 Rebecca K. Bubenheimer1,2
Rebecca K. Bubenheimer1,2 Isola A. M. Brown2,3
Isola A. M. Brown2,3 David E. Fried2Jonathon L. McClain2
David E. Fried2Jonathon L. McClain2 Brian D. Gulbransen1,2*
Brian D. Gulbransen1,2*- 1Neuroscience Program, Michigan State University, East Lansing, MI, USA
- 2Department of Physiology, Michigan State University, East Lansing, MI, USA
- 3Pharmacology and Toxicology Program, Michigan State University, East Lansing, MI, USA
Gut inflammation contributes to the development of gut motility disorders in part by disrupting the function and survival of enteric neurons through mechanisms that involve oxidative stress. How enteric neurons regulate oxidative stress is still poorly understood. Importantly, how neuron autonomous antioxidant mechanisms contribute to the susceptibility of enteric neurons to oxidative stress in disease is not known. Here, we discover that sirtuin-3 (Sirt3), a key regulator of oxidative stress and mitochondrial metabolism, is expressed by neurons in the enteric nervous system (ENS) of the mouse colon. Given the important role of Sirt3 in the regulation of neuronal oxidative stress in the central nervous system (CNS), we hypothesized that Sirt3 plays an important role in the cell autonomous regulation of oxidative stress by enteric neurons and that a loss of Sirt3 increases neuronal vulnerability during intestinal inflammation. We tested our hypothesis using a combination of traditional immunohistochemistry, oxidative stress measurements and in vivo and ex vivo measures of GI motility in healthy and inflamed wild-type (wt) and Sirt3 null (Sirt3−/−) mice. Our results show that Sirt3 is widely expressed by neurons throughout the myenteric plexus of the mouse colon. However, the deletion of Sirt3 had surprisingly little effect on gut function and susceptibility to inflammation. Likewise, neither the genetic ablation of Sirt3 nor the inhibition of Sirt3 with antagonists had a significant effect on neuronal oxidative stress. Therefore, we conclude that Sirt3 contributes very little to the overall regulation of neuronal oxidative stress in the ENS. The functional relevance of Sirt3 in enteric neurons is still unclear but our data show that it is an unlikely candidate to explain neuronal vulnerability to oxidative stress during inflammation.
Introduction
The enteric nervous system (ENS) controls essential gastrointestinal (GI) reflexes such as patterns of movement (i.e., peristalsis), fluid exchange across the intestinal mucosa and local blood flow (Furness, 2012). This becomes most evident in disease when gut functions are impaired by changes to the structure or neurochemistry of the ENS. Indeed, alterations to the survival or function of enteric neurons are associated with impaired gut function in a number of conditions such as old age (Wade and Cowen, 2004; Camilleri et al., 2008), diabetes (Chandrasekharan et al., 2011), inflammatory bowel diseases (IBD; Gulbransen et al., 2012), achalasia (De Giorgio et al., 1999), chronic intestinal pseudo-obstruction (Antonucci et al., 2008), slow-transit constipation (De Giorgio et al., 2004), Parkinson’s disease (PD; Wakabayashi et al., 1988; Beach et al., 2010; Devos et al., 2013), irritable bowel syndrome (IBS; Lindberg et al., 2009; Ohlsson et al., 2010) and infectious diseases (Coron et al., 2009; Lourenssen et al., 2010).
Oxidative stress is one of the most important mechanisms that contributes to ENS dysfunction in disease (Lih-Brody et al., 1996; Thrasivoulou et al., 2006; Chandrasekharan et al., 2011; Roberts et al., 2013; Brown et al., 2016). For example, the inability to buffer reactive oxygen species is considered an important factor in the susceptibility of inhibitory, nitrergic neurons to injury in conditions such as diabetes (Chandrasekharan et al., 2011) and inflammation (Sido et al., 1998; Esworthy et al., 2001; Rezaie et al., 2007; Zhu and Li, 2012; Roberts et al., 2013) and excitatory, cholinergic neurons in advanced age (Wade, 2002; Camilleri et al., 2008; Korsak et al., 2012; Saffrey, 2013). Neurons continually generate reactive oxygen species during normal functions because of their high energy demand to sustain electrical signaling. Yet neurons express few anti-oxidant defenses and have a low ability to cope with reactive oxygen species. Therefore, even a subtle shift in the limited defense mechanisms that neurons do possess could have major effects on their susceptibility to disease. Despite the importance of oxidative stress in neuronal susceptibility to cell injury in many enteric neurodegenerative conditions, little is known about the factors that regulate oxidative stress in enteric neurons.
Sirtuin-3 (Sirt3) is a member of the sirtuin family of nicotinamide adenine nucleotide (NAD)–dependent deacetylases that has emerged as an important regulator of neuronal survival in times of stress (Schwer et al., 2002; Kim et al., 2011; Weir et al., 2012; Chen et al., 2013; Dai et al., 2014). In cultured neurons, the expression of Sirt3 is both necessary (Kim et al., 2011) and sufficient (Kim et al., 2011; Shulyakova et al., 2014) for neuroprotection in models of oxidant-induced neurotoxicity and excitotoxicity. These neuroprotective actions appear to stem mainly from Sirt3’s ability to activate key scavengers of reactive oxygen species in mitochondria (Shi et al., 2005; Sundaresan et al., 2009; Kim et al., 2010). Indeed, mice deficient in Sirt3 exhibit higher levels of oxidative stress (Chen et al., 2013) and display enhanced susceptibility to metabolic diseases (Hirschey et al., 2011).
Given the important role of Sirt3 in the regulation of neuronal oxidative stress in neurons from the central nervous system (CNS), we hypothesized that Sirt3 is an important regulator of oxidative stress in the ENS. We tested our hypothesis using a combination of traditional immunohistochemistry, oxidative stress measurements and in vivo and ex vivo measures of GI motility in wild-type (wt) and Sirt3 null (Sirt3−/−) mice. Our results show that Sirt3 is widely expressed by myenteric neurons in the mouse colon. However, the deletion of Sirt3 has surprisingly little effect on gut function and the susceptibility to inflammation. Likewise, neither the genetic ablation of Sirt3 nor its inhibition with antagonists has a significant effect on neuronal oxidative stress. Our results provide the first evidence of Sirt3 expression in the ENS. However, the main function of Sirt3 in enteric neurons is unclear because it does not appear to play a major role in the regulation of oxidative stress by enteric neurons.
Materials and Methods
Animals
Experimental protocols were reviewed and approved by the Institutional Animal Care and Use Committee at Michigan State University. Sirt3 null mice (129-SIRT3tm1.1Fwa/J; RRID:IMSR_JAX:012755), hereafter referred to as Sirt3−/−(Lombard et al., 2007), and background controls (129S1/SvImJ; RRID:IMSR_JAX:002448), hereafter referred to as wild-type (wt; The Jackson Laboratory, Bay Harbor, ME, USA), of both sexes (8–10 weeks of age) were used for all experiments unless otherwise stated. Male C57BL/6 mice (8–10 weeks of age) from Charles River Laboratories (Hollister, CA, USA) were used in some experiments to test Sirt3 expression in inflammation and to compare oxidative stress levels of myenteric neurons. Mice were maintained in a temperature controlled 12 h light:12 h dark cycle environment with ad libitum access to food and water. All genotyping was performed by the Research Technology Support Facility at Michigan State University using the standard PCR protocol supplied by Jackson Labs for Sirt3−/− mice (information available at www.jax.org).
In Vivo Measures of Colonic Motility
Pellet Production
Mice were individually housed in clean, empty cages and fecal output was monitored for 1 h beginning at Zeitgeber time + 3 for 5 consecutive days. Mice were acclimatized over the first 2 days and data was recorded over the last 3 days. Pellets were collected as soon as they were produced and counted and weighed to obtain a wet weight. Pellets were then dried overnight to determine dry weight.
Colon Bead
Individually housed animals were fasted overnight and lightly anesthetized with isoflurane. A 2 mm bead was inserted 2 cm into the colon and the time to expulsion recorded.
In Vivo Colitis
We used the dinitrobenzene sulfonic acid (DNBS) model to induce colitis in mice (Gulbransen et al., 2012). Anesthetized mice received an enema of DNBS (5.5 mg/mouse in 0.1 mL 50% ethanol/50% saline) via a gavage needle inserted 3 cm into the colon. Control animals received an enema of saline. We monitored animal weight daily and 48 h after treatment, animals were euthanized via cervical dislocation to collect tissue samples. The extent of inflammation was scored using an established macroscopic damage scoring system (Storr et al., 2009).
Contractility Studies
Longitudinally oriented segments of mouse intestine were mounted between two platinum ring stimulating electrodes in organ baths. Each intestinal segment was secured to a tissue holder and connected to a force-displacement transducer (Grass Instruments, Quincy, MA, USA). Frequency-response curves (2–30 Hz) for neurogenic contractions and relaxations were generated by electrical field stimulation (EFS) supplied through the electrodes by a GRASS stimulator (S88, GRASS telefactor, West Warwick, RI, USA) and data was charted with Labscribe (iWorx, Dover, NH, USA). Tissue segments were placed under 0.5 g initial tension and allowed to equilibrate for 20 min. Neurogenic relaxations were studied in tissues precontracted with 5 μM prostaglandin F2-α (PGF2α). Bethanechol (BCH, 10 μM) was used to assess the maximum contractile response and tetrodotoxin (TTX, 0.3 μM) was applied to block neurogenic responses.
Whole-Mount Immunohistochemistry
Whole-mount preparations of colonic myenteric plexus and longitudinal muscle (LMMP) were prepared from tissue fixed in Zamboni’s fixative (Nasser et al., 2007) or 4% paraformaldehyde (PFA; Joseph et al., 2011). Nonspecific labeling was blocked by incubating tissue for 45 min in a solution containing 4% normal donkey serum or goat serum, 0.4% Triton X-100, and 1% bovine serum albumin. Primary antibodies were applied overnight at room temperature and secondary antibodies were applied for 2 h at room temperature. Antibody details are supplied in Table 1. Images were acquired through the 40X objective (Planfluor, 0.75 numerical aperture) of an upright epifluorescence microscope (Nikon Eclipse Ni, Melville, NY, USA) with a Retiga 2000R camera (QImaging, Surrey, BC, Canada) controlled by QCapture Pro 7.0 software (QImaging) or by confocal imaging using the Plan-Aprochromat 60X oil immersion objective (1.42 numerical aperture) of an inverted Olympus Fluoview FV1000 microscope (Olympus, Center Valley, PA, USA). Software settings were established using the immunoreactivity of each antibody in control tissues and then kept constant for all remaining tissues.
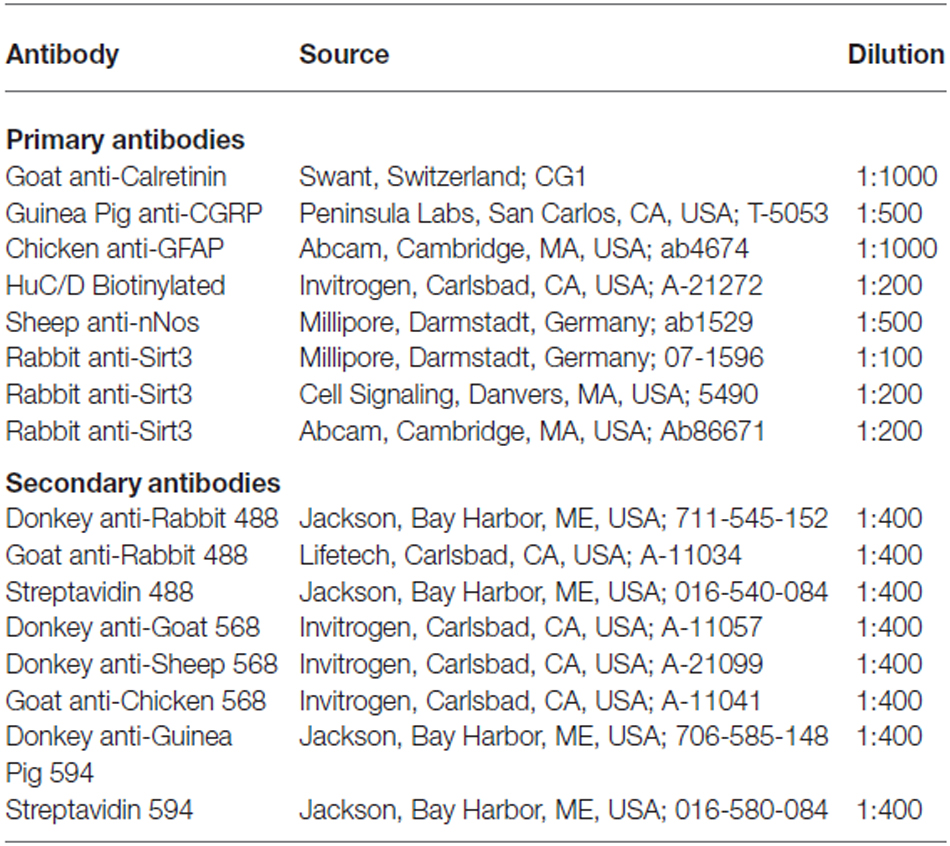
Table 1. Antibodies used in this study.
Thiol Oxidation
We quantified neuronal thiol oxidation as a measure of oxidative stress as previously described (Mullett et al., 2013). Briefly, reduced (-SH) and oxidized (-SS) thiols were labeled with 1 μM Alexa Fluor 680 C2 maleimide and 1 μM Alexa Fluor 546 C5 maleimide, respectively. Dyes were dissolved in 4% PFA, 0.02% Triton X-100 and 1 mM N-ethylymaleimide in PBS. Tissue was washed in 5 mM tris(2-carboxyethyl)phosphine hydrochloride to convert oxidized thiols to reduced thiols. Images were obtained by epifluorescence microscopy as described above and the ratio of 546-maleimide/680-maleimide (SS/SH) calculated with ImageJ software (NIH).
Drugs/Chemicals
Tenovin-6 was purchased from Cayman Chemicals (Ann Arbor, MI, USA). All other reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Data Analysis
Cell counts were performed using the cell counter plug-in of ImageJ software. Individual neuron subpopulations are presented as a percentage of total enteric neuron numbers defined by HuC/D-immunoreactivity. Enteric neuron numbers are presented as the ganglionic neuronal packing density. We calculated the neuron packing density per ganglion by using GFAP-immunoreactivity to demarcate the borders of each ganglion and HuC/D-immunoreactivity to count the number of neurons within that two-dimensional area. Ganglionic GFAP fluorescence density was quantified in ganglia from confocal z-stack images. Each image was imported into ImageJ and mean gray values of GFAP fluorescence and ganglionic area were recorded for each ganglion. GFAP fluorescence density (integrated density) was calculated as the product of area and mean gray value (McCloy et al., 2014) and is reported as the intensity in arbitrary fluorescence units (AFU) per μm2 of ganglionic area. Counts were performed on a minimum of 10 ganglia per animal and averaged to obtain an average packing density for that animal. N values represent the number of animals in each experiment.
Statistical Analysis
Prism 6 software (Graphpad, La Jolla, CA, USA) was used for all statistical analyses and the generation of graphs. Data was analyzed using either a two-way ANOVA (with Bonferroni post hoc test) or a Student’s t-test, where appropriate. A P value less than 0.05 was considered statistically significant. All data are presented as mean ± standard error of the mean (SEM).
Results
Expression of Sirtuin-3 in the Mouse Intestine
Sirtuin-3 (Sirt3) is highly expressed in metabolically active tissues that are rich in mitochondria such as the brain and the heart (Lombard et al., 2007). Sirt3 is also expressed in the stomach (Shi et al., 2005), but whether Sirt3 expression extends to the distal regions of the gastrointestinal tract is not known. Therefore, we began by exploring the expression of Sirt3 in samples of mouse colon using PCR to assess the presence of Sirt3 mRNA and western blots to determine the presence of Sirt3 protein (Figure 1). Our results show robust expression of both Sirt3 message (Figure 1A) and protein (Figure 1B) in the colons of wt mice. Human Sirt3 is expressed in two forms: a long form of ~44 kDa that localizes to the nucleus and a short form of ~28 kDa that is localized to mitochondria (Schwer et al., 2002). Most available evidence shows that mice only express one form of Sirt3 that is equivalent to the human truncated form (~28 kDa; Scher et al., 2007). However, some evidence suggests that mice express both long and short forms of Sirt3 (Sundaresan et al., 2009). Our results show clear bands at both 44 and 28 kDa in samples from wt mouse colon (Figure 1B). However, only the band at 28 kDa was absent in samples from Sirt3−/− mice. These results agree with the conclusion that only one form of Sirt3 is present in the mouse intestine and that this form corresponds to the truncated (28 kDa) product in humans.
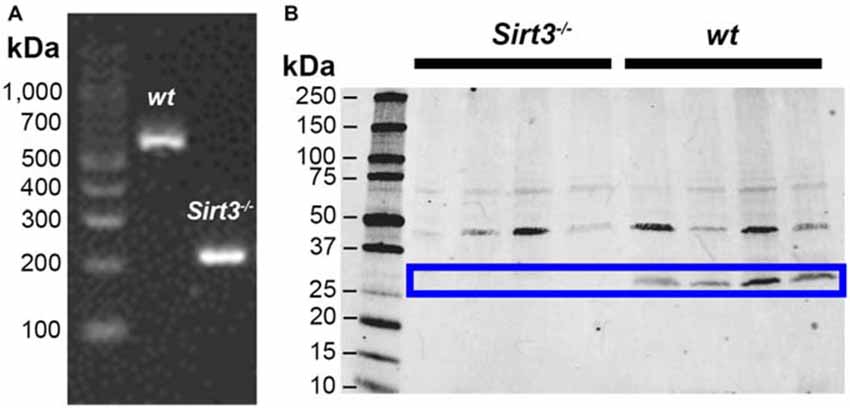
Figure 1. Representative images showing detection of Sirt3 in the mouse colon by RT-PCR (A) and western blot (B). Tissue from Sirt3−/− mice was used as a negative control. PCR amplification of Sirt3 mRNA (A) shows the expected products at 562 bp for wt Sirt3 and 200 bp for mutant Sirt3 (Sirt3−/−). Western blots for Sirt3 protein (B) show two bands potentially corresponding to the full-length nuclear protein at 44 kDa and the processed mitochondrial form at 28 kDa. However, only the band at 28 kDa is absent in Sirt3−/− mice and this band corresponds to the single known form of mouse Sirt3.
Localization of Sirtuin-3 within the Enteric Nervous System
Given that Sirt3 is highly expressed in the brain (Lombard et al., 2007) and our results show that Sirt3 is expressed in the colon (Figure 1), we reasoned that enteric neurons would be one site of high Sirt3 expression within the colon. To explore this possibility, we performed dual-label immunohistochemistry experiments with antibodies against HuC/D (to identify enteric neurons) and Sirt3 (to localize Sirt3 protein). In all, we evaluated the labeling of three commercial antibodies against Sirt3 in the ENS (see Table 1 for antibody details). The pattern of labeling produced by each of the three antibodies against Sirt3 that we tested was consistent with neuronal labeling in the ENS (for example, see Figure 2). However, given the questionable specificity of some antibodies (Baker, 2015), we performed careful control experiments with control peptides and tissue from Sirt3 null animals to validate the labeling of each antibody. Labeling was completely abolished in all cases by preadsorption with the corresponding peptide (data not shown); suggesting that each of the three antibodies is specific against the peptide that it was raised against. Despite all being specific for the corresponding peptides, only one antibody from Cell Signaling (Danvers, MA, USA) was specific for Sirt3 protein when we evaluated labeling in tissue from Sirt3−/− mice using immunohistochemistry and western blot analyses (western blot data shown in Figure 1B). The other two antibodies still exhibited strong labeling in knockout tissue. Based on these results, we concluded that the other two antibodies were not specific for Sirt3 protein and/or not suitable for our application and we chose to perform the remainder of our experiments with the antibody from Cell Signaling.
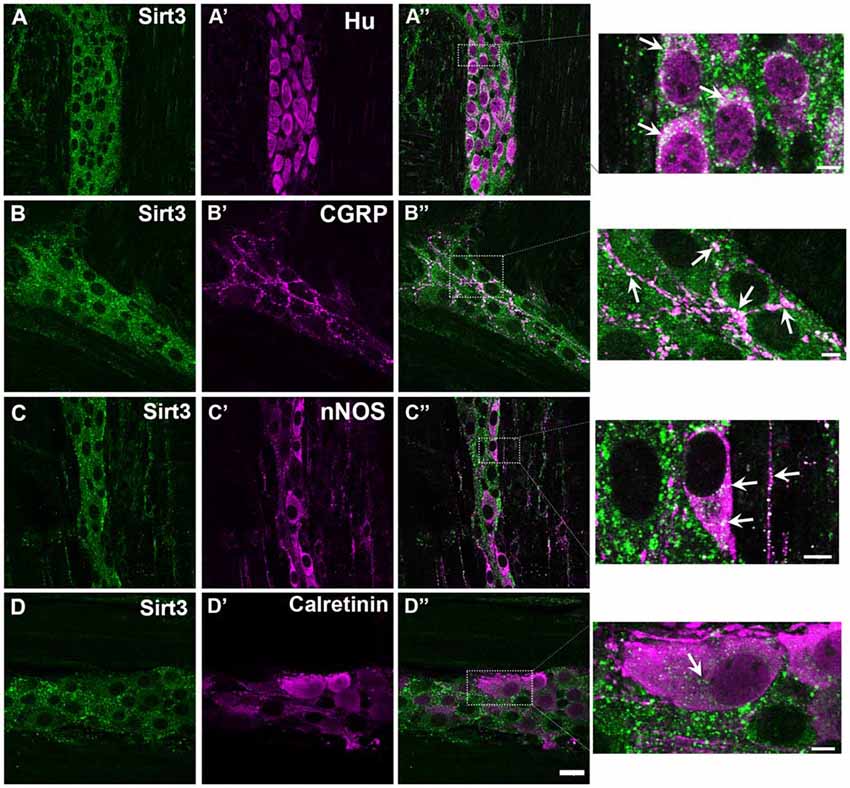
Figure 2. Myenteric neurons in the mouse colon express Sirt3. Panels show confocal fluorescence microscopy images of dual-label immunohistochemistry for Sirt3 (green, A–D) and either the neuronal marker HuC/D (magenta, A’), calcitonin gene-related peptide (CGRP; magenta, B’), nNOS (magenta, C’) or calretinin (magenta, D’). Overlays of each combination are shown in (A”–D”). The areas demarcated by the dashed boxes in the overlay images are shown as enlarged images in the panels at far right. Arrows in these images highlight several regions of co-localization. The scale bar in D” = 20 μm and applies to (A–D”). All scale bars in the enlarged images at right = 5 μm. Labeling is representative of experiments performed on tissue from a minimum of three mice.
Our results show widespread Sirt3 immunoreactivity in myenteric neurons in the mouse colon (Figure 2). In our study, we were unable to find any HuC/D-immunoreactive neurons that did not also display some level of Sirt3 immunoreactivity (Figures 2A–A”). The most intense Sirt3 immunoreactivity that we observed was present in neuronal varicosities and co-localized well with labeling for calcitonin gene-related peptide (CGRP; Figures 2B–B”). Sirt3 immunoreactivity was present in neurons immunoreactive for nNOS and neurons immunoreactive for calretinin (Figures 2C,D). These results suggest that Sirt3 is broadly expressed by myenteric neurons and not confined to a particular neuronal subtype. However, Sirt3 may be expressed at different levels in subpopulations of enteric neurons because Sirt3 immunoreactivity appeared to label nitrergic neurons more intensely than calretinin-immunoreactive neurons (compare Figures 2C”,D”) and nitrergic nerve fibers at the level of the smooth muscle displayed strong immunoreactivity for Sirt3 (see magnification image of Figure 2C”).
Effect of the Ablation of Sirtuin-3 on Gut Function
The results of our expression analysis above show that Sirt3 is highly expressed by myenteric neurons in the mouse colon. Based on these data, we hypothesized that a loss of Sirt3 would disrupt the normal neural control of gut function and/or the susceptibility of enteric neurons to pathological conditions that involve oxidative stress, such as inflammation. We began to test our hypothesis by assessing how the loss of Sirt3 in Sirt3−/− mice impacts the neuronal control of gut function. We assessed gut function in two ways: first, by performing isometric muscle tension recordings in isolated segments of intestine and second, by measuring colonic output in vivo.
We began our muscle tension recordings by assessing the contractile ability of each segment of colon with the muscarinic agonist, bethanechol (BCH, 10 μM). BCH elicited similar contractile responses in tissue segments from both wt and Sirt3−/− mice (Figure 3A). On average, these responses were 0.214 ± 0.026 g in wt and 0.218 ± 0.017 g in Sirt3−/− tissues (P > 0.05). These data show that the contractile capacity of the intestinal smooth muscle is not affected by the ablation of Sirt3. Next, we tested whether the ability of enteric neurons to drive contractions is affected by the ablation of Sirt3 by assessing neurogenic contractions driven by EFS. EFS elicited contractile responses of equal magnitude in tissue segments from both wt and Sirt3−/− mice (Figure 3A). On average, EFS-evoked responses were 0.296 ± 0.048 g in wt and 0.294 ± 0.032 g in Sirt3−/− tissues (P > 0.05). Overall, we found no evidence of altered neurogenic contractions between wt and Sirt3−/− tissue (Figure 3A). Next, we assessed neurogenic relaxations provoked by EFS in tissue samples pre-contracted with prostaglandin F2α (PGF2α, 5 μM; Figure 3B). These experiments revealed a significant defect in neurogenic relaxations driven by frequencies between 3–25 Hz in tissues from Sirt3−/− mice (Data shown: EFS at 20 Hz elicited relaxation responses that averaged 0.132 ± 0.015 g in wt and 0.074 ± 0.013 g in Sirt3−/− tissues; n = 5, P = 0.0192; Figure 3B). These results suggest that Sirt3 is important in the normal functions of inhibitory neurons in the ENS but is dispensable in the normal functions of excitatory neurons.
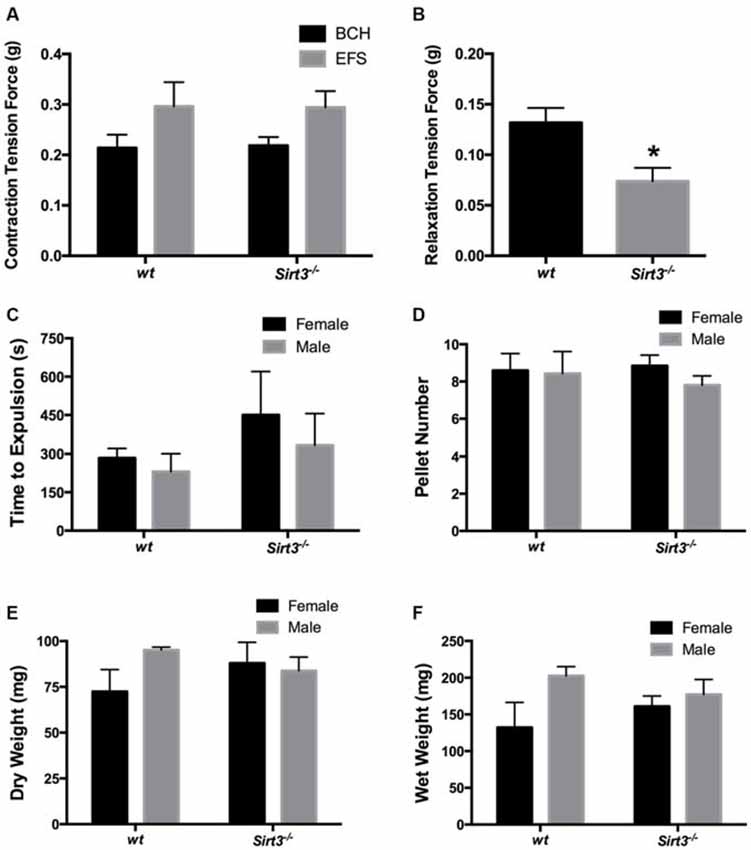
Figure 3. Effect of the ablation of Sirt3 on gut function in healthy animals. (A) Contractile responses of longitudinal segments of colon driven by bethanechol (BCH, 10 μM) or electrical field stimulation (EFS) are similar in mice with wild type Sirt3 (wt) and Sirt3 null mice (Sirt3−/−; n = 5 animals per group, P > 0.05, Student’s t-test). (B) Relaxations driven by EFS (at 20 Hz) in pre-contracted (PGF2α, 5 μM) longitudinally-oriented segments of colon from Sirt3−/− mice and their respective wt controls. EFS produced significantly smaller relaxations in tissues from Sirt3−/− mice than wt controls (n = 5 animals per group, *P = 0.0192, Student’s t-test). Distal colonic transport time (C), the number of fecal pellets produced per hour (D) and pellet composition (E,F) are comparable between male and female agouti wt and Sirt3−/− mice (n = 4 animals per group; P > 0.05, 2-way ANOVA).
Next, we tested the hypothesis that the deficit in the inhibitory component of neuromuscular control that we observed in vitro would translate to poor colonic motility in vivo. We tested our hypothesis by assessing distal colonic transit time and by measuring the fecal pellet output rate and content in Sirt3−/− and wt mice (Figures 3C,D). Our results show that colonic transit time is not significantly affected by the deletion of Sirt3 in either male or female mice (Figure 3C). Likewise, Sirt3−/− mice produce an equal number of pellets per hour as wt mice (wt female 9 ± 0.9 pellets, Sirt3−/− female 9 ± 0.6 pellets, wt male 8 ± 1.2 pellets, Sirt3−/− male 8 ± 0.5 pellets; P = 0.833; 2-way ANOVA; Figure 3D). The pellets produced contained a similar content of fecal matter and liquid (total dry weight of fecal matter: wt female 72.4 ± 12 mg, Sirt3−/− female 87.9 ± 11 mg, wt male 95.1 ± 1.6 mg, Sirt3−/− male 83.8 ± 7.5 mg; P = 0.390; 2-way ANOVA; total wet weight of fecal matter: wt female 132 ± 34 mg, Sirt3−/− female 160 ± 14 mg, wt male 203 ± 12 mg, Sirt3−/− male 177 ± 20 mg; P = 0.940; 2-way ANOVA; Figures 3E,F). These results show that the defect in neurogenic relaxations that we observed in vitro is not a significant barrier to normal colonic transit in vivo.
Effects of the Ablation of Sirtuin-3 on Enteric Neurodegeneration During Gut Inflammation
Colonic inflammation increases oxidative stress in myenteric neurons and myenteric oxidative stress contributes to neurodegeneration during inflammation (Brown et al., 2016). Sirt3 plays a prominent role in the protection of central neurons from oxidative stress and decreasing Sirt3 expression increases their susceptibility to apoptosis (Kim et al., 2011; Dai et al., 2014). Therefore, we expected that mice deficient in Sirt3 would be more susceptible to myenteric neurodegeneration during inflammation. We tested our hypothesis using the DNBS model of colitis because we, and others, have shown that this model drives myenteric neurodegeneration through mechanisms that involve neuronal oxidative stress (Gulbransen et al., 2012; Roberts et al., 2013; Brown et al., 2016). DNBS drove colitis to an equal extent in both wt and Sirt3−/− mice (Figure 4). Loss of Sirt3 did not affect the extent of macroscopic tissue damage (Figure 4A) nor did it significantly change the pattern of weight loss experienced by the mice during the acute episode of colitis (Figure 4B). We did observe greater weight gain in non-inflamed Sirt3−/− mice than healthy wt mice over the course of the experiment (Figure 4B) and this observation is in line with published data showing that Sirt3−/− mice gain weight more rapidly than wt mice (Hirschey et al., 2011). We also observed enlarged stomachs in all mice exposed to DNBS regardless of their sex or genotype (data not shown). This observation was surprising because we have never observed this phenomenon in DNBS-colitis experiments with C57BL/6 mice (see Gulbransen et al., 2012; Brown et al., 2016). However, we did not pursue this finding further because it was independent of Sirt3 and beyond the scope of the current study.
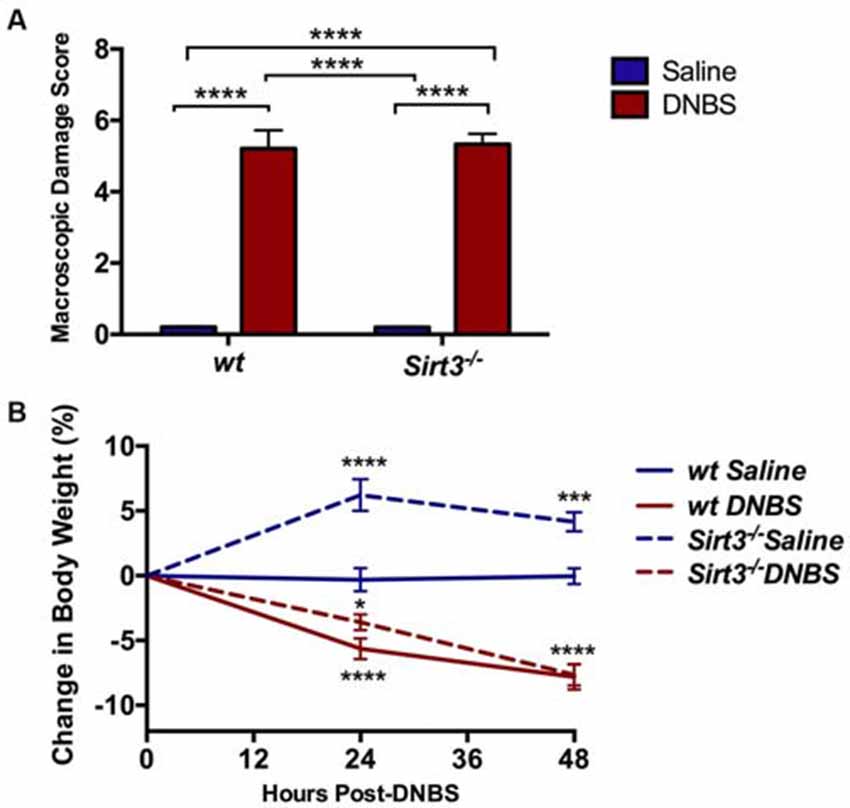
Figure 4. Assessment of gross inflammation driven by dinitrobenzene sulfonic acid (DNBS)-colitis in wt or Sirt3−/− mice as measured by macroscopic damage scoring (A) and monitoring changes in body weight (B; n = 5–7 per group, *P < 0.05, ***P < 0.005, ****P < 0.001, 2-way ANOVA).
The results above suggest that Sirt3 is not an essential protective factor against the gross inflammatory insult driven by DNBS colitis in mice. To gain more specific insight into the role of Sirt3 in the protection of neurons, we performed whole-mount immunohistochemistry to determine if neuronal Sirt3 expression changes during inflammation and if a loss of Sirt3 alters the susceptibility to neurodegeneration and neurochemical remodeling in the myenteric plexus during inflammation. We did not detect any obvious changes in Sirt3 expression or localization within the myenteric plexus during DNBS colitis in wt mice (Figure 5). This outcome suggests that inflammation does not cause overt changes in Sirt3 that could account for neuronal susceptibility in the intestine. Rather, the maintenance of Sirt3 expression could be important to protect the remaining neurons. To test if the maintenance of Sirt3 expression is necessary to protect enteric neurons, we conducted inflammation studies in Sirt3−/− mice. The packing density of HuC/D-immunoreactive myenteric neurons was not significantly different between healthy Sirt3−/− and wt animals (2043 ± 85 neurons/mm2 in wt vs. 1709 ± 153 neurons/mm2 in Sirt3−/−, Figure 6). Likewise, the neurochemical composition of myenteric neurons was not significantly affected by the deletion of Sirt3 because we observed equal percentages of nNOS-immunoreactive neurons (Figure 7) and calretinin-immunoreactive neurons (Figure 8) in healthy Sirt3−/− and wt animals. DNBS-colitis drove myenteric neurodegeneration to a similar extent in Sirt3−/− animals as wt mice (31% neuron loss in wt vs. 25% neuron loss in Sirt3−/−, Figure 6). This was an indiscriminate loss of myenteric neurons in both wt and Sirt3−/− mice because the neurochemical makeup of the remaining HuC/D-immunoreactive neurons was not significantly different from healthy mice (Figures 7, 8). Interestingly, glia failed to up-regulate glial fibrillary acidic protein (GFAP) expression during inflammation in Sirt3−/− mice (Figure 6F). Together, these results show that the loss of Sirt3 does not increase the susceptibility of myenteric neurons to neurodegeneration during inflammation. However, the lack of glial GFAP up-regulation suggests that Sirt3−/− mice may experience less neuroinflammation in the myenteric plexus. Alternatively, our results indicate that Sirt3 molecular signaling might be necessary for reactive gliosis. This hypothesis should be tested in the future with cell-specific knockouts of Sirt3.
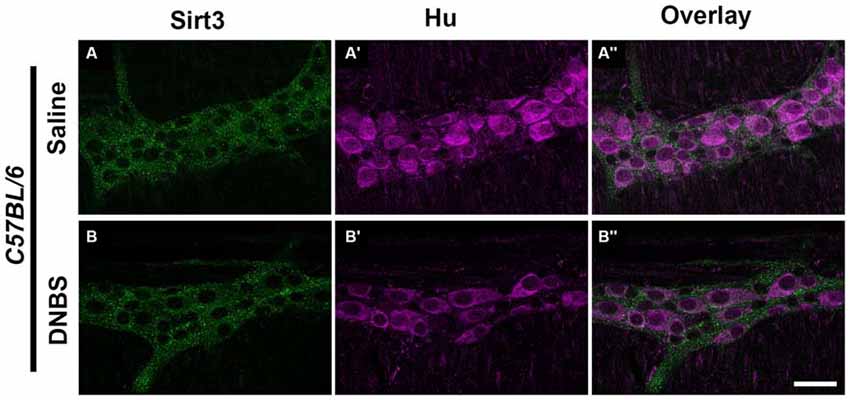
Figure 5. Sirt3 expression in healthy and inflamed C57BL/6 mice. Panels show confocal fluorescence microscopy images of dual-label immunohistochemistry for Sirt3 (green, A,B) and the neuronal marker HuC/D (magenta, A’,B’). Overlays of each combination are shown in (A”,B”). The scale bar in panel B” = 50 μm and applies to (A–B”). Labeling is representative of experiments performed on tissue from a minimum of four mice.
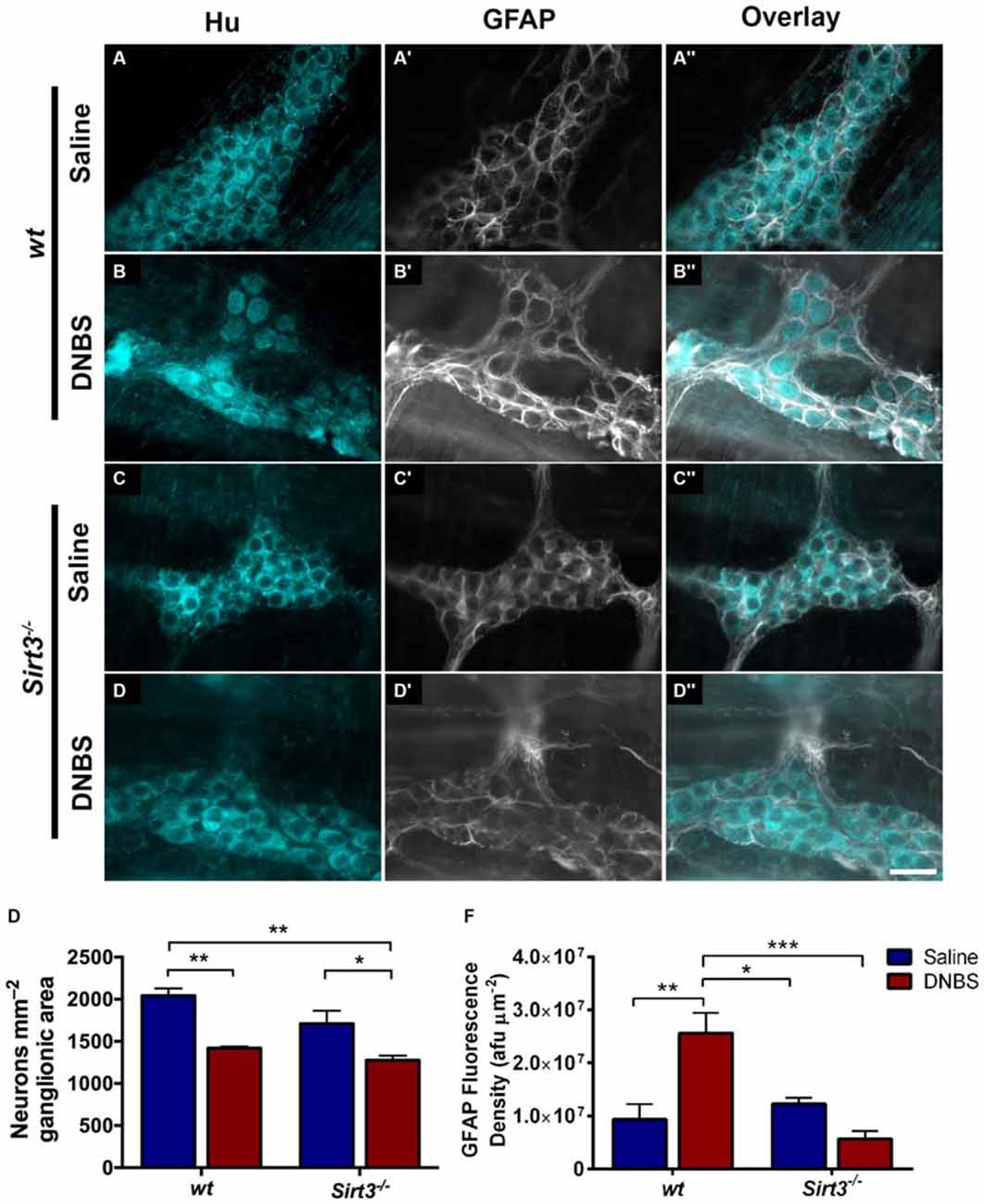
Figure 6. Enteric neuron survival and glial reactivity in healthy and inflamed wt and Sirt3−/− mice. (A–D”) show dual-label fluorescence immunohistochemistry for HuC/D (cyan, A–D) and GFAP (grayscale, A’–D’) in myenteric ganglia from saline- (A–A”, C–C”) or DNBS-treated (B–B”, D–D”) wt (A–B”) or Sirt3−/− (C–D”) mice. Overlays are shown in (A”–D”). Scale bar in D” = 50 μm and applies to all images. (E) Quantification of myenteric neuron packing density. (F) Quantification of ganglionic GFAP fluorescence density (afu/μm2). n = 3–5 animals per group, *P < 0.05, **P < 0.01, ***P < 0.001, 2-way ANOVA.
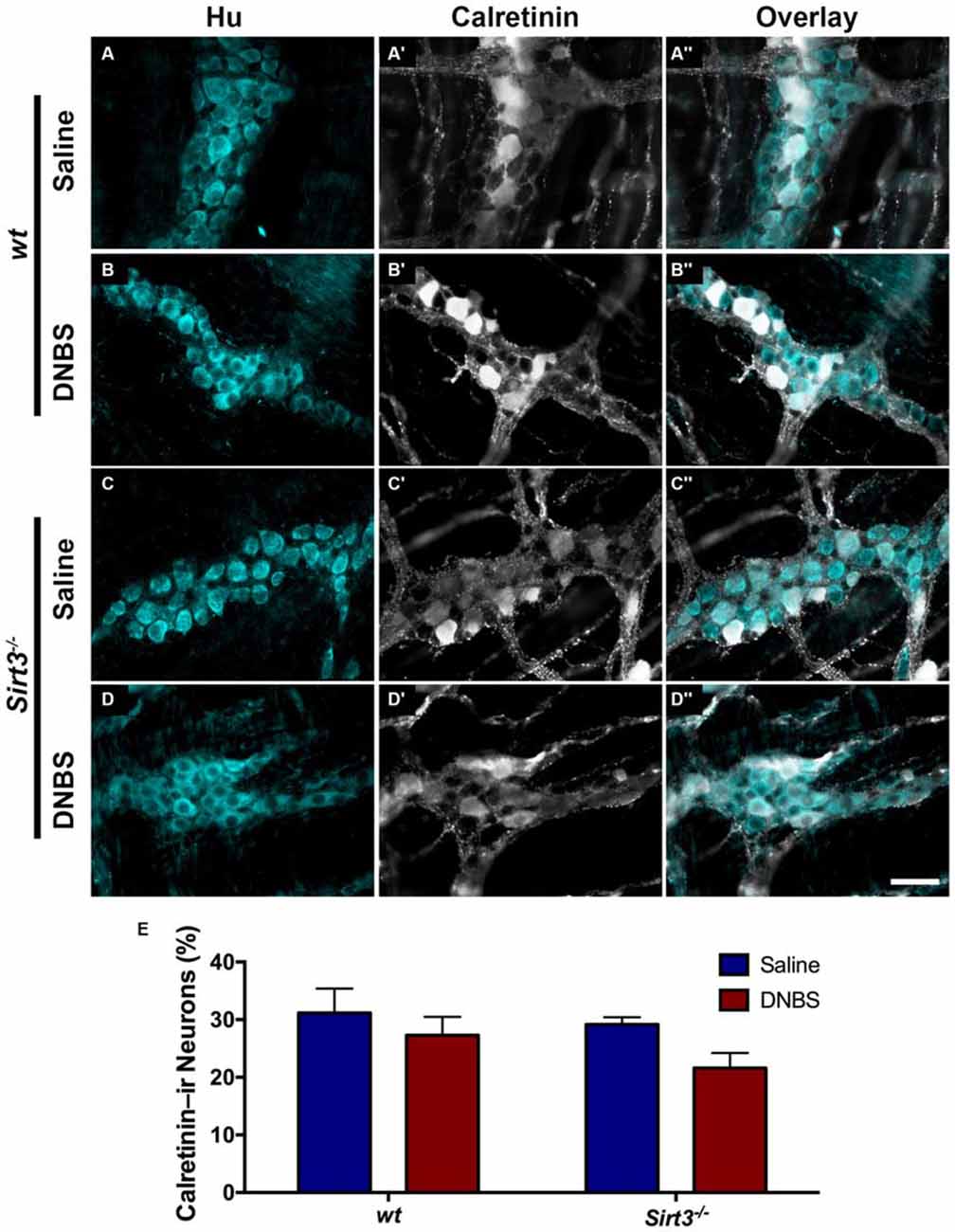
Figure 7. The percentage of myenteric neurons that express calretinin is not altered by DNBS-colitis in wt or Sirt3−/− mice. (A–D”) show dual-label fluorescence immunohistochemistry for HuC/D (cyan, A–D) and calretinin (grayscale, A’–D’) in myenteric ganglia from saline- (A–A”, C–C”) or DNBS-treated (B–B”, D–D”) mice. Overlays are shown in (A”–D”). Scale bar in D” = 50 μm and applies to all images. (E) The percentage of HuC/D immunoreactive neurons that also display immunoreactivity for calretinin in healthy and inflamed mice with wt or Sirt3−/− genotypes (n = 4–5 animals per group, P > 0.05, 2-way ANOVA).
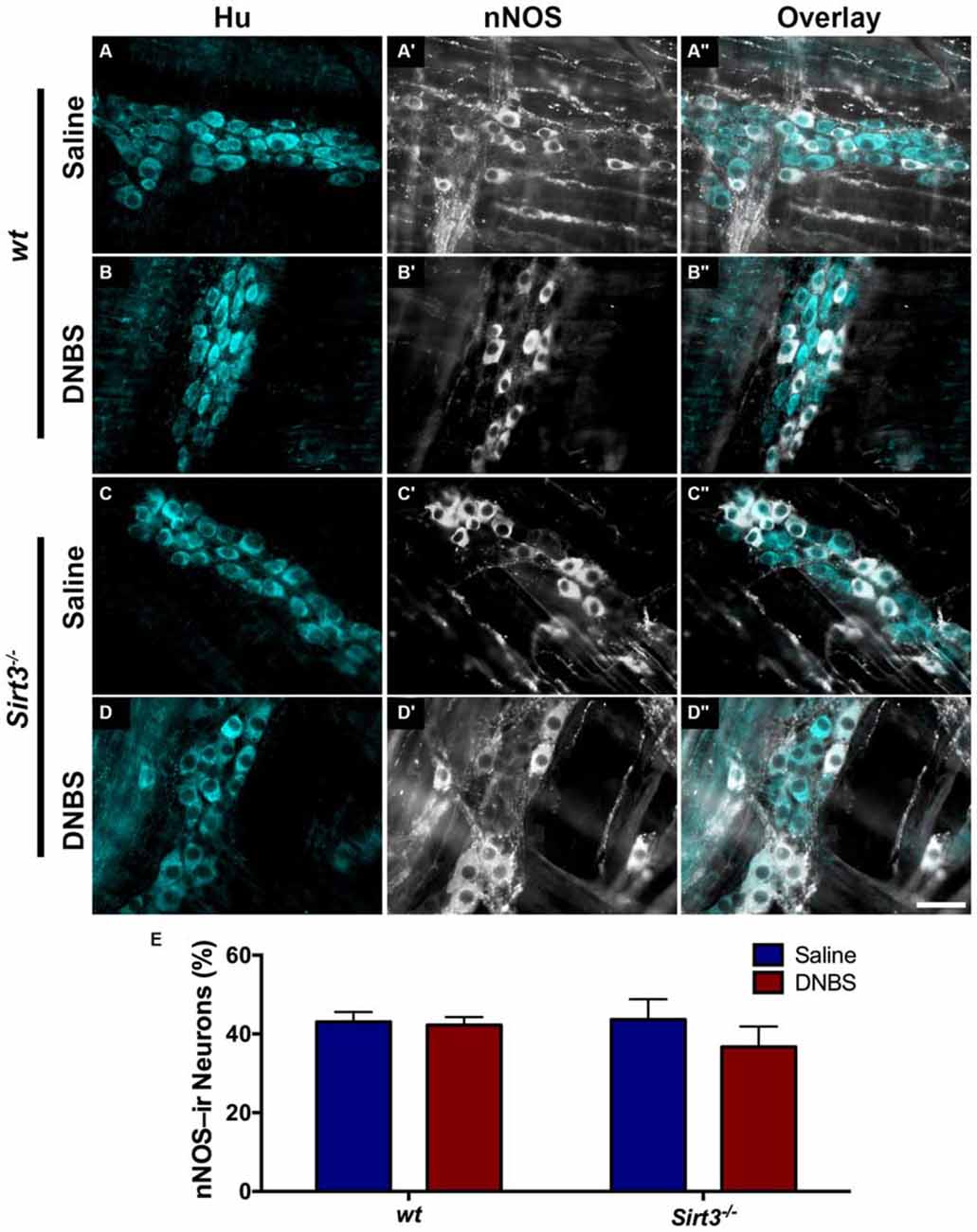
Figure 8. The percentage of myenteric neurons that express nNOS is not altered by DNBS-colitis in wt or Sirt3−/− mice. (A–D”) show dual-label fluorescence immunohistochemistry for HuC/D (cyan, A–D) and nNOS (grayscale, A’–D’) in myenteric ganglia from saline- (A–A”,C–C”) or DNBS-treated (B–B”,D–D”) mice. Overlays are shown in (A”–D”). Scale bar in D” = 50 μm and applies to all images. (E) Quantification of the percentage of HuC/D immunoreactive neurons that also display immunoreactivity for nNOS in healthy and inflamed mice with wt or Sirt3−/− genotypes (n = 4–5 animals per group, P > 0.05, 2-way ANOVA).
Effects of Sirtuin-3 Inhibition on the Redox State of Enteric Neurons
The fact that the deletion of Sirt3 did not affect the susceptibility of enteric neurons to inflammation was very surprising because reducing Sirt3 dramatically increases neuronal vulnerability in neurons isolated from the brain (Kim et al., 2011; Dai et al., 2014; Shulyakova et al., 2014). This difference suggests that there may be important differences in the function of Sirt3 between enteric and central neurons or important differences between the roles of Sirt3 in neurons in vivo vs. in vitro. One interpretation of the above data is that Sirt3 plays a much less prominent role in the regulation of oxidative stress in enteric neurons. We tested this hypothesis by measuring how the deletion of Sirt3 affects the redox state of enteric neurons in healthy and inflamed animals. Our results show that the ablation of Sirt3 in Sirt3−/− mice does not affect the oxidative balance of myenteric neurons in healthy animals (Figure 9A). In agreement with our previous work (Brown et al., 2016), myenteric neurons in tissue from wt mice with active DNBS-colitis exhibit higher levels of oxidative stress. However, the ablation of Sirt3 did not exacerbate oxidative stress in enteric neurons during inflammation (Figure 9A). To the contrary, myenteric neurons from inflamed Sirt3−/− mice exhibited less oxidative stress than neurons in tissue from healthy wt mice, healthy Sirt3−/− mice or inflamed wt mice (Figure 9A).
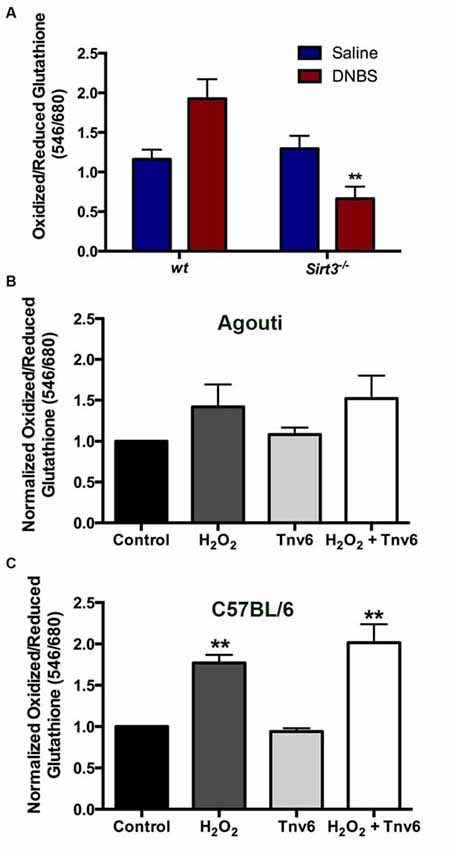
Figure 9. Neither the ablation of Sirt3 nor the inhibition of Sirt3 has a significant effect on myenteric neuron redox balance. (A) Thiol oxidation measurements (ratio of oxidized/reduced glutathione) in myenteric neurons from saline or DNBS-treated wild type (wt) and Sirt3−/− animals (n = 3–5 animals, **P < 0.01 vs. wt DNBS-treated, ANOVA). (B,C) Neuronal thiol oxidation measurements in whole-mount preparations from (B) wt agouti mice and (C) wt C57BL/6 mice treated with the oxidative stressor hydrogen peroxide (H2O2) and/or the Sirt3 inhibitor Tenovin-6 (Tnv-6, 100μm; n = 3 animals, **P < 0.01, ANOVA).
The above findings were entirely unexpected and puzzling. One possible explanation for these results could be that compensatory mechanisms are enacted in the constitutive knockout animal that efficiently buffer reactive oxygen species in the absence of Sirt3. Therefore, we used a pharmacological approach to assess the effects of acutely blocking Sirt3 function with the drug Tenovin-6 (Tnv6, 100 μm). In agreement with our in vivo work with Sirt3−/− mice, we found that blocking Sirt3 function acutely had no effect on the oxidative balance of myenteric neurons in tissue (Figures 9B,C). Interestingly, myenteric neurons in tissue from the wt agouti background mouse strain were less sensitive to oxidative stress induced by H2O2 (0.1 M) than myenteric neurons in tissue from wt C57BL/6 mice (Figures 9B,C). This may suggest that the handling of oxidative stress by myenteric neurons is fundamentally different in these two common mouse strains. Despite this difference, the inhibition of Sirt3 had absolutely no effect on baseline neuronal redox state or their susceptibility to an oxidative insult (driven by H2O2) in either strain. In all, these results show that Sirt3 does not play a major role in the antioxidant defenses of enteric neurons.
Discussion
The cell autonomous mechanisms that enteric neurons utilize to regulate oxidative stress are still poorly understood. Here, we show that enteric neurons express Sirt3; a key regulator of oxidative stress in CNS neurons. Sirt3 is widely expressed by neurons throughout the myenteric plexus of the mouse colon and its expression is potentially enriched in nitrergic neurons. Our in vivo experiments show that Sirt3 does play a role in the normal functions of nitrergic neurons because we observed a deficit in neurogenic relaxations in tissue from Sirt3−/− mice. However, Sirt3 seems to play a minor role, if any, in the regulation of oxidative stress by enteric neurons. Mice deficient in Sirt3 have an incredibly unremarkable GI phenotype and do not exhibit gross abnormalities in GI function or ENS structure. Likewise, the ablation of Sirt3 does not affect the susceptibility of enteric neurons to degeneration during colitis or their redox state.
The minor functional role of Sirt3 in the regulation of oxidative stress by enteric neurons is surprising given the major role that Sirt3 plays in the handling of oxidative stress by other populations of neurons in the CNS (Kim et al., 2011; Weir et al., 2012; Dai et al., 2014; Shulyakova et al., 2014). For example, Sirt3 expression is rapidly up-regulated by cortical neurons during excitotoxic insults and the increase in Sirt3 is an important defensive mechanism against oxidative stress that aids neuron survival (Kim et al., 2011). Sirt3 is also thought to play an important role in the regulation of oxidative stress by non-neuronal cells such as microglia (Rangarajan et al., 2015), skeletal myocytes (Jing et al., 2011), cardiomyocytes (Chen et al., 2013) and brown adipocytes (Shi et al., 2005). We did not observe any evidence of a similar role of Sirt3 in enteric neurons in our study. Enteric neurons did not exhibit higher levels of oxidative stress in Sirt3−/− mice and the acute inhibition of Sirt3 with the antagonist, tenovin-6, also had no effect on neuronal oxidative stress. These results suggest that Sirt3 fulfills a much different function in enteric neurons than in other cells and possibly that enteric neurons are fundamentally unique in the way that they handle oxidative stress. Importantly, the lack of functional Sirt3 neuroprotective mechanisms may partially explain why enteric neurons are highly susceptible to oxidative injury and death in many enteric neurodegenerative conditions (Wade and Cowen, 2004; Thrasivoulou et al., 2006; Korsak et al., 2012; Roberts et al., 2013; Brown et al., 2016).
An alternate explanation for our results is that Sirt3 is more important for the regulation of neuronal oxidative stress in culture conditions than it is in vivo. Indeed, the notion that Sirt3 plays a major role in the handling of neuronal oxidative stress has largely been based on data collected from cell culture model systems (Kim et al., 2011; Dai et al., 2014; Shulyakova et al., 2014). We assessed neuronal oxidative stress in tissue samples shortly after collecting them from the animal. Thus, our data exclude the possible confounding effects of the culture environment. If Sirt3 does in fact play a major role in the regulation of neuronal oxidative stress, mice lacking Sirt3 should be incredibly susceptible to neurodegeneration and would be expected to exhibit phenotypic abnormalities that reflect deficits in nervous system function. This is not the case. To the contrary, Sirt3 null mice have a surprisingly unremarkable phenotype despite having mitochondrial protein hyperacetylation at the biochemical level (Lombard et al., 2007). These discrepancies raise some interesting questions about what the primary role of Sirt3 is in the nervous system. Sirt3 undoubtedly plays an important role in the regulation of oxidative stress under some conditions but whether these conditions reflect the circumstances under which Sirt3 would normally function remains unclear.
If Sirt3 does contribute to the regulation of oxidative stress by enteric neurons, our data would suggest that this is a very minor contribution. Indeed, non-cell autonomous mechanisms mediated by enteric glia may play a more major role in the regulation of the overall redox state of enteric neurons than cell autonomous mechanism under normal conditions (Abdo et al., 2010, 2012). This major influence of glia likely masks any minor effects of the ablation of Sirt3 on the oxidative balance of enteric neurons in vivo. It is interesting to speculate that the same would be true of central neurons if they were assessed in their native environment where astrocytes regulate their oxidative stress vs. in culture when neuron autonomous mechanisms would be isolated and appear more prominent.
In conclusion, our study provides the first evidence of Sirt3 expression by enteric neurons. Sirt3 has received a great deal of attention as a key regulator of metabolism (Nogueiras et al., 2012) and neuroprotection against oxidative stress (Yin et al., 2015). Indeed, decreased Sirt3 expression is now being proposed by some as a potential biomarker for disease (Yu and Sun, 2015). However, we find very little evidence that would support the conclusion that low Sirt3 expression increases the susceptibility of the ENS, or the colon in general, to disease. Rather, our results are more in line with the conclusion that neuronal Sirt3 contributes very little to the overall regulation of neuronal oxidative stress and that non-cell autonomous mechanism are more important. Understanding the precise function of Sirt3 in enteric neurons will be an important issue for future molecular studies to dissect because we show that it plays a role in the normal functions of inhibitory neurons. Given the important role of Sirt3 in mitochondrial protein acetylation, a more thorough understanding of the targets and activity of Sirt3 in enteric neurons might be important to understand neuronal vulnerability or function in the gut.
Author Contributions
BDG was responsible for the overall project conception, design and supervision. Experiments performed by RKB, IAMB, DEF and JLM. Manuscript written by RKB and BDG with all authors contributing to editing.
Funding
This work was funded by the Crohn’s and Colitis Foundation of America (CCFA Senior Research Award to BDG), the National Institutes of Health (R01DK103723 to BDG), start-up funds from the Michigan State University Neuroscience Program to BDG and undergraduate stipends to RKB from the Michigan State University College of Natural Science and Neuroscience Program.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Drs. Mohr and Leinninger (Michigan State University) for their assistance with experiments involving western blot and PCR assays, respectively.
Abbreviations
Sirt, Sirtuin; GFAP, glial fibrillary acidic protein; IBD, Inflammatory Bowel Disease; DNBS, dinitrobenzene sulfonic acid; ENS, enteric nervous system; ROS, reactive oxygen species; wt, wildtype.
References
Abdo, H. H., Derkinderen, P., Gomes, P., Chevalier, J., Aubert, P., Masson, D., et al. (2010). Enteric glial cells protect neurons from oxidative stress in part via reduced glutathione. FASEB J. 24, 1082–1094. doi: 10.1096/fj.09-139519
Abdo, H. H., Mahé M. M., Derkinderen, P., Bach-Ngohou, K., Neunlist, M., and Lardeux, B. (2012). The omega-6 fatty acid derivative 15-deoxy-Δ12,14-prostaglandin J2 is involved in neuroprotection by enteric glial cells against oxidative stress. J. Physiol. 590, 2739–2750. doi: 10.1113/jphysiol.2011.222935
Antonucci, A., Fronzoni, L., Cogliandro, L., Cogliandro, R. F., Caputo, C., De Giorgio R., et al. (2008). Chronic intestinal pseudo-obstruction. World J. Gastroenterol 14, 2953–2961. doi: 10.1007/springerreference_109610
Baker, M. (2015). Reproducibility crisis: Blame it on the antibodies. Nature 521, 274–276. doi: 10.1038/521274a
Beach, T. G., Adler, C. H., Sue, L. I., Vedders, L., Lue, L., White Iii, C. L., et al. (2010). Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta. Neuropathol. 119, 689–702. doi: 10.1007/s00401-010-0664-3
Brown, I. A., McClain, J. L., Watson, R. E., Patel, B. A., and Gulbransen B. D. (2016). Enteric glia mediate neuron death in colitis through purinergic pathways that require connexin-43 and nitric oxide. Cell. Mol. Gastroenterol. Hepatol. 2, 77–91. doi: 10.1016/j.jcmgh.2015.08.007
Camilleri, M., Cowen, T., and koch T. R. (2008). Enteric neurodegeneration in ageing. Neurogastroenterol. Motil. 20, 185–196. doi: 10.1111/j.1365-2982.2007.01072.x
Chandrasekharan, B., Anitha, M., Blatt, R., Shahnavaz, N., Kooby, D., Staley, C., et al. (2011). Colonic motor dysfunction in human diabetes is associated with enteric neuronal loss and increased oxidative stress. Neurogastroenterol. Motil. 23, 131–138. doi: 10.1111/j.1365-2982.2010.01611.x
Chen, C. J., Fu, Y. C., Yu, W., and Wang, W. (2013). SIRT3 protects cardiomyocytes from oxidative stress-mediated cell death by activating NF-κB. Biochem. Biophys. Res. Commun. 430, 798–803. doi: 10.1016/j.bbrc.2012.11.066
Coron, E., Flamant, M., Aubert, P., Wedel, T., Pedron, T., Letessier, E., et al. (2009). Characterisation of early mucosal and neuronal lesions following Shigella flexneri infection in human colon. PLoS One 4:e4713. doi: 10.1371/journal.pone.0004713
Dai, S. H., Chen, T., Wang, Y. H., Zhu, J., Luo, P., Rao, W., et al. (2014). Sirt3 protects cortical neurons against oxidative stress via regulating mitochondrial Ca2+ and mitochondrial biogenesis. Int. J. Mol. Sci. 15, 14591–14609. doi: 10.3390/ijms150814591
De Giorgio, R., Di Simone, M. P., Stanghellini, V., Barbara, G., Tonini, M., Salvioli, B., et al. (1999). Esophageal and gastric nitric oxide synthesizing innervation in primary achalasia. Am. J. Gastroenterol. 94, 2357–2362. doi: 10.1016/s0002-9270(99)00413-x
De Giorgio, R., Guerrini, S., Barbara, G., Stanghellini, V., De Ponti, F., Corinaldesi, R., et al. (2004). Inflammatory neuropathies of the enteric nervous system. Gastroenterology 126, 1872–1883. doi: 10.1053/j.gastro.2004.02.024
Devos, D., Lebouvier, T., Lardeux, B., Biraud, M., Rouaud, T., Pouclet, H., et al. (2013). Colonic inflammation in Parkinson’s disease. Neurobiol. Dis. 50, 42–48. doi: 10.1007/978-3-319-08046-8_2
Esworthy, R. S., Aranda, R., Martín, M. G., Doroshow, J. H., Binder, S. W., and Chu, F. F. (2001). Mice with combined disruption of Gpx1 and Gpx2 genes have colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 281, G848–G855.
Furness, J. B. (2012). The enteric nervous system and neurogastroenterology. Nat. Rev. Gastroenterol. Hepatol. 9, 286–294. doi: 10.1038/nrgastro.2012.32
Gulbransen, B. D., Bashashati, M., Hirota, S. A., Gui, X., Roberts, J. A., MacDonald, J. A., et al. (2012). Activation of neuronal P2X7 receptor-pannexin-1 mediates death of enteric neurons during colitis. Nat. Med. 18, 600–604. doi: 10.1038/nm.2679
Hirschey, M. D., Shimazu, T., Jing, E., Grueter, C. A., Collins, A. M., Aouizerat, B. (2011). SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol. Cell. 44, 177–190. doi: 10.1016/j.molcel.2011.07.019
Jing, E., Emanuelli, B., Hirschey, M. D., Boucher, J., Lee, K. Y., Lombard, D., et al. (2011). Sirtuin-3 (Sirt3) regulates skeletal muscle metabolism and insulin signaling via altered mitochondrial oxidation and reactive oxygen species production. Proc. Natl. Acad. Sci. U S A. 108, 14608–14613. doi: 10.1073/pnas.1111308108
Joseph, N. M., He, S., Quintana, E., Kim, Y. G., Núñez, G., and Morrison, S. J. (2011). Enteric glia are multipotent in culture but primarily form glia in the adult rodent gut. J. Clin. Invest. 121, 3398–3411. doi: 10.3410/f.13357404.14726824
Kim, H. S. M., Patel, K., Muldoon-Jacobs, K., Bisht, K. S., Aykin-Burns, N., Pennington, J. D., et al. (2010). SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell 17, 41–52. doi: 10.1016/j.ccr.2009.11.023
Kim, S. H., Lu, H. F., and Alano, C. C. (2011). Neuronal Sirt3 Protects against Excitotoxic Injury in Mouse Cortical Neuron Culture. C. Combs. PLoS One 6:e14731. doi: 10.1371/journal.pone.0014731
Korsak, K., Dolatshad, N. F., Silva, A. T., and Saffrey, M. J. (2012). Ageing of enteric neurons: oxidative stress, neurotrophic factors and antioxidant enzymes. Chem. Cent. J. 6:80. doi: 10.1186/1752-153x-6-80
Lih-Brody, L., Powell, S. R., Collier, K. P., Reddy, G. M., Cerchia, R., Kahn, E., et al. (1996). Increased oxidative stress and decreased antioxidant defenses in mucosa of inflammatory bowel disease. Dig. Dis. Sci. 41, 2078–2086. doi: 10.1007/bf02093613
Lindberg, G., Törnblom, H., Iwarzon, M., Nyberg, B., Martin, J. E., and Veress, B. (2009). Full-thickness biopsy findings in chronic intestinal pseudo-obstruction and enteric dysmotility. Gut 58, 1084–1090. doi: 10.1136/gut.2008.148296
Lombard, D. B., Alt, F. W., Cheng, H. L., Bunkenborg, J., Streeper, R. S., Mostoslavsky, R., et al. (2007). Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell. Biol. 27, 8807–8814. doi: 10.1128/mcb.01636-07
Lourenssen, S., Houpt, E. R., Chadee, K., and Blennerhassett, M. G. (2010). Entamoeba histolytica infection and secreted proteins proteolytically damage enteric neurons. Infect. Immun. 78, 5332–5340. doi: 10.1128/iai.00699-10
McCloy, R. A., Rogers, S., Caldon, C. E., Lorca, T., Castro, A., and Burgees, A. (2014). Partial inhibition of Cdk1 in G2 phase overrides the SAC and decouples mitotic events. Cell Cycle 13, 1400–1412. doi: 10.4161/cc.28401
Mullett, S. J., Di Maio, R., Greenamyre, J. T., and Hinkle, D. A. (2013). DJ-1 expression modulates astrocyte-mediated protection against neuronal oxidative stress. J. Mol. Neurosci. 49, 507–511. doi: 10.1007/s12031-012-9904-4
Nasser, Y., Keenan, C. M., Ma, A. C., and McCafferty, D. M., Sharkey, K. A. (2007). Expression of a functional metabotropic glutamate receptor 5 on enteric glia is altered in states of inflammation. Glia 55, 859–872. doi: 10.1002/glia.20507
Nogueiras, R., Habegger, K. M., Chaudhary, N., Finan, B., Banks, A. S., Dietrich, M. O., et al. (2012). Sirtuin 1 and Sirtuin 3: physiological modulators of metabolism. Physiol. Rev. 92, 1479–1514. doi: 10.1152/physrev.00022.2011
Ohlsson, B., Ekblad, E., Veress, B., Montgomery, A., and Janciauskiene, S. (2010). Antibodies against gonadotropin-releasing hormone (GnRH) and destruction of enteric neurons in 3 patients suffering from gastrointestinal dysfunction. BMC Gastroenterol. 10:48. doi: 10.1186/1471-230x-10-48
Rangarajan, P., Karthikeyan, A., Lu, J., and Ling, E. A., Dheen, S. T. (2015). Sirtuin 3 regulates Foxo3a-mediated antioxidant pathway in microglia. Neuroscience 311, 398–414. doi: 10.1016/j.neuroscience.2015.10.048
Rezaie, A., Parker, R. D., and Abdollahi, M. M. (2007). Oxidative stress and pathogenesis of inflammatory bowel disease: an epiphenomenon or the cause? Dig. Dis. Sci. 52, 2015–2021. doi: 10.1007/s10620-006-9622-2
Roberts, J. A., Durnin, L., Sharkey, K. A., Mutafova-Yambolieva, V. N., and Mawe, G. M. (2013). Oxidative stress disrupts purinergic neuromuscular transmission in the inflamed colon. J. Physiol. 591, 3725–3737. doi: 10.1113/jphysiol.2013.254136
Saffrey, M. J. (2013). Cellular changes in the enteric nervous system during ageing. Dev. Biol. 382, 344–355. doi: 10.1016/j.ydbio.2013.03.015
Scher, M. B., Vaquero, A., and Reinberg, D. (2007). SirT3 is a nuclear NAD+-dependent histone deacetylase that translocates to the mitochondria upon cellular stress. Genes. Dev. 21, 920–928. doi: 10.1101/gad.1527307
Schwer, B., North, B. J., Frye, R. A., Ott, M., and Verdin, E. (2002). The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. J. Cell. Biol. 158, 647–657. doi: 10.1083/jcb.200205057
Shi, T., Wang, F., Stieren, E., and Tong, Q. (2005). SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J. Biol. Chem. 280, 13560–13567. doi: 10.1074/jbc.m414670200
Shulyakova, N., Sidorova-Darmos, E., Fong, J., Zhang, G., Mills, L. R., and Eubanks, J. H. (2014). Over-expression of the Sirt3 sirtuin Protects neuronally differentiated PC12 Cells from degeneration induced by oxidative stress and trophic withdrawal. Brain Res. 1587, 40–53. doi: 10.1016/j.brainres.2014.08.066
Sido, B., Hack, V., Hochlehnert, A., Lipps, H., Herfarth, C., and Dröge, W. (1998). Impairment of intestinal glutathione synthesis in patients with inflammatory bowel disease. Gut 42, 485–492. doi: 10.1136/gut.42.4.485
Storr, M. A., Keenan, C. M., Zhang, H., Patel, K. D., Makriyannis, A., and Sharkey, K. A. (2009). Activation of the cannabinoid 2 receptor (CB2) protects against experimental colitis. Inflamm. Bowel. Dis. 15, 1678–1685. doi: 10.1002/ibd.20960
Sundaresan, N. R., Gupta, M., Kim, G., Rajamohan, S. B., Isbatan, A., and Gupta, M. P. (2009). Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Invest. 119, 2758–2771. doi: 10.1172/jci39162
Thrasivoulou, C., Soubeyre, V., Ridha, H., Giuliani, D., Giaroni, C., Michael, G. J., et al. (2006). Reactive oxygen species, dietary restriction and neurotrophic factors in age-related loss of myenteric neurons. Aging Cell 5, 247–257. doi: 10.1111/j.1474-9726.2006.00214.x
Wade, P. R. (2002). Aging and neural control of the GI tract. I. Age-related changes in the enteric nervous system. Am. J. Physiol. Gastrointest. Liver Physiol. 283, G489–G495. doi: 10.1002/9780470988756.ch5
Wade, P. R., and Cowen, T. (2004). Neurodegeneration: a key factor in the ageing gut. Neurogastroenterol. Motil. 16, 19–23. doi: 10.1111/j.1743-3150.2004.00469.x
Wakabayashi, K., Takahashi, H., Takeda, S., Ohama, E., and Ikuta, F. (1988). Parkinson’s disease: the presence of Lewy bodies in Auerbach‘s and Meissner’s plexuses. Acta. Neuropathol. 76, 217–221. doi: 10.4324/9780203313909
Weir, H. J., Murray, T. K., Kehoe, P. G., Love, S., Verdin, E. M., O’Neill, M. J., et al. (2012). CNS SIRT3 expression is altered by reactive oxygen species and in Alzheimer’s disease. PLoS One 7:e48225. doi: 10.1371/journal.pone.0048225
Yin, J., Han, P., Tang, Z., Liu, Q., and Shi, J. (2015). Sirtuin 3 mediates neuroprotection of ketones against ischemic stroke. J. Cereb. Blood Flow Metab. 35, 1783–1789. doi: 10.1038/jcbfm.2015.123
Yu, W. L., Sun, Y. (2015). Letter to the editor: decreased SIRT3 expression is a good potential biomarker associated with diseases. Am. J. Physiol. Heart Circ. Physiol. 308:H258. doi: 10.1152/ajpheart.00829.2014
Keywords: Sirt3, sirtuin, autonomic, intestine, peripheral nervous system, oxidative stress
Citation: Bubenheimer RK, Brown IAM, Fried DE, McClain JL and Gulbransen BD (2016) Sirtuin-3 Is Expressed by Enteric Neurons but It Does not Play a Major Role in Their Regulation of Oxidative Stress. Front. Cell. Neurosci. 10:73. doi: 10.3389/fncel.2016.00073
Received: 08 January 2016; Accepted: 08 March 2016;
Published: 22 March 2016.
Edited by:
Pier Giorgio Mastroberardino, University Medical Center Rotterdam, NetherlandsReviewed by:
Lei Liu, University of Florida College of Medicine, USAOnesmo B. Balemba, University of Idaho, USA
Copyright © 2016 Bubenheimer, Brown, Fried, McClain and Gulbransen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brian D. Gulbransen, Z3VsYnJhbnNAbXN1LmVkdQ==