 Elena Agostoni1‡
Elena Agostoni1‡ Silvia Michelazzi1‡
Silvia Michelazzi1‡ Marta Maurutto1Alisia Carnemolla1
Marta Maurutto1Alisia Carnemolla1 Yari Ciani2,3
Yari Ciani2,3 Paolo Vatta1Paola Roncaglia1†
Paolo Vatta1Paola Roncaglia1† Silvia Zucchelli1,4
Silvia Zucchelli1,4 Giampiero Leanza3Fiamma Mantovani2,3
Giampiero Leanza3Fiamma Mantovani2,3 Stefano Gustincich1,5Claudio Santoro4
Stefano Gustincich1,5Claudio Santoro4 Silvano Piazza2,3
Silvano Piazza2,3 Giannino Del Sal2,3
Giannino Del Sal2,3 Francesca Persichetti4*
Francesca Persichetti4*- 1International School for Advanced Studies (SISSA), Area of Neuroscience, Trieste, Italy
- 2Laboratorio Nazionale del Consorzio Interuniversitario per le Biotecnologie (LNCIB), Area Science Park, Trieste, Italy
- 3Department of Life Sciences, University of Trieste, Trieste, Italy
- 4Department of Health Sciences, University of Piemonte Orientale, Novara, Italy
- 5Department of Neuroscience and Brain Technologies, Italian Institute of Technology, Genova, Italy
Huntington’s disease (HD) is a fatal, dominantly inherited, neurodegenerative disorder due to a pathological expansion of the CAG repeat in the coding region of the HTT gene. In the quest for understanding the molecular basis of neurodegeneration, we have previously demonstrated that the prolyl isomerase Pin1 plays a crucial role in mediating p53-dependent apoptosis triggered by mutant huntingtin (mHtt) in vitro. To assess the effects of the lack of Pin1 in vivo, we have bred Pin1 knock-out mice with HdhQ111 knock-in mice, a genetically precise model of HD. We show that Pin1 genetic ablation modifies a portion of HdhQ111 phenotypes in a time-dependent fashion. As an early event, Pin1 activity reduces the DNA damage response (DDR). In midlife mice, by taking advantage of next-generation sequencing technology, we show that Pin1 activity modulates a portion of the alterations triggered by mHtt, extending the role of Pin1 to two additional HdhQ111 phenotypes: the unbalance in the “synthesis/concentration of hormones”, as well as the alteration of “Wnt/β-catenin signaling”. In aging animals, Pin1 significantly increases the number of mHtt-positive nuclear inclusions while it reduces gliosis. In summary, this work provides further support for a role of Pin1 in HD pathogenesis.
Introduction
Huntington’s disease (HD) is a dominantly inherited neurodegenerative disorder, characterized by motor impairment and cognitive decline. Neuropathologically, HD presents selective neuronal loss in the striatum associated to astrocytic gliosis that increases with disease progression (Vonsattel et al., 1985). In the later stage of the disease, pathologic alterations have been described in other brain regions, including cerebral cortex, thalamus, and subthalamic nucleus (Vonsattel and DiFiglia, 1998).
The HD mutation alters a polymorphic CAG trinucleotide repeat in the first exon of HTT, the gene encoding for huntingtin (The Huntington’s Disease Collaborative Research Group, 1993). The number of CAG repeats varies between 6 and 35 units on normal chromosomes, whereas on HD chromosomes the repeat is expanded above the pathological threshold of 36 CAGs. This leads to the synthesis of a mutant protein containing an expanded polyglutamine tract at the N-terminus that confers to the protein the tendency to aggregate. Neuronal intranuclear inclusions (NIIs) and neuropil aggregates that stain positive for huntingtin are a clear histopathological marker of the disease (DiFiglia et al., 1997; Gutekunst et al., 1999).
Mutant huntingtin (mHtt) triggers a large plethora of cellular dysfunctions, as reviewed by Zuccato et al. (2010) and Labbadia and Morimoto (2013). Among them, there is transcriptional deregulation as well as induction of nucleolar and endoplasmic reticulum stress (Sugars and Rubinsztein, 2003; Cha, 2007; Carnemolla et al., 2009; Lee et al., 2014). mHtt expression is also associated with extensive Deoxyribonucleic acid (DNA) lesions and the activation of the ATM/ATR DNA damage response (DDR) pathway with consequent phosphorylation of H2AX at the damaged sites and stabilization of p53, which in turn mediates mitochondrial dysfunction and cytotoxicity (Bae et al., 2005; Anne et al., 2007; Illuzzi et al., 2009).
Recently, a genome-scale RNAi screen to search for pathways interfering with mHtt toxicity has identified peptidyl-prolyl cis/trans isomerase NIMA-interacting 1 (Pin1; Miller et al., 2012). siRNA reduction of Pin1 was found to decrease caspase-3 activation induced by the expression of a truncated N-terminal fragment of mHtt in HEK293T cells. Similarly, in mutant STHdhQ111 striatal cell line, knock-down of Pin1 reduced caspase activation triggered by serum deprivation to a level comparable to controls. Furthermore, the motor performance defect in a Drosophila HD model was significantly rescued upon decreased expression of the Pin1 homolog dodo (Miller et al., 2012). Intriguingly, juglone, a potent Pin1 inhibitor, was previously found to reverse the abnormal cellular localization of full-length mHtt in STHdhQ111 striatal cells (Wang et al., 2005).
Phosphorylation—dependent prolyl isomerization triggered by Pin1 represents an essential mechanism in modulating many cellular processes. Pin1 is highly expressed in the brain where it plays a critical role in healthy aging and prevention of age-related neurodegeneration (Liou et al., 2003; Sorrentino et al., 2014). Detailed molecular analyses in mice and cells led to the findings that in Alzheimer’s disease Pin1 regulates amyloid precursor protein (APP) processing and amyloid-beta production (Pastorino et al., 2006) while it is required for Tau dephosphorylation and correct neurofibrillary organization (Liou et al., 2003). Furthermore, in Parkinson’s disease Pin1 facilitates formation of alpha-synuclein inclusions by regulating its binding partner synphilin (Ryo et al., 2006).
We have previously shown that in cell culture and in mouse brain Pin1 activity is required for p53 activation in response to mHtt (Grison et al., 2011). In HD cellular models and human post-mortem brains, p53 is phosphorylated on Ser46, a modification that triggers the interaction with Pin1 and activates p53 apoptotic functions. As a consequence, the genetic ablation of Pin1 prevents the expression of p53 apoptotic target genes in the striatum of HdhQ111 mice (Grison et al., 2011).
HdhQ111 knock-in mice (White et al., 1997) display molecular and behavioral phenotypes that recapitulate, in a time-dependent fashion, many features of the human disease (Fossale et al., 2002; Wheeler et al., 2002; Gines et al., 2003; Lloret et al., 2006; Lynch et al., 2007; Carnemolla et al., 2009; Giralt et al., 2012; Hölter et al., 2013). In the first months of life, initial molecular changes (here referred to as “early phenotypes”) include the activation of DDR and the induction of ribosome biogenesis regulator 1 (Rrs1) expression, a nucleolar protein involved in rRNA biogenesis and endoplasmic reticulum stress. At a later age (“intermediate phenotypes”), mHtt alters the expression of several pathways and genes with a plethora of different functions. In aging animals (“late phenotypes”), NIIs decorate the large majority of striatal neurons while gliosis infiltrate the nervous system as evidenced in human post-mortem brains.
Since Pin1 activity is involved in DDR and formation of protein aggregates, here we have taken advantage of HdhQ111 mice in a Pin1 knock-out genetic background (Grison et al., 2011) to prove that Pin1 modulates multiple HdhQ111 phenotypes throughout the lifespan of the rodent.
Materials and Methods
Mice Strain
Heterozygous (HdhQ7/Q111) knock-in mice in C57BL/6 background (Lloret et al., 2006) were provided by M. MacDonald (Massachusetts General Hospital, Boston, MA, USA). Pin1 knock-out mice in C57BL/6 background (Atchison et al., 2003) were provided by A. Means (Duke University, Durham, NC, USA). HdhQ7/Q111 and Pin1WT/KO mice were crossed to generate double heterozygous mice. These mice were then crossed to generate littermates of different genotypes. Mice used in this study were HdhQ7/Q7, HdhQ111/Q111 (3.5 and 12 months) or HdhQ7/Q111 (24 months) either wild-type (Pin1WT/WT) or homozygous knock-out (Pin1KO/KO) for Pin1. Genotyping for both loci was performed by PCR on tail DNA as described previously (White et al., 1997; Atchison et al., 2003). Mice were balanced with respect to gender except for the gene expression profiling experiments where only female mice were used. HdhQ111:Pin1KO mice appear healthy, do not display any gross physical or behavioral abnormalities and have a normal life span. Animal care, handling and subsequent procedures were performed in accordance with the European Community Council Directive of November 24, 1986 (86/609EEC) and following SISSA Ethical Committee permissions.
RNA Isolation and Real Time PCR
Total RNA was extracted from mouse striata using TRIzol® reagent (Invitrogen) according to manufacturer’s instructions. RNA was quantified (Qubit, Life Technologies) and quality was monitored using a Bioanalyzer (Agilent Technologies).
Rrs1 RT-qPCR was performed with CFX96 TouchTM Real-Time PCR instrument (Bio-Rad), using the iQ SYBR Green Supermix (Bio-Rad) as previously described (Carnemolla et al., 2009). Normalized expression values were calculated using β-actin as the endogenous control.
RNA-Seq and Data Processing
RNAseq was performed by IGA Technology Services, Udine, Italy. Libraries were prepared using the TruSeq RNA Kit (Illumina) and sequenced with a HiSeq2000 sequencer (Illumina) to generate 50bp single reads. For each library, more than 40 million of reads were obtained.
Quality control of RNA-seq reads was performed using FastQC1. Genome mapping was carried out with STAR aligner (v2.3.0, ENSEMBL.mus_musculus.release-75 as reference genome, Dobin et al., 2013). Mapped reads were counted with HTSeq (v0.6,2 Anders et al., 2015). Differential gene expression analysis was performed using the gene raw counts, within the R/Bioconductor edgeR package (Robinson et al., 2010). The bioinformatic pipeline was customized to: (1) estimate the dispersion parameter for each library using the biological group dispersion; (2) identify differentially expressed genes between the animal models (HdhQ111 vs. HdhQ7 in Pin1WT background, Pin1WT vs. Pin1KO in HdhQ111 background); (3) consider log2 (fold-change) ≥0.7 as threshold; and (4) adjust the P-value for multiple testing using the Benjamini-Hochberg correction with a false discovery rate (FDR) ≤0.05. Functional annotation, category and pathway analysis of the differential gene lists was carried out using the R/Bioconductor ClusterProfiler (Yu et al., 2012) package and Ingenuity Pathway Analysis tool (IPA; QIAGEN Silicon Valley, Redwood City, CA, USA3). For ClusterProfiler we calculated the enrichment value (p < 0.05, additional parameters as standard) for biological process, molecular function and cellular component. For IPA, transcripts were associated with biological functions/transcriptional regulators in the Ingenuity Knowledge Base. Right-tailed Fisher’s exact test was used to calculate a p-value determining the probability that each biological function/transcriptional regulator assigned to that data set is due to chance alone. All sequencing data were submitted to the Gene Expression Omnibus (GEO; accession number GSE64478).
Western Blot
Total protein lysates from mouse striatum were extracted using TRIzol® reagent (Invitrogen) according to manufacturer’s instructions, after RNA extraction. Ten microgram (10 μg) of protein lysates were analyzed by western blot. Primary antibodies were: anti γ-H2AX (1:1000, Millipore), anti-β-actin (1:5000, A1978, Sigma) and anti-Pin1 (1:2000, G-8, Santa Cruz Biotech).
Relative quantification of protein bands from western blot scans was performed with ImageJ Software.
Immunohistochemistry
Animals were transcardially perfused with 4% paraformaldehyde in Phosphate-Buffered Saline (PBS) solution (pH 7.4). The brain was rapidly dissected out, post-fixed o/n at 4°C in the same fixative and then kept at 4°C in 30% sucrose/PBS until it had sunk. Using a vibratome (Vibratome 1000 Plus Sectioning System, St Louis, MO, USA), coronal sections of 40 μm thickness were cut throughout the striatum (−3 mm to +3 mm from Bregma) and every 10th sections samples were processed for immunohistochemistry. To block endogenous peroxidase free-floating sections were treated for 10 min with Tris-Buffered Saline (TBS) solution containing 10% methanol and 3% H2O2. After washing in TBS, sections were incubated for 1 h in blocking solution (1% BSA, 10% FCS, 1% fish gelatin) and then o/n at 4°C with the primary antibodies in TBS containing 1% BSA, 0.3% Triton X-100 and 0.1% fish gelatin. Sections were washed and incubated for 1 h with secondary anti-mouse Alexa-488 conjugated antibody (1:2000). Images were captured using a Leica TCS SP2 confocal microscope.
Primary antibodies were: EM48 anti-huntingtin (1:100, MAB5374, Chemicon), anti-glial fibrillary acidic protein (GFAP; 1:1000, G6171, Sigma) and anti-γH2AX (1:500, 05–636, Millipore).
Huntingtin Inclusions and GFAP Fluorescence Quantification
Quantification of nuclear inclusions in 24 month old mice was performed as follow. Three serial coronal sections (0.78–0 mm from bregma) were chosen for each mouse. In each section two areas of the striatum (375 μm2 each), one dorsal and one ventral, were identified using a 40× oil objective. Confocal photomicrograhs of six Z-sections (5 μm) of the selected areas were taken and the total number of nuclear inclusions was counted in the Z-stack images using eCELLence Software (Glance Vision Technologies, Trieste, Italy). The total number of inclusions was counted per mouse and the arithmetic mean of four mice for each genotype was calculated and expressed as number of NI/100 μm2. The same method was applied to evaluate the number of inclusions in piriform cortex and olfactory tubercles regions (40× objective, one area of 375 μm2 per section). GFAP fluorescence intensity was evaluated in the same striatal areas described above with a 20× objective (one area of 750 μm2 per section, three sections per mice, four mice per genotype) using the Leica Confocal Software Histogram Quantification Tool. Pixel mean fluorescence intensity (expressed in arbitrary units) of the selected area was normalized to background fluorescence (region without cells). A paired two-tailed Student’s t-test (Microsoft Excel Software) was used for statistical analysis. Values with p < 0.05 were considered significant.
Statistical Analysis
Depending on the number of groups within the data set, data were analyzed using either the Student’s t-test (two groups) or one-way ANOVA with Tukey’s HSD post hoc test (three groups). The p values < 0.05 were considered statistically significant. Calculated means and standard errors were plotted using the graph tool of Microsoft Excel.
Results
Effects of Loss of Pin1 on HdhQ111 “Early Phenotypes”
A number of alterations have been identified so far as early events in HdhQ111 mice. Among them we have studied the effects of Pin1 activity on DDR and Rrs1 induction. These phenotypes were assessed by analyzing homozygous HdhQ111 mice that were either wild-type (HdhQ111:Pin1WT) or knock-out for Pin1 (HdhQ111:Pin1KO) at 3.5 months of age.
Pin1 Modulates DDR Induced by mHtt in Mouse Striatum
Many studies in cell lines, transgenic mouse models and HD post-mortem brains have shown that mHtt can induce DNA damage (Giuliano et al., 2003; Anne et al., 2007; Stack et al., 2008; Illuzzi et al., 2009). Importantly, Pin1 has also been linked to DDR, particularly in the regulation of double strand break repair (Steger et al., 2013). Therefore, we compared the level of DNA damage in HdhQ111:Pin1WT and HdhQ111:Pin1KO mice. For this purpose we assayed the DNA damage marker γH2AX by immunofluorescence and western blot analysis (Figures 1A–C). Phosphorylation of the histone variant H2AX, forming γH2AX-labeled foci, represents the earliest visible response to the induction of DNA double-strand breaks. As expected, protein extracts from striatum of HdhQ111 mice showed high levels of phosphorylated H2AX compared to wild-type littermates, supporting the presence of DNA damage in neurons of 3.5 month old HdhQ111 mice (Figure 1A). Interestingly, the lack of Pin1 expression decreased the intensity of γH2AX signal (Figures 1B–C). Western blot quantification of γH2AX relative to β-actin indicated that, in absence of Pin1, the expression of phosphorylated H2AX is significantly reduced by about 20% (Figure 1B). These results suggest that Pin1 modulates striatal DNA damage induced by mHtt.
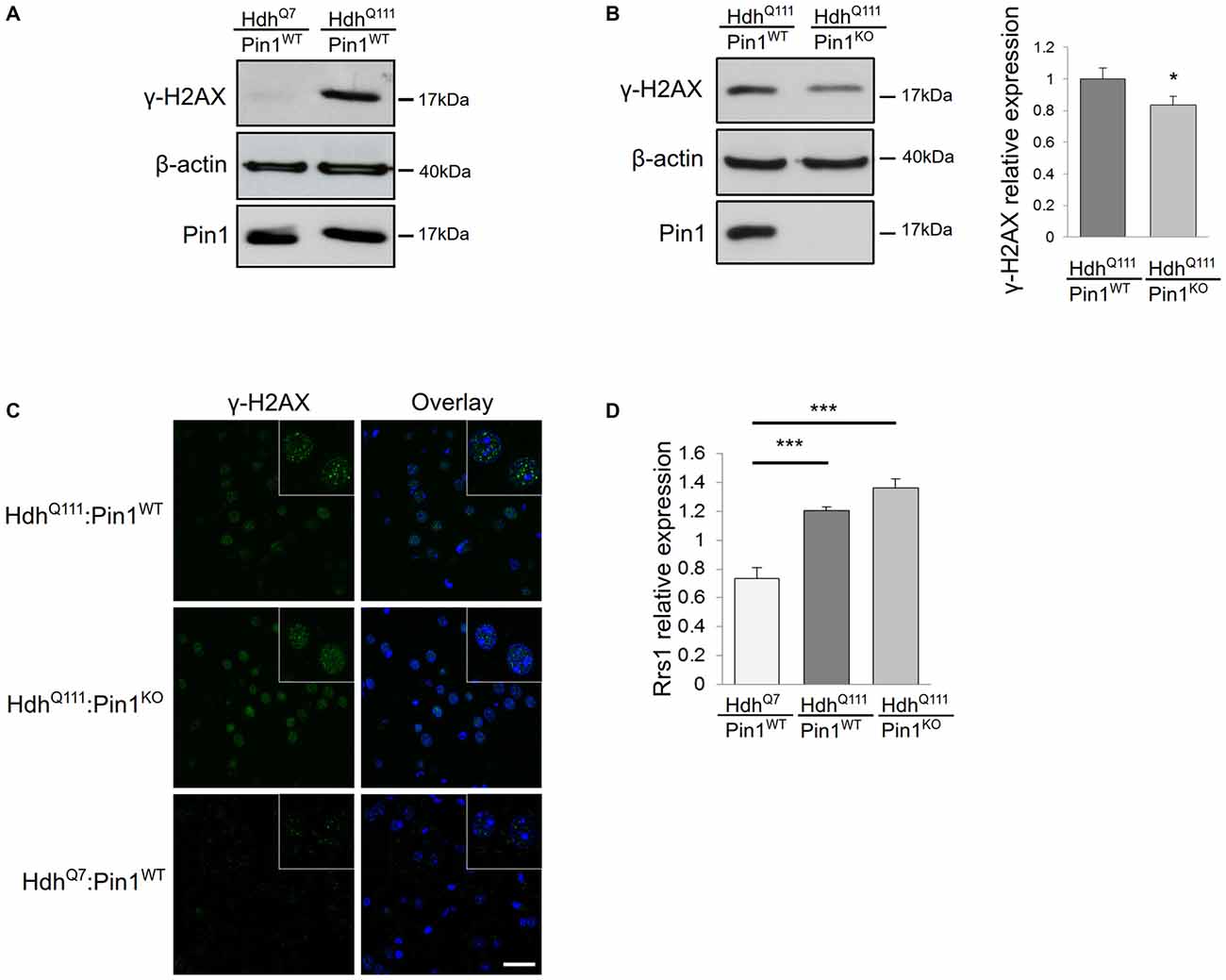
Figure 1. Consequences of Pin1 depletion on HdhQ111 “early phenotypes”. (A–C) Pin1 modulates mutant huntingtin (mHtt)-induced DNA damage. (A) Representative western blot showing γH2AX protein levels in lysates from striatum of homozygous HdhQ111 and HdhQ7 mice at 3.5 months of age. β-actin antibody was used as control of sample loading. (B) Representative western blot showing comparison of γH2AX protein levels in striatum of HdhQ111 mice on wild-type (n = 6) or knock-out (n = 6) Pin1 genetic backgrounds at 3.5 months of age. β-actin was used as a loading control. The histogram on the right shows relative quantifications of γH2AX signals between the two genotypes. Histogram bars represent mean ± standard error (*Two tail paired t-test p < 0.05). (C) Comparison of immunofluorescence staining of γH2AX foci in coronal brain sections from striatum of 3.5 months mice of the indicated genotypes. The nuclear staining with 4′,6-diamidino-2-phenylindole (DAPI) is shown in blue. Scale bar, 30 μm. Inset shows γH2AX foci at higher magnification. Scale bar, 10 μm. (D) Rrs1 mRNA induction is not modulated by Pin1. RT-qPCR analysis was performed to detect levels of Rrs1 relative to β-actin mRNAs in striatum of HdhQ7 (n = 6) and homozygous HdhQ111 mice on wild type (n = 6) or knock out (n = 6) Pin1 genetic background at 3.5 months of age. A significant increase of Rrs1 mRNA is found in HdhQ111:Pin1WT and HdhQ111:Pin1KO mice compared to HdhQ7:Pin1WT mice. No significant difference in the level of Rrs1 mRNA is detected in HdhQ111:Pin1WT compared to HdhQ111:Pin1KO mice. Histogram bars represent mean ± standard error (***one-way ANOVA Tukey HSD post hoc test, p < 0.001).
Rrs1 Induction by mHtt is not Modulated by Pin1
We have previously demonstrated that Rrs1 mRNA expression is increased in post-mortem brain of HD patients (Fossale et al., 2002). Rrs1 is a nucleolar protein involved in rRNA biogenesis and a component of the endoplasmic reticulum stress response (Carnemolla et al., 2009). Its induction is one of the earliest events occurring in HdhQ111 mice that persist throughout the life span of the rodent (Fossale et al., 2002; Carnemolla et al., 2009). To test whether Pin1 could modulate Rrs1 expression in HdhQ111 mice, we compared the expression level of Rrs1 by RT-qPCR in HdhQ111:Pin1WT and HdhQ111:Pin1KO at 3.5 months of age. As expected, Rrs1 is induced by 1.6 fold in HdhQ111 compared to wild type littermates in a Pin1 wild type background (Figure 1D). No significant difference was detectable between HdhQ111:Pin1WT and HdhQ111:Pin1KO mice.
In summary these results suggest that Pin1 may modify some but not all of the “early phenotypes” selected for assessment in HdhQ111 mice.
Effects of Lack of Pin1 on HdhQ111 “Intermediate Phenotypes”
A plethora of genes and pathways are known to be altered in the striatum of midlife HD mouse models. To estimate the portion of HdhQ111 “intermediate phenotypes” that requires a functional Pin1, we decided to carry out an unbiased transcriptome-wide approach by taking advantage of next generation sequencing technology.
We thus carried out a search for differentially expressed transcripts in the following genotypes: HdhQ7/Q7:Pin1WT/WT, HdhQ111/Q111:Pin1WT/WT, HdhQ7/Q7:Pin1KO/KO and HdhQ111/Q111:Pin1KO/KO. RNAs from the striatum of three mice of 12 months of age were purified for each genotype to carry out gene expression profiling with next-generation sequencing technology with the MiSeq Illumina platform and analyzed following standard best practices.
First we compared HdhQ111 with HdhQ7 mice on a Pin1 wild-type background to describe differences in gene expression triggered by mHtt. Overall we obtained a list of 621 genes significantly altered by mHtt (Figure 2A, Supplementary Table T1). We then asked whether lack of a functional Pin1 modifies the expression pattern of HdhQ111 mice identifying 145 genes as differentially regulated between Pin1 wild-type and knock-out mice on a mHtt background.

Figure 2. Transcriptome functional analysis in the striatum of midlife mice suggests that loss of Pin1 reverts biological functions altered by mutant huntingtin (mHtt). (A) Venn diagram showing the overlap of differentially expressed gene lists among the comparisons of the indicated genotypes (Supplementary Table T2). (B) Venn diagram showing the overlap of the biological terms among the comparisons of the indicated genotypes (Supplementary Table T4). (C) Table showing the predicted activation and repression of different biological terms using Ingenuity Pathway Analysis (IPA). Activation Z-score and –log (p-value) are shown. Activation Z-score makes predictions about potential regulators by using information about the direction of gene regulation. Red or blue box correspond to activation or repression prediction of the biological pathways.
Interestingly, 44% of them (64/145, overlap p-value = 10−64) were commonly targeted by mHtt in a wild-type genotype for Pin1 suggesting that Pin1 may act on pathways shared with mHtt. In the same stage and tissue, loss of Pin1 in a wild-type Htt background was altering the expression of 213 genes.
In summary, 29 genes (out of 64) were differentially regulated in all three comparisons while 35 of the genes altered in HdhQ111 mice were changed by loss of Pin1 activity exclusively in mHtt but not in HdhQ7 mice (Figure 2A, Supplementary Tables T1, T2).
We then analyzed the enrichment for Gene Ontology biological terms in the three differentially expressed gene lists using ClusterProfiler (Supplementary Table T3). While mHtt changed the expression of genes associated to 1493 biological terms, the effects of Pin1 loss on mHtt were described with 194 terms. Importantly, 156 of them were common in the two lists (Figure 2B). Loss of Pin1 in a wild-type Htt background was involving 242 biological terms.
In summary, loss of Pin1 in HdhQ111 background affected only 10% (156/1493) of the biological terms targeted by mHtt alone. These represented 80% (156/194) of all the biological terms associated to loss of Pin1 in mHtt background. Seventy five (out of 156) were common among all three comparisons and 81 were part of a Pin1-dependent gene expression pattern exclusively triggered by mHtt (Supplementary Table T4).
Since this type of analysis does not allow the prediction of the activation/inhibition status for each biological term, we took advantage of the IPA Downstream Effects Analysis tool to identify biological functions that are expected to increase or decrease according to gene expression changes. By comparing the gene lists in Supplementary Table T1 we observed enriched biological functions in opposite directions (activation/inhibition) when induced by mHtt on a Pin1 wild-type background compared to loss of Pin1 in a mHtt background (Pearson’s Correlation = 0.69, p-value = 10−10; Figure 2C). Among them, the biological functions “quantity/synthesis of steroid” and “synthesis/concentration of hormone” triggered by mHtt were reverted by loss of Pin1. These two pathways were not significantly affected in Pin1 knock-out mice in a wild-type Htt background. Loss of Pin1 in HdhQ111 mice was able to counteract changes in “Wnt/β-catenin signaling”, “PCP pathway” and “Role of Wnt/GSK-3β signaling in the pathogenesis of influenza” as triggered by mHtt. These pathways were also altered in Pin1 knock-out mice that were wild-type for Htt (Figure 2C). In Supplementary Table T5 gene lists associated to each pathways and biological functions are shown.
Effects of Lack of Pin1 on HdhQ111 “Late Phenotypes”
Aging HdhQ111 mice recapitulate two neuropathological hallmarks of human HD post-mortem brains: the presence of NIIs in striatal neurons and the occurrence of massive gliosis. Therefore we investigate these phenotypes in heterozygous HdhQ111 mice either wild-type (HdhQ111:Pin1WT) or knock-out (HdhQ111:Pin1KO) for Pin1 at 24 months of age.
Mutant Huntingtin NIIs in Mouse Striatum are Increased in the Absence of Pin1
NIIs of amino-terminal fragments of mHtt are a well-known histopathological marker of HD (DiFiglia et al., 1997; Gutekunst et al., 1999). Pin1 has been previously found to modulate protein aggregate formation in other neurodegenerative diseases (Lu et al., 1999; Ryo et al., 2006; Kesavapany et al., 2007). Coronal sections of mouse brains were immunostained with EM48 antibody that selectively labels mHtt. As shown in Figure 3A, mHtt was detectable in striatum of both genotypes as nuclear inclusions. Interestingly, the number of NIIs was significantly increased in HdhQ111 mice lacking Pin1 compared to HdhQ111:Pin1WT mice (Figure 3B). At 24 months of age, NIIs were also detectable in brain area other than the striatum including the olfactory tuberculus and piriform cortex (Supplementary Figure S1). However, no differences were detected in these regions between HdhQ111:Pin1WT and HdhQ111:Pin1KO mice (Figure 3C). In summary, Pin1 loss increases NIIs formation selectively in the striatum proving that Pin1 plays a role in NIIs formation in vivo.
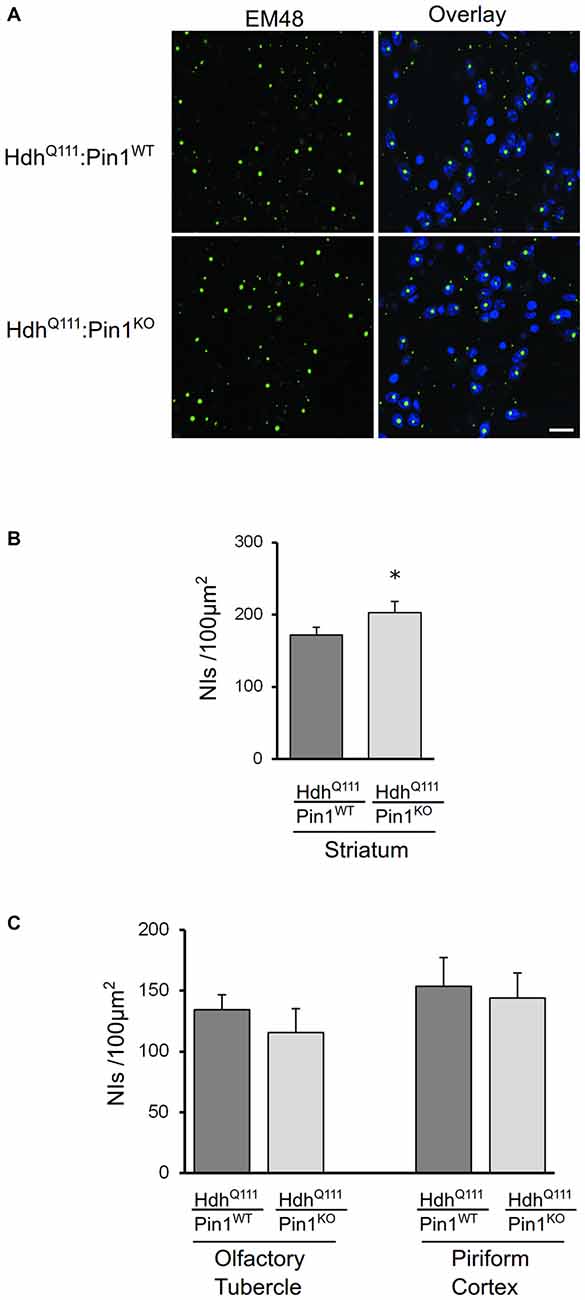
Figure 3. The number of mHtt intranuclear inclusions in aged mice is modulated by Pin1 in striatum but not in olfactory tubercle and piriform cortex. (A) Representative confocal images of EM48 positive neuronal intranuclear inclusions (NIIs) from striatal sections of 24 months mice (genotypes as indicated). The nuclear staining with 4′, 6-diamidino-2-phenylindole (DAPI) is shown in blue. Scale bar, 20 μm. (B) The number of NIIs in striatum of heterozygous HdhQ111 mice lacking of Pin1 is significantly increased compared to HdhQ111:Pin1WT mice. The histogram shows quantification of EM48-positive inclusions in striatum of HdhQ111 mice on wild-type or knock-out Pin1 genotype. Histogram bars represent mean ± standard error of the number of inclusions per 100 μm2, three sections per mouse and four mice per genotype. (*Two tail paired t-test p < 0.05). (C) Histograms showing quantification of EM48-positive inclusions in olfactory tubercle (left) and piriform cortex (right) of HdhQ111 mice on wild-type (n = 4) or knock-out (n = 4) Pin1 genotype at 24 months of age. No significant differences are observed in these brain regions between the two genotypes.
Astrocytic Gliosis in Striatum of HdhQ111 Mice is Reduced in the Absence of Pin1
Astrocytic gliosis, an important hallmark of HD pathogenesis, is recapitulated in aged HdhQ111 mice (Wheeler et al., 2002). To assess whether Pin1 may modulate the proliferation of astrocytes in HdhQ111 mice, we carried out immunofluorescence analysis using the GFAP antibody in mouse brain sections. First we compared HdhQ111 mice with HdhQ7 wild-type littermates, both on a Pin1 wild-type background, at 24 months of age. As expected, a fibrillary network intensely stained by GFAP antibody is shown in the striatum of mutant knock-in mice, but not in their wild-type counterpart (Figure 4A). Interestingly, GFAP immunoreactivity was markedly reduced in striatum of HdhQ111:Pin1KO mice compared to littermates HdhQ111 expressing Pin1 (Figure 4B). To better assess these differences we quantified the fluorescence intensity of GFAP. As shown in Figure 4C we found a significant reduction of GFAP immunoreactivity in HhdQ111 mice lacking Pin1.
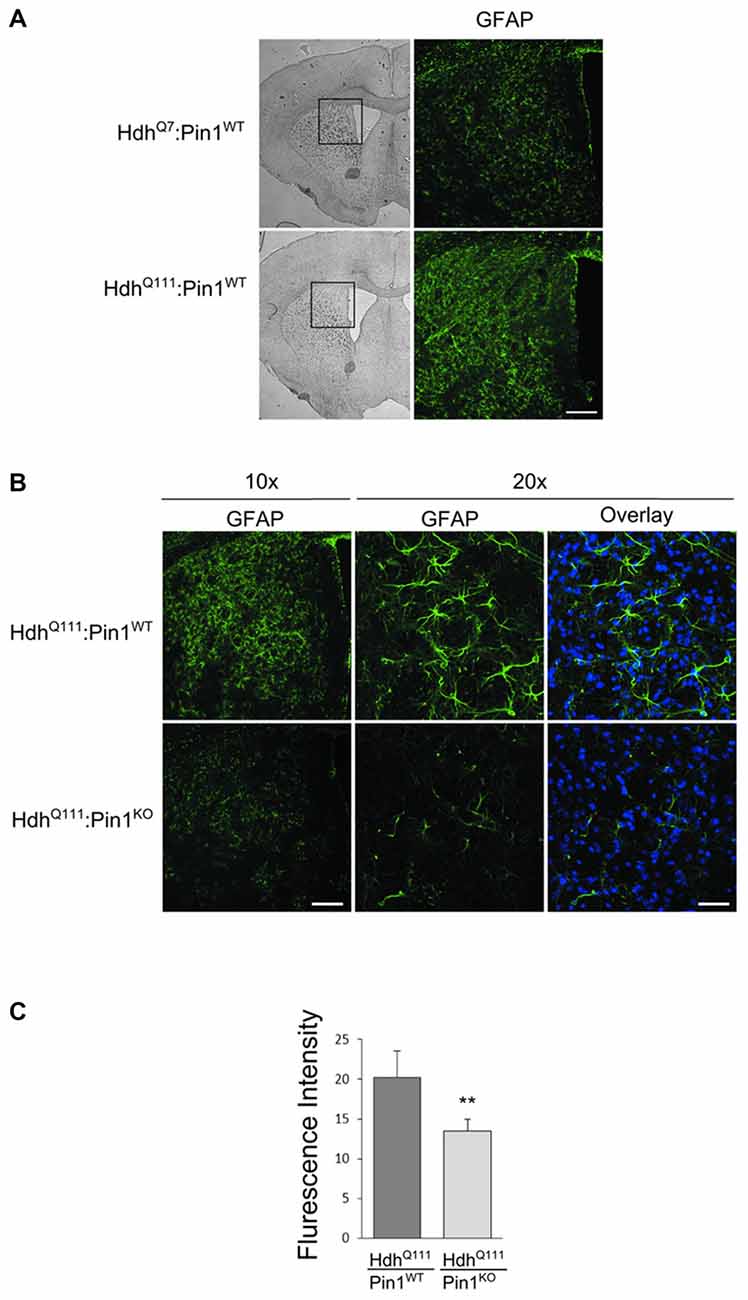
Figure 4. Lack of Pin1 reduces astrocytic gliosis in striatum of aged HdhQ111 mice. (A) Comparison of glial fibrillary acidic protein (GFAP) immunostaining between striatum of heterozygous HdhQ111 and wild-type HdhQ7 mice at 24 months of age. Left panels show representative bright-field images of mouse brain coronal sections (2.5x). The region of the striatum highlighted in the box is shown in the right panels (10x) after immunostaining with GFAP antibody. GFAP reactivity indicates reactive astrocytic gliosis in mutant compared to wild-type striatum. Scale bar, 100 μm. (B) Representative confocal images of GFAP staining at two different magnification (10x − 20x) of striatal sections from heterozygous HdhQ111 mice on wild-type or knock-out Pin1 genetic background. GFAP reactivity reveals reduced astrocytic gliosis in HdhQ111 mice depleted of Pin1 compared to Pin1 wild type mice. The nuclear staining with 4′,6-diamidino-2-phenylindole (DAPI) is shown in blue. (Scale bars: panel 10x = 100 μm, panel 20x = 40 μm). (C) Quantification (arbitrary units) of GFAP fluorescent intensity in striatum of HdhQ111 mice on wild-type or knock-out Pin1 genetic background. Histogram bars represent mean ± standard error, three sections per mouse and four mice per genotype. Data indicate significant reduction of GFAP reactivity in HdhQ111 mice lacking of Pin1 (**Two tail paired t-test p < 0.01).
These results suggest that Pin1 is involved in astrocytosis triggered by mHtt.
Discussion
In the quest for understanding HD pathogenesis, the use of animal models has been instrumental in evaluating the impact of manipulating the expression of a single gene or the activity of entire pathways on the hallmarks of disease progression.
HdhQ111 mice present several advantages. This genetically precise HD mouse model shows different molecular phenotypes at different ages. The main interest resides in the identification of early alterations (“early phenotypes”) that cannot be studied in human tissues. “Late phenotypes” such as the formation of NIIs and the presence of massive gliosis mimic neuro-pathological data in human post-mortem brains.
By breeding HdhQ111 knock-in mice with a knock-out line for Pin1, we assessed the effects of the lack of a functional Pin1 on selected phenotypes evident at early, mid and late stage of life of diseased mice. The selection of phenotypes was based on different objectives according to the age of the rodent.
As “early phenotypes” we specifically analyzed two events: the induction of DDR and the expression of Rrs1. mHtt is associated with DNA damage and its phosphorylation status is crucial for cellular handling of genomic stress (Anne et al., 2007). DDR leads to ATM kinase activation followed by phosphorylation of H2AX at genomic sites of double strand breaks and consequent stabilization and activation of p53. This pathway is a hallmark of the disease since H2AX-positive sites have been found in the striatum of HD human post-mortem brains and recapitulated in PC12 cells expressing a pathogenic fragment of mHtt, in HD striatal cell lines (STHdhQ111) and in the striatum of R6/2 mice (Anne et al., 2007; Illuzzi et al., 2009). Here we provide evidence that DDR in the striatum of HdhQ111 knock-in mice is significantly reduced in the absence of Pin1. In this context a crucial Pin1 target is p53. We previously showed that in the striatum of 12 month old HdhQ111 mice the p53 protein is increased and phosphorylated on Ser46, mimicking p53 status in HD post-mortem brains. This promotes p53/Pin1 interaction and its transcriptional activity targeted to p21WAF and PUMA. p53 transcriptional property is strictly dependent on a functional Pin1 since HdhQ111 mice that are knock-out for Pin1 lack p21WAF and PUMA induction (Grison et al., 2011). Apoptosis induced by mHtt is mediated at least in part by p53 since it was reduced by 50% upon p53 silencing (Bae et al., 2005). A similar effect was observed upon silencing of Pin1 in the same experimental settings. Importantly, neuronal loss observed at 24 months in Pin1 knock-out mice was not increased in HdhQ111:Pin1KO mice suggesting that in mice devoid of Pin1 the lack of p53 activation prevents mHtt-dependent neurodegeneration (Grison et al., 2011).
We also analyzed the effects of Pin1 loss on the induction of Rrs1. We chose this molecular event since Rrs1 is a nucleolar protein involved in rRNA biogenesis and associated to endoplasmic reticulum stress. Both pathways are altered in several neurodegenerative diseases suggesting they might play an important role in neurodegeneration. We previously reported that Rrs1 induction was evident for the entire first year of life (both “early” and “intermediate phenotypes”; Carnemolla et al., 2009). Here we assess that Pin1 is dispensable for Rrs1 overexpression both at 3.5 (Figure 1D) and at 12 months (data not shown).
In summary these results suggest that Pin1 is involved in some but not all pathways altered early in HdhQ111 mice.
This observation led us to ask a crucial question: given that Pin1 participates in selected pathways triggered by mHtt, may we estimate and identify the fraction of HdhQ111 phenotypes that depend on a functional Pin1? We reasoned that the best strategy to assess this fundamental feature was to take advantage of next generation sequencing technology to carry out an unbiased transcriptome-wide approach. To study the largest number of pathways in a single experiment we choose to analyze 12 months old mice (“intermediate phenotype”). At this age mHtt can trigger a large number of alterations in the striatum while events are still probably far from being at the final irreversible disease stage.
We thus compared genes and associated biological terms that are altered in mHtt with those changing in absence of Pin1. By this approach we obtained two pieces of information. First, 10% (156/1493) of the biological terms affected by mHtt are common with those targeted by deleting Pin1 in HdhQ111 mice. This suggests that Pin1 is involved in a small portion of mHtt phenotypes. However, 44% (64/145) of the genes and 80% (156/194) of biological terms associated to loss of Pin1 are contained in the list of those targeted by mHtt, proving that the majority of pathways regulated by Pin1 in HdhQ111 mice are common with those targeted by mHtt in a Pin1 wild-type background. Half of these genes and biological terms were also changed in Pin1 knock-out mice with a wild-type Htt background.
We then showed that loss of Pin1 activity in HdhQ111 mice exerts opposite effects to those triggered by mHtt for selected pathways and functions. Among them there are “quantity/synthesis of steroids” and “synthesis/concentration of hormones”, two previously known pathways altered in HD. Concerning hormones, prolactin (PRL) and growth hormone (GH) have been found down-regulated in the plasma of HD patients (Markianos et al., 2009; Saleh et al., 2009). They are part of the hypothalamic-pituitary system that is targeted in HD affecting food intake and energy balance. Other genes decreased in HdhQ111 mice include arginine vasopressin (AVP) and proopiomelanocortin (POMC), a precursor protein of multiple peptide hormones including adrenocorticotropic hormone (ACTH) and melanocyte-stimulating hormone (MSH). While these pathways and functions are not significantly altered in Pin1 knock-out mice in a Htt wild-type background, selected genes such as PRL and GH are among the most differentially expressed genes in this comparison. Overall, these results may lead to further studies concerning the still unexplored role of Pin1 as a regulator of neuropeptides and hormone synthesis/release in health and diseased conditions.
“Wnt/β-catenin signaling”, “PCP pathway” and “Role of Wnt/GSK-3β signaling in the pathogenesis of influenza” are all changed in mHtt mice (repression) but in opposite direction when Pin1 is depleted (activation). Several evidences support the notion that Wnt/β-catenin signaling is altered in HD although contrasting data and interpretations have been proposed (Carmichael et al., 2002; Gines et al., 2003; Godin et al., 2010; Valencia et al., 2010; Dupont et al., 2012; Lim et al., 2014). In our study Wnt9b, Wnt8b and Wnt4 levels are all decreased in the striatum of HdhQ111 mice. Furthermore, there is a significant reduction in the expression of the receptor Fzd10 and of the transcription factor Lef1. Interestingly, one of the genes induced at the highest level is Wilms tumor suppressor gene (Wt1), a well-known regulator of the β-catenin pathway. Pin1 has been shown to regulate Wnt/β-catenin signaling pathways at least in two regulatory steps. In neuronal progenitor cells, Pin1 binds and stabilizes its substrate β-catenin and this interaction is required for appropriate neuronal differentiation (Nakamura et al., 2012). In the brain Pin1 also binds GSK-3β inhibiting its kinase activity and this is believed to play a fundamental role in promoting APP turnover (Lu et al., 1999). In the differential gene expression analysis of Pin1 knock-out mice in a Htt wild-type background all three pathways related to Wnt signaling were significantly enriched. Loss of Pin1 might thus interfere with Wnt signaling pathways at distinct regulatory steps both in wild-type and HdhQ111 mice.
As “late phenotypes” we focused our attention on the formation of NIIs and gliosis in HdhQ111 mice that both mimic neuropathological data in human post-mortem brains. It is therefore particularly interesting that both of them are modified by Pin1 loss.
NIIs are produced from the monomeric forms of mHtt through the generation of various intermediate species. The diverse toxicity of these structures is however still controversial and is discussed in several reviews (Hands and Wyttenbach, 2010; Arrasate and Finkbeiner, 2012). In brief, it has been suggested that intermediate aggregates are more toxic than inclusions. In turn these might represent a protective mechanism to sequester toxic misfolded species of mHtt that would otherwise impair cellular viability. However, in the long run, inclusions may become toxic as well, by disrupting cellular transport or sequestering protective soluble proteins.
Here we show that loss of Pin1 significantly enhances the number of NIIs specifically in the striatum of HdhQ111 mice. The mechanism that promotes the formation of inclusions in striatal neurons when Pin1 is depleted remains unclear. Pin1 exerts its activity on different substrates in different physiological and/or pathological conditions while multiple cellular pathways are involved in the aggregation process of proteins with an expanded polyglutamine tract. Indeed, conformational changes and aggregation of mHtt may be modulated by interacting partners, posttranslational modifications of the protein, as well as by alteration in the homeostasis of protein degradation systems such as UPS and autophagy. In this context, in Parkinson’s disease Pin1 has been shown to regulate alpha-synuclein aggregation by modifying the activity of the alpha-synuclein binding partner synphilin (Ryo et al., 2006). In Alzheimer’s disease Pin1 participates in APP processing and neurofibrillary tangle formation (Pastorino et al., 2006). Importantly, in Hdh knock-in mice we found the Pin1 effect visible only in striatal cells suggesting a cell-type specific activity in the very same neurons that undergo degeneration.
Proliferation of glial cells is a prominent feature of HD pathology. Reactive astrocytic gliosis is selectively detected in HD striatum starting from Grade 0 of pathology (Vonsattel et al., 1985). Several mechanisms could account for the loss of gliosis in a Pin1 knock-out background. This phenotype may depend on the established role of Pin1 in cell proliferation control and therefore be due to a cell autonomous function of Pin1 in astrocytes. However we may also speculate that this may be a secondary effect of Pin1 function in neurons. Therefore, the role of Pin1 in gliosis remains a very interesting topic to be further investigated.
It is not clear whether Pin1 effects in NIIs formation and gliosis are the effects of loss of Pin1 activity throughout the lifespan of the rodent or are the consequence of specific functions in aging mice. This observation is indeed valid for “intermediate phenotypes” as well. The use of Pin1 inhibitors in selected time windows may provide the correct answer to this important question.
In summary, we have shown that Pin1 participates in a portion of the processes at the core of HD pathogenesis in vivo. Further studies will evaluate the impact of these molecular phenotypes on the behavioral alterations observed in HD mice models.
Author Contributions
EA, SG, GDS, and FP designed research; EA, SM, MM, AC, PV, PR, and SZ performed research; EA, YC, SP, CS and FP analyzed data; FM, GL and GDS contributed reagents, methods and analytic tools; SG, GDS and FP wrote the manuscript; FP supervised the entire project; EA and SM have contributed equally to this work. All authors listed, have made substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by the Telethon Foundation (Grant No. GGP07185) to FP and GDS and by the Italian Ministry of Education, University and Research (FIRB Grant prot. RBAP11FRE9) to FP and SG.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Cristina Leonesi for technical support and Valter Vanzella (GlanceVision Technologies S.r.l.) for the help with the eCELLence Software. We are grateful to SISSA technical and administrative staff.
Footnotes
- ^ www.bioinformatics.babraham.ac.uk/projects/fastqc/
- ^ http://www-huber.embl.de/users/anders/HTSeq
- ^ www.ingenuity.com.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fncel.2016.00110/abstract
Supplementary Figure S1 | Representative confocal images of EM48 positive NIIs from piriform cortex (A) and olfactory tubercle (B) of 24 months mice (genotypes as indicated). The nuclear staining with 4′,6-diamidino-2-phenylindole (DAPI) is shown in blue. Scale bar, 20 μm.
Supplementary Table T1 | Differential gene expression in the striatum of HdhQ111 vs. HdhQ7 (Pin1WT background), Pin1WT vs. Pin1KO (HdhQ111 background) and Pin1WT vs. Pin1KO (HdhQ7 background) mice. Significantly differentially expressed gene lists were computed using EdgeR software (see methods). For each gene table we selected the genes with adjust p value < 0.001 and log fold change (logFC) higher than 0.7 or smaller than −0.7. A list of 621 genes (116 increase and 505 decreased) are identified as significantly altered in HdhQ111 vs. HdhQ7 (Pin1WT background) and a list of 145 genes (48 increased and 97 decreased) are identified as differentially regulated in Pin1WT vs. Pin1KO (HdhQ111 background). By comparing Pin1WT vs. Pin1KO (HdhQ7 background) 213 genes (121 increased and 92 decreased) are identified as significantly altered.
Supplementary Table T2 | Comparison of differentially expressed gene lists. By comparing the gene lists in Table T1 we obtained seven new list of genes. The column “n=29 Overlaps” contains the genes that are common among the three gene lists of Table T1. The column “n=85 Overlaps” contains the genes that are common between HdhQ111 vs. HdhQ7 (Pin1WT) and Pin1WT vs. Pin1KO (HdhQ7) lists. The column “n=35 Overlaps” contains the genes that are common between HdhQ111 vs. HdhQ7 (Pin1WT) and Pin1WT vs. Pin1KO (HdhQ111) lists. The column “n=33 Overlaps” contains the genes that are common between Pin1WT vs. Pin1KO (HdhQ111) and Pin1WT vs. Pin1KO (HdhQ7). The column “n=472 Specific” contains the genes that are specific for mHtt. The column “n=48 Specific” contains the genes that are specific for loss of Pin1 in a mHtt background. The column “n=66 Specific” contains the genes that are specific for loss of Pin1 in a wild-type Htt background.
Supplementary Table T3 | Functional analysis of differentially expressed genes lists. Differentially expressed gene lists in Table T1 were analyzed for functional associations with ClusterProfiler. All mouse genes from “org.Mm.eg.db” package were used as background population. Each table reports statistical data and the gene list associated with each GO term. GO terms are based on the Gene Ontology Consortium available at: http://www.geneontology.org/.
Supplementary Table T4 | Comparison of functional analysis lists. By comparing the functional analysis lists in Table T3 we obtained seven new lists: 1) “n=75 Overlaps” contains 75 GO terms that are common among the three lists of Table T3; 2) “n=135 Overlaps” contains 135 GO terms that are common between HdhQ111 vs. HdhQ7 (Pin1WT) and Pin1WT vs. Pin1KO (HdhQ7) lists; 3) “n=81 Overlaps” contains 135 GO terms that are common between HdhQ111 vs. HdhQ7 (Pin1WT) and Pin1WT vs. Pin1KO (HdhQ111) lists; 4) “n=3 Overlaps” contains 3 GO terms that are common between Pin1WT vs. Pin1KO (HdhQ111) and Pin1WT vs. Pin1KO (HdhQ7); 5) “n=1202 Specific” contains contains 1202 GO terms specific for HdhQ111 vs. HdhQ7 (Pin1WT); 6) “n=35 Specific” contains 35 GO terms specific for Pin1WT vs. Pin1KO (HdhQ111); 7) “n=29 Specific” contains 29 GO terms specific for Pin1WT vs. Pin1KO (HdhQ7).
Supplementary Table T5 | Activation and repression of biological terms with Ingenuity Pathway Analysis. This table shows the predicted activation and repression of different biological terms obtained from Table T1 using IPA. For each table, the gene list associated to each pathway or function is reported.
References
Anders, S., Pyl, P. T., and Huber, W. (2015). HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. doi: 10.1093/bioinformatics/btu638
Anne, S. L., Saudou, F., and Humbert, S. (2007). Phosphorylation of huntingtin by cyclin-dependent kinase 5 is induced by DNA damage and regulates wild-type and mutant huntingtin toxicity in neurons. J. Neurosci. 27, 7318–7328. doi: 10.1523/jneurosci.1831-07.2007
Arrasate, M., and Finkbeiner, S. (2012). Protein aggregates in Huntington’s disease. Exp. Neurol. 238, 1–11. doi: 10.1016/j.expneurol.2011.12.013
Atchison, F. W., Capel, B., and Means, A. R. (2003). Pin1 regulates the timing of mammalian primordial germ cell proliferation. Development 130, 3579–3586. doi: 10.1242/dev.00584
Bae, B.-I., Xu, H., Igarashi, S., Fujimuro, M., Agrawal, N., Taya, Y., et al. (2005). p53 mediates cellular dysfunction and behavioral abnormalities in Huntington’s disease. Neuron 47, 29–41. doi: 10.1016/j.neuron.2005.06.005
Carmichael, J., Sugars, K. L., Bao, Y. P., and Rubinsztein, D. C. (2002). Glycogen synthase kinase-3β inhibitors prevent cellular polyglutamine toxicity caused by the Huntington’s disease mutation. J. Biol. Chem. 277, 33791–33798. doi: 10.1074/jbc.m204861200
Carnemolla, A., Fossale, E., Agostoni, E., Michelazzi, S., Calligaris, R., De Maso, L., et al. (2009). Rrs1 is involved in endoplasmic reticulum stress response in Huntington disease. J. Biol. Chem. 284, 18167–18173. doi: 10.1074/jbc.m109.018325
Cha, J.-H. J. (2007). Transcriptional signatures in Huntington’s disease. Prog. Neurobiol. 83, 228–248. doi: 10.1016/j.pneurobio.2007.03.004
DiFiglia, M., Sapp, E., Chase, K. O., Davies, S. W., Bates, G. P., Vonsattel, J. P., et al. (1997). Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277, 1990–1993. doi: 10.1126/science.277.5334.1990
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. doi: 10.1093/bioinformatics/bts635
Dupont, P., Besson, M.-T., Devaux, J., and Liévens, J.-C. (2012). Reducing canonical Wingless/Wnt signaling pathway confers protection against mutant Huntingtin toxicity in Drosophila. Neurobiol. Dis. 47, 237–247. doi: 10.1016/j.nbd.2012.04.007
Fossale, E., Wheeler, V. C., Vrbanac, V., Lebel, L.-A., Teed, A., Mysore, J. S., et al. (2002). Identification of a presymptomatic molecular phenotype in Hdh CAG knock-in mice. Hum. Mol. Genet. 11, 2233–2241. doi: 10.1093/hmg/11.19.2233
Gines, S., Seong, I. S., Fossale, E., Ivanova, E., Trettel, F., Gusella, J. F., et al. (2003). Specific progressive cAMP reduction implicates energy deficit in presymptomatic Huntington’s disease knock-in mice. Hum. Mol. Genet. 12, 497–508. doi: 10.1093/hmg/ddg046
Giralt, A., Puigdellívol, M., Carretón, O., Paoletti, P., Valero, J., Parra-Damas, A., et al. (2012). Long-term memory deficits in Huntington’s disease are associated with reduced CBP histone acetylase activity. Hum. Mol. Genet. 21, 1203–1216. doi: 10.1093/hmg/ddr552
Giuliano, P., De Cristofaro, T., Affaitati, A., Pizzulo, G. M., Feliciello, A., Criscuolo, C., et al. (2003). DNA damage induced by polyglutamine-expanded proteins. Hum. Mol. Genet. 12, 2301–2309. doi: 10.1093/hmg/ddg242
Godin, J. D., Poizat, G., Hickey, M. A., Maschat, F., and Humbert, S. (2010). Mutant huntingtin-impaired degradation of β-catenin causes neurotoxicity in Huntington’s disease. EMBO J. 29, 2433–2445. doi: 10.1038/emboj.2010.117
Grison, A., Mantovani, F., Comel, A., Agostoni, E., Gustincich, S., Persichetti, F., et al. (2011). Ser46 phosphorylation and prolyl-isomerase Pin1-mediated isomerization of p53 are key events in p53-dependent apoptosis induced by mutant huntingtin. Proc. Natl. Acad. Sci. U S A. 108, 17979–17984. doi: 10.1073/pnas.1106198108
Gutekunst, C. A., Li, S. H., Yi, H., Mulroy, J. S., Kuemmerle, S., Jones, R., et al. (1999). Nuclear and neuropil aggregates in Huntington’s disease: relationship to neuropathology. J. Neurosci. 19, 2522–2534. doi: 10.1093/hmg/8.7.1227
Hands, S. L., and Wyttenbach, A. (2010). Neurotoxic protein oligomerisation associated with polyglutamine diseases. Acta. Neuropathol. 120, 419–437. doi: 10.1007/s00401-010-0703-0
Hölter, S. M., Stromberg, M., Kovalenko, M., Garrett, L., Glasl, L., Lopez, E., et al. (2013). A broad phenotypic screen identifies novel phenotypes driven by a single mutant allele in Huntington’s disease CAG knock-in mice. PLoS One 8:e80923. doi: 10.1371/journal.pone.0080923
Illuzzi, J., Yerkes, S., Parekh-Olmedo, H., and Kmiec, E. B. (2009). DNA breakage and induction of DNA damage response proteins precede the appearance of visible mutant Huntingtin aggregates. J. Neurosci. Res. 87, 733–747. doi: 10.1002/jnr.21881
Kesavapany, S., Patel, V., Zheng, Y.-L., Pareek, T. K., Bjelogrlic, M., Albers, W., et al. (2007). Inhibition of Pin1 reduces glutamate-induced perikaryal accumulation of phosphorylated neurofilament-H in neurons. Mol. Biol. Cell 18, 3645–3655. doi: 10.1091/mbc.e07-03-0237
Labbadia, J., and Morimoto, R. I. (2013). Huntington’s disease: underlying molecular mechanisms and emerging concepts. Trends Biochem. Sci. 38, 378–385. doi: 10.1016/j.tibs.2013.05.003
Lee, J., Hwang, Y. J., Ryu, H., Kowall, N. W., and Ryu, H. (2014). Nucleolar dysfunction in Huntington’s disease. Biochim. Biophys. Acta 1842, 785–790. doi: 10.1016/j.bbadis.2013.09.017
Lim, N. K. H., Hung, L. W., Pang, T. Y., Mclean, C. A., Liddell, J. R., Hilton, J. B., et al. (2014). Localized changes to glycogen synthase kinase-3 and collapsin response mediator protein-2 in the Huntington’s disease affected brain. Hum. Mol. Genet. 23, 4051–4063. doi: 10.1093/hmg/ddu119
Liou, Y.-C., Sun, A., Ryo, A., Zhou, X. Z., Yu, Z.-X., Huang, H.-K., et al. (2003). Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Nature 424, 556–561. doi: 10.1038/nature01832
Lloret, A., Dragileva, E., Teed, A., Espinola, J., Fossale, E., Gillis, T., et al. (2006). Genetic background modifies nuclear mutant huntingtin accumulation and HD CAG repeat instability in Huntington’s disease knock-in mice. Hum. Mol. Genet. 15, 2015–2024. doi: 10.1093/hmg/ddl125
Lu, P. J., Wulf, G., Zhou, X. Z., Davies, P., and Lu, K. P. (1999). The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature 399, 784–788.
Lynch, G., Kramar, E. A., Rex, C. S., Jia, Y., Chappas, D., Gall, C. M., et al. (2007). Brain-derived neurotrophic factor restores synaptic plasticity in a knock-in mouse model of Huntington’s disease. J. Neurosci. 27, 4424–4434. doi: 10.1523/jneurosci.5113-06.2007
Markianos, M., Panas, M., Kalfakis, N., and Vassilopoulos, D. (2009). Plasma homovanillic acid and prolactin in Huntington’s disease. Neurochem. Res. 34, 917–922. doi: 10.1007/s11064-008-9851-1
Miller, J. P., Yates, B. E., Al-Ramahi, I., Berman, A. E., Sanhueza, M., Kim, E., et al. (2012). A genome-scale RNA-interference screen identifies RRAS signaling as a pathologic feature of Huntington’s disease. PLoS Genet. 8:e1003042. doi: 10.1371/journal.pgen.1003042
Nakamura, K., Kosugi, I., Lee, D. Y., Hafner, A., Sinclair, D. A., Ryo, A., et al. (2012). Prolyl isomerase Pin1 regulates neuronal differentiation via β-catenin. Mol. Cell. Biol. 32, 2966–2978. doi: 10.1128/mcb.05688-11
Pastorino, L., Sun, A., Lu, P.-J., Zhou, X. Z., Balastik, M., Finn, G., et al. (2006). The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-β production. Nature 440, 528–534. doi: 10.1038/nature04543
Robinson, M. D., McCarthy, D. J., and Smyth, G. K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi: 10.1093/bioinformatics/btp616
Ryo, A., Togo, T., Nakai, T., Hirai, A., Nishi, M., Yamaguchi, A., et al. (2006). Prolyl-isomerase Pin1 accumulates in lewy bodies of Parkinson disease and facilitates formation of alpha-synuclein inclusions. J. Biol. Chem. 281, 4117–4125. doi: 10.1074/jbc.m507026200
Saleh, N., Moutereau, S., Durr, A., Krystkowiak, P., Azulay, J.-P., Tranchant, C., et al. (2009). Neuroendocrine disturbances in Huntington’s disease. PLoS One 4:e4962. doi: 10.1371/journal.pone.0004962
Sorrentino, G., Comel, A., Mantovani, F., and Del Sal, G. (2014). Regulation of mitochondrial apoptosis by Pin1 in cancer and neurodegeneration. Mitochondrion 19, 88–96. doi: 10.1016/j.mito.2014.08.003
Stack, E. C., Matson, W. R., and Ferrante, R. J. (2008). Evidence of oxidant damage in Huntington’s disease: translational strategies using antioxidants. Ann. N. Y. Acad. Sci. 1147, 79–92. doi: 10.1196/annals.1427.008
Steger, M., Murina, O., Hühn, D., Ferretti, L. P., Walser, R., Hänggi, K., et al. (2013). Prolyl isomerase PIN1 regulates DNA double-strand break repair by counteracting DNA end resection. Mol. Cell 50, 333–343. doi: 10.1016/j.molcel.2013.03.023
Sugars, K. L., and Rubinsztein, D. C. (2003). Transcriptional abnormalities in Huntington disease. Trends Genet. 19, 233–238. doi: 10.1016/s0168-9525(03)00074-x
The Huntington’s Disease Collaborative Research Group. (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72, 971–983. doi: 10.1016/0092-8674(93)90585-e
Valencia, A., Reeves, P. B., Sapp, E., Li, X., Alexander, J., Kegel, K. B., et al. (2010). Mutant huntingtin and glycogen synthase kinase 3-β accumulate in neuronal lipid rafts of a presymptomatic knock-in mouse model of Huntington’s disease. J. Neurosci. Res. 88, 179–190. doi: 10.1002/jnr.22184
Vonsattel, J. P., and DiFiglia, M. (1998). Huntington disease. J. Neuropathol. Exp. Neurol. 57, 369–384.
Vonsattel, J. P., Myers, R. H., Stevens, T. J., Ferrante, R. J., Bird, E. D., and Richardson, E. P. (1985). Neuropathological classification of Huntington’s disease. J. Neuropathol. Exp. Neurol. 44, 559–577.
Wang, J., Gines, S., MacDonald, M. E., and Gusella, J. F. (2005). Reversal of a full-length mutant Huntingtin neuronal cell phenotype by chemical inhibitors of polyglutamine-mediated aggregation. BMC Neurosci. 6:1. doi: 10.1186/1471-2202-6-1
Wheeler, V. C., Gutekunst, C.-A., Vrbanac, V., Lebel, L.-A., Schilling, G., Hersch, S., et al. (2002). Early phenotypes that presage late-onset neurodegenerative disease allow testing of modifiers in Hdh CAG knock-in mice. Hum. Mol. Genet. 11, 633–640. doi: 10.1093/hmg/11.6.633
White, J. K., Auerbach, W., Duyao, M. P., Vonsattel, J. P., Gusella, J. F., Joyner, A. L., et al. (1997). Huntingtin is required for neurogenesis and is not impaired by the Huntington’s disease CAG expansion. Nat. Genet. 17, 404–410. doi: 10.1038/ng1297-404
Yu, G., Wang, L.-G., Han, Y., and He, Q.-Y. (2012). ClusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. doi: 10.1089/omi.2011.0118
Keywords: Huntington’s disease, Pin1, DNA damage response, gliosis, neuronal intranuclear inclusions
Citation: Agostoni E, Michelazzi S, Maurutto M, Carnemolla A, Ciani Y, Vatta P, Roncaglia P, Zucchelli S, Leanza G, Mantovani F, Gustincich S, Santoro C, Piazza S, Del Sal G and Persichetti F (2016) Effects of Pin1 Loss in HdhQ111 Knock-in Mice. Front. Cell. Neurosci. 10:110. doi: 10.3389/fncel.2016.00110
Received: 17 December 2015; Accepted: 18 April 2016;
Published: 02 May 2016.
Edited by:
Marco Martina, Northwestern University, USAReviewed by:
Anthony John Hannan, University of Melbourne, AustraliaSimonetta Sipione, University of Alberta, Canada
Copyright © 2016 Agostoni, Michelazzi, Maurutto, Carnemolla, Ciani, Vatta, Roncaglia, Zucchelli, Leanza, Mantovani, Gustincich, Santoro, Piazza, Del Sal and Persichetti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesca Persichetti, ZnJhbmNlc2NhLnBlcnNpY2hldHRpQHVuaXVwby5pdA==
†Present address: Paola Roncaglia, European Molecular Biology Laboratory, European Bioinformatics Institute (EMBL-EBI), CB10 1SD Hinxton, Cambridge, UK
‡ These authors have contributed equally to this work.