 Qian Cai
Qian Cai Prasad Tammineni
Prasad Tammineni- Department of Cell Biology and Neuroscience, Rutgers, The State University of New Jersey, Piscataway, NJ, USA
Mitochondrial dysfunction is one of the earliest and most prominent features in the brains of Alzheimer’s disease (AD) patients. Recent studies suggest that mitochondrial dysfunction plays a pivotal role in the pathogenesis of AD. Neurons are metabolically active cells, causing them to be particularly dependent on mitochondrial function for survival and maintenance. As highly dynamic organelles, mitochondria are characterized by a balance of fusion and fission, transport, and mitophagy, all of which are essential for maintaining mitochondrial integrity and function. Mitochondrial dynamics and mitophagy can therefore be identified as key pathways in mitochondrial quality control. Tremendous progress has been made in studying changes in these key aspects of mitochondrial biology in the vulnerable neurons of AD brains and mouse models, and the potential underlying mechanisms of such changes. This review highlights recent findings on alterations in the mitochondrial dynamics and mitophagy in AD and discusses how these abnormalities impact mitochondrial quality control and thus contribute to mitochondrial dysfunction in AD.
Introduction
Mitochondria are organelles essential for neuronal function and survival (Nicholls and Budd, 2000; Sheng and Cai, 2012; Mishra and Chan, 2014). Their key role is ATP production, which is vital for maintaining neuronal integrity and function (Verstreken et al., 2005; Sun et al., 2013; Rangaraju et al., 2014). Mitochondria also play a critical role in buffering intracellular Ca2+ levels by taking up and releasing Ca2+. At synaptic terminals, mitochondria take up excess intracellular Ca2+ and release Ca2+ to prolong residual levels, maintaining Ca2+ homeostasis (Tang and Zucker, 1997). Through this mechanism, synaptic mitochondria are thought to be involved in the regulation of neurotransmission (Billups and Forsythe, 2002; David and Barrett, 2003) or certain types of short-term synaptic plasticity (Levy et al., 2003; Kang et al., 2008). Dysfunctional mitochondria not only produce energy and buffer Ca2+ less efficiently, but also release harmful reactive oxygen species (ROS; Court and Coleman, 2012; Sheng and Cai, 2012). As a result, mitochondrial oxidative stress triggers leakage of mitochondrial intermembranous contents, such as cytochrome c, into the cytosol, causing caspase activation, DNA damage, and apoptosis (Mishra and Chan, 2014). The progressive accumulation of these damaged mitochondria in axons and synapses over the lifetime of neurons is thought to contribute to the pathogenesis of Alzheimer’s disease (AD; Du et al., 2012; Reddy, 2013).
The half-life of neuronal mitochondria is estimated to be ~30 days (Gross et al., 1969; Menzies and Gold, 1971). Throughout a neuron’s lifetime, aged and damaged mitochondria undergo dynamic recycling via fusion and fission or elimination via mitophagy, a cargo-selective autophagy that degrades mitochondria within lysosomes after their transport back to the soma. These dynamic processes of mitochondrial fusion, fission, transport, and turnover constitute the elaborate system of mitochondrial quality control, which regulates mitochondrial function by enabling recruitment of healthy mitochondria to subcellular compartments with high demands for ATP, such as synaptic terminals or axonal branches (Bogan and Cabot, 1991); content exchange between mitochondria; mitochondrial shape control; and mitochondrial turnover via mitophagy (Chen and Chan, 2009; Sheng and Cai, 2012). Disruptions in any of these processes lead to mitochondrial pathology, cellular dysfunction, and neurological defects (Chen and Chan, 2009).
Mitochondrial dysfunction is a significant concern in the nervous system of aging and it has been associated with major neurodegenerative disorders including the devastating AD that now affects 50% of individuals over 85 years old (Chen and Chan, 2009; Sheng and Cai, 2012). Mitochondrial deficiency has been suggested to be a hallmark of AD as the patients display early metabolic changes prior to the emergence of any histopathological or clinical abnormalities (Gibson and Shi, 2010). Mitochondrial pathology is also thought to contribute to synaptic deficits, an early pathophysiological feature of AD (Du et al., 2012; Reddy, 2013). Other prominent mitochondria-related anomalies in AD include the accumulation of damaged mitochondria in both familial and sporadic forms of the disease (Swerdlow et al., 2010), as well as changes in mitochondrial structure, dynamics, and motility in vulnerable neurons of affected brain regions (Baloyannis, 2006; Chen and Chan, 2009; Sheng and Cai, 2012). Here, we provide an overview of the underlying mechanisms regulating mitochondrial dynamics and transport and discuss how abnormalities in these mechanisms compromise mitochondrial quality control, thus contributing to mitochondrial dysfunction in AD.
Mitochondrial Fission
Mitochondria constantly change their shape through continuous fusion and fission events (Figure 1). Mitochondrial fission in mammals requires Drp1, a dynamin-like protein (Detmer and Chan, 2007; Reddy et al., 2011; Kandimalla and Hemachandra Reddy, 2015). Drp1 is a cytosolic protein recruited to mitochondria during fission, forming a ring-like higher order structure that physically pinches the mitochondria into two daughter mitochondria. Drp1-mediated mitochondrial fission in yeast requires an additional mitochondrial outer membrane (OM) protein, Fis1 (Okamoto and Shaw, 2005). Knocking down Fis1 in mammalian cells was also found to block mitochondrial fission without affecting Drp1 localization to mitochondria (Lee et al., 2004). Posttranslational modifications of Drp1 appear to be important for regulating Drp1 activity. As a proposed substrate of Sumo 1, Sumo 1-dependent sumoylation protects Drp1 from degradation, thus promoting mitochondrial fission (Harder et al., 2004). Loss of Parkin or PINK1 function in human SH-SY5Y cells also resulted in increased Drp1-dependent mitochondrial fragmentation (Lutz et al., 2009). Consistently, a study showed that Parkin ubiquitinates Drp1 for its degradation within the proteasome system (Wang et al., 2011a). However, it was also found that loss of PINK1 in Drosophila impairs mitochondrial fission (Liu et al., 2011). Another mitochondrial E3 ubiquitin ligase known to regulate Drp1-mediated fission is MARCH-V, which mediates ubiquitination of Drp1 (Nakamura et al., 2006; Karbowski et al., 2007). Phosphorylation of Drp1 at different sites, which functions variably to either promote or inhibit its activity, is another mechanism regulating mitochondrial morphology (Chang and Blackstone, 2007; Cribbs and Strack, 2007; Taguchi et al., 2007). Although no inherited diseases caused by mutations of Drp1 have been reported, Drp1 is necessary for embryonic development of the mouse brain and for synapse formation in cultured neurons (Ishihara et al., 2009). In addition, Drp1 maintains the proper distribution of mitochondria near Drosophila neuromuscular junctions (Verstreken et al., 2005). It was shown that defects in Drp1-mediated mitochondrial fission result in the accumulation of mitochondria in the cell body and reduced dendritic mitochondrial content (Li et al., 2004).
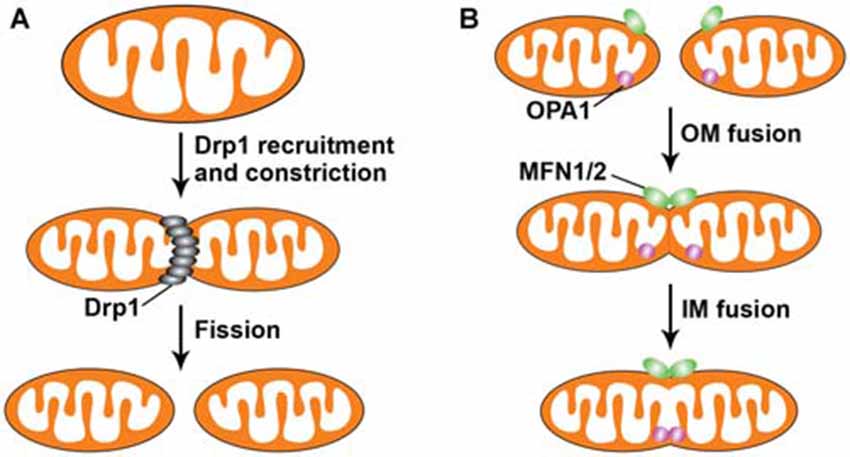
Figure 1. Mitochondrial fission and fusion. Mitochondria are dynamic organelles that undergo continuous fusion and fission events to intermix their lipids and contents. (A) Dynamin-related protein 1 (DRP1) regulates mitochondrial fission, which consists of two steps: first, DRP1 is recruited from the cytosol to the mitochondrial outer membrane (OM); second, its assemblage on the mitochondrial surface results in constriction of the mitochondria, leading to the separation of one mitochondrion into two entities. (B) Mitofusins 1 and 2 (MFN1/2) at the OM and optic atrophy 1 (OPA1) at the inner membrane (IM) orchestrate mitochondrial fusion, which involves MFN1/2-mediated OM fusion of two mitochondria, followed by OPA1-directed IM fusion. Mitochondrial fusion leads to elongated and highly interconnected mitochondria.
Mitochondrial Fusion
Mitochondrial fusion in mammals is mainly mediated by three proteins: Mfn1, Mfn2 and OPA1 (Mishra and Chan, 2014; Roy et al., 2015). Mfn1 and Mfn2 are required for OM fusion, whereas OPA1 is involved in inner membrane (IM) fusion (Meeusen et al., 2006; Song et al., 2009). Recent studies propose that long forms of OPA1 is sufficient for mediating efficient fusion and that short OPA1 forms may act independently to promote mitochondrial fission (Ishihara et al., 2006; Anand et al., 2014). OPA1 proteolysis itself has been reported to be a profusion event that operates at the time of fusion (Mishra et al., 2014). Mutations of each of these genes have been linked to neurological disorders. Mutations in Mfn2 cause Charcot-Marie-Tooth type 2A (CMT2A), a peripheral neuropathy affecting sensory and motor neurons (Zuchner et al., 2004), while mutations in OPA1 are the primary cause of autosomal dominant optic atrophy (DOA), a degeneration of retinal ganglia cells that results in atrophy of the optic nerve (Alexander et al., 2000; Delettre et al., 2000). Deletion of each of these genes in mice leads to mitochondrial dysfunction and embryonic lethality (Chen et al., 2003; Alavi et al., 2007; Davies et al., 2007). In addition to regulating mitochondrial shape, mitochondrial fusion affects the distribution and transport of mitochondria in neurons. Mitochondria in Mfn2 conditional knockout mice are fragmented and absent from distal neurites, impeding dendritic outgrowth, formation of axonal projections, and neuronal survival (Chen et al., 2007; Lee et al., 2012; Pham et al., 2012). Moreover, motor neurons derived from transgenic mice expressing mutant Mfn2 showed improper mitochondrial distribution characterized by tight clusters of mitochondria within axons (Detmer et al., 2008). Recent studies suggest that mitochondrial transport is regulated through fusion machinery by showing that Mfn1 and Mfn2 interact with Miro and Milton, members of the adaptor complex linking mitochondria to kinesin motors. Loss of Mfn2 or expression of Mfn2 disease mutants in neurons was shown to affect axonal mitochondrial fusion and transport and produce classic features of segmental axonal degeneration (Misko et al., 2010, 2012). A study in living zebrafish revealed the contribution of mitofusins, and thus fusion, to mitochondrial transport, which is essential for maintaining motor function (Chapman et al., 2013). Moreover, the motor neurons obtained from differentiation of CMT patient-derived iPS (induced pluripotent stem cells) presented abnormalities in mitochondrial trafficking and abnormal electrophysiological properties (Saporta et al., 2015). Thus, these studies provide evidence that a clear link exists between mitochondrial fusion and transport in neurons.
Mitochondrial fission and fusion ensure optimal functioning of mitochondria. Mitochondrial fusion allows the exchange of mitochondrial metabolites, mitochondrial DNA (mtDNA), and oxidative phosphorylation components within a mitochondrial network. While damaged mitochondria can be repaired through fusion with healthy mitochondria for mixture of contents, mitochondrial elongation upon nutrient deprivation maximizes energy production and promotes cell survival by preventing autophagy-mediated degradation of mitochondria (Gomes et al., 2011; Rambold et al., 2011). On the other hand, mitochondrial fission enables the segregation of mitochondria that become severely and irreversibly damaged or are fusion-incompetent, thereby leading to their subsequent elimination via mitophagy (Twig et al., 2008). Defects in either mitochondrial fusion or fission also lead to impaired mitochondrial motility and distribution in neurons, resulting in reduced mitochondrial content in distal neurites. Together, mitochondrial fusion and fission are required for the maintenance of mitochondrial shape, integrity, and distribution.
Mitochondrial Transport in Neurons
Altered mitochondrial transport is one of the pathogenic changes in major neurodegenerative diseases (Sheng and Cai, 2012). In mature neurons, ~20–30% of axonal mitochondria are motile while the remaining two thirds are stationary (Cai et al., 2005; Kang et al., 2008). Long-distance transport of mitochondria along microtubules (MTs) between the soma and distal processes or synapses is dependent on MT-based motor proteins, which drive their cargoes via mechanisms requiring ATP hydrolysis (Martin et al., 1999; Hirokawa et al., 2010). MTs are uniformly organized in axons, with the plus end pointed away from the soma and the minus end directed toward the soma (Hirokawa et al., 2010). Thus, kinesin motors, the plus-end driven motors, participate in anterograde axonal transport from the soma to nerve terminals, whereas the minus-end directed dynein motors mediate retrograde transport back to the soma. Efficient regulation of mitochondrial transport is essential for recruiting and redistributing mitochondria to specific domains with high-energy demands, such as synaptic terminals. Mitochondrial transport also plays a critical role in removing aged and damaged mitochondria, and replenishing them with healthy ones at distal regions of neurons.
The kinesin-1 family (KIF5) is the major motor that drives mitochondrial transport (Hurd and Saxton, 1996; Tanaka et al., 1998; Górska-Andrzejak et al., 2003; Cai et al., 2005; Pilling et al., 2006). KIF5 heavy chain (KHC) has a motor domain with ATPase activity located at the N terminus and a C-terminal tail domain required for binding its cargo. Adaptor proteins such as Drosophila protein Milton enable KIF5 motors to attach to mitochondria. Milton acts as a KIF5 motor adaptor by binding to both the KIF5 C-terminal tail domain and the mitochondrial OM receptor Miro (Stowers et al., 2002; Glater et al., 2006). Consistently, Milton mutation in Drosophila reduces mitochondrial trafficking into synapses. Two Milton orthologues, Trak1 and Trak2, are found in mammals (Smith et al., 2006; MacAskill et al., 2009; Koutsopoulos et al., 2010). Trak2 overexpression in cultured hippocampal neurons robustly enhances axonal mitochondrial motility (Chen and Sheng, 2013). In contrast, loss of Trak1 or expression of its mutants leads to a reduction in mitochondrial transport along axons (Brickley and Stephenson, 2011). A recent study showed that mammalian Trak1 and Trak2 each contain one N-terminal KIF5B binding site and two dynein/dynactin-binding sites, one located at the N-terminus and the other at the C-terminus (van Spronsen et al., 2013). This suggests that the Trak proteins can mediate both KIF5- and dynein-driven bi-directional mitochondrial transport.
Miro or MIRO, the mitochondrial OM receptor that binds to Trak/Milton, is a Rho-GTPase that consists of two Ca2+-binding EF-hand motifs and two GTPase domains (Frederick et al., 2004; Fransson et al., 2006). In Drosophila, mutation of the miro gene impairs mitochondrial anterograde transport and depletes the supply of mitochondria in distal synaptic terminals (Guo et al., 2005). Mammalian Miro has two isoforms, Miro1 and Miro2. The Miro1-Trak2 adaptor complex regulates mitochondrial transport in hippocampal neurons (MacAskill et al., 2009). A recent study showed that elevated Miro1 expression increased mitochondrial transport, likely by recruiting more Trak2 and KIF5 motors to mitochondria (Chen and Sheng, 2013). KIF5, Milton (Trak), and Miro are thus assembled into the transport machinery that drives mitochondrial anterograde transport. Syntabulin similarly serves as a KIF5 adaptor for neuronal mitochondria (Cai et al., 2005). Loss of syntabulin or interruption of KIF5-syntabulin coupling reduces mitochondrial anterograde transport in axons. In addition, a number of other proteins have been suggested as KIF5 motor adaptor candidates for mitochondrial transport, including FEZ1 (fasciculation and elongation protein zeta-1) and RAN-binding protein 2 (RANBP2; Cho et al., 2007; Fujita et al., 2007; Figure 2).
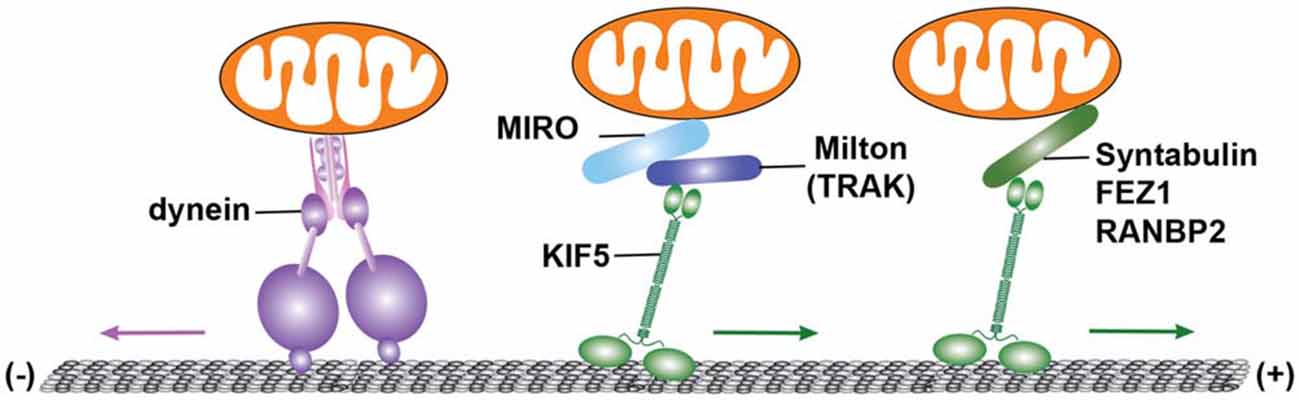
Figure 2. Microtubule-dependent motor-driven mitochondrial transport. Cytoplasmic dynein motors and kinesin-1 family (KIF5) of the KIF5 mediate mitochondrial transport. Dynein motors, the minus-end driven motors, carry out retrograde transport of mitochondria toward the soma of neurons. By contrast, KIF5 selectively moves mitochondria toward the plus-end of microtubules (MTs), and participates in anterograde transport from the soma to distal axons and synaptic terminals. Mitochondrial transport driven by KIF5 requires the mitochondrial rho (Miro)-Milton (or Miro-Trak) motor-adaptor complex. MIiro or MIRO is a mitochondrial outer membrane (OM) protein of the Rho GTPase family. In Drosophila melanogaster, Milton recruits KIF5 to mitochondria by binding to Miro. In a similar way, Trak1 and Trak2 (mammalian Milton orthologues) can bind to Miro1 and Miro2 (mammalian orthologues of Miro). The Miro1-Trak2 complex is an important regulator of mitochondrial transport in hippocampal neurons. KIF5 also associates with mitochondria and mediates mitochondrial anterograde transport via syntabulin, a KIF5 adaptor that binds to mitochondria via its carboxy-terminal transmembrane domain. Fasciculation and elongation protein zeta-1 (FEZ1), as well as RAN-binding protein 2 (RANBP2), are additional kinesin adaptors that may contribute to mitochondrial transport. Figure is modified from Sheng (2014).
Cytoplasmic dynein motors are composed of multiple subunits including heavy chains (DHC) that function as the motor domain for force production, and intermediate (DIC), light intermediate (DLIC), and light chains (DLC) that function in cargo attachment and motility regulation. Dynein motors associate with Drosophila mitochondria, and mutations of DHC alter both velocity and run length of retrograde transport of axonal mitochondria (Pilling et al., 2006). In addition to its role as a KIF5 motor adaptor, Miro may also serve as an adaptor for dynein motors in Drosophila (Guo et al., 2005; Russo et al., 2009). Loss of dMiro impairs both kinesin- and dynein-driven transport, while dMiro overexpression alters mitochondrial transport in both directions. It is likely that regulation of the opposite-direction motor activity is achieved through modulation of the cargo-adaptor proteins. The existence of multiple motor adaptors suggests that complex mechanisms regulate mitochondrial motility in response to various physiological and pathological conditions.
Mitophagy
Mitophagy, a selective autophagy for the removal of dysfunctional mitochondria, constitutes a key cellular pathway in mitochondrial quality control. It involves sequestering damaged mitochondria into autophagosomes and subsequently degrading them within lysosomes. Recent studies indicate that PINK1/Parkin-mediated mitophagy ensures mitochondrial integrity and function (Clark et al., 2006; Gautier et al., 2008; Narendra et al., 2008). This type of mitophagy is initiated with stable accumulation of PTEN-induced putative kinase protein 1 (PINK1) on the surface of damaged mitochondria, followed by recruitment of Parkin from the cytosol to the mitochondria. Parkin, an E3 ubiquitin ligase, then ubiquitinates a number of mitochondrial OM proteins and activates the ubiquitin-proteasome system (Geisler et al., 2010; Poole et al., 2010; Ziviani et al., 2010; Chan et al., 2011; Yoshii et al., 2011), which works in conjunction with the AAA ATPase p97 to degrade mitochondrial OM proteins (Tanaka et al., 2010; Chan et al., 2011). This triggers engulfment of damaged mitochondria by isolation membranes to form autophagosomes. Loss-of-function mutations of PINK1 and Parkin are associated with autosomal recessive forms of Parkinson’s disease. While the functions of PINK1 and Parkin in mitophagy have been proposed mostly from in vitro studies, their involvement in this pathway has recently been supported by proteomic analyses in Drosophila under normal physiological conditions (Vincow et al., 2013). The PINK1/Parkin pathway thus promotes both mitophagy in vivo and the selective turnover of mitochondrial respiratory chain subunits.
Other PINK1/Parkin-independent mitophagy pathways have been identified as well. For instance, a mitochondrial OM protein, the BCL-2 homology 3 (BH3)-containing protein NIP3-like X (NIX, also known as BNIP3L), was shown to play an important role in the elimination of mitochondria in erythrocytes (Sandoval et al., 2008). NIX contains an amino-terminal LC3-interacting region (LIR) that binds to LC3 on isolation membranes (Novak et al., 2010). This enables NIX to act as a selective mitophagy receptor that physically connects the autophagy machinery to the mitochondrial surface in erythroid cells. Another mitochondrial OM protein that has been proposed as a mitophagy receptor is FUN14 domain containing 1 (FUNDC1), which regulates the autophagic degradation of mitochondria in response to hypoxia. FUNDC1 has a LIR required for recruitment of LC3 (Liu et al., 2012a). Under hypoxic conditions, this LIR is dephosphorylated by the mitochondrial phosphatase phosphoglycerate mutase family member 5 (PGAM5), thus increasing its physical association with LC3 and promoting mitophagy (Chen et al., 2014). In addition, a recent study reported that in neuronal cells, cardiolipin—an inner mitochondrial membrane phospholipid—externalizes to the OM upon mitochondrial damage. LC3 has been shown to contain cardiolipin-binding sites, which suggests that externalized cardiolipin acts as an elimination signal for neuronal mitophagy (Chu et al., 2013). Altogether, these observations indicate that specific mitophagy receptors on the mitochondrial OM play an essential role in mitochondrial degradation by recruiting autophagy machinery to mitochondria (Figure 3).
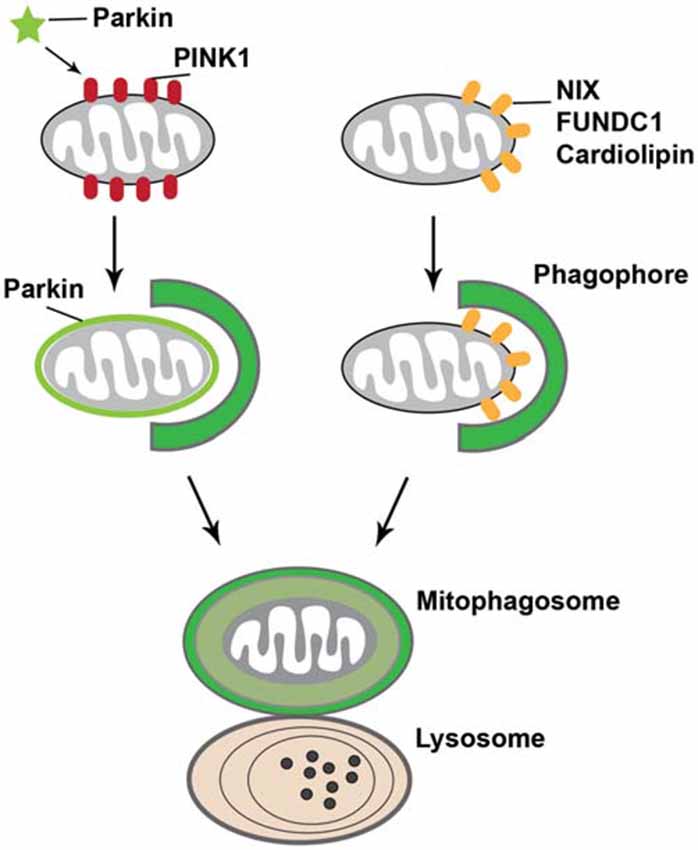
Figure 3. Mitophagy. Mitophagy, which is initiated when damaged mitochondria are labeled for their subsequent recruitment into phagophore or isolation membranes, occurs through two mechanisms. First, upon loss of mitochondrial membrane potential, the E3 ubiquitin ligase Parkin is recruited from the cytosol to damaged mitochondria in a PTEN-induced putative kinase protein 1 (PINK1)-dependent manner. Parkin ubiquitinates mitochondrial proteins and causes mitochondria to become engulfed by phagophore or isolation membranes that then fuse with lysosomes. Second, outer mitochondrial membrane proteins, such as NIP3-like protein X (NIX; also known as BNIP3L), FUN14 domain containing 1 (FUNDC1), or cardiolipin externalized from the inner mitochondrial membrane phospholipid upon mitochondrial damage, bind to LC3 on the phagophore or isolation membranes, which mediate the sequestration of damaged mitochondria into mitophagosomes for lysosomal degradation.
As a highly dynamic process, mitophagy needs to be evaluated using several complementary assays: (1) cellular analysis of depolarization of mitochondrial membrane potential with Parkin translocation, co-localization with autophagy markers (mitophagosome formation), and lysosomal sequestration of defective mitochondria; (2) ultrastructural analysis by electron microscopy based on alterations in mitochondrial structure and morphological features of autophagic vacuoles (AVs) containing mitochondrial profiles; and (3) western blot analysis of increased association of mitophagy machinery with mitochondria and reduced levels of mitochondrial proteins upon mitophagy induction. These assays need to be combined with the application of mitochondrial membrane potential dissipating agents and a flux inhibitor to trap newly formed autophagosomes.
Mitochondrial Quality Control
Mitochondrial quality control involves surveillance and protective strategies at multiple levels in order to limit mitochondrial damage and ensure mitochondrial integrity. The quality control occurs at the molecular, organellar, and cellular levels. At the molecular level, a proteolytic system, involving molecular chaperones and ATP-dependent proteases in the matrix and the inner membrane (IM) of mitochondria, degrades damaged and misfolded proteins and/or dissolves protein aggregates for proteolysis (de Castro et al., 2010). The ubiquitin-proteasome system in the cytosolic additionally participates in the quality control of mitochondrial proteins (Tatsuta and Langer, 2008). At the organellar level, mitochondrial fusion and fission provide additional protection against mitochondrial damage. Dysfunctional mitochondria can be repaired by fusion with healthy mitochondria, which allows for mixture of the contents of healthy and defective mitochondria (Detmer and Chan, 2007; Chen and Chan, 2009; Westermann, 2010). In other cases, severely damaged mitochondria are segregated by fission, ultimately leading to their elimination within the autophagy-lysosomal system through mitophagy (Youle and Narendra, 2011). However, if the quality control pathways at the molecular and organellar levels are defective or if the levels of damage exceed the capacity of these two pathways, damaged mitochondria can rupture and release pro-apoptotic factors, leading to activation of apoptosis and cell death (Figure 4).
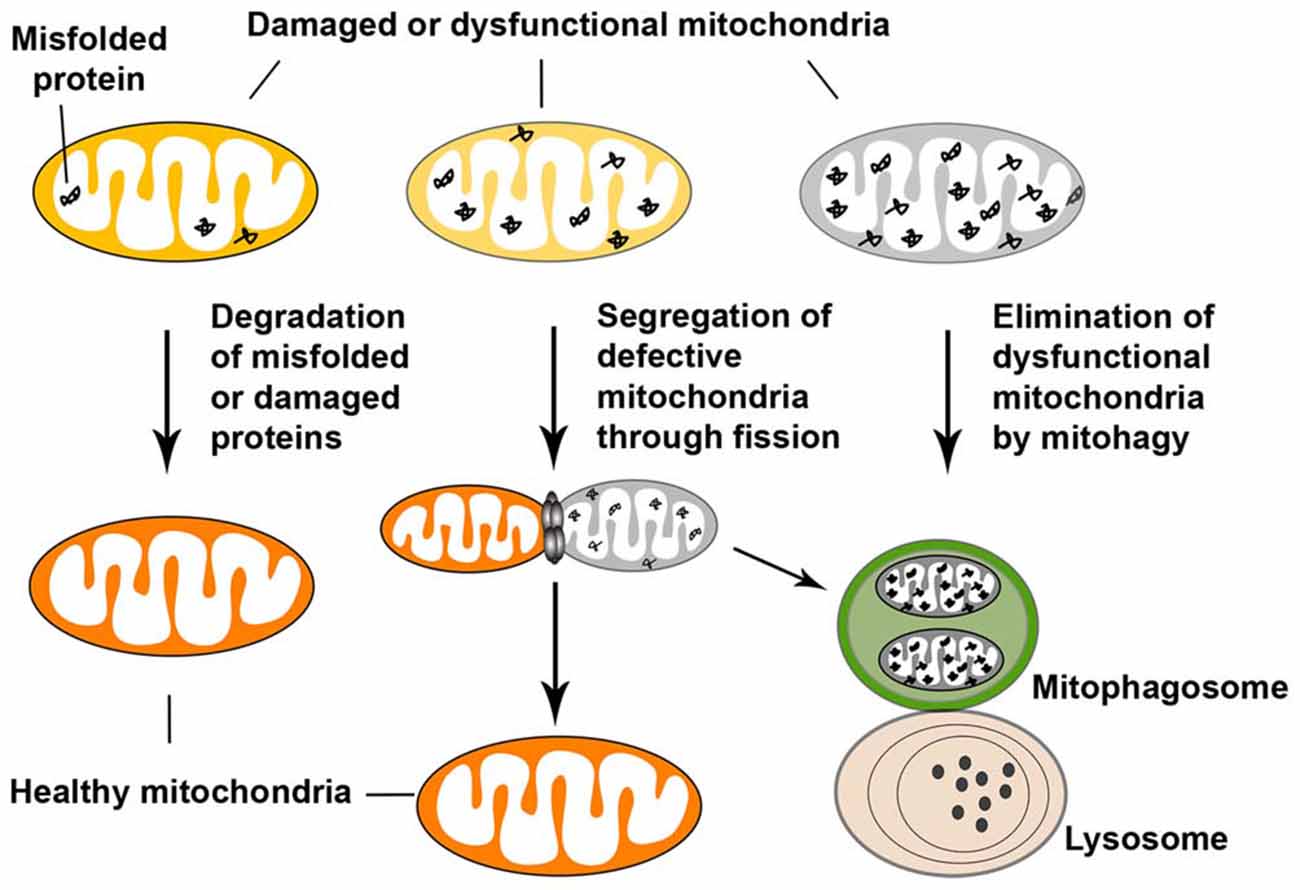
Figure 4. Mitochondrial quality control. Mitochondrial quality control occurs at multiple levels in order to limit mitochondrial damage and ensure mitochondrial integrity. At the molecular level of defense, degradation of misfolded or damaged mitochondrial proteins is supported by the proteolytic system. Molecular chaperones and ATP-dependent proteases in the matrix and inner membrane (IM) of mitochondria degrade damaged proteins, stabilize misfolded proteins (thus preventing their aggregation), and/or dissolve protein aggregates, and thereby promote proteolysis. In addition, the cytosolic ubiquitin-proteasome system can participate in the quality control of mitochondrial proteins. At the organellar level, mitochondrial fusion and fission provide additional protection against mitochondrial damage. Damaged mitochondria can be repaired by fusion with healthy mitochondria, which allows the contents of healthy and dysfunctional mitochondria to be mixed. Fission, on the other hand, segregates mitochondria that have become irreversibly damaged or are fusion-incompetent and results in their subsequent elimination by autophagy. If the two quality control pathways described above are ineffective, dysfunctional mitochondria are eliminated by autophagy. One type of cargo-specific autophagy is mitophagy, which selectively removes damaged mitochondria. Figure is modified from Sheng and Cai (2012).
Mitochondrial Dynamics and Mitophagy
Mitochondrial dynamics regulate mitochondrial elimination through the autophagy-lysosomal system. Mitochondrial elongation protects mitochondria from autophagy-mediated degradation (Gomes et al., 2011; Rambold et al., 2011). Parkin-mediated mitophagy can also be prevented by inhibition of Drp1-mediated fission prevents (Tanaka et al., 2010). Studies have indicated that mitochondrial fission yields uneven products, leaving one hyperpolarized mitochondrion and one depolarized daughter mitochondrion (Twig et al., 2008). Such depolarized mitochondria display reduced levels of OPA1 protein, causing them to become much less likely to fuse and to eventually undergo autophagocytosis. Additional evidence supports the notion that mitophagy relies on the loss of fusion and the presence of fission, as OPA1-overexpression, Fis1 RNAi, and Drp1 dominant-negative expression all reduce mitophagy levels (Narendra et al., 2008; Twig et al., 2008). Moreover, a study in yeast indicates that dynamin-related 1 (Dnm1) and other components of the mitochondrial fission machinery are dispensable for mitophagy (Mendl et al., 2011). However, it should be noted that mitochondrial fragmentation facilitates, but is not itself sufficient for, mitophagy. Reciprocally, mitophagy influences mitochondrial dynamics. Upon mitophagy induction, Parkin induces ubiquitination of Mfn1 and Mfn2, which leads to their degradation in a proteasome- and p97-dependent manner (Gegg et al., 2010; Poole et al., 2010; Tanaka et al., 2010; Ziviani et al., 2010; Chan et al., 2011). A recent study further showed that PINK1 phosphorylates Mfn2 and thereby promotes Parkin-mediated Mfn2 ubiquitination, which is required for the quality control of cardiac mitochondria (Chen and Dorn, 2013). Through this interplay between mitophagy and mitochondrial dynamics, Parkin-mediated mitophagy inhibits the fusion of defective mitochondria with healthy ones, thus promoting the segregation of dysfunctional mitochondria for degradation via the autophagy-lysosomal pathway.
Mitochondrial Transport and Mitophagy
The mechanism coordinating mitochondrial motility and mitophagy represents an important emerging area. It has been suggested that a likely relationship between mitochondrial membrane potential and the direction of mitochondrial movement exists. One study showed that mitochondria with high membrane potentials exhibit anterograde transport towards distal processes, whereas depolarized mitochondria return to the soma following acute membrane potential dissipation (Miller and Sheetz, 2004). These findings suggest that damaged or defective mitochondria are delivered to the soma for repair and/or degradation, which is consistent with the observation that somatodendritic regions and the proximal axon are relatively enriched with mature lysosomes (Overly and Hollenbeck, 1996; Cai et al., 2010; Lee et al., 2011). Consistently, one recent study revealed that depolarized mitochondria in PINK1 mutant Drosophila neurons showed reduced anterograde axonal transport and altered morphology in the soma, indicating that mitochondrial turnover might be restricted to the cell body in vivo in the intact nervous system (Devireddy et al., 2015). Another study reported that axonal mitochondria displayed reduced anterograde transport and relatively increased retrograde transport upon mitophagy induction, promoting the accumulation of Parkin-targeted mitochondria in the soma and proximal regions (Cai et al., 2012a,b). The altered motility may serve as a protective mechanism: healthy mitochondria remain in distal areas whereas aged and defective mitochondria are returned to the soma. This dynamic process allows neurons to efficiently remove defective mitochondria from distal regions, and then eliminate them within mature lysosomes in the soma.
Reduced anterograde transport of depolarized mitochondria is consistent with the results of many studies showing that Miro is degraded in response to mitophagy induction. During the early phase of mitophagy, the Parkin-activated ubiquitin-proteasome system was shown to mediate widespread degradation of Miro1 and Miro2 (Chan et al., 2011; Yoshii et al., 2011). In addition to binding to KIF5-Trak motor complex, Miro has been found to interact with Parkin and is ubiquitinated by Parkin following mitochondrial depolarization (Weihofen et al., 2009; Chan et al., 2011; Wang et al., 2011b; Liu et al., 2012b; Sarraf et al., 2013; Birsa et al., 2014). Degradation of Miro on the surfaces of defective mitochondria may not only suppress mitochondrial anterograde transport, but also favor their retrograde transport to the soma or immobilization at distal regions in preparation for mitophagy.
Once mitochondria are immobilized at distal axons by Miro degradation or by syntaphilin, the axonal mitochondrial-anchoring protein (Kang et al., 2008; Chen et al., 2009), damaged mitochondria may also recruit Parkin or be engulfed by autophagosomes (Cai et al., 2012a,b; Ashrafi et al., 2014). Recent studies indicate that autophagosomes, including those containing engulfed mitochondria, predominantly undergo retrograde transport from distal axons to the soma, which allows for autophagosome maturation and subsequent degradation within mature lysosomes in the proximal regions and soma of neurons (Maday and Holzbaur, 2014; Maday et al., 2012; Cheng et al., 2015). This functional interplay between mitochondrial motility and mitophagy is a particularly important mechanism by which the PINK1/Parkin pathway governs mitochondrial quality control for the proper removal of aged and defective mitochondria from distal axons and nerve terminals (Figure 5).
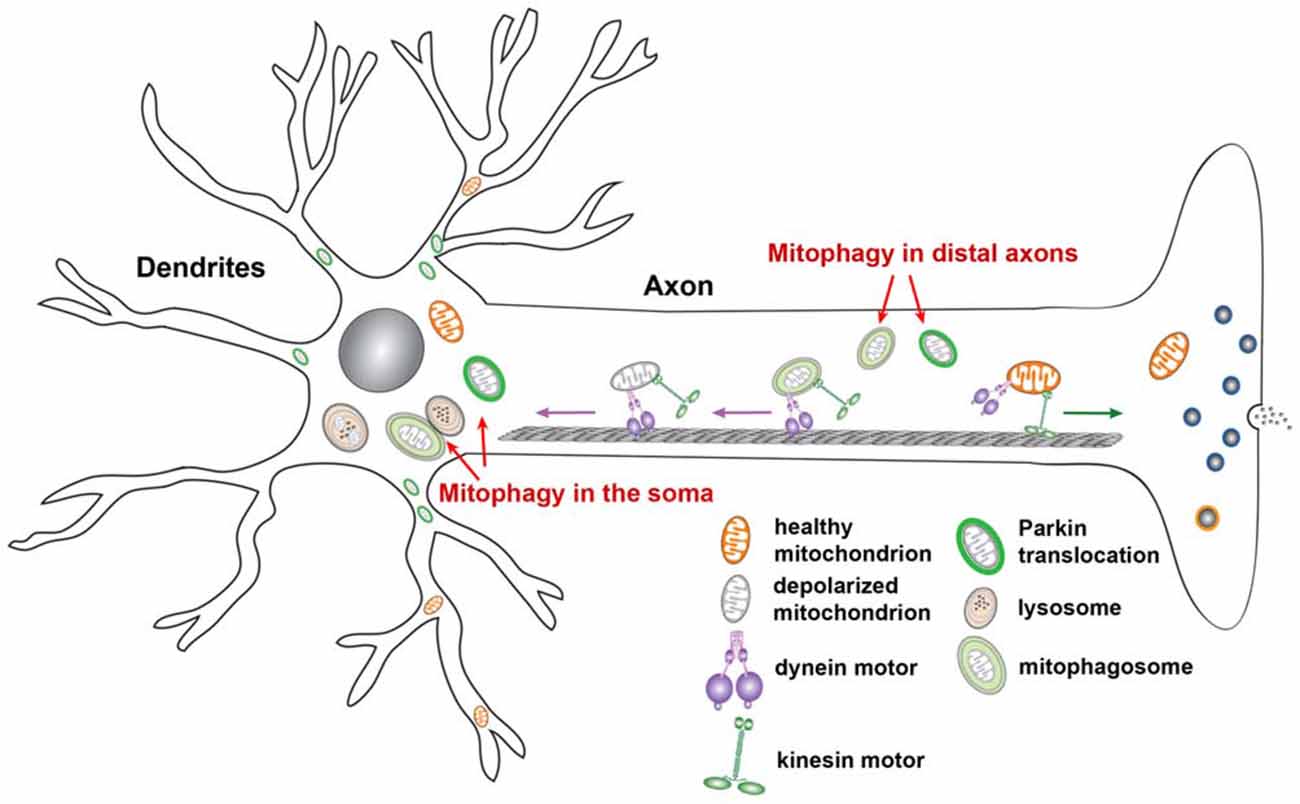
Figure 5. Functional interplay of mitochondrial transport and mitophagy in neurons. Upon mitochondrial membrane potential dissipation, Parkin-targeted mitochondria accumulate in the soma and proximal regions. Such compartmental restriction is attributed to altered motility of depolarized mitochondria, which exhibit reduced anterograde and relatively enhanced retrograde transport, thus reducing anterograde flux of damaged mitochondria into distal processes. This spatial process allows neurons to efficiently remove dysfunctional mitochondria from distal axons via the autophagy-lysosomal pathway in the soma, where mature lysosomes are mainly located. Damaged mitochondria at axonal terminals can also recruit Parkin for mitophagy once they are anchored by syntaphilin, or immobilized by turnover of the motor adaptor Miro on the mitochondrial surface. Autophagosomes containing engulfed mitochondria at axonal terminals are predominantly transported to the soma for maturation and for more efficient cargo degradation within acidic lysosomes. Figure is modified from Sheng (2014).
Mitochondrial Abnormalities in Alzheimer’s Disease
In AD brains, mitochondria display multiple abnormalities, including impaired mitochondrial function, altered mitochondrial dynamics and transport, increased mtDNA mutations, defective mitochondrial enzyme activities, and abnormal expression of mitochondrial genes (Reddy et al., 2011). A growing body of evidence suggests that accumulation of amyloid precursor protein (APP) and amyloid β (Aβ) peptides plays a central role in mediating mitochondrial toxicity.
APP and Aβ have been found in the purified mitochondria from patient brains and AD mouse models (Anandatheerthavarada et al., 2003; Lustbader et al., 2004; Caspersen et al., 2005; Crouch et al., 2005; Devi et al., 2006; Manczak et al., 2006; Du et al., 2008; Yao et al., 2009). Aβ has been shown to interact with mitochondrial matrix proteins ABAD and cyclophilin D, which has been suggested to induce cytotoxic effects (Lustbader et al., 2004; Du et al., 2008). Aβ was also found to affect mitochondrial fusion and fission (Wang et al., 2008b; Manczak et al., 2011; Manczak and Reddy, 2012b), alter mitochondrial motility (Rui et al., 2006; Du et al., 2010; Calkins et al., 2011), disrupt function of the electron transfer chain, increase ROS production (Keller et al., 1997; Abramov et al., 2004; Manczak et al., 2006), and impair mitochondrial function (Mattson et al., 1998; Lustbader et al., 2004; Du et al., 2008). Neurons derived from mutant APP transgenic (Tg) mouse models display altered mitochondrial dynamics, impaired trafficking, and reduced biogenesis (Calkins et al., 2011; Trushina et al., 2012). Abnormal accumulation of Aβ within synaptic mitochondria was proposed to contribute to early deficits in synaptic function in AD (Du et al., 2010). Moreover, the degree of cognitive impairment in AD brains has been linked to the extent of mitochondrial dysfunction and mitochondrial Aβ accumulation (Dragicevic et al., 2010).
While it remains to be experimentally proven whether intramitochondrial production of Aβ occurs (Pinho et al., 2014), studies have shown possible routes for Aβ entry into mitochondria—via mitochondrial-associated endoplasmic reticulum membrane (MAM; Pinho et al., 2014) or via the translocase of the outer mitochondrial membrane (TOM) complex (Hansson Petersen et al., 2008). Extracellular Aβ was also shown to be internalized and taken up by mitochondria (Hansson Petersen et al., 2008; Hedskog et al., 2013). One recent study reported that reduction in mitochondrial membrane potential and the emergence of dystrophic and fragmented mitochondria were limited to the vicinity of Aβ plaques in a live AD mouse model, suggesting that Aβ plaques likely serve as focal sources that promote mitochondrial Aβ accumulation and thus Aβ-mediated toxicity (Xie et al., 2013). Altogether, these pieces of evidences indicate that the accumulation of APP and Aβ in the mitochondrial compartment likely has a causative role in altering mitochondrial dynamics and transport, thereby leading to mitochondrial dysfunction.
Abnormal Mitochondrial Dynamics in Alzheimer’s Disease
Mitochondrial fragmentation and reduced mitochondrial density in neuronal processes have been consistently observed in neurons exposed to Aβ-derived diffusible ligands (ADDLs) or oligomeric Aβ, as well as in primary neurons cultured from AβPP mice (Wang et al., 2009a; Du et al., 2010; Calkins et al., 2011). One recent study provided in vivo evidence of the emergence of dystrophic and fragmented mitochondria and a reduction in the total number of mitochondria near amyloid plaques in living AD mouse brains (Xie et al., 2013). In another study, neuronal cells incubated with conditional medium from cells stably expressing mutant forms of APP showed an increase in mitochondrial fission, caused by elevated levels of S-nitrosylated Drp1 (SNO-Drp1; Cho et al., 2009). Enhanced dimerization of SNO-Drp1, which was found in AD patient brains and mouse models, likely contributes to increased fission activity. These observations suggest that Aβ-mediated cytotoxic effects lead to enhanced Drp1 activity through the generation of nitric oxide. One study found the contrary result of reduced levels of Drp1 in fibroblasts from sporadic AD patients and AD patient brains (Wang et al., 2008a, 2009a). The same group provided further evidence that overexpression of APP in M17 neuroblastoma cells results in predominant mitochondrial fragmentation and decreased levels of Drp1 and OPA1, while overexpression of Drp1 or OPA1 could partially rescue some of these defects (Wang et al., 2008b). However, more recent studies found increased levels of Drp1 and Fis1 and reduced expression of Mfn1, Mfn2 and OPA1 in AD patient brains. Moreover, increased Aβ production and the interactions of Drp1 with Aβ and phosphorylated tau lead to abnormal mitochondrial fragmentation. These abnormal interactions are increased as AD progresses (Manczak et al., 2011; Reddy et al., 2011; Manczak and Reddy, 2012a; Kandimalla and Hemachandra Reddy, 2015). Therefore, impaired balance in mitochondrial fusion and fission in AD neurons likely interferes with mitochondrial motility and mitophagy, thereby compromising mitochondrial quality control.
Alterations in Mitochondrial Transport in Alzheimer’S Disease
Several lines of evidence support the hypothesis that impaired axonal transport plays an important role in the pathogenesis of AD (Stokin and Goldstein, 2006; Wang et al., 2009b). Axonal degeneration in patients with AD is characterized by swollen regions where abnormal amounts of organelles (including mitochondria) accumulate (Stokin et al., 2005). It was reported that presenilin 1 (PS1) mutations impair kineisin-1 based axonal transport by increased GSK3β activity and thereby enhancing the phosphorylation of kinesin-1 light chains (KLC). This defect leads to a reduction in the densities of APP, synaptic vesicles and mitochondria in the neuronal processes of hippocampal neurons and sciatic nerves from mutant PS1 knock-in mice (Pigino et al., 2003). Consistent with this finding, a study utilizing real-time analysis of vesicle motility demonstrated that isolated axoplasms perfused with soluble intracellular oligomeric Aβ exhibited inhibition of bidirectional axonal transport as a result of increased phosphorylation of KLC and subsequent release of kinesin from its cargoes (Pigino et al., 2009). These observations suggest that defects in axonal transport may compromise neuronal function by interfering with both trafficking and distribution densities of important cargoes, including mitochondria, in distal axons and at nerve terminals.
Exposure of cultured neurons to Aβ or ADDLs consistently results in decreased mitochondrial motility and reduced mitochondrial density in axons (Rui et al., 2006; Du et al., 2010; Vossel et al., 2010; Wang et al., 2010). One study showed than overexpression of Aβ in Drosophila slowed down bidirectional transport of axonal mitochondria and depleted presynaptic mitochondria, leading to presynaptic dysfunction (Zhao et al., 2010). Primary neurons derived from AβPP Tg mice also exhibited impaired anterograde transport of axonal mitochondria, mitochondrial dysfunction, and synaptic deficiency, which could be attributed to the accumulation of oligomeric Aβ in mitochondria (Calkins et al., 2011). Mitophagy induction is associated with alterations in mitochondrial motility. As a result of Parkin-mediated degradation of Miro, mitophagy is accompanied by reduced anterograde mitochondrial transport (Chan et al., 2011; Wang et al., 2011b; Cai et al., 2012a,b; Liu et al., 2012b; Bingol et al., 2014; Birsa et al., 2014). Our recent study showed that Parkin-mediated mitophagy is robustly induced in AD neurons of mouse models and patient brains. Consequently, these neurons display reduced anterograde transport of axonal mitochondria (Ye et al., 2015). Application of a mitochondria-targeted antioxidant was shown to reverse these defects and restore mitochondrial motility in AD neurons (Calkins et al., 2011), likely by attenuating mitophagy and Miro degradation. These studies show that abnormal mitochondrial motility in AD neurons may be attributed to the induction of mitophagy. Together, defective mitochondrial transport and reduced mitochondrial density in distal axons and at synaptic terminals may cause local energy depletion and toxic changes in Ca2+ buffering, thus triggering synaptic dysfunction and loss in the pathogenesis of AD.
Abnormal Autophagy and Mitophagy in Alzheimer’S Disease
Mounting evidence has implicated defective autophagy in the pathogenesis of AD (Nixon, 2007, 2013; Nixon and Yang, 2011). This is evidenced in AD brains by the massive accumulation of AVs in the soma and within large swellings along dystrophic and degenerating neurites. These serve as major reservoirs of intracellular Aβ and create conditions favorable for Aβ accumulation (Nixon et al., 2005; Yu et al., 2005; Nixon, 2007). However, it is largely unknown what cellular basis contributes to altered autophagy in affected AD neurons. Previous studies reported prominent autophagic accumulation of mitochondria in AD patient brains, suggesting increased mitochondrial turnover by autophagy (Hirai et al., 2001; Moreira et al., 2007a,b). These findings suggest that altered autophagy or mitophagy contributes to mitochondrial defects in AD brains. Despite enhanced autophagy induction, AD brains still exhibit aberrant accumulation of ultrastructurally altered mitochondria with reduced size and broken internal membrane cristae (Hirai et al., 2001; Baloyannis, 2006; Moreira et al., 2007a,b; Trushina et al., 2012).
Our recent study showed that enhanced mitophagy induction contributes to increased autophagic flux in AD (Ye et al., 2015). In both mutant hAPP neurons and AD patient brains, robust induction of Parkin-mediated mitophagy was observed. In the absence of mitochondrial membrane potential dissipation reagents, hAPP neurons exhibit increased recruitment of cytosolic Parkin to depolarized mitochondria. Under AD-linked pathophysiological conditions, Parkin translocation predominantly occurs in the somatodendritic regions, coupled with reduced anterograde and increased retrograde transport of axonal mitochondria. As the disease progresses, this enhancement of mitophagy in AD patient brains is accompanied by a depletion of cytosolic Parkin. Consistent with this, overexpression of Parkin in an AD mouse model was shown to enhance autophagic clearance of defective mitochondria and prevent mitochondrial dysfunction (Khandelwal et al., 2011; Martín-Maestro et al., 2015). Thus, these data indicate that aberrant accumulation of defective mitochondria in AD-affected neurons is likely caused by inadequate mitophagy capabilities for eliminating the increased numbers of damaged mitochondria.
Deficits in lysosomal degradation capacity have been implicated in AD as well, as it leads to the accumulation of proteolytic substrates in affected neurons (Boland et al., 2008; Lee et al., 2010, 2011; Yang et al., 2011). One recent study provided evidence showing that endolysosomal deficits augment mitochondrial pathology in the motor neurons of an ALS mouse model (Xie et al., 2015). Defective lysosomal proteolysis likely impairs turnover of mitophagosomes, thereby contributing to their aberrant accumulation in AD brains. While enhanced mitophagy induction contributes to increased autophagic flux, autophagy failure as a result of lysosomal deficits leads to inadequate mitophagic clearance and therefore mitochondrial pathology observed in AD.
Abnormal Mitochondrial Quality Control in Alzheimer’S Disease
Mitochondrial quality control, essential for limiting mitochondrial damage and maintaining mitochondrial integrity and function, occurs at the molecular, organellar, and cellular levels. Defects in each level interfere with the efficiency of quality control, thus leading to mitochondrial dysfunction or abnormal accumulation of damaged mitochondria. AD neurons have been shown to exhibit mitochondrial abnormalities including an imbalance in mitochondrial dynamics, impaired axonal transport, and inadequate mitophagy capacity, all of which may disrupt efficient elimination of dysfunctional mitochondria. Thus, mitochondrial pathology in AD can likely be attributed to impaired quality control (Figure 6).
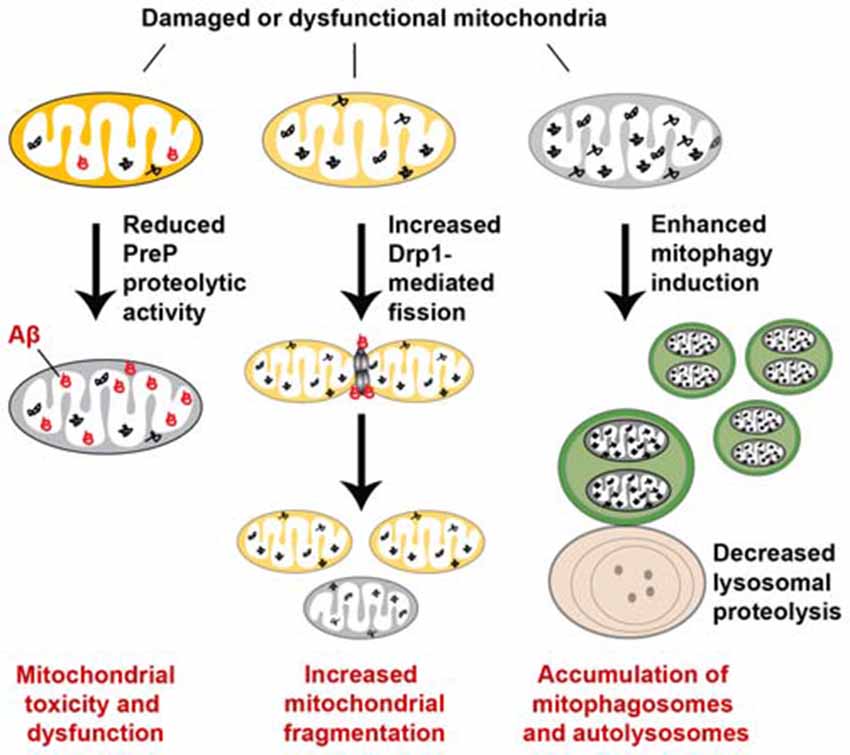
Figure 6. Abnormal mitochondrial quality control in Alzheimer’s disease (AD). Mitochondrial quality control is impaired at multiple levels in AD. At the molecular level, the mitochondrial presequence protease (PreP) degrades Aβ within mitochondria and thus reduces its toxic effects on mitochondria. However, the proteolytic activity of PreP is impaired as a result of increased reactive oxygen species (ROS) production in AD, thereby promoting accumulation of Aβ and Aβ-mediated mitochondrial toxicity. A decrease in proteasome activity could contribute to altered quality control of other presequence peptides and mitochondrial proteins. Mitochondrial quality control at the organellar level is also disrupted in AD. Elevated Drp1 levels or activity and reduced levels of Mfn1 and Mfn2 result in mitochondrial fragmentation and reduced fusion, thus preventing damaged mitochondria from being repaired through the fusion-mediated mixture of contents with healthy mitochondria. Enhanced mitophagy induction and defective lysosomal proteolysis result in aberrant accumulation of damaged mitochondria within mitophagosomes and autolysosomes, which also contribute to impaired mitochondrial quality control in AD. Moreover, defects in axonal transport and mitochondrial motility compromise mitochondrial quality control by hindering dysfunctional mitochondria from being returned to the soma for lysosomal degradation.
Mitochondrial quality control at the molecular level is altered in AD. The mitochondrial presequence protease (PreP) was shown to degrade Aβ within mitochondria and thus reduce its toxic effects on mitochondrial function (Falkevall et al., 2006; Alikhani et al., 2011). However, in AD patient brains and mouse models, studies showed that increased ROS production impairs the proteolytic activity of PreP, thereby promoting accumulation of Aβ and Aβ-mediated mitochondrial toxicity. A decrease in proteasome activity could also compromise the quality control of other presequence peptides and mitochondrial proteins (Gregori et al., 1995; Keller et al., 2000; Tseng et al., 2008). As a result, the potential toxic effect caused by accumulation of damaged presequence peptides and mitochondrial proteins could further exacerbate mitochondrial dysfunction in AD (Teixeira and Glaser, 2013).
Mitochondrial quality control at the organellar level—involving mitochondrial dynamics, motility, and mitophagy—is also disrupted in AD. The important process of mitochondrial fusion typically allows damaged mitochondria to be repaired through fusion with healthy mitochondria, while mitochondrial fission segregates severely and irreversibly damaged mitochondria for their clearance via the autophagy-lysosomal pathway. AD brains exhibit an imbalance in mitochondrial fusion and fission, showing mitochondrial fragmentation and reduced fusion (Cho et al., 2009; Du et al., 2010; Calkins et al., 2011; Manczak et al., 2011; Manczak and Reddy, 2012a). Mitochondrial fragmentation is likely attributed to elevated Drp1 levels or activity and reduced levels of Mfn1 and Mfn2. Its enhanced occurrence promotes mitochondrial elimination via mitophagy, causing this process to become dominant. Meanwhile, suppressed fusion may prevent damaged mitochondria from being repaired through the fusion-mediated mixture of contents with healthy mitochondria. Thus, altered mitochondrial dynamics in AD neurons will interfere with mitochondrial quality control.
Retrograde transport of axonal mitochondria facilitates removal of aged and defective mitochondria from distal axons and nerve terminals, since mature lysosomes are mainly located in the soma (Overly and Hollenbeck, 1996; Cai et al., 2010, 2012a,b; Lee et al., 2011; Ye et al., 2015). Decreased mitochondrial motility has been observed in neurons exposed to Aβ or ADDLs, or in neurons cultured from AβPP Tg mice, and in Drosophila following Aβ overexpression (Rui et al., 2006; Du et al., 2010; Vossel et al., 2010; Wang et al., 2010; Zhao et al., 2010; Calkins et al., 2011). Such defects in axonal transport and mitochondrial motility compromise mitochondrial quality control by hindering dysfunctional mitochondria from being returned to the soma for lysosomal degradation. As a result, defective mitochondria are abnormally accumulated in the distal regions of AD neurons.
The key pathway of mitochondrial quality control, mitophagy, is also altered in AD. Among the various types of mitophagy, the PINK1/Parkin-mediated mitophagy in AD has been the focus of most current studies. Earlier studies revealed autophagic accumulation of mitochondria in AD patient brains (Hirai et al., 2001; Moreira et al., 2007a,b), suggesting enhanced turnover of damaged mitochondria via the autophagy-lysosomal pathway. We further showed that Parkin-mediated mitophagy, is robustly induced in AD patient brains and mouse models (Ye et al., 2015). However, AD brains exhibit depletion of cytosolic Parkin throughout the disease’s progression, which is consistent with a recent study’s findings that Parkin is diminished in the fibroblasts and brain biopsies of AD patients, leading to PINK1 accumulation (Martín-Maestro et al., 2015). Overexpression of Parkin in these cells rescues mitophagy failure by inducing recovery of mitochondrial membrane potential, as well as reducing PINK1 levels and the accumulation of abnormal mitochondria. These observations indicate that defective mitophagy in AD causes the aberrant accumulation of dysfunctional mitochondria. In addition, degradation of mitophagosomes relies on proper lysosomal function. Deficits in lysosomal proteolysis of autophagic substrates may further compromise mitophagic elimination of defective mitochondria, thus exacerbating AD pathology. Given that endolysosomal deficits were shown to augment mitochondrial pathology in ALS (Xie et al., 2015), it is possible that similar deficits may contribute to mitochondrial dysfunction in AD. Moreover, altered mitochondrial dynamics and reduced mitochondrial motility may interfere with mitophagy in AD, although no studies have yet confirmed this relationship. Therefore, the molecular interplay between abnormal mitochondrial fission, fusion, transport, and defective mitophagy must be further investigated to advance our understanding of mitochondrial quality control in AD. This represents an important research field, as a variety of neurodegenerative diseases are associated with mitochondrial dysfunction.
Altogether, alterations in mitochondrial fusion and fission, transport, mitophagy, and lysosomal proteolysis lead to disrupted mitochondrial quality control, thus augmenting mitochondrial pathology in AD brains.
Tau-Mediated Mitochondrial Defects
Neurofibrillary tangle formation, composed mainly of hypophosphorylated tau, is also a pathological hallmark of AD. There is little information regarding tau-mediated regulation of mitochondria, and it remains largely unknown whether tauopathy contributes to mitochondrial dysfunction in AD. It has been shown that expression of human tau results in elongation of mitochondria in both Drosophila and mouse neurons by blocking recruitment of Drp1 to mitochondria, which is accompanied by mitochondrial dysfunction and cell death (DuBoff et al., 2012). As these phenotypes can be rescued by genetically restoring the proper balance of mitochondrial fusion and fission, it suggests that tau plays a role in regulating mitochondrial dynamics.
As for disease-related pathogenic tau mutants, overexpression of mutant tau—causing frontotemporal dementia with Parkinsonism linked to chromosome 17 (FTDP-17)—decreased fusion and fission rates due to reduced levels of OPA-1 and Drp-1. In contrast, overexpressing wild-type tau was shown to have protective effects on both mitochondrial dynamics and function, including the enhancement of complex I activity (Schulz et al., 2012). One study reported that hypophosphorylated tau induced mitochondrial fission and excessive mitochondrial fragmentation in postmortem brain tissues from patients with AD and mouse models by directly interacting with Drp1 (Manczak and Reddy, 2012a). In addition to its effects on fission, phosphorylated tau was shown to interact with VDAC in AD brains, leading to mitochondrial dysfunction likely by blocking mitochondrial pores (Manczak and Reddy, 2012b). These studies indicate that pathogenic forms of tau affect mitochondrial function directly through interaction with VDAC or indirectly through interference with Drp1-mediated fission.
Tau is a microtubule-associated protein (MAP) responsible for stabilizing axonal MTs. It has also been shown to regulate axonal transport of membranous organelles, including mitochondria (Stamer et al., 2002). Overexpressing tau selectively inhibits kinesin-driven anterograde mitochondrial transport in N2a and NB2a/d1 neuroblastoma cell lines, primary cortical neurons, and retinal ganglion neurons (Stamer et al., 2002; Dubey et al., 2008; Stoothoff et al., 2009). These studies suggest that tau preferentially competes with kinesin motors for binding to MTs. One study showed that the binding of tau to MTs can reverse the direction in which the dynein motor moves, whereas the same process tended to cause kinesin to detach from the MTs (Dixit et al., 2008). Interestingly, complete or partial loss of tau expression in mutant neurons prevents Aβ-mediated defects in axonal transport of mitochondria (Vossel et al., 2010). This suggests that the ability of Aβ to inhibit mitochondrial motility is dependent on tau expression levels. Thus, perturbing tau expression and distribution in axons would impair axonal transport and lead to neurodegeneration.
While mutant tau expression results in mitochondrial dysfunction, deregulation of mitochondrial dynamics, and impaired mitochondrial transport, one recent study reported that pathogenic tau truncation might also contribute to abnormal mitophagy in AD. A 20–22 kDa NH2-tau fragment detectable in cellular and animal AD models and human AD subjects was shown to be stably associated with Parkin and UCHL-1. As a result, it led to aberrant recruitment of Parkin and UCHL-1 to mitochondria and excessive Parkin-dependent mitochondrial turnover (Corsetti et al., 2015). Suppression of this improper mitophagy restores mitochondrial content at synapses and results in partial, but significant, protection against the NH2-htau-induced neuronal death. Altogether, the pathogenic forms of tau-mediated mitochondrial alterations—including impaired balance of mitochondrial fusion and fission, reduced mitochondrial motility, and excessive mitophagy—ultimately interfere with mitochondrial quality control, thus contributing to mitochondrial dysfunction in AD.
Concluding Remarks
Mitochondrial dysfunction is a prominent feature in AD, but it remains unclear whether the cellular mechanisms maintaining mitochondrial quality are defective and further augments mitochondrial pathology. Mitochondrial quality control involves multiple levels of surveillance and protective strategies to limit mitochondrial damage and efficiently eliminate defective mitochondria for the maintenance of mitochondrial homeostasis. Mitochondrial fusion and fission control mitochondrial shape and function, while mitochondrial transport plays a critical role in regulating mitochondrial distribution and removing aged and damaged mitochondrial from distal axons and synapses for lysosomal degradation in the soma. These key features of mitochondria operate strongly in conjunction with mitophagy as the key pathways of mitochondrial quality control. Studies reviewed here suggest that a complex network of dynamic and reciprocal interactions among mitochondrial fusion, fission, transport, and mitophagy governs mitochondrial integrity and function. Accumulating evidence notes that perturbed mitochondrial dynamics and abnormal mitophagy exist in AD brains (Chen and Chan, 2009; Sheng and Cai, 2012), which may directly or indirectly interfere with the quality control of mitochondria. Further investigation into these mechanisms would advance our understanding of mitochondrial dysfunction in AD. Given that manipulation of genes controlling mitophagy can ameliorate some phenotypes (Khandelwal et al., 2011; Martín-Maestro et al., 2015), there is compelling reason to hope that efforts to artificially manipulate mitochondrial dynamics, motility, and mitophagy will enhance mitochondrial surveillance mechanisms and attenuate the neuropathology of AD, ultimately yielding new therapeutic approaches for the devastating disease.
Author Contributions
QC conceived ideas and wrote the manuscript. PT contributed to some sections related with AD.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank J. Lam, J. Sheu, and D. Aikal for editing and other members of the Cai laboratory for their assistance and discussion. This work was supported by the National Institutes of Health [R00AG033658 and R01NS089737 to QC]; the Alzheimer’s Association [NIRG-14-321833 to QC]; and the Charles and Johanna Busch Biomedical Award [to QC].
References
Abramov, A. Y., Canevari, L., and Duchen, M. R. (2004). β-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J. Neurosci. 24, 565–575. doi: 10.1523/jneurosci.4042-03.2004
Alavi, M. V., Bette, S., Schimpf, S., Schuettauf, F., Schraermeyer, U., Wehrl, H. F., et al. (2007). A splice site mutation in the murine Opa1 gene features pathology of autosomal dominant optic atrophy. Brain 130, 1029–1042. doi: 10.1093/brain/awm005
Alexander, C., Votruba, M., Pesch, U. E., Thiselton, D. L., Mayer, S., Moore, A., et al. (2000). OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 26, 211–215. doi: 10.1038/79944
Alikhani, N., Guo, L., Yan, S., Du, H., Pinho, C. M., Chen, J. X., et al. (2011). Decreased proteolytic activity of the mitochondrial amyloid-β degrading enzyme, PreP peptidasome, in Alzheimer’s disease brain mitochondria. J. Alzheimers Dis. 27, 75–87. doi: 10.3233/JAD-2011-101716
Anand, R., Wai, T., Baker, M. J., Kladt, N., Schauss, A. C., Rugarli, E., et al. (2014). The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J. Cell Biol. 204, 919–929. doi: 10.1083/jcb.201308006
Anandatheerthavarada, H. K., Biswas, G., Robin, M. A., and Avadhani, N. G. (2003). Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells. J. Cell Biol. 161, 41–54. doi: 10.1083/jcb.200207030
Ashrafi, G., Schlehe, J. S., LaVoie, M. J., and Schwarz, T. L. (2014). Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J. Cell Biol. 206, 655–670. doi: 10.1083/jcb.201401070
Baloyannis, S. J. (2006). Mitochondrial alterations in Alzheimer’s disease. J. Alzheimers Dis. 9, 119–126.
Billups, B., and Forsythe, I. D. (2002). Presynaptic mitochondrial calcium sequestration influences transmission at mammalian central synapses. J. Neurosci. 22, 5840–5847.
Bingol, B., Tea, J. S., Phu, L., Reichelt, M., Bakalarski, C. E., Song, Q., et al. (2014). The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 510, 370–375. doi: 10.1038/nature13418
Birsa, N., Norkett, R., Wauer, T., Mevissen, T. E., Wu, H. C., Foltynie, T., et al. (2014). Lysine 27 ubiquitination of the mitochondrial transport protein Miro is dependent on serine 65 of the Parkin ubiquitin ligase. J. Biol. Chem. 289, 14569–14582. doi: 10.1074/jbc.m114.563031
Bogan, N., and Cabot, J. B. (1991). Light and electron microscopic analyses of intraspinal axon collaterals of sympathetic preganglionic neurons. Brain Res. 541, 241–251. doi: 10.1016/0006-8993(91)91024-u
Boland, B., Kumar, A., Lee, S., Platt, F. M., Wegiel, J., Yu, W. H., et al. (2008). Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J. Neurosci. 28, 6926–6937. doi: 10.1523/JNEUROSCI.0800-08.2008
Brickley, K., and Stephenson, F. A. (2011). Trafficking kinesin protein (TRAK)-mediated transport of mitochondria in axons of hippocampal neurons. J. Biol. Chem. 286, 18079–18092. doi: 10.1074/jbc.m111.236018
Cai, Q., Gerwin, C., and Sheng, Z. H. (2005). Syntabulin-mediated anterograde transport of mitochondria along neuronal processes. J. Cell Biol. 170, 959–969. doi: 10.1083/jcb.200506042
Cai, Q., Lu, L., Tian, J. H., Zhu, Y. B., Qiao, H., and Sheng, Z. H. (2010). Snapin-regulated late endosomal transport is critical for efficient autophagy-lysosomal function in neurons. Neuron 68, 73–86. doi: 10.1016/j.neuron.2010.09.022
Cai, Q., Zakaria, H. M., and Sheng, Z. H. (2012a). Long time-lapse imaging reveals unique features of PARK2/Parkin-mediated mitophagy in mature cortical neurons. Autophagy 8, 976–978. doi: 10.4161/auto.20218
Cai, Q., Zakaria, H. M., Simone, A., and Sheng, Z. H. (2012b). Spatial parkin translocation and degradation of damaged mitochondria via mitophagy in live cortical neurons. Curr. Biol. 22, 545–552. doi: 10.1016/j.cub.2012.02.005
Calkins, M. J., Manczak, M., Mao, P., Shirendeb, U., and Reddy, P. H. (2011). Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 20, 4515–4529. doi: 10.1093/hmg/ddr381
Caspersen, C., Wang, N., Yao, J., Sosunov, A., Chen, X., Lustbader, J. W., et al. (2005). Mitochondrial Aβ: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 19, 2040–2041. doi: 10.1096/fj.05-3735fje
Chan, N. C., Salazar, A. M., Pham, A. H., Sweredoski, M. J., Kolawa, N. J., Graham, R. L., et al. (2011). Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum. Mol. Genet. 20, 1726–1737. doi: 10.1093/hmg/ddr048
Chang, C. R., and Blackstone, C. (2007). Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J. Biol. Chem. 282, 21583–21587. doi: 10.1074/jbc.c700083200
Chapman, A. L., Bennett, E. J., Ramesh, T. M., De Vos, K. J., and Grierson, A. J. (2013). Axonal transport defects in a mitofusin 2 loss of function model of charcot-marie-tooth disease in zebrafish. PLoS One 8:e67276. doi: 10.1371/journal.pone.0067276
Chen, H., and Chan, D. C. (2009). Mitochondrial dynamics-fusion, fission, movement and mitophagy-in neurodegenerative diseases. Hum. Mol. Genet. 18, R169–R176. doi: 10.1093/hmg/ddp326
Chen, H., Detmer, S. A., Ewald, A. J., Griffin, E. E., Fraser, S. E., and Chan, D. C. (2003). Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 160, 189–200. doi: 10.1083/jcb.200211046
Chen, Y., and Dorn, G. W. (2013). PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 340, 471–475. doi: 10.1126/science.1231031
Chen, Y. M., Gerwin, C., and Sheng, Z. H. (2009). Dynein light chain LC8 regulates syntaphilin-mediated mitochondrial docking in axons. J. Neurosci. 29, 9429–9438. doi: 10.1523/jneurosci.1472-09.2009
Chen, G., Han, Z., Feng, D., Chen, Y., Chen, L., Wu, H., et al. (2014). A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol. Cell 54, 362–377. doi: 10.1016/j.molcel.2014.02.034
Chen, H., McCaffery, J. M., and Chan, D. C. (2007). Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 130, 548–562. doi: 10.1016/j.cell.2007.06.026
Chen, Y., and Sheng, Z. H. (2013). Kinesin-1-syntaphilin coupling mediates activity-dependent regulation of axonal mitochondrial transport. J. Cell Biol. 202, 351–364. doi: 10.1083/jcb.201302040
Cheng, X. T., Zhou, B., Lin, M. Y., Cai, Q., and Sheng, Z. H. (2015). Axonal autophagosomes recruit dynein for retrograde transport through fusion with late endosomes. J. Cell Biol. 209, 377–386. doi: 10.1083/jcb.201412046
Cho, K. I., Cai, Y., Yi, H., Yeh, A., Aslanukov, A., and Ferreira, P. A. (2007). Association of the kinesin-binding domain of RanBP2 to KIF5B and KIF5C determines mitochondria localization and function. Traffic 8, 1722–1735. doi: 10.1111/j.1600-0854.2007.00647.x
Cho, D. H., Nakamura, T., Fang, J., Cieplak, P., Godzik, A., Gu, Z., et al. (2009). S-nitrosylation of Drp1 mediates β-amyloid-related mitochondrial fission and neuronal injury. Science 324, 102–105. doi: 10.1126/science.1171091
Chu, C. T., Ji, J., Dagda, R. K., Jiang, J. F., Tyurina, Y. Y., Kapralov, A. A., et al. (2013). Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol. 15, 1197–1205. doi: 10.1038/ncb2837
Clark, I. E., Dodson, M. W., Jiang, C., Cao, J. H., Huh, J. R., Seol, J. H., et al. (2006). Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441, 1162–1166. doi: 10.1038/nature04779
Corsetti, V., Florenzano, F., Atlante, A., Bobba, A., Ciotti, M. T., Natale, F., et al. (2015). NH2-truncated human tau induces deregulated mitophagy in neurons by aberrant recruitment of Parkin and UCHL-1: implications in Alzheimer’s disease. Hum. Mol. Genet. 24, 3058–3081. doi: 10.1093/hmg/ddv059
Court, F. A., and Coleman, M. P. (2012). Mitochondria as a central sensor for axonal degenerative stimuli. Trends Neurosci. 35, 364–372. doi: 10.1016/j.tins.2012.04.001
Cribbs, J. T., and Strack, S. (2007). Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 8, 939–944. doi: 10.1038/sj.embor.7401062
Crouch, P. J., Blake, R., Duce, J. A., Ciccotosto, G. D., Li, Q. X., Barnham, K. J., et al. (2005). Copper-dependent inhibition of human cytochrome coxidase by a dimeric conformer of amyloid-β1–42. J. Neurosci. 25, 672–679. doi: 10.1523/jneurosci.4276-04.2005
David, G., and Barrett, E. F. (2003). Mitochondrial Ca2+ uptake prevents desynchronization of quantal release and minimizes depletion during repetitive stimulation of mouse motor nerve terminals. J. Physiol. 548, 425–438. doi: 10.1113/jphysiol.2002.035196
Davies, V. J., Hollins, A. J., Piechota, M. J., Yip, W., Davies, J. R., White, K. E., et al. (2007). Opa1 deficiency in a mouse model of autosomal dominant optic atrophy impairs mitochondrial morphology, optic nerve structure and visual function. Hum. Mol. Genet. 16, 1307–1318. doi: 10.1093/hmg/ddm079
de Castro, I. P., Martins, L. M., and Tufi, R. (2010). Mitochondrial quality control and neurological disease: an emerging connection. Expert Rev. Mol. Med. 12:e12. doi: 10.1017/s1462399410001456
Delettre, C., Lenaers, G., Griffoin, J. M., Gigarel, N., Lorenzo, C., Belenguer, P., et al. (2000). Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 26, 207–210. doi: 10.1038/79936
Detmer, S. A., and Chan, D. C. (2007). Functions and dysfunctions of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol. 8, 870–879. doi: 10.1038/nrm2275
Detmer, S. A., Vande Velde, C., Cleveland, D. W., and Chan, D. C. (2008). Hindlimb gait defects due to motor axon loss and reduced distal muscles in a transgenic mouse model of Charcot-Marie-Tooth type 2A. Hum. Mol. Genet. 17, 367–375. doi: 10.1093/hmg/ddm314
Devi, L., Prabhu, B. M., Galati, D. F., Avadhani, N. G., and Anandatheerthavarada, H. K. (2006). Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J. Neurosci. 26, 9057–9068. doi: 10.1523/jneurosci.1469-06.2006
Devireddy, S., Liu, A., Lampe, T., and Hollenbeck, P. J. (2015). The organization of mitochondrial quality control and life cycle in the nervous system in vivo in the absence of PINK1. J. Neurosci. 35, 9391–9401. doi: 10.1523/jneurosci.1198-15.2015
Dixit, R., Ross, J. L., Goldman, Y. E., and Holzbaur, E. L. (2008). Differential regulation of dynein and kinesin motor proteins by tau. Science 319, 1086–1089. doi: 10.1126/science.1152993
Dragicevic, N., Mamcarz, M., Zhu, Y., Buzzeo, R., Tan, J., Arendash, G. W., et al. (2010). Mitochondrial amyloid-β levels are associated with the extent of mitochondrial dysfunction in different brain regions and the degree of cognitive impairment in Alzheimer’s transgenic mice. J. Alzheimers Dis. 20, S535–S550. doi: 10.3233/JAD-2010-100342
Du, H., Guo, L., Fang, F., Chen, D., Sosunov, A. A., McKhann, G. M., et al. (2008). Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat. Med. 14, 1097–1105. doi: 10.1038/nm.1868
Du, H., Guo, L., and Yan, S. S. (2012). Synaptic mitochondrial pathology in Alzheimer’s disease. Antioxid. Redox Signal. 16, 1467–1475. doi: 10.1089/ars.2011.4277
Du, H., Guo, L., Yan, S., Sosunov, A. A., McKhann, G. M., and Yan, S. S. (2010). Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc. Natl. Acad. Sci. U S A 107, 18670–18675. doi: 10.1073/pnas.1006586107
Dubey, M., Chaudhury, P., Kabiru, H., and Shea, T. B. (2008). Tau inhibits anterograde axonal transport and perturbs stability in growing axonal neurites in part by displacing kinesin cargo: neurofilaments attenuate tau-mediated neurite instability. Cell Motil. Cytoskeleton 65, 89–99. doi: 10.1002/cm.20243
DuBoff, B., Götz, J., and Feany, M. B. (2012). Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron 75, 618–632. doi: 10.1016/j.neuron.2012.06.026
Falkevall, A., Alikhani, N., Bhushan, S., Pavlov, P. F., Busch, K., Johnson, K. A., et al. (2006). Degradation of the amyloid β-protein by the novel mitochondrial peptidasome, PreP. J. Biol. Chem. 281, 29096–29104. doi: 10.1074/jbc.m602532200
Fransson, S., Ruusala, A., and Aspenström, P. (2006). The atypical Rho GTPases Miro-1 and Miro-2 have essential roles in mitochondrial trafficking. Biochem. Biophys. Res. Commun. 344, 500–510. doi: 10.1016/j.bbrc.2006.03.163
Frederick, R. L., McCaffery, J. M., Cunningham, K. W., Okamoto, K., and Shaw, J. M. (2004). Yeast Miro GTPase, Gem1p, regulates mitochondrial morphology via a novel pathway. J. Cell Biol. 167, 87–98. doi: 10.1083/jcb.200405100
Fujita, T., Maturana, A. D., Ikuta, J., Hamada, J., Walchli, S., Suzuki, T., et al. (2007). Axonal guidance protein FEZ1 associates with tubulin and kinesin motor protein to transport mitochondria in neurites of NGF-stimulated PC12 cells. Biochem. Biophys. Res. Commun. 361, 605–610. doi: 10.1016/j.bbrc.2007.07.050
Gautier, C. A., Kitada, T., and Shen, J. (2008). Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc. Natl. Acad. Sci. U S A 105, 11364–11369. doi: 10.1073/pnas.0802076105
Gegg, M. E., Cooper, J. M., Chau, K. Y., Rojo, M., Schapira, A. H., and Taanman, J. W. (2010). Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum. Mol. Genet. 19, 4861–4870. doi: 10.1093/hmg/ddq419
Geisler, S., Holmström, K. M., Skujat, D., Fiesel, F. C., Rothfuss, O. C., Kahle, P. J., et al. (2010). PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 12, 119–131. doi: 10.1038/ncb2012
Gibson, G. E., and Shi, Q. (2010). A mitocentric view of Alzheimer’s disease suggests multi-faceted treatments. J. Alzheimers Dis. 20, S591–S607. doi: 10.3233/JAD-2010-100336
Glater, E. E., Megeath, L. J., Stowers, R. S., and Schwarz, T. L. (2006). Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J. Cell Biol. 173, 545–557. doi: 10.1083/jcb.200601067
Gomes, L. C., Di Benedetto, G., and Scorrano, L. (2011). During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 13, 589–598. doi: 10.1038/ncb2220
Górska-Andrzejak, J., Stowers, R. S., Borycz, J., Kostyleva, R., Schwarz, T. L., and Meinertzhagen, I. A. (2003). Mitochondria are redistributed in Drosophila photoreceptors lacking milton, a kinesin-associated protein. J. Comp. Neurol. 463, 372–388. doi: 10.1002/cne.10750
Gregori, L., Fuchs, C., Figueiredo-Pereira, M. E., Van Nostrand, W. E., and Goldgaber, D. (1995). Amyloid β-protein inhibits ubiquitin-dependent protein degradation in vitro. J. Biol. Chem. 270, 19702–19708. doi: 10.1074/jbc.270.34.19702
Gross, N. J., Getz, G. S., and Rabinowitz, M. (1969). Apparent turnover of mitochondrial deoxyribonucleic acid and mitochondrial phospholipids in the tissues of the rat. J. Biol. Chem. 244, 1552–1562.
Guo, X., Macleod, G. T., Wellington, A., Hu, F., Panchumarthi, S., Schoenfield, M., et al. (2005). The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron 47, 379–393. doi: 10.1016/j.neuron.2005.06.027
Hansson Petersen, C. A., Alikhani, N., Behbahani, H., Wiehager, B., Pavlov, P. F., Alafuzoff, I., et al. (2008). The amyloid β-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. U S A 105, 13145–13150. doi: 10.1073/pnas.0806192105
Harder, Z., Zunino, R., and McBride, H. (2004). Sumo1 conjugates mitochondrial substrates and participates in mitochondrial fission. Curr. Biol. 14, 340–345. doi: 10.1016/j.cub.2004.02.004
Hedskog, L., Pinho, C. M., Filadi, R., Rönnbäck, A., Hertwig, L., Wiehager, B., et al. (2013). Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’s disease and related models. Proc. Natl. Acad. Sci. U S A 110, 7916–7921. doi: 10.1073/pnas.1300677110
Hirai, K., Aliev, G., Nunomura, A., Fujioka, H., Russell, R. L., Atwood, C. S., et al. (2001). Mitochondrial abnormalities in Alzheimer’s disease. J. Neurosci. 21, 3017–3023.
Hirokawa, N., Niwa, S., and Tanaka, Y. (2010). Molecular motors in neurons: transport mechanisms and roles in brain function, development and disease. Neuron 68, 610–638. doi: 10.1016/j.neuron.2010.09.039
Hurd, D. D., and Saxton, W. M. (1996). Kinesin mutations cause motor neuron disease phenotypes by disrupting fast axonal transport in Drosophila. Genetics 144, 1075–1085.
Ishihara, N., Fujita, Y., Oka, T., and Mihara, K. (2006). Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 25, 2966–2977. doi: 10.1038/sj.emboj.7601184
Ishihara, N., Nomura, M., Jofuku, A., Kato, H., Suzuki, S. O., Masuda, K., et al. (2009). Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat. Cell Biol. 11, 958–966. doi: 10.1038/ncb1907
Kandimalla, R., and Hemachandra Reddy, P. (2015). Multiple faces of dynamin-related protein 1 and its role in Alzheimer’s disease pathogenesis. Biochim. Biophys. Acta doi: 10.1016/j.bbadis.2015.12.018 [Epub ahead of print].
Kang, J. S., Tian, J. H., Pan, P. Y., Zald, P., Li, C., Deng, C., et al. (2008). Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell 132, 137–148. doi: 10.1016/j.cell.2007.11.024
Karbowski, M., Neutzner, A., and Youle, R. J. (2007). The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. J. Cell Biol. 178, 71–84. doi: 10.1083/jcb.200611064
Keller, J. N., Hanni, K. B., and Markesbery, W. R. (2000). Impaired proteasome function in Alzheimer’s disease. J. Neurochem. 75, 436–439. doi: 10.1046/j.1471-4159.2000.0750436.x
Keller, J. N., Pang, Z., Geddes, J. W., Begley, J. G., Germeyer, A., Waeg, G., et al. (1997). Impairment of glucose and glutamate transport and induction of mitochondrial oxidative stress and dysfunction in synaptosomes by amyloid β-peptide: role of the lipid peroxidation product 4-hydroxynonenal. J. Neurochem. 69, 273–284. doi: 10.1046/j.1471-4159.1997.69010273.x
Khandelwal, P. J., Herman, A. M., Hoe, H. S., Rebeck, G. W., and Moussa, C. E. (2011). Parkin mediates beclin-dependent autophagic clearance of defective mitochondria and ubiquitinated Aβ in AD models. Hum. Mol. Genet. 20, 2091–2102. doi: 10.1093/hmg/ddr091
Koutsopoulos, O. S., Laine, D., Osellame, L., Chudakov, D. M., Parton, R. G., Frazier, A. E., et al. (2010). Human Miltons associate with mitochondria and induce microtubule-dependent remodeling of mitochondrial networks. Biochim. Biophys. Acta 1803, 564–574. doi: 10.1016/j.bbamcr.2010.03.006
Lee, S., Sato, Y., and Nixon, R. A. (2011). Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer’s-like axonal dystrophy. J. Neurosci. 31, 7817–7830. doi: 10.1523/JNEUROSCI.6412-10.2011
Lee, S., Sterky, F. H., Mourier, A., Terzioglu, M., Cullheim, S., Olson, L., et al. (2012). Mitofusin 2 is necessary for striatal axonal projections of midbrain dopamine neurons. Hum. Mol. Genet. 21, 4827–4835. doi: 10.1093/hmg/dds352
Lee, Y. J., Jeong, S. Y., Karbowski, M., Smith, C. L., and Youle, R. J. (2004). Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1 and Opa1 in apoptosis. Mol. Biol. Cell 15, 5001–5011. doi: 10.1091/mbc.e04-04-0294
Lee, J. H., Yu, W. H., Kumar, A., Lee, S., Mohan, P. S., Peterhoff, C. M., et al. (2010). Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 141, 1146–1158. doi: 10.1016/j.cell.2010.05.008
Levy, M., Faas, G. C., Saggau, P., Craigen, W. J., and Sweatt, J. D. (2003). Mitochondrial regulation of synaptic plasticity in the hippocampus. J. Biol. Chem. 278, 17727–17734. doi: 10.1074/jbc.m212878200
Li, Z., Okamoto, K., Hayashi, Y., and Sheng, M. (2004). The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 119, 873–887. doi: 10.1016/j.cell.2004.11.003
Liu, W., Acin-Peréz, R., Geghman, K. D., Manfredi, G., Lu, B., and Li, C. (2011). Pink1 regulates the oxidative phosphorylation machinery via mitochondrial fission. Proc. Natl. Acad. Sci. U S A 108, 12920–12924. doi: 10.1073/pnas.1107332108
Liu, L., Feng, D., Chen, G., Chen, M., Zheng, Q., Song, P., et al. (2012a). Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 14, 177–185. doi: 10.1038/ncb2422
Liu, S., Sawada, T., Lee, S., Yu, W., Silverio, G., Alapatt, P., et al. (2012b). Parkinson’s disease-associated kinase PINK1 regulates Miro protein level and axonal transport of mitochondria. PLoS Genet. 8:e1002537. doi: 10.1371/journal.pgen.1002537
Lustbader, J. W., Cirilli, M., Lin, C., Xu, H. W., Takuma, K., Wang, N., et al. (2004). ABAD directly links Aβ to mitochondrial toxicity in Alzheimer’s disease. Science 304, 448–452. doi: 10.1126/science.1091230
Lutz, A. K., Exner, N., Fett, M. E., Schlehe, J. S., Kloos, K., Lämmermann, K., et al. (2009). Loss of parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. J. Biol. Chem. 284, 22938–22951. doi: 10.1074/jbc.M109.035774
MacAskill, A. F., Brickley, K., Stephenson, F. A., and Kittler, J. T. (2009). GTPase dependent recruitment of Grif-1 by Miro1 regulates mitochondrial trafficking in hippocampal neurons. Mol. Cell. Neurosci. 40, 301–312. doi: 10.1016/j.mcn.2008.10.016
Maday, S., and Holzbaur, E. L. (2014). Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway. Dev. Cell 30, 71–85. doi: 10.1016/j.devcel.2014.06.001
Maday, S., Wallace, K. E., and Holzbaur, E. L. (2012). Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J. Cell Biol. 196, 407–417. doi: 10.1083/jcb.201106120
Manczak, M., Anekonda, T. S., Henson, E., Park, B. S., Quinn, J., and Reddy, P. H. (2006). Mitochondria are a direct site of Aβ accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 15, 1437–1449. doi: 10.1093/hmg/ddl066
Manczak, M., Calkins, M. J., and Reddy, P. H. (2011). Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: implications for neuronal damage. Hum. Mol. Genet. 20, 2495–2509. doi: 10.1093/hmg/ddr139
Manczak, M., and Reddy, P. H. (2012a). Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet. 21, 2538–2547. doi: 10.1093/hmg/dds072
Manczak, M., and Reddy, P. H. (2012b). Abnormal interaction of VDAC1 with amyloid beta and phosphorylated tau causes mitochondrial dysfunction in Alzheimer’s disease. Hum. Mol. Genet. 21, 5131–5146. doi: 10.1093/hmg/dds360
Martin, M., Iyadurai, S. J., Gassman, A., Gindhart, J. G., Hays, T. S., and Saxton, W. M. (1999). Cytoplasmic dynein, the dynactin complex and kinesin are interdependent and essential for fast axonal transport. Mol. Biol. Cell 10, 3717–3728. doi: 10.1091/mbc.10.11.3717
Martín-Maestro, P., Gargini, R., Perry, G., Avila, J., and García-Escudero, V. (2015). PARK2 enhancement is able to compensate mitophagy alterations found in sporadic Alzheimer’s disease. Hum. Mol. Genet. doi: 10.1093/hmg/ddv616 [Epub ahead of print].
Mattson, M. P., Partin, J., and Begley, J. G. (1998). Amyloid β-peptide induces apoptosis-related events in synapses and dendrites. Brain Res. 807, 167–176. doi: 10.1016/s0006-8993(98)00763-x
Meeusen, S., DeVay, R., Block, J., Cassidy-Stone, A., Wayson, S., McCaffery, J. M., et al. (2006). Mitochondrial inner-membrane fusion and crista maintenance requires the dynamin-related GTPase Mgm1. Cell 127, 383–395. doi: 10.1016/j.cell.2006.09.021
Mendl, N., Occhipinti, A., Müller, M., Wild, P., Dikic, I., and Reichert, A. S. (2011). Mitophagy in yeast is independent of mitochondrial fission and requires the stress response gene WHI2. J. Cell Sci. 124, 1339–1350. doi: 10.1242/jcs.076406
Menzies, R. A., and Gold, P. H. (1971). The turnover of mitochondria in a variety of tissues of young adult and aged rats. J. Biol. Chem. 246, 2425–2429.
Miller, K. E., and Sheetz, M. P. (2004). Axonal mitochondrial transport and potential are correlated. J. Cell Sci. 117, 2791–2804. doi: 10.1242/jcs.01130
Mishra, P., Carelli, V., Manfredi, G., and Chan, D. C. (2014). Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. 19, 630–641. doi: 10.1016/j.cmet.2014.03.011
Mishra, P., and Chan, D. C. (2014). Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 15, 634–646. doi: 10.1038/nrm3877
Misko, A., Jiang, S., Wegorzewska, I., Milbrandt, J., and Baloh, R. H. (2010). Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J. Neurosci. 30, 4232–4240. doi: 10.1523/JNEUROSCI.6248-09.2010
Misko, A. L., Sasaki, Y., Tuck, E., Milbrandt, J., and Baloh, R. H. (2012). Mitofusin2 mutations disrupt axonal mitochondrial positioning and promote axon degeneration. J. Neurosci. 32, 4145–4155. doi: 10.1523/JNEUROSCI.6338-11.2012
Moreira, P. I., Siedlak, S. L., Wang, X., Santos, M. S., Oliveira, C. R., Tabaton, M., et al. (2007a). Autophagocytosis of mitochondria is prominent in Alzheimer disease. J. Neuropathol. Exp. Neurol. 66, 525–532. doi: 10.1097/01.jnen.0000240476.73532.b0
Moreira, P. I., Siedlak, S. L., Wang, X., Santos, M. S., Oliveira, C. R., Tabaton, M., et al. (2007b). Increased autophagic degradation of mitochondria in Alzheimer’s disease. Autophagy 3, 614–615. doi: 10.4161/auto.4872
Nakamura, N., Kimura, Y., Tokuda, M., Honda, S., and Hirose, S. (2006). MARCH-V is a novel mitofusin 2- and Drp1-binding protein able to change mitochondrial morphology. EMBO Rep. 7, 1019–1022. doi: 10.1038/sj.embor.7400790
Narendra, D., Tanaka, A., Suen, D. F., and Youle, R. J. (2008). Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 183, 795–803, doi: 10.1083/jcb.200809125
Nicholls, D. G., and Budd, S. L. (2000). Mitochondria and neuronal survival. Physiol. Rev. 80, 315–360.
Nixon, R. A. (2007). Autophagy, amyloidogenesis and Alzheimer disease. J. Cell Sci. 120, 4081–4091. doi: 10.1242/jcs.019265
Nixon, R. A. (2013). The role of autophagy in neurodegenerative disease. Nat. Med. 19, 983–997. doi: 10.1038/nm.3232
Nixon, R. A., and Yang, D. S. (2011). Autophagy failure in Alzheimer’s disease-locating the primary defect. Neurobiol. Dis. 43, 38–45. doi: 10.1016/j.nbd.2011.01.021
Nixon, R. A., Wegiel, J., Kumar, A., Yu, W. H., Peterhoff, C., Cataldo, A., et al. (2005). Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 64, 113–122. doi: 10.1093/jnen/64.2.113
Novak, I., Kirkin, V., McEwan, D. G., Zhang, J., Wild, P., Rozenknop, A., et al. (2010). Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 11, 45–51. doi: 10.1038/embor.2009.256
Okamoto, K., and Shaw, J. M. (2005). Mitochondrial morphology and dynamics in yeast and multicellular eukaryotes. Annu. Rev. Genet. 39, 503–536. doi: 10.1146/annurev.genet.38.072902.093019
Overly, C. C., and Hollenbeck, P. J. (1996). Dynamic organization of endocytic pathways in axons of cultured sympathetic neurons. J. Neurosci. 16, 6056–6064.
Pham, A. H., Meng, S., Chu, Q. N., and Chan, D. C. (2012). Loss of Mfn2 results in progressive, retrograde degeneration of dopaminergic neurons in the nigrostriatal circuit. Hum. Mol. Genet. 21, 4817–4826. doi: 10.1093/hmg/dds311
Pigino, G., Morfini, G., Atagi, Y., Deshpande, A., Yu, C., Jungbauer, L., et al. (2009). Disruption of fast axonal transport is a pathogenic mechanism for intraneuronal amyloid beta. Proc. Natl. Acad. Sci. U S A 106, 5907–5912. doi: 10.1073/pnas.0901229106
Pigino, G., Morfini, G., Pelsman, A., Mattson, M. P., Brady, S. T., and Busciglio, J. (2003). Alzheimer’s presenilin 1 mutations impair kinesin-based axonal transport. J. Neurosci. 23, 4499–4508.
Pilling, A. D., Horiuchi, D., Lively, C. M., and Saxton, W. M. (2006). Kinesin-1 and Dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Mol. Biol. Cell 17, 2057–2068. doi: 10.1091/mbc.e05-06-0526
Pinho, C. M., Teixeira, P. F., and Glaser, E. (2014). Mitochondrial import and degradation of amyloid-β peptide. Biochim. Biophys. Acta 1837, 1069–1074. doi: 10.1016/j.bbabio.2014.02.007
Poole, A. C., Thomas, R. E., Yu, S., Vincow, E. S., and Pallanck, L. (2010). The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PLoS One 5:e10054. doi: 10.1371/journal.pone.0010054
Rambold, A. S., Kostelecky, B., Elia, N., and Lippincott-Schwartz, J. (2011). Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. U S A 108, 10190–10195. doi: 10.1073/pnas.1107402108
Rangaraju, V., Calloway, N., and Ryan, T. A. (2014). Activity-driven local ATP synthesis is required for synaptic function. Cell 156, 825–835. doi: 10.1016/j.cell.2013.12.042
Reddy, P. H. (2013). Amyloid beta-induced glycogen synthase kinase 3β phosphorylated VDAC1 in Alzheimer’s disease: implications for synaptic dysfunction and neuronal damage. Biochim. Biophys. Acta 1832, 1913–1921. doi: 10.1016/j.bbadis.2013.06.012
Reddy, P. H., Reddy, T. P., Manczak, M., Calkins, M. J., Shirendeb, U., and Mao, P. (2011). Dynamin-related protein 1 and mitochondrial fragmentation in neurodegenerative diseases. Brain Res. Rev. 67, 103–118. doi: 10.1016/j.brainresrev.2010.11.004
Roy, M., Reddy, P. H., Iijima, M., and Sesaki, H. (2015). Mitochondrial division and fusion in metabolism. Curr. Opin. Cell Biol. 33, 111–118. doi: 10.1016/j.ceb.2015.02.001
Rui, Y., Tiwari, P., Xie, Z., and Zheng, J. Q. (2006). Acute impairment of mitochondrial trafficking by β-amyloid peptides in hippocampal neurons. J. Neurosci. 26, 10480–10487. doi: 10.1523/JNEUROSCI.3231-06.2006
Russo, G. J., Louie, K., Wellington, A., Macleod, G. T., Hu, F., Panchumarthi, S., et al. (2009). Drosophila Miro is required for both anterograde and retrograde axonal mitochondrial transport. J. Neurosci. 29, 5443–5455. doi: 10.1523/JNEUROSCI.5417-08.2009
Sandoval, H., Thiagarajan, P., Dasgupta, S. K., Schumacher, A., Prchal, J. T., Chen, M., et al. (2008). Essential role for Nix in autophagic maturation of erythroid cells. Nature 454, 232–235. doi: 10.1038/nature07006
Saporta, M. A., Dang, V., Volfson, D., Zou, B., Xie, X. S., Adebola, A., et al. (2015). Axonal Charcot-Marie-Tooth disease patient-derived motor neurons demonstrate disease-specific phenotypes including abnormal electrophysiological properties. Exp. Neurol. 263, 190–199. doi: 10.1016/j.expneurol.2014.10.005
Sarraf, S. A., Raman, M., Guarani-Pereira, V., Sowa, M. E., Huttlin, E. L., Gygi, S. P., et al. (2013). Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 496, 372–376. doi: 10.1038/nature12043
Schulz, K. L., Eckert, A., Rhein, V., Mai, S., Haase, W., Reichert, A. S., et al. (2012). A new link to mitochondrial impairment in tauopathies. Mol. Neurobiol. 46, 205–216. doi: 10.1007/s12035-012-8308-3
Sheng, Z. H. (2014). Mitochondrial trafficking and anchoring in neurons: new insight and implications. J. Cell. Biol. 204, 1087–1098. doi: 10.1083/jcb.201312123
Sheng, Z. H., and Cai, Q. (2012). Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 13, 77–93. doi: 10.1038/nrn3156
Smith, M. J., Pozo, K., Brickley, K., and Stephenson, F. A. (2006). Mapping the GRIF-1 binding domain of the kinesin, KIF5C, substantiates a role for GRIF-1 as an adaptor protein in the anterograde trafficking of cargoes. J. Biol. Chem. 281, 27216–27228. doi: 10.1074/jbc.m600522200
Song, Z., Ghochani, M., McCaffery, J. M., Frey, T. G., and Chan, D. C. (2009). Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol. Biol. Cell 20, 3525–3532. doi: 10.1091/mbc.E09-03-0252
Stamer, K., Vogel, R., Thies, E., Mandelkow, E., and Mandelkow, E. M. (2002). Tau blocks traffic of organelles, neurofilaments and APP vesicles in neurons and enhances oxidative stress. J. Cell Biol. 156, 1051–1063. doi: 10.1083/jcb.200108057
Stokin, G. B., and Goldstein, L. S. (2006). Axonal transport and Alzheimer’s disease. Annu. Rev. Biochem. 75, 607–627. doi: 10.1146/annurev.biochem.75.103004.142637
Stokin, G. B., Lillo, C., Falzone, T. L., Brusch, R. G., Rockenstein, E., Mount, S. L., et al. (2005). Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science 307, 1282–1288. doi: 10.1126/science.1105681
Stoothoff, W., Jones, P. B., Spires-Jones, T. L., Joyner, D., Chhabra, E., Bercury, K., et al. (2009). Differential effect of three-repeat and four-repeat tau on mitochondrial axonal transport. J. Neurochem. 111, 417–427. doi: 10.1111/j.1471-4159.2009.06316.x
Stowers, R. S., Megeath, L. J., Górska-Andrzejak, J., Meinertzhagen, I. A., and Schwarz, T. L. (2002). Axonal transport of mitochondria to synapses depends on milton, a novel Drosophila protein. Neuron 36, 1063–1077. doi: 10.1016/s0896-6273(02)01094-2
Sun, T., Qiao, H., Pan, P. Y., Chen, Y., and Sheng, Z. H. (2013). Motile axonal mitochondria contribute to the variability of presynaptic strength. Cell Rep. 4, 413–419. doi: 10.1016/j.celrep.2013.06.040
Swerdlow, R. H., Burns, J. M., and Khan, S. M. (2010). The Alzheimer’s disease mitochondrial cascade hypothesis. J. Alzheimers Dis. 20, S265–S279. doi: 10.3233/JAD-2010-100339
Taguchi, N., Ishihara, N., Jofuku, A., Oka, T., and Mihara, K. (2007). Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 282, 11521–11529. doi: 10.1074/jbc.m607279200
Tanaka, A., Cleland, M. M., Xu, S., Narendra, D. P., Suen, D. F., Karbowski, M., et al. (2010). Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 191, 1367–1380. doi: 0.1083/jcb.201007013
Tanaka, Y., Kanai, Y., Okada, Y., Nonaka, S., Takeda, S., Harada, A., et al. (1998). Targeted disruption of mouse conventional kinesin heavy chain, kif5B, results in abnormal perinuclear clustering of mitochondria. Cell 93, 1147–1158. doi: 10.1016/s0092-8674(00)81459-2
Tang, Y., and Zucker, R. S. (1997). Mitochondrial involvement in post-tetanic potentiation of synaptic transmission. Neuron 18, 483–491. doi: 10.1016/s0896-6273(00)81248-9
Tatsuta, T., and Langer, T. (2008). Quality control of mitochondria: protection against neurodegeneration and ageing. EMBO J. 27, 306–314. doi: 10.1038/sj.emboj.7601972
Teixeira, P. F., and Glaser, E. (2013). Processing peptidases in mitochondria and chloroplasts. Biochim. Biophys. Acta 1833, 360–370. doi: 10.1016/j.bbamcr.2012.03.012
Trushina, E., Nemutlu, E., Zhang, S., Christensen, T., Camp, J., Mesa, J., et al. (2012). Defects in mitochondrial dynamics and metabolomic signatures of evolving energetic stress in mouse models of familial Alzheimer’s disease. PLoS One 7:e32737. doi: 10.1371/journal.pone.0032737
Tseng, B. P., Green, K. N., Chan, J. L., Blurton-Jones, M., and LaFerla, F. M. (2008). Aβ inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiol. Aging 29, 1607–1618. doi: 10.1016/j.neurobiolaging.2007.04.014
Twig, G., Elorza, A., Molina, A. J., Mohamed, H., Wikstrom, J. D., Walzer, G., et al. (2008). Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 27, 433–446. doi: 10.1038/sj.emboj.7601963
van Spronsen, M., Mikhaylova, M., Lipka, J., Schlager, M. A., van den Heuvel, D. J., Kuijpers, M., et al. (2013). TRAK/Milton motor-adaptor proteins steer mitochondrial trafficking to axons and dendrites. Neuron 77, 485–502. doi: 10.1016/j.neuron.2012.11.027
Verstreken, P., Ly, C. V., Venken, K. J., Koh, T. W., Zhou, Y., and Bellen, H. J. (2005). Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron 47, 365–378. doi: 10.1016/j.neuron.2005.06.018
Vincow, E. S., Merrihew, G., Thomas, R. E., Shulman, N. J., Beyer, R. P., MacCoss, M. J., et al. (2013). The PINK1-Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proc. Natl. Acad. Sci. U S A 110, 6400–6405. doi: 10.1073/pnas.1221132110
Vossel, K. A., Zhang, K., Brodbeck, J., Daub, A. C., Sharma, P., Finkbeiner, S., et al. (2010). Tau reduction prevents Aβ-induced defects in axonal transport. Science 330:198. doi: 10.1126/science.1194653
Wang, X., Perry, G., Smith, M. A., and Zhu, X. (2010). Amyloid-beta-derived diffusible ligands cause impaired axonal transport of mitochondria in neurons. Neurodegener. Dis. 7, 56–59. doi: 10.1159/000283484
Wang, H., Song, P., Du, L., Tian, W., Yue, W., Liu, M., et al. (2011a). Parkin ubiquitinates Drp1 for proteasome-dependent degradation: implication of dysregulated mitochondrial dynamics in Parkinson disease. J. Biol. Chem. 286, 11649–11658. doi: 10.1074/jbc.M110.144238
Wang, X., Winter, D., Ashrafi, G., Schlehe, J., Wong, Y. L., Selkoe, D., et al. (2011b). PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147, 893–906. doi: 10.1016/j.cell.2011.10.018
Wang, X., Su, B., Fujioka, H., and Zhu, X. (2008a). Dynamin-like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic Alzheimer’s disease patients. Am. J. Pathol. 173, 470–482. doi: 10.2353/ajpath.2008.071208
Wang, X., Su, B., Siedlak, S. L., Moreira, P. I., Fujioka, H., Wang, Y., et al. (2008b). Amyloid-β overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc. Natl. Acad. Sci. U S A 105, 19318–19323. doi: 10.1073/pnas.0804871105
Wang, X., Su, B., Lee, H. G., Li, X., Perry, G., Smith, M. A., et al. (2009a). Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 29, 9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009
Wang, X., Su, B., Zheng, L., Perry, G., Smith, M. A., and Zhu, X. (2009b). The role of abnormal mitochondrial dynamics in the pathogenesis of Alzheimer’s disease. J. Neurochem. 109(Suppl. 1), 153–159. doi: 10.1111/j.1471-4159.2009.05867.x
Weihofen, A., Thomas, K. J., Ostaszewski, B. L., Cookson, M. R., and Selkoe, D. J. (2009). Pink1 forms a multiprotein complex with Miro and Milton, linking Pink1 function to mitochondrial trafficking. Biochemistry 48, 2045–2052. doi: 10.1021/bi8019178
Westermann, B. (2010). Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 11, 872–884. doi: 10.1038/nrm3013
Xie, H., Guan, J., Borrelli, L. A., Xu, J., Serrano-Pozo, A., and Bacskai, B. J. (2013). Mitochondrial alterations near amyloid plaques in an Alzheimer’s disease mouse model. J. Neurosci. 33, 17042–17051. doi: 10.1523/JNEUROSCI.1836-13.2013
Xie, Y., Zhou, B., Lin, M. Y., Wang, S., Foust, K. D., and Sheng, Z. H. (2015). Endolysosomal deficits augment mitochondria pathology in spinal motor neurons of Asymptomatic fALS mice. Neuron 87, 355–370. doi: 10.1016/j.neuron.2015.06.026
Yang, D. S., Stavrides, P., Mohan, P. S., Kaushik, S., Kumar, A., Ohno, M., et al. (2011). Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer’s disease ameliorates amyloid pathologies and memory deficits. Brain 134, 258–277. doi: 10.1093/brain/awq341
Yao, J., Irwin, R. W., Zhao, L., Nilsen, J., Hamilton, R. T., and Brinton, R. D. (2009). Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 106, 14670–14675. doi: 10.1073/pnas.0903563106
Ye, X., Sun, X., Starovoytov, V., and Cai, Q. (2015). Parkin-mediated mitophagy in mutant hAPP neurons and Alzheimer’s disease patient brains. Hum. Mol. Genet. 24, 2938–2951. doi: 10.1093/hmg/ddv056
Yoshii, S. R., Kishi, C., Ishihara, N., and Mizushima, N. (2011). Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J. Biol. Chem. 286, 19630–19640. doi: 10.1074/jbc.M110.209338
Youle, R. J., and Narendra, D. P. (2011). Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 12, 9–14. doi: 10.1038/nrm3028
Yu, W. H., Cuervo, A. M., Kumar, A., Peterhoff, C. M., Schmidt, S. D., Lee, J. H., et al. (2005). Macroautophagy-a novel β-amyloid peptide-generating pathway activated in Alzheimer’s disease. J. Cell Biol. 171, 87–98. doi: 10.1083/jcb.200505082
Zhao, X. L., Wang, W. A., Tan, J. X., Huang, J. K., Zhang, X., Zhang, B. Z., et al. (2010). Expression of β-amyloid induced age-dependent presynaptic and axonal changes in Drosophila. J. Neurosci. 30, 1512–1522. doi: 10.1523/JNEUROSCI.3699-09.2010
Ziviani, E., Tao, R. N., and Whitworth, A. J. (2010). Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc. Natl. Acad. Sci. U S A 107, 5018–5023. doi: 10.1073/pnas.0913485107
Keywords: mitochondrial quality control, mitochondrial dynamics, mitophagy, mitochondrial transport, mitophagosome, Alzheimer’s disease, axonal transport
Citation: Cai Q and Tammineni P (2016) Alterations in Mitochondrial Quality Control in Alzheimer’s Disease. Front. Cell. Neurosci. 10:24. doi: 10.3389/fncel.2016.00024
Received: 30 November 2015; Accepted: 25 January 2016;
Published: 09 February 2016.
Edited by:
Chao Deng, University of Wollongong, AustraliaReviewed by:
P. Hemachandra Reddy, Texas Tech University, USAGiovanna Cenini, Universität Bonn, Germany
Copyright © 2016 Cai and Tammineni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qian Cai, Y2FpQGJpb2xvZ3kucnV0Z2Vycy5lZHU=