 Juhyun Song
Juhyun Song Seong-Min Choi
Seong-Min Choi Byeong C. Kim
Byeong C. Kim- 1Department of Biomedical Sciences, Center for Creative Biomedical Scientists at Chonnam National University, Gwangju, South Korea
- 2Department of Neurology, Chonnam National University Medical School, Gwangju, South Korea
Alzheimer’s disease (AD), characterized by the abnormal accumulation of amyloid beta (Aβ), is gradually increasing globally. Given that AD is considered a neuroinflammatory disease, recent studies have focused on the cellular mechanisms in brain inflammatory conditions that underlie AD neuropathology. Microglia are macrophage cells in the central nervous system (CNS) that are activated in response to Aβ condition. The function of microglia contributes to the neuroinflammation in AD brain, suggesting that microglia regulate the production of inflammatory mediators and contribute to the regeneration of damaged tissues. Adiponectin, an adipokine derived from adipose tissue, has been known to regulate inflammation and control macrophages during oxidative stress conditions. In present study, we investigated whether adiponectin influences the polarization and function of microglia under Aβ toxicity by examining alterations of BV2 microglia function and polarization by Acrp30 (a globular form of adiponectin) treatment using reverse transcription PCR, western blotting and immunofluorescence staining. Acrp30 promoted the induction of the M2 phenotype, and regulated the inflammatory responses through peroxisome proliferator-activated receptor (PPAR)-γ signaling under Aβ toxicity. In addition, Acrp30 boosted the capacity of Aβ scavenging in microglia. Taken together, we suggest that adiponectin may control the function of microglia by promoting anti-inflammatory responses through PPAR- γ signaling. Hence, we conclude that adiponectin may act as a critical controller of microglia function in the AD brain.
Introduction
Alzheimer’s disease (AD) is the most common cause of dementia, manifesting as memory decline and cognitive dysfunction (Bubu et al., 2016; De Strooper and Karran, 2016; Tarantini et al., 2016). AD is defined by two pathological characteristics: amyloid β-protein (Aβ) deposition in senile plaques and phosphorylated tau in neurofibrillary tangles (Goate, 2006). Excessive accumulation of Aβ has been shown to lead to the cognitive impairments in AD (Hardy and Selkoe, 2002; Hardy et al., 2014), and has been shown to trigger inflammation (Del Bo et al., 1995; Combs et al., 2001; Takata et al., 2003; Lindberg et al., 2005; Kempuraj et al., 2016; Stamouli and Politis, 2016) through the production of cytokines and other inflammatory mediators. Several studies of AD brains reported the presence of activated glial cells around the Aβ plaques (Mehlhorn et al., 2000; Rogers and Lue, 2001); (uroff, 2017 #365). Microglia, the resident macrophages of the central nervous system (CNS; Nakamichi et al., 2013; Ginhoux and Prinz, 2015), constitute 10%–15% of brain cells and play a critical role in CNS homeostasis (Solé-Domènech et al., 2016). Previous studies demonstrated that microglia polarizes into two states, M1 like phenotype and M2 like phenotypes (Francos-Quijorna et al., 2016; Lee et al., 2016). M1 like phenotype microglia is related with pro inflammatory response’s induction, whereas M2 like phenotype microglia is associated with neuroprotective roles (Eggen et al., 2013; Gertig and Hanisch, 2014; Natoli and Monticelli, 2014; Plastira et al., 2016; Xu et al., 2016). The polarization of microglia is important in CNS homeostasis as it has been shown to affect learning and memory (Tremblay et al., 2011, 2015; Parkhurst et al., 2013). Numerous studies have suggested that microglia are activated in the AD brain and contribute to the prevention of Aβ formation through phagocytosis of Aβ (Rogers et al., 2002; Doens and Fernández, 2014). Several studies have demonstrated that M2 type microglia reduce inflammation in the brain (Weekman et al., 2014; Latta et al., 2015) and ameliorate cognitive dysfunction during Aβ toxicity (Zhu et al., 2016). These data highlight the necessity to elucidate the mechanisms underlying Aβ clearance by microglia, thereby attenuating AD progression. Adiponectin is encoded by the ADIPOQ gene (Maeda et al., 1996) and exerts multiple effects by binding to two specific receptors, AdipoR1 and AdipoR2 (Yamauchi et al., 2003; Hug et al., 2004). Adiponectin receptors exist in various organs, including the liver and brain (Kim et al., 2013; Kaminski et al., 2014). Previous researches have demonstrated that adiponectin acts as an insulin sensitizer, an anti-inflammatory regulator and an anti-atherosclerotic molecule (Kadowaki et al., 2006; Renaldi et al., 2009). Recent research has focused on the effect of adiponectin on macrophages (Elfeky et al., 2016; Masamoto et al., 2016; Wang et al., 2016); specifically, that adiponectin may not only act as a key regulator of inflammatory response in metabolic diseases, but also in CNS diseases (Wang et al., 2016). In the present study, we investigate whether adiponectin contributes to the polarization and the scavenger capacity of microglia under Aβ toxicity. The results provide significant evidence for the therapeutic potential of adiponectin to mitigate AD pathology by regulating the function of microglia.
Materials and Methods
Animal Experiments
5xFAD male transgenic mice (5 months of age) with detectable Aβ42 production in the brain were purchased from The Jackson Laboratory (Bar Harbor, ME). Wild-type male C57BL/6 mice (25 g, 5 months of age) were provided by Koatech (Koatech, Pyeongtaek, South Korea). In the present study, we used four mice in each groups. This study was carried out in accordance with the recommendations of “96 Guidance for Animal Experiments”, established by the “Animal Ethics Committee” at Chonnam National University. The protocol was approved by the “Animal Ethics Committee” at Chonnam National University, approved study number 2016-84. The animals were housed in an air-conditioned room at 25°C with a 12 h dark/light cycle. All animals received human care with unlimited access to chow and water.
Cell Culture and Drug Treatment
Murine BV2 microglial cells were cultured in Dulbecco Modified Eagle Medium (DMEM; Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (Gibco), 100 μg/ml penicillin (Gibco) and 100 μg/ml streptomycin (Gibco). BV2 cells were grown in a humidified incubator at 37°C with 5% CO2. The cells were pretreated with Acrp30 (5 μg/ml; Sigma-Aldrich, St. Louis, MO, USA) for 24 h and then treated with Aβ (10 μM) for 24 h. GW9662 was used as the antagonist for PPAR-γ (Chen et al., 2016). BV2 cells were pretreated with GW9662 (10 μM) for 3 h.
Aβ Oligomer Preparation
Oligomeric Aβ was prepared as previously described (An et al., 2013). One milligram of synthetic Aβ 42 peptide (American Peptide, Sunnyvale, CA, USA) was dissolved into 1 ml HFIP (1,1,1,3,3,3-hexafluoro-2-propanol). The solution should then be dried under a nitrogen stream. Re-dissolved the remaining film in 100% HFIP to a concentration of 1 mg/ml. Sonicated in bath sonicator for 5 min and dried under a nitrogen stream. Repeated the HFIP treatment twice. Then re-dissolved in 1 ml HFIP and dried for 2 h. After that the film was resuspended in 200 ul DMSO to obtain a 1 mM Aβ stock solution. For 10 μM Aβ 42 treatment, 10 ul of Aβ 42 1 mM was dilute into 10 ml DMEM cell culture media and incubated for 12 h at 37°C.
Preparation of the AdipoR1 and AdipoR2 Targeting siRNA
The siRNA for AdipoR1 and AdipoR2 (5 μM) was prepared to silence AdipoR1 siRNA sc-60123 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and AdipoR2 siRNA sc-46756 (Santa Cruz Biotechnology). For the transfection of siRNA, a 5 μM final concentration of siRNA AdipoR1 and siRNA AdipoR2 were combined with lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) in Opti-MEM medium, and incubated at room temperature for 20 min. The mixture was added to the BV2 cells in six well plates. After 2 days, the cells were collected for total protein or RNA extraction.
Determination of Nitrite
BV2 microglia were seeded onto 96-well plates and pre-treated with Acrp30 (5 μg/ml) for 24 h prior to stimulation with 10 μM of Aβ. Supernatants in all groups were measured by nitric oxide (NO) production using Griess reagent (Sigma-Aldrich). The reagents (100 μl) were added to the plate and incubated for 30 min. The absorbance of supernatants was measured at 540 nm using an ELISA reader (Bio-Rad, Irvine, CA, USA).
ELISA Assay
BV2 cells were plated in 6-well plates (5 × 105 cells/ml) and incubated with Acrp30 (5 μg/ml) in the presence of Aβ 10 μM for 24 h. The production of TNF-α was checked by Cymax Mouse TNF-α ELISA kit (AbFrontier, Seoul, Korea) per manufacturer’s instructions. The absorbance at 450 nm was assessed by ELISA (Bio-Rad).
Phagocytosis Assay
The phagocytosis assay was conducted using the Cayman chemical phagocytosis kit (Cayman Chemical, Ann Arbor, MI, USA). To check opsonization of target particles, all phagocytosis assays were conducted in complete DMEM media. Additionally, in some experiments, labeled particles were preopsonized by incubation at 37°C for 3 h with 5 mg/mL immunoglobulin G (IgG; Beletskii et al., 2005; Cayman Chemical).
Western Blot Analysis
The BV2 cells were washed with phosphate-buffered saline (PBS) and collected. Cell pellets were lysed with ice-cold RIPA buffer (Sigma-Aldrich). The lysates were centrifuged at 15,900 g for 30 min at 4°C to produce whole-cell extracts. Protein (30 μg) in cells was separated on a 10% SDS-polyacrylamide gel and transferred onto a polyvinylidene difluoride membrane. After blocking with 5% skim milk prepared in Tris-buffered saline-Tween (20 nM Tris [pH 7.2], 150 mM NaCl, 0.1% Tween 20) for 1 h, 30 min at room temperature, immunoblots were incubated for 18 h at 4°C with primary antibodies that detect AdipoR1 (1:1000, Abcam, Cambridge, MA, USA), AdipoR2 (1:1000, Abcam, Cambridge, MA, USA), CD86 (1:1000, Cell Signaling, Danvers, MA, USA), p-NF-κB (1:1000, Cell Signaling), PPAR-γ (1:1000, Cell Signaling, Danvers, MA), p-PPAR-γ (1:1000, Santa Cruz Biotechnology, Santa Cruz, CA, USA) and β-actin (1:1000; Millipore, Billerica, MA, USA). All blots were then incubated with appropriate secondary antibodies (Abcam, Cambridge, MA, USA) for 1 h 30 min at room temperature. All blots were visualized using ECL solution (Millipore, Billerica, MA, USA).
Quantitative Real-Time PCR
To examine the amount of CD86, CD206, iNOS and TNF-α mRNA in BV2 cells, quantitative real-time PCR was performed using each primer. Cellular RNA was extracted from BV2 cells using Trizol reagent (Invitrogen). RNA was mixed with One Step SYBR® Prime Script TM RT-PCR Kit II (Takara, Otsu, Shiga, Japan) and specific primers in a total reaction volume of 50 μl. PCR was performed using the following each primers (5′ to 3′): CD86 (F): GTA TTT TGG CAG GAC CAG GA, (R): GCC GCT TCT TCT TCT TCC AT, CD206 (F): GAG GGA AGC GAG AGA TTA TGG A, (R): GCC TGA TGC CAG GTT AAA GCA, iNOS (F): GGG AAT CTT GGA GCG AGT TG, (R): GTG AGG GCT TGG CTG AGT GA, TNF-α (F): CGT CAG CCG ATT TGC TAT CT, (R): CGG ACT CCG CAA AGT CTA AG, GAPDH (F): GAC AAG CTT CCC GTT CTC AG, (R): GAG TCA ACG GAT TTG GTC GT. Amplification cycles were carried out at 52°C for 5 min, 95°C for 10 s, 95°C for 5 s, 58°C for 35 s and 65°C for 15 s. Quantitative SYBR Green real time PCR was performed with Takara PCR System (Takara, Otsu, Shiga, Japan). GAPDH was used as an internal control. The ΔCt values of treated cells were compared to those of normal, control cells (Popivanova et al., 2008).
Reverse Transcription PCR
Total RNA in BV2 cells was isolated using Trizol Reagent (Gibco). RT-PCR reaction was conducted by using the Invitrogen One step IIITM Reverse Transcription PCR kit (Invitrogen). cDNA synthesis from mRNA and sample normalization were conducted. PCR was performed using the following thermal cycling conditions: 95°C for 10 min;, 40 cycles of denaturing at 95°C for 15 s, annealing at 60°C for 30 s, elongation at 72°C for 30 s, final extension at 72°C for 5 min and holding at 4°C. PCR was performed using the following primers (5′ to 3′): AdipoR1 (F): CCCACCATGCACTTTACTAT, (R) CACCATAGAAGTGGACGAAA, AdipoR2 (F): CAACCTTGCTTCATCTACCT, (R): CTAGCCATAAGCATTAGCCA, CD36 (F): GAG CCA TCT TTG AGC CTT CA, (R): TCA GAT CCG AAC ACA GCG TA, PPAR- γ (F): CAA TCC GAA TTT TTC AAG GGT GCC A, (R): GAG CAC CTT GGC GAA CAG CTG AGA G, iNOS (F): GGG AAT CTT GGA GCG AGT TG, (R): GTG AGG GCT TGG CTG AGT GA, TNF-α (F): CGT CAG CCG ATT TGC TAT CT, (R): CGG ACT CCG CAA AGT CTA AG, GAPDH (F): GAC AAG CTT CCC GTT CTC AG, (R): GAG TCA ACG GAT TTG GTC GT. PCR products were electrophoresed in 1% agarose gels. All samples were normalized with GAPDH.
Immunocytochemistry
BV2 cells were washed with PBS and permeabilized for 20 min with 4% paraformaldehyde (Sigma-Aldrich). The cells were incubated with the primary antibodies overnight at 4°C. The primary antibody anti-rabbit CD86 (1:500, Cell Signaling) as a marker of M1 phenotype microglia (Gong et al., 2016) was used. After incubation, BV2 cells were washed twice with PBS and incubated with specific secondary antibody for 1 h 30 min at room temperature. The cells were counterstained with 1 μg/ml 4′,6-diamidino-2-phenylindole (DAPI, 1:100, Invitrogen) for 10 min at room temperature. Images were obtained using an LSM 520 confocal microscope (Carl Zeiss, Thornwood, NY, USA).
Immunohistochemistry
Brain sections (20 μm) were mounted onto coated glass slides (Thermo Scientific, Waltham, MA, USA), and fixed in cold acetone for 10 min at −20°C. The slides were washed in Tris-buffered saline and incubated with 0.3% H2O2 in methanol. To block nonspecific labeling, sections were incubated in 5% bovine serum albumin (Sigma-Aldrich) and diluted in PBS for 80 min before the addition of primary and secondary antibody. Primary antibodies for anti-rabbit CD86 (1:100, Cell Signaling) and anti-rabbit Iba-1 (1:100, Cell Signaling) were applied to the samples overnight at 4°C, followed by 1 h 30 min incubation with appropriate florescence secondary antibody (1:100, Millipore), and three washes in PBS for 5 min each. After three washes in 0.1% PBS with Tween-20 (PBST), the sections were incubated with secondary antibody for 1 h 30 min in the dark at room temperature. After three washes in PBS, all sections were incubated with 1 μg/ml DAPI (Sigma-Aldrich, St. Louis, MO, USA) and visualized under a LSM 520 confocal microscope (Carl Zeiss).
Statistical Analysis
The data were analyzed by SPSS 18.0 software (IBM Corp., Armonk, NY, USA). All results are expressed as mean ± standard deviation (SD). Statistical significance was determined using one-way analysis of variance (ANOVA) followed by Turkey’s multiple comparison and Student’s t-test. Differences were considered statistically significant at p < 0.05.
Results
The Activation and Polarization of Microglia in AD Mouse Brain
To confirm the activation of microglia in the mouse brain, we detected the protein level of CD86 as a marker of activated microglia using western blot (Figure 1A), and the expression of CD86 and Iba-1 using immunohistochemistry (Figures 1B,C). Our data showed higher activation of microglia (increased CD86 protein level and a greater percentage of CD86 and Iba-1 positive cells) in the AD compared to the control mouse brain (Figures 1A–C). These results suggest that microglia are activated in an AD brain with Aβ accumulation. To assess the polarization of microglia with Aβ treatment and Acrp30 (a globular form of adiponectin) pretreatment, we assessed the mRNA level of CD86 as a marker of M1 phenotype microglia and of CD206 as a marker of M2 phenotype microglia (Figure 2). The mRNA level of CD206 was reduced while the mRNA level of CD86 was markedly increased in Aβ-exposed microglia (Figures 2A,B). Acrp30 reduced the expression of CD86 and increased the expression of CD206 in BV2 microglia (Figures 2A,B). Furthermore, Acrp30 pretreatment reduced CD86 mRNA level and increased CD206 mRNA level in spite of Aβ treatment (Figures 2A,B). Immunostaining data showed a decrease in CD86 expression in Aβ-exposed microglia with Acrp30 pretreatment (Figure 2C).
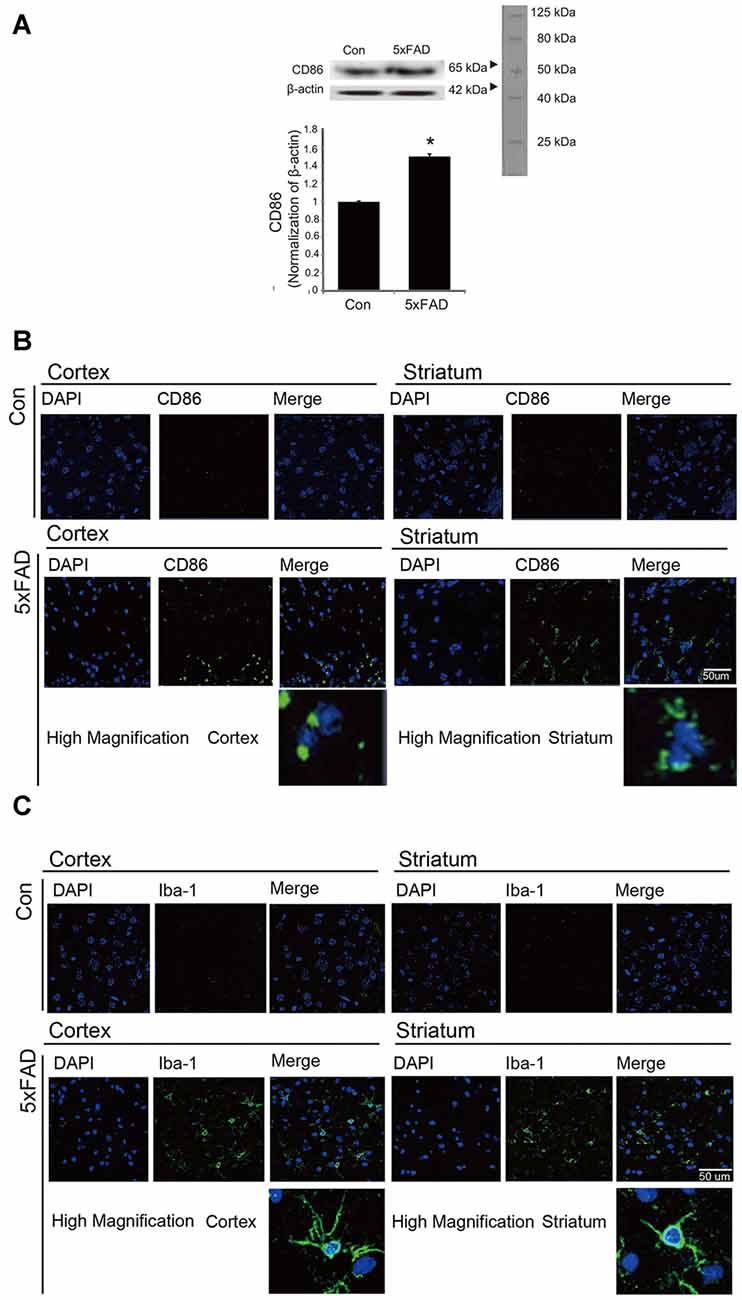
Figure 1. The expression of CD86 and Iba-1 in 5xFAD mouse brain. (A) Western blotting band showed increased CD86 protein level in the 5xFAD mouse brain. Data are expressed as mean ± SEM. Each experiment was conducted three times per condition. β-actin served as a control. Differences were considered statistically significant at *p < 0.05 (compared to control). Immunofluorescence images showed increased expression of CD86 (B) and Iba-1 (C) in the cortex and striatum of the 5xFAD mouse brain. Scale bar: 50 μm, Con: normal mouse, 5xFAD: 5xFAD mouse, CD86: green, Iba-1: green, 4′,6-diamidino-2-phenylindole (DAPI): blue.
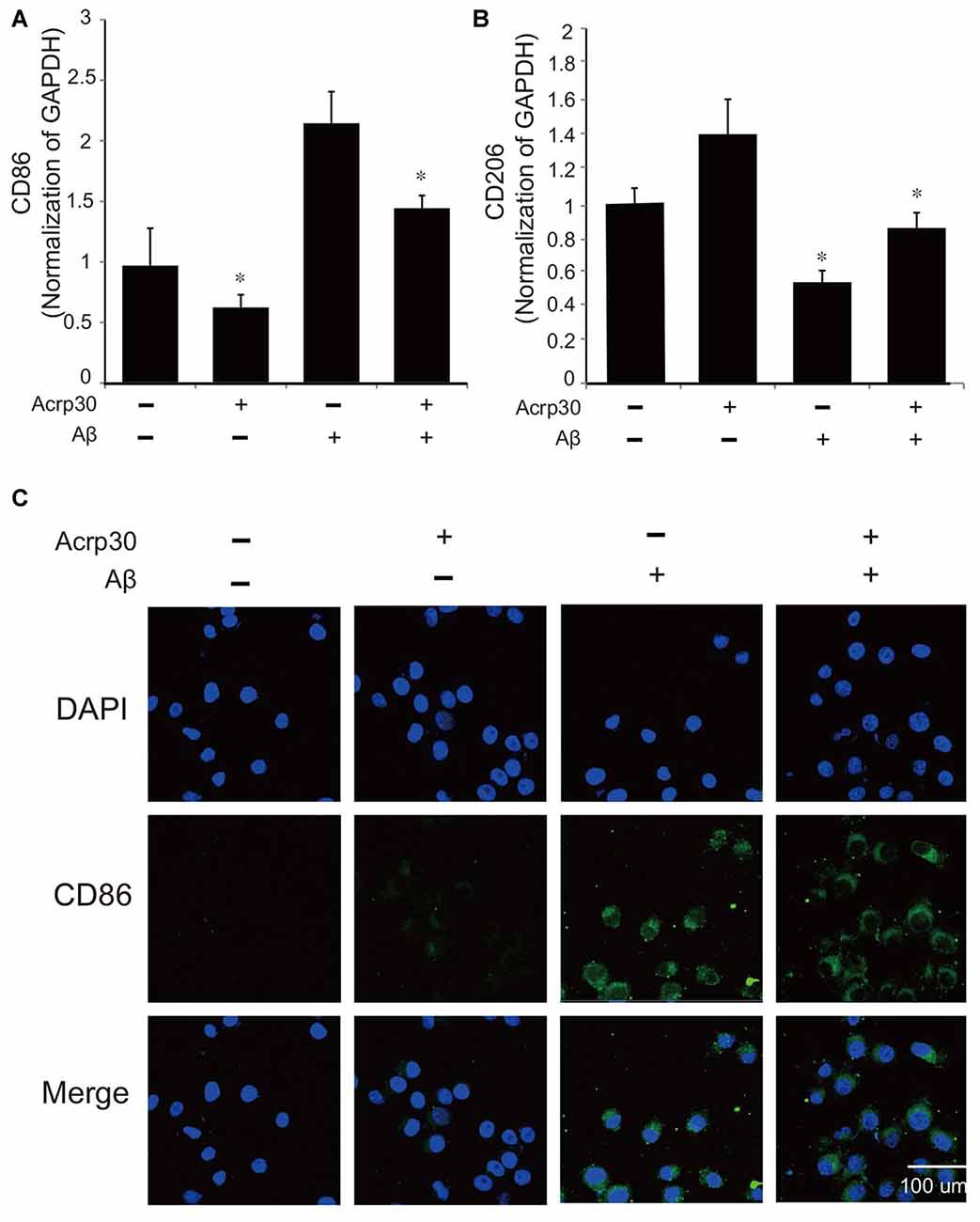
Figure 2. The expression of CD86 and CD206 in amyloid beta (Aβ)-treated BV2 microglia. The mRNA level of CD86 (A) and CD206 (B) was detected by RT PCR. Data are expressed as mean ± SEM. Each experiment was conducted three times per condition. GAPDH served as a control. Differences were considered statistically significant at *p < 0.05 (compared to control). (C) Immunofluorescence images showed the reduction of CD86 by Acrp30 pretreatment in Aβ- exposed BV2 microglia. Acrp30: Acrp30 5 μg/ml pretreatment, Aβ: Aβ 10 μM treatment for 24 h, Scale bar: 100 μm, CD86: green, 4′,6-diamidino-2-phenylindole (DAPI): blue.
The Activation and Polarization of Microglia in AD Mouse Brain
To examine the changes of adiponectin receptors in microglia under Aβ toxicity, we conducted reverse transcriptional PCR (Figures 3A,B) and western blotting (Figures 3C,D). The mRNA levels (Figures 3A,B) and protein levels (Figures 3C,D) of AdipoR1 and AdipoR2 were reduced in Aβ-treated microglia. Acrp30 promoted the expression of AdipoR1, whereas Acrp30 did not meaningfully change the expression of AdipoR2, in Aβ-treated microglia (Figures 3A,B). Notably, the protein level of AdipoR1 in microglia was decreased by Aβ treatment (Figure 3C).
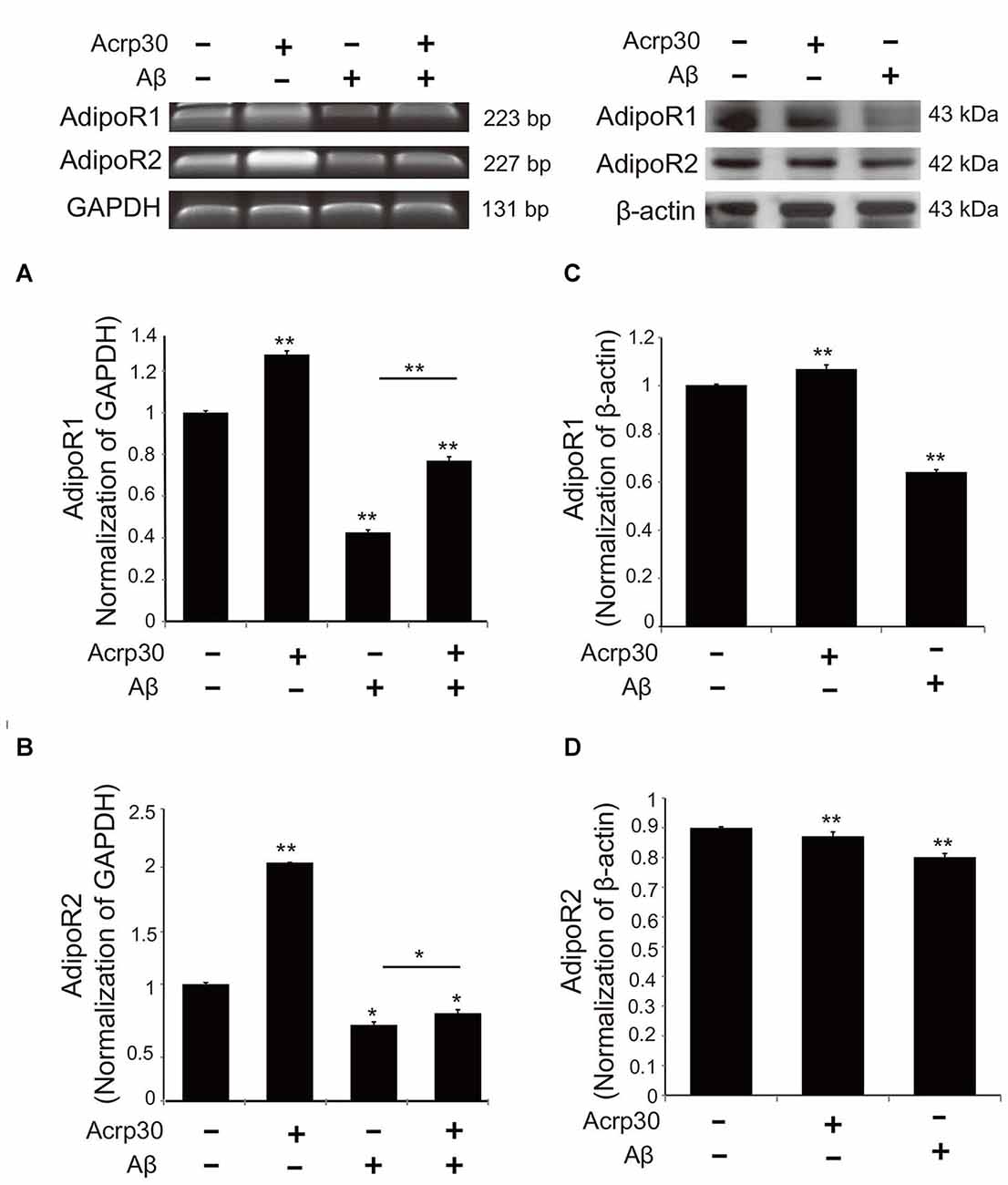
Figure 3. The expression of adiponectin receptors in BV2 microglia. The mRNA levels of AdipoR1 (A) and AdipoR2 (B) were measured by RT-PCR. The protein levels of AdipoR1 (C) and AdipoR2 (D) were detected by western blotting analysis. Data are expressed as mean ± SEM. Each experiment was conducted three times per condition. β- actin served as a control. Differences were considered statistically significant at *p < 0.05, **p < 0.001 (compared to control). Acrp30: Acrp30 5 μg/ml pretreatment, Aβ: Aβ 10 μM treatment for 24 h.
Acrp30 Treatment Leads to the Reduced Expression of Pro-Inflammatory Mediators in Aβ-Exposed Microglia
To examine alterations in the expression of pro-inflammatory mediators by Acrp30, we assessed the production of NO using the Griess assay (Figure 4A) and the mRNA of iNOS (Figure 4B). Moreover, we detected the mRNA level of IL-6 and TNF-α as pro-inflammatory cytokines using real time PCR in microglia (Figures 4C,D). Acrp30 treatment did not change the level of NO, iNOS, IL-6, or TNF-α in BV2 microglia, whereas the expression of these pro-inflammatory mediators were increased in Aβ-treated microglia. Acrp30 pretreatment led to a decrease in NO, iNOS, IL-6 and TNF-α in Aβ-treated microglia (Figures 4A–D).
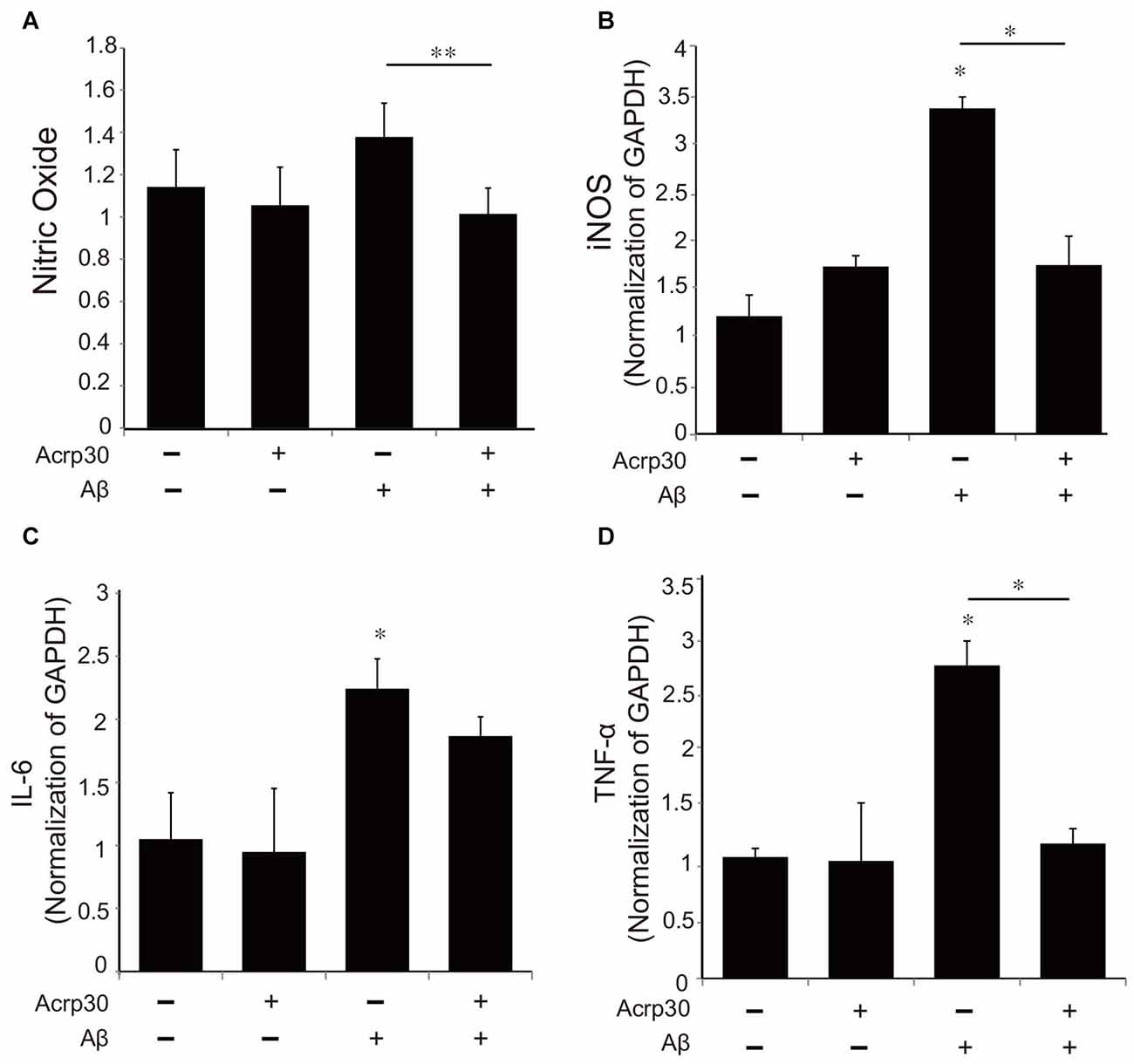
Figure 4. The expression of pro-inflammatory mediators in Aβ-treated BV2 microglia. (A) The production of nitric oxide (NO) was assessed by Griess reagent assay. Acrp30 reduced the production of NO in Aβ-treated microglia. The increased mRNA level of iNOS (B) and IL-6 (C) and TNF-α (D) were measured by RT PCR. Data are expressed as mean ± SEM. Each experiment was conducted four times per condition. GAPDH served as a control. Differences were considered statistically significant at *p < 0.05. **p < 0.001 (compared to control). Acrp30: Acrp30 5 μg/ml pretreatment, Aβ: Aβ 10 μM treatment for 24 h.
Acrp30 Reduced the Expression of TNF-α and iNOS in Aβ-Exposed Microglia
To test for the expression of TNF-α and iNOS in Aβ-exposed microglia, we detected mRNA levels using reverse transcription PCR (Figure 5). The mRNA level of TNF-α in microglia under Aβ toxicity was significantly increased, while the mRNA level of TNF-α in Aβ-treated microglia was considerably reduced by Acrp30 pretreatment (Figure 5A). When we inhibited the expression of AdipoR1 using siRNA AdipoR1, we observed an increase in TNF-α mRNA level in Aβ-exposed microglia in spite of Acrp30 pretreatment (Figure 5A). In addition, the mRNA levels of iNOS and TNF-α showed the same expression patterns (Figure 5B), and the secretion of TNF-α and TNF-α mRNA level demonstrated similar patterns (Figure 5C). The knockdown of AdipoR1 did not reduce the expression of pro-inflammatory mediators (Figure 5).
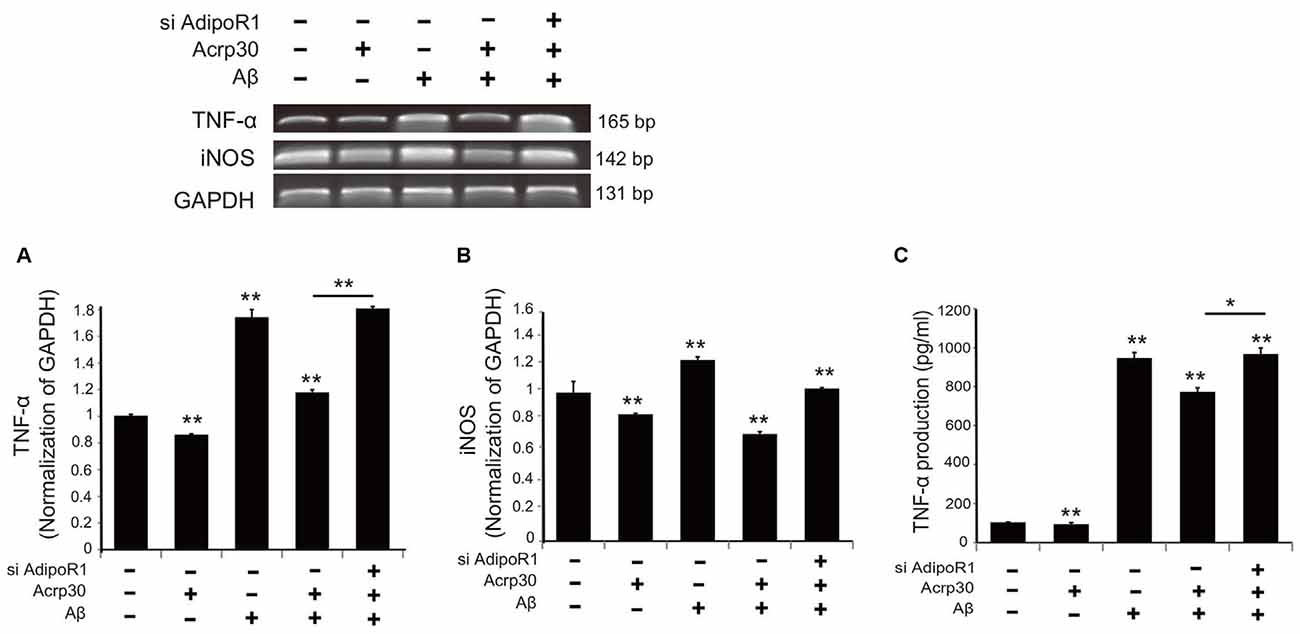
Figure 5. The expression of pro-inflammatory mediators by suppressing AdipoR1 in Aβ-treated BV2 microglia. The reduced mRNA level of TNF-α (A) and iNOS (B) and TNF-α (C) in Aβ-treated microglia were assessed by RT PCR. (C) The reduced production of TNF-α by Acrp30 in Aβ-treated microglia was increased by blocking AdipoR1. Data are expressed as mean ± SEM. Each experiment was conducted four times per condition. GAPDH served as a control. Differences were considered statistically significant at *p < 0.05. **p < 0.001 (compared to control). Acrp30: Acrp30 5 μg/ml pretreatment, Aβ: Aβ 10 μM treatment for 24 h.
Acrp30 Increases the Ability of Phagocytosis in Aβ Exposed Microglia
To examine the phagocytotic ability of microglia, we conducted the phagocytosis assay (Figure 6). Aβ-exposed microglia showed that the green beads were phagocytosed in many microglia. Acrp30 were increased the phagocytosis ability of microglia in Aβ toxicity (Figure 6). Thus, adiponectin promoted the phagocytosis ability of microglia in Aβ toxicity.
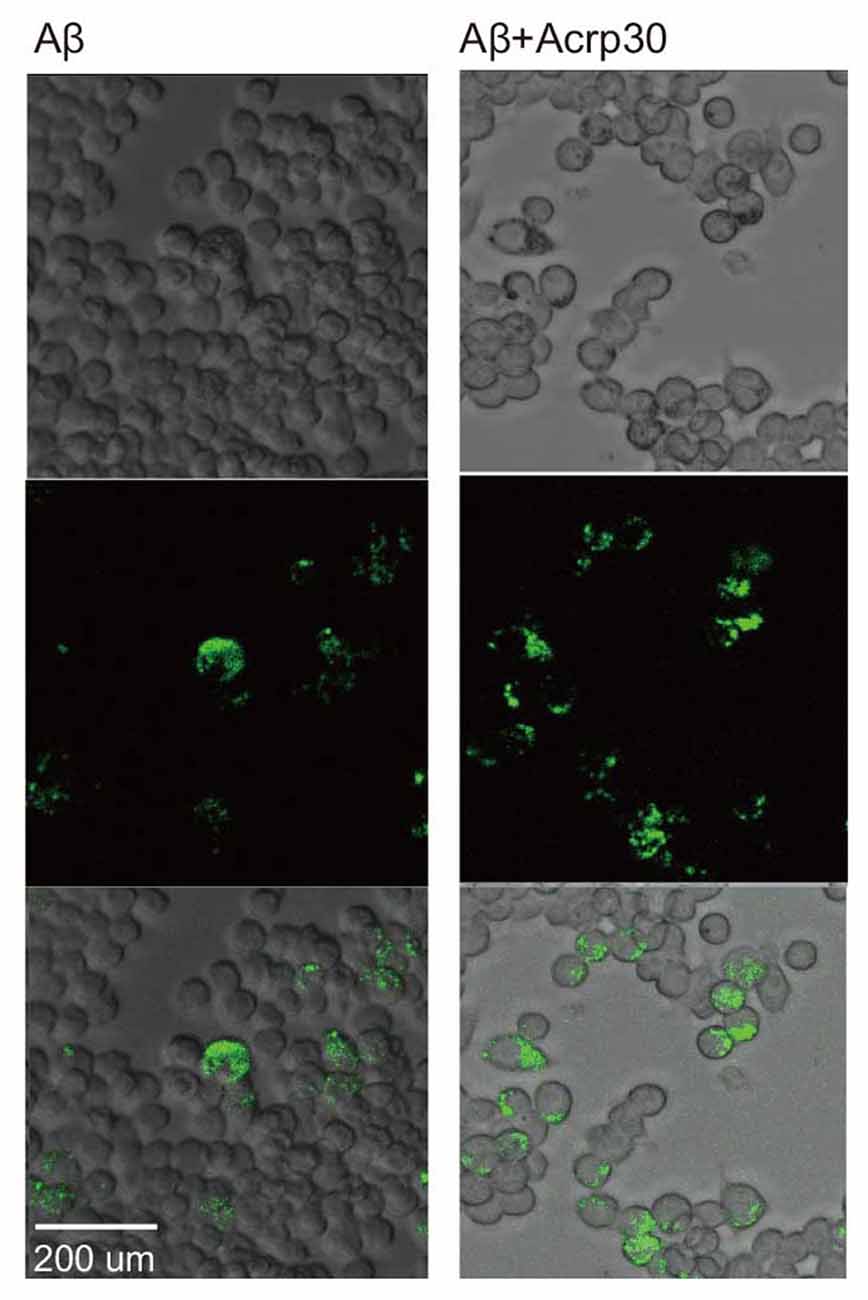
Figure 6. The measurement of phagocytosis ability in Aβ-treated BV2 microglia. Images showing the ability of phagocytosis in BV2 microglia. The green color shows the beads for determining the ability of phagocytosis in Aβ-treated microglia. Scale bar: 200 μm, Bead particles: green.
Acrp30 Controls PPAR-γ Signaling in Aβ Exposed Microglia
To examine whether Acrp30 influences the expression of PPAR-γ in Aβ-treated microglia, we conducted real time PCR (Figure 7). The mRNA level of CD36 (a scavenger receptor for phagocytosis) was markedly increased in Aβ-treated microglia (Figure 7A). Acrp30 attenuated the expression of CD36 in microglia under Aβ toxicity (Figure 7A). The mRNA level of PPAR-γ was increased in Aβ treated microglia, while Acrp30 reduced the expression of PPAR-γ in microglia under Aβ toxicity (Figure 7B). To test whether Acrp30 directly regulates the PPAR-γ signaling in Aβ treated microglia, we examined CD86, p- NF-κB and PPAR-γ protein levels using western blotting analysis (Figure 8) and tested the effect of the PPAR-γ antagonist GW9662 in Aβ-treated microglia (Figure 8). Our results showed that GW9662 reduces the protein level of CD86 in spite of Aβ-treatment (Figure 8A). Reduced CD86 protein level in Aβ treated microglia with GW9662 was increased by inhibiting AdipoR1 (Figure 8A). The blocking of AdipoR1 reduced the function of PPAR-γ (decreased the expression of CD86) in Aβ-treated microglia (Figure 8A). In addition, GW9662 reduced the phosphorylation of NF-κB in spite of Aβ-treatment (Figure 8B). Reduced protein level of p-NF-κB in Aβ-treated microglia with GW9662 was increased by inhibiting AdipoR1 (Figure 8B). The protein level of PPAR-γ in Aβ-treated microglia was reduced by Acrp30 and GW 9662, whereas the protein level of PPAR-γ in Aβ-treated microglia with GW9662 was not changed by inhibition of AdipoR1 (Figure 8C). Based on our results, Acrp30 attenuated PPAR-γ signaling in microglia by mediating AdipoR1.
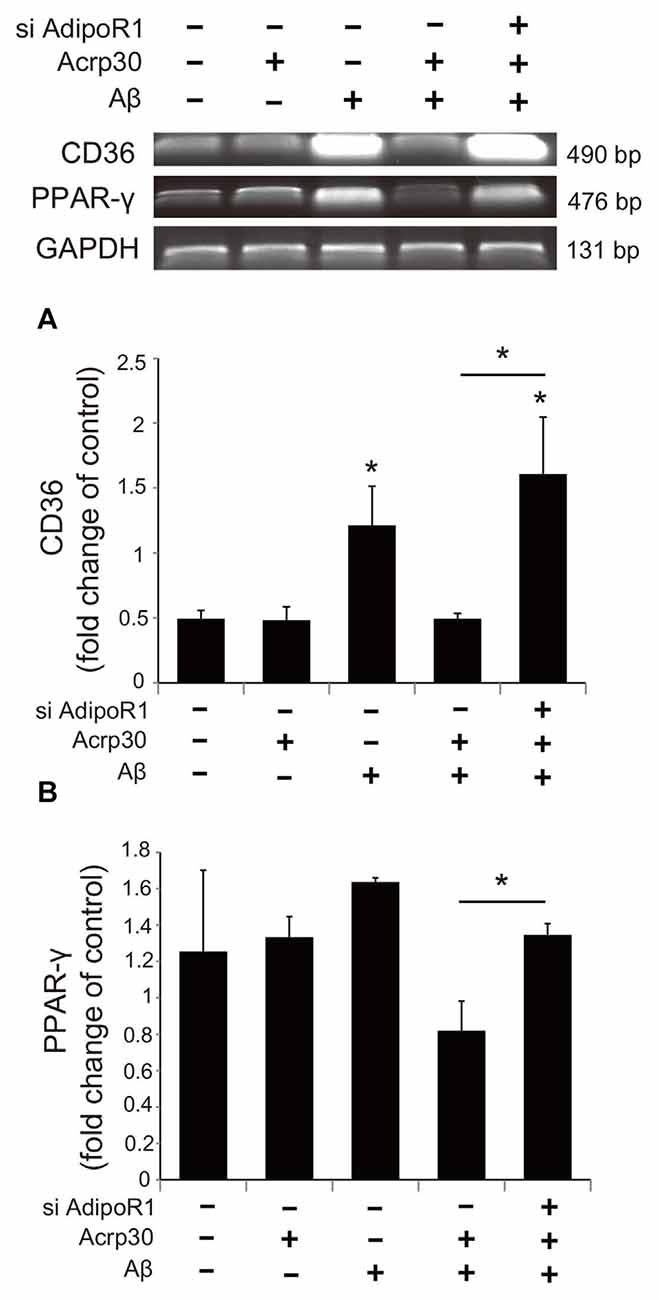
Figure 7. The expression of CD36 and PPAR-γ in Aβ treated BV2 microglia. The mRNA levels of CD36 (A) and PPAR-γ (B) were checked by RT PCR. The reduced mRNA levels of CD36 and PPAR-γ by Acrp30 in Aβ treated microglia were increased by inhibiting AdipoR1. Data are expressed as mean ± SEM. Each experiment was conducted four times per condition. GAPDH served as a control. Differences were considered statistically significant at *p < 0.05 (compared to control). Acrp30: Acrp30 5 μg/ml pretreatment, Aβ: Aβ 10 μM treatment for 24 h, siAdipoR1: siRNA AdipoR1 transfection.
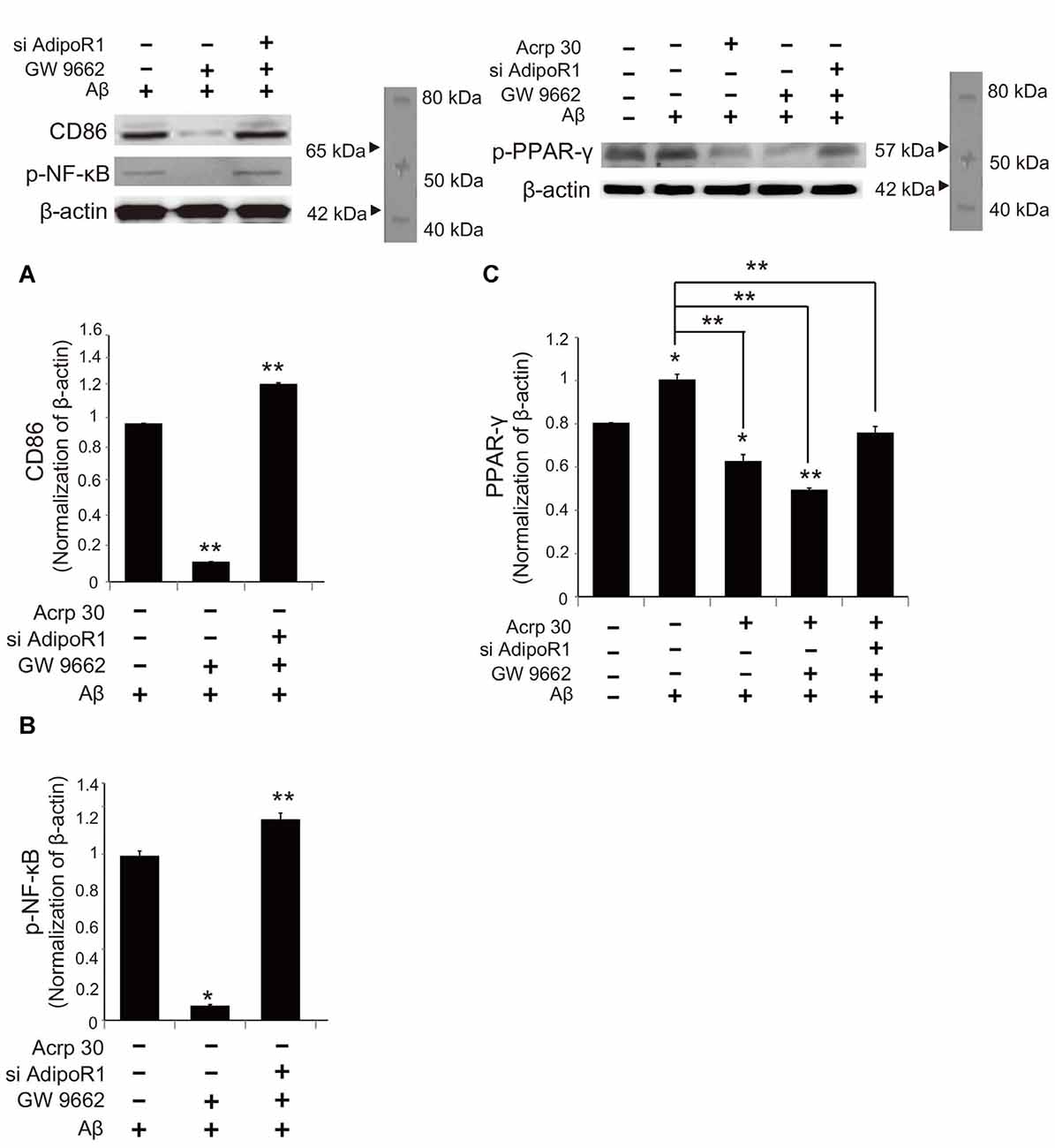
Figure 8. The change in PPAR-γ signaling by inhibiting AdipoR1 in Aβ-treated BV2 microglia. The protein levels of CD86 (A) and p-NF-κB (B) p-PPAR-γ (C) protein were detected by western blotting analysis. (C) The decreased p-PPAR-γ protein level by Acrp30 and GW9662 in Aβ-treated microglia was not changed by suppressing AdipoR1. Data are expressed as mean ± SEM. Each experiment was conducted four times per condition. GAPDH served as a control. Differences were considered statistically significant at *p < 0.05 (compared to control). Acrp30: Acrp30 5 μg/ml pretreatment, Aβ: Aβ 10 μM treatment for 24 h, siAdipoR1: siRNA AdipoR1 transfection, GW9662: PPAR-γ antagonist 10 μM. **p < 0.001.
Discussion
AD is characterized by the abnormal accumulation of Aβ and chronic neuroinflammation (Ramanathan et al., 2015; Scheltens et al., 2016). Clearance of Aβ and chronic inflammation are related to the polarization of and phagocytotic ability of microglia (Ide et al., 1996; Heese et al., 1998; Hutchinson et al., 2009; Tang and Le, 2016). The M2 phenotype microglia have been demonstrated to have a neuroprotective function in AD (Zhu et al., 2016). A recent study showed that M2 macrophage transplantation reduces neuroinflammation in the brain, promotes clearance of Aβ, and improves cognitive impairment (Zhu et al., 2016). Aβ and tau oligomers activate M1 pro-inflammatory responses and induce irreversible neuron loss (Tang and Le, 2016). The M1 phenotype microglia secrete pro-inflammatory mediators including TNF-α, IL-6 and NO, whereas anti-inflammatory M2 phenotype microglia produce anti-inflammatory mediators such as IL-10 and IL-4 under oxidative stress conditions (Tang and Le, 2016). In this study, we examined the alteration of cytokine expression by Acrp30 in Aβ-treated microglia. We found that adiponectin attenuates the expression of pro-inflammatory cytokines, and AdipoR1 may mediate the function of adiponectin secretion of inflammatory cytokines, considering that the expression of pro-inflammatory cytokines was not reduced in Aβ-treated microglia despite Acrp30 pretreatment.
In the present study, we found activated microglia in vivo in the AD mouse brain and in vitro under Aβ–induced inflammatory conditions. Increased expression of CD86, a known M1 receptor marker (Gong et al., 2016) was found in the cortex and striatum of 5xFAD mice, and in Aβ-treated BV2 microglia. Based on our results, we suggest that adiponectin might promote the polarization of M2 type microglia under Aβ toxicity conditions. Moreover, our results showed that adiponectin activates M2 phenotype microglia (Lovren et al., 2010). Increased CD206 and reduced M1 polarization was demonstrated by the reduction of pro-inflammatory cytokines such as TNF-α and IL-6, and the reduction of CD86 and iNOS expression.
Adiponectin regulates many processes by binding with specific receptors, AdipoR1 and AdipoR2 (Hug et al., 2004) and exists in various organs, including the brain (Kaminski et al., 2014). We observed more expression of AdipoR1 than AdipoR2 in BV2 microglia under Aβ induced toxicity. Previous studies demonstrated that downregulation of AdipoR1 attenuates anti-inflammatory responses (Zhang et al., 2016) and reduces the adiponectin’s beneficial actions in macrophages (Luo et al., 2013). We observed that the downregulation of AdipoR1 by siRNA AdipoR1 increases pro-inflammatory cytokines including TNF-α and iNOS production under Aβ toxicity in spite of Acrp30 pretreatment. Judging from our results, we assume that the inhibitory effect of adiponectin in pro-inflammatory cytokine production may be mediated by AdipoR1.
Fibrillar Aβ has been reported to interact with the cell surface receptor and leads to intracellular signaling, and then stimulates phagocytosis (D’Andrea et al., 2004; Mohamed and Posse de Chaves, 2011). CD36, one of the class B scavenger receptors, has been shown to be expressed in various cell types and bind with diverse ligands (Febbraio et al., 2001), and to participate in the internalization of numerous small particles, including Aβ (Koenigsknecht and Landreth, 2004) and apoptotic cells (Fadok et al., 1998). CD36 has been shown to uptake Aβ (Moore et al., 2002), and is involved in the production of pro-inflammatory mediators (Janabi et al., 2000; Stuart et al., 2005; Baranova et al., 2008). Several studies have demonstrated that CD36 boosts pro-inflammatory responses (Janabi et al., 2000; Moore et al., 2002; Okamura et al., 2009; Silverstein and Febbraio, 2009) and phagocytosis (Erdman et al., 2009). Studies have also shown that the activation of macrophages promotes the expression of CD36 by generation of ligands of PPAR-γ (Huang et al., 1999; Berry et al., 2007) and adiponectin inhibits the expression of scavenger receptor CD36 (Ouchi et al., 2001). Based on the current results, we hypothesize that adiponectin can attenuate the expression of CD36, leading to the production of pro-inflammatory cytokines.
Moreover, previous studies have demonstrated that PPAR-γ can mediate the conversion of the microglia phenotype (Yamanaka et al., 2012; Saijo et al., 2013). Our results indicate that PPAR-γ may induce M2 phenotype microglia under Aβ toxicity via adiponectin treatment. Several studies have demonstrated that PPAR-γ agonists alleviate neuroinflammation in AD models by acting as anti-inflammatory regulators (Ghisletti et al., 2007; Saijo et al., 2013) while others have shown that PPAR-γ can regulate the secretion of inflammatory cytokines in microglia, ultimately promoting tissue repair (Chawla, 2010; Chinetti-Gbaguidi et al., 2011).
In the present study, we found that adiponectin may contribute to the function and polarization of microglia through PPAR-γ signaling. In particular, adiponectin may play a pro- inflammatory role through PPAR-γ by mediating AdipoR1. Even though we did not present evidence on memory improvement by adiponectin through PPAR-γ in AD brain, we speculate that adiponectin may improve memory in AD by attenuating PPAR-γ signaling in microglia, and regulate the function of microglia under Aβ toxicity by regulating PPAR-γ through AdipoR1.
Taken together, we conclude that: (1) adiponectin may induce the M2 polarization of microglia in Aβ toxicity; (2) adiponectin may regulate the production of pro-inflammatory cytokines by mediating AdipoR1 under Aβ toxicity; and (3) adiponectin may exert anti-inflammatory functions in microglia and regulate the polarization of microglia through PPAR-γ signaling under Aβ toxicity. Thus, we suggest that adiponectin is a crucial adipokine to attenuate AD pathology by regulating the function of microglia.
Author Contributions
JS conducted the experiments and contributed to the writing of the preliminary draft. S-MC conducted the experiments. BCK designed the study and wrote the manuscript, and provided overall supervision for the project.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by the Brain Research Program through the National Research Foundation of Korea funded by the Ministry of Science, ICT and Future Planning NRF-2016M3C7A1905469 (BCK) and a grant from 2016R1D1A1B03930394 (JS).
References
An, K., Klyubin, I., Kim, Y., Jung, J. H., Mably, A. J., O’Dowd, S. T., et al. (2013). Exosomes neutralize synaptic-plasticity-disrupting activity of Aβ assemblies in vivo. Mol. Brain 6:47. doi: 10.1186/1756-6606-6-47
Baranova, I. N., Kurlander, R., Bocharov, A. V., Vishnyakova, T. G., Chen, Z., Remaley, A. T., et al. (2008). Role of human CD36 in bacterial recognition, phagocytosis and pathogen-induced JNK-mediated signaling. J. Immunol. 181, 7147–7156. doi: 10.4049/jimmunol.181.10.7147
Beletskii, A., Cooper, M., Sriraman, P., Chiriac, C., Zhao, L., Abbot, S., et al. (2005). High-throughput phagocytosis assay utilizing a pH-sensitive fluorescent dye. Biotechniques 39, 894–897. doi: 10.2144/000112001
Berry, A., Balard, P., Coste, A., Olagnier, D., Lagane, C., Authier, H., et al. (2007). IL-13 induces expression of CD36 in human monocytes through PPARγ activation. Eur. J. Immunol. 37, 1642–1652. doi: 10.1002/eji.200636625
Bubu, O. M., Brannick, M., Mortimer, J., Umasabor-Bubu, O., Sebastião, Y. V., Wen, Y., et al. (2016). Sleep, cognitive impairment and Alzheimer’s disease: a systematic review and meta-analysis. Sleep doi: 10.1093/sleep/zsw032 [Epub ahead of print].
Chawla, A. (2010). Control of macrophage activation and function by PPARs. Circ. Res. 106, 1559–1569. doi: 10.1161/CIRCRESAHA.110.216523
Chen, M., Li, H., Wang, G., Shen, X., Zhao, S., and Su, W. (2016). Atorvastatin prevents advanced glycation end products (AGEs)-induced cardiac fibrosis via activating peroxisome proliferator-activated receptor gamma (PPAR-γ). Metabolism 65, 441–453. doi: 10.1016/j.metabol.2015.11.007
Chinetti-Gbaguidi, G., Baron, M., Bouhlel, M. A., Vanhoutte, J., Copin, C., Sebti, Y., et al. (2011). Human atherosclerotic plaque alternative macrophages display low cholesterol handling but high phagocytosis because of distinct activities of the PPARγ and LXRα pathways. Circ. Res. 108, 985–995. doi: 10.1161/CIRCRESAHA.110.233775
Combs, C. K., Karlo, J. C., Kao, S. C., and Landreth, G. E. (2001). β-Amyloid stimulation of microglia and monocytes results in TNFα-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J. Neurosci. 21, 1179–1188.
D’Andrea, M. R., Cole, G. M., and Ard, M. D. (2004). The microglial phagocytic role with specific plaque types in the Alzheimer disease brain. Neurobiol. Aging 25, 675–683. doi: 10.1016/j.neurobiolaging.2003.12.026
Del Bo, R., Angeretti, N., Lucca, E., De Simoni, M. G., and Forloni, G. (1995). Reciprocal control of inflammatory cytokines, IL-1 and IL-6 and β-amyloid production in cultures. Neurosci. Lett. 188, 70–74. doi: 10.1016/0304-3940(95)11384-9
De Strooper, B., and Karran, E. (2016). The cellular phase of Alzheimer’s disease. Cell 164, 603–615. doi: 10.1016/j.cell.2015.12.056
Doens, D., and Fernández, P. L. (2014). Microglia receptors and their implications in the response to amyloid β for Alzheimer’s disease pathogenesis. J. Neuroinflammation 11:48. doi: 10.1186/1742-2094-11-48
Eggen, B. J. L., Raj, D., Hanisch, U.-K., and Boddeke, H. W. G. (2013). Microglial phenotype and adaptation. J. Neuroimmune Pharmacol. 8, 807–823. doi: 10.1007/s11481-013-9490-4
Elfeky, M., Kaede, R., Okamatsu-Ogura, Y., and Kimura, K. (2016). Adiponectin inhibits LPS-induced HMGB1 release through an AMP kinase and heme oxygenase-1-dependent pathway in RAW 264 macrophage cells. Mediators Inflamm. 2016:5701959. doi: 10.1155/2016/5701959
Erdman, L. K., Cosio, G., Helmers, A. J., Gowda, D. C., Grinstein, S., and Kain, K. C. (2009). CD36 and TLR interactions in inflammation and phagocytosis: implications for malaria. J. Immunol. 183, 6452–6459. doi: 10.4049/jimmunol.0901374
Fadok, V. A., Warner, M. L., Bratton, D. L., and Henson, P. M. (1998). CD36 is required for phagocytosis of apoptotic cells by human macrophages that use either a phosphatidylserine receptor or the vitronectin receptor (αvβ3). J. Immunol. 161, 6250–6257.
Febbraio, M., Hajjar, D. P., and Silverstein, R. L. (2001). CD36: a class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation and lipid metabolism. J. Clin. Invest. 108, 785–791. doi: 10.1172/JCI14006
Francos-Quijorna, I., Amo-Aparicio, J., Martinez-Muriana, A., and López-Vales, R. (2016). IL-4 drives microglia and macrophages toward a phenotype conducive for tissue repair and functional recovery after spinal cord injury. Glia 64, 2079–2092. doi: 10.1002/glia.23041
Gertig, U., and Hanisch, U. K. (2014). Microglial diversity by responses and responders. Front. Cell. Neurosci. 8:101. doi: 10.3389/fncel.2014.00101
Ghisletti, S., Huang, W., Ogawa, S., Pascual, G., Lin, M. E., Willson, T. M., et al. (2007). Parallel SUMOylation-dependent pathways mediate gene- and signal-specific transrepression by LXRs and PPARγ. Mol. cell 25, 57–70. doi: 10.1016/j.molcel.2006.11.022
Ginhoux, F., and Prinz, M. (2015). Origin of microglia: current concepts and past controversies. Cold Spring Harb. Perspect. Biol. 7:a020537. doi: 10.1101/cshperspect.a020537
Goate, A. (2006). Segregation of a missense mutation in the amyloid β-protein precursor gene with familial Alzheimer’s disease. J. Alzheimers Dis. 9, 341–347. doi: 10.1007/978-3-540-37652-1_16
Gong, L., Wang, H., Sun, X., Liu, C., Duan, C., Cai, R., et al. (2016). Toll-Interleukin 1 Receptor domain-containing adaptor protein positively regulates BV2 cell M1 polarization. Eur. J. Neurosci. 43, 1674–1682. doi: 10.1111/ejn.13257
Hardy, J., Bogdanovic, N., Winblad, B., Portelius, E., Andreasen, N., Cedazo-Minguez, A., et al. (2014). Pathways to Alzheimer’s disease. J. Intern. Med. 275, 296–303. doi: 10.1111/joim.12192
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
Heese, K., Hock, C., and Otten, U. (1998). Inflammatory signals induce neurotrophin expression in human microglial cells. J. Neurochem. 70, 699–707. doi: 10.1046/j.1471-4159.1998.70020699.x
Huang, J. T., Welch, J. S., Ricote, M., Binder, C. J., Willson, T. M., Kelly, C., et al. (1999). Interleukin-4-dependent production of PPAR-γ ligands in macrophages by 12/15-lipoxygenase. Nature 400, 378–382. doi: 10.1038/22572
Hug, C., Wang, J., Ahmad, N. S., Bogan, J. S., Tsao, T.-S., and Lodish, H. F. (2004). T-cadherin is a receptor for hexameric and high-molecular-weight forms of Acrp30/adiponectin. Proc. Natl. Acad. Sci. U S A 101, 10308–10313. doi: 10.1073/pnas.0403382101
Hutchinson, A. J., Chou, C. L., Israel, D. D., Xu, W., and Regan, J. W. (2009). Activation of EP2 prostanoid receptors in human glial cell lines stimulates the secretion of BDNF. Neurochem. Int. 54, 439–446. doi: 10.1016/j.neuint.2009.01.018
Ide, C. F., Scripter, J. L., Coltman, B. W., Dotson, R. S., Snyder, D. C., and Jelaso, A. (1996). Cellular and molecular correlates to plasticity during recovery from injury in the developing mammalian brain. Prog. Brain Res. 108, 365–377. doi: 10.1016/s0079-6123(08)62552-2
Janabi, M., Yamashita, S., Hirano, K., Sakai, N., Hiraoka, H., Matsumoto, K., et al. (2000). Oxidized LDL-induced NF-κB activation and subsequent expression of proinflammatory genes are defective in monocyte-derived macrophages from CD36-deficient patients. Arterioscler. Thromb. Vasc. Biol. 20, 1953–1960. doi: 10.1161/01.atv.20.8.1953
Kadowaki, T., Yamauchi, T., Kubota, N., Hara, K., Ueki, K., and Tobe, K. (2006). Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J. Clin. Invest. 116, 1784–1792. doi: 10.1172/jci29126
Kaminski, T., Smolinska, N., Maleszka, A., Kiezun, M., Dobrzyn, K., Czerwinska, J., et al. (2014). Expression of adiponectin and its receptors in the porcine hypothalamus during the oestrous cycle. Reprod. Domest. Anim. 49, 378–386. doi: 10.1111/rda.12282
Kempuraj, D., Thangavel, R., Natteru, P. A., Selvakumar, G. P., Saeed, D., Zahoor, H., et al. (2016). Neuroinflammation induces neurodegeneration. J. Neurol. Neurosurg. Spine 1.
Kim, J., Zheng, W., Grafer, C., Mann, M. L., and Halvorson, L. M. (2013). GnRH decreases adiponectin expression in pituitary gonadotropes via the calcium and PKA pathways. Reprod. Sci. 20, 937–945. doi: 10.1177/1933719112468947
Koenigsknecht, J., and Landreth, G. (2004). Microglial phagocytosis of fibrillar β-amyloid through a β1 integrin-dependent mechanism. J. Neurosci. 24, 9838–9846. doi: 10.1523/JNEUROSCI.2557-04.2004
Latta, C. H., Sudduth, T. L., Weekman, E. M., Brothers, H. M., Abner, E. L., Popa, G. J., et al. (2015). Determining the role of IL-4 induced neuroinflammation in microglial activity and amyloid-β using BV2 microglial cells and APP/PS1 transgenic mice. J. Neuroinflammation 12:41. doi: 10.1186/s12974-015-0243-6
Lee, J. H., Wei, Z. Z., Cao, W., Won, S., Gu, X., Winter, M., et al. (2016). Regulation of therapeutic hypothermia on inflammatory cytokines, microglia polarization, migration and functional recovery after ischemic stroke in mice. Neurobiol. Dis. 96, 248–260. doi: 10.1016/j.nbd.2016.09.013
Lindberg, C., Selenica, M. L., Westlind-Danielsson, A., and Schultzberg, M. (2005). β-amyloid protein structure determines the nature of cytokine release from rat microglia. J. Mol. Neurosci. 27, 1–12. doi: 10.1385/jmn:27:1:001
Lovren, F., Pan, Y., Quan, A., Szmitko, P. E., Singh, K. K., Shukla, P. C., et al. (2010). Adiponectin primes human monocytes into alternative anti-inflammatory M2 macrophages. Am. J. Physiol. Heart Circ. Physiol. 299, H656–H663. doi: 10.1152/ajpheart.00115.2010
Luo, N., Chung, B. H., Wang, X., Klein, R. L., Tang, C. K., Garvey, W. T., et al. (2013). Enhanced adiponectin actions by overexpression of adiponectin receptor 1 in macrophages. Atherosclerosis 228, 124–135. doi: 10.1016/j.atherosclerosis.2013.02.026
Maeda, K., Okubo, K., Shimomura, I., Funahashi, T., Matsuzawa, Y., and Matsubara, K. (1996). cDNA cloning and expression of a novel adipose specific collagen-like factor, apM1 (AdiPose Most abundant Gene transcript 1). Biochem. Biophys. Res. Commun. 221, 286–289. doi: 10.1006/bbrc.1996.0587
Masamoto, Y., Arai, S., Sato, T., Yoshimi, A., Kubota, N., Takamoto, I., et al. (2016). Adiponectin enhances antibacterial activity of hematopoietic cells by suppressing bone marrow inflammation. Immunity 44, 1422–1433. doi: 10.1016/j.immuni.2016.05.010
Mehlhorn, G., Hollborn, M., and Schliebs, R. (2000). Induction of cytokines in glial cells surrounding cortical β-amyloid plaques in transgenic Tg2576 mice with Alzheimer pathology. Int. J. Dev. Neurosci. 18, 423–431. doi: 10.1016/s0736-5748(00)00012-5
Mohamed, A., and Posse de Chaves, E. (2011). Aβ internalization by neurons and glia. Int. J. Alzheimers Dis. 2011:127984. doi: 10.4061/2011/127984
Moore, K. J., El Khoury, J., Medeiros, L. A., Terada, K., Geula, C., Luster, A. D., et al. (2002). A CD36-initiated signaling cascade mediates inflammatory effects of β-amyloid. J. Biol. Chem. 277, 47373–47379. doi: 10.1074/jbc.M208788200
Nakamichi, Y., Udagawa, N., and Takahashi, N. (2013). IL-34 and CSF-1: similarities and differences. J. Bone Miner. Metab. 31, 486–495. doi: 10.1007/s00774-013-0476-3
Natoli, G., and Monticelli, S. (2014). Macrophage activation: glancing into diversity. Immunity 40, 175–177. doi: 10.1016/j.immuni.2014.01.004
Okamura, D. M., Pennathur, S., Pasichnyk, K., López-Guisa, J. M., Collins, S., Febbraio, M., et al. (2009). CD36 regulates oxidative stress and inflammation in hypercholesterolemic CKD. J. Am. Soc. Nephrol. 20, 495–505. doi: 10.1681/ASN.2008010009
Ouchi, N., Kihara, S., Arita, Y., Nishida, M., Matsuyama, A., Okamoto, Y., et al. (2001). Adipocyte-derived plasma protein, adiponectin, suppresses lipid accumulation and class A scavenger receptor expression in human monocyte-derived macrophages. Circulation 103, 1057–1063. doi: 10.1161/01.cir.103.8.1057
Parkhurst, C. N., Yang, G., Ninan, I., Savas, J. N., Yates, J. R. III, Lafaille, J. J., et al. (2013). Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155, 1596–1609. doi: 10.1016/j.cell.2013.11.030
Plastira, I., Bernhart, E., Goeritzer, M., Reicher, H., Kumble, V. B., Kogelnik, N., et al. (2016). 1-Oleyl-lysophosphatidic acid (LPA) promotes polarization of BV-2 and primary murine microglia towards an M1-like phenotype. J. Neuroinflammation 13:205. doi: 10.1186/s12974-016-0701-9
Popivanova, B. K., Kitamura, K., Wu, Y., Kondo, T., Kagaya, T., Kaneko, S., et al. (2008). Blocking TNF-α in mice reduces colorectal carcinogenesis associated with chronic colitis. J. Clin. Invest. 118, 560–570. doi: 10.1172/JCI32453
Ramanathan, A., Nelson, A. R., Sagare, A. P., and Zlokovic, B. V. (2015). Impaired vascular-mediated clearance of brain amyloid beta in Alzheimer’s disease: the role, regulation and restoration of LRP1. Front. Aging Neurosci. 7:136. doi: 10.3389/fnagi.2015.00136
Renaldi, O., Pramono, B., Sinorita, H., Purnomo, L. B., Asdie, R. H., and Asdie, A. H. (2009). Hypoadiponectinemia: a risk factor for metabolic syndrome. Acta Med. Indones. 41, 20–24.
Rogers, J., and Lue, L. F. (2001). Microglial chemotaxis, activation and phagocytosis of amyloid β-peptide as linked phenomena in Alzheimer’s disease. Neurochem. Int. 39, 333–340. doi: 10.1016/s0197-0186(01)00040-7
Rogers, J., Strohmeyer, R., Kovelowski, C. J., and Li, R. (2002). Microglia and inflammatory mechanisms in the clearance of amyloid β peptide. Glia 40, 260–269. doi: 10.1002/glia.10153
Saijo, K., Crotti, A., and Glass, C. K. (2013). Regulation of microglia activation and deactivation by nuclear receptors. Glia 61, 104–111. doi: 10.1002/glia.22423
Scheltens, P., Blennow, K., Breteler, M. M., de Strooper, B., Frisoni, G. B., Salloway, S., et al. (2016). Alzheimer’s disease. Lancet 388, 505–517. doi: 10.1016/S0140-6736(15)01124-1
Silverstein, R. L., and Febbraio, M. (2009). CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis and behavior. Sci. Signal. 2:re3. doi: 10.1126/scisignal.272re3
Solé-Domènech, S., Cruz, D. L., Capetillo-Zarate, E., and Maxfield, F. R. (2016). The endocytic pathway in microglia during health, aging and Alzheimer’s disease. Ageing Res. Rev. 32, 89–103. doi: 10.1016/j.arr.2016.07.002
Stamouli, E. C., and Politis, A. M. (2016). Pro-inflammatory cytokines in Alzheimer’s disease. Psychiatrike 27, 264–275. doi: 10.22365/jpsych.2016.274.264
Stuart, L. M., Deng, J., Silver, J. M., Takahashi, K., Tseng, A. A., Hennessy, E. J., et al. (2005). Response to Staphylococcus aureus requires CD36-mediated phagocytosis triggered by the COOH-terminal cytoplasmic domain. J. Cell Biol. 170, 477–485. doi: 10.1083/jcb.200501113
Takata, K., Kitamura, Y., Umeki, M., Tsuchiya, D., Kakimura, J., Taniguchi, T., et al. (2003). Possible involvement of small oligomers of amyloid-β peptides in 15-deoxy-δ 12,14 prostaglandin J2-sensitive microglial activation. J. Pharmacol. Sci. 91, 330–333. doi: 10.1254/jphs.91.330
Tang, Y., and Le, W. (2016). Differential Roles of M1 and M2 microglia in neurodegenerative diseases. Mol. Neurobiol. 53, 1181–1194. doi: 10.1007/s12035-014-9070-5
Tarantini, S., Tran, C. H., Gordon, G. R., Ungvari, Z., and Csiszar, A. (2016). Impaired neurovascular coupling in aging and Alzheimer’s disease: contribution of astrocyte dysfunction and endothelial impairment to cognitive decline. Exp. Gerontol. doi: 10.1016/j.exger.2016.11.004 [Epub ahead of print].
Tremblay, M. È., Lecours, C., Samson, L., Sánchez-Zafra, V., and Sierra, A. (2015). From the Cajal alumni Achucarro and Rio-Hortega to the rediscovery of never-resting microglia. Front. Neuroanat. 9:45. doi: 10.3389/fnana.2015.00045
Tremblay, M. È., Stevens, B., Sierra, A., Wake, H., Bessis, A., and Nimmerjahn, A. (2011). The role of microglia in the healthy brain. J. Neurosci. 31, 16064–16069. doi: 10.1523/JNEUROSCI.4158-11.2011
Wang, X., Buechler, N. L., Yoza, B. K., McCall, C. E., and Vachharajani, V. (2016). Adiponectin treatment attenuates inflammatory response during early sepsis in obese mice. J. Inflamm. Res. 9, 167–174. doi: 10.2147/jir.s119021
Weekman, E. M., Sudduth, T. L., Abner, E. L., Popa, G. J., Mendenhall, M. D., Brothers, H. M., et al. (2014). Transition from an M1 to a mixed neuroinflammatory phenotype increases amyloid deposition in APP/PS1 transgenic mice. J. Neuroinflammation 11:127. doi: 10.1186/1742-2094-11-127
Xu, X., Gao, Y., Wen, L., Zhai, Z., Zhang, S., Shan, F., et al. (2016). Methionine enkephalin regulates microglia polarization and function. Int. Immunopharmacol. 40, 90–97. doi: 10.1016/j.intimp.2016.08.037
Yamanaka, M., Ishikawa, T., Griep, A., Axt, D., Kummer, M. P., and Heneka, M. T. (2012). PPARγ/RXRα-induced and CD36-mediated microglial amyloid-β phagocytosis results in cognitive improvement in amyloid precursor protein/presenilin 1 mice. J. Neurosci. 32, 17321–17331. doi: 10.1523/JNEUROSCI.1569-12.2012
Yamauchi, T., Kamon, J., Ito, Y., Tsuchida, A., Yokomizo, T., Kita, S., et al. (2003). Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 423, 762–769. doi: 10.1038/nature01705
Zhang, P., Huang, C., Li, J., Li, T., Guo, H., Liu, T., et al. (2016). Globular CTRP9 inhibits oxLDL-induced inflammatory response in RAW 264.7 macrophages via AMPK activation. Mol. Cell. Biochem. 417, 67–74. doi: 10.1007/s11010-016-2714-1
Keywords: adiponectin, Acrp30, microglia, inflammation, amyloid beta (Aβ), cytokines, peroxisome proliferator-activated receptor (PPAR)-γ
Citation: Song J, Choi S-M and Kim BC (2017) Adiponectin Regulates the Polarization and Function of Microglia via PPAR-γ Signaling Under Amyloid β Toxicity. Front. Cell. Neurosci. 11:64. doi: 10.3389/fncel.2017.00064
Received: 22 December 2016; Accepted: 23 February 2017;
Published: 07 March 2017.
Edited by:
Edna Grünblatt, University of Zurich, SwitzerlandReviewed by:
Markus P. Kummer, University of Bonn, GermanyKempuraj Duraisamy, University of Missouri, USA
Copyright © 2017 Song, Choi and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Byeong C. Kim, Ynllb25nLmtpbTdAZ21haWwuY29t