 Yoshito Mizoguchi
Yoshito Mizoguchi Akira Monji
Akira Monji- Department of Psychiatry, Faculty of Medicine, Saga University, Saga, Japan
Autism spectrum disorders (ASDs) are neurodevelopmental disorders characterized by deficits in social interaction, difficulties with language and repetitive/restricted behaviors. Microglia are resident innate immune cells which release many factors including proinflammatory cytokines, nitric oxide (NO) and brain-derived neurotrophic factor (BDNF) when they are activated in response to immunological stimuli. Recent in vivo imaging has shown that microglia sculpt and refine the synaptic circuitry by removing excess and unwanted synapses and be involved in the development of neural circuits or synaptic plasticity thereby maintaining the brain homeostasis. BDNF, one of the neurotrophins, has various important roles in cell survival, neurite outgrowth, neuronal differentiation, synaptic plasticity and the maintenance of neural circuits in the CNS. Intracellular Ca2+ signaling is important for microglial functions including ramification, de-ramification, migration, phagocytosis and release of cytokines, NO and BDNF. BDNF induces a sustained intracellular Ca2+ elevation through the upregulation of the surface expression of canonical transient receptor potential 3 (TRPC3) channels in rodent microglia. BDNF might have an anti-inflammatory effect through the inhibition of microglial activation and TRPC3 could play important roles in not only inflammatory processes but also formation of synapse through the modulation of microglial phagocytic activity in the brain. This review article summarizes recent findings on emerging dual, inflammatory and non-inflammatory, roles of microglia in the brain and reinforces the importance of intracellular Ca2+ signaling for microglial functions in both normal neurodevelopment and their potential contributing to neurodevelopmental disorders such as ASDs.
Introduction
Autism spectrum disorders (ASDs) are neurodevelopmental disorders characterized by deficits in social interaction, difficulties with language, and repetitive/restricted behaviors (Lai et al., 2014). The etiology of ASDs is still largely unclear, but both immune dysfunction and abnormalities in synaptogenesis have repeatedly been implicated as contributing to the disease phenotype (Edmonson et al., 2016).
Microglia are immune cells which are derived from progenitors that have migrated from the periphery and are from mesodermal/mesenchymal origin (Kettenmann et al., 2011). After invading the brain parenchyma, microglia transform into the “resting” ramified phenotype and are distributed in the whole brain. However, microglia revert to an ameboid appearance when they are activated in the disturbances including infection, trauma, ischemia, neurodegenerative diseases or any loss of brain homeostasis (Aguzzi et al., 2013; Cunningham, 2013). Microglia are the most active cytokine producing cells in the brain and can release many factors including pro-inflammatory cytokines (such as TNFα, IL-6), nitric oxide (NO) and neurotrophic factors (such as brain-derived neurotrophic factor, BDNF) when they are activated in response to immunological stimuli (Kato et al., 2013; Monji et al., 2013, 2014; Mizoguchi et al., 2014a; Smith and Dragunow, 2014). However, recent in vivo imaging has shown that microglia constantly use highly motile processes to survey their assigned brain regions and phagocyte pathogens and cellular debris even in their resting state, and are ready to transform to “activated” state in responses to injury, ischemia or autoimmune challenges in the brain (Wake et al., 2013). Microglia are also shown to sculpt and refine the synaptic circuitry by removing excess and unwanted synapses and be involved in the development of neural circuits or synaptic plasticity thereby maintaining the brain homeostasis (Schwartz et al., 2013; Hong et al., 2016). By extension, neurodevelopmental disorders such as ASDs might not need to involve a pathological gain in microglial function but simply a disruption of their physiological functioning in the regulation of synaptic circuits (Salter and Beggs, 2014; Ziats et al., 2015; Macht, 2016). This review article summarizes recent findings on emerging dual, inflammatory and non-inflammatory, roles of microglia in the brain and reinforces the importance of intracellular Ca2+ signaling for microglial functions in both normal neurodevelopment and their potential contributing to neurodevelopmental disorders such as ASDs.
Inflammatory and Non-Inflammatory Roles of Microglia in ASDs
Numerous studies of serum cytokines demonstrated lower levels of transforming growth factor-β (TGF-β) and higher levels of macrophage inhibitory factor, leptin, interleukin 1β (IL-1β), IL-6, interferon-γ (IFN-γ) and IL-12 in various age groups of patients with ASDs (Goines and Ashwood, 2013). Cerebrospinal fluid (CSF) samples from patients with autism also showed an increase in pro-inflammatory macrophage chemoattractant protein 1 (MCP-1; Vargas et al., 2005) and TNF-α (Chez et al., 2007). Microglial abnormality could result from CNS or peripheral immune signals, such as auto-antibodies (Wills et al., 2009), or by peripheral chemokines/cytokines (such as IL-1β, IL-6 and TNF-α) up-regulated in autism (Gupta et al., 1998; Vargas et al., 2005).
The pioneer work by Vargas et al. (2005) and subsequent studies revealed an active neuroinflammatory phenotype of microglia in the post-mortem brains of patients with autism (Morgan et al., 2010). Marked changes in microglial morphology, accompanied by a unique profile of pro-inflammatory cytokines were seen in the cerebral cortex, white matter and cerebellum of patients with autism. Excessive microglia activation in young adults (age 18–31 years) affected by ASDs was also confirmed with PET using [11C]-(R)-PK11195. In this study, ASD brain regions showing increased binding potentials of the radiotracer included the cerebellum, midbrain, pons, fusiform gyri, and the anterior cingulate and orbitofrontal cortices. The most prominent increase was observed in the cerebellum (Suzuki et al., 2013). In the cerebellum, activated microglia were observed to be intimately associated with Purkinje cells undergoing apoptosis in cerebellar organotypic cultures during normal development. This could be consistent with a role for microglia in developmentally regulated neuronal death by promoting Purkinje cell apoptosis (Marín-Teva et al., 2004), an important physiological activity that could be impaired in autism.
A deficit in microglia/complement-mediated synaptic pruning might be fundamental to the cognitive effects associated with ASDs (Voineagu et al., 2011). The chemotactic/phagocytic activity of microglia could also be impaired, further aggravating the symptoms by insufficient clearance of debris (Derecki et al., 2013). The complement cascade, normally associated with removal of pathogens and cellular debris, is also crucial to microglial-mediated synaptic pruning and refinement of neuronal connectivity in the normal brain (Stephan et al., 2012). Evidence points to convergence on C3 and its microglial receptor C3 receptors (C3R). The initiator of the complement cascade is C1q, which induces C3 secretion via C4. The presence of C3 on unwanted synapses “tags” them for recognition by microglia to be eliminated. In addition, decreased C4 leading to reduced synaptic pruning in early life, mediated through reduced C3 synaptic tagging, is implicated in ASD-like behaviors (Estes and McAllister, 2015). Furthermore, mice deficient in the CX3CR1, a chemokine receptor expressed in the brain exclusively by microglia, have increased densities of immature synapses caused by delayed synaptic pruning, resulting in excessive and electrophysiologically immature synapses and deficits in functional connectivity (Zhan et al., 2014). Altogether, recent findings on emerging dual, inflammatory and non-inflammatory, roles of microglia in the brain suggest that abnormal secretion of inflammatory cytokines and abnormal or exaggerated execution of normal developmental microglial functions, including incorrect synaptic pruning, failure of phagocytosis of apoptotic neurons might be underlying mechanisms of neurodevelopmental disorders such as ASDs (Edmonson et al., 2016).
Neuronal Intracellular Ca2+ Signaling Mediated by VGCCs and ASDs
The electrical activity of neurons (i.e., excitable cells) depends on a number of different types of voltage- or ligand-gated ion channels that are permeable to inorganic ions such as sodium, potassium, chloride and calcium. While the former three ions predominantly support the electrogenic roles, Ca2+ are different in that they can not only alter the membrane potential but also serve as important intracellular signaling entities by themselves. In the CNS, intracellular Ca2+ signaling regulates many different neuronal functions, such as cell proliferation, gene transcription and exocytosis at synapses (Berridge, 1998). In neurons, because the prolonged elevation of intracellular Ca2+ concentration ([Ca2+]i) is cytotoxic, [Ca2+]i is tightly regulated by intrinsic gating processes mediated by voltage-gated calcium channels (VGCCs) and NMDA receptors (NMDARs; Simms and Zamponi, 2014). In addition, dysregulation of neuronal Ca2+ signaling have been linked to neurodevelopmental disorders including ASDs (Krey and Dolmetsch, 2007). CaV1.3 channels are a major class of L-type VGCCs which constitute an important calcium entry pathway implicated in the regulation of spine morphology and then contribute to the rhythmicity of brain (Stanika et al., 2016). In the brain, VGCCs are vital for coupling of neuronal excitation-transcription, synaptic plasticity and neuronal firing, and de novo missense mutation A760G of CaV1.3 channels has been linked to ASDs (Pinggera et al., 2015). CaV1.3 channels employ two major forms of feedback regulation, voltage-dependent inactivation (VDI) and Ca2+-dependent inactivation (CDI). Limpitikul et al. (2016) recently found that introduction of missense mutation A760G to CaV1.3 severely suppressed the CDI but also potentiated the VDI of CaV1.3 channels, suggesting that deficits of these two feedback regulation appear to increase the [Ca2+]i, thus potentially disrupting both neuronal development and synapse formation, ultimately leading to ASDs. There are many other reports showing that functional mutations in genes encoding VGCCs can lead to ASDs (Splawski et al., 2004; Li et al., 2015). In addition, disruption of the BKCa gene KCNMA1 which encodes the α-subunit of the large conductance Ca2+-activated K+ channel (BKCa) led to both haplo-insufficiency and reduced BKCa activity (Laumonnier et al., 2006). Thus, the reported decrease in BKCa channel activity, together with the reduced inactivation of VGCCs in autistic patients, suggests that ASDs are caused by abnormally sustained increases in intracellular Ca2+ levels (Krey and Dolmetsch, 2007).
Microglial Intracellular Ca2+ Signaling and Importance of TRP Channels
Elevation of [Ca2+]i is also important for the activation of microglia, including proliferation, migration, ramification, de-ramification and release of NO, proinflammatory cytokines and BDNF (Kettenmann et al., 2011). In addition, disruption of microglial Ca2+ homeostasis triggers activation of death programs, which are regulated by the microglia activation status. Treatment of primary cultured microglial cells with thapsigargin or ionomycin induced apoptosis, whereas the same agents applied to lipopolysaccharide (LPS)-activated microglia resulted in necrotic cell death (Nagano et al., 2006). Both apoptotic and necrotic pathways are regulated by [Ca2+]i because the treatment of cultures with BAPTA-AM reduced microglial cell death (Nagano et al., 2006). However, in microglial cells, an application of high [K+]out or glutamate does not elevate [Ca2+]i. This observation is supported by the fact that both VGCCs and NMDARs are not expressed in microglia (Kettenmann et al., 2011). For electrically non-excitable cells including microglia, the primary source of intracellular Ca2+ is the release from intracellular Ca2+ stores and the entry through the ligand-gated and/or store operated Ca2+ channels (Möller, 2002). Microglia contain at least two types of intracellular Ca2+ stores: the endoplasmic reticulum (ER) and mitochondria. The main route for the generation of intracellular Ca2+ signaling is associated with inositol 1,4,5-trisphosphate (InsP3) receptors on the ER membrane. Stimulation of G protein-coupled metabotropic or tyrosine kinase receptors results in the activation of the phospholipase C (PLC), production of two second messengers including the diacylglycerol (DAG) and the InsP3 and the release of Ca2+ from the ER. Importantly, the depletion of ER activates the store-operated Ca2+ entry (SOCE), known as a capacitative Ca2+ influx, mediated by plasmalemmal channels such as calcium release-activated Ca2+ (CRAC) channels and/or transient receptor potential (TRP) channels (Parekh and Putney, 2005). In addition, STIM1, one of ER membrane proteins, senses the filling state of ER Ca2+ and delivers the ER to the plasma membrane where it directly activates Orai1/CRAC channels, thereby facilitating the re-uptake of Ca2+ to ER through the sarco(endo)plasmic reticulum Ca2+-ATPases (SERCA). The concentration of Ca2+ in the ER is precisely controlled by SERCA. Recently, Schmunk et al. (2015) found that dysregulation of InsP3/ER signaling in primary, untransformed skin fibroblasts derived from patients with Fragile X (FXS) or tuberous sclerosis syndromes. This suggests that ASDs might also affect the status of the ER-Ca2+ store in microglial cells.
The influx of Ca2+ through the TRP channels could play some important roles in many inflammatory processes including the activation of microglia (Nilius and Szallasi, 2014). There are seven transient receptor potential canonical (TRPC) channels in mammalian species. Among them, TRPC2 is a pseudogene in humans. The remaining members of the TRPC subfamily are classified into three groups according to sequence homology, TRPC1, canonical TRPC3/C6/C7 and TRPC4/C5. Quantitative comparisons of mRNA expression using real-time RT-PCR showed that TRPM7 > TRPC6 > TRPM2 > TRPC1 > TRPC3 ≥ TRPC4 > TRPC7 > TRPC5 > TRPC2, where “>” denotes a significant difference from the preceding gene, and “≥” indicates a non-significant difference, in microglial cells cultured from rats (Ohana et al., 2009).
Importance of BDNF Signaling in ASDs
BDNF, one of the neurotrophins, has various important roles in cell survival, neurite outgrowth, neuronal differentiation and gene expression in the brain (Thoenen, 1995; Park and Poo, 2013). BDNF is most abundantly expressed in the hippocampus and cerebral cortex and is also involved in the pathophysiology of psychiatric disorders (Sen et al., 2008). Two meta-analysis studies recently showed that neonates diagnosed with ASDs later in life had no association with blood levels of BDNF, while children in the ASD group demonstrated significantly increased BDNF levels compared with healthy controls (Qin et al., 2016; Zheng et al., 2016). These suggest that peripheral BDNF levels might serve as a potential biomarker for the diagnosis of ASDs and further studies are needed to clarify the causal relationship between the symptoms of ASDs and peripheral levels of BDNF.
BDNF binds to the tropomyosin-related kinase B (TrkB) receptor and induces the activation of intracellular signaling pathways, including PLC-γ, phosphatidylinositol 3-kinase (PI3K) and mitogen activated protein kinase-1/2 (MAPK-1/2; Patapoutian and Reichardt, 2001). BDNF rapidly activates the PLC pathway, leading to the generation of InsP3 and the mobilization of intracellular Ca2+ from the ER (Mizoguchi et al., 2003a,b). TRPC3 channels are shown to be necessary for BDNF to increase the density of dendritic spines in rodent hippocampal CA1 pyramidal neurons (Amaral and Pozzo-Miller, 2007). Rett syndrome (RTT) is caused by loss-of-function mutations in MECP2, encoding methyl-CpG-binding protein 2 (Amir et al., 1999). The TRPC3 mRNA and protein levels are lower in CA3 pyramidal neurons of symptomatic Mecp2 mutant mice and chromatin immunoprecipitation (ChIP) identified Trpc3 as a target of MeCP2 transcriptional regulation. BDNF mRNA and protein levels are also lower in Mecp2 mutant hippocampus and dentate gyrus granule cells, which is reflected in impaired activity-dependent release of endogenous BDNF. These results identify the gene encoding TRPC3 channels as a MeCP2 target and suggest a potential therapeutic strategy to boost impaired BDNF signaling in RTT (Li et al., 2012).
Possible Involvement of Microglial Intracellular Ca2+ Signaling Modulated by BDNF in ASDs
In the rodent brain, microglial cells express BDNF mRNA (Elkabes et al., 1996) and secrete BDNF following stimulation with LPS (Nakajima et al., 2001). BDNF released from activated microglia then induces the sprouting of nigrostriatal dopaminergic neurons (Batchelor et al., 1999), causing a shift in the neuronal anion gradient (Coull et al., 2005), or promotes the proliferation and survival of microglia themselves (Zhang et al., 2003). In addition, Parkhurst et al. (2013) showed that the Cre-dependent removal of BDNF from microglia induces deficits in multiple learning tasks mediated by reduction in learning-dependent spine elimination/formation. These suggest that microglia serve important physiological functions in learning and memory by promoting learning-related synapse formation through the BDNF signaling.
We have reported that BDNF induced a sustained increase in [Ca2+]i through binding with the truncated tropomyosin-related kinase B receptor (TrkB-T1), resulting in activation of the PLC pathway and SOCE in rodent microglial cells. Sustained activation of SOCE occurred after a brief BDNF application and contributed to the maintenance of sustained [Ca2+]i elevation. Pretreatment with BDNF significantly suppressed the release of NO from activated microglia. Additionally, pretreatment of BDNF suppressed the IFN-γ-induced increase in [Ca2+]i, along with a rise in basal levels of [Ca2+]i in rodent microglial cells (Mizoguchi et al., 2009). Thereafter, we observed that TRPC3 channels contribute to the maintenance of BDNF-induced sustained intracellular Ca2+ elevation. Immunocytochemical technique and flow cytometry also revealed that BDNF rapidly up-regulated the surface expression of TRPC3 channels in rodent microglial cells. BDNF-induced up-regulation of surface expression of TRPC3 channels also depends on activation of the PLC pathway, as previously shown by others (van Rossum et al., 2005). In addition, pretreatment with BDNF suppressed the production of NO induced by TNFα, which was prevented by co-administration of a selective TRPC3 inhibitor, Pyr3. These suggest that TRPC3 channels could be important for the BDNF-induced suppression of the NO production in activated microglia. We first showed direct evidence that rodent microglial cells are able to respond to BDNF and TRPC3 channels could also play important roles in microglial functions. Hall et al. (2009) have previously demonstrated the implication of the basal level of [Ca2+]i in the activation of rodent microglia, including NO production. BDNF-induced elevation of basal levels of [Ca2+]i could regulate the microglial intracellular signal transduction to suppress the release of NO induced by IFN-γ (Hoffmann et al., 2003; Mizoguchi et al., 2009). We observed that pretreatment with BDNF also suppressed the production of NO in murine microglial cells activated by TNFα, which was prevented by co-administration of Pyr3. We also found that pretreatment with both BDNF and Pyr3 did not elevate the basal [Ca2+]i in rodent microglial cells. These suggest that BDNF-induced elevation of basal levels of [Ca2+]i mediated by TRPC3 channels could be important for the BDNF-induced suppression of NO production in rodent microglial cells. Although the mechanism underlying the activation of TRPCs via PLC stimulation is still not completely resolved, TRPC3, like TRPC6 and TRPC7, can be activated directly by DAG. The trafficking of TRPC3 channels to the plasma membrane depends on interactions with Cav-1, Homer1, PLC-γ, VAMP2 and RFN24 (de Souza and Ambudkar, 2014). In addition, phagocytic activity is suppressed by pharmacological inhibitors of SOCE in murine microglial cells (Heo et al., 2015). Altogether, these suggest that BDNF might have an anti-inflammatory effect through the inhibition of microglial activation and TRPC3 could play important roles in not only inflammatory processes but also formation of synapse through the modulation of microglial phagocytic activity in the brain. We need additional studies to identify the molecular mechanisms that determine the trafficking and activity of TRPC3 channels and what underlies the BDNF-induced up-regulation of surface TRPC3 channels in these mechanisms (Mizoguchi et al., 2014a,b).
Using multiple models including individual’s dental pulp cells (DPCs), neural cells derived from induced pluripotent stem cell (iPSCs) and mouse models, Griesi-Oliveira et al. (2015) recently reported that loss-of-function mutations of TRPC6 is a novel predisposing factor for ASDs, suggesting that dysfunction of Ca2+ signaling mediated by TRPC6 contributes to altered neuronal development, neuronal morphology and synaptic function in ASDs. It is not well known whether TRPC6 channels of microglia also serve important physiological roles in the alteration of synaptic function in ASDs.
Future Prospects
Elevation of intracellular Ca2+ is important for the activation of microglial cell functions, including proliferation, release of NO, cytokines and BDNF. It has been shown that alteration of intracellular Ca2+ signaling underlies the pathophysiology of neurodevelopmental disorders including ASDs. BDNF induces a sustained intracellular Ca2+ elevation through the upregulation of the surface expression of TRPC3 channels in rodent microglial cells. Microglial cells are able to respond to BDNF, which may be important for the regulation of inflammatory responses, and may also be involved in the normal development of CNS. BDNF is first synthesized as proBDNF protein. ProBDNF is then either proteolytically cleaved intracellularly or by extracellular proteases, such as metalloproteinases and plasmin, to mature BDNF. Interestingly, interaction of mature neurotrophins with Trk receptors leads to cell survival, whereas binding of proBDNF to p75NTR leads to apoptosis. In addition, mature BDNF and proBDNF facilitates long-term potentiation (LTP) and long-term depression (LTD) at the hippocampal CA1 synapses, respectively. Thus, Trk and p75NTR preferentially bind mature- and pro-neurotrophins, respectively, to elicit opposing biological responses in the CNS (Greenberg et al., 2009). Indeed, a recently published report shows that pruning of spines promoted by proBDNF is mediated by the p75NTR-RhoA, while maturation of spines induced by BDNF is through the stimulation of TrkB-Rac1 signaling (Orefice et al., 2016). However, the effects of proBDNF on microglial cells are not fully understood. Thus, further work will be needed to elucidate the role of proBDNF on microglial cells by focusing on TRPC channels (Figure 1).
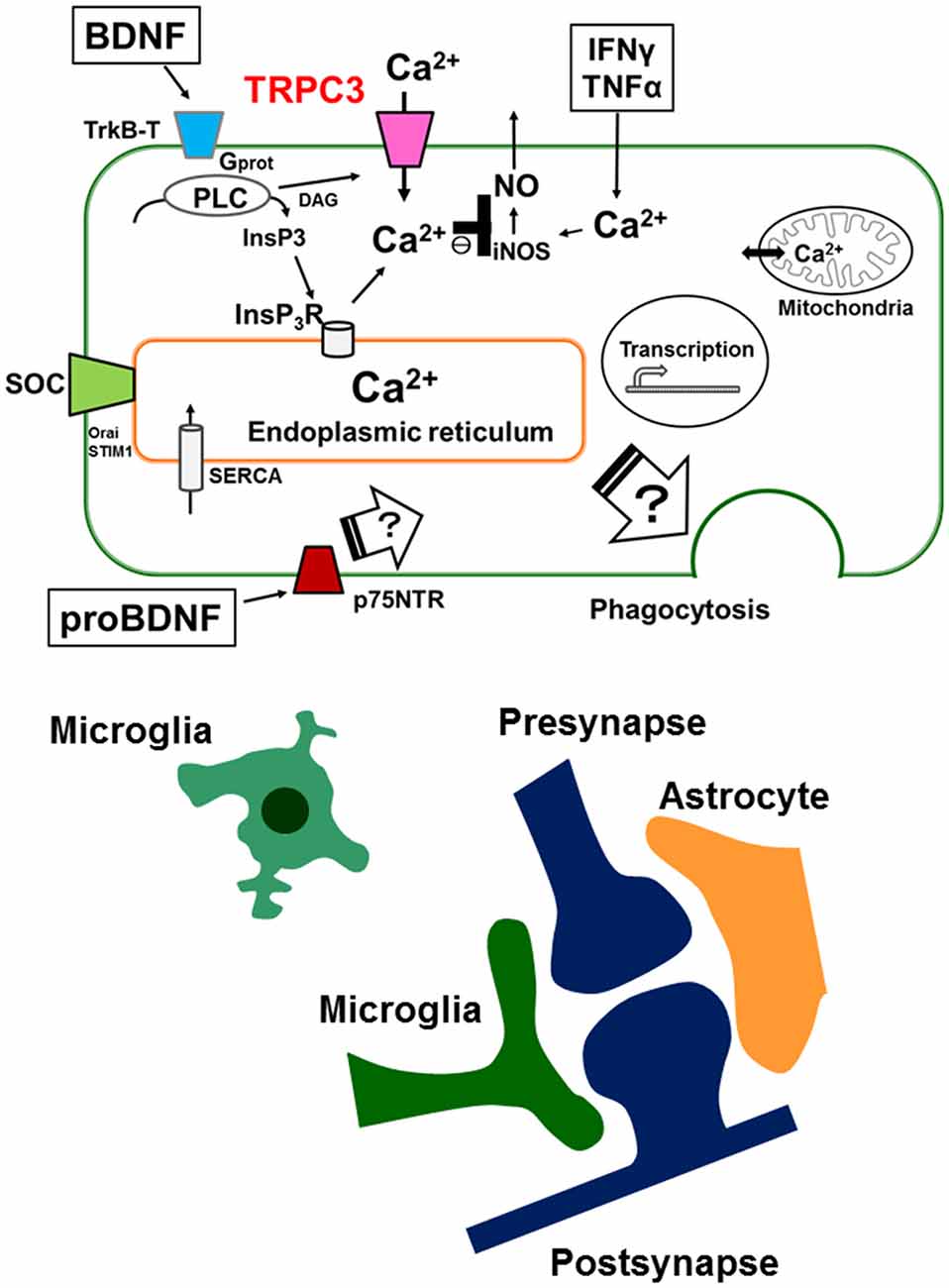
Figure 1. Schematic illustration representing the microglial intracellular Ca2+ signaling mediated by canonical transient receptor potential 3 (TRPC3) channels and the tripartite synapse. In microglia, brain-derived neurotrophic factor (BDNF) induces a sustained increase in [Ca2+]i through binding of the truncated TrkB receptors (TrkB-T), resulting in activation of the phospholipase C (PLC) pathway. Up-regulation of cell surface TRPC3 channels occurs after a brief treatment with BDNF and contributes to the maintenance of BDNF-induced sustained intracellular Ca2+ elevation. BDNF-induced elevation of basal levels of [Ca2+]i mediated by TRPC3 channels could be important for the BDNF-induced suppression of nitric oxide (NO) production induced by TNFα or IFNγ. Microglial intracellular Ca2+ signaling is also important for microglial functions such as phagocytosis in the brain. The tripartite synapse consists of the presynaptic (glutamatergic) terminal, postsynaptic terminal, astrocytes and microglia. Dysregulation of normal microglial functions including incorrect synaptic pruning, failure of phagocytosis of apoptotic neurons and abnormal secretion of inflammatory cytokines might be underlying mechanisms of neurodevelopmental disorders such as autism spectrum disorders (ASDs). On the other hand, the effects of proBDNF on microglial functions are not fully understood. Further work will be needed to elucidate the role of proBDNF on microglial cells by focusing on intracellular Ca2+ signaling mediated by TRPC channels.
Oxytocin (OT) is a pituitary neuropeptide hormone synthesized from the paraventricular and supra-optic nuclei within the hypothalamus. Like other neuropeptides, OT can modulate a wide range of neurotransmitter and neuromodulator activities. OT is secreted into the systemic circulation to act as a hormone, thereby influencing several body functions. OT plays a pivotal role in parturition, milk let-down and maternal behavior and has been demonstrated to be important in the formation of pair bonding between mother and infants as well as in mating pairs. Furthermore, OT has been proven to play a key role in the regulation of several behaviors associated with neuropsychiatric disorders, including social interactions, social memory response to social stimuli, decision-making in the context of social interactions, feeding behavior, emotional reactivity, etc. An increasing body of evidence suggests that dysregulation of the oxytocinergic system might be involved in the pathophysiology of neurodevelopmental disorders such as ASDs (Romano et al., 2016). Using functional magnetic resonance imaging, single-dose intranasal administration of OT was shown to improve the frequency of the nonverbal information-based judgments with a shorter response time and the brain activity of the medial prefrontal cortex in participants with ASDs (Watanabe et al., 2014). Thus, there is a significant potential for OT to ameliorate some aspects of the persistent and debilitating social impairments in individuals with ASDs (Alvares et al., 2016). Although there is no clinical use of minocycline in ASDs, prenatal minocycline treatment can alter the expression of PSD-95 and ameliorate abnormal mother-infant communication in oxytocin receptor (Oxtr)-deficient mice (Miyazaki et al., 2016). This finding suggests that minocycline has a therapeutic potential for the development of OT/Oxtr-mediated ASD-like phenotypes (Nakagawa and Chiba, 2016). In addition, OT suppressed both the mRNA expression of TNFα, IL-1β, COX-2 and iNOS and the elevation of [Ca2+]i in LPS-stimulated microglia cells (Yuan et al., 2016). These suggest that OT would be a potential therapeutic agent for alleviating neuro-inflammatory processes in ASDs. However, the effects of OT on microglial intracellular Ca2+ signaling are not fully understood. Thus, it will be important to study the effect of OT on microglial cells especially by focusing on TRPC channels.
Conclusions
There is increasing evidence suggesting that pathophysiology of neurodevelopmental disorders is related to the inflammatory responses mediated by microglial cells. In addition, recent advances in the understanding of microglial functions suggest an important role for these cells in the normal development of CNS in addition to their traditional role as immune cells of the brain. Dysregulation of normal microglial functions such as regulation of programmed cell death and/or synaptic pruning is supposed to be increasingly implicated in ASDs associated with cognitive deficits. These findings have resulted in a new model of the synapse as “tripartite,” recognizing the important role of not just neurons and astrocytes but also microglia in the normal physiological function of the brain. We now need to explore the emerging dual, inflammatory and non-inflammatory, roles of microglia in the brain and recent findings reinforce the importance of intracellular Ca2+ signaling for microglial functions in both normal neurodevelopment and their potential contributing to neurodevelopmental disorders such as ASDs.
Author Contributions
YM and AM wrote this article.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by grants from SENSHIN Medical Research Foundation (to YM), the Japan Agency for Medical Research and Development (AMED; to YM and AM), the Japan Society for the Promotion of Science—KAKENHI ((C) to YM and AM).
References
Aguzzi, A., Barres, B. A., and Bennett, M. L. (2013). Microglia: scapegoat, saboteur, or something else? Science 339, 156–161. doi: 10.1126/science.1227901
Alvares, G. A., Quintana, D. S., and Whitehouse, A. J. (2016). Beyond the hype and hope: critical considerations for intranasal oxytocin research in autism spectrum disorder. Autism Res. 10, 25–41. doi: 10.1002/aur.1692
Amaral, M. D., and Pozzo-Miller, L. (2007). TRPC3 channels are necessary for brain-derived neurotrophic factor to activate a nonselective cationic current and to induce dendritic spine formation. J. Neurosci. 27, 5179–5189. doi: 10.1523/JNEUROSCI.5499-06.2007
Amir, R. E., Van den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188. doi: 10.1038/13810
Batchelor, P. E., Liberatore, G. T., Wong, J. Y., Porritt, M. J., Frerichs, F., Donnan, G. A., et al. (1999). Activated macrophages and microglia induce dopaminergic sprouting in the injured striatum and express brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor. J. Neurosci. 19, 1708–1716.
Berridge, M. J. (1998). Neuronal calcium signaling. Neuron 21, 13–26. doi: 10.1016/s0896-6273(00)80510-3
Chez, M. G., Dowling, T., Patel, P. B., Khanna, P., and Kominsky, M. (2007). Elevation of tumor necrosis factor-α in cerebrospinal fluid of autistic children. Pediatr. Neurol. 36, 361–365. doi: 10.1016/j.pediatrneurol.2007.01.012
Coull, J. A., Beggs, S., Boudreau, D., Boivin, D., Tsuda, M., Inoue, K., et al. (2005). BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 438, 1017–1021. doi: 10.1038/nature04223
Cunningham, C. (2013). Microglia and neurodegeneration: the role of systemic inflammation. Glia 61, 71–90. doi: 10.1002/glia.22350
Derecki, N. C., Cronk, J. C., and Kipnis, J. (2013). The role of microglia in brain maintenance: implications for Rett syndrome. Trends Immunol. 34, 144–150. doi: 10.1016/j.it.2012.10.002
de Souza, L. B., and Ambudkar, I. S. (2014). Trafficking mechanisms and regulation of TRPC channels. Cell Calcium 56, 43–50. doi: 10.1016/j.ceca.2014.05.001
Edmonson, C. A., Ziats, M. N., and Rennert, O. M. (2016). A non-inflammatory role for microglia in autism spectrum disorders. Front. Neurol. 7:9. doi: 10.3389/fneur.2016.00009
Elkabes, S., DiCicco-Bloom, E. M., and Black, I. B. (1996). Brain microglia/macrophages express neurotrophins that selectively regulate microglial proliferation and function. J. Neurosci. 16, 2508–2521.
Estes, M. L., and McAllister, A. K. (2015). Immune mediators in the brain and peripheral tissues in autism spectrum disorder. Nat. Rev. Neurosci. 16, 469–486. doi: 10.1038/nrn3978
Goines, P. E., and Ashwood, P. (2013). Cytokine dysregulation in autism spectrum disorders (ASD): possible role of the environment. Neurotoxicol. Teratol. 36, 67–81. doi: 10.1016/j.ntt.2012.07.006
Greenberg, M. E., Xu, B., Lu, B., and Hempstead, B. L. (2009). New insights in the biology of BDNF synthesis and release: implications in CNS function. J. Neurosci. 29, 12764–12767. doi: 10.1523/JNEUROSCI.3566-09.2009
Griesi-Oliveira, K., Acab, A., Gupta, A. R., Sunaga, D. Y., Chailangkarn, T., Nicol, X., et al. (2015). Modeling non-syndromic autism and the impact of TRPC6 disruption in human neurons. Mol. Psychiatry 20, 1350–1365. doi: 10.1038/mp.2014.141
Gupta, S., Aggarwal, S., Rashanravan, B., and Lee, T. (1998). Th1- and Th2-like cytokines in CD4+ and CD8+ T cells in autism. J. Neuroimmunol. 85, 106–109. doi: 10.1016/s0165-5728(98)00021-6
Hall, A. A., Herrera, Y., Ajmo, C. T. Jr., Cuevas, J., and Pennypacker, K. R. (2009). Sigma receptors suppress multiple aspects of microglial activation. Glia 57, 744–754. doi: 10.1002/glia.20802
Heo, D. K., Lim, H. M., Nam, J. H., Lee, M. G., and Kim, J. Y. (2015). Regulation of phagocytosis and cytokine secretion by store-operated calcium entry in primary isolated murine microglia. Cell. Signal. 27, 177–186. doi: 10.1016/j.cellsig.2014.11.003
Hoffmann, A., Kann, O., Ohlemeyer, C., Hanisch, U. K., and Kettenmann, H. (2003). Elevation of basal intracellular calcium as a central element in the activation of brain macrophages (microglia): suppression of receptor-evoked calcium signaling and control of release function. J. Neurosci. 23, 4410–4419.
Hong, S., Beja-Glasser, V. F., Nfonoyim, B. M., Frouin, A., Li, S., Ramakrishnan, S., et al. (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. doi: 10.1126/science.aad8373
Kato, T. A., Watabe, M., and Kanba, S. (2013). Neuron-glia interaction as a possible glue to translate the mind-brain gap: a novel multi-dimensional approach toward psychology and psychiatry. Front. Psychiatry 4:139. doi: 10.3389/fpsyt.2013.00139
Kettenmann, H., Hanisch, U.-K., Noda, M., and Verkhratsky, A. (2011). Physiology of microglia. Physiol. Rev. 91, 461–553. doi: 10.1152/physrev.00011.2010
Krey, J. F., and Dolmetsch, R. E. (2007). Molecular mechanisms of autism: a possible role for Ca2+ signaling. Curr. Opin. Neurobiol. 17, 112–119. doi: 10.1016/j.conb.2007.01.010
Lai, M. C., Lombardo, M. V., and Baron-Cohen, S. (2014). Autism. Lancet 383, 896–910. doi: 10.1016/S0140-6736(13)61539-1
Laumonnier, F., Roger, S., Guérin, P., Molinari, F., M’Rad, R., Cahard, D., et al. (2006). Association of a functional deficit of the BKCa channel, a synaptic regulator of neuronal excitability, with autism and mental retardation. Am. J. Psychiatry 163, 1622–1629. doi: 10.1176/ajp.2006.163.9.1622
Li, W., Calfa, G., Larimore, J., and Pozzo-Miller, L. (2012). Activity-dependent BDNF release and TRPC signaling is impaired in hippocampal neurons of Mecp2 mutant mice. Proc. Natl. Acad. Sci. U S A 109, 17087–17092. doi: 10.1073/pnas.1205271109
Li, J., You, Y., Yue, W., Jia, M., Yu, H., Lu, T., et al. (2015). Genetic evidence for possible involvement of the calcium channel gene CACNA1A in Autism pathogenesis in chinese han population. PLoS One 10:e0142887. doi: 10.1371/journal.pone.0142887
Limpitikul, W. B., Dick, I. E., Ben-Johny, M., and Yue, D. T. (2016). An autism-associated mutation in CaV1.3 channels has opposing effects on voltage- and Ca2+-dependent regulation. Sci. Rep. 6:27235. doi: 10.1038/srep27235
Macht, V. A. (2016). Neuro-immune interactions across development: a look at glutamate in the prefrontal cortex. Neurosci. Biobehav. Rev. 71, 267–280. doi: 10.1016/j.neubiorev.2016.08.039
Marín-Teva, J. L., Dusart, I., Colin, C., Gervais, A., van Rooijen, N., and Mallat, M. (2004). Microglia promote the death of developing Purkinje cells. Neuron 41, 535–547. doi: 10.1016/s0896-6273(04)00069-8
Miyazaki, S., Hiraoka, Y., Hidema, S., and Nishimori, K. (2016). Prenatal minocycline treatment alters synaptic protein expression and rescues reduced mother call rate in oxytocin receptor-knockout mice. Biochem. Biophys. Res. Commun. 472, 319–323. doi: 10.1016/j.bbrc.2016.02.109
Mizoguchi, Y., Ishibashi, H., and Nabekura, J. (2003a). The action of BDNF on GABAA currents changes from potentiating to suppressing during maturation of rat hippocampal CA1 pyramidal neurons. J. Physiol. 548, 703–709. doi: 10.1113/jphysiol.2003.038935
Mizoguchi, Y., Kanematsu, T., Hirata, M., and Nabekura, J. (2003b). A rapid increase in the total number of cell surface functional GABAA receptors induced by brain-derived neurotrophic factor in rat visual cortex. J. Biol. Chem. 278, 44097–44102. doi: 10.1074/jbc.M305872200
Mizoguchi, Y., Monji, A., Kato, T., Seki, Y., Gotoh, L., Horikawa, H., et al. (2009). Brain-derived neurotrophic factor induces sustained elevation of intracellular Ca2+ in rodent microglia. J. Immunol. 183, 7778–7786. doi: 10.4049/jimmunol.0901326
Mizoguchi, Y., Kato, T. A., Horikawa, H., and Monji, A. (2014a). Microglial intracellular Ca2+ signaling as a target of antipsychotic actions for the treatment of schizophrenia. Front. Cell. Neurosci. 8:370. doi: 10.3389/fncel.2014.00370
Mizoguchi, Y., Kato, T. A., Seki, Y., Ohgidani, M., Sagata, N., Horikawa, H., et al. (2014b). BDNF induces sustained intracellular Ca2+ elevation through the upregulation of surface TRPC3 channels in rodent microglia. J. Biol. Chem. 289, 18549–18555. doi: 10.1074/jbc.M114.555334
Monji, A., Kato, T. A., Mizoguchi, Y., Horikawa, H., Seki, Y., Kasai, M., et al. (2013). Neuroinflammation in schizophrenia especially focused on the role of microglia. Prog. Neuropsychopharmacol. Biol. Psychiatry 42, 115–121. doi: 10.1016/j.pnpbp.2011.12.002
Monji, A., Maezawa, I., Mizoguchi, Y., Kato, T. A., and Jin, L.-W. (2014). “Neurodevelopmental and neuropsychiatric disorders,” in Microglia in Health and Disease, eds M.-E. Tremblay and A. Sierra (New York, NY: Springer Science + Business Media), 345–372.
Morgan, J. T., Chana, G., Pardo, C. A., Achim, C., Semendeferi, K., Buckwalter, J., et al. (2010). Microglial activation and increased microglial density observed in the dorsolateral prefrontal cortex in autism. Biol. Psychiatry 68, 368–376. doi: 10.1016/j.biopsych.2010.05.024
Nagano, T., Kimura, S. H., Takai, E., Matsuda, T., and Takemura, M. (2006). Lipopolysaccharide sensitizes microglia toward Ca2+-induced cell death: mode of cell death shifts from apoptosis to necrosis. Glia 53, 67–73. doi: 10.1002/glia.20260
Nakagawa, Y., and Chiba, K. (2016). Involvement of neuroinflammation during brain development in social cognitive deficits in autism spectrum disorder and schizophrenia. J. Pharmacol. Exp. Ther. 358, 504–515. doi: 10.1124/jpet.116.234476
Nakajima, K., Honda, S., Tohyama, Y., Imai, Y., Kohsaka, S., and Kurihara, T. (2001). Neurotrophin secretion from cultured microglia. J. Neurosci. Res. 65, 322–331. doi: 10.1002/jnr.1157
Nilius, B., and Szallasi, A. (2014). Transient receptor potential channels as drug targets: from the science of basic research to the art of medicine. Pharmacol. Rev. 66, 676–814. doi: 10.1124/pr.113.008268
Ohana, L., Newell, E. W., Stanley, E. F., and Schlichter, L. C. (2009). The Ca2+ release-activated Ca2+ current (ICRAC) mediates store-operated Ca2+ entry in rat microglia. Channels 3, 129–139. doi: 10.4161/chan.3.2.8609
Orefice, L. L., Shih, C. C., Xu, H., Waterhouse, E. G., and Xu, B. (2016). Control of spine maturation and pruning through proBDNF synthesized and released in dendrites. Mol. Cell. Neurosci. 71, 66–79. doi: 10.1016/j.mcn.2015.12.010
Parekh, A. B., and Putney, J. W. Jr. (2005). Store-operated calcium channels. Physiol. Rev. 85, 757–810. doi: 10.1152/physrev.00057.2003
Pinggera, A., Lieb, A., Benedetti, B., Lampert, M., Monteleone, S., Liedl, K. R., et al. (2015). CACNA1D de novo mutations in autism spectrum disorders activate Cav1.3 L-type calcium channels. Biol. Psychiatry 77, 816–822. doi: 10.1016/j.biopsych.2014.11.020
Park, H., and Poo, M. M. (2013). Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 14, 7–23. doi: 10.1038/nrn3379
Parkhurst, C. N., Yang, G., Ninan, I., Savas, J. N., Yates, J. R. III., Lafaille, J. J., et al. (2013). Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155, 1596–1609. doi: 10.1016/j.cell.2013.11.030
Patapoutian, A., and Reichardt, L. F. (2001). Trk receptors: mediators of neurotrophin action. Curr. Opin. Neurobiol. 11, 272–280. doi: 10.1016/s0959-4388(00)00208-7
Qin, X. Y., Feng, J. C., Cao, C., Wu, H. T., Loh, Y. P., and Cheng, Y. (2016). Association of peripheral blood levels of brain-derived neurotrophic factor with autism spectrum disorder in children: a systematic review and meta-analysis. JAMA Pediatr. 170, 1079–1086. doi: 10.1001/jamapediatrics.2016.1626
Romano, A., Tempesta, B., Micioni Di Bonaventura, M. V., and Gaetani, S. (2016). From autism to eating disorders and more: the role of oxytocin in neuropsychiatric disorders. Front. Neurosci. 9:497. doi: 10.3389/fnins.2015.00497
Salter, M. W., and Beggs, S. (2014). Sublime microglia: expanding roles for the guardians of the CNS. Cell 158, 15–24. doi: 10.1016/j.cell.2014.06.008
Sen, S., Duman, R., and Sanacora, G. (2008). Serum brain-derived neurotrophic factor, depression and antidepressant medications: meta-analyses and implications. Biol. Psychiatry 64, 527–532. doi: 10.1016/j.biopsych.2008.05.005
Schmunk, G., Boubion, B. J., Smith, I. F., Parker, I., and Gargus, J. J. (2015). Shared functional defect in IP3R-mediated calcium signaling in diverse monogenic autism syndromes. Transl. Psychiatry 5:e643. doi: 10.1038/tp.2015.123
Schwartz, M., Kipnis, J., Rivest, S., and Prat, A. (2013). How do immune cells support and shape the brain in health, disease, and aging? J. Neurosci. 33, 17587–17596. doi: 10.1523/JNEUROSCI.3241-13.2013
Simms, B. A., and Zamponi, G. W. (2014). Neuronal voltage-gated calcium channels: structure, function and dysfunction. Neuron 82, 24–45. doi: 10.1016/j.neuron.2014.03.016
Smith, A. M., and Dragunow, M. (2014). The human side of microglia. Trends Neurosci. 37, 125–135. doi: 10.1016/j.tins.2013.12.001
Splawski, I., Timothy, K. W., Sharpe, L. M., Decher, N., Kumar, P., Bloise, R., et al. (2004). CaV1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119, 19–31. doi: 10.1016/j.cell.2004.09.011
Stanika, R., Campiglio, M., Pinggera, A., Lee, A., Striessnig, J., Flucher, B. E., et al. (2016). Splice variants of the CaV1.3 L-type calcium channel regulate dendritic spine morphology. Sci. Rep. 6:34528. doi: 10.1038/srep34528
Stephan, A. H., Barres, B. A., and Stevens, B. (2012). The complement system: an unexpected role in synaptic pruning during development and disease. Annu. Rev. Neurosci. 35, 369–389. doi: 10.1146/annurev-neuro-061010-113810
Suzuki, K., Sugihara, G., Ouchi, Y., Nakamura, K., Futatsubashi, M., Takebayashi, K., et al. (2013). Microglial activation in young adults with autism spectrum disorder. JAMA Psychiatry 70, 49–58. doi: 10.1001/jamapsychiatry.2013.272
Thoenen, H. (1995). Neurotrophins and neuronal plasticity. Science 270, 593–598. doi: 10.1126/science.270.5236.593
van Rossum, D. B., Patterson, R. L., Sharma, S., Barrow, R. K., Kornberg, M., Gill, D. L., et al. (2005). Phospholipase Cγ1 controls surface expression of TRPC3 through an intermolecular PH domain. Nature 434, 99–104. doi: 10.1038/nature03340
Vargas, D. L., Nascimbene, C., Krishnan, C., Zimmerman, A. W., and Pardo, C. A. (2005). Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. 57, 67–81. doi: 10.1002/ana.20315
Voineagu, I., Wang, X., Johnston, P., Lowe, J. K., Tian, Y., Horvath, S., et al. (2011). Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474, 380–384. doi: 10.1038/nature10110
Wake, H., Moorhouse, A. J., Miyamoto, A., and Nabekura, J. (2013). Microglia: actively surveying and shaping neuronal circuit structure and function. Trends Neurosci. 36, 209–217. doi: 10.1016/j.tins.2012.11.007
Watanabe, T., Abe, O., Kuwabara, H., Yahata, N., Takano, Y., Iwashiro, N., et al. (2014). Mitigation of sociocommunicational deficits of autism through oxytocin-induced recovery of medial prefrontal activity: a randomized trial. JAMA Psychiatry 71, 166–175. doi: 10.1001/jamapsychiatry.2013.3181
Wills, S., Cabanlit, M., Bennett, J., Ashwood, P., Amaral, D. G., and Van de Water, J. (2009). Detection of autoantibodies to neural cells of the cerebellum in the plasma of subjects with autism spectrum disorders. Brain. Behav. Immun. 23, 64–74. doi: 10.1016/j.bbi.2008.07.007
Yuan, L., Liu, S., Bai, X., Gao, Y., Liu, G., Wang, X., et al. (2016). Oxytocin inhibits lipopolysaccharide-induced inflammation in microglial cells and attenuates microglial activation in lipopolysaccharide-treated mice. J. Neuroinflammation 13:77. doi: 10.1186/s12974-016-0541-7
Zhang, J., Geula, C., Lu, C., Koziel, H., Hatcher, L. M., and Roisen, F. J. (2003). Neurotrophins regulate proliferation and survival of two microglial cell lines in vitro. Exp. Neurol. 183, 469–481. doi: 10.1016/s0014-4886(03)00222-x
Zhan, Y., Paolicelli, R. C., Sforazzini, F., Weinhard, L., Bolasco, G., Pagani, F., et al. (2014). Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat. Neurosci. 17, 400–406. doi: 10.1038/nn.3641
Zheng, Z., Zhang, L., Zhu, T., Huang, J., Qu, Y., and Mu, D. (2016). Peripheral brain-derived neurotrophic factor in autism spectrum disorder: a systematic review and meta-analysis. Sci. Rep. 6:31241. doi: 10.1038/srep31241
Keywords: microglia, calcium signaling, BDNF, TRPC3 channels, proBDNF, oxytocin, synapse development, autism spectrum disorders
Citation: Mizoguchi Y and Monji A (2017) Microglial Intracellular Ca2+ Signaling in Synaptic Development and its Alterations in Neurodevelopmental Disorders. Front. Cell. Neurosci. 11:69. doi: 10.3389/fncel.2017.00069
Received: 25 November 2016; Accepted: 24 February 2017;
Published: 17 March 2017.
Edited by:
Mark Nicholas Ziats, University of Michigan Health System, USAReviewed by:
Andrew MacLean, Tulane University School of Medicine, USAChristiane Charriaut-Marlangue, Institut National de la Santé et de la Recherche Médicale (INSERM), France
Copyright © 2017 Mizoguchi and Monji. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yoshito Mizoguchi, eW1pem9AY2Muc2FnYS11LmFjLmpw