 Simona Pellacani1†
Simona Pellacani1† Federico Sicca1,2*†Cherubino Di Lorenzo3Gaetano S. Grieco4Giulia Valvo1
Federico Sicca1,2*†Cherubino Di Lorenzo3Gaetano S. Grieco4Giulia Valvo1 Cristina Cereda4Anna Rubegni2
Cristina Cereda4Anna Rubegni2 Filippo M. Santorelli2*
Filippo M. Santorelli2*- 1Clinical Neurophysiology Laboratory, IRCCS Stella Maris Foundation, Pisa, Italy
- 2Molecular Medicine, IRCCS Stella Maris Foundation, Pisa, Italy
- 3Don Carlo Gnocchi Onlus Foundation, Milan, Italy
- 4Genomic and Post-Genomic Center, C. Mondino National Institute of Neurology, Pavia, Italy
Channelopathies are a heterogeneous group of neurological disorders resulting from dysfunction of ion channels located in cell membranes and organelles. The clinical scenario is broad and symptoms such as generalized epilepsy (with or without fever), migraine (with or without aura), episodic ataxia and periodic muscle paralysis are some of the best known consequences of gain- or loss-of-function mutations in ion channels. We review the main clinical effects of ion channel mutations associated with a significant impact on migraine headache. Given the increasing and evolving use of genetic analysis in migraine research—greater emphasis is now placed on genetic markers of dysfunctional biological systems—we also show how novel information in rare monogenic forms of migraine might help to clarify the disease mechanisms in the general population of migraineurs. Next-generation sequencing (NGS) and more accurate and precise phenotyping strategies are expected to further increase understanding of migraine pathophysiology and genetics.
Introduction
The channelopathies are a heterogeneous group of neurological disorders that result from genetic dysfunction of ion channels located in cell membranes and organelles. Similarly to ion pumps and transporters, ion channels are highly selective and coordinate ion fluxes during the generation of action potentials, or following neurotransmitter release, in the nervous system and muscles (Spillane et al., 2016). Their dysfunction may therefore impair neuronal excitability and synaptic transmission, thus constituting a key pathophysiological element of a wide range of disorders.
Generally, the symptoms of channelopathies appear early in life and are typically paroxysmal or episodic. Defects in a single channel may lead to different neurological manifestations, e.g., seizures, paroxysmal movement disorders/periodic paralyzes, and migraine. However, despite the variable presentations, certain trigger factors (i.e., sleep, stress, hormonal fluctuations), patterns of age dependence of manifestations, and treatment modalities may overlap, suggesting the existence of common pathogenic substrates. Conversely, defects in different ion channels, or transporters, can often underpin the single neurological picture. It is therefore difficult to predict the clinical consequences of ion channel dysfunctions, and to establish clear pathophysiological explanations in episodic neurological channelopathies. The reason why defects in single channels can lead to seizures, episodic ataxia, movement disorders or migraine, or to a combination of these, is only partially understood and possibly dependent on diverse molecular mechanisms that affect channel function (i.e., gain- or loss-of-function effects of mutations), and on the specific neuronal circuitry involved.
In this mini review, we focus on the complex pathophysiological relationship underlying migraine disorders, in which an array of genetic and environmental components strongly contributes to variable individual susceptibility and clinical manifestations (i.e., migraine with aura [MA] or without aura [MO]). Indeed, largely because of the phenotypic heterogeneity and genetic pleiotropy and variability of migraine disorders (de Vries et al., 2009), investigation of the common forms of migraine has, to date, provided only limited insight into the underlying genetics and pathophysiology. Studies on rarer monogenic forms of migraine (termed familial hemiplegic migraine [FHM] syndromes), on the other hand, have identified new genes pinpointing fundamental disease mechanisms that possibly also contribute to the common forms of migraine in the general population.
Migraine: Clinical and Pathophysiological Aspects
Migraine affects about 15% of the general population, and women three times more often than men (Launer et al., 1999; Jensen and Stovner, 2008). It is characterized by episodic and disabling attacks of headache, often accompanied by nausea, vomiting, photophobia and/or phonophobia, which may, or may not, be preceded by an aura. Aura is a transient neurological symptom, lasting 5–60 min, that usually includes visual, sensory and/or aphasic features. Even though MA and MO are considered distinct disorders, increasing evidence suggests that the two conditions are, in fact, variable clinical expressions of substantially the same genetic disease. Indeed, the fact that the prevailing migraine form in a single patient might vary over time suggests that the main pathophysiological pathways are essentially the same in MO and MA, and that external modulating factors might favor the switching on/off of one of the two migraine types (Ferrari et al., 2015).
Auras are likely caused by the phenomenon of cortical spreading depression (CSD), namely a wave of neuronal and glial depolarization that starts in visual cortical areas and moves slowly (2–6 mm/min) over the cortex (Lauritzen, 1994). CSD is thought to be the consequence of noxious stimuli that alter the neuronal environment, leading to glutamate-induced toxicity (Kramer et al., 2016). Glutamate activates cation currents, particularly through the N-methyl-D-aspartate receptors, leading to near breakdown of neuronal transmembrane ion gradients (Ca2+, Na+, Cl-, and K+). This loss of potential, which is normally reinstated by Na+/K+ pumps, is not recovered immediately, resulting in long-lasting inhibition of spontaneous and evoked neuronal activity (Dreier and Reiffurth, 2015). Although the mechanism of CSD has been extensively investigated in animal models (Charles and Baca, 2013), experimental evidence in humans is still scarce. In MA, functional magnetic resonance imaging findings have revealed a local increase in blood oxygen level-dependent signals, which were found to spread through the visual cortex at a rate similar to what is seen in experimentally induced CSD in animals (Hadjikhani et al., 2001). Magnetoencephalography studies have also shown that large cortical areas are activated in spontaneous and visually induced migraine auras, producing a spreading depression-like neuroelectric event that may be likened to CSD (Bowyer et al., 2001).
The pain in migraine headache results from activation of the trigeminovascular system (Noseda and Burstein, 2013). Indeed, signals from activated nociceptors located on large cranial vessels and the dura mater are transmitted to the trigeminal bipolar neurons, and further relayed, through extensive connections with brainstem regions (i.e., the periaqueductal gray and locus coeruleus), to thalamic and cortical areas, ultimately producing the sensation of pain (Ferrari et al., 2015).
Multiple evidences suggest that CSD might not only cause migraine auras, but also, by itself, trigger the mechanisms underlying the headache and associated symptoms. These mechanisms consist mainly of the release, by neurons, glia and vascular cells, of pro-inflammatory peptides, such as substance P and calcitonin gene-related peptide, but also adenosine triphosphate (ATP), glutamate and potassium, and the resulting local increase in neuroactive inflammatory mediators and sensitization of pain-relevant brainstem regions (Zhang et al., 2007; Levy, 2012). The opening of neuronal Panx1 channels in response to the CSD stimulus also helps to trigger an inflammatory cascade by releasing HMGB1 proteins, which activate neighboring astrocytes leading to sustained release of inflammatory mediators (Karatas et al., 2013). Although definitive proof is lacking, drugs preventing CSD may be effective in treating migraine attacks (Costa et al., 2013). Pain is only the tip of iceberg of a complex chronic disease in which several molecular mechanisms lead to increased susceptibility to CSD (Antal et al., 2008) and the release of soluble mediators, and thus to long lasting neuronal sensitization, amplified nociceptive signaling by trigeminal sensory neurons, and stable neuroinflammatory tissue responses (Franceschini et al., 2013). Indeed, clinical and neurophysiological studies have confirmed that individuals suffering from migraine display chronic hypersensitivity to sensory stimuli or abnormal processing of sensory information (Aurora et al., 2007; Vecchia and Pietrobon, 2012), which may be reflected in more frequent premonitory symptoms (e.g., speech/reading difficulties, sensory hypersensitivity) preceding the attacks (Pietrobon and Moskowitz, 2013).
Familial Hemiplegic Migraine
Molecular insights into the rare monogenic FHM syndromes have highlighted the central role of calcium (Ca2+) and sodium (Na+) channels, and of sodium-potassium (Na+/K+) ATPase, in the etiology and pathophysiology of migraine. In FHM, migraine attacks are associated with transient hemiparesis, lasting minutes to hours or days, or alternatively may present as episodes of “regular” MO or MA without major motor weakness. Patients may also suffer from a variety of symptoms that include cerebellar ataxia, seizures and even mild head trauma-induced brain edema that can be fatal (Kaja et al., 2010). Three FHM genes have been identified: CACNA1A (FHM1; Ophoff et al., 1996), ATP1A2 (FHM2; De Fusco et al., 2003), and SCN1A (FHM3; Dichgans et al., 2005).
CACNA1A codes for the alpha subunit of the neuronal voltage-gated Ca2+ channel Cav2.1 (Diriong et al., 1995). Cav2.1 channels are predominantly expressed at the presynaptic terminals of glutamatergic and GABAergic neurons in the cerebral cortex, trigeminal ganglia, brainstem nuclei and cerebellum (Catterall, 1998), where they play a crucial role in neurotransmitter release. The clinical features of Cav2.1 channelopathies range from pure FHM1 to forms that include episodic or progressive ataxia (Jouvenceau et al., 2001; Imbrici et al., 2004) and seizures. In FHM1, CACNA1A mutations typically lead to a gain of Cav2.1 channel function, although in model organisms this seems have the effect of enhancing only glutamatergic neurotransmission, whereas inhibitory synapses remain unaffected (Tottene et al., 2009). This differential effect at excitatory and inhibitory synapses suggests that altered regulation of cortical excitatory-inhibitory balance may be a likely pathomechanism in FHM1. The gain of Cav2.1 channel function may indeed favor glutamate release, and consequently the induction and propagation of CSD (Vecchia and Pietrobon, 2012; Pietrobon and Moskowitz, 2013).
The second FHM gene (FHM2), ATP1A2, encodes the alpha-2 subunit of a Na+/K+ pump (De Fusco et al., 2003). This catalytic subunit utilizes ATP hydrolysis to exchange Na+ ions (leaving the cell) for K+ ions (entering the cell) and is present in the membrane of astrocytes at tripartite synapses, where it contributes to K+ and glutamate re-uptake. More than 30 FHM2 mutations have been identified (de Vries et al., 2009) and associated with pure disease (De Fusco et al., 2003; Riant et al., 2005; Vanmolkot et al., 2006), or with a combination of FHM and cerebellar ataxia (Spadaro et al., 2004), recurrent encephalopathy (Ducros et al., 2001; Spacey et al., 2005), impaired hearing and vertigo (Jurkat-Rott et al., 2004), or epilepsy (Roth et al., 2014). Some ATP1A2 mutations have also been associated with non-FHM phenotypes, such as basilar migraine (Ambrosini et al., 2005) or common migraine (de Vries et al., 2009). Defective function of glial Na+/K+-ATPase at tripartite synapses may interfere with glutamate clearance by astrocytes, leading to increased cortical excitatory neurotransmission which facilitates CSD.
The third FHM gene (FHM3), SCN1A, encodes the alpha-1 pore-forming subunit of the neuronal voltage-gated Na+ channel Nav1.1 (Dichgans et al., 2005). Voltage-gated sodium channels have a crucial role in cellular excitability and are essential for the initiation of action potentials in the brain. Mutations in SCN1A are associated with a wide spectrum of epilepsy phenotypes (e.g., severe myoclonic epilepsy; Marini et al., 2007; Dravet and Oguni, 2013). More rarely, SCN1A mutations lead to pure FHM (Dichgans et al., 2005; Vanmolkot et al., 2007), or to FHM associated either with generalized seizures (Castro et al., 2009), or with a stereotyped phenotype (elicited repetitive transient daily blindness) that suggests a retinal form of spreading depression (Vahedi et al., 2009; Fan et al., 2016). Epileptogenic Nav1.1 mutations cause loss of channel function of variable degrees, leading to reduced Na+ currents in GABAergic inhibitory interneurons (Yu et al., 2006), thus defining an interneuron-specific generalized defect in action potential initiation which results in multisystem disinhibition and network hyperexcitability (Hedrich et al., 2014). Mutated Nav1.1 channels in FHM3 instead exhibit a broad range of abnormalities, including gain of function and partial or complete loss of function, confirming the complex relationship between clinical and biophysical phenotypes in SCN1A-related pathology (Kahlig et al., 2008). Regardless of the molecular mechanism, however, the high-frequency firing of mutant Nav1.1 channels, by producing a rise in extracellular K+ concentration and consequent further depolarization, may enhance the release of glutamate and sustain CSD mechanisms.
Taken together, the three different forms of FHM indicate the existence of a main pathophysiological pathway that, starting from excessive neuronal release of glutamate (CACNA1A), impaired glutamate reuptake by glial cells (ATP1A2), or enhanced glutamatergic activity due to impaired GABAergic inhibition (SCN1A), ultimately leads to altered glutamatergic neurotransmission, with consequent neuronal hyperexcitability and increased susceptibility to CSD (Ferrari et al., 2015). The three major genes, however, do not account for all affected cases, and at least three additional genes (SLC1A3, PRRT2 and SLC4A4) have been suggested, albeit on the basis of limited evidence, to be associated with FHM in a minority of cases. Notably, defects in all of these genes lead to enhanced excitatory neurotransmission and cortical excitability. Mutations in SLC1A3, encoding the excitatory amino acid transporter 1 (EAAT1; Jen et al., 2005), cause decreased glutamate reuptake, whereas PRRT2, through defective interaction with SNAP25 and GRIA1 proteins (Li et al., 2015), affects the glutamate signaling pathway and results in increased glutamate release. Finally, the sodium bicarbonate cotransporter NBCe1 (SLC4A4) may derange synaptic pH regulation in astrocytes, leading to neuronal hyperexcitability predisposing to migraine (Suzuki et al., 2010).
Main Genetic Issues in the Study of Migraine
Migraine is a multifactorial disorder resulting from complex interactions between multiple predisposing genes and environmental factors (Russell et al., 1995; Mulder et al., 2003). The latter include hormone fluctuations, and this may explain the increased prevalence of migraine in females, and its variability across the individual life span (MacGregor, 2004). The clinical presentation is also variable, making the pathogenesis of migraine particularly difficult to unravel. Furthermore, it is not clear whether, from the perspective of genetic study designs, the two forms (MO and MA) should be considered the same disease, given that findings from epidemiological and clinical studies are still conflicting (Russell et al., 2002; Ligthart et al., 2006). Many clinical aspects contribute to the extreme variability of migraine phenotypes: for example, the severity and frequency of the attacks, the attack triggers, and the neuropsychiatric comorbidities possibly involved (depression, epilepsy, etc.) are all highly variable. This strong heterogeneity, together with the lack of any genetic biomarker, makes it difficult to stratify patients for genetic studies and consequently to identify strong genotype-phenotype correlations. Previous linkage analyses on large pedigrees, and screening of candidate genes, including more than 150 ion transporter genes (Nyholt et al., 2008), in several thousand migraineurs were largely unsuccessful (de Vries et al., 2009). Similarly, testing of the three major FHM genes in patients with common migraine has shown no evidence to support their involvement in the disorder. It is indeed possible that disease risk in common migraine may be conferred by multiple genes and their variants (each with a small effect size) that control neurotransmitter and ion pathways through complex interactions and regulatory mechanisms (Eising et al., 2013b). Moreover, none of the three major FHM genes has been identified in unbiased genome-wide association (GWA) studies, which until now have constituted the most robust approach for identifying genetic factors underlying complex disorders (Spain and Barrett, 2015). Overall, GWA studies have uncovered 13 susceptibility loci that involve a set of genes clustering into clear pathways likely related to migraine (Freilinger et al., 2012). Notably, several of these genes (i.e., MTDH, LRP1, PRDM16, MEF2D, ASTN2, PHACTR1, FHL5, MMP16) are involved in glutamatergic neurotransmission and synaptic function/development, whose impairment may therefore be considered a main dysfunctional mechanism underlying susceptibility to common forms of migraine. Pain-sensing mechanisms, metalloproteinases and vessel metabolism seem likely to be additional migraine-related pathways (Tolner et al., 2015). The strong association between candidate genes emerging from GWA studies and glutamate metabolism is in line with evidence from FHM, which suggests that impaired glutamatergic neurotransmission is a key disease mechanism underlying the abnormal cortical excitability that favors the initiation and propagation of CSD and the recurrence of migraine attacks. However, none of these candidate genes can conclusively be regarded as a genetic biomarker of the disease; each has limited predictive value given their small effect size (Di Lorenzo et al., 2015). It is possible that multiple gene variants affecting protein-to-protein interactions play an important role in disease mechanisms in specific clinical conditions. For example, analysis of GWA study data, looking for specific disease-relevant functional gene sets, i.e., lists of genes related to glial metabolism or synaptic function (Eising et al., 2015), has disclosed a role for astrocyte- and oligodendrocyte-related genes in MO and MA.
It is to be hoped that thanks to the advent of novel and cost-effective genetic technologies, known collectively as next-generation sequencing (NGS), it will be possible to overcome some limitations of the more traditional approaches used to investigate the genetic basis of migraine. NGS will serve not only as a tool for identifying new genes responsible for monogenic forms of the disorder (i.e., FHM), but also, quite probably, for identifying low-frequency variants that have moderate effects in more common forms of migraine. The combination of data from monogenic migraine and GWA studies, will make it possible to pinpoint the biological systems crucially involved (e.g., glutamatergic neurotransmission and metabolism at the tripartite synapse, Figure 1), and to use this information to design specific customized gene panels allowing thorough investigation of the contribution made by each single molecular player to the dysfunctional protein networks underlying this complex polygenic disease.
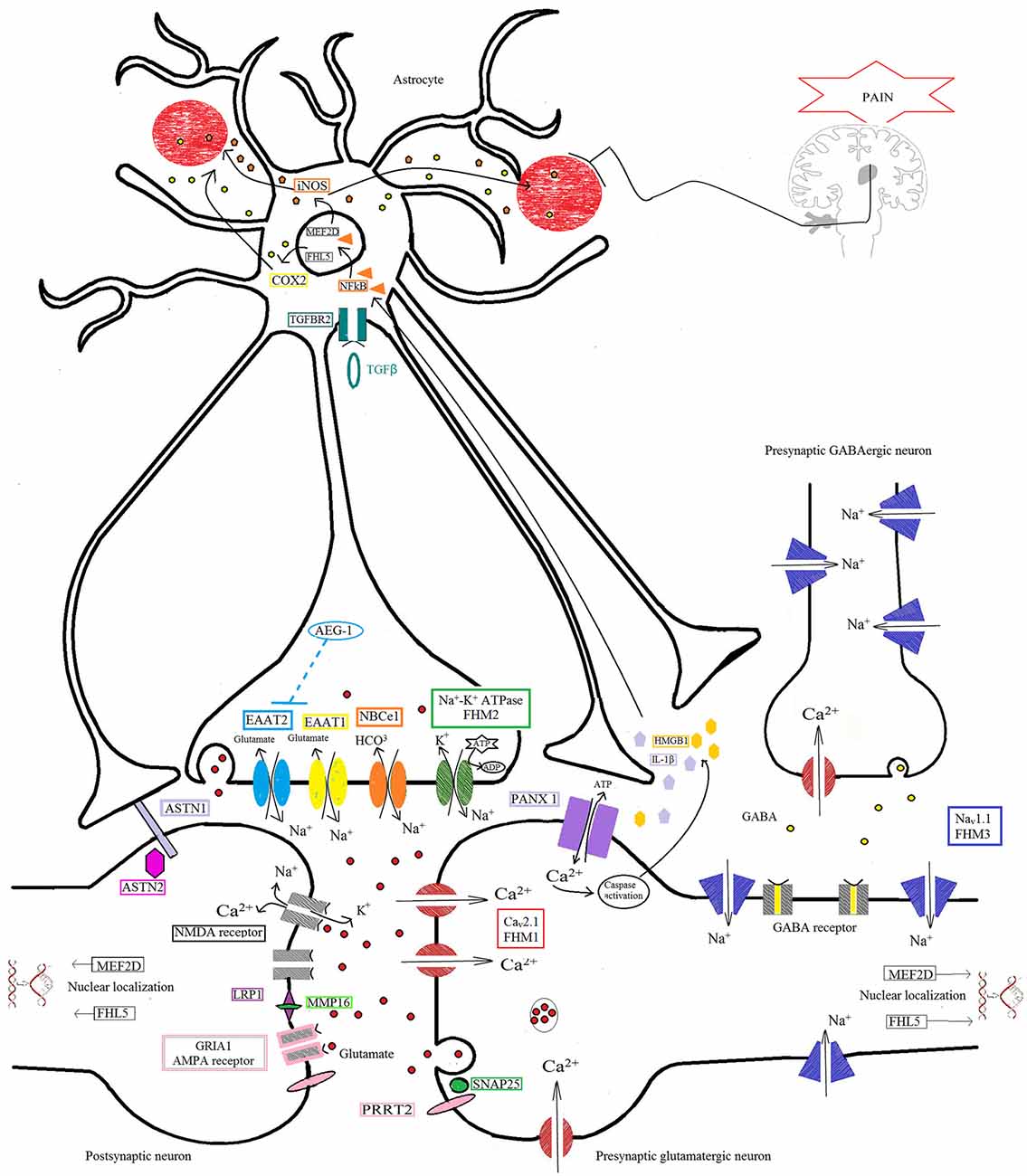
Figure 1. Protein pathway driving the migraine process at the tripartite synapse. The illustration depicts different proteins at the tripartite synapse possibly involved in glutamatergic dysfunction in migraine (see text for details). Cav2.1 (CACNA1A; red) dysfunction at presynaptic terminals of glutamatergic neurons leads to altered Ca2+ influx and enhanced glutamate release by vesicles into the synaptic cleft, favoring the activation and propagation of cortical spreading depression (CSD) in familial hemiplegic migraine 1 (FHM1). Na+/K+-ATPase (ATP1A2; green) at the astrocyte plasma membrane utilizes ATP hydrolysis to exchange Na+ for K+ ions, generating a Na+ gradient that helps to modulate the glutamate re-uptake by glial excitatory amino acid transporter 1 (EAAT1; SLC1A3; yellow) and EAAT2 (SLC1A2; cyan). Loss-of-function of Na+/K+-ATPase (FHM2), as well as of EAAT1, slows the clearance of glutamate leading to increased cortical excitability that favors the initiation and propagation of CSD. The activity of EAAT2 also contributes to glutamate clearance, and is downregulated by mutations in astrocyte elevated gene-1 (AEG-1) (MTDH; dashed line), one of the candidate genes emerging from genome-wide association (GWA) studies. NaV1.1 channels (SCN1A, FHM3; purple) are essential for the generation and propagation of action potentials. FHM3-associated mutations can reduce firing of inhibitory interneurons, or accelerate the recovery of the channel after fast inactivation, causing high-frequency firing of presynaptic glutamatergic neurons. PRRT2 (pink) also affects the glutamate signaling pathway, through defective interaction with SNAP25 (forest green) and the ionotropic glutamate receptor AMPA1 (termed GRIA1; gray with pale pink border), resulting in increased glutamate release. Defective membrane expression of the Na(+)-HCO(3)(-) cotransporter NBCe1 (SLC4A4; orange) may affect the uptake of HCO3- into astrocytes leading to altered activity of pH-sensitive NMDA receptors (gray). Both AMPA and NMDA receptors are also directly modulated by LRP1 (dark pink), which is cleaved by a metalloproteinase that is encoded by another migraine-susceptibility gene, MMP16 (fluorescent green). Synaptic activity is also influenced by other proteins thought to contribute to migraine pathophysiology, such as the nuclear transcription factors MEF2D and FHL5, the serine-threonine kinase TGFBR2 (aquamarine), and ASTN2 (fuchsia), a protein related to ASTN1 (pale lilac) and thought to influence neuronal migration. All these mechanisms, when defective, may affect the glutamate signaling pathway, possibly leading to neuronal hyperexcitability predisposing to migraine. The illustration also shows the pathway that starts from the CSD-driven opening of PANX1 channels (lilac) and triggers the inflammatory cascade and subsequent trigeminovascular sensitization. Signaling to PANX1 leads to caspase 1 activation that, in turn, stimulates the release of high-mobility group box 1 (HMGB1) proteins and the activation of the transcription factor nuclear factor κB (NF-κB) in astrocytes. This may lead to local increase in vasoactive inflammatory mediators and sensitization of pain-relevant brainstem regions.
Conclusions and Future Directions
The emerging NGS techniques are seen as the most promising resource for overcoming gene-finding problems in future migraine genetic research. The various approaches tried to date, using linkage, candidate gene and GWA studies (Figure 2), have not been sufficient to unravel the complex genetic background of MA and MO. In addition, other mechanisms, such as gene-gene or gene-environment interactions and epigenetics, further complicate the already complex picture of the heritability of migraine syndromes (Rudkjobing et al., 2012), suggesting that genotyping data need to be integrated with the results of deep clinical stratification, gene expression data, and proteomics/metabolomics studies in order to fully understand the effects of genetic variability (Bras et al., 2012). The process of deep phenotyping is expected to be a crucial tool for future research in migraine genetics. Focusing genetic analyses on groups with more homogeneous presentations will help in investigating the function and pathogenic relevance of gene variants emerging from NGS studies, strongly increasing the power of genetic information and the strength of genotype-phenotype correlations (Hennekam and Biesecker, 2012) and paving the way for more personalized/“precision” medicine (Zhang et al., 2016). Evaluation of migraine comorbidities, in particular, should be regarded as a pivotal part of the stratification process. It is well established, for example, that adults and children with migraine may have increased susceptibility to seizures (Rajapakse and Buchhalter, 2016). Identifying this potential comorbidity may allow efforts to dissect the genetic basis of the condition to be targeted toward specific sets of genes that may have a role in both migraine and epilepsy pathophysiology, such as the ion channels enhancing excitatory neurotransmitter release (i.e., Cav2.1) or dendrite neuronal excitability and firing (Nav1.1), and their molecular interactors. The use of metabolic parameters will also help the stratification process, possibly favoring the discovery of new genetic biomarkers based on NGS analyses. Gene expression alterations could be used as markers of epigenetic mechanisms (DNA methylation, histone tail modifications, noncoding RNA metabolism) thought to play a role in the development of migraine (Eising et al., 2013a). Finally, data from studies of proteomics and metabolomics might make it possible to define the full metabolic profile of individuals suffering from migraine, fostering efforts to arrive at a phenotypic dissection at molecular level. Although we still do not know whether metabolic changes can be detected in peripheral fluids of migraine patients, data on animal models are promising. Indeed, Cav2.1 transgenic mice have shown measurable metabolic changes in plasma after experimentally induced CSD (Shyti et al., 2015). Defining clinical phenotypes and detectable biomarkers in humans might enable a better understanding of the molecular pathways involved in migraine, and thus allow more accurate understanding of the bulk of data emerging from NGS.
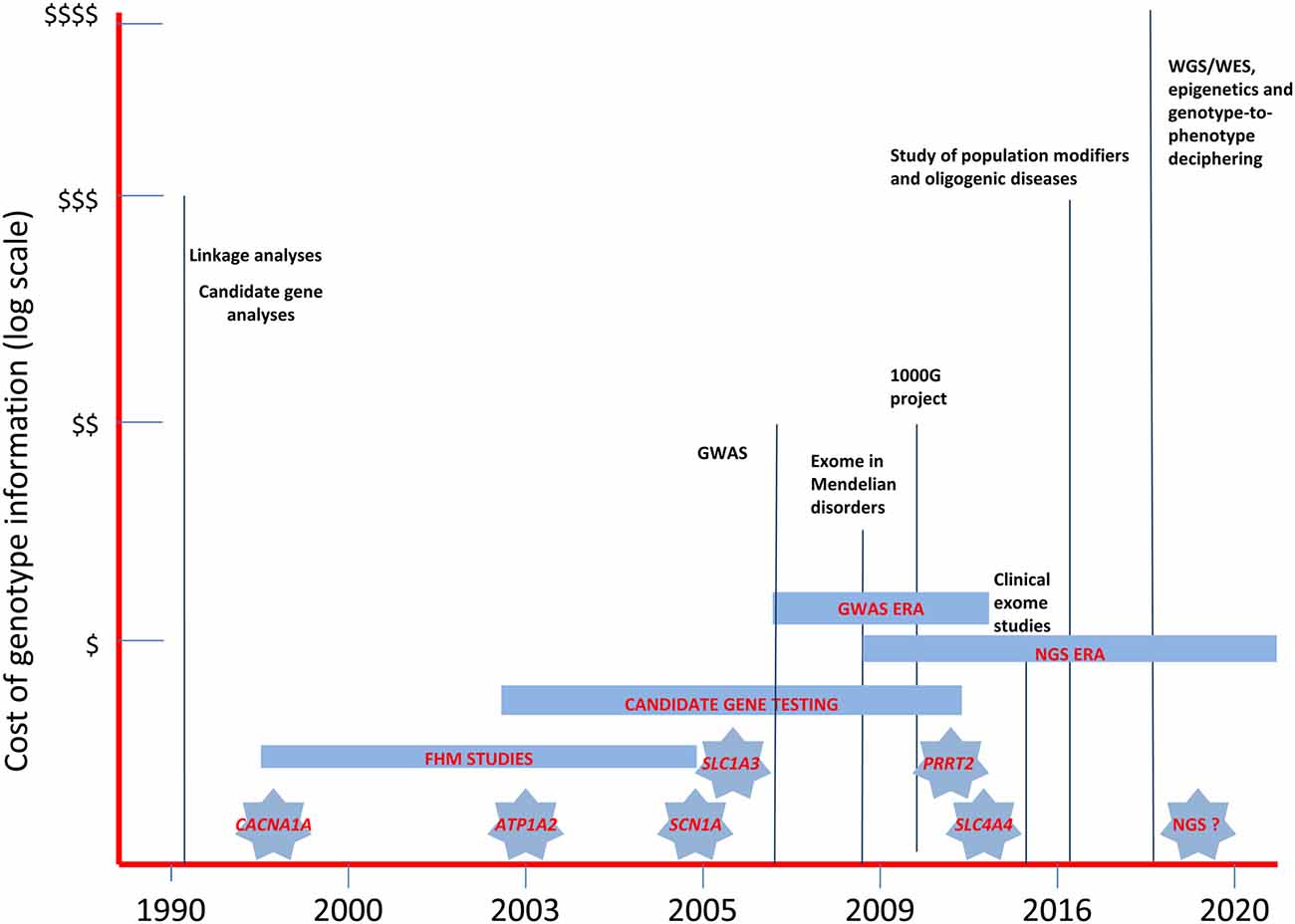
Figure 2. The landscape of past and near future genetic research in migraine headache disorders. The diagram illustrates the relative costs of the molecular methodologies employed in the study of migraine genetics and the timing of the discovery of the major genes involved in FHM and in related disorders. WES, whole-exome sequencing; WGS, whole-genome sequencing; GWAS, genome-wide association studies; NGS, next-generation sequencing.
To date, very little research using NGS methods in migraine has been published, and that which can be found is limited to the sequencing of very few familial cases (Nagata et al., 2014; Jiang et al., 2015). There are, indeed, some technical problems that limit the use of NGS in complex polygenic disorders (Topper et al., 2011), and translation of data into diagnostic information often requires further validation through functional assays, even using unanticipated new tools (Doğanli et al., 2013). As already established in relation to other neurodevelopmental disorders (e.g., autism, intellectual disability, etc.; Hoischen et al., 2014), targeted resequencing approaches may be a valid strategy for reducing the costs and improving the specificity of analyses. The application of pathway-focused large gene panels or biomarker-driven genomic investigations, combined with a stringent endophenotype-oriented approach, may allow a deeper assessment of the role of specific proteins presumably involved in migraine pathomechanisms (for example, those belonging to the dysfunctional pathway at the astrocyte-neuron synaptic cleft, Figure 1). This could also lead to the discovery of new biologically relevant checkpoints in the pathways crucially involved in aura and pain mechanisms underlying migraine disorders. As the costs associated with genome-scale sequencing progressively fall, and new tools for high-throughput functional assays are developed, NGS techniques will gradually become a more feasible clinical option for the decoding of complex polygenic conditions such as migraine, revealing previously unexpected opportunities for personalized medicine.
Author Contributions
All the authors have contributed substantially to the writing and revising of the manuscript. SP, FS, and FMS participated in the conception and design of the work, collected the literature, prepared the figures and wrote the manuscript. CDL, CC, GSG, GV, and AR reviewed and edited the manuscript, and approved the final version.
Funding
Research in our laboratories is partially supported by grants from the Italian Ministry of Health (Ricerca Corrente to FMS) and Telethon-Italy (http://www.telethon.it/en; Grant no. GGP11188 to FS).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We also thank Catherine J. Wrenn for providing expert editing.
Abbreviations
MA, migraine with aura; MO, migraine without aura.
References
Ambrosini, A., D’Onofrio, M., Grieco, G. S., Di Mambro, A., Montagna, G., Fortini, D., et al. (2005). Familial basilar migraine associated with a new mutation in the ATP1A2 gene. Neurology 65, 1826–1828. doi: 10.1212/01.wnl.0000187072.71931.c0
Antal, A., Lang, N., Boros, K., Nitsche, M., Siebner, H. R., and Paulus, W. (2008). Homeostatic metaplasticity of the motor cortex is altered during headache-free intervals in migraine with aura. Cereb. Cortex 18, 2701–2705. doi: 10.1093/cercor/bhn032
Aurora, S. K., Barrodale, P. M., Tipton, R. L., and Khodavirdi, A. (2007). Brainstem dysfunction in chronic migraine as evidenced by neurophysiological and positron emission tomography studies. Headache 47, 996–1003; discussion 1004–1007. doi: 10.1111/j.1526-4610.2007.00853.x
Bowyer, S. M., Aurora, K. S., Moran, J. E., Teplet, N., and Welch, K. M. (2001). Magnetoencephalographic fields from patients with spontaneous and induced migraine aura. Ann. Neurol. 50, 582–587. doi: 10.1002/ana.1293
Bras, J., Guerreiro, R., and Hardy, J. (2012). Use of next-generation sequencing and other whole-genome strategies to dissect neurological disease. Nat. Rev. Neurosci. 13, 453–464. doi: 10.1038/nrn3271
Castro, M. J., Stam, A. H., Lemos, C., de Vries, B., Vanmolkot, K. R., Barros, J., et al. (2009). First mutation in the voltage-gated Nav1.1 subunit gene SCN1A with co-occurring familial hemiplegic migraine and epilepsy. Cephalalgia 29, 308–313. doi: 10.1111/j.1468-2982.2008.01721.x
Catterall, W. A. (1998). Structure and function of neuronal Ca2+ channels and their role in neurotransmitter release. Cell Calcium 24, 307–323. doi: 10.1016/s0143-4160(98)90055-0
Charles, A. C., and Baca, S. M. (2013). Cortical spreading depression and migraine. Nat. Rev. Neurol. 9, 637–644. doi: 10.1038/nrneurol.2013.192
Costa, C., Tozzi, A., Rainero, I., Cupini, L. M., Calabresi, P., Ayata, C., et al. (2013). Cortical spreading depression as a target for anti-migraine agents. J. Headache Pain 33, 18631–18640. doi: 10.1186/1129-2377-14-62
De Fusco, M., Marconi, R., Silvestri, L., Atorino, L., Rampoldi, L., Morgante, L., et al. (2003). Haploinsufficiency of ATP1A2 encoding the Na+/K+ pump α2 subunit associated with familial hemiplegic migraine type 2. Nat. Genet. 33, 192–196. doi: 10.1038/ng1081
de Vries, B., Frants, R. R., Ferrari, M. D., and van den Maagdenberg, A. M. (2009). Molecular genetics of migraine. Hum. Genet. 126, 115–132. doi: 10.1007/s00439-009-0684-z
Di Lorenzo, C., Santorelli, F. M., and van den Maagdenberg, A. M. (2015). “Genetics of headache,” in Pathophysiology of Headaches: From Molecule to Man, eds M. Ashina and P. Geppetti (Switzerland: Springer International Publishing), 83–99.
Dichgans, M., Freilinger, T., Eckstein, G., Babini, E., Lorenz-Depiereux, B., Biskup, S., et al. (2005). Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet 366, 371–377. doi: 10.1016/s0140-6736(05)66786-4
Diriong, S., Lory, P., Williams, M. E., Ellis, S. B., Harpold, M. M., and Taviaux, S. (1995). Chromosomal localization of the human genes for α1A, α1B and α1E voltage-dependent Ca2+ channel subunits. Genomics 30, 605–609. doi: 10.1006/geno.1995.1284
Doğanli, C., Oxvig, C., and Lykke-Hartmann, K. (2013). Zebrafish as a novel model to assess Na+/K+-ATPase-related neurological disorders. Neurosci. Biobehav. Rev. 37, 2774–2787. doi: 10.1016/j.neubiorev.2013.09.013
Dravet, C., and Oguni, H. (2013). Dravet syndrome (severe myoclonic epilepsy in infancy). Handb. Clin. Neurol. 111, 627–633. doi: 10.1016/B978-0-444-52891-9.00065-8
Dreier, J. P., and Reiffurth, C. (2015). The stroke-migraine depolarization continuum. Neuron 86, 902–922. doi: 10.1016/j.neuron.2015.04.004
Ducros, A., Denier, C., Joutel, A., Cecillon, M., Lescoat, C., Vahedi, K., et al. (2001). The clinical spectrum of familial hemiplegic migraine associated with mutations in a neuronal calcium channel. N. Engl. J. Med. 345, 17–24. doi: 10.1056/nejm200107053450103
Eising, E., A Datson, N., van den Maagdenberg, A. M., and Ferrari, M. D. (2013a). Epigenetic mechanisms in migraine: a promising avenue? BMC Med. 11:26. doi: 10.1186/1741-7015-11-26
Eising, E., de Vries, B., Ferrari, M. D., Terwindt, G. M., and van den Maagdenberg, A. M. (2013b). Pearls and pitfalls in genetic studies of migraine. Cephalalgia 33, 614–625. doi: 10.1177/0333102413484988
Eising, E., de Leeuw, C., Min, J. L., Anttila, V., Verheijen, M. H., Terwindt, G. M., et al. (2015). Involvement of astrocyte and oligodendrocyte gene sets in migraine. Cephalalgia doi: 10.1177/0333102415618614 [Epub ahead of print].
Fan, C., Wolking, S., Lehmann-Horn, F., Hedrich, U. B., Freilinger, T., Lerche, H., et al. (2016). Early-onset familial hemiplegic migraine due to a novel SCN1A mutation. Cephalalgia doi: 10.1177/0333102415608360 [Epub ahead of print].
Ferrari, M. D., Klever, R. R., Terwindt, G. M., Ayata, C., and van den Maagdenberg, A. M. (2015). Migraine pathophysiology: lessons from mouse models and human genetics. Lancet Neurol. 14, 65–80. doi: 10.1016/s1474-4422(14)70220-0
Franceschini, A., Vilotti, S., Ferrari, M. D., van den Maagdenberg, A. M., Nistri, A., and Fabbretti, E. (2013). TNFα levels and macrophages expression reflect an inflammatory potential of trigeminal ganglia in a mouse model of familial hemiplegic migraine. PLoS One 8:e52394. doi: 10.1371/journal.pone.0052394
Freilinger, T., Anttila, V., de Vries, B., Malik, R., Kallela, M., Terwindt, G. M., et al. (2012). Genome-wide association analysis identifies susceptibility loci for migraine without aura. Nat. Genet. 44, 777–782. doi: 10.1038/ng.2307
Hadjikhani, N., Sanchez Del Rio, M., Wu, O., Schwartz, D., Bakker, D., Fischl, B., et al. (2001). Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc. Natl. Acad. Sci. U S A 98, 4687–4692. doi: 10.1073/pnas.071582498
Hedrich, U. B., Liautard, C., Kirschenbaum, D., Pofahl, M., Lavigne, J., Liu, Y., et al. (2014). Impaired action potential initiation in GABAergic interneurons causes hyperexcitable networks in an epileptic mouse model carrying a human Na(V)1.1 mutation. J. Neurosci. 34, 14874–14889. doi: 10.1523/JNEUROSCI.0721-14.2014
Hennekam, R. C., and Biesecker, L. G. (2012). Next-generation sequencing demands next-generation phenotyping. Hum. Mutat. 33, 884–886. doi: 10.1002/humu.22048
Hoischen, A., Krumm, N., and Eichler, E. E. (2014). Prioritization of neurodevelopmental disease genes by discovery of new mutations. Nat. Neurosci. 17, 764–772. doi: 10.1038/nn.3703
Imbrici, P., Jaffe, S. L., Eunson, L. H., Davies, N. P., Herd, C., Robertson, R., et al. (2004). Dysfunction of the brain calcium channel CaV2.1 in absence epilepsy and episodic ataxia. Brain 127, 2682–2692. doi: 10.1093/brain/awh301
Jen, J. C., Wan, J., Palos, T. P., Howard, B. D., and Baloh, R. W. (2005). Mutation in the glutamate transporter EAAT1 causes episodic ataxia, hemiplegia and seizures. Neurology 65, 529–534. doi: 10.1212/01.wnl.0000172638.58172.5a
Jensen, R., and Stovner, L. J. (2008). Epidemiology and comorbidity of headache. Lancet Neurol. 7, 354–361. doi: 10.1016/S1474-4422(08)70062-0
Jiang, Y., Wu, R., Chen, C., You, Z. F., Luo, X., and Wang, X. P. (2015). Six novel rare non-synonymous mutations for migraine without aura identified by exome sequencing. J. Neurogenet. 29, 188–194. doi: 10.3109/01677063.2015.1122787
Jouvenceau, A., Eunson, L. H., Spauschus, A., Ramesh, V., Zuberi, S. M., Kullmann, D. M., et al. (2001). Human epilepsy associated with dysfunction of the brain P/Q-type calcium channel. Lancet 358, 801–807. doi: 10.1016/s0140-6736(01)05971-2
Jurkat-Rott, K., Freilinger, T., Dreier, J. P., Herzog, J., Göbel, H., Petzold, G. C., et al. (2004). Variability of familial hemiplegic migraine with novel A1A2 Na+/K+-ATPase variants. Neurology 62, 1857–1861. doi: 10.1212/01.WNL.0000127310.11526.fd
Kahlig, K. M., Rhodes, T. H., Pusch, M., Freilinger, T., Pereira-Monteiro, J. M., Ferrari, M. D., et al. (2008). Divergent sodium channel defects in familial hemiplegic migraine. Proc. Natl. Acad. Sci. U S A 105, 9799–9804. doi: 10.1073/pnas.0711717105
Kaja, S., Van de Ven, R. C., Broos, L. A., Frants, R. R., Ferrari, M. D., Van den Maagdenberg, A. M., et al. (2010). Severe and progressive neurotransmitter release aberrations in familial hemiplegic migraine type 1 Cacna1a S218L knock-in mice. J. Neurophysiol. 104, 1445–1455. doi: 10.1152/jn.00012.2010
Karatas, H., Erdener, S. E., Gursoy-Ozdemir, Y., Lule, S., Eren-Koçak, E., Sen, Z. D., et al. (2013). Spreading depression triggers headache by activating neuronal Panx1 channels. Science 339, 1092–1095. doi: 10.1126/science.1231897
Kramer, D. R., Fujii, T., Ohiorhenuan, I., and Liu, C. Y. (2016). Cortical spreading depolarization: pathophysiology, implications and future directions. J. Clin. Neurosci. 87, 37–48. doi: 10.1016/j.jocn.2015.08.004
Launer, L. J., Terwindt, G. M., and Ferrari, M. D. (1999). The prevalence and characteristics of migraine in a population-based cohort: the GEM study. Neurology 53, 537–542. doi: 10.1212/wnl.53.3.537
Lauritzen, M. (1994). Pathophysiology of the migraine aura. The spreading depression theory. Brain 117, 199–210. doi: 10.1093/brain/117.1.199
Levy, D. (2012). Endogenous mechanisms underlying the activation and sensation of meningeal nociceptors: the role of immune-vascular interactions and cortical spreading depression. Curr. Pain Headache Rep. 16, 270–277. doi: 10.1007/s11916-012-0255-1
Li, M., Niu, F., Zhu, X., Wu, X., Shen, N., Peng, X., et al. (2015). PRRT2 mutant leads to dysfunction of glutamate signaling. Int. J. Mol. Sci. 16, 9134–9151. doi: 10.3390/ijms16059134
Ligthart, L., Boomsma, D. I., Martin, N. G., Stubbe, J. H., and Nyholt, D. R. (2006). Migraine with aura and migraine without aura are not distinct entities: further evidence from a large Dutch population study. Twin Res. Hum. Genet. 9, 54–63. doi: 10.1375/twin.9.1.54
MacGregor, E. A. (2004). Oestrogen and attacks of migraine with and without aura. Lancet Neurol. 3, 354–361. doi: 10.1016/s1474-4422(04)00768-9
Marini, C., Mei, D., Temudo, T., Ferrari, A. R., Buti, D., Dravet, C., et al. (2007). Idiopathic epilepsies with seizures precipitated by fever and SCN1A abnormalities. Epilepsia 48, 1678–1685. doi: 10.1111/j.1528-1167.2007.01122.x
Mulder, E. J., Van Baal, C., Gaist, D., Kallela, M., Kaprio, J., Svensson, D. A., et al. (2003). Genetic and environmental influences on migraine: a twin study across six countries. Twin Res. 6, 422–431. doi: 10.1375/136905203770326420
Nagata, E., Fujii, N., Hosomichi, K., Mitsunaga, S., Suzuki, Y., Mashimo, Y., et al. (2014). Possible association between dysfunction of vitamin D binding protein (GC Globulin) and migraine attacks. PLoS One 9:e105319. doi: 10.1371/journal.pone.0105319
Noseda, R., and Burstein, R. (2013). Migraine pathophysiology: anatomy of the trigeminovascular pathway and associated neurological symptoms, CSD, sensitization and modulation of pain. Pain 154, S44–S53. doi: 10.1016/j.pain.2013.07.021
Nyholt, D. R., LaForge, K. S., Kallela, M., Alakurtti, K., Anttila, V., Färkkilä, M., et al. (2008). A high-density association screen of 155 ion transport genes for involvement with common migraine. Hum. Mol. Genet. 17, 3318–3331. doi: 10.1093/hmg/ddn227
Ophoff, R. A., Terwindt, G. M., Vergouwe, M. N., van Eijk, R., Oefner, P. J., Hoffman, S. M., et al. (1996). Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell 87, 543–552. doi: 10.1016/s0092-8674(00)81373-2
Pietrobon, D., and Moskowitz, M. A. (2013). Pathophysiology of migraine. Annu. Rev. Physiol. 75, 365–391. doi: 10.1146/annurev-physiol-030212-183717
Rajapakse, T., and Buchhalter, J. (2016). The borderland of migraine and epilepsy in children. Headache doi: 10.1111/head.12827 [Epub ahead of print].
Riant, F., De Fusco, M., Aridon, P., Ducros, A., Ploton, C., Marchelli, F., et al. (2005). ATP1A2 mutations in 11 families with familial hemiplegic migraine. Hum. Mutat. 26:281. doi: 10.1002/humu.9361
Roth, C., Freilinger, T., Kirovski, G., Dunkel, J., Shah, Y., Wilken, B., et al. (2014). Clinical spectrum in three families with familial hemiplegic migraine type 2 including a novel mutation in the ATP1A2 gene. Cephalalgia 34, 183–190. doi: 10.1177/0333102413506128
Rudkjobing, L. A., Esserlind, A. L., and Olesen, J. (2012). Future possibilities in migraine genetics. J. Headache Pain 13, 505–511. doi: 10.1007/s10194-012-0481-2
Russell, M. B., Iselius, L., and Olesen, J. (1995). Inheritance of migraine investigated by complex segregation analysis. Hum. Genet. 96, 726–730. doi: 10.1007/bf00210307
Russell, M. B., Ulrich, V., Gervil, M., and Olesen, J. (2002). Migraine without aura and migraine with aura are distinct disorders. A population-based twin survey. Headache 42, 332–336. doi: 10.1046/j.1526-4610.2002.02102.x
Shyti, R., Kohler, I., Schoenmaker, B., Derks, R. J., Ferrari, M. D., Tolner, E. A., et al. (2015). Plasma metabolic profiling after cortical spreading depression in a transgenic mouse model of hemiplegic migraine by capillary electrophoresis–mass spectrometry. Mol. Biosyst. 11, 1462–1471. doi: 10.1039/c5mb00049a
Spacey, S. D., Vanmolkot, K. R., Murphy, C., van den Maagdenberg, A. M., and Hsiung, R. G. (2005). Familial hemiplegic migraine presenting as recurrent encephalopathy in a Native Indian family. Headache 45, 1244–1249. doi: 10.1111/j.1526-4610.2005.00249.x
Spadaro, M., Ursu, S., Lehmann-Horn, F., Veneziano, L., Antonini, G., Giunti, P., et al. (2004). A G301R Na+/K+-Atpase mutation causes familial hemiplegic migraine type 2 with cerebellar signs. Neurogenetics 5, 177–185. doi: 10.1007/s10048-004-0183-2
Spain, S. L., and Barrett, J. C. (2015). Strategies for fine-mapping complex traits. Hum. Mol. Genet. 24, R111–R119. doi: 10.1093/hmg/ddv260
Spillane, J., Kullmann, D. M., and Hanna, M. G. (2016). Genetic neurological channelopathies: molecular genetics and clinical phenotypes. J. Neurol. Neurosurg. Psychiatry 87, 37–48. doi: 10.1136/jnnp-2015-311233
Suzuki, M., Van Paesschen, W., Stalmans, I., Horita, S., Yamada, H., Bergmans, B. A., et al. (2010). Defective membrane expression of the Na(+)-HCO(3)(-) cotransporter NBCe1 is associated with familial migraine. Proc. Natl. Acad. Sci. U S A 107, 15963–15968. doi: 10.1073/pnas.1008705107
Tolner, E. A., Houben, T., Terwindt, G. M., de Vries, B., Ferrari, M. D., and van den Maagdenberg, A. M. (2015). From migraine genes to mechanisms. Pain 156, S64–S74. doi: 10.1097/01.j.pain.0000460346.00213.16
Topper, S., Ober, C., and Das, S. (2011). Exome sequencing and the genetics of intellectual disability. Clin. Genet. 80, 117–126. doi: 10.1111/j.1399-0004.2011.01720.x
Tottene, A., Conti, R., Fabbro, A., Vecchia, D., Shapovalova, M., Santello, M., et al. (2009). Enhanced excitatory transmission at cortical synapses as the basis for facilitated spreading depression in Cav2.1 knockin migraine mice. Neuron 61, 762–773. doi: 10.1016/j.neuron.2009.01.027
Vahedi, K., Depienne, C., Le Fort, D., Riant, F., Chaine, P., Trouillard, O., et al. (2009). Elicited repetitive daily blindness: a new phenotype associated with hemiplegic migraine and SCN1A mutations. Neurology 72, 178–183. doi: 10.1212/01.wnl.0000345393.53132.8c
Vanmolkot, K. R., Babini, E., de Vries, B., Stam, A. H., Freilinger, T., Terwindt, G. M., et al. (2007). The novel p.L1649Q mutation in the SCN1A epilepsy gene is associated with familial hemiplegic migraine: genetic and functional studies. Hum. Mutat. 28:522. doi: 10.1002/humu.9486
Vanmolkot, K. R., Kors, E. E., Turk, U., Turkdogan, D., Keyser, A., Broos, L. A., et al. (2006). Two de novo mutations in the Na, K-ATPase gene ATP1A2 associated with pure familial hemiplegic migraine. Eur. J. Hum. Genet. 14, 555–560. doi: 10.1038/sj.ejhg.5201607
Vecchia, D., and Pietrobon, D. (2012). Migraine: a disorder of brain excitatory-inhibitory balance? Trends Neurosci. 35, 507–520. doi: 10.1016/j.tins.2012.04.007
Yu, F. H., Mantegazza, M., Westenbroek, R. E., Robbins, C. A., Kalume, F., Burton, K. A., et al. (2006). Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci. 9, 1142–1149. doi: 10.1038/nn1754
Zhang, L. M., Dong, Z., and Yu, S. Y. (2016). Migraine in the era of precision medicine. Ann. Transl. Med. 4:105. doi: 10.21037/atm.2016.03.13
Keywords: migraine, calcium channel, sodium channel, tripartite synapse, astrocyte, glutamate
Citation: Pellacani S, Sicca F, Di Lorenzo C, Grieco GS, Valvo G, Cereda C, Rubegni A and Santorelli FM (2016) The Revolution in Migraine Genetics: From Aching Channels Disorders to a Next-Generation Medicine. Front. Cell. Neurosci. 10:156. doi: 10.3389/fncel.2016.00156
Received: 10 March 2016; Accepted: 30 May 2016;
Published: 13 June 2016.
Edited by:
Antonio Gambardella, University Magna Graecia Catanzaro, ItalyReviewed by:
Elsa Fabbretti, University of Nova Gorica, SloveniaEleonora Palma, Università di Roma “La Sapienza”, Italy
Copyright © 2016 Pellacani, Sicca, Di Lorenzo, Grieco, Valvo, Cereda, Rubegni and Santorelli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Federico Sicca, ZmVkZXJpY28uc2ljY2FAZnNtLnVuaXBpLml0;
Filippo M. Santorelli, ZmlsaXBwbzMzNjRAZ21haWwuY29t
† These authors have contributed equally to this work.