate for changes in astrocyte passive conductance.
TREK-1 Gene Knockout does not Alter the Basic Electrophysiological Properties of Mature Hippocampal Astrocytes
TREK-1 follows the GHK prediction for a leak type K+ channel by showing an outwardly rectifying current profile in an asymmetrical physiological K+ gradient (Fink et al., 1996; Enyedi and Czirják, 2010). As a leak type K+ channel, the contribution of TREK-1 to the resting VM was unequivocally demonstrated in a human adrenal cell line (Brenner and O’Shaughnessy, 2008).
To determine the contribution of TREK-1 to astrocyte membrane conductance and resting VM, whole-cell patch clamp recordings were made from mature hippocampal CA1 astrocytes from WT and TREK-1−/− mice (Figure 2A). The whole-cell resting VM was comparable between the two genotypes: −75.9 ± 0.17 mV (n = 103) in WT vs. −76.0 ± 0.15 mV (n = 152) in TREK-1−/− (P > 0.05, Figure 2B), so was the Rin: 13.6 ± 0.45 MΩ (n = 47) in WT vs. 13.2 ± 0.40 MΩ (n = 64) in TREK-1−/− (P > 0.05, Figure 2C). We have previously shown that passive membrane conductance exhibits a weak inward rectification with a rectification index (RI) around 0.9 (Wang et al., 2013). Should the outwardly rectifying TREK-1 contribute substantially to passive conductance, TREK-1−/− would increase the inward rectification of whole-cell current with a further reduced RI value. However, the RI was essentially unchanged in TREK-1−/− astrocytes: 0.93 ± 0.02 (n = 14) in WT vs. 0.93 ± 0.01 (n = 19) in TREK-1−/− (P > 0.05, Figures 2D–F). Altogether, basic electrophysiological features remain intact in TREK-1−/− astrocytes.
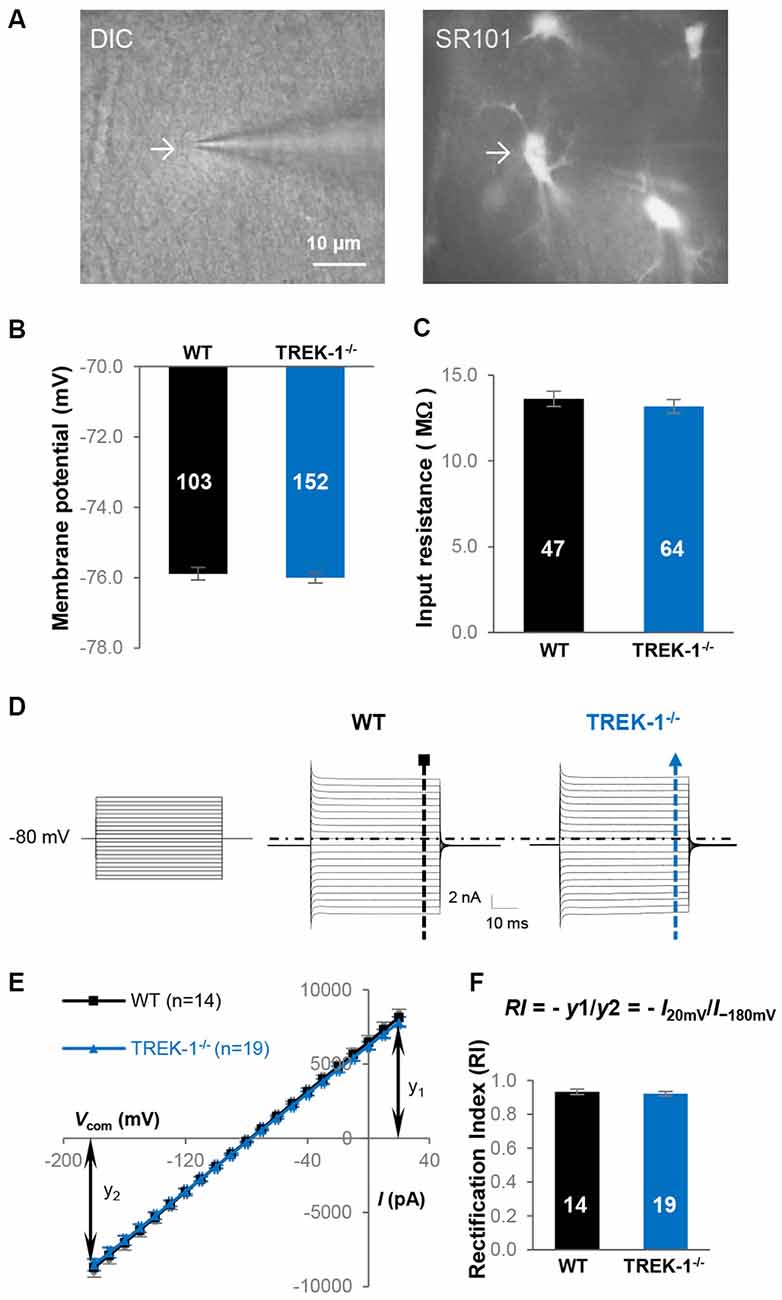
Figure 2. TREK-1 gene deletion does not alter the electrophysiological properties of astrocytes. (A) Identification of astrocyte in hippocampal slices based on cellular morphology and positive SR101 staining in CA1 region. (B,C) Membrane potential (VM), and input resistance (Rin) in WT and TREK-1−/− astrocytes. (D) Representative astrocyte whole-cell passive conductance from a WT and a TREK-1−/− astrocyte are shown separately as indicated. The voltage commands used for whole-cell current induction are displayed on the left panel. (E) I-V plots show the averaged current amplitudes from WT and TREK-1−/− astrocytes. The equation for rectification index (RI) is also illustrated and the resulted RI values are summarized in (F).
In hippocampal astrocytes, Kir4.1 contributes to 48% of passive membrane conductance (Ma et al., 2014), therefore inhibition of Kir4.1 should improve voltage clamping quality for a higher resolution examination of TREK-1 conductance using RI analysis. Accordingly, Kir4.1 was inhibited by 100 μM BaCl2 (IC50 ~ 3.5 μM; Ransom and Sontheimer, 1995; Seifert et al., 2009), and RI was compared again between WT and TREK-1−/− astrocytes.
In current clamp recording, 100 μM BaCl2-induced VM depolarization did not differ significantly between WT, 3.5 ± 0.23 mV (n = 10), and TREK-1−/− astrocytes, 3.8 ± 0.29 mV (n = 24; P > 0.05, Figure 3B). This result was consistent with an unchanged Kir4.1 expression at mRNA levels in TREK-1−/− mice (Figure 1C). In voltage clamp recording, 100 μM BaCl2 suppressed the whole-cell passive conductance comparably in WT and TREK-1−/− astrocytes, and the resulting Ba2+-sensitive currents were also comparable (Figures 3C,D). However, inhibition of Kir4.1 shifted RI toward a linear level, i.e., approach to the value 1, in both WT and TREK-1−/− astrocytes: 0.98 ± 0.03 (n = 5) in WT vs. 1.00 ± 0.03 (n = 5) in TREK-1−/− astrocytes (P > 0.05, Figure 3E), which further corroborates the view that TREK-1 shows no detectable contribution to passive conductance.
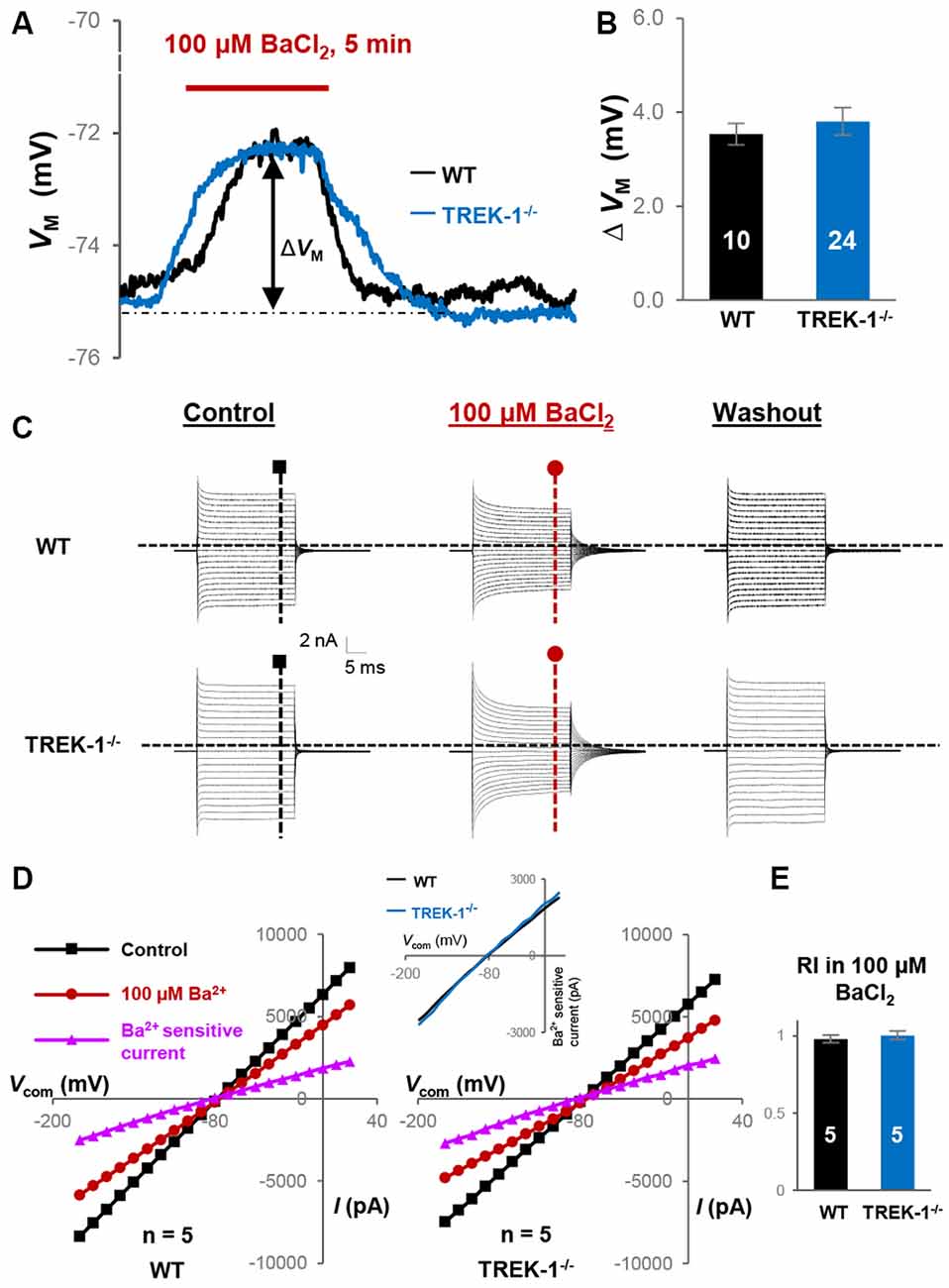
Figure 3. Kir4.1 inhibition does not reveal the functional contribution of TREK-1 in TREK-1−/− astrocytes. (A) Representative VM response to Kir4.1 inhibitor 100 μM BaCl2 from a WT and a TREK-1−/− astrocyte, as indicated in situ. ΔVM indicates the peak VM depolarization during a 5 min BaCl2 bath application. (B) Summary of 100 μM BaCl2-induced VM depolarization, where the VM depolarization was comparable between WT and TREK-1−/− astrocytes. (C) Representative whole-cell current recorded first in control, then 5 min in 100 μM BaCl2, and washout. (D) I-V plots derived from recordings in (C). The Ba2+-sensitive currents in I-V plots were obtained from sweep subtraction. The Ba2+-sensitive currents were shown in expanded y-axis in the inset that showed a moderate inward rectification in both WT, RI = 0.91, and TREK-1 KO, RI = 0.90, respectively. (E) Summary of RI values from WT and TREK-1−/− astrocytes obtained from recordings in the presence of 100 μM BaCl2 for Kir4.1 inhibition; the RI values were comparable between the two groups.
Interestingly, Ba2+-induced linear shift in RI differed significantly when compared with control groups both in WT and TREK-1−/− astrocytes (P < 0.05, data not shown), indicating elimination of a significant portion of Kir4.1-mediated inward currents from the overall passive conductance.
The activity of TREK-1 channel can be positively modulated by hypoosmotic extracellular solution and warmer temperature in the heterologous expression system (Maingret et al., 1999, 2000). To explore whether the same conditions could facilitate identification of functional TREK-1 in hippocampal astrocytes, we first applied hypoosmotic aCSF, −90 mOsm, to astrocyte recordings. However, the RI remained comparable between WT (0.95 ± 0.01, n = 3) and TREK-1−/− astrocytes (0.95 ± 0.02, n = 4; P > 0.05). Raising the temperature of the bath aCSF to 32 ± 1°C did not alter the RI: 0.95 ± 0.01 (n = 6) in WT vs. 0.95 ± 0.02 (n = 3) in TREK-1−/− astrocytes (P > 0.05). Both in WT and in TREK-1−/− astrocytes, the RI values in high temperature were comparable to the RI in room temperature. Additionally, arachidonic acid, halothane and angiotensin II have been shown as TREK-1 modulators (Fink et al., 1998; Patel et al., 1999; Enyeart et al., 2005). However, none of these TREK-1 modulators induced noticeable change in astrocyte RI, VM, and the amplitude of passive conductance in WT astrocytes (data not shown).
Together, these results show that functional Kir4.1 expression is largely intact in TREK-1−/− astrocytes, and there is no detectable contribution of TREK-1 to astrocyte passive conductance.
TREK-1 Channels are Predominantly Located in Cytoplasm in Hippocampus
Because TREK-1 gene knockout does not affect the basic properties of astrocytes, we explored further the mechanism underlying this observation. We have previously shown that TWIK-1 proteins are mainly located in the intracellular compartments of hippocampal astrocytes (Wang et al., 2013), whereas the subcellular distribution of TREK-1 in hippocampal astrocytes is yet unknown. Differing from TWIK-1, TREK-1 is widely expressed in neurons and astrocytes in the CNS, and the subcellular distribution of this channel varies substantially depending on different species, tissues and cell types (Hervieu et al., 2001; Medhurst et al., 2001; Talley et al., 2001). To address whether lack of functional TREK-1 contribution in astrocytes is also attributable to retention of channels in the intracellular compartments, cytoplasmic and membrane proteins were extracted separately from hippocampus in fractionation western blot analysis as we reported previously (Wang et al., 2013). In this analysis, the antibodies against ATP1α2 and GFAP were used as astrocyte-specific membrane and cytoplasmic markers (Eng et al., 2000; Dinuzzo et al., 2012; Wang et al., 2013). Similar to TWIK-1 proteins, TREK-1 proteins showed a predominant location in the cytoplasmic fraction (Figure 4A). Specifically, 81.4 ± 3.3% of the total TREK-1 proteins were located in the cytoplasmic fraction in contrast with 18.6 ± 3.3% of channel proteins in the membrane fraction (n = 6, P < 0.01, Figure 4B). As TREK-1 is expressed both in astrocytes and neurons, it remained unknown whether TREK-1 expression in the membrane fraction is astrocytic, neuronal, or shared by both. Nevertheless, the results suggest that retention of TREK-1 in the intracellular compartments is likely accountable for the limited functional contribution of TREK-1 to astrocyte membrane conductance and VM.
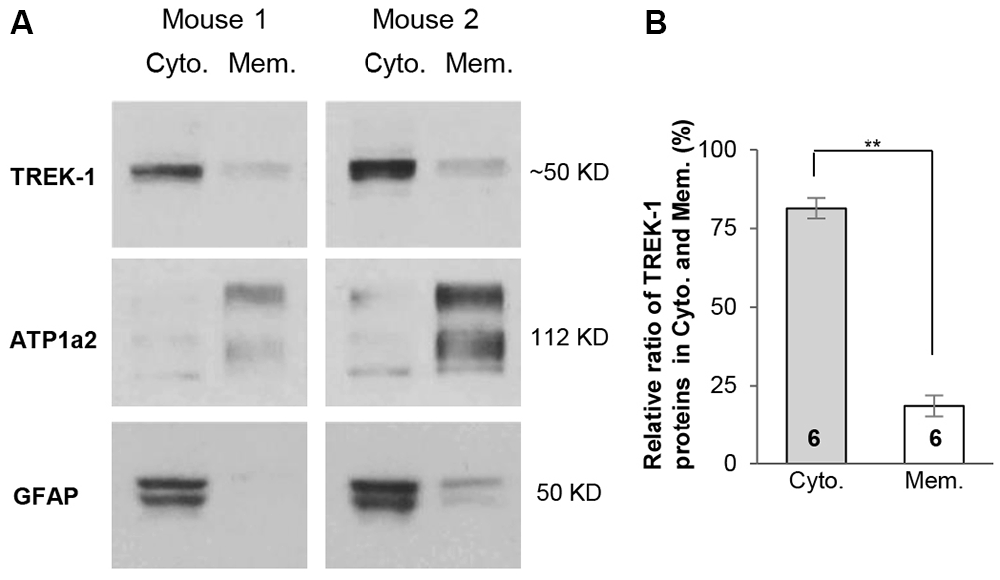
Figure 4. TREK-1 channels are predominantly located in cytoplasm. (A) Fractionation western blot results revealed the subcellular distribution of TREK-1 channels in cytoplasmic fraction vs. membrane fraction in two independent tests of mice hippocampal samples. Anti-glial fibrillary acidic protein (GFAP) (50 kDa) and ATP1α2 (112 kDa) were markers for cytoplasmic and membrane fractions, respectively. The blots shown in (A) were first incubated with anti-TREK-1 antibody and then re-probed with the rest of other primary antibodies sequentially after the original membranes were stripped with stripping buffer (see “Materials and Methods” Section). (B) Bar graph summary showing the relative ratio of TREK-1 proteins located in cytoplasmic vs. membrane fractions. Data are shown as mean ± SEM. Numbers indicate the times of observations. **p < 0.01.
TWIK-1/TREK-1 Double Gene Knockout does not Alter Astrocyte Passive Conductance
We have previously shown that membrane TWIK-1 functions as a non-selective cation channel with no detectable contribution to passive conductance (Wang et al., 2013). Here, we show no detectable functional contribution of TREK-1 to passive conductance. However, formation of a TWIK-1/TREK-1 heterodimer via a disulfide bridge has been shown as a prerequisite for trafficking of the TWIK-1/TREK-1 heterodimer to the membrane and contribution to astrocyte passive conductance (Hwang et al., 2014). In the very same study, in vivo delivery of shRNA for knockdown of either TWIK-1 or TREK-1 was sufficient enough to eliminate passive conductance (Hwang et al., 2014). To answer whether co-expression of TWIK-1 and TREK-1 is required for astrocytes to show passive conductance, we examined the impact of TWIK-1−/−/TREK-1−/− on the basic electrophysiological properties of astrocytes.
We show that the VM was comparable between the two experimental groups: −75.9 ± 0.17 mV (n = 103) in WT vs. −75.7 ± 0.18 mV (n = 94) in TWIK-1−/−/TREK-1−/− astrocytes (P > 0.05; Figure 5A), indicating a TWIK-1/TREK-1 heterodimer is not required for the highly negative VM that is critical for the homeostatic function of astrocytes (Figure 5A). Second, the Rin was similar between WT, 13.6 ± 0.45 MΩ (n = 47), and TWIK-1−/−/TREK-1−/−, 14.1 ± 0.80 MΩ (n = 27) (P > 0.05; Figure 5B), indicating no change in the amount of background leak channels in the absence of two highly expressed leak type K+ channels, TWIK-1 and TREK-1, in astrocytes. Third, in the absence of TWIK-1 and TREK-1, the former a weakly inward rectifier and the latter a GHK outwardly rectifier channel (Fink et al., 1996; Lesage et al., 1996a), the RI was essentially unchanged in astrocytes: 0.94 ± 0.01 (n = 15) in TWIK-1−/−/TREK-1−/− compared to 0.93 ± 0.02 (n = 14) in WT (P > 0.05, Figures 5C–E).
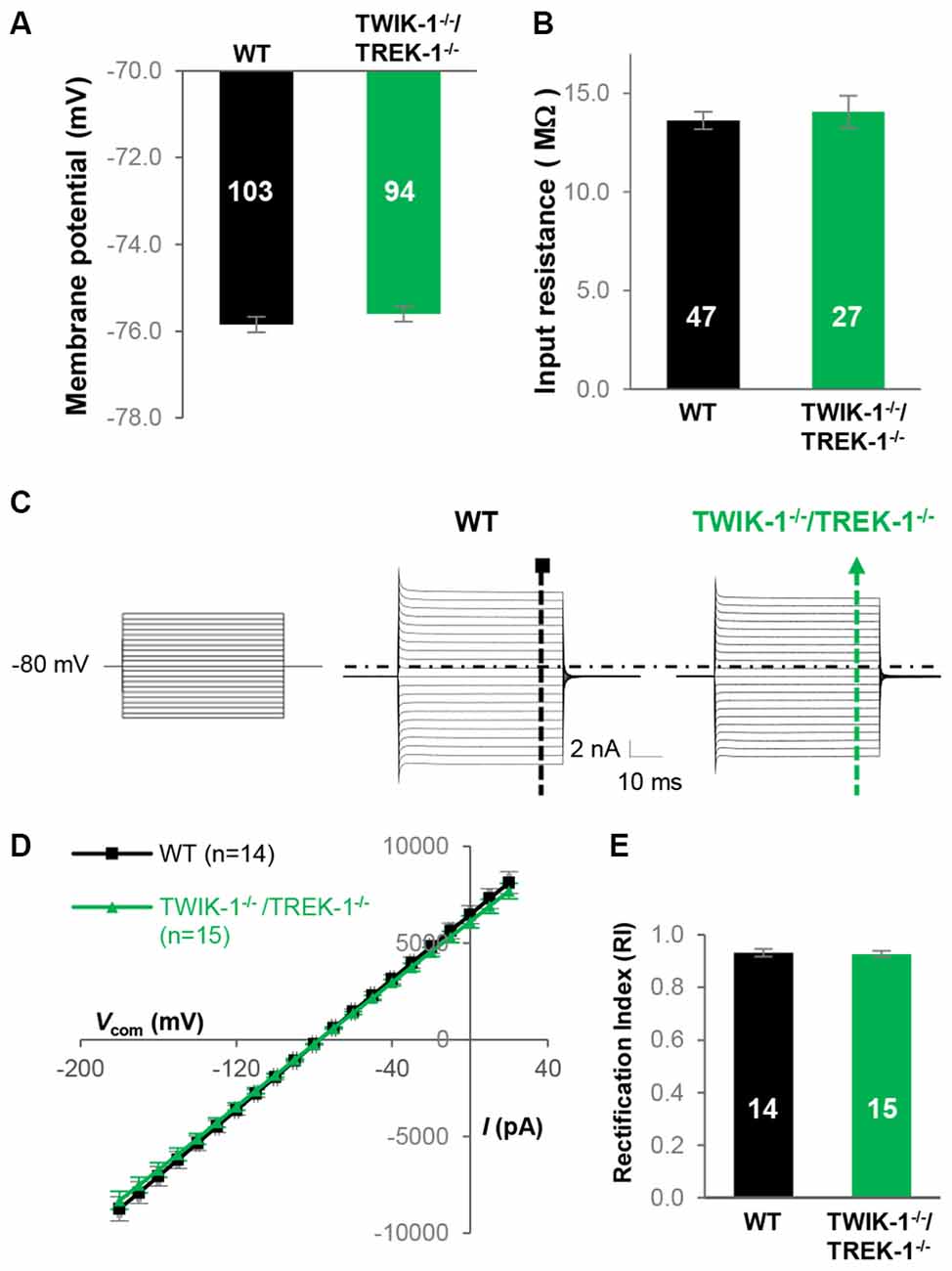
Figure 5. Genetic deletion of TWIK-1 and TREK-1 genes together does not alter the electrophysiological properties of astrocytes. (A,B) Bar graph summary of the VM and Rin from WT and TWIK-1−/−/TREK-1−/− astrocytes. (C) Representative whole-cell current profiles from WT and TWIK-1−/−/TREK-1−/− astrocytes, respectively. (D) Averaged I-V plots from these two genotypes, where the whole-cell current amplitudes in both inward and outward directions were comparable. (E) The RI values were also comparable between the two genotypes.
Consistent with our mRNA analysis, where Kir4.1 expression was unchanged in TWIK-1−/−/TREK-1−/− astrocytes (Figure 1C), 100 μM BaCl2-induced VM depolarization was comparable between WT, 3.5 ± 0.23 mV (n = 10), and TWIK-1−/−/TREK-1−/− astrocytes, 3.9 ± 0.23 mV (n = 32; P > 0.05, Figures 6A,B). In voltage clamp recording, 100 μM BaCl2 produced similar inhibition of passive conductance as compared with WT and TWIK-1−/−/TREK-1−/− astrocytes (Figures 6C,D). Additionally, in the presence of 100 μM BaCl2, the RI of Ba2+-insensitive currents was comparable between WT and TWIK-1−/−/TREK-1−/− astrocytes: 0.98 ± 0.03 (n = 5) in WT vs. 0.97 ± 0.01 (n = 5) in TWIK-1−/−/TREK-1−/− astrocytes (P > 0.05, Figure 6E). These results indicated that neither TWIK-1 nor TREK-1 has a significant contribution to the remaining whole-cell currents in the absence of Kir4.1 channels.
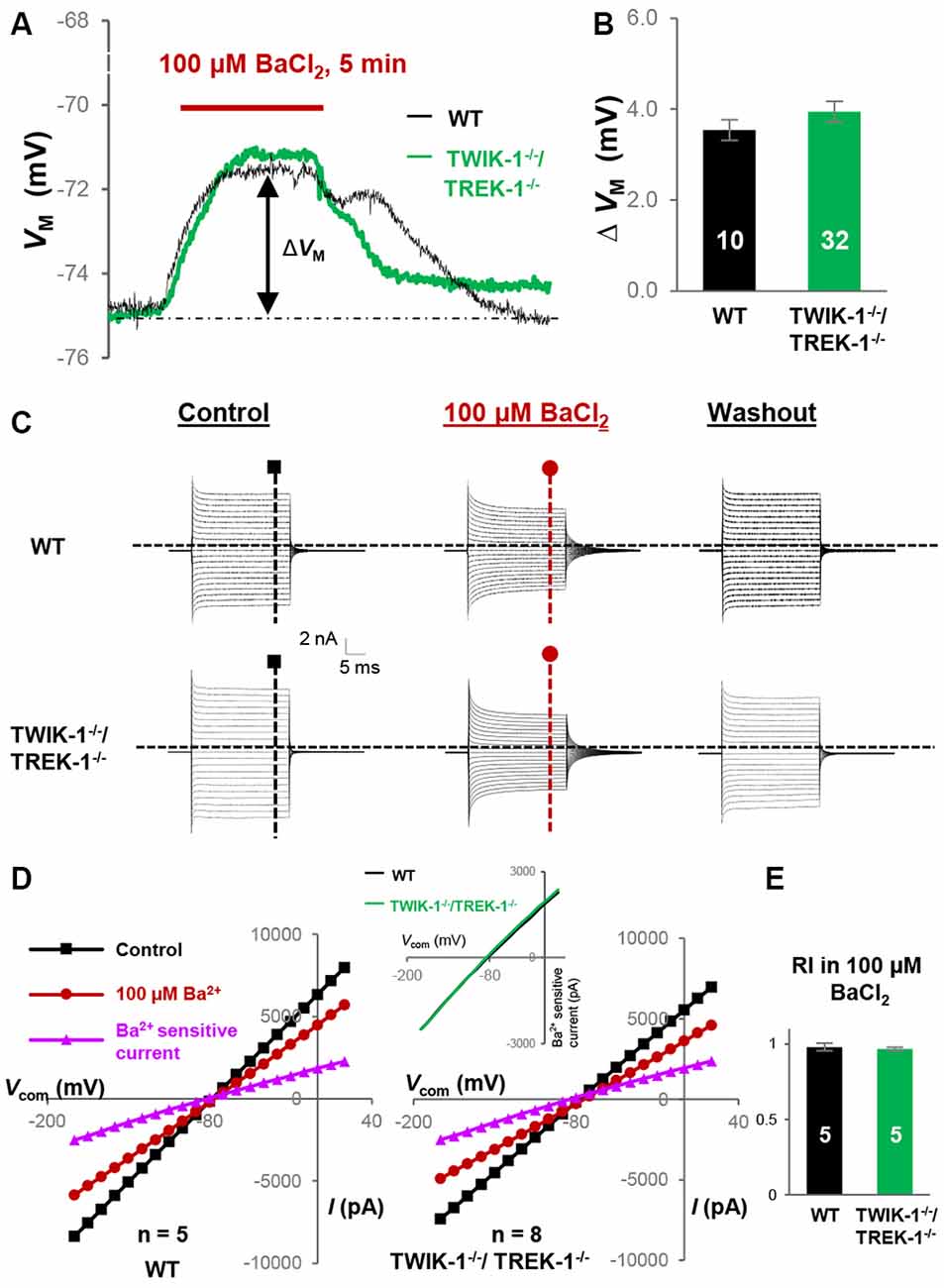
Figure 6. Kir4.1 inhibition does not reveal the functional contribution of TWIK-1/TREK-1 in double gene knockout mice. (A) Representative VM response to Kir4.1 inhibitor, 100 μM BaCl2, from a WT and a TWIK-1−/−/TREK-1−/− astrocyte as indicated in situ. (B) Summary of 100 μM BaCl2-induced VM depolarization, where the VM depolarization was comparable between WT and TWIK-1−/−/TREK-1−/− astrocytes. (C) Representative whole-cell current recorded first in control, then 5 min in 100 μM BaCl2, and washout. I–V relationships were shown in (D). (D) I–V plots derived from recordings in (C). The Ba2+-sensitive currents, in I-V plots were obtained from sweep subtraction. The Ba2+- sensitive currents were shown in expanded y-axis in the inset that showed a moderate inward rectification in both WT, RI = 0.91, and double gene knockout mice, RI = 0.90, respectively. (E) Summary of RI values from WT and TWIK-1−/−/TREK-1−/− astrocytes obtained from recordings in the presence of 100 μM BaCl2 for Kir4.1 inhibition; the RI values were comparable between the two groups.
Quinine is a potent TWIK-1 and TREK-1 inhibitor (Zhou et al., 2009). To examine and compare K2P conductance selectively in gene knockout astrocytes, the Kir4.1 was first inhibited by 100 μM BaCl2, and that was followed by addition of 400 μM quinine. Under these conditions, quinine further depolarized astrocyte VM (ΔVM1) by 7.4 ± 0.50 mV (n = 14) in WT, 7.4 ± 1.30 mV (n = 7) in TREK-1−/−, 7.2 ± 0.44 mV (n = 16) in TWIK-1−/− and 7.4 ± 0.68 mV (n = 11) in TWIK-1−/−/TREK-1−/− astrocytes (P > 0.05, Figures 7A,B). Total VM depolarization (ΔVM2) induced by 100 μM BaCl2 and 400 μM quinine was also comparable in the four genotypes: 10.6 ± 0.62 mV (n = 14) in WT, 10.6 ± 1. 20 mV (n = 7) in TREK-1−/−, 10.6 ± 0.62 mV (n = 16) in TWIK-1−/− and 11.0 ± 0.69 mV (n = 11) in TWIK-1−/−/TREK-1−/− astrocytes (P > 0.05, Figures 7A,B). In voltage clamp recording, 400 μM quinine produced similar inhibition of passive conductance as compared with WT, TREK-1−/−, TWIK-1−/− and TWIK-1−/−/TREK-1−/− astrocytes (Figure 7C). Specifically, in the presence of 400 μM quinine and 100 μM BaCl2, the RI of quinine and Ba2+-insensitive currents was comparable between WT, TREK-1−/−, TWIK-1−/− and TWIK-1−/−/TREK-1−/− astrocytes: 1.04 ± 0.01 (n = 5) in WT, 1.04 ± 0.06 (n = 5) in TREK-1−/−, 1.03 ± 0.02 (n = 6) in TWIK-1−/− and 1.04 ± 0.01 (n = 8) in TWIK-1−/−/TREK-1−/− astrocytes (P > 0.05, Figure 7D). These results strongly indicate that quinine sensitive K2P conductance is not altered in gene knockout astrocytes.
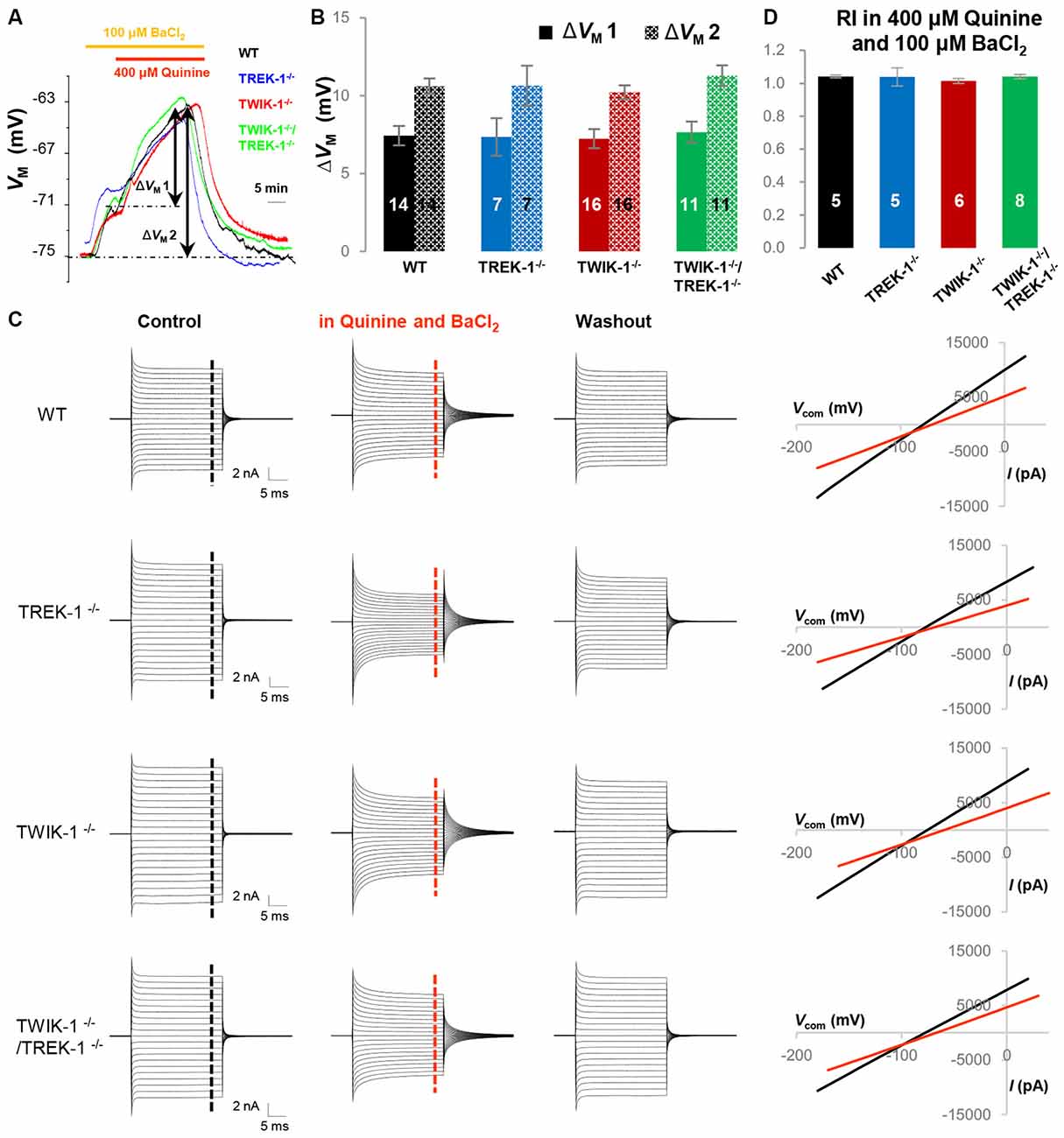
Figure 7. Quinine does not reveal the functional contribution of TWIK-1 and TREK-1 in single or double gene knockout mice. (A) Representative astrocyte VM recordings first in 100 μM BaCl2 bath application for 5 min, followed by addition of 400 μM quinine for 20 min, from a WT, TREK-1−/−, TWIK-1−/− and TWIK-1−/−/TREK-1−/− astrocyte as indicated in situ. (B) Summary of 400 μM quinine-induced VM depolarization (ΔVM 1) and the total VM depolarization induced by BaCl2 plus quinine from all four genotypes. (C) Representative whole-cell current recordings first in aCSF as control, then in 100 μM BaCl2 plus 400 μM quinine, and washout. Representative I–V relationships were shown in the right panel. (D) Summary of RI values from four genotypes obtained from astrocyte recordings in the presence of 400 μM quinine and 100 μM BaCl2 together in bath. The RI values were comparable among the four genotypes.
In summary, by using genetic gene knockout approach, our study found no evidence in support of the notion that the TWIK-1/TREK-1 heterodimer contributes to the passive conductance of mature hippocampal astrocytes.
Discussion
The present study was conceived to determine the contribution of TREK-1 to astrocyte passive conductance and resting VM, and to answer whether co-expression of TWIK-1/TREK-1, presumably as heterodimer channel, is required for their functional contribution to passive conductance. To address these questions, TREK-1 single- and TWIK-1/TREK-1 double-gene knockout mice were used in this study. We found that the passive conductance and the resting VM of mature hippocampal astrocytes were not noticeably altered in these gene knockout mice. Additionally, similar to TWIK-1(Wang et al., 2013), TREK-1 proteins were retained predominantly in the intracellular compartments, which likely accounts for the limited TREK-1 and TWIK-1 contribution, either alone or together, to the basic electrophysiological properties of mature hippocampal astrocytes.
Passive Conductance and Astrocyte Function
Increasing evidence shows that astrocytes act as the most versatile cell type in various CNS functions such as CNS homeostasis, synaptogenesis, synaptic transmission modulation, and neurovascular signaling (Nedergaard et al., 2003; Haydon and Carmignoto, 2006; Barres, 2008; Wang and Bordey, 2008; Zolessi, 2009; Kimelberg, 2010). Among them, the homeostatic support function was the earliest role assigned to astrocytes. The discovery of ohmic behavior passive conductance in amphibian optic nerve neuroglia was one of the cornerstones that led to this notion (Kuffler et al., 1966) and also promoted the “K+ spatial buffering hypothesis” (Orkand et al., 1966). Afterward, the importance of this conductance in astrocyte homeostatic functions, such as neurotransmitter uptake and pH regulation, has been increasingly appreciated (Wang and Bordey, 2008; Kimelberg, 2010).
At the mRNA expression level, TWIK-1, Kir4.1 and TREK-1 are the highly expressed K+ channels in isolated cortical astrocytes (Cahoy et al., 2008). While the inwardly rectifying Kir4.1 has been a well-recognized and studied astrocytic K+ channel (Olsen et al., 2015) at both protein and functional levels, Kir4.1 expression is relatively low in mouse hippocampal astrocytes compared to its expression in the spinal cord and brainstem (Ma et al., 2014; Nwaobi et al., 2014). This leaves the molecular identities of other K+ channels in hippocampal astrocytes to be further determined.
In the heterologous system, constitutive endocytosis restrains TWIK-1 to the recycling endosomes, and membrane TWIK-1 is able to conduct Na+ in the event of a fall in extracellular pH or K+ concentration (Feliciangeli et al., 2010; Chatelain et al., 2012; Ma et al., 2012c). In native cells, i.e., astrocytes, pancreatic β cells, and kidney tubular cells, TWIK-1 behaves as a non-selective cation channel (Millar et al., 2006; Chatelain et al., 2012; Wang et al., 2013). Because the channel is also highly permeable to (Ma et al., 2012b), a critical role of astrocytic TWIK-1 in homeostasis has been recently identified (Wang et al., 2015). These findings make it less likely that TWIK-1 could act as a classic background K+ channel in astrocytes. Thus, here we extended the search to another K2P channel, TREK-1.
Absence of Fucntional Trek-1 Contribution to Astrocyte Passive Conductance and Resting VM
TREK-1 is highly expressed in astrocytes and neurons in the mouse CNS (Meadows et al., 2000; Nicolas et al., 2004). Although TREK-1 shares a similar overall structural arrangement with TWIK-1, consisting of two pore-forming domains and four transmembrane segments (TMS), this structural similarity does not give rise to similar electrophysiological characteristics. Specifically, TWIK-1 in heterologous systems is characterized by a weakly inward rectification, whereas TREK-1 is an outward rectifier following GHK constant field rectification in physiological K+ solutions (Fink et al., 1996; Lesage et al., 1996a). TREK-1 is responsive to a variety of physiochemical stimuli and pharmacological agents, such as changes in temperature, osmolarity, mechanical force and anesthetic. The channel activity can also be modulated by activation of Gs and Gq coupled receptors (Patel et al., 1998; Maingret et al., 1999; Lesage and Lazdunski, 2000; Chemin et al., 2005; Murbartián et al., 2005). Thus, TREK-1 has the potential to enable astrocytes to engage in astrocyte-neuron crosstalk through a unique channel mechanism.
In the present study, TREK-1 gene knockout resulted in no detectable change in the basic electrophysiological properties in hippocampal astrocytes. First, the macroscopic whole-cell passive conductance and resting VM were not altered in mature astrocytes of TREK-1−/− mice. Second, the Rin, a functional readout of the quantity of resting open leak channels, did not differ between WT and TREK-1−/− astrocytes. Third, the RI remained comparable between WT and TREK-1−/− astrocytes when Kir4.1 was pharmacologically inhibited by 100 μM Ba2+. Fourth, when Kir4.1 was inhibited by 100 μM Ba2+, the putative quinine-sensitive K2P conductance and quinine induced VM depolarization remained comparable between WT and TREK-1−/− astrocytes.
Interestingly, qRT-PCR analysis from other K+ channels showing variable expression levels in WT astrocytes (Cahoy et al., 2008) resulted in no detectable change in those candidate channels in TREK-1−/− astrocytes; thus compensation by other K+ channels for the loss of TREK-1 is unlikely to occur in knockout astrocytes.
Subcellular Location of Trek-1 Protein in Hippocampus
The subcellular distribution of TREK-1 appears to be quite diverse. In the rat brain TREK-1 is predominantly expressed in the plasma membrane of projection neurons and interneurons (Hervieu et al., 2001), whereas the same channel in human HaCaT cell line is widespread in various subcellular locations, including the perinuclear, nuclear compartments and other intracellular organelles (Kang et al., 2007).
Interestingly, TREK-1e, a nonfunctional splice variant of the TREK-1 channel resulting from the skipping of exon 5 in gene transcription and subsequent loss of the second pore-forming domain between the transmembrane domains M3 and M4, was identified from human and rat brain and kidney (Rinne et al., 2014). In transfected COS-7 cells, the TREK-1e expression appears to be retained in the endoplasmic reticulum (ER) with a potential role of modulating vesicular traffic of full-length TREK-1 channels from the ER to the plasma membrane (Rinne et al., 2014).
In the present study, a preferential cytoplasmic expression was observed for TREK-1 channels expressed in the hippocampus (Figures 3A,B). The antibody used in our western blot analysis was generated to recognize the N-terminus of TREK-1, which includes the splice variant TREK-1e. Although more precise analysis with the TREK-1e specific antibody is needed to explore this issue further, should TREK-1e be expressed in astrocytes, one explanation for lack of functional TREK-1 contribution to passive conductance would be the retention of TREK-1 in the ER.
As noted, 18.6% of TREK-1 protein in the membrane fraction could be neuronal, astrocytic or both. Therefore, we are yet unable to determine the precise membrane presence of TREK-1 in astrocytes. Nevertheless, passive conductance and astrocyte VM was unchanged in the presence of several TREK-1 activators, such as arachidonic acid, halothane (data not shown); thus it is possible that the membrane expression of TREK-1 proteins may be predominantly neuronal in mice hippocampus, which offers another plausible explanation for lack of functional TREK-1 contribution.
TREK-1 trafficking and targeting to the plasma membrane is subject to regulation through several identified mechanisms. First, TREK-1e might modulate vesicular traffic of full-length TREK-1 channels from the ER to the plasma membrane in transfected COS-7 cells (Rinne et al., 2014). Second, TREK-1 has a distinct C terminus binding site with microtubule associated protein (Mtap2) in neurons, resulting in an enhanced membrane TREK-1 expression and functional currents (Sandoz et al., 2008). Third, βIV-spectrin, an actin-associated protein, is required for the membrane targeting and activity of TREK-1 in the cardiomyocytes (Hund et al., 2014). It remains to be determined whether any of these mechanisms exist in astrocytes.
Astrocyte Passive Conductance Remains Intact in the Absence of TWIK-1/TREK-1 K+ Channels
Most of the functional K2P channels are formed by covalent association of two K2P subunits, and each subunit contributes to two pore-forming regions of K+ selective filter (Lesage et al., 1996b; Doyle et al., 1998). It has been shown that formation of a TWIK-1/TREK-1 heterodimeric channel is the mechanism underlying astrocytic passive conductance (Hwang et al., 2014). In the present study, we have addressed this possibility through a different approach; by genetic elimination of either TREK-1 alone or of TREK-1 and TWIK-1 together. We show that neither passive conductance nor VM were altered in the double gene knockout mice. Meanwhile, the quantity and expression pattern of other known K+ channels were unchanged in TWIK-1−/−/TREK-1−/− astrocytes; thus compensatory K+ channel expression was unlikely a cause for our functional observations. Our results are inconsistent with the conclusion in a previous study, where virus shRNA delivery was used as gene knockdown approach (Hwang et al., 2014). However, because different experimental approaches are subject to different limitations, such as potential alternation in other gene expression in gene knockout mice and potential off-target effect in shRNA gene knockdown method (Toro Cabrera and Mueller, 2016), caution should be exercised in viewing the discrepancy resulting from different experimental approaches, and a more definite experimental approach would be highly valuable to resolve this inconsistency in the future.
Nevertheless, the function of the highly expressed K2Ps, TWIK-1 and TREK-1 channels in astrocytes remains to be precisely determined. The present study also calls attention to explore alternative channels contributing to the important but still mysterious passive conductance.
Author Contributions
YD, WW, BM and MZ conceived the project, YD, CMK, WW, QW, BM and CCA conducted the research. CH contributed TWIK-1 mice study, participated in experimental design and discussion. EEM, RMB created TREK-1 knockout mice and provided consultation to this project. YD and MZ wrote the manuscript. MZ supervised the project. All authors listed, have made substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work is sponsored by a grant from National Institute of Neurological Disorders and Stroke RO1NS062784 (MZ), a start-up fund from The Ohio State University College of Medicine (to MZ), and the National Natural Science Foundation of China (No. 81400973) (WW).
References
Barres, B. A. (2008). The mystery and magic of glia: a perspective on their roles in health and disease. Neuron 60, 430–440. doi: 10.1016/j.neuron.2008.10.013
Brenner, T., and O’Shaughnessy, K. M. (2008). Both TASK-3 and TREK-1 two-pore loop K channels are expressed in H295R cells and modulate their membrane potential and aldosterone secretion. Am. J. Physiol. Endocrinol. Metab. 295, E1480–1486. doi: 10.1152/ajpendo.90652.2008
Bushong, E. A., Martone, M. E., Jones, Y. Z., and Ellisman, M. H. (2002). Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J. Neurosci. 22, 183–192.
Butt, A. M., and Kalsi, A. (2006). Inwardly rectifying potassium channels (Kir) in central nervous system glia: a special role for Kir4.1 in glial functions. J. Cell. Mol. Med. 10, 33–44. doi: 10.1111/j.1582-4934.2006.tb00289.x
Cahoy, J. D., Emery, B., Kaushal, A., Foo, L. C., Zamanian, J. L., Christopherson, K. S., et al. (2008). A transcriptome database for astrocytes, neurons and oligodendrocytes: a new resource for understanding brain development and function. J. Neurosci. 28, 264–278. doi: 10.1523/JNEUROSCI.4178-07.2008
Chatelain, F. C., Bichet, D., Douguet, D., Feliciangeli, S., Bendahhou, S., Reichold, M., et al. (2012). TWIK1, a unique background channel with variable ion selectivity. Proc. Natl. Acad. Sci. U S A 109, 5499–5504. doi: 10.1073/pnas.1201132109
Chemin, J., Patel, A. J., Duprat, F., Lauritzen, I., Lazdunski, M., and Honore, E. (2005). A phospholipid sensor controls mechanogating of the K+ channel TREK-1. EMBO J. 24, 44–53. doi: 10.1038/sj.emboj.7600494
Dinuzzo, M., Mangia, S., Maraviglia, B., and Giove, F. (2012). The role of astrocytic glycogen in supporting the energetics of neuronal activity. Neurochem. Res. 37, 2432–2438. doi: 10.1007/s11064-012-0802-5
Djukic, B., Casper, K. B., Philpot, B. D., Chin, L. S., and McCarthy, K. D. (2007). Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake and enhanced short-term synaptic potentiation. J. Neurosci. 27, 11354–11365. doi: 10.1523/jneurosci.0723-07.2007
Doyle, D. A., Morais Cabral, J., Pfuetzner, R. A., Kuo, A., Gulbis, J. M., Cohen, S. L., et al. (1998). The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 280, 69–77. doi: 10.1126/science.280.5360.69
Du, Y., Ma, B., Kiyoshi, C. M., Alford, C. C., Wang, W., and Zhou, M. (2015). Freshly dissociated mature hippocampal astrocytes exhibit passive membrane conductance and low membrane resistance similarly to syncytial coupled astrocytes. J. Neurophysiol. 113, 3744–3750. doi: 10.1152/jn.00206.2015
Eng, L. F., Ghirnikar, R. S., and Lee, Y. L. (2000). Glial fibrillary acidic protein: GFAP-thirty-one years (1969–2000). Neurochem. Res. 25, 1439–1451. doi: 10.1023/A:1007677003387
Enyeart, J. J., Danthi, S. J., Liu, H., and Enyeart, J. A. (2005). Angiotensin II inhibits bTREK-1 K+ channels in adrenocortical cells by separate Ca2+- and ATP hydrolysis-dependent mechanisms. J. Biol. Chem. 280, 30814–30828. doi: 10.1074/jbc.m504283200
Enyedi, P., and Czirják, G. (2010). Molecular background of leak K+ currents: two-pore domain potassium channels. Physiol. Rev. 90, 559–605. doi: 10.1152/physrev.00029.2009
Feliciangeli, S., Tardy, M. P., Sandoz, G., Chatelain, F. C., Warth, R., Barhanin, J., et al. (2010). Potassium channel silencing by constitutive endocytosis and intracellular sequestration. J. Biol. Chem. 285, 4798–4805. doi: 10.1074/jbc.M109.078535
Fink, M., Duprat, F., Lesage, F., Reyes, R., Romey, G., Heurteaux, C., et al. (1996). Cloning, functional expression and brain localization of a novel unconventional outward rectifier K+ channel. EMBO J. 15, 6854–6862.
Fink, M., Lesage, F., Duprat, F., Heurteaux, C., Reyes, R., Fosset, M., et al. (1998). A neuronal two P domain K+ channel stimulated by arachidonic acid and polyunsaturated fatty acids. EMBO J. 17, 3297–3308. doi: 10.1093/emboj/17.12.3297
Haydon, P. G., and Carmignoto, G. (2006). Astrocyte control of synaptic transmission and neurovascular coupling. Physiol. Rev. 86, 1009–1031. doi: 10.1152/physrev.00049.2005
Hervieu, G. J., Cluderay, J. E., Gray, C. W., Green, P. J., Ranson, J. L., Randall, A. D., et al. (2001). Distribution and expression of TREK-1, a two-pore-domain potassium channel, in the adult rat CNS. Neuroscience 103, 899–919. doi: 10.1016/s0306-4522(01)00030-6
Hibino, H., Fujita, A., Iwai, K., Yamada, M., and Kurachi, Y. (2004). Differential assembly of inwardly rectifying K+ channel subunits, Kir4.1 and Kir5.1, in brain astrocytes. J. Biol. Chem. 279, 44065–44073. doi: 10.1074/jbc.m405985200
Hund, T. J., Snyder, J. S., Wu, X., Glynn, P., Koval, O. M., Onal, B., et al. (2014). βIV-Spectrin regulates TREK-1 membrane targeting in the heart. Cardiovasc. Res. 102, 166–175. doi: 10.1093/cvr/cvu008
Hwang, E. M., Kim, E., Yarishkin, O., Woo, D. H., Han, K. S., Park, N., et al. (2014). A disulphide-linked heterodimer of TWIK-1 and TREK-1 mediates passive conductance in astrocytes. Nat. Commun. 5:3227. doi: 10.1038/ncomms4227
Kafitz, K. W., Meier, S. D., Stephan, J., and Rose, C. R. (2008). Developmental profile and properties of sulforhodamine 101—Labeled glial cells in acute brain slices of rat hippocampus. J. Neurosci. Methods 169, 84–92. doi: 10.1016/j.jneumeth.2007.11.022
Kang, D., Kim, S. H., Hwang, E. M., Kwon, O. S., Yang, H. Y., Kim, E. S., et al. (2007). Expression of thermosensitive two-pore domain K+ channels in human keratinocytes cell line HaCaT cells. Exp. Dermatol. 16, 1016–1022. doi: 10.1111/j.1600-0625.2007.00626.x
Kelly, T., Kafitz, K. W., Roderigo, C., and Rose, C. R. (2009). Ammonium-evoked alterations in intracellular sodium and pH reduce glial glutamate transport activity. Glia 57, 921–934. doi: 10.1002/glia.20817
Kimelberg, H. K. (2010). Functions of mature mammalian astrocytes: a current view. Neuroscientist 16, 79–106. doi: 10.1177/1073858409342593
Kucheryavykh, Y. V., Kucheryavykh, L. Y., Nichols, C. G., Maldonado, H. M., Baksi, K., Reichenbach, A., et al. (2007). Downregulation of Kir4.1 inward rectifying potassium channel subunits by RNAi impairs potassium transfer and glutamate uptake by cultured cortical astrocytes. Glia 55, 274–281. doi: 10.1002/glia.20455
Kuffler, S. W., Nicholls, J. G., and Orkand, R. K. (1966). Physiological properties of glial cells in the central nervous system of amphibia. J. Neurophysiol. 29, 768–787.
Lesage, F., Guillemare, E., Fink, M., Duprat, F., Lazdunski, M., Romey, G., et al. (1996a). TWIK-1, a ubiquitous human weakly inward rectifying K+ channel with a novel structure. EMBO J. 15, 1004–1011.
Lesage, F., and Lazdunski, M. (2000). Molecular and functional properties of two-pore-domain potassium channels. Am. J. Physiol. Renal Physiol. 279, F793–F801.
Lesage, F., Reyes, R., Fink, M., Duprat, F., Guillemare, E., and Lazdunski, M. (1996b). Dimerization of TWIK-1 K+ channel subunits via a disulfide bridge. EMBO J. 15, 6400–6407.
Ma, B. F., Xie, M. J., and Zhou, M. (2012a). Bicarbonate efflux via GABA(A) receptors depolarizes membrane potential and inhibits two-pore domain potassium channels of astrocytes in rat hippocampal slices. Glia 60, 1761–1772. doi: 10.1002/glia.22395
Ma, L., Xie, Y. P., Zhou, M., and Chen, H. (2012b). Silent TWIK-1 potassium channels conduct monovalent cation currents. Biophys. J. 102, L34–L36. doi: 10.1016/j.bpj.2012.03.011
Ma, L., Zhang, X., Zhou, M., and Chen, H. (2012c). Acid-sensitive TWIK and TASK two-pore domain potassium channels change ion selectivity and become permeable to sodium in extracellular acidification. J. Biol. Chem. 287, 37145–37153. doi: 10.1074/jbc.M112.398164
Ma, B., Xu, G., Wang, W., Enyeart, J. J., and Zhou, M. (2014). Dual patch voltage clamp study of low membrane resistance astrocytes in situ. Mol. Brain 7:18. doi: 10.1186/1756-6606-7-18
Maingret, F., Lauritzen, I., Patel, A. J., Heurteaux, C., Reyes, R., Lesage, F., et al. (2000). TREK-1 is a heat-activated background K(+) channel. EMBO J. 19, 2483–2491. doi: 10.1093/emboj/19.11.2483
Maingret, F., Patel, A. J., Lesage, F., Lazdunski, M., and Honoré, E. (1999). Mechano- or acid stimulation, two interactive modes of activation of the TREK-1 potassium channel. J. Biol. Chem. 274, 26691–26696. doi: 10.1074/jbc.274.38.26691
Meadows, H. J., Benham, C. D., Cairns, W., Gloger, I., Jennings, C., Medhurst, A. D., et al. (2000). Cloning, localisation and functional expression of the human orthologue of the TREK-1 potassium channel. Pflugers. Arch. 439, 714–722. doi: 10.1007/s004249900235
Medhurst, A. D., Rennie, G., Chapman, C. G., Meadows, H., Duckworth, M. D., Kelsell, R. E., et al. (2001). Distribution analysis of human two pore domain potassium channels in tissues of the central nervous system and periphery. Brain Res. Mol. Brain Res. 86, 101–114. doi: 10.1016/s0169-328x(00)00263-1
Millar, I. D., Taylor, H. C., Cooper, G. J., Kibble, J. D., Barhanin, J., and Robson, L. (2006). Adaptive downregulation of a quinidine-sensitive cation conductance in renal principal cells of TWIK-1 knockout mice. Pflugers. Arch. 453, 107–116. doi: 10.1007/s00424-006-0107-0
Murbartián, J., Lei, Q., Sando, J. J., and Bayliss, D. A. (2005). Sequential phosphorylation mediates receptor- and kinase-induced inhibition of TREK-1 background potassium channels. J. Biol. Chem. 280, 30175–30184. doi: 10.1074/jbc.m503862200
Namiranian, K., Lloyd, E. E., Crossland, R. F., Marrelli, S. P., Taffet, G. E., Reddy, A. K., et al. (2010). Cerebrovascular responses in mice deficient in the potassium channel, TREK-1. Am. J. Physiol. Regul. Integr. Comp. Physiol. 299, R461–R469. doi: 10.1152/ajpregu.00057.2010
Nedergaard, M., Ransom, B., and Goldman, S. A. (2003). New roles for astrocytes: redefining the functional architecture of the brain. Trends Neurosci. 26, 523–530. doi: 10.1016/j.tins.2003.08.008
Neusch, C., Papadopoulos, N., Müller, M., Maletzki, I., Winter, S. M., Hirrlinger, J., et al. (2006). Lack of the Kir4.1 channel subunit abolishes K+ buffering properties of astrocytes in the ventral respiratory group: impact on extracellular K+ regulation. J. Neurophysiol. 95, 1843–1852. doi: 10.1152/jn.00996.2005
Nicolas, M. T., Lesage, F., Reyes, R., Barhanin, J., and Dememes, D. (2004). Localization of TREK-1, a two-pore-domain K+ channel in the peripheral vestibular system of mouse and rat. Brain Res. 1017, 46–52. doi: 10.1016/j.brainres.2004.05.012
Nie, X., Arrighi, I., Kaissling, B., Pfaff, I., Mann, J., Barhanin, J., et al. (2005). Expression and insights on function of potassium channel TWIK-1 in mouse kidney. Pflugers. Arch. 451, 479–488. doi: 10.1007/s00424-005-1480-9
Nimmerjahn, A., Kirchhoff, F., Kerr, J. N., and Helmchen, F. (2004). Sulforhodamine 101 as a specific marker of astroglia in the neocortex in vivo. Nat. Methods 1, 31–37. doi: 10.1038/nmeth706
Nwaobi, S. E., Lin, E., Peramsetty, S. R., and Olsen, M. L. (2014). DNA methylation functions as a critical regulator of Kir4.1 expression during CNS development. Glia 62, 411–427. doi: 10.1002/glia.22613
Olsen, M. L., Khakh, B. S., Skatchkov, S. N., Zhou, M., Lee, C. J., and Rouach, N. (2015). New insights on astrocyte ion channels: critical for homeostasis and neuron-glia signaling. J. Neurosci. 35, 13827–13835. doi: 10.1523/JNEUROSCI.2603-15.2015
Olsen, M. L., and Sontheimer, H. (2008). Functional implications for Kir4.1 channels in glial biology: from K+ buffering to cell differentiation. J. Neurochem. 107, 589–601. doi: 10.1111/j.1471-4159.2008.05615.x
Orkand, R. K., Nicholls, J. G., and Kuffler, S. W. (1966). Effect of nerve impulses on the membrane potential of glial cells in the central nervous system of amphibia. J. Neurophysiol. 29, 788–806.
Patel, A. J., Honoré, E., Lesage, F., Fink, M., Romey, G., and Lazdunski, M. (1999). Inhalational anesthetics activate two-pore-domain background K+ channels. Nat. Neurosci. 2, 422–426. doi: 10.1038/8084
Patel, A. J., Honoré, E., Maingret, F., Lesage, F., Fink, M., Duprat, F., et al. (1998). A mammalian two pore domain mechano-gated S-like K+ channel. EMBO J. 17, 4283–4290. doi: 10.1093/emboj/17.15.4283
Ransom, C. B., and Sontheimer, H. (1995). Biophysical and pharmacological characterization of inwardly rectifying K+ currents in rat spinal cord astrocytes. J. Neurophysiol. 73, 333–346.
Rinne, S., Renigunta, V., Schlichthorl, G., Zuzarte, M., Bittner, S., Meuth, S. G., et al. (2014). A splice variant of the two-pore domain potassium channel TREK-1 with only one pore domain reduces the surface expression of full-length TREK-1 channels. Pflugers. Arch. 466, 1559–1570. doi: 10.1007/s00424-013-1384-z
Sandoz, G., Tardy, M. P., Thümmler, S., Feliciangeli, S., Lazdunski, M., and Lesage, F. (2008). Mtap2 is a constituent of the protein network that regulates twik-related K+ channel expression and trafficking. J. Neurosci. 28, 8545–8552. doi: 10.1523/JNEUROSCI.1962-08.2008
Seifert, G., Hüttmann, K., Binder, D. K., Hartmann, C., Wyczynski, A., Neusch, C., et al. (2009). Analysis of astroglial K+ channel expression in the developing hippocampus reveals a predominant role of the Kir4.1 subunit. J. Neurosci. 29, 7474–7488. doi: 10.1523/JNEUROSCI.3790-08.2009
Talley, E. M., Solorzano, G., Lei, Q., Kim, D., and Bayliss, D. A. (2001). Cns distribution of members of the two-pore-domain (KCNK) potassium channel family. J. Neurosci. 21, 7491–7505.
Tong, X., Ao, Y., Faas, G. C., Nwaobi, S. E., Xu, J., Haustein, M. D., et al. (2014). Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nat. Neurosci. 17, 694–703. doi: 10.1038/nn.3691
Toro Cabrera, G., and Mueller, C. (2016). Design of shRNA and miRNA for Delivery to the CNS. Methods Mol. Biol. 1382, 67–80. doi: 10.1007/978-1-4939-3271-9_5
Verkhratsky, A., and Steinhauser, C. (2000). Ion channels in glial cells. Brain Res. Brain Res. Rev. 32, 380–412. doi: 10.1016/S0165-0173(99)00093-4
Walz, W. (2000). Role of astrocytes in the clearance of excess extracellular potassium. Neurochem. Int. 36, 291–300. doi: 10.1016/s0197-0186(99)00137-0
Wang, D. D., and Bordey, A. (2008). The astrocyte odyssey. Prog. Neurobiol. 86, 342–367. doi: 10.1016/j.pneurobio.2008.09.015
Wang, W., Kiyoshi, C. M., Du, Y., Ma, B., Alford, C. C., Chen, H., et al. (2015). mGluR3 activation recruits cytoplasmic TWIK-1 channels to membrane that enhances ammonium uptake in hippocampal astrocytes. Mol. Neurobiol. doi: 10.1007/s12035-015-9496-4 [Epub ahead of print].
Wang, W., Putra, A., Schools, G. P., Ma, B., Chen, H., Kaczmarek, L. K., et al. (2013). The contribution of TWIK-1 channels to astrocyte K(+) current is limited by retention in intracellular compartments. Front. Cell. Neurosci. 7:246. doi: 10.3389/fncel.2013.00246
Xie, M., Lynch, D. T., Schools, G. P., Feustel, P. J., Kimelberg, H. K., and Zhou, M. (2007). Sodium channel currents in rat hippocampal NG2 glia: characterization and contribution to resting membrane potential. Neuroscience 150, 853–862. doi: 10.1016/j.neuroscience.2007.09.057
Zhou, M., and Kimelberg, H. K. (2001). Freshly isolated hippocampal CA1 astrocytes comprise two populations differing in glutamate transporter and AMPA receptor expression. J. Neurosci. 21, 7901–7908.
Zhou, M., Schools, G. P., and Kimelberg, H. K. (2006). Development of GLAST(+) astrocytes and NG2(+) glia in rat hippocampus CA1: mature astrocytes are electrophysiologically passive. J. Neurophysiol. 95, 134–143. doi: 10.1152/jn.00570.2005
Zhou, M., Xu, G., Xie, M., Zhang, X., Schools, G. P., Ma, L., et al. (2009). TWIK-1 and TREK-1 are potassium channels contributing significantly to astrocyte passive conductance in rat hippocampal slices. J. Neurosci. 29, 8551–8564. doi: 10.1523/JNEUROSCI.5784-08.2009
Keywords: astrocytes, TREK-1 potassium channel, passive membrane conductance, patch-clamp recording, hippocampus
Citation: Du Y, Kiyoshi CM, Wang Q, Wang W, Ma B, Alford CC, Zhong S, Wan Q, Chen H, Lloyd EE, Bryan RM Jr. and Zhou M (2016) Genetic Deletion of TREK-1 or TWIK-1/TREK-1 Potassium Channels does not Alter the Basic Electrophysiological Properties of Mature Hippocampal Astrocytes In Situ. Front. Cell. Neurosci. 10:13. doi: 10.3389/fncel.2016.00013
Received: 01 September 2015; Accepted: 14 January 2016;
Published: 03 February 2016.
Edited by:
Marco Martina, Northwestern University, USAReviewed by:
Douglas A. Bayliss, University of Virginia, USAJulien Dine, Max Planck Institut of Psychiatry, Germany
Copyright © 2016 Du, Kiyoshi, Wang, Wang, Ma, Alford, Zhong, Wan, Chen, Lloyd, Bryan and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min Zhou, emhvdS43ODdAb3N1LmVkdQ==