 Radhika Rastogi1
Radhika Rastogi1 Fengwu Li
Fengwu Li Yuchuan Ding
Yuchuan Ding- 1Department of Neurosurgery, Wayne State University School of Medicine, Detroit, MI, USA
- 2China-America Institute of Neuroscience, Beijing Luhe Hospital, Capital Medical University, Beijing, China
- 3Department of Neurology, Beijing Luhe Hospital, Capital Medical University, Beijing, China
Nicotinamide adenine dinucleotide phosphate (NAPDH) oxidase (NOX) is an enzyme complex with the sole function of producing superoxide anion and reactive oxygen species (ROS) at the expense of NADPH. Vital to the immune system as well as cellular signaling, NOX is also involved in the pathologies of a wide variety of disease states. Particularly, it is an integral player in many neurological diseases, including stroke, TBI, and neurodegenerative diseases. Pathologically, NOX produces an excessive amount of ROS that exceed the body’s antioxidant ability to neutralize them, leading to oxidative stress and aberrant signaling. This prevalence makes it an attractive therapeutic target and as such, NOX inhibitors have been studied and developed to counter NOX’s deleterious effects. However, recent studies of NOX have created a better understanding of the NOX complex. Comprised of independent cytosolic subunits, p47-phox, p67-phox, p40-phox and Rac, and membrane subunits, gp91-phox and p22-phox, the NOX complex requires a unique activation process through subunit interaction. Of these subunits, p47-phox plays the most important role in activation, binding and translocating the cytosolic subunits to the membrane and anchoring to p22-phox to organize the complex for NOX activation and function. Moreover, these interactions, particularly that between p47-phox and p22-phox, are dependent on phosphorylation initiated by upstream processes involving protein kinase C (PKC). This review will look at these interactions between subunits and with PKC. It will focus on the interaction involving p47-phox with p22-phox, key in bringing the cytosolic subunits to the membrane. Furthermore, the implication of these interactions as a target for NOX inhibitors such as apocynin will be discussed as a potential avenue for further investigation, in order to develop more specific NOX inhibitors based on the inhibition of NOX assembly and activation.
Introduction
Reactive oxygen species (ROS) have been identified as essential players in an increasing number of disease states, exacerbating oxidative stress and facilitating tissue dysfunction. However, the origin of ROS varies widely. They originate from both exogenous sources, such as radiation or drugs, and endogenous sources, including mitochondria, peroxisomes, and a host of enzymes which include xanthine oxidase, nitric oxide synthetase, p450 cytochrome, and most importantly, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) (Brieger et al., 2012). Of these, NOX is the only one with the exclusive role in producing ROS and is the fundamental source of ROS in our body.
NOX is a family of integral enzyme complexes present prevalently throughout the body. Classically known for its role in phagocytic leukocytes, NOX generates superoxide anion (O2•–) from molecular oxygen at the expense of NADPH when activated in these cells in order to destroy phagocytosed organisms and facilitate their anti-microbial function. However, its presence is not limited to leukocytes but has also been found in most tissues of the body. Its production of ROS has also been implicated in biosignaling and cell function as well as in apoptotic regulation (Sumimoto et al., 2005; D’Autréaux and Toledano, 2007). To prevent ROS overproduction, NOX activation is heavily modulated through its activation via phosphorylation. Such phosphorylation is facilitated by stimuli such as activation via phagocytic particles or by physiological or pathological cues, particularly cellular stresses (Jiang et al., 2011). These cues include altered nutritional status, such as hyperglycemia which can lead to protein kinase C (PKC)-activated NOX activation, altered cellular environments, such as hypoxia, and altered chemical environments due to inflammatory and stressed states (Jiang et al., 2011; Shao and Bayraktutan, 2014).
Beyond its role in defense and signaling, NOX is a primary player in oxidative stress, the condition where ROS levels exceed the body’s antioxidant defense mechanisms. With uncompensated increases in ROS, increases in lipid peroxidation, cell death, matrix metalloprotease (MMP) production, DNA damage and deleterious signaling cascades ensue. Such pathologic production of ROS can lead to a host of disease states. Excessive NOX production of ROS as well as upregulation of NOX is seen in numerous disease states, including those involving inflammation, diabetes, and cancer (Tang et al., 2012). Moreover, it plays a key role in neurological pathologies. As a main producer of ROS within the central nervous system (CNS), NOX contributes greatly to subsequent brain damage resulting from traumatic brain injury, ischemia/reperfusion injury in stroke, and to the pathologies of neurodegenerative diseases (Tang et al., 2011). As a result of its pervasive action, NOX has become an attractive target for therapies and drugs, but understanding its assembly and activation process is critical to developing therapeutic systems with appropriate specificity.
NOX Structure, Subunit Interactions, and Activation
Structure and Subunits
With its prevalence in disease, it is important to characterize NOX in its structure. The classical NOX found in neutrophils is an enzyme complex that is comprised of several subunits, including two membrane subunits (gp91-phox (NOX2) and p22-phox), three cytosolic subunits (p47-phox, p67-phox, p40-phox), and the G-protein Rac (Figure 1). More recently, several NOX complexes have been found with homologs of the gp91-phox (NOX2) subunit, which are NOX1, NOX3 to NOX5, DUOX1, and DUOX2. The enzyme complexes take the name of their catalytic homolog. These alternative subunits have unique roles in their respective NOX complexes and will be discussed below.
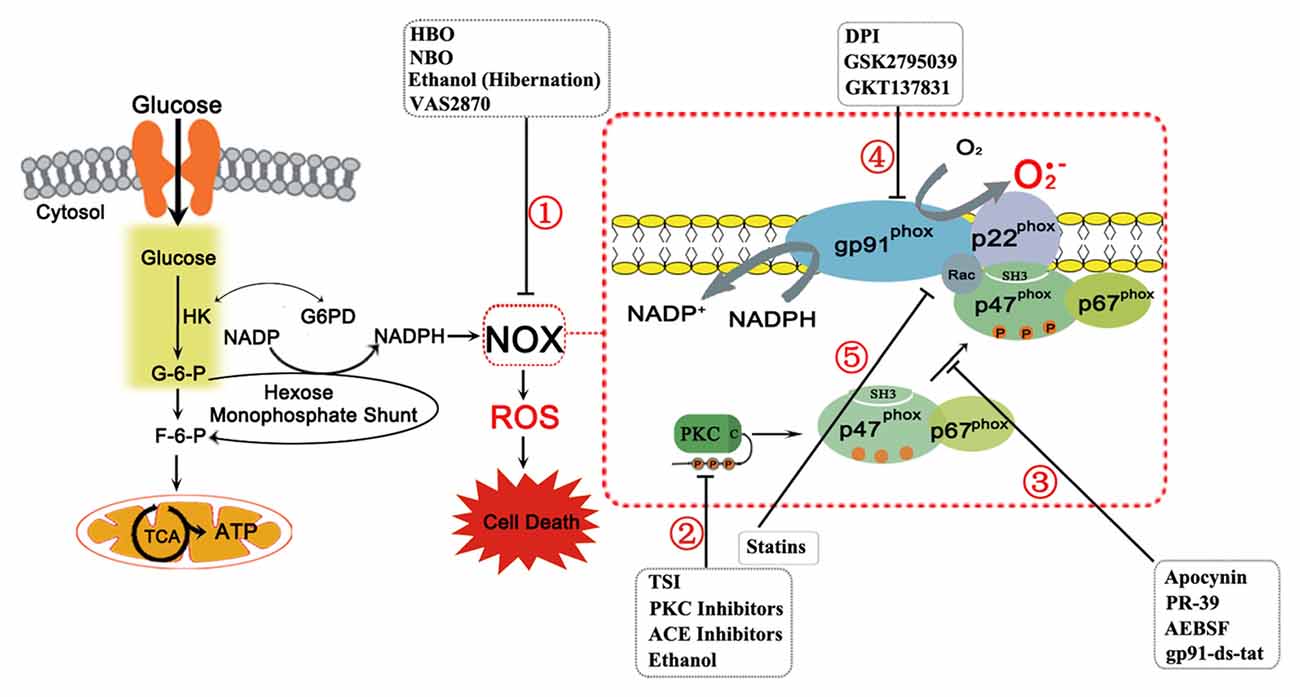
Figure 1. The nicotinamide adenine dinucleotide phosphate (NAPDH) oxidase (NOX) complex and its subunits are shown above with details pertaining to its activation sequence. The NOX complex consists of two membrane subunits (gp91-phox, or its homologs, and p22-phox) that form the catalytic core of NOX, several cytosolic subunits (p47-phox, p67-phox, p40-phox (not shown)), and the G-protein Rac, which are required for assembly and activation. In NOX activation, stimuli induce Protein Kinase C (PKC) activation, which phosphorylates p47-phox, already complexed to p67-phox, and reveals the p47-phox SH3 domain. The cytosolic subunits then translocate to the membrane due to interactions between the SH3 domains of p47-phox with the proline rich region of p22-phox. Rac independently translocates to the complex to activate NOX. When activated, NOX produces superoxide ion through a redox reaction with molecular oxygen and NADPH. The latter is produced from glucose, which enters the cell and as an intermediate of glycolysis, produces glucose-6-phosphate (G-6-P). This substrate may continue through glycolysis or may be shunted to the hexose monophosphate shunt to produce NADPH by reducing NADP+. The remaining carbon backbone is shunted back to the glycolytic process downstream of G-6-P to fructose-6-phosphate (F-6-P) and then enters the tricarboxylic acid (TCA) cycle and electron transport chain to produce energy as adenosine triphosphate (ATP) in mitochondria. However, the NADPH produced, with molecular oxygen, acts as a substrate for NOX to produce reactive oxygen species (ROS). In disease states, ROS overproduction leads to cell death. NOX inhibitors targeted to prevent this are also shown with their location of action. NOX inhibition can function through several pathways: (1) By acting on NOX via an unspecified mechanism; (2) By acting on the PKC isoforms or upstream to PKC to prevent NOX activation by inhibiting phosphorylation; (3) By inhibiting the interactions of p47-phox with NOX subunits, particularly p22-phox, preventing NOX activation by inhibiting assembly; (4) By acting directly on gp91-phox or its homologs, preventing NOX catalytic activity; and (5) By preventing Rac translocation to the NOX complex to prevent NOX activation.
The NOX complex itself is split between the membrane compartment and the cytosolic compartment at rest. The membrane compartment forms the catalytic core of NOX, the flavocytochrome b558. In the classical NOX complex, this is comprised of the gp91-phox subunit (NOX2) and p22-phox. The gp91-phox subunit is the main catalytic subunit that transfers NADPH electrons via FAD and heme to molecular oxygen through coupled redox reactions, producing superoxide anion. It constitutively forms a heterodimer with p22-phox on the membrane (Yu et al., 1998). However, activation is dependent on the translocation of the cytosolic subunits to the membrane subunits as well as independent activation of Rac to fully assemble the complex. The cytosolic subunits depend on phosphorylation for activation. At rest, p40-phox and p67-phox subunits are commonly complexed in the cytosol and may be associated with p47-phox as well. Phosphorylation activates p47-phox and unmasks a region to allow it to definitely bind p67-phox and form a trimeric cytosolic complex (Tsunawaki and Yoshikawa, 2000; Lapouge et al., 2002). Subsequently, p47-phox facilitates their translocation to the membrane, binding primarily to p22-phox and assembling the active NOX complex (Ago et al., 2003). Though a complex process, this unique activation process allows for specific modulation at many levels of the NOX complex both prior to activation and in the active state (Groemping and Rittinger, 2005; Sumimoto et al., 2005).
With seven different isoforms, NOX may contain homologs instead of the gp91-phox (NOX2) subunit. As the catalytic core of NOX, catalytic function is preserved through structural homology within these homologs NOX1, NOX 3–5, DUOX1, and DUOX2. All have six or seven transmembrane domains, with two heme binding regions containing histidine residues and a NADPH binding region on the intracellular C-terminus to facilitate superoxide production. However, regulation, localization, and function differ slightly across the isoforms. NOX1 is commonly found in the colon, vascular smooth muscle cells, and the stomach (Sumimoto et al., 2005). It is constitutively active due to the subunits with which it interacts. It is found with the homologs NOXO1 and NOXA1 rather than p47-phox and p67-phox, respectively, which contribute to its constitutive activity, as unlike p47-phox, NOXO1 does not need phosphorylation to translocate to and activate NOX1 (Takeya et al., 2003; Sumimoto et al., 2005). Aside from phagocytic cells, including microglia, NOX2 is also found in endothelial cells and neurons. NOX3 is mainly in fetal tissues of the kidney and liver. It is also implicated in the development of otoconia in the inner ear of mice and is localized to adult cochlear and vestibular systems of the inner ear (Bánfi et al., 2004). Its presence in the human ear has yet to be established. NOX4 is localized to the kidneys, cardiovascular endothelial cells and many other cells, as it is also constitutively active. It does not need cytosolic subunits for activation, though they are able to modulate NOX4 activity. It also produces hydrogen peroxide rather than superoxide anion as its product. NOX1–4 depend on complexing with the p22-phox subunit and NOX1–3 depend on cytosolic subunits to function. NOX5, found in lymphocytes of the spleen and lymph nodes and in the testes, does not need p22-phox to function and has a calcium-dependent activation mechanism facilitated via additional EF-hand motifs in its structure. DUOX1 and 2, dual oxidases, similarly have EF-hand motifs and subsequently, calcium-dependent activation. However, they also have a peroxidase-like ectodomain and are mainly localized to thyrocytes (Sumimoto et al., 2005). With such variety in structure, regulation, activation and localization, the different NOX complex isoforms have differing roles across disease states. Interestingly, however, the NOX2 isoform has been greatly implicated in most diseases due to its association with inflammatory cells migrating to disease sites.
p47-phox and p22-phox
Of the NOX subunits, p47-phox plays a central role as the organizer and translocator of the subunits for activation. It targets the cytosolic subunits to the membrane via association with p22-phox. In several NOX isoforms, it is integral in facilitating NOX activation and function. While the other components (p67-phox, p40-phox, gp91-phox, and Rac) are also important in NOX function, their role in NOX assembly and activation is limited, and thus, will not be elucidated further in this review. The function of p47-phox is enabled by its structure, with two adjacent src-homology 3 (SH3) domains, a polybasic region c-terminal to them, a proline rich region, and a PX (phox) domain for interactions. The proline rich region binds p67-phox while the SH3 domains of p47-phox interact with proline rich regions of p22-phox to create the complex (DeLeo et al., 1995; Huang and Kleinberg, 1999). However, in the resting state, p47-phox SH3 domains are hidden due to internal interactions with a polybasic region C-terminal to the SH3 domains, stretching from residues 286 to 340 (Leusen et al., 1996; Ago et al., 1999; Huang and Kleinberg, 1999). This internal interaction basally inhibits NOX activation by preventing the translocation of the cytosolic subunits to the flavocytochrome b558 (Ago et al., 1999). The autoinhibited region is unmasked through phosphorylation of several key residues in the C-terminus region from Ser303 to Ser379, inducing a conformational change that disrupts the SH3 domain and C-terminus internal binding and allowing for translocation and binding to the phosphorylated p22-phox (Faust et al., 1995; Leusen et al., 1996; Ago et al., 1999; Huang and Kleinberg, 1999; Sumimoto, 2008). Its structure forms a highly regulated and interactive subunit, reflecting its role as the main organizer of NOX.
The p22-phox subunit, the target subunit of p47-phox, has 195 amino acids with hydrophobic helices in its N-terminus anchoring it to the membrane. It is constitutively associated with gp91-phox or its analog in NOX1 to NOX4 to anchor the membrane subunits. Structurally, it contains a proline rich region in its C-terminal tail stretching from residues 149 to 162 that binds to the SH3 domains on the p47-phox subunit to allow for the necessary interactions for NOX activation (Leusen et al., 1996). The interaction between the two subunits will be further elucidated below.
NOX Activation Through Phosphorylation by Protein Kinase C (PKC)
Phosphorylation is a vital part of NOX activation in all NOX isoforms at various stages of activation, even in constitutively active forms. However, subunit phosphorylation is necessary for activation in NOX1 to NOX3. In those isoforms, phosphorylation of p47-phox is of particular interest in order to uncover its SH3 domains and facilitate translocation of the cytosolic subunits for activation. However, the mechanism of phosphorylation provides potential for further therapeutic targets and elucidates mechanisms through which disease states potentially progress. Phosphorylation of p47-phox may occur through several pathways involving serine kinases, including through PKC isoforms (δ, β, α, ζ), mitogen-activated protein kinases (MAPK), cAMP dependent kinases, p21-activated kinases (PAK), and PKB/AKT (Leusen et al., 1996; Groemping and Rittinger, 2005). However, a primary pathway implicates PKC isoforms in its phosphorylation and subsequent activation, particularly in situations of cellular stress and phagocytic NOX activation (Bey et al., 2004; Jiang et al., 2011). The p47-phox subunit is a particular target of PKC and has been found to be phosphorylated with pure PKC lysate but not in mutated versions from patients with autosomal chronic granulomatous disease (CGD) (Kramer et al., 1988). PMA (phorbol 12-myristate 13-acetate), a potent PKC activator, has been shown to induce a respiratory burst of ROS generation by NOX through the stimulation of PKC (Cox et al., 1985). This behavior is the foundation for subsequent studies of NOX activation as well. Furthermore, inhibiting PKC activation, both broadly with staurosporine in PMA-stimulated cells and specifically with PKC-δ inhibitors, prevented NOX activation and ROS generation (Nauseef et al., 1991; Bey et al., 2004).
As most NOX subunits require phosphorylation to become activated and complex together, the targeting of PKC to the p47-phox subunit has been demonstrated as well. The key serine residues for activation on p47-phox, Ser303, 304 and 328, are residues that are specifically phosphorylated by PKC isoforms. Staurosporine inhibition of PKC resulted in p47-phox and p67-phox subunits localized exclusively in the cytosol and also attenuated p47-phox phosphorylation (Nauseef et al., 1991). PKC-β and PKC-δ isoforms were specifically implicated in p47-phox phosphorylation in neutrophils, as both were co-immunoprecipitated with p47-phox in PMA-treated cells (Reeves et al., 1999). Moreover, while PKC-δ inhibition prevented p47-phox phosphorylation and translocation in monocytes in vitro, PKC-α and PKC-ε inhibition had no effect, indicating PKC specificity with activation (Bey et al., 2004). PKC, particularly the α and δ isoforms, was found to additionally phosphorylate the p22-phox subunit at its Thr147 position to further promote interaction with p47-phox (Lewis et al., 2010). Thus, PKC plays a critical role in activation.
PKC’s role in NOX activation extends to upstream stimuli and pathways that activate PKC itself. Hyperglycemia-stimulated PKC expression affected NOX production of ROS in the blood brain barrier. PKC-α and –β isoforms were elevated and resulted in increased p47-phox phosphorylation and NOX activation under hyperglycemic conditions with brain microvascular endothelial cells, astrocytes, and pericytes simulating the blood brain barrier. Inhibition of the PKC isoforms reduced p47-phox phosphorylation accordingly (Shao and Bayraktutan, 2013). In hyperglycemic stimulation of PKC, particularly PKC-β1, NOX activation was found to potentiate blood brain barrier breakdown and apoptosis. Such states are common in stroke injury (Shao and Bayraktutan, 2013, 2014).
Other upstream pathways activating PKC and subsequently NOX include those of diabetic nephropathy, where advanced glycation end products (AGEs) or advanced oxidation protein products (AOPPs) increased PKC activity, particularly of PKC-α, with resulting increases in NOX (Williams, 2007; Thallas-Bonke et al., 2008; Wei et al., 2009). The PKC-α inhibitor Ro-32-0432 reduced superoxide production and NOX activation, demonstrating PKC’s role in activating NOX (Thallas-Bonke et al., 2008; Wei et al., 2009). Moreover, the angiotensin II (ANGII) axis of ROS production via endothelial NOX similarly implicates PKC. ANGII significantly increased p47-phox phosphorylation, increased p47-phox interaction with p22-phox, and subsequent increased superoxide production (Landmesser et al., 2002; Li and Shah, 2003). In vascular smooth muscle cells, ANGII increased PKC-δ association with p47-phox and p47-phox translocation to the membrane, inducing ROS production via an ANGII/PKC-δ/p47-phox pathway of NOX activation (Lv et al., 2012).
Thus, PKC plays an essential role in activating NOX and PKC itself is activated by several upstream stimuli to facilitate NOX activation. Interestingly, different PKC isoforms act as the main NOX activator in different tissues. PKC-δ is the dominant isoform in leukocytes, associated particularly with NOX2. PKC-β also played a role, but PKC-α did not act in leukocytes (Reeves et al., 1999; Bey et al., 2004). However, PKC-α appears to have a large role in NOX activation in the kidney, where NOX2 and NOX4 dominate. Alternate NOX isoforms may account for the difference in activating PKC isoforms across studies, though definitive preference or association between PKC and NOX isoforms is not yet clear. Moreover, different instigating stimuli, such as hyperglycemia, AGEs, and ANGII, may also affect different PKC isoforms and cause the subsequently observed differences in NOX activation across different PKC isoforms. Elucidating which isoforms are dominant in neurological conditions requires further study, though the inflammatory pathology behind many diseases suggests a larger role of PKC δ and PKC-β.
Lastly, arachidonic acid was found to act synergistically with PKC activation of NOX to increase activation, especially by potentiating the p47-phox binding to p22-phox. It likely functions through disrupting the p47-phox SH3 masking by the polybasic C-terminal region in conjunction with phosphorylation of Ser residues by PKC (Shiose and Sumimoto, 2000). Overall, PKC plays a crucial role in phosphorylation of NOX subunits. This suggests that there is therapeutic potential in manipulating NOX activation through modulating upstream PKC pathways in pathologic states. However, to achieve this therapeutic goal, more specificity regarding the action of specific PKC isoforms and how they affect specific NOX isoforms must be determined.
Interactions of p47-phox and p22-phox
NOX activation is heavily modulated, especially through protein-protein interactions. The cytosolic subunits complex through such interactions and subsequently translocate to the membrane subunits to activate the catalytic core of gp91-phox (Sumimoto, 2008). One key protein-protein interaction is the binding of p47-phox to p22-phox during translocation, to anchor the complex of cytosolic subunits to the membrane and provide the correct enzyme configuration for activation (Sumimoto, 2008). As mentioned, this is facilitated by the interaction between the SH3 domains of p47-phox and the proline rich regions of p22-phox. Unmasking the SH3 domain is necessary to this interaction and is facilitated by phosphorylation, most commonly through the PKC isoforms as previously explained (Groemping and Rittinger, 2005). In the activated NOX complex, the p47-phox subunit is the most phosphorylated subunit. Thus far, 11 phosphorylation sites have been identified: serine residues 303, 304, 310, 315, 320, 328, 345, 348, 359, 370, and 379 (Faust et al., 1995).
Only the most phosphorylated p47-phox subunits translocate to the membrane, but the residues themselves have been found to play different roles in the process. An initial study found that independent phosphorylation of Ser379 is necessary for p47-phox translocation (Faust et al., 1995). It was also found that the residues at Ser359 and Ser370 were necessary in facilitating phosphorylation of subsequent residues, allowing translocation and enabling oxidase activity (Johnson et al., 1998). However, these three residues (Ser379, Ser359 and Ser370) are not sufficient to interact with the p22-phox subunit. These residues are also located outside the region interacting with the SH3 domains, from 286 to 340 (Ago et al., 1999). Thus, the Ser359 and Ser370 residues are likely involved in separate aspects of translocation and NOX activation, likely supporting superoxide production rather than unmasking the SH3 domain (Johnson et al., 1998; Ago et al., 1999). Instead, simultaneous phosphorylation of only Ser303, Ser304, and Ser328 was found to be sufficient in inducing the conformational change needed to reveal the SH3 domains for p22-phox (Ago et al., 1999). As a result, Ser303, Ser304, and Ser328 are the key serines on p47-phox that facilitate targeting of the cytosolic subunits to the membrane at p22-phox for proper NOX assembly.
NOXO1 (NOX organizer 1) further demonstrates the functionality of the p47-phox subunit. It encodes for a homolog of p47-phox and conserves the main interactive domains of p47-phox but lacks the polybasic region that masks the SH3 domains in p47-phox. Thus, it is constitutively active and able to bind p22-phox without phosphorylation. Moreover, superoxide production in cells transfected with NOXO1 was elevated compared to those with p47-phox in the absence of any stimulants, demonstrating that the unmasking of SH3 domains is key in organizing and activating NOX. Moreover, this explains the constitutive activity of NOX1, as it is commonly found with NOXO1 (Takeya et al., 2003).
Once exposed, the SH3 domain binds to the proline rich region of p22-phox, which anchors the cytosolic p47-p67-p40 complex to the membrane complex for activation (Leusen et al., 1996). Moreover, phosphorylation of the p22-phox subunit at the Thr147 position is also integral to the p47-phox/p22-phox interaction. When replaced with alanine, NOX activity was reduced significantly and phosphorylation of p22-phox by the PKC-α and –δ isoforms was prevented altogether (Lewis et al., 2010). Thus, this interaction is key to the recruitment of cytosolic subunits and NOX function.
The importance of the proline rich region in p47-phox and p22-phox interaction has been demonstrated clinically in a patient with CGD who had a single mutation of proline to glutamine at residue 156 of p22-phox. p47-phox translocation and association with the membrane was nearly absent in cells stimulated with PMA. The recruitment of p67-phox was similarly absent in this patient’s cells without p47-phox to organize it. Dysfunction of NOX in the patient resulted from the lack of activation as the catalytic b558 unit functioned normally (Leusen et al., 1994a).
For further membrane stabilization, p47-phox also has a PX (phox) domain which binds phosphoinositides in the membrane. This domain is similarly masked by interactions with the SH3 domains internal to p47-phox and unmasked by phosphorylation, crucial for targeting to the membrane (Ago et al., 2003). p47-phox also has multiple binding sites on the gp91-phox subunit for further NOX complex stabilization, located along the C-terminal tail of gp91-phox as well as on sites more N terminal in the peptide. The region around 500 of gp91-phox is clinically critical. A patient suffering from CGD had an Asp to Gly mutation at residue 500 and subsequent p47-phox and p67-phox recruitment to the membrane fraction was strongly disrupted due to disruption of p47-phox binding at the gp91-phox region (Leusen et al., 1994b). Thus, these binding sites play additional supplementary roles in p47-phox translocation to the flavocytochrome b558.
Once bound, the p47-phox subunit permits for electron transfers to proceed from the FAD to the heme groups in flavocytochrome b558 in order to reach oxygen (DeLeo et al., 1995). The initial transfer from NADPH to FAD only requires the p67-phox isoform, the activator subunit (Leusen et al., 1996).
NOX in Disease
An attractive target for therapeutic inhibition, NOX has been greatly implicated in varied disease processes. Interestingly, it has been upregulated and/or activated in several pathological states such as hyperglycemia, inflammation, cancer, and vascular cases. This review will focus on its role in neurological diseases. A summary of these effects are provided in Table 1.
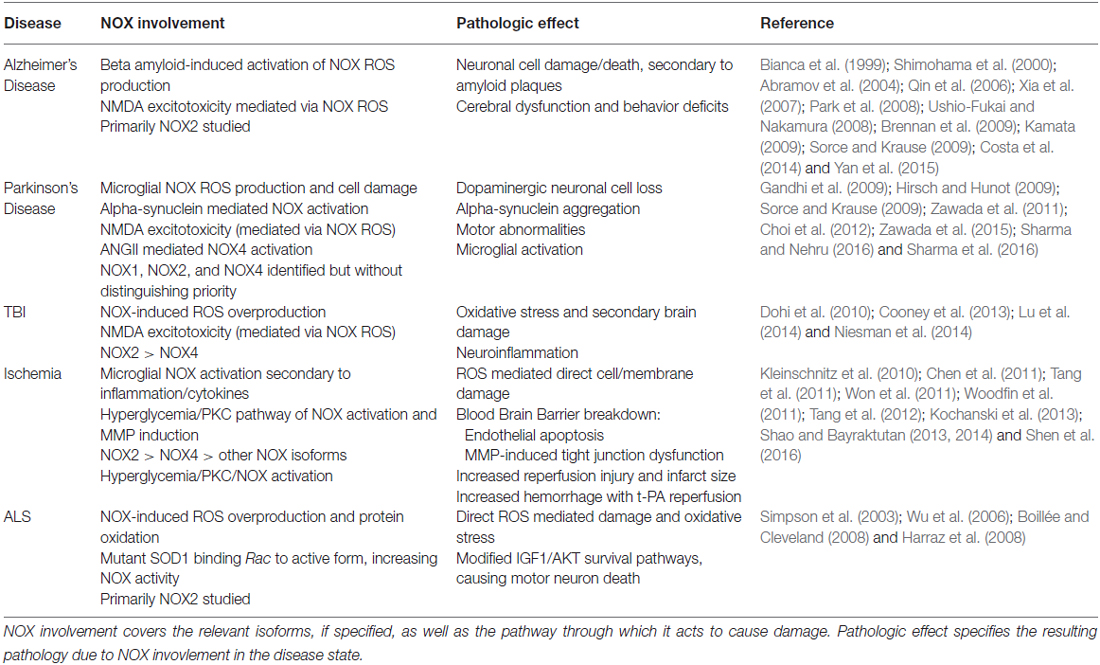
Table 1. This table summarizes the involvement of nicotinamide adenine dinucleotide phosphate (NAPDH) oxidase (NOX) in the various disease states discussed in the article and the pathologic effect mediated by NOX.
Neurodegenerative Diseases
A particular field of interest is targeting NOX in the CNS as it is implicated in numerous diseases of the CNS and acts as its main contributor of ROS. Through its inflammatory actions and oxidative stress, it is also involved in neurodegenerative diseases such as Alzheimer’s disease (AD) and Parkinson’s Disease (PD). Moreover, NMDA receptor activation leads to superoxide production, primarily via NOX. Thus, sustained NMDA activation can lead to NMDA excitotoxicity, mediated by NOX-induced ROS overproduction. As a core mechanism of several neurodegenerative diseases as well as stroke, this aspect adds to the potential protection of NOX inhibition (Brennan et al., 2009).
With differing isoforms present in all neuronal and glial cells, NOX plays a role in exacerbating AD presentation by worsening the effects of neurofibrillary tangles and β-amyloid plaques, though they develop without NOX involvement (Sorce and Krause, 2009). Beta-amyloid plaques have been found to activate NOX in astrocytes and microglia, with the cytosolic p47-phox fraction migrating to the membrane (Bianca et al., 1999; Shimohama et al., 2000; Abramov et al., 2004; Qin et al., 2006). NOX inhibition of a neuron-astrocyte culture and a neuron-microglia culture was found to protect against neuronal cell damage, implicating both cell types in AD pathology (Abramov et al., 2004; Qin et al., 2006). Thus, the plaques themselves lead to the production of toxic ROS and exacerbate both neuronal death and the symptomatic presentation and progression of AD. AD animal models deficient in NOX2 were protected against damage caused by beta-amyloid and did not develop oxidative stress, cerebral dysfunction or behavior deficits of AD (Park et al., 2008). Thus, NOX activation secondary to disease development can exacerbate the disease, providing a potential pathway to mitigate AD progression through NOX inhibition.
Though the etiology of PD is still relatively unclear, oxidative stress and neuroinflammation have been strongly implicated in PD’s loss of dopaminergic cells. This, in part, has been attributed to microglial NOX ROS production and cell damage (Hirsch and Hunot, 2009). Several in vitro and in vivo models have demonstrated the role of NOX in PD development. In genetic PD in vitro and in vivo models with knockdown/knockout PINK1, increases in ROS from both mitochondrial and NOX sources were observed, with both the nonspecific NOX inhibitor DPI and NOX-siRNA reducing cytosolic ROS (Gandhi et al., 2009). In in vitro rat nigral dopaminergic cells with PD induced by MPTP toxin injections, elevated levels of NOX2 and ROS were observed, both mitigated by NOX inhibition or NOX2 knockdown (Zawada et al., 2011). Similarly NOX1 and ROS were elevated in in vitro and in vivo PD models induced by 6-OHDA injections, and NOX inhibition reduced cell death (Choi et al., 2012). In PD models in rats, apocynin, a nonspecific NOX inhibitor, was capable of preventing alpha-synuclein aggregation as well as alpha-synuclein mediated NOX activation and ROS production (Sharma et al., 2016). Moreover, NOX inhibition was protective and reduced dopaminergic neuronal death, motor abnormalities, microglial activation and subsequent neuroinflammation after PD was induced via LPS injection into the rat substantia nigra (Sharma and Nehru, 2016; Sharma et al., 2016). Interestingly, a postmortem examination of a small sample of human prePD and PD substantia nigra brain sections suggest that there may be an ANGII/AT1 (ANGII Type 1 receptor)/NOX4 mediated axis of dopaminergic nigral dysfunction. In the brain samples, nuclear AT1 was elevated, though the total AT1 decreased, with increasing disease severity. This accompanied an elevation in NOX4 and caspase-3, suggesting ANGII-activated NOX4 as a possible pathway of dopaminergic cell death in clinical PD (Zawada et al., 2015). Thus, NOX appears to play a key role in both PD development and progression and is a promising therapeutic target, both independently and via upstream signaling pathways such as ANGII.
Traumatic Brain Injury (TBI)
In TBI, multiple mechanisms cause the final injury, of which oxidative stress and ROS play a pivotal role. NOX acts as a key producer of ROS in TBI, mediating further injury. Though NOX1 to NOX4 are found in the brain, NOX2 increased most after TBI, though an increase in NOX4 was also observed within microglial cells. Moreover, NOX inhibition in TBI models was found to reduce inflammatory injury as well (Cooney et al., 2013). Other models have also demonstrated increases in NOX activity post injury (Lu et al., 2014; Niesman et al., 2014). In TBI models, increased gp91-phox (NOX2) was also observed and mice deficient for NOX2 genes were found to have less ROS and oxidative damage, inflammation and secondary damage at the site of injury (Dohi et al., 2010). Moreover, post injury, the brain exhibited NMDA excitotoxicity, which may cause further NOX activation and damage (Niesman et al., 2014). As TBI derives from direct injury and the resulting neuroinflammation responses that occur, the oxidative damage done is likely due to direct ROS damage from NOX rather than through signaling pathways. NOX has emerged as one of the main contributors of ROS in TBI, making it a potent neuroprotective target.
Stroke
Of CNS disease states, ischemic stroke is one of the most studied conditions for NOX activity. Stroke-induced increases in NOX expression, activity, and ROS-mediated damage have been widely observed post ischemia (Tang et al., 2012; Kochanski et al., 2013; Shen et al., 2016). As part of stroke pathology, the lack of blood flow causes injury to mitochondria to the affected regions of the brain. Once reperfusion occurs, the influx of oxygen cannot be detoxified by the mitochondria. As shown in Figure 1, the influx of excess glucose with reperfusion causes increased production of NADPH via the hexose monophosphate shunt, using the glycolytic intermediate glucose 6-phosphate and producing downstream glycolytic intermediates such as fructose 6-phosphate that go on to enter the tricarboxylic acid (TCA) cycle and produce adenosine triphosphate (ATP) in mitochondria. Thus, with plentiful substrates and a hyperglycemic state, NOX is activated and generates an increased amount of ROS, resulting in subsequent injury. The ROS directly damage cells by compromising membrane integrity via lipid peroxidation and organelle damage. However, they also cause injury by upregulating several inflammatory factors and cytokines, including IL-1β, TNF-α, iNOS, and bradykinin (Chen et al., 2011; Tang et al., 2012). Moreover, bradykinin and IL-1β activate NOX in order to cause blood brain barrier breakdown (Woodfin et al., 2011). NOX2 knockout mice demonstrated reduced inflammation with a reduced elevation of these inflammatory factors, reduced microglial activation and overall reduced infarct size, greatly implicating NOX in both inflammation and infarct progression (Chen et al., 2011).
A hyperglycemic state has been associated with acute stroke and hyperglycemia has independently been found to induce NOX activation as well (Wang et al., 2013; Shao and Bayraktutan, 2014) Hyperglycemic states have been found to potentiate blood-brain barrier breakdown through endothelial apoptosis facilitated by PKC-induced NOX activation of apoptotic pathways and through tight junction dysfunction due to NOX-induced MMP induction (Shao and Bayraktutan, 2013, 2014). Rat models have also shown increased blood-brain barrier permeability and increased infarct and hemorrhage with t-PA reperfusion in hyperglycemic rat models that was reversed with NOX inhibition with apocynin (Won et al., 2011). Thus, hyperglycemia may exacerbate stroke and reperfusion injury.
Moreover, stroke pathogenesis is attributed more to the NOX (NOX2) of circulating immune cells compared to the endogenous microglial and neuronal cells, though both contribute to the stroke pathology (Tang et al., 2011; Kahles and Brandes, 2013). However, a growing body of evidence shows that targeting NOX4 is effective as well, with deletion of NOX4 or targeted inhibition with VAS2870, a NOX4 inhibitor, resulting in reduced oxidative stress, blood brain barrier disruption, neuronal death, and mortality (Kleinschnitz et al., 2010). The prominence of NOX in ischemia/reperfusion pathology is critical to its understanding and to potential treatment options of the disease.
Cerebrovascular conditions potentiate the risk of stroke as well, and these conditions are further mediated by NOX. ROS increases hypertension and atherosclerosis by inducing endothelial dysfunction, vascular remodeling and increased stiffness as well as altering the availability of vasoactive compounds such as nitric oxide to reduce vasodilation capabilities (Williams, 2007; Kinkade et al., 2013). Moreover, NOX is inextricably linked to the inflammatory response, which exacerbates atherosclerosis further (Arenillas, 2015). The isoforms located within blood vessels vary widely, with a widespread presence of NOX4 as well as NOX1, NOX2 and NOX5 in different vessel layers (Takac et al., 2012). However NOX 1 and 2 are the main mediators of vascular damage (Chrissobolis et al., 2012; Takac et al., 2012; Gray and Jandeleit-Dahm, 2015). With such increases in vascular damage, increases in thrombus formation and thromboembolic stroke can increase. Moreover, atherosclerosis and hypertension are associated risk factors and potential causative agents for the disease (Ay et al., 2005; Goldstein et al., 2011). Thus, NOX acts on both the initial cause of stroke and mediates its injurious effects afterwards.
Amyotrophic Lateral Sclerosis (ALS)
Neurodegeneration seen in ALS has also been attributed largely to oxidative stress. In ALS, neurodegeneration of motor neurons occurs, resulting in muscle weakness, paralysis and death. Oxidative stress markers, including lipid peroxidation products malondialdehyde and 4-hydroxynonenal, were elevated in both patient CSF and serum as well as in animal models of ALS (Simpson et al., 2003). Moreover, in both sporadic ALS patients and animal models, NOX2 was elevated, particularly in microglia in the spinal cord. It was found to mediate protein oxidation, and particularly, modify IGF1/AKT survival pathways in motor neurons to facilitate damage (Wu et al., 2006). In familial ALS, a toxic gain of function mutation in superoxide dismutase 1 (SOD1) is the main cause in 25% of cases. SOD1 regulates NOX by binding Rac and inhibiting its GTPase activity and maintaining its active GTP bound form. Normally, it uncouples from Rac in the presence of H2O2. With the mutation, the redox uncoupling is defective and leads to increases in Rac/NOX activity and activation as Rac remains in its active form (Simpson et al., 2003; Harraz et al., 2008). Moreover, NOX deletion or treatment with apocynin to inhibit NOX prolonged survival and reduced neurodegeneration in mice models and glial cells (Wu et al., 2006; Boillée and Cleveland, 2008; Harraz et al., 2008). Overall, the pathology of ALS implicates NOX-mediated ROS production and damage and provides a novel therapeutic target for the condition.
NOX-Induced ROS Mechanisms in Cellular Damage
In these disease states, despite normal NOX function acting through protective or biologically necessary mechanisms, the pathological and unbalanced ROS production can damage tissue though multiple pathways. Primarily, it contributes to direct damage. This damage is through direct ROS damage to lipid membranes, causing lipid peroxidation breakdown and potential necrotic death. This can also lead to apoptotic pathway initiation with the release of cytochrome C from the mitochondrial membrane, through mitochondrial damage or upregulation of pro-apoptotic BAX and cause both endothelial and neuronal cell death (Hampton and Orrenius, 1997; Brieger et al., 2012). Necrotic death can also ensue, as seen in ischemia/reperfusion injury. Further damage can be caused through direct damage to DNA itself, further inducing either apoptosis or carcinogenesis if the cell cannot repair itself (Brieger et al., 2012). Moreover, ROS can induce autophagy and at excessive levels cause autophagic cell death, as seen in nerve growth factor deprived sympathetic neurons (Scherz-Shouval and Elazar, 2007).
However, damage is also done through auxiliary signaling pathways. In neuronal damage, cell death results from both direct damage, with ROS and mitochondrial dysfunction, and through the induced inflammation (Lambeth, 2007). ROS can upregulate the production of toxic factors, such as IL-1, TNF-α and more superoxide causing neurodegeneration (Block and Hong, 2005). Several other pathways are involved in a more disease specific manner that allow for disease progression outside of simple cell death, as mentioned in the previous disease states. However, NOX overproduction of ROS seen in pathology can be very cytotoxic through numerous mechanisms.
Potential Treatments on NOX Activation
NOX inhibition through therapy and targeted drugs has great potential for mitigating disease progression, especially given the widespread involvement of NOX in various disease states. However, targeting NOX specifically is a goal in therapy, given the different roles, particularly in immunity, that NOX has physiologically and the different roles each isoform has in disease. NOX inhibition itself can function through several pathways: (1) To inhibit upstream activators of NOX; (2) To interrupt subunit interactions for activation, preventing assembly; (3) To block the flavocytochrome itself, particularly targeting gp91-phox (NOX2) or its catalytic homolog; and (4) To interfere with NOX activity through indirect pathways. However, in many cases, the mechanism of inhibition is unclear or acts on a transcriptional/translational level prior to activation. A general summary of the inhibitors’ location of action is shown in Figure 1.
A General Inhibition of NOX Action
Several therapeutic interventions are available, include normobaric oxygen (NBO), hyperbaric oxygen (HBO), and hibernation therapy, which inhibit NOX via unspecified mechanisms (Figure 1①). In the cases of NBO and HBO, there is the contradicting thought that increased oxygen would exacerbate NOX-mediated ROS production. However, both NBO and HBO have been found to be paradoxically protective in cases of stroke without contributing to further oxidative stress (Singhal et al., 2002; Schäbitz et al., 2004). In one study, NBO reduced blood brain barrier disruption through reduced levels of NOX2 and NOX-induced MMP-9 while preventing occludin degradation in cerebral ischemia (Liu et al., 2011). HBO also downregulated NOX2 expression and NOX activity and improved functional outcome and neuronal survival due to the decrease in oxidative stress in a subarachnoid hemorrhage model (Ostrowski et al., 2006). Hibernation therapies, such as hypothermia or drug-induced hibernation such as ethanol, are also protective in stroke (Kim and Yenari, 2015; Forreider et al., 2016). Of these, ethanol, by itself and in conjunction with NBO, has been shown to decrease NOX levels and activity in a model of ischemic stroke. It likely acts through an upstream mechanism involving PKC and AKT. It also exhibited reductions in infarct volume, neurological deficit, and ROS levels in a stroke model (Kochanski et al., 2013; Cai et al., 2016). These broad therapies however lack specificity in NOX action and act on multiple levels.
VAS2870 (Vasopharm), 3-benzyl-7-(2-benzoxazolyl)thio-1, 2,3-triazolo[4, 5-d]pyrimidine, is another inhibitor and has been found to reduce ROS increases in oxidized LDL stimulated endothelial cells (Stielow et al., 2006). Though its mechanism of action is not certain, recent studies have implicated NOX2 and NOX4. One study demonstrated a reduction in ROS and NOX4 expression in skeletal muscle with VAS2870 and suggested a thiol alkylation and modification mechanism of NOX4 inhibition (Sun et al., 2012). A more recent study with a stroke model suggests it acts by upregulation of microRNA that target NOX2 and NOX4 genes and suppress them in stroke. VAS2870 was also found to reduce infarct volume in stroke as well, providing potential for future use (Liu et al., 2016). While promising, off target effects via thiol alkylation on the ryanodine receptor-Ca2+ channel were found, eliminating its activation and potentially causing muscle dysfunction (Sun et al., 2012).
Inhibition of Upstream Activators of NOX
The first type of inhibitors utilizes upstream targeting of PKC and PKC activation pathways for NOX inhibition as a potential therapy (Figure 1②). PKC inhibitors such as Calphostin C, chelerythrine and ruboxistaurin mesylate (RBX) have all been shown to reduce hypertension. RBX is currently in clinical trials for use in diabetic complications, but it has additional function in NOX inhibition (Williams, 2007). In several studies, high concentrations of PKC inhibitors have reduced ischemic damage in vivo or cell death in vitro, though more work needs to be done to assess its use for ischemia or neurodegenerative diseases therapeutically (Bright and Mochly-Rosen, 2005). However, PKC inhibitors lack specificity to NOX activation, and PKC is implicated in numerous cellular functions and signaling, creating a potential for varied and severe side effects. Other upstream targets also have shown potential. As previously described, ethanol potentially acts through a PKC/AKT pathway of NOX inhibition (Cai et al., 2016). Total salivanolic acid injection (TSI), an injection derived from Salvia miltiorrhiza, is a therapy targeting a PKC activation pathway. It is currently approved in China for treatment of ischemic stroke, particularly with thrombolytic therapy. Through its use, AMPK expression is enhanced, resulting in the subsequent observed reduction of AKT activation, PKC translocation to the membrane, and overall NOX activation with the failure of migration of the cytosolic subunits to the membrane. It acts via the AMPK/AKT/PKC signaling pathway (Tang et al., 2014). By defining the pathological upstream activators of NOX in specific disease states, NOX inhibition can be facilitated by targeting these upstream players, such as PKC and ANGII.
NOX Subunit Interactions and their Therapeutic Perspectives
The second type of NOX inhibitors acts via inhibition of NOX assembly (Figure 1③). Understanding NOX activation via p47-phox phosphorylation by PKC isoforms and subsequent interaction with the p22-phox subunit has opened up new areas of therapeutic investigation. While many of the inhibitors or therapies mentioned previously function by inhibiting upstream NOX activators or through unspecified overall inhibition, greater specificity can be achieved through targeting therapies at subunit interactions.
One of the most promising NOX inhibitors is apocynin, or 4-hydrozy-3-methoxy acetophenone, which blocks migration of p47-phox to the membrane and prevents NOX assembly and activation. In ischemic injury, it protected against early lipid peroxidation, neuronal degeneration and death, and glial cell activation as a pretreatment and, at low doses, was found to be effective in improving neurological outcomes, reducing infarct volume and hemorrhage (Wang et al., 2006; Tang et al., 2008). It was also found to reduce glial death and promote survival in ALS mice models (Boillée and Cleveland, 2008; Harraz et al., 2008). However, apocynin acts on NOX1 and 2 more than NOX4, the constitutively active NOX isoform. It also requires myeloperoxidase (MPO) for function as it must dimerize to act on NOX. When acting on NOX4 and in the absence of MPO, apocynin was found to act as an antioxidant scavenger (Heumüller et al., 2008). Thus, its scope of use must be more definitively established.
Another promising drug is gp91-ds-tat, a NOX inhibitor specific to NOX2, and potentially NOX1 (Williams, 2007). It functions through inhibition of p47-phox binding to gp91-phox through emulating the binding region of gp91-phox. As a peptide, it internalizes via a sequence corresponding to the HIV viral coat. In mice with AD, it was shown to reduce ROS as well as cerebrovascular dysfunction that contributes to AD pathology (Park et al., 2008). It potentially has other cerebrovascular benefits, as it has been shown to reduce hypertension in ANGII stimulated models as well (Rey et al., 2001). However, it requires intravenous injection due to limited oral bioavailability as a peptide, restricting its clinical potential (Williams, 2007).
While potential pharmaceuticals and their mechanisms are still being investigated, the uniquely modulated activation of NOX allows for increased specificity to NOX isoforms controlled by cytosolic subunits, primarily NOX1–3. Moreover, understanding the biochemical interactions between subunits opens the field to finding inhibitors that target sequences specific to NOX subunits, lowering the widespread side effects of affecting similar groups on other molecules or upstream activators that are not specific to NOX.
NOX Flavocytochrome Inhibitors
Certain inhibitors, the third type, target the flavocytochrome itself to inhibit NOX (Figure 1④). Of these, diphenyleneiodonium (DPI) is the most commonly used. It forms an irreversible redox adduct with reduced FAD of the NOX catalytic core. Thus, it halts superoxide production altogether upon NOX activation through uncompetitive inhibition (O’Donnell et al., 1993). In conjunction with dimethylsulfoxide, a free radical scavenger, it was found to reduce infarct size, blood brain barrier disruption, and neurological damage in a stroke model (Nagel et al., 2007). However, its safety is questionable as other flavoenzymes necessary to metabolism may also be inhibited (O’Donnell et al., 1993; Tang et al., 2012). More recent inhibitors are focusing on generating inhibitors specific to NOX isoforms when targeting the catalytic core. GSK2795039, a small molecular inhibitor, has been recently developed and was found to be protective in paw inflammation and acute pancreatitis mouse models. Unlike DPI, it is specific to NOX2, demonstrating reduction in NOX2 activity via competitive inhibition with little to no inhibition of other NOX isoforms and other flavoenzymes, such as xanthine oxidase or nitric oxide synthase (Hirano et al., 2015). Given the increasing evidence for NOX2’s role in such conditions as stroke and the moderate bioavailability of the drug in the brain, the inhibitor has potential in treating ischemia and neurodegenerative disorders as well (Kahles and Brandes, 2013; Hirano et al., 2015). GKT137831 is another such inhibitor but it is specific to NOX 1/4, allowing for varying specificity and targeting. It has shown potential in cases of fibrotic and inflammatory diseases, including liver fibrosis, pulmonary fibrosis, inflammation, and ischemic retinopathy (Aoyama et al., 2012; Deliyanti and Wilkinson-Berka, 2015). Given its role in inflammation and ischemic retinopathy, it may show potential in cerebrovascular conditions like stroke and in neurodegenerative diseases where neuroinflammation plays a core role in the pathology, but further studies need to be done to investigate its use in neurological diseases. Unlike with NOX2 inhibition, little immunosuppression is observed and no spontaneous pathologies are found upon NOX4 inhibition or deletion (Aoyama et al., 2012). Moreover, it has progressed to clinical trials, showing great promise (Teixeira et al., 2016).
Indirect NOX Inhibitors
Finally, the fourth type of inhibitors use indirect pathways of action and include angiotensin converting enzyme (ACE) inhibitors and angiotensin receptor blockers, which would function by reducing NOX activation induced by the ANGII/PKC pathway. Statins also inhibit NOX by preventing geranylgeranylation, necessary for Rac translocation to NOX (Figure 1⑤ Wassmann et al., 2002; Williams, 2007). They have been shown to reduce ROS production, atherosclerotic lesion size, and even endothelial dysfunction in mild essential hypertension in a small human study (Williams, 2007). Though they have not been studied with regard to neurological diseases, they play an important role through reducing cerebrovascular complications that may ensue from such conditions, including stroke.
Conclusion and Prospective Studies
NOX plays a detrimental role in a widespread range of diseases through its overproduction of ROS, which then damage tissue directly and initiate deleterious signaling cascades within the body. Thus, NOX inhibition has great potential as a therapeutic treatment that can act across several disease states. The potential for further refinement of NOX inhibition is in understanding the key role of p47-phox and its translocation to the membrane subunit p22-phox via phosphorylation by PKC. The p47-phox subunit SH3 domains are masked at rest, and through serine phosphorylation by PKC isoforms at multiple sites, the polybasic masking domain changes conformation to allow for the SH3 domains of p47-phox to interact with the proline rich region of p22-phox. It translocates to the membrane to anchor the other cytosolic subunits to the membrane catalytic core and facilitate NOX activation. Through understanding this process, the potential of NOX inhibitors of NOX assembly, function, and upstream activation, as a clinical therapy for neurological, vascular, and metabolic conditions has been unveiled. Particularly, NOX activation can be prevented through the inhibition of subunit assembly by, surprisingly, blocking the interaction between two key subunits, p47-phox and p22-phox. Further research into finding new potential inhibitors and enhancement of the current ones is still necessary, but investigation into inhibitors of NOX assembly allows for a unique avenue of exploration.
Author Contributions
YD and RR conceived the study, RR drafted the manuscript, FL made the figure, and all authors, YD, XG, FL and RR critically revised and approved the final manuscript. YD and XG provided research funding.
Funding
This work was partially supported by American Heart Association Grant-in-Aid (14GRNT20460246; YD), Merit Review Award (I01RX-001964-01) from the US Department of Veterans Affairs Rehabilitation R&D Service (YD), National Natural Science Foundation of China (81501141; XG), and Beijing NOVA program (xx2016061; XG).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abramov, A. Y., Canevari, L., and Duchen, M. R. (2004). β-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J. Neurosci. 24, 565–575. doi: 10.1523/jneurosci.4042-03.2004
Ago, T., Kuribayashi, F., Hiroaki, H., Takeya, R., Ito, T., Kohda, D., et al. (2003). Phosphorylation of p47phox directs phox homology domain from SH3 domain toward phosphoinositides, leading to phagocyte NADPH oxidase activation. Proc. Natl. Acad. Sci. U S A 100, 4474–4479. doi: 10.1073/pnas.0735712100
Ago, T., Nunoi, H., Ito, T., and Sumimoto, H. (1999). Mechanism for phosphorylation-induced activation of the phagocyte NADPH oxidase protein p47phox. Triple replacement of serines 303, 304, and 328 with aspartates disrupts the SH3 domain-mediated intramolecular interaction in p47phox, thereby activating the oxidase. J. Biol. Chem. 274, 33644–33653. doi: 10.1074/jbc.274.47.33644
Aoyama, T., Paik, Y.-H., Watanabe, S., Laleu, B., Gaggini, F., Fioraso-Cartier, L., et al. (2012). Nicotinamide adenine dinucleotide phosphate oxidase (NOX) in experimental liver fibrosis: GKT137831 as a novel potential therapeutic agent. Hepatology 56, 2316–2327. doi: 10.1002/hep.25938
Arenillas, J. F. (2015). Intracranial atherosclerosis and inflammation: lessons from the East and the West. Brain Circ. 1, 47–52. doi: 10.4103/2394-8108.162531
Ay, H., Furie, K. L., Singhal, A., Smith, W. S., Sorensen, A. G., and Koroshetz, W. S. J. (2005). An evidence-based causative classification system for acute ischemic stroke. Ann. Neurol. 58, 688–697. doi: 10.1002/ana.20617
Bánfi, B., Malgrange, B., Knisz, J., Steger, K., Dubois-Dauphin, M., and Krause, K. H. (2004). NOX3, a superoxide-generating NADPH oxidase of the inner ear. J. Biol. Chem. 279, 46065–46072. doi: 10.1074/jbc.m403046200
Bey, E. A., Xu, B., Bhattacharjee, A., Oldfield, C. M., Zhao, X., Li, Q., et al. (2004). Protein kinase Cδ is required for p47phox phosphorylation and translocation in activated human monocytes. J. Immunol. 173, 5730–5738. doi: 10.4049/jimmunol.173.9.5730
Bianca, V. D., Dusi, S., Bianchini, E., Dal Prà, I., and Rossi, F. (1999). β-amyloid activates the O-2 forming NADPH oxidase in microglia, monocytes and neutrophils. A possible inflammatory mechanism of neuronal damage in Alzheimer’s disease. J. Biol. Chem. 274, 15493–15499. doi: 10.1074/jbc.274.22.15493
Block, M. L., and Hong, J.-S. (2005). Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog. Neurobiol. 76, 77–98. doi: 10.1016/j.pneurobio.2005.06.004
Boillée, S., and Cleveland, D. W. (2008). Revisiting oxidative damage in ALS: microglia, Nox and mutant SOD1. J. Clin. Invest. 118, 474–478. doi: 10.1172/JCI34613
Brennan, A. M., Suh, S. W., Won, S. J., Narasimhan, P., Kauppinen, T. M., Lee, H., et al. (2009). NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat. Neurosci. 12, 857–863. doi: 10.1038/nn.2334
Brieger, K., Schiavone, S., Miller, F. J. Jr., and Krause, K. H. (2012). Reactive oxygen species: from health to disease. Swiss Med. Wkly. 142:w13659. doi: 10.4414/smw.2012.13659
Bright, R., and Mochly-Rosen, D. (2005). The role of protein kinase C in cerebral ischemic and reperfusion injury. Stroke 36, 2781–2790. doi: 10.1161/01.STR.0000189996.71237.f7
Cai, L., Stevenson, J., Geng, X., Peng, C., Ji, X., Xin, R., et al. (2016). Combining normobaric oxygen with ethanol or hypothermia prevents brain damage from thromboembolic stroke via PKC-Akt-NOX modulation. Mol. Neurobiol. doi: 10.1007/s12035-016-9695-7 [Epub ahead of print].
Chen, H., Kim, G. S., Okami, N., Narasimhan, P., and Chan, P. H. (2011). NADPH oxidase is involved in post-ischemic brain inflammation. Neurobiol. Dis. 42, 341–348. doi: 10.1016/j.nbd.2011.01.027
Choi, D. H., Cristóvão, A. C., Guhathakurta, S., Lee, J., Joh, T. H., Beal, M. F., et al. (2012). NADPH oxidase 1-mediated oxidative stress leads to dopamine neuron death in Parkinson’s disease. Antioxid. Redox Signal. 16, 1033–1045. doi: 10.1089/ars.2011.3960
Chrissobolis, S., Banfi, B., Sobey, C. G., and Faraci, F. M. (2012). Role of Nox isoforms in angiotensin II-induced oxidative stress and endothelial dysfunction in brain. J. Appl. Physiol. (1985) 113, 184–191. doi: 10.1152/japplphysiol.00455.2012
Cooney, S. J., Bermudez-Sabogal, S. L., and Byrnes, K. R. (2013). Cellular and temporal expression of NADPH oxidase (NOX) isotypes after brain injury. J. Neuroinflammation 10:155. doi: 10.1186/1742-2094-10-155
Costa, A., Scholer-Dahirel, A., and Mechta-Grigoriou, F. (2014). The role of reactive oxygen species and metabolism on cancer cells and their microenvironment. Semin. Cancer Biol. 25, 23–32. doi: 10.1016/j.semcancer.2013.12.007
Cox, J. A., Jeng, A. Y., Sharkey, N. A., Blumberg, P. M., and Tauber, A. I. (1985). Activation of the human neutrophil nicotinamide adenine dinucleotide phosphate (NADPH)-oxidase by protein kinase C. J. Clin. Invest. 76, 1932–1938. doi: 10.1172/jci112190
D’Autréaux, B., and Toledano, M. B. (2007). ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 8, 813–824. doi: 10.1038/nrm2256
DeLeo, F. R., Yu, L., Burritt, J. B., Loetterle, L. R., Bond, C. W., Jesaitis, A. J., et al. (1995). Mapping sites of interaction of p47-phox and flavocytochrome b with random-sequence peptide phage display libraries. Proc. Natl. Acad. Sci. U S A 92, 7110–7114. doi: 10.1073/pnas.92.15.7110
Deliyanti, D., and Wilkinson-Berka, J. L. (2015). Inhibition of NOX1/4 with GKT137831: a potential novel treatment to attenuate neuroglial cell inflammation in the retina. J. Neuroinflammation 12:136. doi: 10.1186/s12974-015-0363-z
Dohi, K., Ohtaki, H., Nakamachi, T., Yofu, S., Satoh, K., Miyamoto, K., et al. (2010). Gp91phox (NOX2) in classically activated microglia exacerbates traumatic brain injury. J. Neuroinflammation 7:41. doi: 10.1186/1742-2094-7-41
Faust, L. R., el Benna, J., Babior, B. M., and Chanock, S. J. (1995). The phosphorylation targets of p47phox, a subunit of the respiratory burst oxidase. Functions of the individual target serines as evaluated by site-directed mutagenesis. J. Clin. Invest. 96, 1499–1505. doi: 10.1172/jci118187
Forreider, B., Pozivilko, D., Kawaji, Q., Geng, X., and Ding, Y. (2016). Hibernation-like neuroprotection in stroke by attenuating brain metabolic dysfunction. Prog. Neurobiol. doi: 10.1016/j.pneurobio.2016.03.002 [Epub ahead of print].
Gandhi, S., Wood-Kaczmar, A., Yao, Z., Plun-Favreau, H., Deas, E., Klupsch, K., et al. (2009). PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Mol. Cell 33, 627–638. doi: 10.1016/j.molcel.2009.02.013
Goldstein, L. B., Bushnell, C. D., Adams, R. J., Appel, L. J., Braun, L. T., Chaturvedi, S., et al. (2011). Guidelines for the primary prevention of stroke: a guideline for healthcare professionals from the American heart Association/American stroke association. Stroke 42, 517–584. doi: 10.1161/STR.0b013e3181fcb238
Gray, S. P., and Jandeleit-Dahm, K. A. (2015). The role of NADPH oxidase in vascular disease—hypertension, atherosclerosis and stroke. Curr. Pharm. Des. 21, 5933–5944. doi: 10.2174/1381612821666151029112302
Groemping, Y., and Rittinger, K. (2005). Activation and assembly of the NADPH oxidase: a structural perspective. Biochem. J. 386, 401–416. doi: 10.1042/bj20041835
Hampton, M. B., and Orrenius, S. (1997). Dual regulation of caspase activity by hydrogen peroxide: implications for apoptosis. FEBS Lett. 414, 552–556. doi: 10.1016/s0014-5793(97)01068-5
Harraz, M. M., Marden, J. J., Zhou, W., Zhang, Y., Williams, A., Sharov, V. S., et al. (2008). SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model. J. Clin. Invest. 118, 659–670. doi: 10.1172/JCI34060
Heumüller, S., Wind, S., Barbosa-Sicard, E., Schmidt, H. H., Busse, R., Schroder, K., et al. (2008). Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension 51, 211–217. doi: 10.1161/HYPERTENSIONAHA.107.100214
Hirano, K., Chen, W. S., Chueng, A. L., Dunne, A. A., Seredenina, T., Filippova, A., et al. (2015). Discovery of GSK2795039, a novel small molecule NADPH oxidase 2 inhibitor. Antioxid. Redox Signal. 23, 358–374. doi: 10.1089/ars.2014.6202
Hirsch, E. C., and Hunot, S. (2009). Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol. 8, 382–397. doi: 10.1016/S1474-4422(09)70062-6
Huang, J., and Kleinberg, M. E. (1999). Activation of the phagocyte NADPH oxidase protein p47phox. Phosphorylation controls SH3 domain-dependent binding to p22phox. J. Biol. Chem. 274, 19731–19737. doi: 10.1074/jbc.274.28.19731
Jiang, F., Zhang, Y., and Dusting, G. J. (2011). NADPH oxidase-mediated redox signaling: roles in cellular stress response, stress tolerance and tissue repair. Pharmacol. Rev. 63, 218–242. doi: 10.1124/pr.110.002980
Johnson, J. L., Park, J.-W., Benna, J. E., Faust, L. P., Inanami, O., and Babior, B. M. (1998). Activation of p47phox, a cytosolic subunit of the leukocyte NADPH oxidase. Phosphorylation of ser-359 or ser-370 precedes phosphorylation at other sites and is required for activity. J. Biol. Chem. 273, 35147–35152. doi: 10.1074/jbc.273.52.35147
Kahles, T., and Brandes, R. P. (2013). Which NADPH oxidase isoform is relevant for ischemic stroke? The case for NOX 2. Antioxid. Redox Signal. 18, 1400–1417. doi: 10.1089/ars.2012.4721
Kamata, T. (2009). Roles of Nox1 and other Nox isoforms in cancer development. Cancer Sci. 100, 1382–1388. doi: 10.1111/j.1349-7006.2009.01207.x
Kim, J., and Yenari, M. (2015). Hypothermia for treatment of stroke. Brain Circ. 1, 14–25. doi: 10.4103/2394-8108.164997
Kinkade, K., Streeter, J., and Miller, F. J. (2013). Inhibition of NADPH oxidase by apocynin attenuates progression of atherosclerosis. Int. J. Mol. Sci. 14, 17017–17028. doi: 10.3390/ijms140817017
Kleinschnitz, C., Grund, H., Wingler, K., Armitage, M. E., Jones, E., Mittal, M., et al. (2010). Post-stroke inhibition of induced NADPH oxidase type 4 prevents oxidative stress and neurodegeneration. PLoS Biol. 8:e1000479. doi: 10.1371/journal.pbio.1000479
Kochanski, R., Peng, C., Higashida, T., Geng, X., Hüttemann, M., Guthikonda, M., et al. (2013). Neuroprotection conferred by post-ischemia ethanol therapy in experimental stroke: an inhibitory effect on hyperglycolysis and NADPH oxidase activation. J. Neurochem. 126, 113–121. doi: 10.1111/jnc.12169
Kramer, I. M., Verhoeven, A. J., van der Bend, R. L., Weening, R. S., and Roos, D. (1988). Purified protein kinase C phosphorylates a 47-kDa protein in control neutrophil cytoplasts but not in neutrophil cytoplasts from patients with the autosomal form of chronic granulomatous disease. J. Biol. Chem. 263, 2352–2357.
Lambeth, J. D. (2007). Nox enzymes, ROS and chronic disease: an example of antagonistic pleiotropy. Free Radic. Biol. Med. 43, 332–347. doi: 10.1016/j.freeradbiomed.2007.03.027
Landmesser, U., Cai, H., Dikalov, S., McCann, L., Hwang, J., Jo, H., et al. (2002). Role of p47phox in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension 40, 511–515. doi: 10.1161/01.hyp.0000032100.23772.98
Lapouge, K., Smith, S. J., Groemping, Y., and Rittinger, K. (2002). Architecture of the p40–p47-p67phox complex in the resting state of the NADPH oxidase. A central role for p67phox. J. Biol. Chem. 277, 10121–10128. doi: 10.1074/jbc.M112065200
Leusen, J. H., Bolscher, B. G., Hilarius, P. M., Weening, R. S., Kaulfersch, W., Seger, R. A., et al. (1994a). 156Pro–>Gln substitution in the light chain of cytochrome b558 of the human NADPH oxidase (p22-phox) leads to defective translocation of the cytosolic proteins p47-phox and p67-phox. J. Exp. Med. 180, 2329–2334. doi: 10.1084/jem.180.6.2329
Leusen, J. H., de Boer, M., Bolscher, B. G., Hilarius, P. M., Weening, R. S., Ochs, H. D., et al. (1994b). A point mutation in gp91-phox of cytochrome b558 of the human NADPH oxidase leading to defective translocation of the cytosolic proteins p47-phox and p67-phox. J. Clin. Invest. 93, 2120–2126. doi: 10.1172/jci117207
Leusen, J. H., Verhoeven, A. J., and Roos, D. (1996). Interactions between the components of the human NADPH oxidase: intrigues in the phox family. J. Lab. Clin. Med. 128, 461–476. doi: 10.2741/a117
Lewis, E. M., Sergeant, S., Ledford, B., Stull, N., Dinauer, M. C., and McPhail, L. C. (2010). Phosphorylation of p22phox on threonine 147 enhances NADPH oxidase activity by promoting p47phox binding. J. Biol. Chem. 285, 2959–2967. doi: 10.1074/jbc.M109.030643
Li, J. M., and Shah, A. M. (2003). Mechanism of endothelial cell NADPH oxidase activation by angiotensin II. Role of the p47phox subunit. J. Biol. Chem. 278, 12094–12100. doi: 10.1074/jbc.M209793200
Liu, W., Chen, Q., Liu, J., and Liu, K. J. (2011). Normobaric hyperoxia protects the blood brain barrier through inhibiting Nox2 containing NADPH oxidase in ischemic stroke. Med. Gas Res. 1:22. doi: 10.1186/2045-9912-1-22
Liu, Z., Tuo, Y. H., Chen, J. W., Wang, Q. Y., Li, S., Li, M. C., et al. (2016). NADPH oxidase inhibitor regulates microRNAs with improved outcome after mechanical reperfusion. J. Neurointerv. Surg. doi: 10.1136/neurintsurg-2016-012463 [Epub ahead of print].
Lu, X. Y., Wang, H. D., Xu, J. G., Ding, K., and Li, T. (2014). NADPH oxidase inhibition improves neurological outcome in experimental traumatic brain injury. Neurochem. Int. 69, 14–19. doi: 10.1016/j.neuint.2014.02.006
Lv, P., Miao, S. B., Shu, Y. N., Dong, L. H., Liu, G., Xie, X. L., et al. (2012). Phosphorylation of smooth muscle 22α facilitates angiotensin II-induced ROS production via activation of the PKCδ-P47phox axis through release of PKCδ and actin dynamics and is associated with hypertrophy and hyperplasia of vascular smooth muscle cells in vitro and in vivo. Circ. Res. 111, 697–707. doi: 10.1161/CIRCRESAHA.112.272013
Nagel, S., Genius, J., Heiland, S., Horstmann, S., Gardner, H., and Wagner, S. (2007). Diphenyleneiodonium and dimethylsulfoxide for treatment of reperfusion injury in cerebral ischemia of the rat. Brain Res. 1132, 210–217. doi: 10.1016/j.brainres.2006.11.023
Nauseef, W. M., Volpp, B. D., McCormick, S., Leidal, K. G., and Clark, R. A. (1991). Assembly of the neutrophil respiratory burst oxidase. Protein kinase C promotes cytoskeletal and membrane association of cytosolic oxidase components. J. Biol. Chem. 266, 5911–5917.
Niesman, I. R., Schilling, J. M., Shapiro, L. A., Kellerhals, S. E., Bonds, J. A., Kleschevnikov, A. M., et al. (2014). Traumatic brain injury enhances neuroinflammation and lesion volume in caveolin deficient mice. J. Neuroinflammation 11:39. doi: 10.1186/1742-2094-11-39
O’Donnell, B. V., Tew, D. G., Jones, O. T., and England, P. J. (1993). Studies on the inhibitory mechanism of iodonium compounds with special reference to neutrophil NADPH oxidase. Biochem. J. 290, 41–49. doi: 10.1042/bj2900041
Ostrowski, R. P., Tang, J., and Zhang, J. H. (2006). Hyperbaric oxygen suppresses NADPH oxidase in a rat subarachnoid hemorrhage model. Stroke 37, 1314–1318. doi: 10.1161/01.STR.0000217310.88450.c3
Park, L., Zhou, P., Pitstick, R., Capone, C., Anrather, J., Norris, E. H., et al. (2008). Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc. Natl. Acad. Sci. U S A 105, 1347–1352. doi: 10.1073/pnas.0711568105
Qin, B., Cartier, L., Dubois-Dauphin, M., Li, B., Serrander, L., and Krause, K.-H. (2006). A key role for the microglial NADPH oxidase in APP-dependent killing of neurons. Neurobiol. Aging 27, 1577–1587. doi: 10.1016/j.neurobiolaging.2005.09.036
Reeves, E. P., Dekker, L. V., Forbes, L. V., Wientjes, F. B., Grogan, A., Pappin, D. J., et al. (1999). Direct interaction between p47phox and protein kinase C: evidence for targeting of protein kinase C by p47phox in neutrophils. Biochem. J. 344, 859–866. doi: 10.1042/0264-6021:3440859
Rey, F. E., Cifuentes, M. E., Kiarash, A., Quinn, M. T., and Pagano, P. J. (2001). Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O2- and systolic blood pressure in mice. Circ. Res. 89, 408–414. doi: 10.1161/hh1701.096037
Schäbitz, W. R., Schade, H., Heiland, S., Kollmar, R., Bardutzky, J., Henninger, N., et al. (2004). Neuroprotection by hyperbaric oxygenation after experimental focal cerebral ischemia monitored by MRI. Stroke 35, 1175–1179. doi: 10.1161/01.STR.0000125868.86298.8e
Scherz-Shouval, R., and Elazar, Z. (2007). ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. 17, 422–427. doi: 10.1016/j.tcb.2007.07.009
Shao, B., and Bayraktutan, U. (2013). Hyperglycaemia promotes cerebral barrier dysfunction through activation of protein kinase C-β. Diabetes Obes. Metab. 15, 993–999. doi: 10.1111/dom.12120
Shao, B., and Bayraktutan, U. (2014). Hyperglycaemia promotes human brain microvascular endothelial cell apoptosis via induction of protein kinase C-βI and prooxidant enzyme NADPH oxidase. Redox Biol. 2, 694–701. doi: 10.1016/j.redox.2014.05.005
Sharma, N., Kapoor, M., and Nehru, B. (2016). Apocyanin, NADPH oxidase inhibitor prevents lipopolysaccharide induced α-synuclein aggregation and ameliorates motor function deficits in rats: possible role of biochemical and inflammatory alterations. Behav. Brain Res. 296, 177–190. doi: 10.1016/j.bbr.2015.09.012
Sharma, N., and Nehru, B. (2016). Apocyanin, a microglial NADPH oxidase inhibitor prevents dopaminergic neuronal degeneration in lipopolysaccharide-induced Parkinson’s disease model. Mol. Neurobiol. 53, 3326–3337. doi: 10.1007/s12035-015-9267-2
Shen, J., Huber, M., Zhao, E. Y., Peng, C., Li, F., Li, X., et al. (2016). Early rehabilitation aggravates brain damage after stroke via enhanced activation of nicotinamide adenine dinucleotide phosphate oxidase (NOX). Brain Res. 1648, 266–276. doi: 10.1016/j.brainres.2016.08.001
Shimohama, S., Tanino, H., Kawakami, N., Okamura, N., Kodama, H., Yamaguchi, T., et al. (2000). Activation of NADPH oxidase in Alzheimer’s disease brains. Biochem. Biophys. Res. Commun. 273, 5–9. doi: 10.1006/bbrc.2000.2897
Shiose, A., and Sumimoto, H. (2000). Arachidonic acid and phosphorylation synergistically induce a conformational change of p47phox to activate the phagocyte NADPH oxidase. J. Biol. Chem. 275, 13793–13801. doi: 10.1074/jbc.275.18.13793
Simpson, E. P., Yen, A. A., and Appel, S. H. (2003). Oxidative Stress: a common denominator in the pathogenesis of amyotrophic lateral sclerosis. Curr. Opin. Rheumatol. 15, 730–736. doi: 10.1097/00002281-200311000-00008
Singhal, A. B., Wang, X., Sumii, T., Mori, T., and Lo, E. H. (2002). Effects of normobaric hyperoxia in a rat model of focal cerebral ischemia-reperfusion. J. Cereb. Blood Flow Metab. 22, 861–868. doi: 10.1097/00004647-200207000-00011
Sorce, S., and Krause, K. H. (2009). NOX enzymes in the central nervous system: from signaling to disease. Antioxid. Redox Signal. 11, 2481–2504. doi: 10.1089/ARS.2009.2578
Stielow, C., Catar, R. A., Muller, G., Wingler, K., Scheurer, P., Schmidt, H. H. H. W., et al. (2006). Novel Nox inhibitor of oxLDL-induced reactive oxygen species formation in human endothelial cells. Biochem. Biophys. Res. Commun. 344, 200–205. doi: 10.1016/j.bbrc.2006.03.114
Sumimoto, H. (2008). Structure, regulation and evolution of Nox-family NADPH oxidases that produce reactive oxygen species. FEBS J. 275, 3249–3277. doi: 10.1111/j.1742-4658.2008.06488.x
Sumimoto, H., Miyano, K., and Takeya, R. (2005). Molecular composition and regulation of the Nox family NAD(P)H oxidases. Biochem. Biophys. Res. Commun. 338, 677–686. doi: 10.1016/j.bbrc.2005.08.210
Sun, Q.-A., Hess, D. T., Wang, B., Miyagi, M., and Stamler, J. S. (2012). Off-target thiol alkylation by the NADPH oxidase inhibitor 3-benzyl-7- (2-benzoxazolyl)thio-1,2,3-triazolo[4,5-d]pyrimidine (VAS2870). Free Radic. Biol. Med. 52, 1897–1902. doi: 10.1016/j.freeradbiomed.2012.02.046
Takac, I., Schröder, K., and Brandes, R. P. (2012). The nox family of NADPH oxidases: friend or foe of the vascular system? Curr. Hypertens. Rep. 14, 70–78. doi: 10.1007/s11906-011-0238-3
Takeya, R., Ueno, N., Kami, K., Taura, M., Kohjima, M., Izaki, T., et al. (2003). Novel human homologues of p47phox and p67phox participate in activation of superoxide-producing NADPH oxidases. J. Biol. Chem. 278, 25234–25246. doi: 10.1074/jbc.M212856200
Tang, X. N., Cairns, B., Cairns, N., and Yenari, M. A. (2008). Apocynin improves outcome in experimental stroke with a narrow dose range. Neuroscience 154, 556–562. doi: 10.1016/j.neuroscience.2008.03.090
Tang, X. N., Cairns, B., Kim, J. Y., and Yenari, M. A. (2012). NADPH oxidase in stroke and cerebrovascular disease. Neurol. Res. 34, 338–345. doi: 10.1179/1743132812Y.0000000021
Tang, H., Pan, C. S., Mao, X. W., Liu, Y. Y., Yan, L., Zhou, C. M., et al. (2014). Role of NADPH oxidase in total salvianolic acid injection attenuating ischemia-reperfusion impaired cerebral microcirculation and neurons: implication of AMPK/Akt/PKC. Microcirculation 21, 615–627. doi: 10.1111/micc.12140
Tang, X. N., Zheng, Z., Giffard, R. G., and Yenari, M. A. (2011). Significance of marrow-derived nicotinamide adenine dinucleotide phosphate oxidase in experimental ischemic stroke. Ann. Neurol. 70, 606–615. doi: 10.1002/ana.22476
Teixeira, G., Szyndralewiez, C., Molango, S., Carnesecchi, S., Heitz, F., Wiesel, P., et al. (2016). Therapeutic potential of NOX1/4 inhibitors. Br. J. Pharmacol. doi: 10.1111/bph.13532 [Epub ahead of print].
Thallas-Bonke, V., Thorpe, S. R., Coughlan, M. T., Fukami, K., Yap, F. Y., Sourris, K. C., et al. (2008). Inhibition of NADPH oxidase prevents advanced glycation end product-mediated damage in diabetic nephropathy through a protein kinase C-α-dependent pathway. Diabetes 57, 460–469. doi: 10.2337/db08-0406
Tsunawaki, S., and Yoshikawa, K. (2000). Relationships of p40phox with p67phox in the activation and expression of the human respiratory burst NADPH oxidase. J. Biochem. 128, 777–783. doi: 10.1093/oxfordjournals.jbchem.a022815
Ushio-Fukai, M., and Nakamura, Y. (2008). Reactive oxygen species and angiogenesis: NADPH oxidase as target for cancer therapy. Cancer Lett. 266, 37–52. doi: 10.1016/j.canlet.2008.02.044
Wang, Y.-Y., Chen, C.-J., Lin, S.-Y., Chuang, Y.-H., Sheu, W. H.-H., and Tung, K.-C. (2013). Hyperglycemia is associated with enhanced gluconeogenesis in a rat model of permanent cerebral ischemia. Mol. Cell. Endocrinol. 367, 50–56. doi: 10.1016/j.mce.2012.12.016
Wang, Q., Tompkins, K. D., Simonyi, A., Korthuis, R. J., Sun, A. Y., and Sun, G. Y. (2006). Apocynin protects against global cerebral ischemia-reperfusion-induced oxidative stress and injury in the gerbil hippocampus. Brain Res. 1090, 182–189. doi: 10.1016/j.brainres.2006.03.060
Wassmann, S., Laufs, U., Muller, K., Konkol, C., Ahlbory, K., Baumer, A. T., et al. (2002). Cellular antioxidant effects of atorvastatin in vitro and in vivo. Arterioscler. Thromb. Vasc. Biol. 22, 300–305. doi: 10.1161/hq0202.104081
Wei, X. F., Zhou, Q. G., Hou, F. F., Liu, B. Y., and Liang, M. (2009). Advanced oxidation protein products induce mesangial cell perturbation through PKC-dependent activation of NADPH oxidase. Am. J. Physiol. Renal. Physiol. 296, F427–F437. doi: 10.1152/ajprenal.90536.2008
Williams, H. C. (2007). NADPH oxidase inhibitors: new antihypertensive agents? J. Cardiovasc. Pharmacol. 50, 9–16. doi: 10.1097/fjc.0b013e318063e820
Won, S. J., Tang, X. N., Suh, S. W., Yenari, M. A., and Swanson, R. A. (2011). Hyperglycemia promotes tissue plasminogen activator-induced hemorrhage by Increasing superoxide production. Ann. Neurol. 70, 583–590. doi: 10.1002/ana.22538
Woodfin, A., Hu, D. E., Sarker, M., Kurokawa, T., and Fraser, P. (2011). Acute NADPH oxidase activation potentiates cerebrovascular permeability response to bradykinin in ischemia-reperfusion. Free Radic. Biol. Med. 50, 518–524. doi: 10.1016/j.freeradbiomed.2010.12.010
Wu, D.-C., Ré, D. B., Nagai, M., Ischiropoulos, H., and Przedborski, S. (2006). The inflammatory NADPH oxidase enzyme modulates motor neuron degeneration in amyotrophic lateral sclerosis mice. Proc. Natl. Acad. Sci. U S A 103, 12132–12137. doi: 10.1073/pnas.0603670103
Xia, C., Meng, Q., Liu, L. Z., Rojanasakul, Y., Wang, X. R., and Jiang, B. H. (2007). Reactive oxygen species regulate angiogenesis and tumor growth through vascular endothelial growth factor. Cancer Res. 67, 10823–10830. doi: 10.1158/0008-5472.can-07-0783
Yan, S., Liu, G., Pei, C., Chen, W., Li, P., Wang, Q., et al. (2015). Inhibition of NADPH oxidase protects against metastasis of human lung cancer by decreasing microRNA-21. Anticancer Drugs 26, 388–398. doi: 10.1097/CAD.0000000000000198
Yu, L., Quinn, M. T., Cross, A. R., and Dinauer, M. C. (1998). Gp91phox is the heme binding subunit of the superoxide-generating NADPH oxidase. Proc. Natl. Acad. Sci. U S A 95, 7993–7998. doi: 10.1073/pnas.95.14.7993
Zawada, W. M., Banninger, G. P., Thornton, J., Marriott, B., Cantu, D., Rachubinski, A. L., et al. (2011). Generation of reactive oxygen species in 1-methyl-4-phenylpyridinium (MPP+) treated dopaminergic neurons occurs as an NADPH oxidase-dependent two-wave cascade. J. Neuroinflammation 8:129. doi: 10.1186/1742-2094-8-129
Zawada, W. M., Mrak, R. E., Biedermann, J., Palmer, Q. D., Gentleman, S. M., Aboud, O., et al. (2015). Loss of angiotensin II receptor expression in dopamine neurons in Parkinson’s disease correlates with pathological progression and is accompanied by increases in Nox4- and 8-OH guanosine-related nucleic acid oxidation and caspase-3 activation. Acta Neuropathol. Commun. 3:9. doi: 10.1186/s40478-015-0189-z
Keywords: NAPDH oxidase, NOX inhibitors, reactive oxygen species, stroke, ischemia/reperfusion, neurodegenerative disease, TBI, PKC
Citation: Rastogi R, Geng X, Li F and Ding Y (2017) NOX Activation by Subunit Interaction and Underlying Mechanisms in Disease. Front. Cell. Neurosci. 10:301. doi: 10.3389/fncel.2016.00301
Received: 20 August 2016; Accepted: 20 December 2016;
Published: 10 January 2017.
Edited by:
Hansen Wang, University of Toronto, CanadaReviewed by:
Carla Cannizzaro, University of Palermo, ItalyElsa Fabbretti, University of Nova Gorica, Slovenia
Sharon DeMorrow, Texas A&M Health Science Center, USA
Copyright © 2017 Rastogi, Geng, Li and Ding. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaokun Geng, eGdlbmdAY2NtdS5lZHUuY24=
Yuchuan Ding, eWRpbmdAbWVkLndheW5lLmVkdQ==