 Sanja Ramljak
Sanja Ramljak Holger Herlyn2
Holger Herlyn2 Inga Zerr
Inga Zerr- 1Sciema UG, Mainz, Germany
- 2Institute of Anthropology, Johannes Gutenberg University of Mainz, Mainz, Germany
- 3Department of Neurology, University Medical Center Göttingen and German Center for Neurodegenerative Diseases, Göttingen, Germany
The cellular prion protein (PrPc) and hypoxia appear to be tightly intertwined. Beneficial effects of PrPc on neuronal survival under hypoxic conditions such as focal cerebral ischemia are strongly supported. Conversely, increasing evidence indicates detrimental effects of increased PrPc expression on cancer progression, another condition accompanied by low oxygen tensions. A switch between anaerobic and aerobic metabolism characterizes both conditions. A cellular process that might unite both is glycolysis. Putative role of PrPc in stimulation of glycolysis in times of need is indeed thought provoking. A significance of astrocytic PrPc expression for neuronal survival under hypoxic conditions and possible association of PrPc with the astrocyte-neuron lactate shuttle is considered. We posit PrPc-induced lactate production via transactivation of lactate dehydrogenase A by hypoxia inducible factor 1α as an important factor for survival of both neurons and tumor cells in hypoxic microenvironment. Concomitantly, we discuss a cross-talk between Wnt/β-catenin and PI3K/Akt signaling pathways in executing PrPc-induced activation of glycolysis. Finally, we would like to emphasize that we see a great potential in joining expertise from both fields, neuroscience and cancer research in revealing the mechanisms underlying hypoxia-related pathologies. PrPc may prove focal point for future research.
Adaptation to Hypoxia
What appears ordinary today for most of the time of the Earth’s history was not: free oxygen. Actually, both aquatic and terrestrial environments were widely devoid of free oxygen for thousands of millions of years. With appearance of photosynthesis about 3.5–3.2 billion years ago (Blankenship, 2010), oxygen was for the first time produced in considerable amounts. Yet, this early oxygen was widely consumed for further hundreds of millions of years through precipitation of Fe2+ ions and the formation of ferrous sulfides. Only after a steady-state between the influx of Fe2+ from the continents and the precipitation in the oceans was reached, about 2 billion years ago, free oxygen could be enriched in noteworthy amounts (Martin and Russell, 2003). This short survey illustrates that early heterotrophs had to sustain eons of oxygen deficiency (anoxia) and limited oxygen availability (hypoxia).
Glycolytic enzymes that capacitate endurance of low oxygen tensions most likely arose some 2 billion years before the emergence of first oxygen-consuming species (Webster, 2003). Indeed these enzymes are evolutionary highly conserved (Martin and Russell, 2003; Webster, 2003). Although vertebrates are generally regarded as highly oxygen-reliant they can efficiently switch from aerobic (oxidative phosphorylation) to “ancestral” anaerobic (anaerobic glycolysis) energy production when oxygen falls under the critical mark (Nilsson and Renshaw, 2004; Jackson and Ultsch, 2010). An extreme example represents a freshwater turtle (Trachemys scripta elegans) which can withstand 24 h of anoxia and subsequent re-oxygenation without any apparent loss of neurons (Kesaraju et al., 2009). On the contrary, only short anoxia is sufficient to cause flat electroencephalogram in the human brain (Rossen et al., 1943). Hence, the ability to sense oxygen deprivation is vital to the survival of all aerobic organisms (Lutz and Prentice, 2002).
To survive every healthy cell has to maintain abundant adenosine triphosphate (ATP) levels and regulated metabolic depression, i.e., hypometabolism seems to be the key to survival under conditions of low oxygen (Boutilier, 2001). Consequently, when ATP levels drop reallocation of cell’s energy supplies between critical and non-critical ATP-consuming processes becomes pivotal. It seems that ATP-driven processes are ordered in hierarchy with protein and DNA/RNA synthesis ranked as low priority processes, therefore inhibited first, and fueling of ATP-dependent membrane pumps such as Na+/K+ ATPase and Ca2+ cycling having the highest operating priority (Buttgereit and Brand, 1995). Keeping the latter processes functional is fundamental within the central nervous system (CNS), especially when oxygen supply is sparse.
Effects of Hypoxia On Neurons And Astrocytes
Within the CNS, neurons are the most susceptible cell type in respect to oxygen deprivation. This is an outcome of their high aerobic metabolism. Approximately 50% of neuronal energy expenditure is committed to preserving high priority processes: ionic gradients and fluxes (Hansen, 1985). As a result, when neuronal ATP production fails to meet energy demands mandatory for sustaining ionic and osmotic equilibrium neuronal cell death follows.
In contrast to neurons, astrocytes possess glycogen stores (Magistretti, 2008) and can increase their glycolytic capacity when oxygen supply is inadequate (anaerobic glycolysis) and ATP generation via oxidative phosphorylation flawed. They are also able to increase glycolysis when oxygen levels are adequate (aerobic glycolysis).
Hence, astrocytes can withstand hypoxia without major morphological changes up to 12 h (Yu et al., 1989). An increase in glycolytic capacity of astrocytes is put into action via up-regulation of anaerobic isoforms of glycolytic enzymes such as lactate dehydrogenase A (LDH-A; Marrif and Juurlink, 1999). In addition, astrocytes are also efficient in decreasing ATP consumption when oxygen- and glucose-deprived (Yager et al., 1994). All these traits of astrocytic adaptation to low oxygen tensions presumably contribute to their role in safeguarding neurons from detrimental effects of anoxia and hypoxia (Vibulsreth et al., 1987; Imuta et al., 2007). Previous studies have demonstrated that after ischemic insult neurons fail to survive if neighboring astrocytes are not viable (Takano et al., 2009). Therefore, one can deduce that oxygen deprivation promotes release of certain astrocytic metabolic products, which are crucial for preserving neuronal vitality.
PrPc-Mediated Neuroprotection Against Hypoxia
Oxidative damage is a common denominator of neurodegenerative disorders (reviewed in Zhang et al., 2011). In prion diseases, which are characterized by neuronal loss and astrogliosis (Belay, 1999), the failure in antioxidant defense seems to be crucial (Brown, 2005). The PrPc, which plays a central role in prion diseases, manifests antioxidant properties (Steele et al., 2007) which are obstructed by its conversion into a misfolded, disease-specific isoform (PrPsc).
Despite the fact that PrPc is highly conserved across mammals (Schätzl et al., 1995), PrPc knockout mice (Prnp-/-) show only subtle phenotypes under physiological conditions. However, when cellular energy requirements increase, as under different stress conditions, PrPc presence becomes critical to the survival (Steele et al., 2007). As PrPc expression level is the highest within the CNS, its functions at this site are presumably of uppermost relevance. Actually, one of the best-supported PrPc functions so far is neuroprotection against hypoxic damage (McLennan et al., 2004; Weise et al., 2004; Mitteregger et al., 2007; Doeppner et al., 2015), implying PrPc capacity for sensing and adequately responding to oxygen deprivation. Thus, PrPc expression is up-regulated following cerebral ischemia, and wild-type (WT) mice display significantly smaller infarct volumes as compared to Prnp-/- mice (McLennan et al., 2004; Weise et al., 2004; Mitteregger et al., 2007). Moreover, considerably increased long-term neuroprotection, neurogenesis and angiogenesis was reported in the ischemic brains of PrPc-overexpressing (Prnp+/+) vs. WT and Prnp-/-mice, accenting the importance of elevated PrPc levels in preventing hypoxia-induced neuronal damage (Doeppner et al., 2015). In other words, it appears that a metabolic switch between oxidative-independent and oxidative-dependent metabolism during hypoxia and subsequent re-oxygenation cannot be efficiently executed when PrPc is absent.
Prior study employing astrocyte-neuron co-cultures showed that PrPc expression in astrocytes is fundamental for neuronal differentiation and survival (Lima et al., 2007). Moreover, astrocytic PrPc expression appears to be important for reduction of hydrogen peroxide toxicity (Bertuchi et al., 2012), a reactive oxygen species whose production in mammalian cells is stimulated by hypoxia (Moller, 2001).
Considering that:
(i) astrocytes predominantly rely on glycolytic metabolism and can successfully endure hypoxic episodes;
(ii) astrocytes protect neuronal integrity from different insults;
(iii) astrocytic PrPc expression is pertinent to neuronal survival and
(iv) PrPc confers neuroprotection in a model of focal cerebral ischemia,
it is conceivable that astrocytic PrPc expression may have a considerable influence on a favorable neurologic outcome under hypoxic conditions. Yet, which molecular scenario could support this concept?
PrPc, Glycolysis, and the Astrocyte-Neuron Lactate Shuttle
Pellerin and Magistretti (1994) proposed a so-called astrocyte-neuron lactate shuttle (ANLS) model postulating that neuronal activity increases extracellular levels of glutamate, which is readily absorbed by astrocytes resulting in stimulation of astrocytic glycolysis and lactate production. Subsequently, lactate is shuttled from astrocytes to neurons via monocarboxylate transporters (MCTs) and further utilized by neurons for oxidative-and non-oxidative-derived ATP production (Bélanger et al., 2011).
Lactate is produced in the last step of the glycolytic pathway by reduction of pyruvate and concomitant oxidation of nicotinamide adenine dinucleotide (NADH) to NAD+ in a reaction catalyzed by the LDH-A isoform, when oxygen supply is low. In the opposite direction, lactate is converted to pyruvate by the LDH-B isoform (Le et al., 2010). Favorable effects of lactate on neuronal survival following hypoxia/ischemia are meanwhile widely recognized (Schurr et al., 1988, 1997, 2001; Berthet et al., 2009). Recently, we demonstrated that PrPc markedly enhances expression of both LDH-A and LDH-B isoenzymes after hypoxia/ischemia in WT primary cortical neurons and in WT ischemic brains as compared to PrPc knockout counterparts (Ramljak et al., 2015). Besides, expression of the LDH-A was significantly elevated upon transfection of Prnp0/0 cells with the vector bearing a cDNA encoding human PRNP (Ramljak et al., 2008). Additionally, LDH-A was not only identified as a PrPc interactor protein, but also as an interactor of Doppel and Shadoo, two mammalian PrPc paralogs (Watts et al., 2009). Earlier study investigating cellular distribution of the LDH isoenzymes in the hippocampus and occipital cortex of the human brain demonstrated a marked enrichment of LDH-A in astrocytes as compared to neurons (Bittar et al., 1996). Therefore, in view of ANLS it would be interesting to elucidate the role that presence/absence of PrPc in astrocytes might have on LDH-A expression level/activity, lactate trafficking from astrocytes to neurons and ultimately on neuronal survival under hypoxic conditions.
Dual Roles Of PrPc In Hypoxia: Neuroprotection vs. Tumor Progression
Promoter region of the LDH-A possesses hypoxia-responsive element (HRE) which is trans-activated under hypoxic conditions by the transcription factor hypoxia-inducible factor 1 alpha (HIF-1α; Semenza et al., 1996). HIF-1 α is one of the two subunits of hypoxia-inducible factor 1 (HIF-1) transcription complex which assimilates information on oxygen availability and cellular redox homeostasis. Stabilization of HIF-1α enables adaptive response to hypoxia and other stress conditions (Semenza, 2000; Dery et al., 2005). Thus, stabilization of HIF-1α protects astrocytes from glutamate-induced damage during severe hypoxia (Badawi et al., 2012). On the contrary, in oxygenated cells, HIF-1α is rapidly degraded via ubiquitin-proteasome pathway (Huang et al., 1998). Expression of HIF-1 target genes, such as for instance LDH-A, correlate with the levels of HIF-1 α (Ke and Costa, 2006). Strikingly, HIF-1α expression is significantly decreased in Prnp-/- and increased in Prnp+/+ mice at 24 h post-stroke (Doeppner et al., 2015) suggesting that PrPc might exert its neuroprotective effects against hypoxic damage in vivo via direct or indirect regulation of HIF-1α and hence LDH-A/lactate.
Kleene et al. (2007) demonstrated that PrPc is involved in regulation of lactate transport of astrocytes via MCT1 in conjunction with Na+/K+ ATP-ase and basigin. Astrocytes generally express MCT1 and MCT4 isoforms, engaged in lactate release, whereas neurons predominantly express MCT2 isoform, which facilitates lactate uptake (Dimmer et al., 2000; Pellerin et al., 2005; Rosafio and Pellerin, 2014). Interestingly, transient overexpression of PrPc in HEK293 cells enhanced MCT1 expression under normoxic conditions (Ramljak et al., 2015). Accordingly, in vivo neurochemical profiling in 12 month old WT and Prnp-/- mice under normoxic conditions revealed 100% increase in lactate content in the hippocampus and cerebellum of Prnp-/- mice (Cudalbu et al., 2015) indicating impaired regulation of lactate in Prnp-/- mice.
To the best of our knowledge so far no report considered the presence of two highly conserved early growth response -1 (EGR-1) consensus binding motifs (5′-GCG(T/G)GGGCG-3′) separated by only 15 bases between introns 1 and 2 of the human PRNP gene. These emerged at least 29.1 million years ago in the common stem lineage of extant Catherrini, as determined by own sequence screening (see Table 1 for accession numbers). Binding of Egr-1 to a conserved intron sequence and consecutive regulation of gene expression has been demonstrated in mouse motor spiny neurons (Keilani et al., 2012). Egr-1 is a transcription factor that is rapidly induced by hypoxia, can directly bind to HIF-1α promoter region and trans-activate it (Sperandio et al., 2009), but it can also function independently of HIF-1 α (Yan et al., 1999).
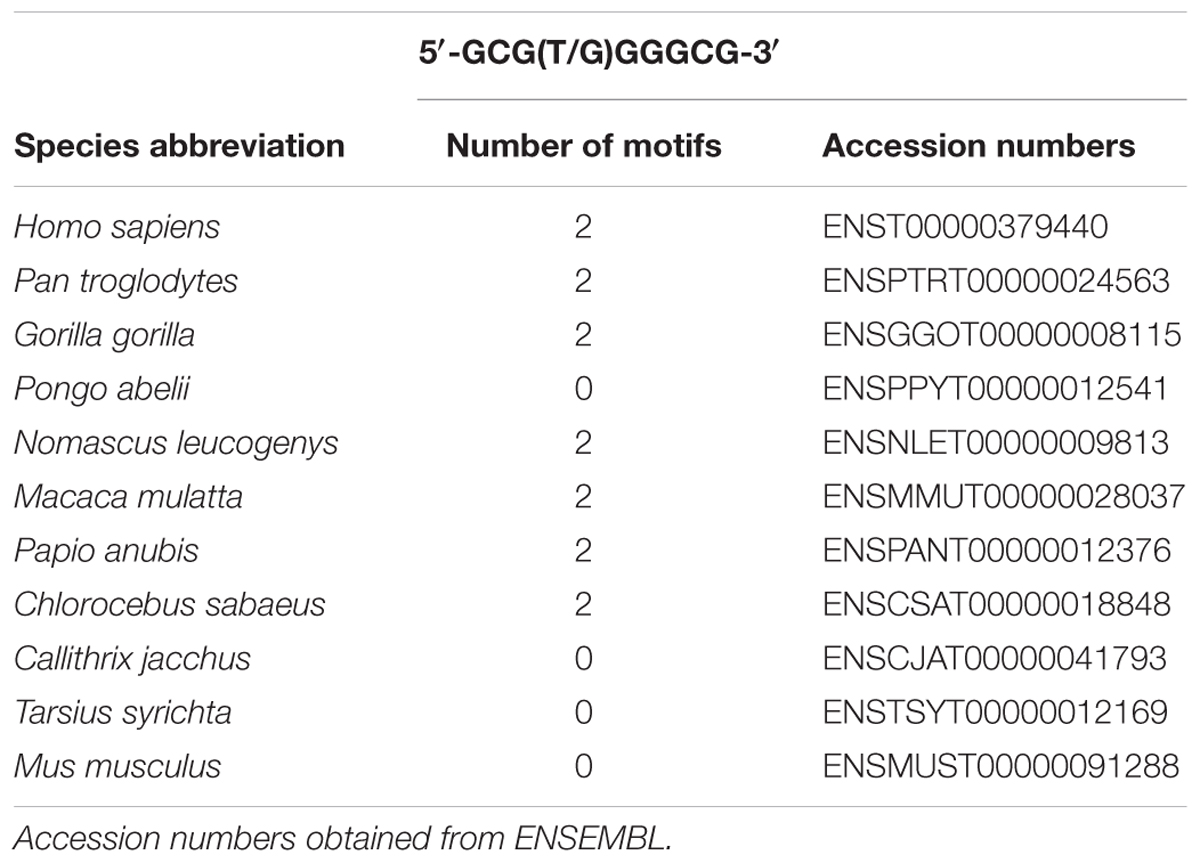
TABLE 1. EGR-1 motif in intron 1/2 of the PRNP gene.
Notably, studies performed on mouse brains suggest that prion diseases deregulate several microRNAs (miRNAs) and one of the gene promotors that were cognate to these miRNAs is Egr-1 (Shapshak, 2013). A so-called neurotoxic peptide PrP(106-126), broadly used as a model of neurotoxicity in prion diseases, induced Egr-1 synthesis in primary cortical neurons just 30 min after the treatment (Gavín et al., 2005) suggesting a hypoxic cellular environment. Furthermore, Seo et al. (2010) showed that low oxygen conditions protect neuroblastoma cells from neurotoxicity of PrP(106-126) by activating Akt signaling pathway and connote an involvement of hypoxia in prion-induced neuronal damage/disease. PrP(106-126) propels aggregation of endogenous PrPc to an amyloidogenic form and shares several properties with the disease-causing PrPsc isoform (Singh et al., 2002).
Intriguingly, distinct protein modifications and formation of detergent-insoluble protein aggregates experimentally induced by proteasome inhibition are oxygen-requiring processes that may be prevented when cells are incubated at 3% instead of 21% oxygen (Demasi and Davies, 2003). Many lines of evidence point to the deficits in cellular protein quality control and hence ubiquitin-proteasome system as central to the pathogenesis of neurodegenerative diseases (Takalo et al., 2013). Therefore, one can conclude that normoxic conditions would favor further formation of aggregates in the brains of individuals affected by neurodegenerative disorders. Contrariwise this finding suggests that hypoxia might be as well regarded as a “survival process” during which cellular machinery maintains only functions of the highest priority (protein synthesis is a low priority process!) in order to survive and concurrently prevent further formation of protein aggregates.
Both Egr-1 and HIF-1α have been associated with neurodegenerative diseases:
(i) Egr-1 is up-regulated in brains of Alzheimer disease patients and regulates transcription of genes involved in synaptic plasticity processes, in particular maintenance of long-term potentiation (Jones et al., 2001; Gómez Ravetti et al., 2010; Lu et al., 2011).
(ii) Increasing HIF-1 activity has been put forward as a potential strategy to alleviate the pathogenesis of Alzheimer’s and other neurodegenerative disorders (Zhang et al., 2011).
A recent study demonstrated that neuronal cells exposed to a highly neurotoxic monomeric misfolded prion protein (TPrP) exhibited profound decline of NAD+ levels followed by diminished ATP production. Neuronal death induced by TPrP could be completely rescued in vitro and in vivo by supplying NAD+ (Zhou et al., 2015). Primary astrocytes subjected to TPrP were not prone to TPrP-mediated toxicity and exhibited even increased NAD+ levels (Zhou et al., 2015). As cytosolic regeneration of NAD+ by LDH-A is necessary for glycolysis to carry on it would be highly interesting to verify if the treatment with TPrP renders the cellular environment hypoxic. It is recognized that diminishing NAD+ levels induce pseudohypoxia by disturbing nuclear-mitochondrial communication during aging (Gomes et al., 2013).
In any case, considering a role of putative synergistic networking between EGR-1-PrPc-HIF-1α-LDH-A under conditions of low oxygen tensions definitely deserves further attention.
Intriguingly, all four members of the above-suggested networking are in one way or another tied to another hypoxia-related disorder: cancer.
(i) EGR-1 directly targets HIF-1 α in hypoxic prostate cancer cells (Sperandio et al., 2009);
(ii) elevated HIF-1 α expression levels are linked to increased risk of mortality in different types of human cancers such as colon, breast, stomach, and other cancer types (Semenza, 2010);
(iii) HIF-1 α activates expression of LDH-A (Semenza et al., 1996);
(iv) inhibition of LDH-A inhibits tumor progression (Le et al., 2010);
(v) PRNP was proved as a prognostic indicator in patients with recurrent colorectal cancers (Antonacopoulou et al., 2010);
(vi)PrPc has a potential as a biomarker of poor prognosis in pancreas ductal adenocarcinoma patients (Sy et al., 2011–2012);
(vii) PrPc-overexpression advances invasive and metastatic features of gastric cancer cell lines (Pan et al., 2006; Liang et al., 2007; Wang et al., 2011) and
(viii) PrPc-overexpression was detected in 90% of prostate tumor biopsies (Yang et al., 2014).
Lately, tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) has been identified as relevant for PrPc-mediated survival of cancer cells. Thus, increase in PrPc expression under hypoxic conditions in human colon carcinoma HCT116 cell line was accompanied with concurrent downregulation of TRAIL (Park et al., 2015). Conversely, down-regulation of PrPc increased TRAIL-induced apoptosis under same experimental conditions (Park et al., 2015). Remarkably, an up-regulation of EGR-1 has also been shown to act as a brake on TRAIL expression (Balzarolo et al., 2013). TRAIL’s ability to selectively induce apoptosis in cancer but not in normal cells is well recognized (Wu, 2009). Considering their effect on TRAIL expression, blocking PrPc, and/or EGR-1 should be further investigated as potentially useful anticancer treatment. Moreover, activation of phosphatidylinositol 3 kinase (PI3K)/Akt survival pathway seems to be critical to TRAIL resistance in human cancer cells whereas its inhibition sensitizes resistant cancer cells to TRAIL (Xu et al., 2010). PrPc is known to modulate PI3K/Akt pathway (Vassallo et al., 2005; Weise et al., 2006).
Cross-Talk Between Wnt/β-Catenin And Pi3K/Akt Signaling Pathways Under Low Oxygen Tensions
We propose a cross-talk between Wnt/β-catenin and PI3K/Akt pathways as underlying PrPc-mediated survival under low oxygen tensions (Figure 1).
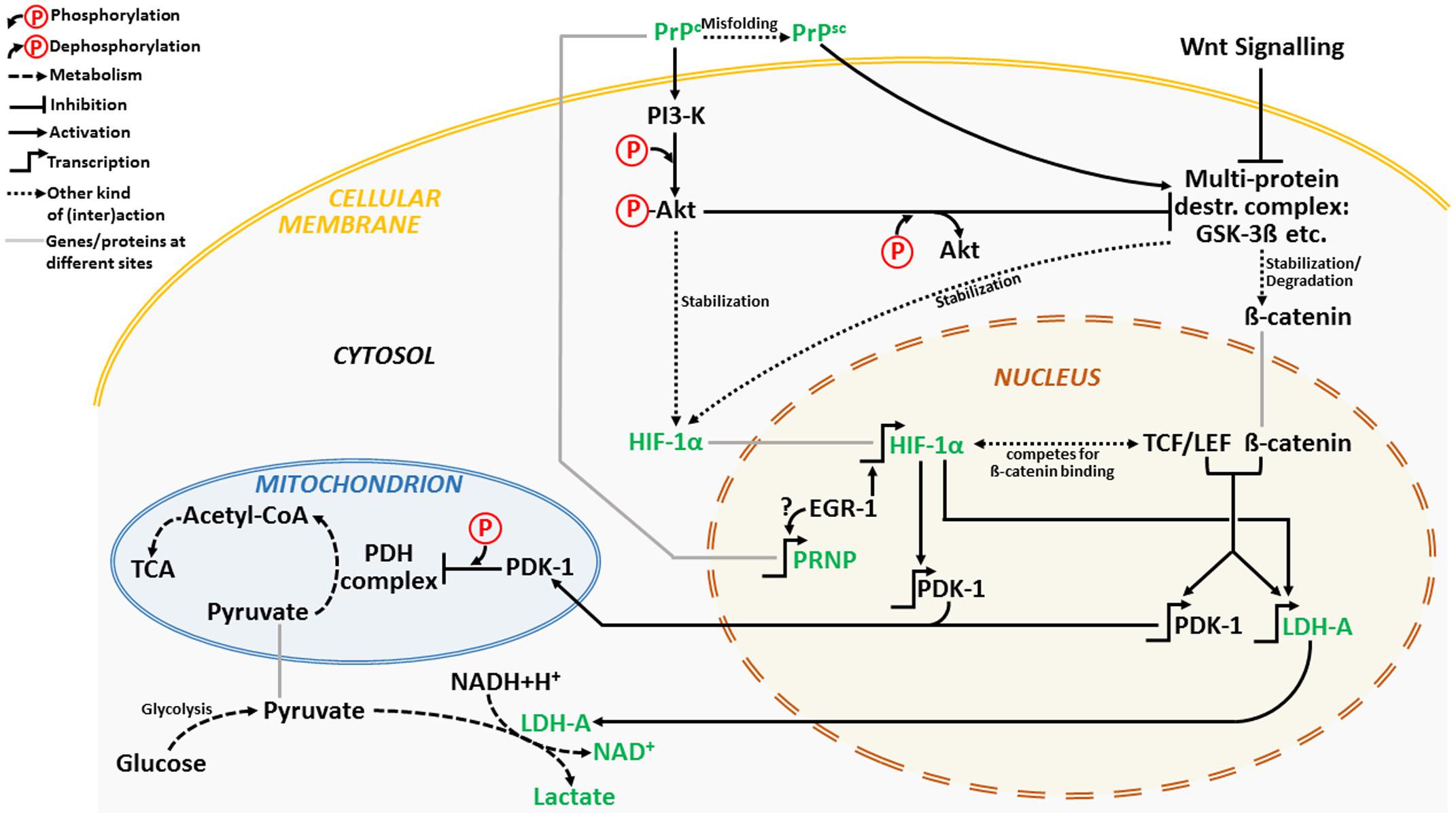
FIGURE 1. A simplified schematic depiction of a hypothetic PrPc-induced signaling cross-talk between PI3K/Akt and Wnt/β-catenin pathway under hypoxic conditions. The major players are depicted in green. For clarification, please see the Section “Cross-Talk between Wnt/β-Catenin and PI3K/Akt Signaling Pathways under Low Oxygen Tensions.”
Cellular prion protein can activate anti-apoptotic PI3K/Akt pathway (Vassallo et al., 2005). Conversely, its deletion impairs the PI3K/Akt pathway by reducing phospho-Akt expression (Weise et al., 2006). Activation of PI3K/Akt pathway seems necessary for HIF-1α stabilization early during hypoxia (Mottet et al., 2003). Besides, inhibition of glycogen synthase kinase-3β (GSK-3β) activity by phospho-Akt leads to stabilization of HIF-1α and increased HIF-1 transcriptional activity (Mottet et al., 2003) (Figure 1).
GSK-3βis a component of the multiprotein destruction complex, a part of the Wnt/β-catenin signaling pathway (MacDonald et al., 2009) which seems pertinent for a cross-talk between the both pathways. Inhibition of GSK-3β activity by phospho-Akt stabilizes β-catenin which in turn together with TCF/LEF transcription factor promotes transcription of Wnt target genes such as: pyruvate dehydrogenase kinase 1 (PDK-1) and LDH. Recently, Wnt/β-catenin signaling was linked to activation of glycolysis in colon cancer via targeting of PDK-1 (Pate et al., 2014). Furthermore, direct targeting of PDK-1 by HIF-1 results in suppression of mitochondrial function by limiting pyruvate entry into the tricarboxylic acid (TCA) cycle (Kim et al., 2006; Papandreou et al., 2006). This kinase phosphorylates and switches off mitochondrial pyruvate dehydrogenase (PDH) complex (Roche et al., 2001) so that the conversion of pyruvate to acetyl-CoA is inhibited and conversion of pyruvate to lactate favored. Intriguingly, Wnt is also capable of enhancing LDH activity thus additionally fostering glycolysis (Chafey et al., 2009).
Cellular prion protein appears to interact with β-catenin and up-regulate transcriptional activity of the β-catenin/TCF complex (Besnier et al., 2015). Moreover, Wnt/β-catenin signaling is impaired in mice infected with scrapie agents with markedly reduced levels of phospho-GSK-3β leading to its enhanced activity (Sun et al., 2015) and degradation of β-catenin. In addition, dysfunctional PI3K-Akt-GSK-3 pathway is common in models of prion diseases (Simon et al., 2014).
If the hypothetic cross-talk between Wnt/β-catenin and PI3K/Akt pathway holds true then the interesting question would be: can PrPsc mice develop cancer?
In summary, it only seems like PrPc has two sides: a “good” one – if not pivotal – for neuroprotection against oxidative stress such as hypoxia and a “bad” one promoting invasiveness of different cancer types. However, these are only two sides of the same medal called: SURVIVAL.
Author Contributions
SR developed the concept and wrote the manuscript; HH analyzed the data and wrote the manuscript; IZ wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Antonacopoulou, A. G., Palli, M., Marousi, S., Dimitrakopoulos, F. I., Kyriakopoulou, U., Tsamandas, A. C., et al. (2010). Prion protein expression and the M129V polymorphism of the PRNP gene in patients with colorectal cancer. Mol. Carcinog. 49, 693–699. doi: 10.1002/mc.20642
Badawi, Y., Ramamoorthy, P., and Shi, H. (2012). Hypoxia-inducible factor 1 protects hypoxic astrocytes against glutamate toxicity. ASN Neuro 4, 231–241. doi: 10.1042/AN20120006
Balzarolo, M., Watzl, C., Medema, J. P., and Wolkers, M. C. (2013). NAB2 and EGR-1 exert opposite roles in regulating TRAIL expression in human Natural Killer cells. Immunol. Lett. 151, 61–67. doi: 10.1016/j.imlet.2013.02.001
Bélanger, M., Allaman, I., and Magistretti, P. J. (2011). Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. 14, 724–738. doi: 10.1016/j.cmet.2011.08.016
Belay, E. D. (1999). Transmissible spongiform encephalopathies in humans. Annu. Rev. Microbiol. 53, 283–314. doi: 10.1146/annurev.micro.53.1.283
Berthet, C., Lei, H., Thevenet, J., Gruetter, R., Magistretti, P. J., and Hirt, L. (2009). Neuroprotective role of lactate after cerebral ischemia. J. Cereb. Blood Flow Metab. 240, 1780–1789. doi: 10.1038/jcbfm.2009.97
Bertuchi, F. R., Bourgeon, D. M., Landemberger, M. C., Martins, V. R., and Cerchiaro, G. (2012). PrPC displays an essential protective role from oxidative stress in an astrocyte cell line derived from PrPC knockout mice. Biochem. Biophys. Res. Commun. 418, 27–32. doi: 10.1016/j.bbrc.2011.12.098
Besnier, L. S., Cardot, P., Da Rocha, B., Simon, A., Loew, D., Klein, C., et al. (2015). The cellular prion protein PrPc is a partner of the Wnt pathway in intestinal epithelial cells. Mol. Biol. Cell 26, 3313–3328. doi: 10.1091/mbc.E14-11-1534
Bittar, P. G., Charnay, Y., Pellerin, L., Bouras, C., and Magistretti, P. J. (1996). Selective distribution of lactate dehydrogenase isoenzymes in neurons and astrocytes of human brain. J. Cereb. Blood Flow Metab. 16, 1079–1089. doi: 10.1097/00004647-199611000-00001
Blankenship, R. E. (2010). Early evolution of photosynthesis. Plant Physiol. 154, 434–438. doi: 10.1104/pp.110.161687
Boutilier, R. G. (2001). Mechanisms of cell survival in hypoxia and hypothermia. J. Exp. Biol. 204, 3171–3181.
Brown, D. R. (2005). Neurodegeneration and oxidative stress: prion disease results from loss of antioxidant defence. Folia Neuropathol. 43, 229–243.
Buttgereit, F., and Brand, M. D. (1995). A hierarchy of ATP-consuming processes in mammalian cells. Biochem. J. 312, 163–167. doi: 10.1042/bj3120163
Chafey, P., Finzi, L., Boisgard, R., Caüzac, M., Clary, G., Broussard, C., et al. (2009). Proteomic analysis of beta-catenin activation in mouse liver by DIGE analysis identifies glucose metabolism as a new target of the Wnt pathway. Proteomics 9, 3889–3900. doi: 10.1002/pmic.200800609
Cudalbu, C., Craveiro, M., Mlynárik, V., Bremer, J., Aguzzi, A., and Gruetter, R. (2015). In Vivo longitudinal (1)H MRS study of transgenic mouse models of prion disease in the hippocampus and cerebellum at 14.1 T. Neurochem. Res. 40, 2639–2646. doi: 10.1007/s11064-015-1643-9
Demasi, M., and Davies, K. J. A. (2003). Proteasome inhibitors induce intracellular protein aggregation and cell death by an oxygen-dependent mechanism. FEBS Lett. 542, 89–94. doi: 10.1016/S0014-5793(03)00353-3
Dery, M. A., Michaud, M. D., and Richard, D. E. (2005). Hypoxia-inducible factor 1: regulation by hypoxic and nonhypoxic activators. Int. J. Biochem. Cell Biol. 37, 535–540. doi: 10.1016/j.biocel.2004.08.012
Dimmer, K. S., Friedrich, B., Lang, F., Deitmer, J. W., and Bröer, S. (2000). The low-affinity monocarboxylate transporter MCT4 is adapted to the export of lactate in highly glycolytic cells. Biochem. J. 350, 219–227. doi: 10.1042/0264-6021:3500219
Doeppner, T. R., Kaltwasser, B., Schlechter, J., Jaschke, J., Kilic, E., Bähr, M., et al. (2015). Cellular prion protein promotes post-ischemic neuronal survival, angioneurogenesis and enhances neural progenitor cell homing via proteasome inhibition. Cell Death Dis. 6:e2024. doi: 10.1038/cddis.2015.365
Gavín, R., Braun, N., Nicolas, O., Parra, B., Ureña, J. M., Mingorance, A., et al. (2005). PrP(106-126) activates neuronal intracellular kinases and Egr1 synthesis through activation of NADPH-oxidase independently of PrPc. FEBS Lett. 579, 4099–4106. doi: 10.1016/j.febslet.2005.06.037
Gomes, A. P., Price, N. L., Ling, A. J., Moslehi, J. J., Montgomery, M. K., Rajman, L., et al. (2013). Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 155, 1624–1638. doi: 10.1016/j.cell.2013.11.037
Gómez Ravetti, M., Rosso, O. A., Berretta, R., and Moscato, P. (2010). Uncovering molecular biomarkers that correlate cognitive decline with the changes of hippocampus’ gene expression profiles in Alzheimer’s disease. PLoS ONE 5:e10153. doi: 10.1371/journal.pone.0010153
Huang, L. E., Gu, J., Schau, M., and Bunn, H. F. (1998). Regulation of hypoxia inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc. Natl. Acad. Sci. U.S.A. 95, 7987–7992. doi: 10.1073/pnas.95.14.7987
Imuta, N., Hori, O., Kitao, Y., Tabata, Y., Yoshimoto, T., Matsuyama, T., et al. (2007). Hypoxia-mediated induction of heme oxygenase type I and carbon monoxide release from astrocytes protects nearby cerebral neurons from hypoxia-mediated apoptosis. Antioxid. Redox Signal. 9, 543–552. doi: 10.1089/ars.2006.1519
Jackson, D. C., and Ultsch, G. R. (2010). Physiology of hibernation under the ice by turtles and frogs. J. Exp. Zool. A Ecol. Genet. Physiol. 313, 311–327. doi: 10.1002/jez.603
Jones, M. W., Errington, M. L., French, P. J., Fine, A., Bliss, T. V., Garel, S., et al. (2001). A requirement for the immediate early gene Zif268 in the expression of late LTP and long-term memories. Nat. Neurosci. 4, 289–296. doi: 10.1038/85138
Ke, Q., and Costa, M. (2006). Hypoxia-inducible factor-1 (HIF-1). Mol. Pharmacol. 70, 1469–1480. doi: 10.1124/mol.106.027029
Keilani, S., Chandwani, S., Dolios, G., Bogush, A., Beck, H., Hatzopoulos, A. K., et al. (2012). Egr-1 induces DARPP-32 expression in striatal medium spiny neurons via a conserved intragenic element. J. Neurosci. 32, 6808–6818. doi: 10.1523/JNEUROSCI.5448-11.2012
Kesaraju, S., Schmidt-Kastner, R., Prentice, H. M., and Milton, S. L. (2009). Modulation of stress proteins and apoptotic regulators in the anoxia tolerant turtle brain. J. Neurochem. 109, 1413–1426. doi: 10.1111/j.1471-4159.2009.06068.x
Kim, J., Tchernyshyov, L., Semenza, G. L., and Dang, C. V. (2006). HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 3, 177–185. doi: 10.1016/j.cmet.2006.02.002
Kleene, R., Loers, G., Langer, J., Frobert, Y., Buck, F., and Schachner, M. (2007). Prion protein regulates glutamate-dependent lactate transport of astrocytes. J. Neurosci. 27, 12331–12340. doi: 10.1523/JNEUROSCI.1358-07.2007
Le, A., Cooper, C. R., Gouw, A. M., Dinavahi, R., Maitra, A., Deck, L. M., et al. (2010). Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. U.S.A. 107, 2037–2042. doi: 10.1073/pnas.0914433107
Liang, J., Pan, Y., Zhang, D., Guo, C., Shi, Y., Wang, J., et al. (2007). Cellular prion protein promotes proliferation and G1/S transition of human gastric cancer cells SGC7901 and AGS. FASEB J. 21, 2247–2256. doi: 10.1096/fj.06-7799com
Lima, F. R., Arantes, C. P., Muras, A. G., Nomizo, R., Brentani, R. R., and Martins, V. R. (2007). Cellular prion protein expression in astrocytes modulates neuronal survival and differentiation. J. Neurochem. 103, 2164–2176. doi: 10.1111/j.1471-4159.2007.04904.x
Lu, Y., Li, T., Qureshi, H. Y., Han, D., and Paudel, H. K. (2011). Early growth response 1 (Egr-1) regulates phosphorylation of microtubule associated protein tau in mammalian brain. J. Biol. Chem. 286, 20569–20581. doi: 10.1074/jbc.M111.220962
Lutz, P. L., and Prentice, H. M. (2002). Sensing and responding to hypoxia, molecular and physiological mechanisms. Integr. Comp. Biol. 42, 463–468. doi: 10.1093/icb/42.3.463
MacDonald, B. T., Tamai, K., and He, X. (2009). Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev. Cell 17, 9–26. doi: 10.1016/j.devcel.2009.06.016
Magistretti, P. J. (2008). “Brain energy metabolism,” in Fundamental Neuroscience, 3rd Edn, eds L. Squire, D. Berg, and F. E. Bloom (San Diego, CA: Academic Press), 271–293.
Marrif, H., and Juurlink, B. H. (1999). Astrocytes respond to hypoxia by increasing glycolytic capacity. J. Neurosci. Res. 57, 255–260. doi: 10.1002/(SICI)1097-4547(19990715)57:2<255::AID-JNR11>3.3.CO;2-Y
Martin, W., and Russell, M. J. (2003). On the origins of cells: a hypothesis for the evolutionary transitions from abiotic geochemistry to chemoautotrophic prokaryotes, and from prokaryotes to nucleated cells. Philos. Trans. R. Soc. Lond. B Biol. Sci. 358, 59–83. doi: 10.1098/rstb.2002.1183
McLennan, N. F., Brennan, P. M., McNeill, A., Davies, I., Fotheringham, A., Rennison, K. A., et al. (2004). Prion protein accumulation and neuroprotection in hypoxic brain damage. Am. J. Pathol. 165, 227–235. doi: 10.1016/S0002-9440(10)63291-9
Mitteregger, G., Vosko, M., Krebs, B., Xiang, W., Kohlmannsperger, V., Nölting, S., et al. (2007). The role of the octarepeat region in neuroprotective function of the cellular prion protein. Brain Pathol. 17, 174–183. doi: 10.1111/j.1750-3639.2007.00061.x
Moller, I. A. (2001). Plant mitochondria and oxidative stress: electron transport, NADPH turnover, and metabolism of reactive oxygen species. Annu. Rev. Plant Mol. Biol. 52, 561–591. doi: 10.1146/annurev.arplant.52.1.561
Mottet, D., Dumont, V., Deccache, Y., Demazy, C., Ninane, N., Raes, M., et al. (2003). Regulation of hypoxia-inducible factor-1 alpha protein level during hypoxic conditions by the phosphatidylinositol 3-kinase/Akt/glycogen synthase kinase 3beta pathway in HepG2 cells. J. Biol. Chem. 278, 31277–31285. doi: 10.1074/jbc.M300763200
Nilsson, G. E., and Renshaw, G. M. (2004). Hypoxic survival strategies in two fishes: extreme anoxia tolerance in the North European crucian carp and natural hypoxic preconditioning in a coral-reef shark. J. Exp. Biol. 207, 3131–3139. doi: 10.1242/jeb.00979
Pan, Y., Zhao, L., Liang, J., Liu, J., Shi, Y., Liu, N., et al. (2006). Cellular prion protein promotes invasion and metastasis of gastric cancer. FASEB J. 20, 1886–1888. doi: 10.1096/fj.06-6138fje
Papandreou, I., Cairns, R. A., Fontana, L., Lim, A. L., and Denko, N. C. (2006). HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 3, 187–197. doi: 10.1016/j.cmet.2006.01.012
Park, J. Y., Jeong, J. K., Lee, J. H., Moon, J. H., Kim, S. W., Lee, Y. J., et al. (2015). Induction of cellular prion protein (PrPc) under hypoxia inhibits apoptosis caused by TRAIL treatment. Oncotarget 10, 5342–5353. doi: 10.18632/oncotarget.3028
Pate, K. T., Stringari, C., Sprowl-Tanio, S., Wang, K., TeSlaa, T., Hoverter, N. P., et al. (2014). Wnt signaling directs a metabolic program of glycolysis and angiogenesis in colon cancer. EMBO J. 33, 1454–1473. doi: 10.15252/embj.201488598
Pellerin, L., Bergersen, L. H., Halestrap, A. P., and Pierre, K. (2005). Cellular and subcellular distribution of monocarboxylate transporters in cultured brain cells and in the adult brain. J. Neurosci. Res. 79, 55–64. doi: 10.1002/jnr.20307
Pellerin, L., and Magistretti, P. J. (1994). Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. U.S.A. 91, 10625–10629. doi: 10.1073/pnas.91.22.10625
Ramljak, S., Asif, A. R., Armstrong, V. W., Wrede, A., Groschup, M. H., Buschmann, A., et al. (2008). Physiological role of the cellular prion protein (PrPc): protein profiling study in two cell culture systems. J. Proteome Res. 7, 2681–2695. doi: 10.1021/pr7007187
Ramljak, S., Schmitz, M., Zafar, S., Wrede, A., Schenkel, S., Asif, A. R., et al. (2015). Cellular prion protein directly interacts with and enhances lactate dehydrogenase expression under hypoxic conditions. Exp. Neurol. 271, 155–167. doi: 10.1016/j.expneurol.2015.04.025
Roche, T. E., Baker, J. C., Yan, Y. H., Hiromasa, Y., Gong, X. M., Peng, T., et al. (2001). Distinct regulatory properties of pyruvate dehydrogenase kinase and phosphatase isoforms. Prog. Nucleic Acid Res. Mol. Biol. 70, 33–75. doi: 10.1016/S0079-6603(01)70013-X
Rosafio, K., and Pellerin, L. (2014). Oxygen tension controls the expression of the monocarboxylate transporter MCT4 in cultured mouse cortical astrocytes via a hypoxia-inducible factor-1α-mediated transcriptional regulation. Glia 62, 477–490. doi: 10.1002/glia.22618
Rossen, R., Kabat, H., and Anderson, J. P. (1943). Acute arrest of cerebral circulation in man. Arch. Neurol. Psychiatry 50, 510–528. doi: 10.1001/archneurpsyc.1943.02290230022002
Schätzl, H. M., Da Costa, M., Taylor, L., Cohen, F. E., and Prusiner, S. B. (1995). Prion protein gene variation among primates. J. Mol. Biol. 245, 362–374. doi: 10.1006/jmbi.1994.0030
Schurr, A., Dong, W. Q., Reid, K. H., West, C. A., and Rigor, B. M. (1988). Lactic acidosis and recovery of neuronal function following cerebral hypoxia in vitro. Brain Res. 438, 311–314. doi: 10.1016/0006-8993(88)91354-6
Schurr, A., Payne, R. S., Miller, J. J., and Rigor, B. M. (1997). Brain lactate, not glucose, fuels the recovery of synaptic function from hypoxia upon re-oxygenation: an in vitro study. Brain Res. 744, 105–111. doi: 10.1016/S0006-8993(96)01106-7
Schurr, A., Payne, R. S., Miller, J. J., Tseng, M. T., and Rigor, B. M. (2001). Blockade of lactate transport exacerbates delayed neuronal damage in a rat model of cerebral ischemia. Brain Res. 895, 268–272. doi: 10.1016/S0006-8993(01)02082-0
Semenza, G. L. (2000). HIF-1: mediator of physiological and pathophysiological responses to hypoxia. J. Appl. Physiol. 88, 1474–1480.
Semenza, G. L. (2010). Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 29, 625–634. doi: 10.1038/onc.2009.441
Semenza, G. L., Jiang, B. H., Leung, S. W., Passantino, R., Concordet, J. P., Maire, P., et al. (1996). Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J. Biol. Chem. 271, 32529–32537. doi: 10.1074/jbc.271.51.32529
Seo, J. S., Seol, J. W., Moon, M. H., Jeong, J. K., Lee, Y. J., and Park, S. Y. (2010). Hypoxia protects neuronal cells from human prion protein fragment-induced apoptosis. J. Neurochem. 112, 715–722. doi: 10.1111/j.1471-4159.2009.06496.x
Shapshak, P. (2013). Molecule of the month: miRNA and Human Prion brain disease. Bioinformation 9, 659–660. doi: 10.6026/97320630009549
Simon, D., Herva, M. E., Benitez, M. J., Garrido, J. J., Rojo, A. I., Cuadrado, A., et al. (2014). Dysfunction of the PI3-Akt-GSK-3 pathway is a common feature in cell culture and in vivo models of prion disease. Neuropathol. Appl. Neurobiol. 40, 311–326. doi: 10.1111/nan.12066
Singh, N., Gu, Y., Bose, S., Kalepu, S., Mishra, R. S., and Verghese, S. (2002). Prion peptide 106–126 as a model for prion replication and neurotoxicity. Front. Biosci. 7:a60–a71. doi: 10.2741/singh
Sperandio, S., Fortin, J., Sasik, R., Robitaille, L., Corbeil, J., and de Belle, I. (2009). The transcription factor Egr1 regulates the HIF-1alpha gene during hypoxia. Mol. Carcinog. 48, 38–44. doi: 10.1002/mc.20454
Steele, A. D., Lindquist, S., and Aguzzi, A. (2007). The prion protein knockout mouse: a phenotype under challenge. Prion 1, 83–93. doi: 10.4161/pri.1.2.4346
Sun, J., Wang, H., Chen, L. N., Wang, J., Lv, Y., Yang, X. D., et al. (2015). Remarkable impairment of Wnt/β-catenin signaling in the brains of the mice infected with scrapie agents. J. Neurochem. doi: 10.1111/jnc.13416 [Epub ahead of print].
Sy, M. S., Altekruse, S. F., Li, C., Lynch, C. F., Goodman M. T., Hernandez, B. Y., et al. (2011–2012). Association of prion protein expression with pancreatic adenocarcinoma survival in the SEER residual tissue repository. Cancer Biomark 10, 251–258. doi: 10.3233/CBM-2012-0256
Takalo, M., Salminen, A., Soininen, H., Hiltunen, M., and Haapasalo, A. (2013). Protein aggregation and degradation mechanisms in neurodegenerative diseases. Am. J. Neurodegener. Dis. 2, 1–14.
Takano, T., Oberheim, N., Cotrina, M. L., and Nedergaard, M. (2009). Astrocytes and ischemic injury. Stroke 40, S8–S12. doi: 10.1161/STROKEAHA.108.533166
Vassallo, N., Herms, J., Behrens, C., Krebs, B., Saeki, K., Onodera, T., et al. (2005). Activation of phosphatidylinositol 3-kinase by cellular prion protein and its role in cell survival. Biochem. Biophys. Res. Commun. 332, 75–82. doi: 10.1016/j.bbrc.2005.04.099
Vibulsreth, S., Hefti, F., Ginsberg, M. D., Dietrich, W. D., and Busto, R. (1987). Astrocytes protect cultured neurons from degeneration induced by anoxia. Brain Res. 422, 303–311. doi: 10.1016/0006-8993(87)90937-1
Wang, J. H., Du, J. P., Zhang, Y. H., Zhao, X. J., Fan, R. Y., Wang, Z. H., et al. (2011). Dynamic changes and surveillance function of prion protein expression in gastric cancer drug resistance. World J. Gastroenterol. 17, 3986–3993. doi: 10.3748/wjg.v17.i35.3986
Watts, J. C., Huo, H., Bai, Y., Ehsani, S., Jeon, A. H., Shi, T., et al. (2009). Interactome analyses identify ties of PrP and its mammalian paralogs to oligomannosidic N-glycans and endoplasmic reticulum-derived chaperons. PLoS Pathog. 5:e1000608. doi: 10.1371/journal.ppat.1000608
Webster, K. A. (2003). Evolution of the coordinate regulation of glycolytic enzyme genes by hypoxia. J. Exp. Biol. 206, 2911–2922. doi: 10.1242/jeb.00516
Weise, J., Crome, O., Sandau, R., Schulz-Schaeffer, W., Bahr, M., and Zerr, I. (2004). Upregulation of cellular prion protein (PrPc) after focal cerebral ischemia and influence of lesion severity. Neurosci. Lett. 372, 146–150. doi: 10.1016/j.neulet.2004.09.030
Weise, J., Sandau, R., Schwarting, S., Crome, O., Wrede, A., Schulz-Schaeffer, W., et al. (2006). Deletion of cellular prion protein results in reduced Akt activation, enhanced postichemic caspase-3 activation, and exacerbation of ischemic brain injury. Stroke 37, 1296–1300. doi: 10.1161/01.STR.0000217262.03192.d4
Wu, G. S. (2009). TRAIL as a target in anti-cancer therapy. Cancer Lett. 285, 1–5. doi: 10.1016/j.canlet.2009.02.029
Xu, J., Zhou, J. Y., Wei, W. Z., and Wu, G. S. (2010). Activation of the Akt survival pathway contributes to TRAIL resistance in cancer cells. PLoS ONE 5:e10226. doi: 10.1371/journal.pone.0010226
Yager, J. Y., Kala, G., Hertz, L., and Juurlink, B. H. (1994). Correlation between content of high-energy phosphates and hypoxic-ischemic damage in immature and mature astrocytes. Brain Res. Dev. Brain Res. 82, 62–68. doi: 10.1016/0165-3806(94)90148-1
Yan, S. F., Lu, J., Zou, Y. S., Soh-Won, J., Cohen, D. M., Buttrick, P. M., et al. (1999). Hypoxia-associated induction of early growth response-1 gene expression. J. Biol. Chem. 274, 15030–15040. doi: 10.1074/jbc.274.21.15030
Yang, X., Zhang, Y., Zhang, L., He, T., Zhang, J., and Li, C. (2014). Prion protein and cancers. Acta Biochim. Biophys Sin. 46, 431–440. doi: 10.1093/abbs/gmu019
Yu, A. C., Gregory, G. A., and Chan, P. H. (1989). Hypoxia-induced dysfunctions and injury of astrocytes in primary cell cultures. J. Cereb. Blood Flow Metab. 9, 20–28. doi: 10.1038/jcbfm.1989.3
Zhang, Z., Yan, J., Chang, Y., ShiDu Yan, S., and Shi, H. (2011). Hypoxia inducible factor-1 as a target for neurodegenerative diseases. Curr. Med. Chem. 18, 4335–4343. doi: 10.2174/092986711797200426
Keywords: PrPc, hypoxia, glycolysis, neuroprotection, cancer
Citation: Ramljak S, Herlyn H and Zerr I (2016) Cellular Prion Protein (PrPc) and Hypoxia: True to Each Other in Good Times and in Bad, in Sickness, and in Health. Front. Cell. Neurosci. 10:292. doi: 10.3389/fncel.2016.00292
Received: 07 October 2016; Accepted: 05 December 2016;
Published: 19 December 2016.
Edited by:
Dirk M. Hermann, University of Duisburg-Essen, GermanyCopyright © 2016 Ramljak, Herlyn and Zerr. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sanja Ramljak, c2FuamEucmFtbGpha0BzY2llbWEuZGU=