 Rana Yadak
Rana Yadak Peter Sillevis Smitt
Peter Sillevis Smitt Marike W. van Gisbergen
Marike W. van Gisbergen Niek P. van Til
Niek P. van Til Irenaeus F. M. de Coo
Irenaeus F. M. de Coo- 1Department of Neurology, Erasmus University Medical Center, Rotterdam, Netherlands
- 2Department of Radiation Oncology (MaastRO-Lab), GROW – School for Oncology and Developmental Biology, Maastricht University Medical Centre, Maastricht, Netherlands
- 3Laboratory of Translational Immunology, University Medical Center Utrecht, Utrecht, Netherlands
Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) is a progressive metabolic disorder caused by thymidine phosphorylase (TP) enzyme deficiency. The lack of TP results in systemic accumulation of deoxyribonucleosides thymidine (dThd) and deoxyuridine (dUrd). In these patients, clinical features include mental regression, ophthalmoplegia, and fatal gastrointestinal complications. The accumulation of nucleosides also causes imbalances in mitochondrial DNA (mtDNA) deoxyribonucleoside triphosphates (dNTPs), which may play a direct or indirect role in the mtDNA depletion/deletion abnormalities, although the exact underlying mechanism remains unknown. The available therapeutic approaches include dialysis and enzyme replacement therapy, both can only transiently reverse the biochemical imbalance. Allogeneic hematopoietic stem cell transplantation is shown to be able to restore normal enzyme activity and improve clinical manifestations in MNGIE patients. However, transplant related complications and disease progression result in a high mortality rate. New therapeutic approaches, such as adeno-associated viral vector and hematopoietic stem cell gene therapy have been tested in Tymp-/-Upp1-/- mice, a murine model for MNGIE. This review provides background information on disease manifestations of MNGIE with a focus on current management and treatment options. It also outlines the pre-clinical approaches toward future treatment of the disease.
Introduction
Mitochondrial diseases represent a genetically and clinically heterogeneous group of disorders caused by mutations in mitochondrial DNA (mtDNA), that affect synthesis and function of mitochondrial proteins, such as tRNA (in MELAS disease) and ND1, 4, 6 (responsible for the majority of cases in LHON disease) (DiMauro, 2004). Another group is caused by mutations in nuclear DNA (nDNA) that lead to defects in nuclear encoded mitochondrial proteins. Part of these proteins exert their effect on mtDNA maintenance, thus known as nuclear-mitochondrial communication disorders. A subtype of the latter is MDS; a group of mainly autosomal recessive disorders caused by defects in nuclear genes involved in mtDNA replication (e.g., POLG and PEO1 causing hepatocerebal MDS), or genes crucial for maintenance of mtDNA including TK2 (responsible for myopathic MDS), RRM2B (encephalomypathic MDS) and thymidine phosphorylase (TYMP) gene mutations associated with MNGIE (El-Hattab and Scaglia, 2013). MNGIE, initially described in Okamura et al. (1976) is a fatal rare inherited metabolic disorder without genetic or ethnic predisposition (Gamez et al., 2005). The estimated rate of occurrence is 1–9:1,000,0001, and as of 2011 fewer than 200 cases have been described in the medical literature (Halter et al., 2011). Due to its variable clinical presentations, MNGIE can be easily overlooked or misdiagnosed as Crohn’s disease, psychiatric disorder, anorexia nervosa, or myasthenia gravis (Rickards et al., 1994; Teitelbaum et al., 2002; Marti et al., 2004).
Genetic Defects, Clinical Manifestations, and Diagnosis
Genetic Defects
Mitochondrial neurogastrointestinal encephalomyopathy is an autosomal recessive inherited disease that is caused by mutations in the nuclear gene TYMP (previously known as ECGF1). TYMP codes for the TP enzyme (EC 2.4.2.4.) and is located on chromosome 22q13.33 (Stenman et al., 1992). TP is a cytoplasmic enzyme expressed in most human tissues, including gastrointestinal tract, central and peripheral nervous system, spleen, liver, bladder, leukocytes and in platelets which account for most of the TP activity in human blood (Fukami and Salganicoff, 1973; Shaw et al., 1988). In contrast, TP is present at low levels in muscles and is lacking in kidney, aorta and fat tissues (Fox et al., 1995; Valentino et al., 2007). MNGIE is caused by a variety of pathogenic homozygous or compound heterozygous mutations in the exons or flanking regions of the TYMP gene. Various mutations are reported to date (Stenson et al., 2014) including deletions, single nucleotide insertions (Nishino et al., 1999), splice site (Kocaefe et al., 2003; Szigeti et al., 2004b) and frameshift mutations (Blazquez et al., 2005) and a homozygous duplication mutation in exon 8 of the TYMP gene (Gamez et al., 2005). The majority of these mutations are loss of function mutations. Heterozygous mutation carriers are asymptomatic with approximately 35% residual TP activity, although the plasma nucleoside levels are similar to healthy controls (Marti et al., 2004).
In addition, non-pathogenic polymorphisms have been described in the TYMP gene. The A465T polymorphism (c.1393G>A) was reported both in subjects with MNGIE like features and control subjects (Vissing et al., 2002; Martin et al., 2004). In some MNGIE cases there is no or mild clinical involvement of gastrointestinal tract or skeletal muscle, despite the presence of mutations in the TYMP gene leading to marked reduction in TP activity, probably indicating that environmental factors contribute to the severity of the clinical symptoms (Martin et al., 2004; Szigeti et al., 2004b). Apart from late-onset forms of the disease (Marti et al., 2005; Massa et al., 2009; Etienne et al., 2012), most patients display typical MNGIE features before the age of 20 years (Nishino et al., 2000; Teitelbaum et al., 2002).
Clinical Manifestations
Gastrointestinal and ocular involvements are usually the first complications in this disease, although neuropathy and hearing loss have been reported as primary symptoms in some cases (Garone et al., 2011). Clinical symptoms are summarized in Table 1.
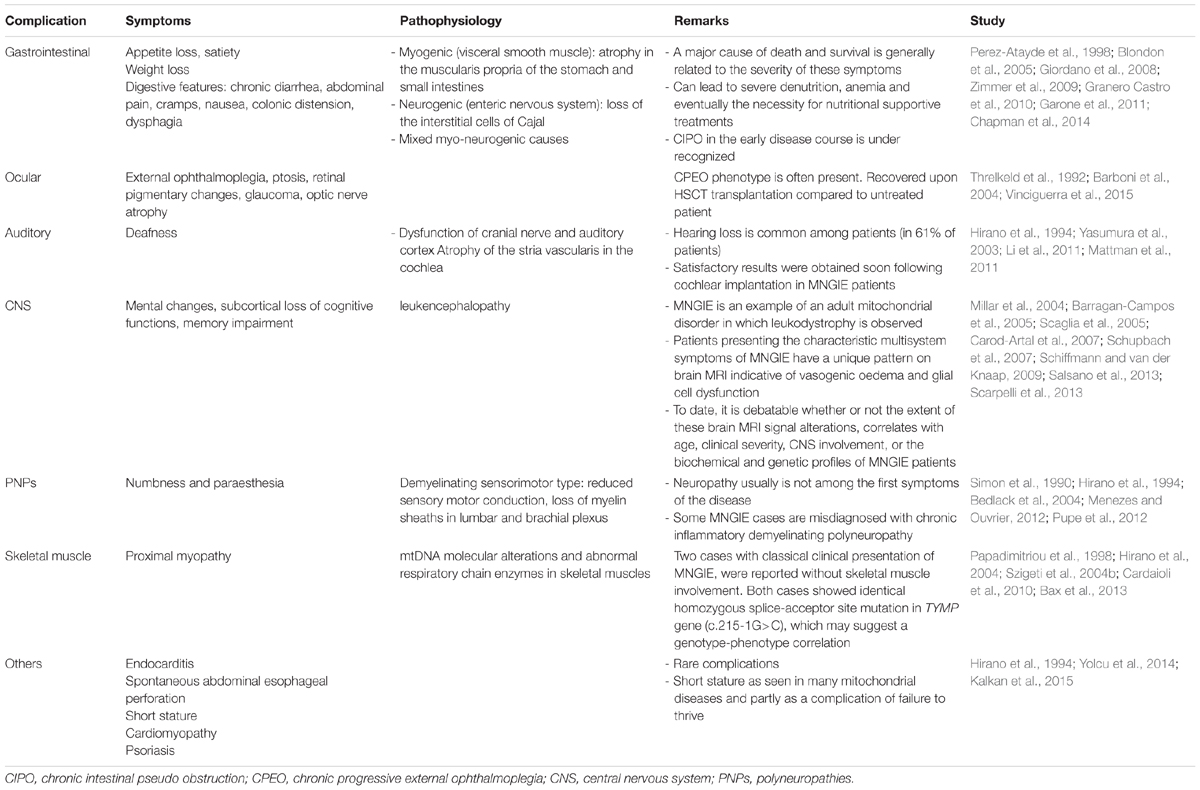
TABLE 1. Common and rare clinical symptoms in MNGIE patients.
Diagnosis
Detailed patient history, thorough clinical examination, particular findings on magnetic resonance imaging (MRI) of the brain (Figure 1), genomic DNA screening for mutations in TYMP gene and biochemical analysis all contribute to the diagnosis of MNGIE. Biochemical diagnosis of MNGIE includes at least one of the following parameters (Marti et al., 2004): (1) Increased blood plasma levels of dThd and dUrd (>3 and >5 μmol/L, respectively). (2) Severely reduced TP enzyme activity in buffy coat leukocytes (<8% of healthy controls; healthy control mean TP activity equivalent to 634 nmol thymine formed/hr/mg protein). Biochemical analysis reduces the risk of missing the diagnosis in case of non-identified mutation sites (Nishino et al., 2000) or in case of unclassified variants (UV). Additionally, biochemical diagnosis contributes to the confirmation or exclusion of the role of a UV as a cause for MNGIE. Similarly, biochemical assessment is preferred over clinical diagnosis since some of the classical symptoms of MNGIE can be absent. Other frequently observed findings in MNGIE patients include metabolic abnormalities such as lactic acidosis, deficiency of mitochondrial respiratory chain enzymes, mainly complex I and IV (Hirano et al., 1994; Debouverie et al., 1997), urinary Thd and dUrd accumulation (Fairbanks et al., 2002; Spinazzola et al., 2002; la Marca et al., 2006) and elevated protein levels in CSF (Bedlack et al., 2004). Infrequently, skeletal muscle biopsies may reveal ragged red fibers, and mtDNA analysis may reveal acquired deletions, depletions or point mutations (Teitelbaum et al., 2002; Nishigaki et al., 2003).
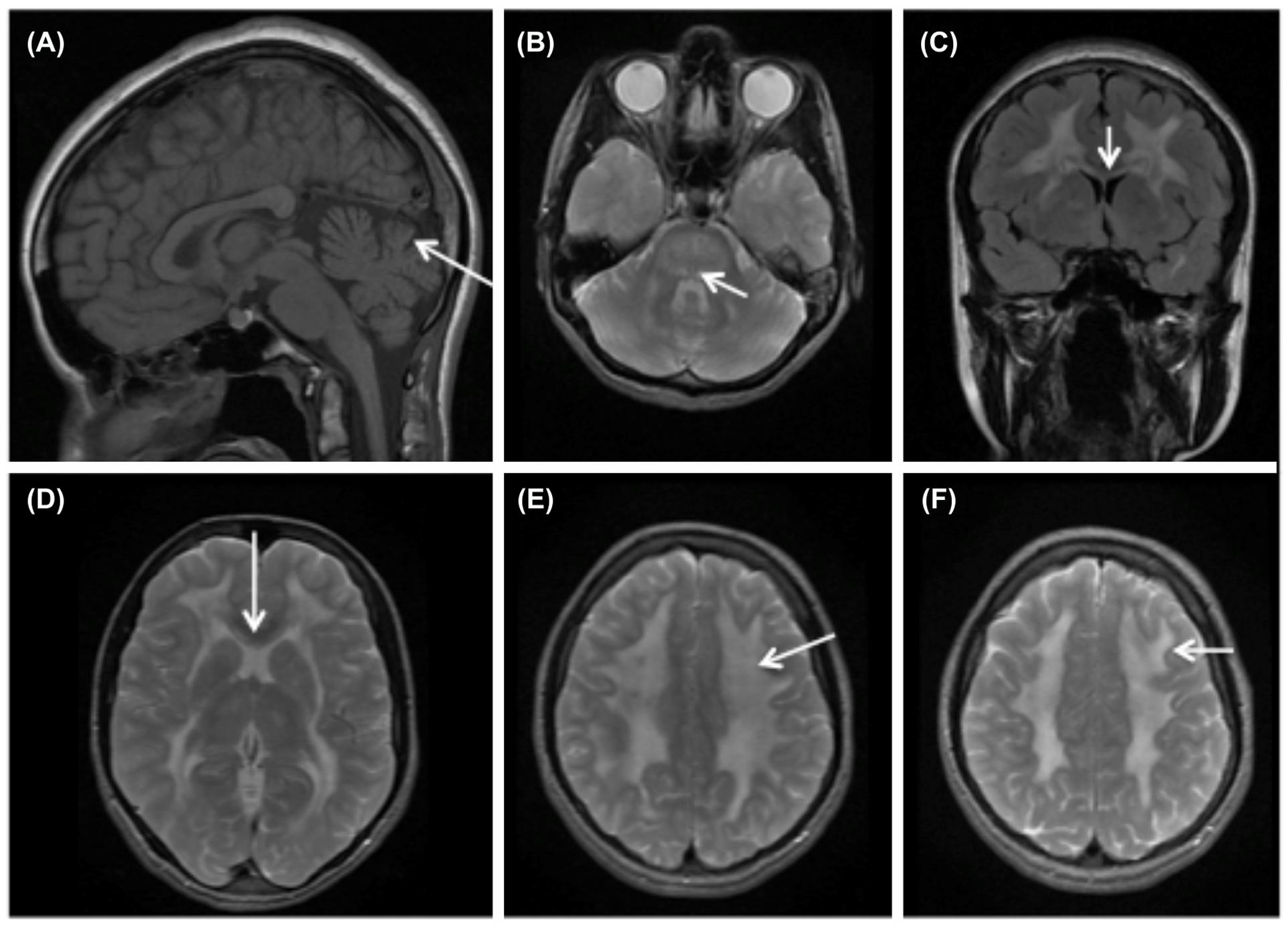
FIGURE 1. Brain MRI findings in MNGIE. MRI of MNGIE patient at age 16 with “typical” MNGIE phenotype. (A) T1 weighted sagital image shows cerebellar vermis atrophy (arrow) and normal gyral pattern. (B) Axial T2 with hyperintensities in the dorsal pons and mesencephalon (arrow). (C coronal flair image, D axial T2) Show extensive signal abnormalities in the cerebral white matter. The external capsule is involved as is the inner blade of the corpus callosum (arrow C,D). (E,F) Extensive white matter involvement with sparing of the U-fibers (arrow).
Pathogenesis
The TP enzyme converts mitochondrial dThd and dUrd to the nucleotide bases thymine and uridine respectively and 2-deoxy ribose 1-phosphate (Friedkin and Roberts, 1954). This occurs in de novo synthesis or via the salvage pathway. dThd and dUrd are homogeneously present in cellular and plasma compartments and they translocate between compartments through NTs. In humans two unrelated protein families have been described (Young et al., 2013), CNTs, an active transport system, and ENTs responsible for passive facilitated diffusion.
The bidirectional ENTs, mainly ENT1, are ubiquitously present on almost all cell types and mediate the uptake and efflux of nucleosides (Figure 2B). Therefore, they are important for cells that rely on the salvage pathway for supply of nucleosides, including bone marrow cells, erythrocytes and leukocytes, brain and muscles (Young et al., 2008). Although TP is not expressed in all tissues, the TP expressed in circulating platelets and leukocytes and some other tissues is essential to degrade the excess amounts of dThd and dUrd nucleosides which are secreted into the blood (Lara et al., 2007).
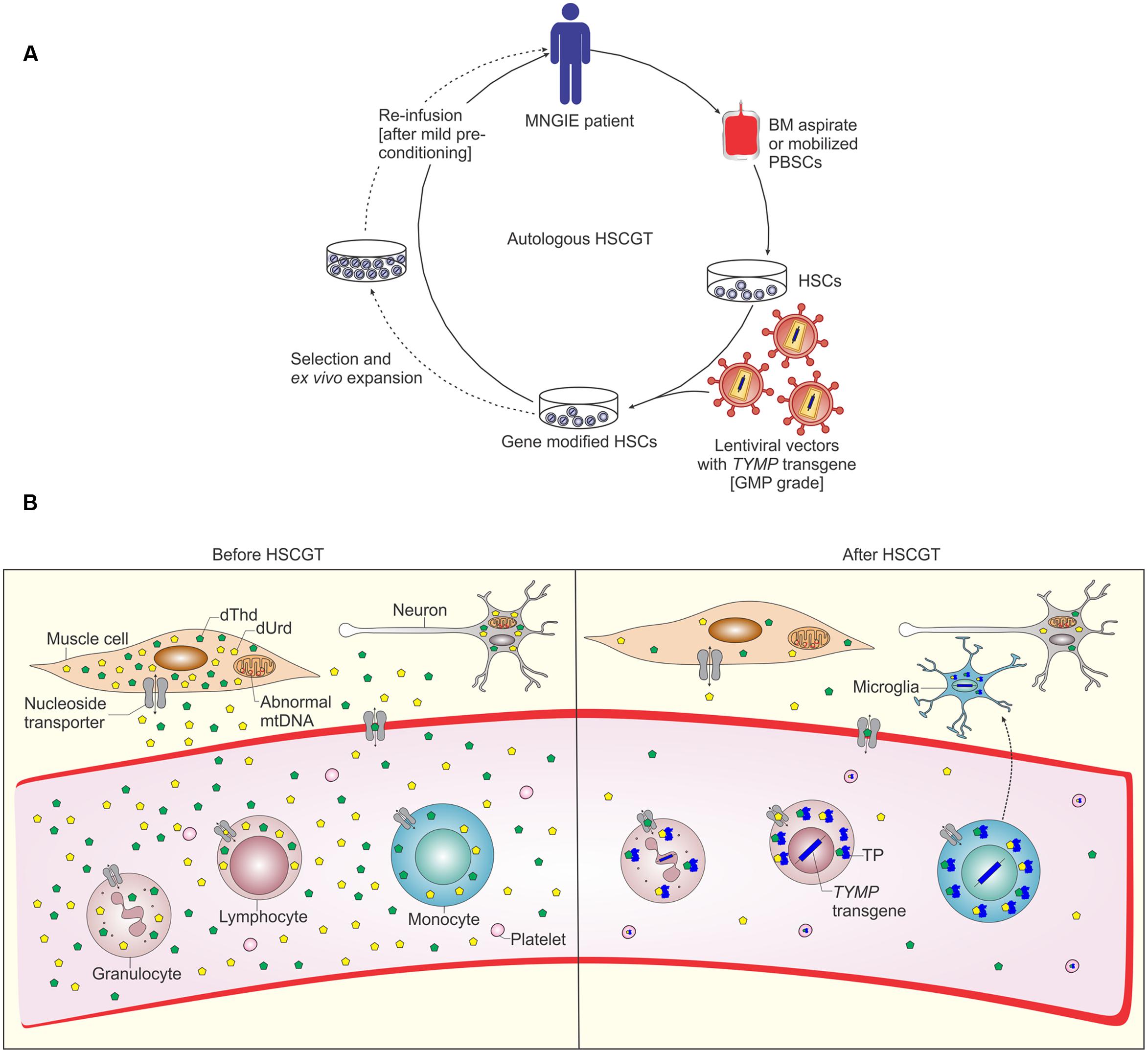
FIGURE 2. Schematic representation of autologous hematopoietic stem cell based gene therapy for MNGIE and possible mechanism of biochemical correction by gene modified HSCs. (A) Autologous bone marrow (BM) aspirates or apheresis of peripheral blood HSCs (PBSCs) after treatment with rh-G-CSF or plerixafor are collected from MNGIE patient. HSCs are ex vivo transduced by GMP grade lentiviral vectors containing the human TYMP transgene. Before infusion of the transduced cells, MNGIE patients are pre-treated with non-myeloablative conditioning to allow minimal engraftment of gene modified HSCs. Selection and ex vivo expansion of gene modified HSCs allows for transplantation of large numbers of gene modified HSCs to obviate the need for myeloablative pre-conditioning and allows (to some degree) for assessment of safety of the gene modified HSCs prior to transplantation, for example by lentiviral vector integration analysis (reviewed in Watts et al., 2011). (B) The enzyme thymidine phosphorylase (TP) is deficient in all tissues of MNGIE patients, which leads to accumulation of the nucleoside substrates dThd and dUrd and depletion of the nucleotide dCTP and finally mtDNA depletion and deletion (Gonzalez-Vioque et al., 2011). Following transplantation of gene modified HSCs and homing to bone marrow, these cells differentiate into all types of blood cells, LV genome and human TYMP transgene are integrated in leukocyte DNA ensuring stable expression of TP. TP catalyzes the chemical reaction which breaks down the nucleosides. This process eventually leads to reduction of systemic nucleosides accumulation. NTs mediate nucleosides transfer via passive facilitated diffusion (ENTs) and active transport (CNTs), the ubiquitous bidirectional ENTs are depicted (Young et al., 2013). In addition, some gene modified HSCs differentiate into monocytes and may migrate to the brain giving rise to microglia which act as a TP reservoir and cross correct the other cells in CNS.
The molecular pathological mechanism in MNGIE involves imbalanced nucleosides and nucleotide pools. Initially, loss of function mutations in TYMP gene were identified resulting in reduced TP activity (Nishino et al., 1999) leading to accumulation of access amounts of the nucleoside substrates in blood plasma, urine and almost all tissues (Spinazzola et al., 2002; Valentino et al., 2007). It has been hypothesized that this biochemical imbalance disturbs the equilibrium of intra-mitochondrial dNTPs pools (Spinazzola et al., 2002) and hence is responsible for mtDNA depletion, multiple deletions, and point mutations associated with MNGIE (Hirano et al., 1994; Papadimitriou et al., 1998; Nishino et al., 2000; Nishigaki et al., 2003). Therefore, recent studies have addressed the relationship between biochemical and dNTP pool imbalances and subsequent mtDNA abnormalities in MNGIE. In vitro, mtDNA point mutations and deletions, similar to those detected in MNGIE patients were reported in cultured HeLa cells after long time culture in the presence of high levels of thymidine in the culture medium. These mtDNA alterations were attributed to expanded levels of dTTP and dGTP and reduced levels of dCTP and dATP. However, no mtDNA depletion was observed in these HeLa cells (Song et al., 2003). Further investigation revealed that this increase in dTTP, under similar culture conditions, was more pronounced in non-cycling skin and lung fibroblasts leading to depletion in mtDNA in a dThd dose and time dependent manner (Pontarin et al., 2006). Interestingly, mtDNA levels were recovered upon removal of the dThd from the culture medium. In order to understand the influence of metabolites accumulation on the creation of mtDNA alterations, an in organello experimental model was used. Excess amounts of dThd were responsible for the significant increase in mitochondrial levels of dTTP, together leading to secondary TK2 inhibition mediated reduction of dCTP nucleotides (Gonzalez-Vioque et al., 2011). Subsequent studies confirmed these findings in in vitro fibroblast cultures and in vivo in the Tymp-/-Upp1-/- mouse model and suggest that the inadequate availability of dCTP accounts for the mtDNA depletion observed in MNGIE (Gonzalez-Vioque et al., 2011; Camara et al., 2014; Torres-Torronteras et al., 2014).
Altogether, these studies demonstrate that indeed it is the nucleoside accumulation and subsequent reduction of dCTP nucleotides, rather than the deficiency of TP per se, that accounts for the molecular and phenotypic alterations in MNGIE. An excellent illustration of this observation is the fact that TP expression in skeletal muscles is absent, nonetheless, some but not all MNGIE cases were reported with skeletal muscle mtDNA deletions, histological and oxidative phosphorylation abnormalities (Papadimitriou et al., 1998; Hirano et al., 2004).
When available, although limited, analysis of postmortem MNGIE samples is relevant and beneficial to gain knowledge about the molecular and pathological basis of the disease. Severe intestinal dysmotility, also known as CIPO, and weight loss are principle presentations of MNGIE. Histopathological analysis of MNGIE gastrointestinal samples revealed depletion in mtDNA and mitochondrial proliferation, and consequently cell atrophy in the muscularis propria layer of the stomach and small intestines (Giordano et al., 2006, 2008). Additionally, loss of interstitial cells of Cajal and morphologically abnormal muscularis propria and ganglion cells have been reported (Zimmer et al., 2009). On the other hand, the study of brain tissues of two MNGIE patients revealed no pathological proliferation of glial cells nor neuronal loss. However, the study suggested a role of TP deficiency in impairment of BBB, which could contribute to the observed hyperintense T2 signals on brain MRI scans (Szigeti et al., 2004a).
Nucleoside accumulation is detrimental probably during the early course of the disease, because nucleoside clearance did not improve mtDNA content per cell or reduce COX deficient fibers after liver transplantation (De Giorgio et al., 2016). Mitochondrial DNA instability is a hallmark for diseases caused by defective nuclear genes essential for mtDNA replication and repair (such as PEO1, POLG1,2) or maintenance of dNTP pools (such as ANT1, TYMP) or others involved in mtDNA homeostasis (such as FBXL4) (Young and Copeland, 2016). Mutations in PEO1, POLG and ANT1 underlie the autosomal dominant form of progressive external ophthalmoplegia (adPEO), a very well-characterized mtDNA disorder involving stalling of mtDNA replication (Van Goethem et al., 2001; Goffart et al., 2009). Therefore, stalling of Twinkle helicase or DNA polymerase γ could be a common pathological mechanism underlying mtDNA instability in MNGIE, PEO and mtDNA depletion syndrome (Hirano et al., 2001; Liu et al., 2008).
Current Treatments For MNGIE
In general, treatment of mitochondrial diseases is mainly based on symptom management and supportive care (Pfeffer et al., 2013). Vitamin and amino acid supplements (Tanaka et al., 1997) and exercise therapy (Taivassalo et al., 1998) aiming to improve mitochondrial functions are recommended for mitochondrial myopathies. Symptomatic management of MNGIE consists of nutritional support (Wang et al., 2015), prevention of infections and pain relief including interventions such as celiac plexus neurolysis and blockage of the splenic nerve (Teitelbaum et al., 2002; Celebi et al., 2006). Since the metabolic and mtDNA abnormalities are attributed to the systemic nucleoside imbalances, clinical interventions focus on direct removal of these metabolites to restore the balance or by introducing the deficient enzyme to reduce the metabolites.
Hemodialysis and Peritoneal Dialysis
The first hemodialysis aiming to remove the excess amounts of nucleosides from the circulation was performed in Spinazzola et al. (2002) in two MNGIE patients followed by another in 2006. In the first two patients, significantly reduced thymidine levels were observed shortly after hemodialysis, however this effect was transient as thymidine levels returned to pre-dialysis levels 3 h after dialysis (Spinazzola et al., 2002). A progressive reduction in Thd levels below the basal levels was observed after repetitive dialysis treatments in the third case (la Marca et al., 2006). A MNGIE patient who had peritoneal dialysis showed an improvement in gastrointestinal symptoms (such as vomiting, anorexia, abdominal pain, and diarrhea) during the continuing peritoneal dialysis for 3 years with body weight gain, although other major symptoms including ocular and neurological abnormalities and brain MRI signals did not change (Yavuz et al., 2007). Another case report noted improvement of the gastrointestinal and neurological symptoms, mainly the mitigation of numbness in the hands, until nucleoside levels increased again 15 months after continuous ambulatory peritoneal dialysis (Ariaudo et al., 2014).
Enzyme Replacement Therapy
Initially, platelet infusions were performed in two MNGIE patients to restore TP enzyme activity in the blood. This approach showed efficient recovery of functional TP enzyme and correction in nucleoside imbalances, however, like dialyses, these improvements were temporary requiring multiple treatment sessions for long-term responses. ERT is a reliable, well-tolerated approach to replace the deficient enzyme in a variety of lysosomal storage disorders including Gaucher, Pompe and Fabry disease, and Sly syndrome (Wilcox et al., 2004; Burrow et al., 2007). For MNGIE, approaches were developed to encapsulate TP in order to prolong the half-life of circulatory TP enzyme and reduce the immunogenic reactions. These include polymeric nanoparticles (De Vocht et al., 2009) and erythrocytes as they are permeable and affect the plasma metabolites, such as in adenosine deaminase deficiency (Bax et al., 2000; Moran et al., 2008). The EE-TP concept is under clinical development as an orphan ERT for MNGIE with the first attempt carried out in a MNGIE patient in Moran et al. (2008). In this approach, autologous erythrocytes were isolated from patients and loaded with recombinant Escherichia coli TP enzyme in vitro via hypo osmotic dialysis. Significant clinical improvements were observed such as the ability to walk and climb and the recovery of sensation and the mitigation of numbness in hands and feet, even after 23 months after termination of multiple cycles of EE-TP (Bax et al., 2013). When using the EE-TP approach, there is a high risk that an immunological reaction is triggered against the bacterial TP, especially if the infusions are repeated several times, although this has not been observed (Levene et al., 2013).
Orthotopic Liver Transplantation
Liver transplantation is a new ERT strategy for treatment of MNGIE patients. TP protein levels are high in healthy human liver tissues and significantly higher than in bone marrow cells (Boschetti et al., 2014). Recently, OLT was successfully applied in a severely affected, 25-years old MNGIE patient (De Giorgio et al., 2016). Steady state nucleoside balance was observed up to13 months post OLT. Slight improvements in lower limb strength and brain metabolism (reduced lactate levels) and structure (reduced cerebellar mean diffusivity values at diffusion MRI), improved quality of life scores and nutritional parameters, but not body weight (40 kg), were observed up to 6 months after OLT. When ileostomy closure was performed, the gastrointestinal (GI) functions and body weight declined at 13 months (37 kg). Therefore, it remains uncertain whether the mild restoration of GI function was due to the decompressive ileostomy, instead of the OLT. In addition, skeletal muscle mtDNA content per cell was slightly increased after OLT. The study suggests that the damage in post-mitotic tissues during late stages of the disease is irreversible despite recovery of nucleoside balance. Therefore, biochemical correction should probably be achieved prior to irreversible damage, preferably before the intestinal symptoms appear. Preoperative conditioning for OLT is not required. However, this approach requires matched organ donors (which are limited), involves transplantation related risks and requires long-term immunosuppression which all can further affect the quality of life of the patients.
Hematopoietic Stem Cell Transplantation
Another possibility to restore TP enzyme activity in the circulation is by HSCT. Recently, a retrospective analysis of all HSC transplanted MNGIE patients between 2005 and 2011 showed that only nine out of 24 patients were alive up to 4 years after transplantation. All nine survivors had normalized TP activity in their blood while seven of them showed improved body mass index, gastrointestinal symptoms and peripheral neuropathy. On the other hand, nine MNGIE patients died mainly due to transplant-related causes such as GVHD and graft failure, including recipients of HLA-mismatched unrelated cord blood transplants, while the remaining six patients died of disease progression. The recommendations of this study included transplantation of a sufficient number of cells, because in some patients the graft was rejected, and to consider more closely HLA matched donor cells, because of the large number of GVHD observed in this retrospective study, and to transplant at an earlier age before major organ damage has occurred (Halter et al., 2011, 2015).
The poor physical state of MNGIE patients when they enroll HSCT trials increases the risk for transplantation related complications caused by conditioning regimens and immune-suppressants. Other problems may arise from the drugs used in HSCT which are potentially harmful to mitochondria such as cyclophosphamide (Mariana Ponte Cardoso et al., 2015). Therefore, MNGIE patients are treated with Busulfan and fludarabine prior to HSCT, following the recommendations of MNGIE consensus meeting in Halter et al. (2011). For MNGIE patients who develop liver cirrhosis, allogeneic HSCT (AHSCT) should be contraindicated and OLT would be the treatment of choice (Finkenstedt et al., 2013). Pre-existing liver cirrhosis complicates liver failure which may develop after AHSCT due to multiple factors such as viral infections in immunocompromised recipients or due to hepatotoxic conditioning drugs.
Pre-Clinical Experimental Approaches Toward Therapy
Models of MNGIE
In organello experiments and human MNGIE fibroblasts were used for highlighting parts of the molecular mechanism of MNGIE (Gonzalez-Vioque et al., 2011; Camara et al., 2014). To study therapeutic interventions, such as gene therapy, in vivo models are required. A mouse model was developed by targeted disruption of exon 4 of the TYMP gene to generate Tp-/- mice. In contrast to human TP, murine TP degrades both dThd and dUrd; Tp-/- mice were crossed with Upp1-/- to generate the Tymp-/-Upp1-/- mice, which are currently the only relevant in vivo animal model (Haraguchi et al., 2002; Lopez et al., 2009). Tymp-/-Upp1-/- mice show increased levels of the purine nucleosides dThd and dUrd in plasma and tissues. Diffuse leukoencephalopathy manifests late during the lifetime of these animals, around the age of 22 months (Lopez et al., 2009). Other symptoms associated with MNGIE, such as decreased motor coordination and gastrointestinal features have not been reported in this mouse model. Brain mtDNA depletion was not consistently found in this mouse model (Lopez et al., 2009; Torres-Torronteras et al., 2011; Camara et al., 2014). Therefore, high doses of exogenous nucleosides were administered to exacerbate the mitochondrial phenotype (Garcia-Diaz et al., 2014), an approach that was rationalized by the lower nucleoside levels in Tymp-/-Upp1-/- mice compared to MNGIE patients. Mice were on an exogenous dThd and dUrd diet for a long time (24 months) before pronounced mtDNA depletion, diffuse leukoencephalopathy and motor abnormalities were observed.
Experimental approaches have been explored for treatment of MNGIE; among which experiments performed by Camara et al. (2014) which suggest that modulation of dNTP metabolism through increasing the availability of dCTP or inhibition of its catabolism can indeed reverse and prevent, at least, dCtd imbalance. A strategy that can be applied for other similar mitochondrial disorders that are caused by altered nucleosides and dNTP metabolism, for example in disorders caused by mutations in TK2 or DGUOK deficiency (Camara et al., 2014).
Gene Therapy
The Tymp-/-Upp1-/- mouse model has also been used for testing potential curative treatments. A recently investigated strategy is the use of gene therapy. Both lentiviral (LV) and adeno- AAV vector mediated TYMP gene transfer have been evaluated in pre-clinical studies for treatment of MNGIE.
AAV-Mediated Liver Directed Gene Therapy
Adeno-associated viral vector gene therapy has been explored in clinical trials for a variety of inherited and acquired diseases (Naldini, 2015). The main limitation of this approach is the human immune response to AAV capsid, as demonstrated in hemophilia B trials. In one of the first AAV trials targeting the liver, therapeutic levels of FIX were achieved at a high vector dose (2 × 1012 vector genomes per kilogram of body weight, vg/kg). Nonetheless, this high vector dose was associated with an early decline of FIX (∼8 weeks after treatment) due to T-cell immunity against AAV capsid antigens eliminating transduced hepatocytes (Manno et al., 2006).
The hybrid vector AAV2/8 with modified molecular configuration (packaged double stranded genome) and improved cassette design (codon optimized hFIX) to enhance transduction and translational efficiency was explored in a hemophilia B trial (Nathwani et al., 2006). Stable FIX expression diminished use of the costly FIX concentrate and importantly, clinical improvement was achieved in a dose dependent manner. The least bleeding episodes were seen in recipients of the highest AAV dose (steady state 5% of normal levels at a single vector peripheral vein infusion of 2 × 1012 vg/kg up to 4 years) (Nathwani et al., 2014). For MNGIE, an AAV2/8 expressing human TYMP under the control of hepatic promoter was used for treatment of Tymp-/-Upp1-/- mice (Torres-Torronteras et al., 2014). Low AAV doses (2 × 1011 vg/kg) were sufficient to reduce nucleoside imbalances to normal levels in liver, skeletal muscle, and brain for up to 8 months, while higher doses reduced nucleosides below detection levels. However, only at higher doses (>2 × 1011 vg/kg) TP activity was increased in the liver (but not in skeletal muscle or brain).
In light of the clinical data of the hemophilia B trial which shows that clinical improvement was AAV dose dependent (Nathwani et al., 2014), the question for MNGIE is whether or not the low AAV dose would be sufficient to reverse a clinical phenotype beyond biochemical correction. MNGIE mouse studies failed to report any relevant clinical phenotype in Tymp-/-Upp1-/- mice, and therefore the potential or required dosage to cure it has not been demonstrated (Torres-Torronteras et al., 2011, 2014). Importantly, upon AAV treatment nucleosides accumulation was not reduced in the intestine of treated mice at the highest dose (1013 vg/kg) administered. Since the intestines are heavily affected in MNGIE patients it is important to obtain evidence of correction in this organ. Preclinical studies in hemophilic dogs and non-human primates could predict the therapeutic dose in human trials (Manno et al., 2006; Nathwani et al., 2014) if that is the case for MNGIE too, biochemical correction in the intestine might require improved expression cassettes to enhance protein production, targeting the expression to major affected organs, or the less favorable option of using higher AAV doses (>1013 vg/kg). High doses, for example 7.2 × 1012 vg/mouse were sufficient to transduce 100% of mouse hepatocytes (Nakai et al., 2005). Such high doses might be required for gene therapy of systemic diseases, i.e., when non-hepatic tissues are also affected, as in MNGIE. However, these high AAV doses are likely to cause hepatocellular toxicity, biodistribution to other unwanted organs and shedding of the AAV, enhanced risk of eliciting immunity toward the viral capsid and increased costs of virus production. Immunity against ectopic TP might be an additional concern for MNGIE patients, therefore prophylactic immunosuppression might be required. Additional pre-clinical studies have to address the possibility of an immune response against ectopic TP in previously untreated patients. In addition to increased liver TP activity correlating with vector dose, an unexpected increase in liver dGTP of Tymp-/-Upp1-/- mice was observed in a dose depend manner as well, although the consequences of this increase are unknown. Together these findings suggest that studies into optimal dosing of AAV may be required for clinical application.
Human AAV trials should be carried out cautiously as they can reveal complications that were not observed during preclinical studies. An example is the early decline in FIX expression (Manno et al., 2006) and hepatotoxicity observed in 4/6 recipients of a high AAV dose (2 × 1012 vg/kg) (Nathwani et al., 2014), due to immunity against AAV capsid. MNGIE patients are often >12 years and probably have been pre-exposed to AAV and consequently can mount strong immune responses to AAV. Therefore, individuals with neutralizing antibodies to AAV should be excluded from clinical trials to avoid an immune response toward AAV. Another concern is the durability of transgene expression considering the longer lifespan of humans, compared with the animals in preclinical studies, and the potential need for recurrent AAV injections, especially at lower vector doses. In this respect HSCGT would provide a preferable option as a single, long lasting intervention method. Additional concerns related to AAV mediated gene therapy include purity of AAV preparations and manufacturing costs (Mingozzi and High, 2013).
LV-Mediated Hematopoietic Stem Cell Gene Therapy (HSCGT)
The encouraging therapeutic outcomes and favorable safety profile renders LV-HSCGT an attractive therapeutic approach for a variety of hereditary metabolic disorders (Wagemaker, 2014), and is potentially advantageous over AHSCT for certain selected diseases (Naldini, 2015). Proof of concept of HSC gene therapy was obtained in Tymp-/-Upp1-/- mice (Torres-Torronteras et al., 2011) using a phosphoglycerate kinase promoter driving native human TYMP cDNA and a GFP reporter in hematopoietic cells resulting in biochemical correction in peripheral blood (Torres-Torronteras et al., 2011). More recently, we developed clinically applicable LVs that carry human TYMP cDNA, and demonstrated long-term biochemical correction in Tymp-/-Upp1-/- mice at low vector copy number (VCN). Our data demonstrates the feasibility to further develop clinical protocols for HSCGT for MNGIE (Yadak et al., 2015). Similar results in a long-term follow up of 20 months confirms the correction of biochemical imbalances which was maintained at low VCN and chimerism (Torres-Torronteras et al., 2016).
In HSCGT for MNGIE, HSCs are isolated from MNGIE patients, transduced ex vivo by LV vectors carrying a functional copy of TYMP and infused back into the patient (Figure 2A). The newly formed HSCs and its progenitors produce TP which catabolize the excess amounts of nucleosides (Figure 2B). Since the patient’s own stem cells are used, GVHD is not a concern. However, mild prophylactic immunosuppression maybe required to prevent possible immune reaction against the TP transgene.
Myeloablative pre-conditioning might be necessary for high levels of engraftment in other metabolic disorders, such as metachromatic leukodystrophy (MLD), due to lack of selective advantage of gene modified cells. In particular, busulfan myeloablative conditioning is used in MLD patients for depletion of endogenous microglia and mobility of gene modified monocytes through the BBB (Capotondo et al., 2012). The contribution of gene modified microglia to correct biochemical imbalances has never been explored in MNGIE. However, murine gene therapy studies using LV and AAV vectors implicate reversal of nucleoside imbalance at low or possibly no increase in brain TP activity (Torres-Torronteras et al., 2014, 2016). In liver directed AAV2/8 gene therapy, it is not expected that brain cells will be transduced. In HSCGT, gene modified monocytes are expected to migrate to brain and differentiate into microglia. Nonetheless, the results of the HSCGT MNGIE mouse study do not rule out the potential that gene modified microglia can contribute to correction of brain biochemistry and phenotype, although this might not be necessary if ectopic expression outside the brain is high enough. When transduction efficiency is high enough, significant TP activity can be measured in the brain, indicating that transduced microglia might reside in the brain after long-term follow-up. To that end, two potential mechanisms might act synergistically to normalize the brain nucleoside levels, a systemic ectopic source and one local contribution of gene-modified cells (Figure 2B).
Potential options and future research for application in MNGIE patients include alternative conditioning strategies to obviate the cytotoxicity related to myeloablative conditioning and strategies to enhance the quality of infused gene modified HSCs. One approach is to mobilize endogenous HSCs into peripheral blood in order to create (space) in the bone marrow for the infused donor HSCs to engraft (Chen et al., 2006). Human G-CSF was sufficient in immunocompromised mice (Huston et al., 2014), probably due to the selective advantage of the gene modified cells, however, more stringent agents might be required in normal immunocompetent mice. A possibility is G-CSF in combination with the more potent HSCs mobilizer plerixafor (a specific CXCR4 antagonist) or the selectins inhibitor fucoidan. Such regimens probably require additional mild chemotherapeutics, in particular if the gene corrected TP-expressing HSCs lack selective growth advantage to overcome host cells. These HSCs mobilizers act via different mechanisms, therefore parameters such as the optimal dose and time frame for transplantation after mobilization need to be established in relevant pre-clinical models. Alternatively, targeting specific endogenous hematopoietic populations might reduce the off-target toxicity related to the common non-specific conditioning (Aiuti and Naldini, 2016). Examples include inhibiting c-kit, a HSC tyrosine kinase cell surface antigen (Xue et al., 2010) and the recently developed immunotoxin against hematopoietic stem cells (CD45-SAP) (Palchaudhuri et al., 2016).
Strategies such as ex vivo expansion of gene modified HSCs can improve the quality of the infused gene modified cells and enhance the outcome of gene therapy (Watts et al., 2011). In particular when combined with additional approaches to enrich for HSCs, preserve stemness of- and enhance homing and engraftment ability of gene modified HSCs (Psatha et al., 2016). Ultimately, this approach combined with improved mild pre-conditioning protocols, could benefit patients in poor health condition at transplantation, such as in MNGIE patients.
A risk of HSC gene therapy is insertional mutagenesis. The first HSC gene therapy trials used gammaretrovirus (γ-RV) based vectors for treatment of SCID-X1 (Gaspar et al., 2004; Hacein-Bey-Abina et al., 2010), adenosine deaminase (ADA-SCID) (Aiuti et al., 2002), CGD (Ott et al., 2006) and WAS (Boztug et al., 2010). Although efficient correction of immunodeficiency was achieved in most patients in SCID-X1, CGD and WAS trials, lympho-proliferative disorders (Hacein-Bey-Abina et al., 2003a,b, 2008; Howe et al., 2008; Braun et al., 2014) and myelodysplasia (Stein et al., 2010) developed secondary to γ-RV vector integrations within or nearby proto-oncogenes. In addition to the preferred integration profiles over γ-RV vector (Deichmann et al., 2007; Cattoglio et al., 2007; Gabriel et al., 2012), LV efficiently transduce non-cycling primitive HSCs and under minimum culture conditions (Naldini et al., 1996; Guenechea et al., 2000). Therefore, attention was focused on development of LV as a relatively safer approach, leading eventually to development of third-generation SIN-LV (Dull et al., 1998; Zufferey et al., 1998). Several pre-clinical studies indicate the reduced genotoxicity of SIN-LV vectors compared with γ-retroviral vectors, in particular SIN-LV CISs revealed no preference of integration near proto-oncogenes (Montini et al., 2006; Modlich et al., 2009; Biffi et al., 2011; Romero et al., 2013; Zhou et al., 2013). Since then, SIN-LV vectors have been applied successfully in ongoing clinical trials for a variety of metabolic (Cartier et al., 2009; Biffi et al., 2013) and immunodeficiency disorders (Aiuti et al., 2013), and no adverse events have yet been reported in these trials. Moreover, the therapeutic benefits without toxicity related to transgene expression and biosafety of SIN-LV vectors has been further validated through a growing body of recent preclinical studies supporting the initiation of clinical trials, for example for β-thalassemia (Negre et al., 2015) and mucopolysaccharidosis I disease (Visigalli et al., 2016).
Furthermore, selective advantage for growth and differentiation conferred by the therapeutic transgene expression increases the potential risk for proliferative disorders, this was reported in some immunodeficiency conditions (Aiuti and Roncarolo, 2009). For metabolic disorders, for instance lysosomal storage disorders, however, most studies show that enzyme positive cells have no selective advantage (Bernardo and Aiuti, 2016), which is most likely the case in MNGIE as well. To improve safety, technologies such as ex vivo expansion of gene modified HSCs may permit for safety assessment (to some degree) prior to transplantation, by analysis of LV integration sites (Watts et al., 2011; Figure 2A).
AAV Mediated GT or HSCGT?
Regardless of the type of viral vector used for gene therapy, the chosen strategy should provide long-term expression of the gene of interest without side effects in the host. It is important to apply a well-defined vector dose that is sufficient to reverse the biochemical and nucleotide imbalance without any potential side effects. In particular, abnormal overexpression of the TP enzyme is detected in different tumor types, including non-small cell lung-, colorectal-, breast-, gastrointestinal-, and hepatic cancers (Koukourakis et al., 1997; Mori et al., 2000; Ikeguchi et al., 2001; Nakayama et al., 2005; Mitselou et al., 2012) and correlates with a worse prognosis in colorectal cancer patients (Takebayashi et al., 1996). Besides, disturbance of dNTP pools can be a trigger for cell cycle arrest and apoptosis (Oliver et al., 1996; Kumar et al., 2010).
The medical condition of the patient can also influence the choice of the vector system for clinical application. For terminally ill patients, the AAV approach could be most suitable to avoid the risks associated with the pre-conditioning for transplantation in autologous HSC gene therapy or if a suitable HSCs donor for AHSCT is lacking.
Concluding Remarks
The lack of mitochondrial histone protection, the limited repair capacity and oxidized dNTPs contributing to mismatch errors (Alexeyev et al., 2013) all make mitochondria more susceptible than nuclear DNA to mutagenesis. It has become evident that it is the systemic accumulation of nucleosides in MNGIE (Di Meo et al., 2015) that causes imbalances in mitochondrial dNTP pools. However, the mechanism by which it causes mtDNA alterations is still unknown. Although the current treatments focus on restoration of TP enzyme activity and/ or elimination of accumulating metabolites, further understanding of cellular mechanisms involved in maintenance of mtDNA integrity and copy number can provide targets for clinical intervention for MNGIE and possibly other mitochondrial disorders.
Platelet infusions, hemato/peritoneal dialysis, and EE-TP ERT could be used to provide biochemical correction. AAV gene therapy and lentiviral HSCGT are potential curative options as evidenced by the promising pre-clinical results in Tymp-/-Upp1-/- mice. OLT is a promising emerging treatment and should currently be the treatment of choice for MNGIE patients with pre-existing liver failure. Allogeneic HSCT has risks of graft failure, GVHD and conditioning-related toxicity. Milder conditioning may be applicable in HSCGT, and treatment should preferably be applied at an early age. Novel strategies are being explored to improve the safety and efficiency of viral based gene therapy, ultimately for MNGIE patients as well. These include strategies to enhance transduction, improve engraftment of gene modified HSCs and limit transplantation related toxicity, and others to overcome the limitation of AAV capsid triggered immunity by means of novel serotypes and improved transcription cassettes.
Mitochondrial neurogastrointestinal encephalomyopathy patients should receive suitable treatment promptly before permanent damage occurs, which can be challenging, as MNGIE patients are often diagnosed late during disease progression in a poor health condition. Because TP activity and nucleoside levels can be routinely measured in blood samples, MNGIE should be considered to be included in newborn screening programs, similar to other (neuro) metabolic disorders for early diagnosis and treatment (Carlson, 2004; McHugh et al., 2011).
Author Contributions
RY framed the structure of the review, analyzed the literature and wrote the manuscript; PS, MvG participated in the literature analysis; NvT, IdC participated in the literature analysis and supervised the writing. All authors discussed the topic and provided intellectual feedback to the article. All the authors read and approved the manuscript.
Funding
We would like to acknowledge the financial support of Join4energy, the Sophia Foundation (SSW0645) and the Stichting NeMo.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AAV, adeno-associated virus; ADA-SCID, adenosine deaminase-severe combined immunodeficiency; AHSCT, allogeneic hematopoietic stem cell transplantation; ANT-1, adenine nucleotide translocase type 1; BBB, blood–brain barrier; CGD, chronic granulomatous disease; CIPO, chronic intestinal pseudo obstruction; CIS, common integration site; CNT, concentrative nucleoside transporter; CPEO, chronic progressive external ophthalmoplegia; dATP, deoxyadenosine triphosphate; dCTP, deoxycytidinetriphosphate; dGTP, deoxyguanosine triphosphate; DGUOK, deoxyguanosine kinase; dNTP, deoxyribonucleoside triphosphates; dThd, thymidine; dTTP, deoxythymidine triphosphate; dUrd, deoxyuridine; ECGF1, platelet-derived endothelial cell growth factor; EE-TP, erythrocyte encapsulated TP; ENT, equilibrative nucleosides transport; ERT, enzyme replacement therapy; FBXL4, F-box and leucine-rich repeat protein 4; FIX, coagulation factor IX; G-CSF, granulocyte colony stimulating factor; GVHD, graft versus host disease; HSCT, hematopoietic stem cell transplantation; HSCGT, hematopoietic stem cell gene therapy; LHON, Leber’s hereditary optic neuropathy; LV, lentivirus; MDS, mitochondrial DNA depletion syndrome; MELAS, mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes; MNGIE, mitochondrial neurogastrointestinal encephalomyopathy; NT, nucleoside transporter; OLT, orthotopic liver transplantation; PEO, progressive external ophthalmoplegia; POLG, DNA polymerase subunit gamma; RRM2B, ribonucleotide reductase M2 B; SCID- X1, X-linked severe combined immunodeficiency; SIN-LV, self-inactivating LV; TK2, thymidine kinase 2; TYMP, thymidine phosphorylase; WAS, Wiskott–Aldrich syndrome; γ-RV, gammaretrovirus.
Footnotes
References
Aiuti, A., Biasco, L., Scaramuzza, S., Ferrua, F., Cicalese, M. P., Baricordi, C., et al. (2013). Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science 341:1233151. doi: 10.1126/science.1233151
Aiuti, A., and Naldini, L. (2016). Safer conditioning for blood stem cell transplants. Nat. Biotechnol. 34, 721–723. doi: 10.1038/nbt.3629
Aiuti, A., and Roncarolo, M. G. (2009). Ten years of gene therapy for primary immune deficiencies. Hematol. Am. Soc. Hematol. Educ. Program 1, 682–689. doi: 10.1182/asheducation-2009.1.682
Aiuti, A., Slavin, S., Aker, M., Ficara, F., Deola, S., Mortellaro, A., et al. (2002). Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science 296, 2410–2413. doi: 10.1126/science.1070104
Alexeyev, M., Shokolenko, I., Wilson, G., and LeDoux, S. (2013). The maintenance of mitochondrial DNA integrity–critical analysis and update. Cold Spring Harb. Perspect. Biol. 5:a012641. doi: 10.1101/cshperspect.a012641
Ariaudo, C., Daidola, G., Ferrero, B., Guarena, C., Burdese, M., Segoloni, G. P., et al. (2014). Mitochondrial neurogastrointestinal encephalomyopathy treated with peritoneal dialysis and bone marrow transplantation. J. Nephrol. 28, 125–127. doi: 10.1007/s40620-014-0069-9
Barboni, P., Savini, G., Plazzi, G., Bellan, M., Valentino, M. L., Zanini, M., et al. (2004). Ocular findings in mitochondrial neurogastrointestinal encephalomyopathy: a case report. Graefes Arch. Clin. Exp. Ophthalmol. 242, 878–880. doi: 10.1007/s00417-004-0914-y
Barragan-Campos, H. M., Vallee, J. N., Lo, D., Barrera-Ramirez, C. F., Argote-Greene, M., Sanchez-Guerrero, J., et al. (2005). Brain magnetic resonance imaging findings in patients with mitochondrial cytopathies. Arch. Neurol. 62, 737–742. doi: 10.1001/archneur.62.5.737.
Bax, B. E., Bain, M. D., Fairbanks, L. D., Simmonds, H. A., Webster, A. D., and Chalmers, R. A. (2000). Carrier erythrocyte entrapped adenosine deaminase therapy in adenosine deaminase deficiency. Adv. Exp. Med. Biol. 486, 47–50. doi: 10.1007/0-306-46843-3_9
Bax, B. E., Bain, M. D., Scarpelli, M., Filosto, M., Tonin, P., and Moran, N. (2013). Clinical and biochemical improvements in a patient with MNGIE following enzyme replacement. Neurology 81, 1269–1271. doi: 10.1212/WNL.0b013e3182a6cb4b
Bedlack, R. S., Vu, T., Hammans, S., Sparr, S. A., Myers, B., Morgenlander, J., et al. (2004). MNGIE neuropathy: five cases mimicking chronic inflammatory demyelinating polyneuropathy. Muscle Nerve 29, 364–368. doi: 10.1002/mus.10546
Bernardo, M. E., and Aiuti, A. (2016). The role of conditioning in hematopoietic stem cell gene therapy. Hum. Gene. Ther. doi: 10.1089/hum.2016.103 [Epub ahead of print].
Biffi, A., Bartolomae, C. C., Cesana, D., Cartier, N., Aubourg, P., Ranzani, M., et al. (2011). Lentiviral vector common integration sites in preclinical models and a clinical trial reflect a benign integration bias and not oncogenic selection. Blood 117, 5332–5339. doi: 10.1182/blood-2010-09-306761.
Biffi, A., Montini, E., Lorioli, L., Cesani, M., Fumagalli, F., Plati, T., et al. (2013). Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 341:1233158. doi: 10.1126/science.1233158
Blazquez, A., Martin, M. A., Lara, M. C., Marti, R., Campos, Y., Cabello, A., et al. (2005). Increased muscle nucleoside levels associated with a novel frameshift mutation in the thymidine phosphorylase gene in a Spanish patient with MNGIE. Neuromuscul. Disord. 15, 775–778. doi: 10.1016/j.nmd.2005.07.008
Blondon, H., Polivka, M., Joly, F., Flourie, B., Mikol, J., and Messing, B. (2005). Digestive smooth muscle mitochondrial myopathy in patients with mitochondrial-neuro-gastro-intestinal encephalomyopathy (MNGIE). Gastroenterol. Clin. Biol. 29, 773–778. doi: 10.1016/S0399-8320(05)86346-8
Boschetti, E., D’Alessandro, R., Bianco, F., Carelli, V., Cenacchi, G., Pinna, A. D., et al. (2014). Liver as a source for thymidine phosphorylase replacement in mitochondrial neurogastrointestinal encephalomyopathy. PLoS ONE 9:e96692. doi: 10.1371/journal.pone.0096692
Boztug, K., Schmidt, M., Schwarzer, A., Banerjee, P. P., Diez, I. A., Dewey, R. A., et al. (2010). Stem-cell gene therapy for the Wiskott-Aldrich syndrome. N. Engl. J. Med. 363, 1918–1927. doi: 10.1056/NEJMoa1003548
Braun, C. J., Boztug, K., Paruzynski, A., Witzel, M., Schwarzer, A., Rothe, M., et al. (2014). Gene therapy for Wiskott-Aldrich syndrome–long-term efficacy and genotoxicity. Sci. Transl. Med. 6:227ra233. doi: 10.1126/scitranslmed.3007280
Burrow, T. A., Hopkin, R. J., Leslie, N. D., Tinkle, B. T., and Grabowski, G. A. (2007). Enzyme reconstitution/replacement therapy for lysosomal storage diseases. Curr. Opin. Pediatr. 19, 628–635. doi: 10.1097/MOP.0b013e3282f161f2.
Camara, Y., Gonzalez-Vioque, E., Scarpelli, M., Torres-Torronteras, J., Caballero, A., Hirano, M., et al. (2014). Administration of deoxyribonucleosides or inhibition of their catabolism as a pharmacological approach for mitochondrial DNA depletion syndrome. Hum. Mol. Genet. 23, 2459–2467. doi: 10.1093/hmg/ddt641
Capotondo, A., Milazzo, R., Politi, L. S., Quattrini, A., Palini, A., Plati, T., et al. (2012). Brain conditioning is instrumental for successful microglia reconstitution following hematopoietic stem cell transplantation. Proc. Natl. Acad. Sci. U.S.A. 109, 15018–15023. doi: 10.1073/pnas.1205858109
Cardaioli, E., Da Pozzo, P., Malfatti, E., Battisti, C., Gallus, G. N., Gaudiano, C., et al. (2010). A second MNGIE patient without typical mitochondrial skeletal muscle involvement. Neurol. Sci. 31, 491–494. doi: 10.1007/s10072-010-0225-5
Carlson, M. D. (2004). Recent advances in newborn screening for neurometabolic disorders. Curr. Opin. Neurol. 17, 133–138. doi: 10.1097/00019052-200404000-00008
Carod-Artal, F. J., Herrero, M. D., Lara, M. C., Lopez-Gallardo, E., Ruiz-Pesini, E., Marti, R., et al. (2007). Cognitive dysfunction and hypogonadotrophic hypogonadism in a Brazilian patient with mitochondrial neurogastrointestinal encephalomyopathy and a novel ECGF1 mutation. Eur. J. Neurol. 14, 581–585. doi: 10.1111/j.1468-1331.2007.01720.x
Cartier, N., Hacein-Bey-Abina, S., Bartholomae, C. C., Veres, G., Schmidt, M., Kutschera, I., et al. (2009). Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science 326, 818–823. doi: 10.1126/science.1171242
Cattoglio, C., Facchini, G., Sartori, D., Antonelli, A., Miccio, A., Cassani, B., et al. (2007). Hot spots of retroviral integration in human CD34+ hematopoietic cells. Blood 110, 1770–1778. doi: 10.1182/blood-2007-01-068759
Celebi, N., Sahin, A., Canbay, O., Uzumcugil, F., and Aypar, U. (2006). Abdominal pain related to mitochondrial neurogastrointestinal encephalomyopathy syndrome may benefit from splanchnic nerve blockade. Paediatr. Anaesth. 16, 1073–1076. doi: 10.1111/j.1460-9592.2006.01918.x
Chapman, T. P., Hadley, G., Fratter, C., Cullen, S. N., Bax, B. E., Bain, M. D., et al. (2014). Unexplained gastrointestinal symptoms: think mitochondrial disease. Dig. Liver Dis. 46, 1–8. doi: 10.1016/j.dld.2013.04.008
Chen, J., Larochelle, A., Fricker, S., Bridger, G., Dunbar, C. E., and Abkowitz, J. L. (2006). Mobilization as a preparative regimen for hematopoietic stem cell transplantation. Blood 107, 3764–3771. doi: 10.1182/blood-2005-09-3593
De Giorgio, R., Pironi, L., Rinaldi, R., Boschetti, E., Caporali, L., Capristo, M., et al. (2016). Liver transplantation for mitochondrial neurogastrointestinal encephalomyopathy. Ann. Neurol. 80, 448–455. doi: 10.1002/ana.24724
De Vocht, C., Ranquin, A., Willaert, R., Van Ginderachter, J. A., Vanhaecke, T., Rogiers, V., et al. (2009). Assessment of stability, toxicity and immunogenicity of new polymeric nanoreactors for use in enzyme replacement therapy of MNGIE. J. Control. Release 137, 246–254. doi: 10.1016/j.jconrel.2009.03.020
Debouverie, M., Wagner, M., Ducrocq, X., Grignon, Y., Mousson, B., and Weber, M. (1997). [MNGIE syndrome in 2 siblings] Le MNGIE syndrome: deux cas dans une meme fratrie. Rev. Neurol. (Paris) 153, 547–553.
Deichmann, A., Hacein-Bey-Abina, S., Schmidt, M., Garrigue, A., Brugman, M. H., Hu, J., et al. (2007). Vector integration is nonrandom and clustered and influences the fate of lymphopoiesis in SCID-X1 gene therapy. J. Clin. Invest. 117, 2225–2232. doi: 10.1172/JCI31659
Di Meo, I., Lamperti, C., and Tiranti, V. (2015). Mitochondrial diseases caused by toxic compound accumulation: from etiopathology to therapeutic approaches. EMBO Mol. Med. 7, 1257–1266. doi: 10.15252/emmm.201505040emmm.201505040
DiMauro, S. (2004). Mitochondrial diseases. Biochim. Biophys. Acta 1658, 80–88. doi: 10.1016/j.bbabio.2004.03.014
Dull, T., Zufferey, R., Kelly, M., Mandel, R. J., Nguyen, M., Trono, D., et al. (1998). A third-generation lentivirus vector with a conditional packaging system. J. Virol. 72, 8463–8471.
El-Hattab, A. W., and Scaglia, F. (2013). Mitochondrial DNA depletion syndromes: review and updates of genetic basis, manifestations, and therapeutic options. Neurotherapeutics 10, 186–198. doi: 10.1007/s13311-013-0177-6
Etienne, G., Shamseddine, K., Pulley, M., and Milfred, F. (2012). Two new gene mutations for late onset mitochondrial neurogastrointestinal encephalopathy (MNGIE). Transl. Neurosci. 3, 413–414. doi: 10.2478/s13380-012-0042-9.
Fairbanks, L. D., Marinaki, A. M., Carrey, E. A., Hammans, S. R., and Duley, J. A. (2002). Deoxyuridine accumulation in urine in thymidine phosphorylase deficiency (MNGIE). J. Inherit. Metab. Dis. 25, 603–604. doi: 10.1023/A:1022007827133
Finkenstedt, A., Schranz, M., Bosch, S., Karall, D., Burgi, S. S., Ensinger, C., et al. (2013). MNGIE syndrome: liver cirrhosis should be ruled out prior to bone marrow transplantation. JIMD Rep. 10, 41–44. doi: 10.1007/8904_2012_199
Fox, S. B., Moghaddam, A., Westwood, M., Turley, H., Bicknell, R., Gatter, K. C., et al. (1995). Platelet-derived endothelial cell growth factor/thymidine phosphorylase expression in normal tissues: an immunohistochemical study. J. Pathol. 176, 183–190. doi: 10.1002/path.1711760212
Friedkin, M., and Roberts, D. (1954). The enzymatic synthesis of nucleosides. I. Thymidine phosphorylase in mammalian tissue. J. Biol. Chem. 207, 245–256.
Fukami, M. H., and Salganicoff, L. (1973). Isolation and properties of human platelet mitochondria. Blood 42, 913–918.
Gabriel, R., Schmidt, M., and von Kalle, C. (2012). Integration of retroviral vectors. Curr. Opin. Immunol. 24, 592–597. doi: 10.1016/j.coi.2012.08.006
Gamez, J., Lara, M. C., Mearin, F., Oliveras-Ley, C., Raguer, N., Olive, M., et al. (2005). A novel thymidine phosphorylase mutation in a Spanish MNGIE patient. J. Neurol. Sci. 228, 35–39. doi: 10.1016/j.jns.2004.09.034
Garcia-Diaz, B., Garone, C., Barca, E., Mojahed, H., Gutierrez, P., Pizzorno, G., et al. (2014). Deoxynucleoside stress exacerbates the phenotype of a mouse model of mitochondrial neurogastrointestinal encephalopathy. Brain 137, 1337–1349. doi: 10.1093/brain/awu068
Garone, C., Tadesse, S., and Hirano, M. (2011). Clinical and genetic spectrum of mitochondrial neurogastrointestinal encephalomyopathy. Brain 134, 3326–3332. doi: 10.1093/brain/awr245
Gaspar, H. B., Parsley, K. L., Howe, S., King, D., Gilmour, K. C., Sinclair, J., et al. (2004). Gene therapy of X-linked severe combined immunodeficiency by use of a pseudotyped gammaretroviral vector. Lancet 364, 2181–2187. doi: 10.1016/S0140-6736(04)17590-9
Giordano, C., Sebastiani, M., De Giorgio, R., Travaglini, C., Tancredi, A., Valentino, M. L., et al. (2008). Gastrointestinal dysmotility in mitochondrial neurogastrointestinal encephalomyopathy is caused by mitochondrial DNA depletion. Am. J. Pathol. 173, 1120–1128. doi: 10.2353/ajpath.2008.080252
Giordano, C., Sebastiani, M., Plazzi, G., Travaglini, C., Sale, P., Pinti, M., et al. (2006). Mitochondrial neurogastrointestinal encephalomyopathy: evidence of mitochondrial DNA depletion in the small intestine. Gastroenterology 130, 893–901. doi: 10.1053/j.gastro.2006.01.004
Goffart, S., Cooper, H. M., Tyynismaa, H., Wanrooij, S., Suomalainen, A., and Spelbrink, J. N. (2009). Twinkle mutations associated with autosomal dominant progressive external ophthalmoplegia lead to impaired helicase function and in vivo mtDNA replication stalling. Hum. Mol. Genet. 18, 328–340. doi: 10.1093/hmg/ddn359
Gonzalez-Vioque, E., Torres-Torronteras, J., Andreu, A. L., and Marti, R. (2011). Limited dCTP availability accounts for mitochondrial DNA depletion in mitochondrial neurogastrointestinal encephalomyopathy (MNGIE). PLoS Genet. 7:e1002035. doi: 10.1371/journal.pgen.1002035
Granero Castro, P., Fernandez Arias, S., Moreno Gijon, M., Alvarez Martinez, P., Granero Trancon, J., Alvarez Perez, J. A., et al. (2010). Emergency surgery in chronic intestinal pseudo-obstruction due to mitochondrial neurogastrointestinal encephalomyopathy: case reports. Int. Arch. Med. 3:35. doi: 10.1186/1755-7682-3-35
Guenechea, G., Gan, O. I., Inamitsu, T., Dorrell, C., Pereira, D. S., Kelly, M., et al. (2000). Transduction of human CD34+ CD38- bone marrow and cord blood-derived SCID-repopulating cells with third-generation lentiviral vectors. Mol. Ther. 1, 566–573. doi: 10.1006/mthe.2000.0077
Hacein-Bey-Abina, S., Garrigue, A., Wang, G. P., Soulier, J., Lim, A., Morillon, E., et al. (2008). Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Invest. 118, 3132–3142. doi: 10.1172/JCI35700.
Hacein-Bey-Abina, S., Hauer, J., Lim, A., Picard, C., Wang, G. P., Berry, C. C., et al. (2010). Efficacy of gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 363, 355–364. doi: 10.1056/NEJMoa1000164
Hacein-Bey-Abina, S., von Kalle, C., Schmidt, M., Le Deist, F., Wulffraat, N., McIntyre, E., et al. (2003a). A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 348, 255–256. doi: 10.1056/nejm200301163480314.
Hacein-Bey-Abina, S., Von Kalle, C., Schmidt, M., McCormack, M. P., Wulffraat, N., Leboulch, P., et al. (2003b). LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 302, 415–419.
Halter, J., Schupbach, W. M., Casali, C., Elhasid, R., Fay, K., Hammans, S., et al. (2011). Allogeneic hematopoietic SCT as treatment option for patients with mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): a consensus conference proposal for a standardized approach. Bone Marrow Transplant. 46, 330–337. doi: 10.1038/bmt.2010.100
Halter, J. P., Michael, W., Schupbach, M., Mandel, H., Casali, C., Orchard, K., et al. (2015). Allogeneic haematopoietic stem cell transplantation for mitochondrial neurogastrointestinal encephalomyopathy. Brain 138, 2847–2858. doi: 10.1093/brain/awv226
Haraguchi, M., Tsujimoto, H., Fukushima, M., Higuchi, I., Kuribayashi, H., Utsumi, H., et al. (2002). Targeted deletion of both thymidine phosphorylase and uridine phosphorylase and consequent disorders in mice. Mol. Cell. Biol. 22, 5212–5221. doi: 10.1128/MCB.22.14.5212-5221.2002
Hirano, M., Marti, R., Ferreiro-Barros, C., Vila, M. R., Tadesse, S., Nishigaki, Y., et al. (2001). Defects of intergenomic communication: autosomal disorders that cause multiple deletions and depletion of mitochondrial DNA. Semin. Cell Dev. Biol. 12, 417–427. doi: 10.1006/scdb.2001.0279
Hirano, M., Nishigaki, Y., and Marti, R. (2004). Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): a disease of two genomes. Neurologist 10, 8–17. doi: 10.1097/01.nrl.0000106919.06469.04
Hirano, M., Silvestri, G., Blake, D. M., Lombes, A., Minetti, C., Bonilla, E., et al. (1994). Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): clinical, biochemical, and genetic features of an autosomal recessive mitochondrial disorder. Neurology 44, 721–727. doi: 10.1212/WNL.44.4.721
Howe, S. J., Mansour, M. R., Schwarzwaelder, K., Bartholomae, C., Hubank, M., Kempski, H., et al. (2008). Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin. Invest. 118, 3143–3150. doi: 10.1172/JCI35798
Huston, M. W., Riegman, A. R., Yadak, R., van Helsdingen, Y., de Boer, H., van Til, N. P., et al. (2014). Pretransplant mobilization with granulocyte colony-stimulating factor improves B-cell reconstitution by lentiviral vector gene therapy in SCID-X1 mice. Hum. Gene Ther. 25, 905–914. doi: 10.1089/hum.2014.101.
Ikeguchi, M., Sakatani, T., Ueda, T., Hirooka, Y., and Kaibara, N. (2001). Thymidine phosphorylase activity in liver tissue and its correlation with multifocal occurrence of hepatocellular carcinomas. In Vivo 15, 265–270.
Kalkan, I. H., Koksal, A. S., Evcimen, S., Sapmaz, F., Oztas, E., Onder, F. O., et al. (2015). Spontaneous abdominal esophageal perforation in a patient with mitochondrial neurogastrointestinal encephalomyopathy. Acta Clin. Belg. 70, 44–45. doi: 10.1179/2295333714Y.0000000053
Kocaefe, Y. C., Erdem, S., Ozguc, M., and Tan, E. (2003). Four novel thymidine phosphorylase gene mutations in mitochondrial neurogastrointestinal encephalomyopathy syndrome (MNGIE) patients. Eur. J. Hum. Genet. 11, 102–104. doi: 10.1038/sj.ejhg.5200908
Koukourakis, M. I., Giatromanolaki, A., O’Byrne, K. J., Comley, M., Whitehouse, R. M., Talbot, D. C., et al. (1997). Platelet-derived endothelial cell growth factor expression correlates with tumour angiogenesis and prognosis in non-small-cell lung cancer. Br. J. Cancer 75, 477–481. doi: 10.1038/bjc.1997.83
Kumar, D., Viberg, J., Nilsson, A. K., and Chabes, A. (2010). Highly mutagenic and severely imbalanced dNTP pools can escape detection by the S-phase checkpoint. Nucleic Acids Res. 38, 3975–3983. doi: 10.1093/nar/gkq128.
la Marca, G., Malvagia, S., Casetta, B., Pasquini, E., Pela, I., Hirano, M., et al. (2006). Pre- and post-dialysis quantitative dosage of thymidine in urine and plasma of a MNGIE patient by using HPLC-ESI-MS/MS. J. Mass Spectrom. 41, 586–592. doi: 10.1002/jms.1013
Lara, M. C., Valentino, M. L., Torres-Torronteras, J., Hirano, M., and Marti, R. (2007). Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): biochemical features and therapeutic approaches. Biosci. Rep. 27, 151–163. doi: 10.1007/s10540-007-9043-2
Levene, M., Coleman, D. G., Kilpatrick, H. C., Fairbanks, L. D., Gangadharan, B., Gasson, C., et al. (2013). Preclinical toxicity evaluation of erythrocyte-encapsulated thymidine phosphorylase in BALB/c mice and beagle dogs: an enzyme-replacement therapy for mitochondrial neurogastrointestinal encephalomyopathy. Toxicol. Sci. 131, 311–324. doi: 10.1093/toxsci/kfs278
Li, J. N., Han, D. Y., Ji, F., Chen, A. T., Wu, N., Xi, X., et al. (2011). Successful cochlear implantation in a patient with MNGIE syndrome. Acta Otolaryngol. 131, 1012–1016. doi: 10.3109/00016489.2011.579623
Liu, Z., Ding, Y., Du, A., Zhang, B., Zhao, G., and Ding, M. (2008). A novel Twinkle (PEO1) gene mutation in a Chinese family with adPEO. Mol. Vis. 14, 1995–2001.
Lopez, L. C., Akman, H. O., Garcia-Cazorla, A., Dorado, B., Marti, R., Nishino, I., et al. (2009). Unbalanced deoxynucleotide pools cause mitochondrial DNA instability in thymidine phosphorylase-deficient mice. Hum. Mol. Genet. 18, 714–722. doi: 10.1093/hmg/ddn401
Manno, C. S., Pierce, G. F., Arruda, V. R., Glader, B., Ragni, M., Rasko, J. J., et al. (2006). Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 12, 342-347. doi: 10.1038/nm1358
Mariana Ponte Cardoso, R., Armanda Emanuela Castro e, S., and José Barata Antunes, C. (2015). Mitochondrial Dysfunction on the Toxic Effects of Anticancer Agents–From Lab Bench to Bedside. Rijeka: InTech.
Marti, R., Spinazzola, A., Tadesse, S., Nishino, I., Nishigaki, Y., and Hirano, M. (2004). Definitive diagnosis of mitochondrial neurogastrointestinal encephalomyopathy by biochemical assays. Clin. Chem. 50, 120–124. doi: 10.1373/clinchem.2003.026179
Marti, R., Verschuuren, J. J., Buchman, A., Hirano, I., Tadesse, S., van Kuilenburg, A. B., et al. (2005). Late-onset MNGIE due to partial loss of thymidine phosphorylase activity. Ann. Neurol. 58, 649–652. doi: 10.1002/ana.20615
Martin, M. A., Blazquez, A., Marti, R., Bautista, J., Lara, M. C., Cabello, A., et al. (2004). Lack of gastrointestinal symptoms in a 60-year-old patient with MNGIE. Neurology 63, 1536–1537. doi: 10.1212/01.WNL.0000141857.37073.97
Massa, R., Tessa, A., Margollicci, M., Micheli, V., Romigi, A., Tozzi, G., et al. (2009). Late-onset MNGIE without peripheral neuropathy due to incomplete loss of thymidine phosphorylase activity. Neuromuscul. Disord. 19, 837–840. doi: 10.1016/j.nmd.2009.08.013
Mattman, A., Sirrs, S., Mezei, M. M., Salvarinova-Zivkovic, R., Alfadhel, M., and Lillquist, Y. (2011). Mitochondrial disease clinical manifestations: an overview. B. C. Med. J. 53, 183–187.
McHugh, D., Cameron, C. A., Abdenur, J. E., Abdulrahman, M., Adair, O., Al Nuaimi, S. A., et al. (2011). Clinical validation of cutoff target ranges in newborn screening of metabolic disorders by tandem mass spectrometry: a worldwide collaborative project. Genet. Med. 13, 230–254. doi: 10.1097/GIM.0b013e31820d5e67
Menezes, M. P., and Ouvrier, R. A. (2012). Peripheral neuropathy associated with mitochondrial disease in children. Dev. Med. Child Neurol. 54, 407–414. doi: 10.1111/j.1469-8749.2012.04271.x
Millar, W. S., Lignelli, A., and Hirano, M. (2004). MRI of five patients with mitochondrial neurogastrointestinal encephalomyopathy. AJR Am. J. Roentgenol. 182, 1537–1541. doi: 10.2214/ajr.182.6.1821537.
Mingozzi, F., and High, K. A. (2013). Immune responses to AAV vectors: overcoming barriers to successful gene therapy. Blood 122, 23–36. doi: 10.1182/blood-2013-01-306647
Mitselou, A., Ioachim, E., Skoufi, U., Tsironis, C., Tsimogiannis, K. E., Skoufi, C., et al. (2012). Predictive role of thymidine phosphorylase expression in patients with colorectal cancer and its association with angiogenesis-related proteins and extracellular matrix components. In Vivo 26, 1057–1067.
Modlich, U., Navarro, S., Zychlinski, D., Maetzig, T., Knoess, S., Brugman, M. H., et al. (2009). Insertional transformation of hematopoietic cells by self-inactivating lentiviral and gammaretroviral vectors. Mol. Ther. 17, 1919–1928. doi: 10.1038/mt.2009.179.
Montini, E., Cesana, D., Schmidt, M., Sanvito, F., Ponzoni, M., Bartholomae, C., et al. (2006). Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nat. Biotechnol. 24, 687–696. doi: 10.1038/nbt1216.
Moran, N. F., Bain, M. D., Muqit, M. M., and Bax, B. E. (2008). Carrier erythrocyte entrapped thymidine phosphorylase therapy for MNGIE. Neurology 71, 686–688. doi: 10.1212/01.wnl.0000324602.97205.ab
Mori, K., Hasegawa, M., Nishida, M., Toma, H., Fukuda, M., Kubota, T., et al. (2000). Expression levels of thymidine phosphorylase and dihydropyrimidine dehydrogenase in various human tumor tissues. Int. J. Oncol. 17, 33–38. doi: 10.1212/01.wnl.0000324602.97205.ab
Nakai, H., Fuess, S., Storm, T. A., Muramatsu, S., Nara, Y., and Kay, M. A. (2005). Unrestricted hepatocyte transduction with adeno-associated virus serotype 8 vectors in mice. J. Virol. 79, 214–224. doi: 10.1128/JVI.79.1.214-224.2005
Nakayama, Y., Inoue, Y., Nagashima, N., Katsuki, T., Matsumoto, K., Kadowaki, K., et al. (2005). Expression levels of thymidine phosphorylase (TP) and dihydropyrimidine dehydrogenase (DPD) in patients with gastrointestinal cancer. Anticancer Res. 25, 3755–3761.
Naldini, L. (2015). Gene therapy returns to centre stage. Nature 526, 351–360. doi: 10.1038/nature15818
Naldini, L., Blomer, U., Gallay, P., Ory, D., Mulligan, R., Gage, F.H., et al. (1996). In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272, 263–267. doi: 10.1126/science.272.5259.263
Nathwani, A. C., Gray, J. T., Ng, C. Y., Zhou, J., Spence, Y., Waddington, S. N., et al. (2006). Self-complementary adeno-associated virus vectors containing a novel liver-specific human factor IX expression cassette enable highly efficient transduction of murine and nonhuman primate liver. Blood 107, 2653–2661. doi: 10.1182/blood-2005-10-4035
Nathwani, A. C., Reiss, U. M., Tuddenham, E. G., Rosales, C., Chowdary, P., McIntosh, J., et al. (2014). Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N. Engl. J. Med. 371, 1994–2004. doi: 10.1056/NEJMoa1407309.
Negre, O., Bartholomae, C., Beuzard, Y., Cavazzana, M., Christiansen, L., Courne, C., et al. (2015). Preclinical evaluation of efficacy and safety of an improved lentiviral vector for the treatment of beta-thalassemia and sickle cell disease. Curr. Gene Ther. 15, 64–81. doi: 10.2174/1566523214666141127095336
Nishigaki, Y., Marti, R., Copeland, W. C., and Hirano, M. (2003). Site-specific somatic mitochondrial DNA point mutations in patients with thymidine phosphorylase deficiency. J. Clin. Invest. 111, 1913–1921. doi: 10.1172/JCI17828
Nishino, I., Spinazzola, A., and Hirano, M. (1999). Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science 283, 689–692. doi: 10.1126/science.283.5402.689
Nishino, I., Spinazzola, A., Papadimitriou, A., Hammans, S., Steiner, I., Hahn, C. D., et al. (2000). Mitochondrial neurogastrointestinal encephalomyopathy: an autosomal recessive disorder due to thymidine phosphorylase mutations. Ann. Neurol. 47, 792–800. doi: 10.1002/1531-8249(200006)47:6<792::AID-ANA12>3.0.CO;2-Y
Okamura, K., Santa, T., Nagae, K., and Omae, T. (1976). Congenital oculoskeletal myopathy with abnormal muscle and liver mitochondria. J. Neurol. Sci. 27, 79–91. doi: 10.1016/0022-510X(76)90236-7
Oliver, F. J., Collins, M. K., and Lopez-Rivas, A. (1996). dNTP pools imbalance as a signal to initiate apoptosis. Experientia 52, 995–1000. doi: 10.1007/BF01920108
Ott, M. G., Schmidt, M., Schwarzwaelder, K., Stein, S., Siler, U., Koehl, U., et al. (2006). Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat. Med. 12, 401–409. doi: 10.1038/nm1393.
Palchaudhuri, R., Saez, B., Hoggatt, J., Schajnovitz, A., Sykes, D. B., Tate, T. A., et al. (2016). Non-genotoxic conditioning for hematopoietic stem cell transplantation using a hematopoietic-cell-specific internalizing immunotoxin. Nat. Biotechnol. 34, 738–745. doi: 10.1038/nbt.3584
Papadimitriou, A., Comi, G. P., Hadjigeorgiou, G. M., Bordoni, A., Sciacco, M., Napoli, L., et al. (1998). Partial depletion and multiple deletions of muscle mtDNA in familial MNGIE syndrome. Neurology 51, 1086–1092. doi: 10.1212/WNL.51.4.1086
Perez-Atayde, A. R., Fox, V., Teitelbaum, J. E., Anthony, D. A., Fadic, R., Kalsner, L., et al. (1998). Mitochondrial neurogastrointestinal encephalomyopathy: diagnosis by rectal biopsy. Am. J. Surg. Pathol. 22, 1141–1147. doi: 10.1097/00000478-199809000-00014
Pfeffer, G., Horvath, R., Klopstock, T., Mootha, V. K., Suomalainen, A., Koene, S., et al. (2013). New treatments for mitochondrial disease-no time to drop our standards. Nat. Rev. Neurol. 9, 474–481. doi: 10.1038/nrneurol.2013.129
Pontarin, G., Ferraro, P., Valentino, M. L., Hirano, M., Reichard, P., and Bianchi, V. (2006). Mitochondrial DNA depletion and thymidine phosphate pool dynamics in a cellular model of mitochondrial neurogastrointestinal encephalomyopathy. J. Biol. Chem. 281, 22720–22728. doi: 10.1074/jbc.M604498200
Psatha, N., Karponi, G., and Yannaki, E. (2016). Optimizing autologous cell grafts to improve stem cell gene therapy. Exp. Hematol. 44, 528–539. doi: 10.1016/j.exphem.2016.04.007
Pupe, C., Nascimento, O. J., Quintanilha, G., Freitas, M. R., Uchoa, E., Matta, A. P., et al. (2012). Mitochondrial neurogastrointestinal encephalomyopathy mimicking chronic inflammatory demyelinating polyradiculoneuropathy. Arq. Neuropsiquiatr. 70, 228–229. doi: 10.1590/S0004-282X2012005000002
Rickards, H., Prendergast, M., and Booth, I. W. (1994). Psychiatric presentation of Crohn’s disease. Diagnostic delay and increased morbidity. Br. J. Psychiatry 164, 256–261. doi: 10.1192/bjp.164.2.256
Romero, Z., Urbinati, F., Geiger, S., Cooper, A. R., Wherley, J., Kaufman, M. L., et al. (2013). beta-globin gene transfer to human bone marrow for sickle cell disease. J. Clin. Invest. doi: 10.1172/jci67930 [Epub ahead of print].
Salsano, E., Farina, L., Lamperti, C., Piscosquito, G., Salerno, F., Morandi, L., et al. (2013). Adult-onset leukodystrophies from respiratory chain disorders: do they exist? J. Neurol. 260, 1617–1623. doi: 10.1007/s00415-013-6844-z
Scaglia, F., Wong, L. J., Vladutiu, G. D., and Hunter, J. V. (2005). Predominant cerebellar volume loss as a neuroradiologic feature of pediatric respiratory chain defects. AJNR Am. J. Neuroradiol. 26, 1675–1680.
Scarpelli, M., Ricciardi, G. K., Beltramello, A., Zocca, I., Calabria, F., Russignan, A., et al. (2013). The role of brain MRI in mitochondrial neurogastrointestinal encephalomyopathy. Neuroradiol. J. 26, 520–530. doi: 10.1177/197140091302600505
Schiffmann, R., and van der Knaap, M. S. (2009). Invited article: an MRI-based approach to the diagnosis of white matter disorders. Neurology 72, 750–759. doi: 10.1212/01.wnl.0000343049.00540.c8
Schupbach, W. M., Vadday, K. M., Schaller, A., Brekenfeld, C., Kappeler, L., Benoist, J. F., et al. (2007). Mitochondrial neurogastrointestinal encephalomyopathy in three siblings: clinical, genetic and neuroradiological features. J. Neurol. 254, 146–153. doi: 10.1007/s00415-006-0255-3
Shaw, T., Smillie, R. H., Miller, A. E., and MacPhee, D. G. (1988). The role of blood platelets in nucleoside metabolism: regulation of platelet thymidine phosphorylase. Mutat. Res. 200, 117–131. doi: 10.1016/0027-5107(88)90075-9
Simon, L. T., Horoupian, D. S., Dorfman, L. J., Marks, M., Herrick, M. K., Wasserstein, P., et al. (1990). Polyneuropathy, ophthalmoplegia, leukoencephalopathy, and intestinal pseudo-obstruction: POLIP syndrome. Ann. Neurol. 28, 349–360. doi: 10.1002/ana.410280308
Song, S., Wheeler, L. J., and Mathews, C. K. (2003). Deoxyribonucleotide pool imbalance stimulates deletions in HeLa cell mitochondrial DNA. J. Biol. Chem. 278, 43893–43896. doi: 10.1074/jbc.C300401200
Spinazzola, A., Marti, R., Nishino, I., Andreu, A. L., Naini, A., Tadesse, S., et al. (2002). Altered thymidine metabolism due to defects of thymidine phosphorylase. J. Biol. Chem. 277, 4128–4133. doi: 10.1074/jbc.M111028200
Stein, S., Ott, M. G., Schultze-Strasser, S., Jauch, A., Burwinkel, B., Kinner, A., et al. (2010). Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat. Med. 16, 198–204. doi: 10.1038/nm.2088
Stenman, G., Sahlin, P., Dumanski, J. P., Hagiwara, K., Ishikawa, F., Miyazono, K., et al. (1992). Regional localization of the human platelet-derived endothelial cell growth factor (ECGF1) gene to chromosome 22q13. Cytogenet. Cell Genet. 59, 22–23. doi: 10.1159/000133191
Stenson, P. D., Mort, M., Ball, E. V., Shaw, K., Phillips, A., and Cooper, D. N. (2014). The human gene mutation database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum. Genet. 133, 1–9. doi: 10.1007/s00439-013-1358-4
Szigeti, K., Sule, N., Adesina, A. M., Armstrong, D. L., Saifi, G. M., Bonilla, E., et al. (2004a). Increased blood-brain barrier permeability with thymidine phosphorylase deficiency. Ann. Neurol. 56, 881–886. doi: 10.1002/ana.20302.
Szigeti, K., Wong, L. J., Perng, C. L., Saifi, G. M., Eldin, K., Adesina, A. M., et al. (2004b). MNGIE with lack of skeletal muscle involvement and a novel TP splice site mutation. J. Med. Genet. 41, 125–129.
Taivassalo, T., De Stefano, N., Argov, Z., Matthews, P. M., Chen, J., Genge, A., et al. (1998). Effects of aerobic training in patients with mitochondrial myopathies. Neurology 50, 1055–1060. doi: 10.1212/WNL.50.4.1055
Takebayashi, Y., Akiyama, S., Akiba, S., Yamada, K., Miyadera, K., Sumizawa, T., et al. (1996). Clinicopathologic and prognostic significance of an angiogenic factor, thymidine phosphorylase, in human colorectal carcinoma. J. Natl. Cancer Inst. 88, 1110–1117. doi: 10.1093/jnci/88.16.1110
Tanaka, J., Nagai, T., Arai, H., Inui, K., Yamanouchi, H., Goto, Y., et al. (1997). Treatment of mitochondrial encephalomyopathy with a combination of cytochrome C and vitamins B1 and B2. Brain Dev. 19, 262–267. doi: 10.1016/S0387-7604(97)00573-1
Teitelbaum, J. E., Berde, C. B., Nurko, S., Buonomo, C., Perez-Atayde, A. R., and Fox, V. L. (2002). Diagnosis and management of MNGIE syndrome in children: case report and review of the literature. J. Pediatr. Gastroenterol. Nutr. 35, 377–383. doi: 10.1097/00005176-200209000-00029
Threlkeld, A. B., Miller, N. R., Golnik, K. C., Griffin, J. W., Kuncl, R. W., Johns, D. R., et al. (1992). Ophthalmic involvement in myo-neuro-gastrointestinal encephalopathy syndrome. Am. J. Ophthalmol. 114, 322–328. doi: 10.1016/S0002-9394(14)71799-4
Torres-Torronteras, J., Cabrera-Perez, R., Barba, I., Costa, C., de Luna, N., Andreu, A. L., et al. (2016). Long-term restoration of thymidine phosphorylase function and nucleoside homeostasis using hematopoietic gene therapy in a murine model of mitochondrial neurogastrointestinal encephalomyopathy. Hum. Gene Ther. 27, 656–667. doi: 10.1089/hum.2015.160
Torres-Torronteras, J., Gomez, A., Eixarch, H., Palenzuela, L., Pizzorno, G., Hirano, M., et al. (2011). Hematopoietic gene therapy restores thymidine phosphorylase activity in a cell culture and a murine model of MNGIE. Gene Ther. 18, 795–806. doi: 10.1038/gt.2011.24
Torres-Torronteras, J., Viscomi, C., Cabrera-Perez, R., Camara, Y., Di Meo, I., Barquinero, J., et al. (2014). Gene therapy using a liver-targeted AAV vector restores nucleoside and nucleotide homeostasis in a murine model of MNGIE. Mol. Ther. 22, 901–907. doi: 10.1038/mt.2014.6
Valentino, M. L., Marti, R., Tadesse, S., Lopez, L. C., Manes, J. L., Lyzak, J., et al. (2007). Thymidine and deoxyuridine accumulate in tissues of patients with mitochondrial neurogastrointestinal encephalomyopathy (MNGIE). FEBS Lett. 581, 3410–3414. doi: 10.1016/j.febslet.2007.06.042
Van Goethem, G., Dermaut, B., Lofgren, A., Martin, J. J., and Van Broeckhoven, C. (2001). Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat. Genet. 28, 211–212. doi: 10.1038/90034.
Vinciguerra, C., Federighi, P., Rosini, F., Pretegiani, E., Cardaioli, E., Dotti, M. T., et al. (2015). Eye movement changes in mitochondrial neurogastrointestinal encephalomyopathy (MNGIE). J. Neurol. Sci. 350, 107–109. doi: 10.1016/j.jns.2015.01.031
Visigalli, I., Delai, S., Ferro, F., Cecere, F., Vezzoli, M., Sanvito, F., et al. (2016). Preclinical testing of the safety and tolerability of LV-mediated above normal alpha-L-iduronidase expression in murine and human hematopoietic cells using toxicology and biodistribution GLP studies. Hum. Gene Ther. doi: 10.1089/hum.2016.068 [Epub ahead of print].
Vissing, J., Ravn, K., Danielsen, E. R., Duno, M., Wibrand, F., Wevers, R. A., et al. (2002). Multiple mtDNA deletions with features of MNGIE. Neurology 59, 926–929.
Wagemaker, G. (2014). Lentiviral hematopoietic stem cell gene therapy in inherited metabolic disorders. Hum. Gene Ther. 25, 862–865. doi: 10.1089/hum.2014.102.
Wang, J., Chen, W., Wang, F., Wu, D., Qian, J., Kang, J., et al. (2015). Nutrition therapy for mitochondrial neurogastrointestinal encephalopathy with homozygous mutation of the TYMP Gene. Clin. Nutr. Res. 4, 132–136. doi: 10.7762/cnr.2015.4.2.132
Watts, K. L., Adair, J., and Kiem, H. P. (2011). Hematopoietic stem cell expansion and gene therapy. Cytotherapy 13, 1164–1171. doi: 10.3109/14653249.2011.620748
Wilcox, W. R., Banikazemi, M., Guffon, N., Waldek, S., Lee, P., Linthorst, G. E., et al. (2004). Long-term safety and efficacy of enzyme replacement therapy for Fabry disease. Am. J. Hum. Genet. 75, 65–74. doi: 10.1086/422366.
Xue, X., Pech, N. K., Shelley, W. C., Srour, E. F., Yoder, M. C., and Dinauer, M. C. (2010). Antibody targeting KIT as pretransplantation conditioning in immunocompetent mice. Blood 116, 5419–5422. doi: 10.1182/blood-2010-07-295949
Yadak, R., Torres-Torronteras, J., Bogaerts, E., de Ruijter, G., Barquinero, J., Marti, R., et al. (2015). OP45 – 3024: efficient lentiviral vector-mediated hematopoietic stem cell gene therapy in MNGIE mice. Eur. J. Paediatr. Neurol. 19(Suppl. 1):S15. doi: 10.1016/s1090-3798(15)30046-5
Yasumura, S., Aso, S., Fujisaka, M., and Watanabe, Y. (2003). Cochlear implantation in a patient with mitochondrial encephalopathy, lactic acidosis and stroke-like episodes syndrome. Acta Otolaryngol. 123, 55–58. doi: 10.1080/0036554021000028081
Yavuz, H., Ozel, A., Christensen, M., Christensen, E., Schwartz, M., Elmaci, M., et al. (2007). Treatment of mitochondrial neurogastrointestinal encephalomyopathy with dialysis. Arch. Neurol. 64, 435–438. doi: 10.1001/archneur.64.3.435
Yolcu, M., Yolcu, C., Kaya, Z., Cakmak, E. O., and Sezen, Y. (2014). Endocarditis in Mitochondrial Neurogastrointestinal Encephalomyopathy (MNGIE) Syndrome: the first in the literature. J. Clin. Diagn. Res. 8, SD01–SD02. doi: 10.7860/JCDR/2014/9528.5016
Young, J. D., Yao, S. Y., Baldwin, J. M., Cass, C. E., and Baldwin, S. A. (2013). The human concentrative and equilibrative nucleoside transporter families, SLC28 and SLC29. Mol. Aspects Med. 34, 529–547. doi: 10.1016/j.mam.2012.05.007
Young, J. D., Yao, S. Y., Sun, L., Cass, C. E., and Baldwin, S. A. (2008). Human equilibrative nucleoside transporter (ENT) family of nucleoside and nucleobase transporter proteins. Xenobiotica 38, 995–1021. doi: 10.1080/00498250801927427
Young, M. J., and Copeland, W. C. (2016). Human mitochondrial DNA replication machinery and disease. Curr. Opin. Genet. Dev. 38, 52–62. doi: 10.1016/j.gde.2016.03.005
Zhou, S., Ma, Z., Lu, T., Janke, L., Gray, J. T., and Sorrentino, B. P. (2013). Mouse transplant models for evaluating the oncogenic risk of a self-inactivating XSCID lentiviral vector. PLoS ONE 8:e62333. doi: 10.1371/journal.pone.0062333.
Zimmer, V., Feiden, W., Becker, G., Zimmer, A., Reith, W., Raedle, J., et al. (2009). Absence of the interstitial cell of Cajal network in mitochondrial neurogastrointestinal encephalomyopathy. Neurogastroenterol. Motil. 21, 627–631. doi: 10.1111/j.1365-2982.2009.01264.x
Keywords: mitochondrial neurogastrointestinal encephalomyopathy, MNGIE, thymidine phosphorylase, metabolic disease, HSCT, HSCGT, lentiviral vector
Citation: Yadak R, Sillevis Smitt P, van Gisbergen MW, van Til NP and de Coo IFM (2017) Mitochondrial Neurogastrointestinal Encephalomyopathy Caused by Thymidine Phosphorylase Enzyme Deficiency: From Pathogenesis to Emerging Therapeutic Options. Front. Cell. Neurosci. 11:31. doi: 10.3389/fncel.2017.00031
Received: 23 December 2016; Accepted: 01 February 2017;
Published: 15 February 2017.
Edited by:
Fabio Blandini, Fondazione Istituto Neurologico Nazionale Casimiro Mondino (IRCCS), ItalyReviewed by:
Robert Weissert, University of Regensburg, GermanyValeria Tiranti, Istituto Neurologico Carlo Besta (IRCCS), Italy
Copyright © 2017 Yadak, Sillevis Smitt, van Gisbergen, van Til and de Coo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rana Yadak, cmFuYS55YWRha0BnbWFpbC5jb20= Irenaeus F. M. de Coo, aS5kZWNvb0BlcmFzbXVzbWMubmw=