 Xuebin Qu1†
Xuebin Qu1† Ruiqin Yao
Ruiqin Yao- 1Department of Neurobiology, Xuzhou Medical University, Xuzhou, China
- 2Department of Neurology, Affiliated Hospital of Xuzhou Medical University, Xuzhou, China
Specific miRNAs are involved in the pathogenesis of multiple sclerosis (MS), during which IL-17-producing CD4+ T helper (Th17) cells accumulate in the central nervous system (CNS). In this study, we identified levels of miR-384 as significantly increased in mice with experimental autoimmune encephalomyelitis (EAE), an animal model of MS. Over-expression of miR-384 in vivo led to severe EAE, characterized by exacerbated demyelination, and increased inflammatory cell infiltration of the spinal cord; inhibition of miR-384 reversed these changes. Both the percentage of Th17, and ratio of Th17/regulatory T (Treg), cells were elevated in miR-384-transfected EAE mice, which was consistent with the observed upregulation of expression of IL-17 and the Th17 lineage-specific transcription factor, RORγt. Importantly, transfer of miR-384 overexpressing naïve T cells from wild-type (WT) to Rag1−/− mice, which are deficient in functional autologous T and B cells, led to aggravated EAE pathogenesis, while an miR-384 inhibited group was protected from EAE. Moreover, miR-384 promoted differentiation of naïve T cells into Th17 cells in vitro. Furthermore, target prediction and dual luciferase reporter assays demonstrated that suppressor of cytokine signaling 3 (SOCS3), a gene encoding protein with an established role in Th17 differentiation, was a direct target of miR-384. Our results demonstrate an important role for miR-384 in regulation of the Th17/Treg ratio during the pathogenesis of EAE, indicating that this molecule may have potential as a biomarker and/or therapeutic target in MS.
Introduction
Multiple sclerosis (MS) in humans, and its animal model, experimental autoimmune encephalomyelitis (EAE), are characterized by demyelination in the central nervous system (CNS), as a result of inappropriate inflammation and infiltration of CD4+ T cells (Jadidi-Niaragh and Mirshafiey, 2011; Rostami and Ciric, 2013). An imbalance between IL-17-producing CD4+ T helper (Th17) cells, which express the lineage-specific transcription factor, RORγt (Ciofani et al., 2012), and activated regulatory T (Treg) cells expressing Foxp3 (Gerriets et al., 2016) has a significant role in induction of inflammatory immune responses (Korn et al., 2009; Liu Y. et al., 2015; Zhu et al., 2016). This Th17/Treg imbalance is associated with various autoimmune and inflammatory diseases, including MS (Tzartos et al., 2008; Jamshidian et al., 2013, 2015; Lochner et al., 2015). An approach to suppress Th17 cell differentiation and correct Th17/Treg imbalances could be an effective treatment for chronic inflammatory diseases, such as MS.
Although many Th17-specific regulators have been investigated, the mechanisms underlying abnormal Th17 generation during MS pathogenesis remain elusive. MicroRNAs (miRNAs) are a class of endogenous non-coding RNAs, of approximately 23 nucleotides in length, that regulate gene expression at the post-transcriptional level (Krol et al., 2010) and are associated with various autoimmune diseases (Singh et al., 2013; Chen et al., 2016). Multiple miRNAs have roles in the development of EAE through targeting specific proteins in relevant Th17 cell pathways. Over-expression of miR-326, which targets the transcription factor, Ets-1, results in increased numbers of Th17 cells and severe EAE in vivo (Du et al., 2009). In mice with EAE, the expression of miR-155 is elevated in CD4+T cells, and mice lacking miR-155 develop milder EAE, and have fewer Th17 cells and less severe CNS inflammation (O’Connell et al., 2010; Murugaiyan et al., 2011). MiR-21 also enhances Th17 generation and mediates EAE development by promoting TGF-β signaling and limiting the inhibitory effects of IL-2 (Murugaiyan et al., 2015). In addition, miR-20b, miR-23b and miR-301a are closely associated with EAE pathogenesis through regulation of factors inducing Th17 generation, such as RORγt, STAT3 (Zhu et al., 2014), TAB2, TAB3, IKK-α (Zhu et al., 2012) and PIAS3 in the IL-6/23-STAT3 pathway (Mycko et al., 2012). Hence there is a clear and close relationship among miRNA expression, Th17 cell differentiation, and EAE pathogenesis, suggesting that specific miRNAs could serve as potential biomarkers or therapeutic targets in EAE and MS (Li et al., 2010; Zhu et al., 2012; Zhernakova et al., 2013).
In this study, we discovered that the expression of a specific miRNA, miR-384, is dramatically upregulated in EAE mice, suggesting a possible role for miR-384 in this disease. Recently, a role for miR-384 in tumor progression and neurotoxicity was identified; however, there are no previous reports of its effects in EAE pathogenesis. We found that over-expression of miR-384 resulted in promotion of Th17 cell differentiation in vitro and severe EAE in vivo, due to an excess of Th17 cells, leading to an imbalance in the Th17/Treg ratio. Hence, our results indicate that miR-384 is involved in the Th17-mediated pathogenesis of EAE.
Materials and Methods
Mice
C57BL/6 wild-type (WT) and Rag1−/− mice were purchased from SLAC Laboratory Animal Co., Ltd. (Shanghai, China) and the Model Animal Research Center of Nanjing University (Nanjing, China), respectively. All experimental mice were maintained under specific-pathogen-free conditions in the animal facility of Xuzhou Medical University (Xuzhou, China). All experiments were performed in accordance with the guidelines set out by the US NIH regarding the care and use of animals for experimental procedures as well as in compliance with the Provisions and General Recommendations of the Chinese Experimental Animal Administration Legislation. The study had institutional approval from the Xuzhou Medical University Experimental Animal Ethics Committee.
Induction of EAE
C57BL/6 WT mice (7–8 weeks old) were immunized subcutaneously with 200 μg myelin oligodendrocyte glycoprotein (MOG) peptide 35–55 (GL Biochem, China) in complete Freund’s adjuvant (Sigma-Aldrich, St. Louis, MO, USA) containing 4 mg/mL heat-killed Mycobacterium tuberculosis H37Ra (Difco, USA). Each mouse received 200 ng pertussis toxin (Sigma-Aldrich, St. Louis, MO, USA) twice, administered intraperitoneally (i.p.) on the day of immunization and 48 h later. Mice immunized without MOG were used as sham controls. Clinical assessment of EAE was performed daily after disease induction, and disease severity scored according to the following criteria (Du et al., 2009): (0) no clinical symptoms; (1) tail paralysis; (2) hind limb weakness or partial paralysis; (3) complete paralysis of two hind limbs; (4) paralysis of both forelimbs and hind limbs; and (5) moribund or dead.
Isolation and Induction of Naïve T Cells
Splenocytes (SPs) or peripheral blood (PB) lymphocytes, isolated from 5- to 6-week-old C57BL/6 mice, were used to generate single-cell suspensions depleted of red-blood-cells. Naïve or CD4+ T cells were purified by magnetic cell sorting according to the manufacturer’s instructions (Miltenyi Biotec, Germany). For adoptive transfer studies, each recipient Rag1−/− mouse was injected intravenously in tail veins with 5 × 106 cells on the day before EAE induction. Successful reconstitution of cells was verified by determining GFP expression from the lentivirus vector (LV).
For Th17 differentiation, purified naïve T cells were cultured for 3 days under Th17-cell polarizing conditions: RPMI-1640 medium containing 10% fetal calf serum, 1 mM glutamine, 0.1 mM beta-mercaptoethanol, 1% nonessential amino acids (Sigma–Aldrich, St. Louis, MO, USA), 5 ng/mL IL-2 (R&D Systems, Minneapolis, MN, USA), 20 ng/mL IL-6, 5 ng/mL transforming growth factor-β, 10 ng/mL IL-23, 2 μg/mL anti-IL-4, 2 μg/mL anti-interferon-γ (BD Pharmingen, San Jose, CA, USA) and anti-CD3 and anti-CD28-coated beads (Invitrogen, Carlsbad, CA, USA).
Lentivirus Preparation and Administration
For lentivirus-mediated over-expression of miR-384 (uguaaacaauuccuaggcaaugu), the genomic sequence spanning the mouse miR-384 coding region and ~70 bp of flanking sequence was cloned into the lentiviral vector, H1-MCS-CMV-EGFP (GV-209, Genechem, Shanghai). For lentivirus-mediated inhibition of miR-384, miR-384 complementary (acauugccuaggaauuguuuaca) or negative control (uucuccgaacgugucacgu) sequences were cloned into the same vector. Lentiviruses were produced by Shanghai Genechem Company. Target naïve T cells were infected at a multiplicity of infection of 3 for 12 h, according to the user’s manual, before induction of Th17 polarization. For administration to mice, approximately 2 × 107 transforming units of recombinant lentiviruses were delivered by injection into tail veins 7 days before EAE induction. GFP+ T cells were collected for further examination.
Flow Cytometric Analyses
PB-, lymph node-, or spinal cord-infiltrating lymphocytes were prepared by Percoll gradient centrifugation and then incubated with Cell Stimulation Cocktail (eBioscience, San Diego, CA, USA) for 5 h. For intracellular staining, single-cell suspensions were prepared, surface-stained with FITC-labeled anti-CD4 antibody (Miltenyi Biotec, Germany), and then fixed and permeabilized using a Fixation/Permeabilization Kit (BD Biosciences, San Jose, CA, USA). Subsequently, the cells were washed and stained with Anti-IL-17-PE antibody (Miltenyi Biotec, Germany). Co-staining with RORγt, Foxp3, suppressor of cytokine signaling 3 (SOCS3) rabbit lgGs (CST, USA) was performed for some experiments, and APC-Goat anti-Rabbit lgG (Miltenyi Biotec, Germany) were used as secondary antibodies. Analyses were performed using a MACSQuantTM Flow Cytometer (Miltenyi Biotec, Germany), and the results were analyzed using FlowJo7.6. Protein expression levels are expressed as mean fluorescence intensity (MFI).
Quantitative RT-PCR
Total RNA, from CD4+ T cells or spinal cords from mice anesthetized by i.p. overdose of pentobarbital (150 mg/kg), was extracted using Trizol reagent (Ambion, USA). Quantitative RT-PCR analyses of miR-384 levels were performed using SYBRGreen miRNA assays (Genechem, Shanghai, China) with U6 small nuclear RNA as an internal reference for normalization. Myelin basic protein (MBP) mRNA was examined using a SYBR Green real-time PCR kit on a LightCycler® 480II System (Roche, Switzerland). Relative expression of miRNA or mRNA was evaluated using the 2−△△ct method and expression levels were normalized relative to those of U6 or β-actin, respectively. The primers used were as follows: MBP, 5′-ACCCTCACAGCGATCCAAGT-3′ and 5′-TACGGCTCGGAGCTCACC-3′; and β-actin, 5′-GAGACCTTCAACACCCCAGCC-3′ and 5′-AATGTCACGCACGATTTCCC-3′.
Histological Analyses
For histological analyses, mice were anesthetized with pentobarbital, and then perfused through the left ventricle, first with normal saline to eliminate the blood and then with buffered 4% paraformaldehyde. Spinal cords were embedded in paraffin, and 4 μm thick sections that had been deparaffinized and rehydrated were stained with hematoxylin and eosin (H&E) or luxol fast blue (LFB) for analysis of inflammation and demyelination. Demyelination was quantified in LFB stained sections by calculation of the area of demyelination relative to the total area analyzed. Spinal cord infiltrates were quantified as infiltrates per mm2 in H&E stained sections.
For transmission electron microscopy analyses, fixed and embedded samples were cut into ultra-thin sections (80 nm) and stained with uranyl acetate and lead citrate. Electron micrographs were captured using an FEI Tecnai G2 T12 transmission electron microscope (USA). The axonal diameter (d) was defined as the shortest distance across the center of the axon, omitting the myelin sheath thickness. Fiber diameter (D) was defined as the axonal diameter plus the total myelin sheath thickness on both sides. The g-ratio was calculated as d/D.
For immunofluorescence analyses, prepared spinal cords were incubated overnight in sodium phosphate buffer containing 30% sucrose, then embedded in Optimal Cutting Temperature medium (Leica, Germany) for sectioning. Cryosections (15 μm) were thawed and washed with 0.01% PBS with 5% BSA and 0.3% Triton X-100, and then incubated with MBP monoclonal antibody (CST, USA) for 1 h at 37°C. Following an additional wash step, specimens were incubated with FITC-Goat IgG (LI-COR, USA) and the cell nuclei counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Images were acquired using a fluorescence microscope system (Olympus, Japan) and MFI values calculated using ImageJ software.
ELISA
Cerebrospinal fluid was collected from LV-transfected mice according to a previously described protocol (Liu and Duff, 2008). An IL-17 ELISA kit was purchased from Westang (Shanghai, China), and ELISA performed according to the manufacturer’s protocol.
Western Blotting
The spinal cord was stripped and then ultrasonically homogenized in RIPA buffer. Protein concentrations were measured using a bicinchoninic acid protein assay kit (Beyotime, Shanghai, China). Protein samples were separated by electrophoresis in SDS denaturing 10% polyacrylamide gels (Beyotime, Shanghai, China) and transferred to nitrocellulose membranes, which were subsequently blocked in 0.01% PBS containing 5% BSA. Then membranes were incubated with MBP and β-actin monoclonal antibodies (CST, USA). After washing, the membranes were incubated with appropriate IRDye-conjugated secondary antibodies (LI-COR, Lincoln, NE, USA), scanned using an Odyssey Infrared Imaging System Scanner (LI-COR, Lincoln, NE, USA), and the results analyzed using ImageJ software.
Dual Luciferase Reporter Assay
A fragment of the 3′-UTR of SOCS3, containing the WT predicted miR-384 binding site, or a mutated form of the site, and flanking sequences containing suitable enzyme cleavage sites, was synthesized by Sangon company (Shanghai, China) and cloned downstream of the luciferase reporter gene. The luciferase reporter vectors, together with plasmids expressing Renilla luciferase, and miR-384 or control miR sequences, were each transfected into 293T cells using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA), following the manufacturer’s instructions. Cells were harvested 24 h after transfection and assayed for luciferase activity using a Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA).
Statistical Analyses
Two-tailed Student’s t tests, two-way analysis of variance followed by Bonferroni’s post hoc test for multiple comparisons, and Mann-Whitney tests (for nonparametric data; EAE score) were used to evaluate differences between groups. The results are expressed as means ± standard deviation (SD). Differences were considered statistically significant when p < 0.05, and statistically significant data are indicated by asterisks (*p < 0.05, **p < 0.01).
Results
miR-384 Regulates EAE Pathogenesis
To identify specific miRs involved in EAE development, we determined the expression levels of miRs in PB CD4+ T cells, which have key roles in EAE pathogenesis. The results demonstrated that the expression of miR-384 was significantly elevated in EAE relative to control mice (Figure 1A). To study the function of miR-384, we injected lentiviral particles expressing the miR-384 sequence (LV-miR-384), an miR-384 inhibitor (LV-inhibitor), or control miR (LV-ctrl) into mice before EAE induction. There were no statistically significant differences in EAE induction in non-infected, empty-vector-infected and LV-ctrl-infected EAE mice (data not shown), indicating that the vector and control sequences had no effect on EAE. Over the 21 days after immunization, clinical assessment of EAE demonstrated that LV-miR-384 mice had earlier onset (day 10) and higher mean disease severity scores (maximum score of 4.0 on day 19) than controls, in which mice began to suffer from EAE on day 11 and the maximum score was 3.0 on day 20. Moreover, deficiency of miR-384 caused by LV-inhibitor led to mild EAE from day 13, with a maximum score of only 1.5, on day 19 (Figure 1B). In the lymph nodes (LN) and PB lymphocytes of mice injected with LV-miR-384, the expression of miR-384 was more than three times that in controls, while levels in LV-inhibitor mice were one-third of those in controls (Figure 1C). Moreover, LV-miR-384 mice suffering from EAE exhibited the greatest weight loss, while LV-inhibitor group mice lost the least weight (Figure 1D). These results imply a potential initiating role for miR-384 in the development of EAE.
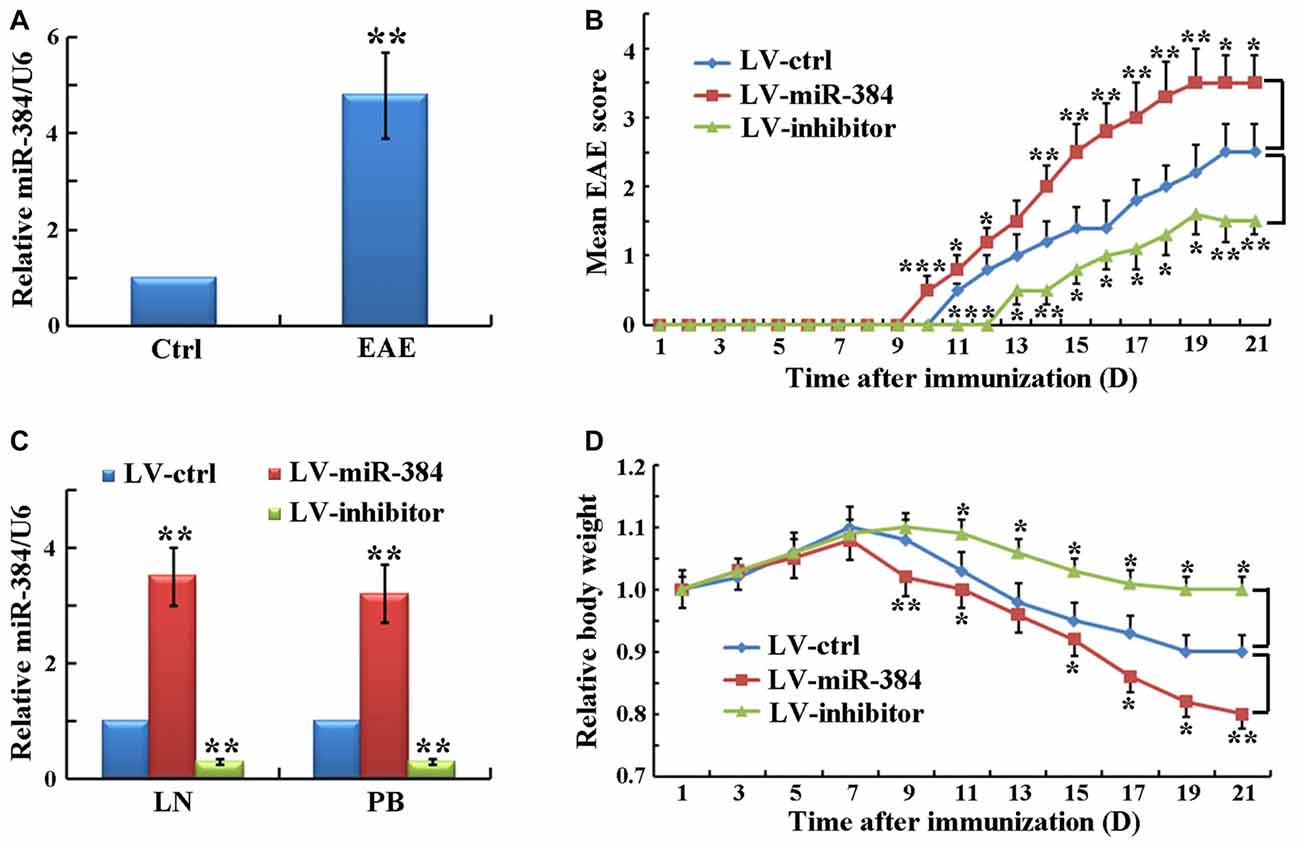
Figure 1. MiR-384 regulates encephalomyelitis (EAE) pathogenesis. (A) Quantitative RT-PCR analyses of miR-384 expression in peripheral blood (PB) naïve CD4+ T cells sorted from control (ctrl) and EAE mice on day 21 after immunization (n = 3 mice per group). Relative miRNA expression levels were evaluated by the 2−△△ct method and normalized to the expression of U6. The control was set as 1. (B) Mean EAE scores of lentivirus vector (LV)-transfected mice were recorded daily (n = 5 mice per group). (C) Quantitative RT-PCR analyses of miR-384 levels in CD4+ T cells from lymph nodes (LN) and PB of LV-transfected mice 7 days later (n = 3 mice per group). (D) Body weights of LV-transfected mice normalized to the initial body weight of each mouse (n = 5 mice per group). Data are presented as means ± standard deviation (SD). *p < 0.05, **p < 0.01 and ***p < 0.001. Data shown are single representative results from three independent experiments. LV-ctrl, control miR; LV-miR-384, miR-384 sequence; and LV-inhibitor, miR-384-inhibitor.
miR-384 Enhances Demyelination in the Spinal Cord during EAE
To further clarify the impact of miR-384 on the pathogenesis of EAE, we performed histological analyses of spinal cord sections by LFB staining. The results indicated that LV-miR-384 mice had the largest areas of demyelination, while the spinal cords of LV-inhibitor mice were relatively intact (Figures 2A,B). Observation by transmission electron microscopy revealed very loose and disintegrated myelin sheaths in the spinal cords of LV-inhibitor and LV-ctrl EAE mice (Figures 2C,D). Next, we examined the expression of MBP, which is indispensable for the process of myelination, and found that it was downregulated in LV-miR-384 mice, and upregulated in the LV-inhibitor group (Figures 2E–G). Similarly, LV-miR-384 mice had severe myelin sheath damage in the spinal cord, with the largest area of MBP loss (Figures 2H,I). Taken together, these findings indicate that miR-384 exacerbates demyelination in the spinal cord during EAE pathogenesis.
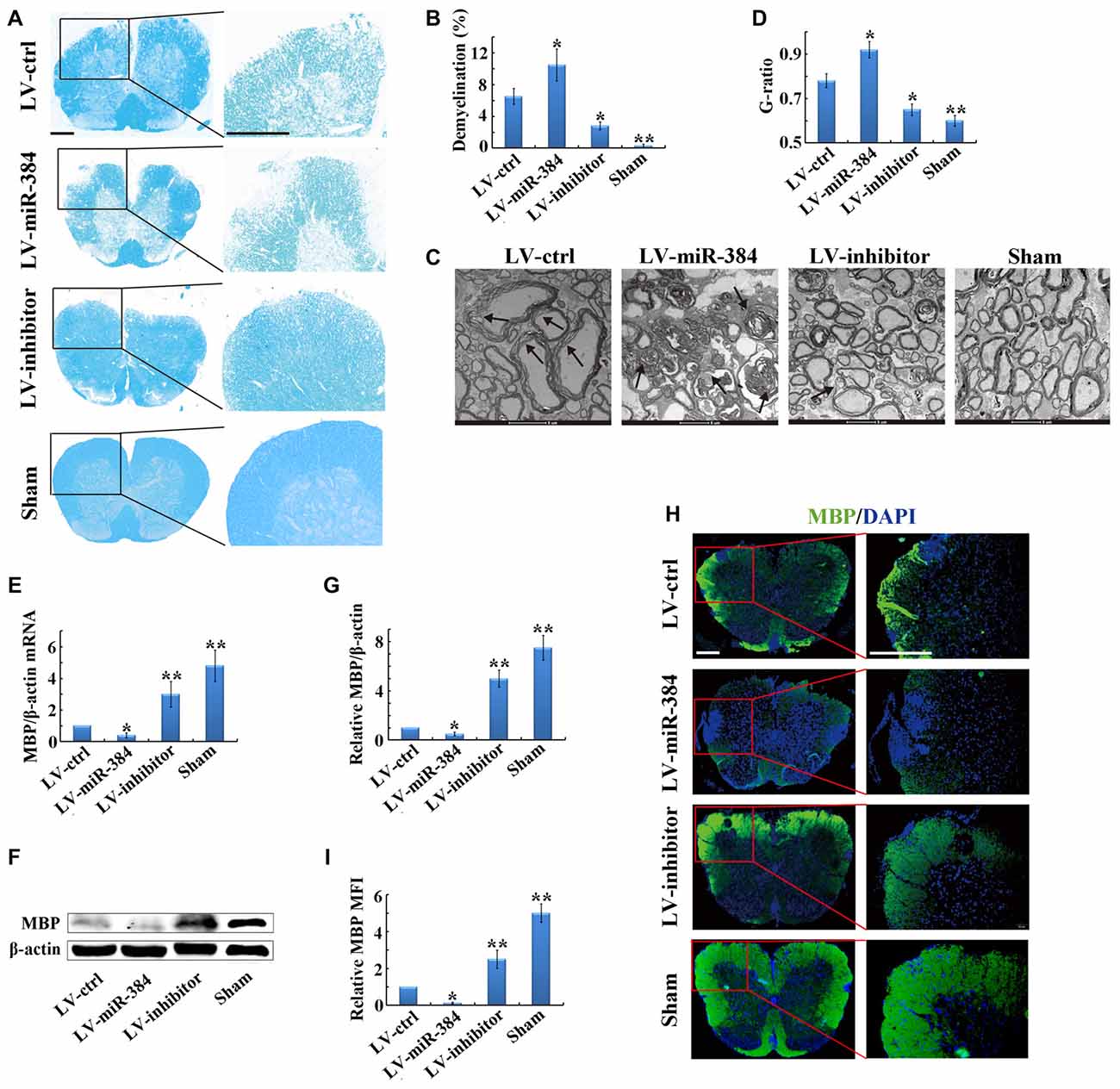
Figure 2. MiR-384 enhances demyelination in the spinal cord during EAE. EAE mice were sacrificed on day 21 after immunization for the following analyses. Sham controls, mice immunized without myelin oligodendrocyte glycoprotein (MOG) peptide. (A) Representative luxol fast blue (LFB) staining of spinal cords. Boxed areas in left images are enlarged in the images on the right. Scale bars, 50 μm. (B) Quantification of demyelination in (A). (C) Transmission electron micrographs of spinal cords. Arrows show myelin sheath damage. Scale bars, 2 μm. (D) Analysis of g-ratios in (C). (E) Quantitative PCR analyses of Myelin basic protein (MBP) mRNA expression in spinal cords (n = 3 mice per group). Relative expression of mRNA was evaluated by the 2−△△ct method and normalized to the expression of β-actin. The control was set as 1. (F,G) Western blotting analyses of MBP in spinal cords (n = 3 mice per group). (H) MBP immunofluorescence staining of spinal cords. Boxed areas in left images are enlarged in right images. Scale bars, 50 μm. (I) Statistical analyses of mean fluorescence intensity (MFI) of MBP in H (n = 5 per group). Data are presented as means ± SD. *p < 0.05 and **p < 0.01. Data shown are single representative results from three independent experiments.
miR-384 Disturbs the Th17/Treg Ratio during EAE
EAE is an autoimmune disease model characterized by severe CNS inflammation. In our study, LV-miR-384 mice developed more extensive inflammatory cell infiltration in the spinal cord (Figures 3A,B), consistent with the observed increases in demyelination and MBP loss (Figure 2). In flow cytometric analyses, we found that the percentage of IL-17+ Th17 cells among PB, lymph node and spinal cord infiltrating (SCI) lymphocytes increased more in LV-miR-384 mice than in LV-ctrl mice (Figures 3C,D), while the percentage of Foxp3+ Treg cells showed no obvious differences among the three groups (Figure 3C). Thus, the relative ratio of Th17 to Treg cells was largest in LV-miR-384 mice (Figure 3E). Moreover, the increased number of Th17 cells in LV-miR-384 mice was confirmed by the observation that the expression of RORγt was upregulated (Figure 3F), along with IL-17 levels (Figure 3G). Therefore, miR-384 appears to impact Th17/Treg ratios in EAE by increasing the number of Th17 cells.
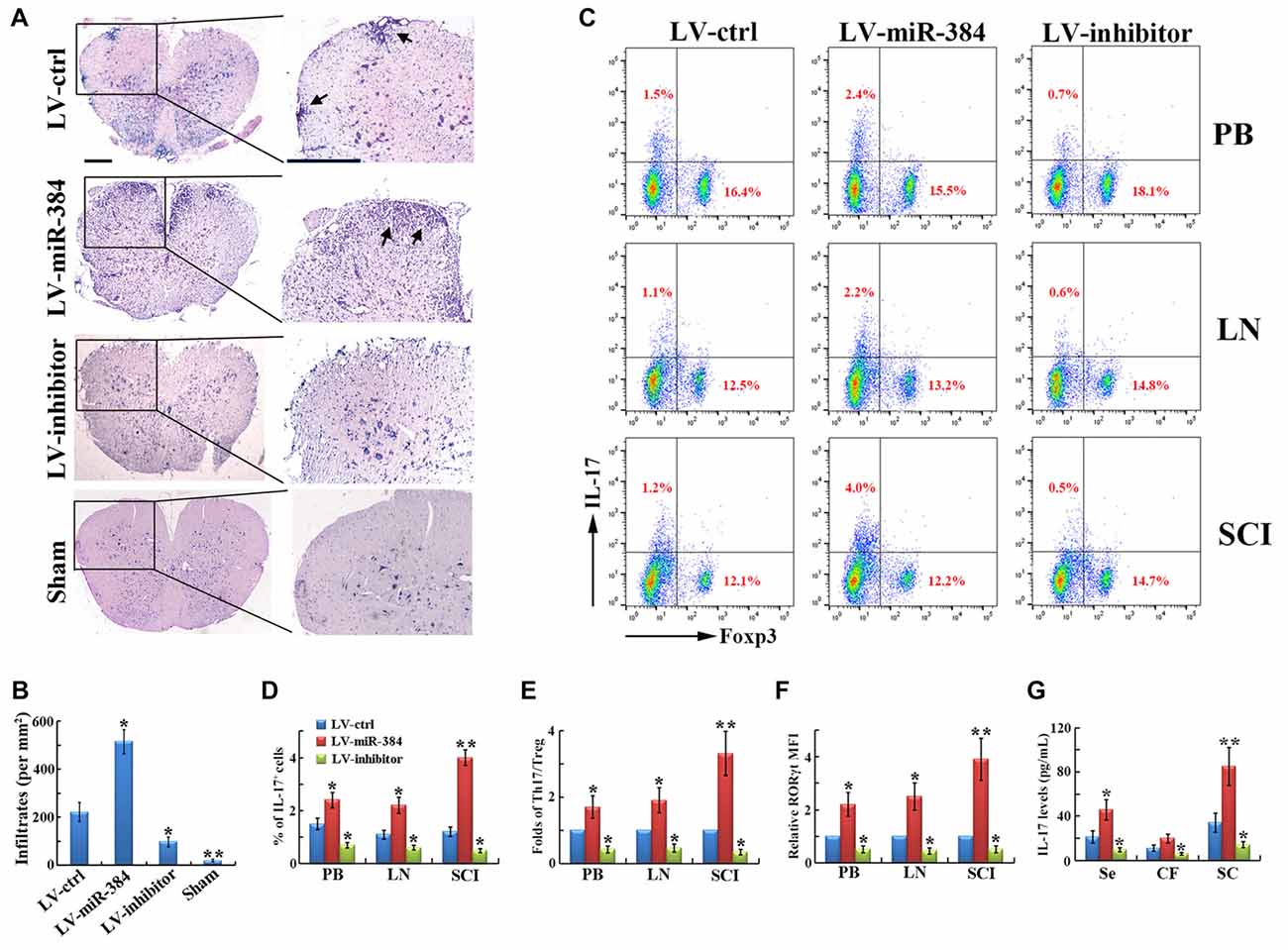
Figure 3. MiR-384 disrupts the Th17/Treg ratio during EAE. (A) Representative hematoxylin and eosin (H&E) staining of spinal cord sections from LV-transfected and sham control mice. Boxed areas in left images are enlarged in right images. Arrows show lymphocyte infiltration. Scale bars, 50 μm. (B) Quantification of spinal cord infiltrates in (A). (C) Representative flow cytometric analyses of IL-17+ and Foxp3+ cells among CD4+ gated cells from stimulated PB, LN and spinal cord infiltrating (SCI) lymphocytes. (D,E) Statistical analyses of the percentage of IL-17+ cells and the ratio between IL-17+ Th17 and Foxp3+ Treg cells in (B; n = 3 per group). (F) MFI analyses of flow cytometry for RORγt (n = 3 per group). (G) The concentration of IL-17 in serum, cerebrospinal fluid and spinal cord homogenate was measured by ELISA (n = 5 per group). Data are presented as means ± SD. *p < 0.05 and **p < 0.01. Data shown are single representative results from three independent experiments.
miR-384 Promotes the Generation of Th17 Cells
To determine the role of miR-384 in Th17 differentiation, naïve T cells were transfected with LV-miR-384, LV-inhibitor, or LV-ctrl and then cultured under Th17-polarizing conditions. Over-expression of miR-384 resulted in a higher percentage of IL-17+ cells (Figure 4A). Accordingly, the expression of RORγt (Figure 4B) and secretion of IL-17 (Figure 4C) were also significantly upregulated by miR-384. Importantly, transfer of the transfected naïve T cells into age-matched Rag1−/− mice, which are deficient in functional T and B cells and hence protected from EAE, restored the susceptibility of these mice to EAE development; furthermore, mice that received LV-miR-384-transfected cells suffered from the most severe EAE, characterized by the earliest onset (on day 11), highest disease severity score (approximately 3.5; Figure 4D), and the greatest number of IL-17+ Th17 cells in spinal cord infiltrates (Figure 4E). Collectively, these data demonstrate that miR-384 promotes the generation of Th17 cells.
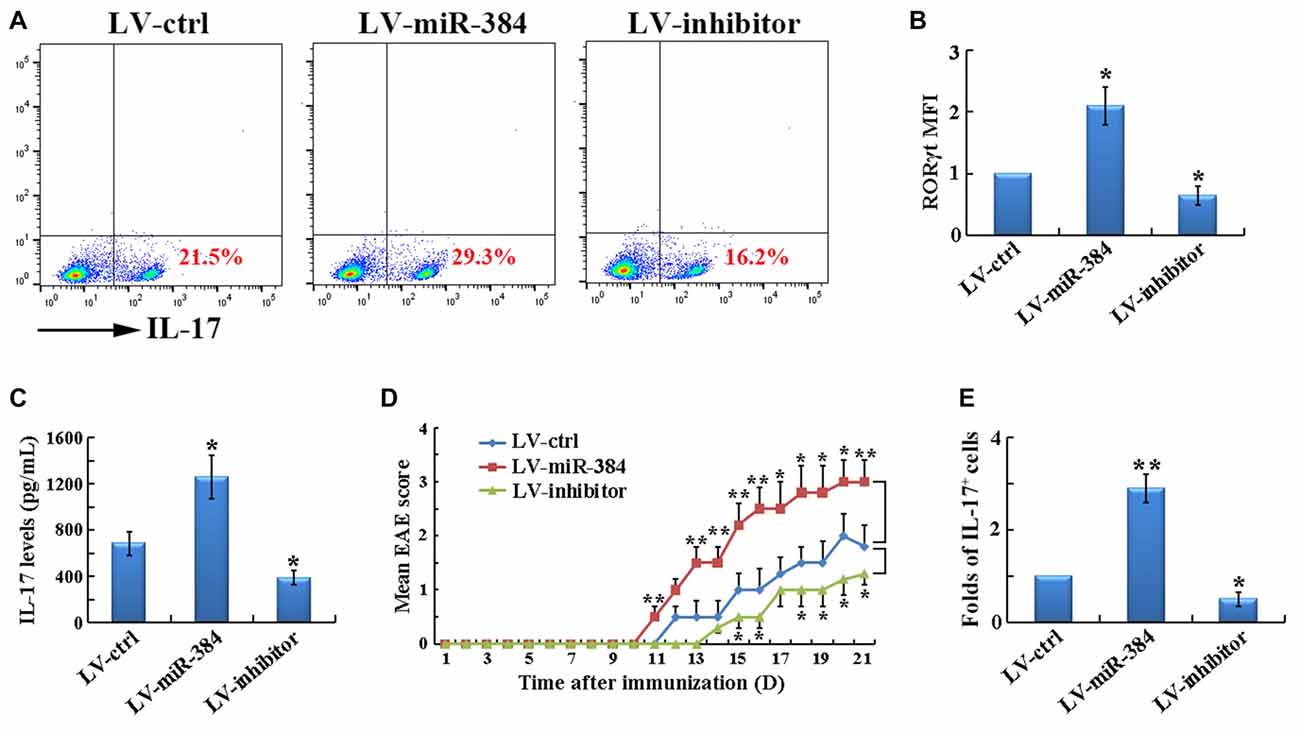
Figure 4. MiR-384 promotes the generation of Th17 cells. For the following analyses, naïve T cells were transfected for 12 h and GFP+ cells collected for tests. (A) Flow cytometric analyses of stimulated IL-17+ cells among the CD4+ gated cells after 3 days of induction under Th17-polarizing conditions. MFI analyses of (B) flow cytometry for RORγt and (C) ELISA of IL-17 in culture supernatants were also performed (n = 3 per group). (D) Mean EAE scores of Rag1−/− mice transfected with naïve T cells (n = 5 mice per group). (E) Relative ratio of Th17 cells in SCI lymphocytes from the mice in (D) on day 21 after immunization, determined by flow cytometric analyses (n = 3 per group). Data are presented as means ± SD. *p < 0.05 and **p < 0.01. Data shown are single representative results from three independent experiments.
miR-384 Directly Targets SOCS3
The identification of gene targets for individual miRs is key to understanding the molecular mechanisms underlying miR-mediated regulation. Among candidates predicted by TargetScan, we identified SOCS3, a gene encoding a protein with an established role in Th17 differentiation, whose 3′-UTR mRNA sequence closely matched the miR-384 target site (Figure 5A). We then conducted luciferase reporter assays, which demonstrated that luciferase activity was reduced by 50% in cells co-transfected with the SOCS3 WT luciferase reporter and miR-384, compared with miR-ctrl (Figure 5B, WT). However, site-directed mutagenesis of the miR-384 recognition site in the SOCS3 3′-UTR abolished the inhibitory effects of miR-384 (Figure 5B, MUT). Consistently, the expression of SOCS3 in naïve T cells over-expressing miR-384 was clearly lower than that in controls, while downregulation of miR-384 led to increased SOCS3 expression levels (Figure 5C). Taken together, these results show that miR-384 can regulate SOCS3 expression through a partially complementary binding site in the 3′-UTR.
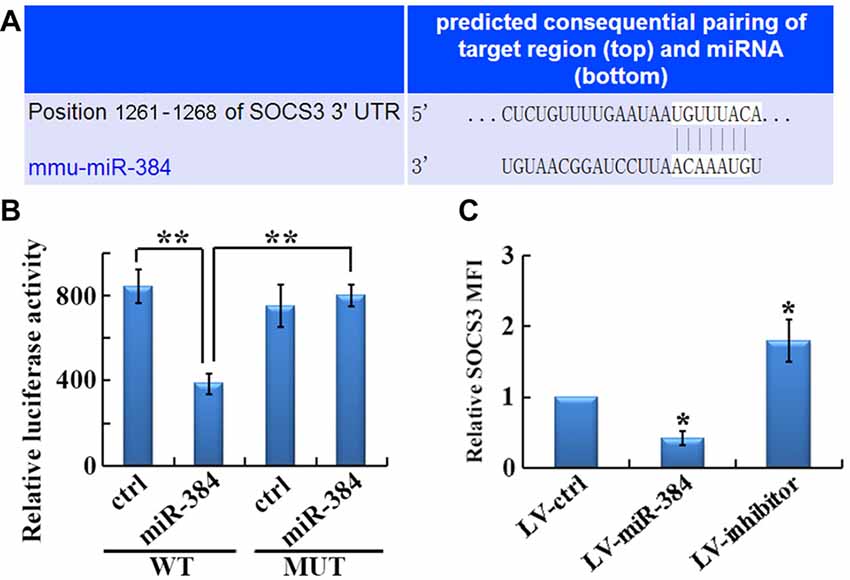
Figure 5. SOCS3 is a direct target of miR-384. (A) Illustration of the sequence match between miR-384 and SOCS3 mRNA determined using TargetScan. (B) Luciferase activity of reporter vectors containing wild-type (WT) or mutated (MUT) SOCS3 3′-UTR co-transfected with miR-384 or control miR (ctrl; n = 3 per group). (C) MFI analyses of flow cytometry for SOCS3 in GFP+ naïve T cells 3 days post-transfection (n = 3 per group). Data are presented as means ± SD. *p < 0.05 and **p < 0.01. Data shown are single representative results from three independent experiments.
Discussion
An appropriate balance between the pro-inflammatory Th17 cell population and immunosuppressive Treg cells is critical for immune homeostasis (Wu et al., 2016); imbalances are associated with various inflammatory and autoimmune diseases, including MS, which is linked to a shift towards increased Th17 cell levels (Venkatesha et al., 2015). Here, we report that miR-384 over-expression leads to an imbalance in Th17/Treg ratios, resulting in exacerbation of EAE with severe demyelination, through the promotion of Th17 cell differentiation by targeting SOCS3. This miR-384-associated Th17/Treg imbalance represents a potential therapeutic target for MS and a novel indicator of disease risk (Yu et al., 2015).
The SOCS family consists of intracellular proteins that are key physiological regulators of the immune system. SOCS3 is an important member of the SOCS family and can negatively regulate the IL-6-JAK-STAT3 pathway, which is indispensable for Th17 cell differentiation (Qu et al., 2012; Liu X. et al., 2015). SOCS3-deficient T cells show hyperactivation of STAT3 and significantly increased secretion of IL-17, induced by IL-6 (Chen et al., 2006). Therefore, miR-384 promotes the differentiation of Th17 cells, which disturbs the Th17/Treg ratio, through targeting of SOCS3. However, there do not appear to be any direct effects of miR-384 on Treg cell development. This may be because SOCS3 is involved in specific inhibition of STAT3, but not STAT5, signaling. The STAT5 pathway is activated by TGF-β to induce Foxp3 expression and Treg cell development (Basu et al., 2015). The mutually inhibitory roles of Th17 and Treg cells may be responsible for the slight decrease in Treg cells when Th17 cell generation is promoted by miR-384 in EAE mice and their slight increase when miR-384 is knocked down (Figure 3C).
miR-384, the role of which has previously been poorly understood, has recently been the focus of research attention. miR-384 has suppressive effects on tumor cell proliferation through the direct inhibition of IRS1 expression (Lai et al., 2016), and contributes to aberrant Runx expression in prostate tumors (Farina et al., 2016). In addition, several studies have demonstrated a relationship between miR-384 and the CNS. Gu et al. (2015) used next-generation sequencing to profile miRNA transcriptomes, and found that miR-384 is required for enduring synaptic and spinal plasticity by regulation of the expression of RSK3. Another study demonstrated that miR-384 is a promising candidate as a biomarker of spinal cord injury, because of a significant increase in miR-384 in the serum, which correlates with injury severity (Hachisuka et al., 2014). In addition, miR-384 is elevated in the hippocampus during neural cell death and is, therefore, considered to have potentially novel roles in neurotoxicity (Ogata et al., 2015) and the development of Parkinson’s disease (Jiang et al., 2016) and Alzheimer’s disease (Liu et al., 2014). In this study, we discovered another novel function of miR-384, namely a critical involvement in the development of the Th17/Treg imbalance and demyelination in the CNS during the pathogenesis of EAE through targeting of SOCS3.
The fact that an increase of the Th17/Treg ratio in MS patients vs. healthy controls (Jamshidian et al., 2013) implies recovering the balance between Th17 and Treg cells may be an important target in the context of inflammation. All trans retinoic acid (ATRA), as a RORγt antagonist, has been proved to ameliorate EAE severity via the following mechanisms: (1) inhibition of Th17 cell differentiation by binding to the RARα sequence to downregulate RORγt expression and then increase Foxp3+ T cells (Hall et al., 2011; Egan et al., 2016); and (2) Th17 cell dysfunction due to the decrease in IL-23 and IL-6 receptor expression accompanied by the promotion of TGF-β signaling through the Smad3 pathway in Treg cells (Klemann et al., 2009; Abdolahi et al., 2015). Recent transcriptional network analysis has identified a core set of transcription factors, such as STAT3, which are crucial for the differentiation of Th17 cell by inducing RORγt expression and DNA binding (Ciofani et al., 2012). In addition, distinct miRNAs, the post-transcriptional modulators of RNA stability, have been reported to regulate the Th17 cell differentiation by modulating the amount of transcripts that encode crucial positive regulators of Th17 cell development (Korn and Kallies, 2017). It is encouraging that the miR-based therapy has been in clinical trials (Gebert et al., 2014), offering a novel therapeutic approach if miRNAs can be effectively applied in vivo.
In conclusion, we found that the expression of miR-384 is greatly increased during the pathogenesis of EAE, and that its over-expression in vivo leads to severe EAE, due to aggravated demyelination caused by the promotion of Th17 cell generation. In addition, this microRNA promotes Th17 cell differentiation through targeting SOCS3. These results facilitate better understanding of the pathogenesis of EAE and imply that miR-384 may be an effective biomarker or potential therapeutic target in MS.
Author Contributions
XQ and RY designed the study, performed experiments, analyzed the data and wrote the article; JH, YZ, YW and JZ performed experiments; HF and RY reviewed the manuscript. All authors read and edited the manuscript.
Funding
This study was supported by grants from the National Natural Science Foundation of China (No.81271345 to RY, 81302519 to XQ), the Natural Science Foundation of Jiangsu Province (No. BK20131132 to RY, BK20130221 to XQ), 333 Project (XQ) and Qing Lan Project (RY) of Jingsu Province, College Graduate Research and Innovation Project of Jingsu Province (KYLX16_1131 to YZ), and Xuzhou Medical University Scientific Research Fund for Talents (2015KJ07 to JH).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abdolahi, M., Yavari, P., Honarvar, N. M., Bitarafan, S., Mahmoudi, M., and Saboor-Yaraghi, A. A. (2015). Molecular mechanisms of the action of Vitamin A in Th17/Treg axis in multiple sclerosis. J. Mol. Neurosci. 57, 605–613. doi: 10.1007/s12031-015-0643-1
Basu, R., Whitley, S. K., Bhaumik, S., Zindl, C. L., Schoeb, T. R., Benveniste, E. N., et al. (2015). IL-1 signaling modulates activation of STAT transcription factors to antagonize retinoic acid signaling and control the TH17 cell-iTreg cell balance. Nat. Immunol. 16, 286–295. doi: 10.1038/ni.3099
Chen, Z., Laurence, A., Kanno, Y., Pacher-Zavisin, M., Zhu, B.-M., Tato, C., et al. (2006). Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. Proc. Natl. Acad. Sci. U S A 103, 8137–8142. doi: 10.1073/pnas.0600666103
Chen, J.-Q., Papp, G., Szodoray, P., and Zeher, M. (2016). The role of microRNAs in the pathogenesis of autoimmune diseases. Autoimmun. Rev. 15, 1171–1180. doi: 10.1016/j.autrev.2016.09.003
Ciofani, M., Madar, A., Galan, C., Sellars, M., Mace, K., Pauli, F., et al. (2012). A validated regulatory network for Th17 cell specification. Cell 151, 289–303. doi: 10.1016/j.cell.2012.09.016
Du, C., Liu, C., Kang, J., Zhao, G., Ye, Z., Huang, S., et al. (2009). MicroRNA miR-326 regulates TH-17 differentiation and is associated with the pathogenesis of multiple sclerosis. Nat. Immunol. 10, 1252–1259. doi: 10.1038/ni.1798
Egan, C. E., Sodhi, C. P., Good, M., Lin, J., Jia, H., Yamaguchi, Y., et al. (2016). Toll-like receptor 4-mediated lymphocyte influx induces neonatal necrotizing enterocolitis. J. Clin. Invest. 126, 495–508. doi: 10.1172/jci83356
Farina, N. H., Zingiryan, A., Akech, J. A., Callahan, C. J., Lu, H., Stein, J. L., et al. (2016). A microRNA/Runx1/Runx2 network regulates prostate tumor progression from onset to adenocarcinoma in TRAMP mice. Oncotarget 7, 70462–70474. doi: 10.18632/oncotarget.11992
Gebert, L. F. R., Rebhan, M. A. E., Crivelli, S. E. M., Denzler, R., Stoffel, M., and Hall, J. (2014). Miravirsen (SPC3649) can inhibit the biogenesis of miR-122. Nucleic Acids Res. 42, 609–621. doi: 10.1093/nar/gkt852
Gerriets, V. A., Kishton, R. J., Johnson, M. O., Cohen, S., Siska, P. J., Nichols, A. G., et al. (2016). Foxp3 and Toll-like receptor signaling balance Treg cell anabolic metabolism for suppression. Nat. Immunol. 17, 1459–1466. doi: 10.1038/ni.3577
Gu, Q.-H., Yu, D., Hu, Z., Liu, X., Yang, Y., Luo, Y., et al. (2015). miR-26a and miR-384–5p are required for LTP maintenance and spine enlargement. Nat. Commun. 6:6789. doi: 10.1038/ncomms7789
Hachisuka, S., Kamei, N., Ujigo, S., Miyaki, S., Yasunaga, Y., and Ochi, M. (2014). Circulating microRNAs as biomarkers for evaluating the severity of acute spinal cord injury. Spinal Cord 52, 596–600. doi: 10.1038/sc.2014.86
Hall, J. A., Grainger, J. R., Spencer, S. P., and Belkaid, Y. (2011). The role of retinoic acid in tolerance and immunity. Immunity 35, 13–22. doi: 10.1016/j.immuni.2011.07.002
Jadidi-Niaragh, F., and Mirshafiey, A. (2011). Th17 cell, the new player of neuroinflammatory process in multiple sclerosis. Scand. J. Immunol. 74, 1–13. doi: 10.1111/j.1365-3083.2011.02536.x
Jamshidian, A., Kazemi, M., Shaygannejad, V., and Salehi, M. (2015). The effect of plasma exchange on the expression of FOXP3 and RORC2 in relapsed multiple sclerosis patients. Iran. J. Immunol. 12, 311–318. doi: IJIv12i4A8
Jamshidian, A., Shaygannejad, V., Pourazar, A., Zarkesh-Esfahani, S. H., and Gharagozloo, M. (2013). Biased Treg/Th17 balance away from regulatory toward inflammatory phenotype in relapsed multiple sclerosis and its correlation with severity of symptoms. J. Neuroimmunol. 262, 106–112. doi: 10.1016/j.jneuroim.2013.06.007
Jiang, M., Yun, Q., Shi, F., Niu, G., Gao, Y., Xie, S., et al. (2016). Downregulation of miR-384–5p attenuates rotenone-induced neurotoxicity in dopaminergic SH-SY5Y cells through inhibiting endoplasmic reticulum stress. Am. J. Physiol. Cell Physiol. 310, C755–C763. doi: 10.1152/ajpcell.00226.2015
Klemann, C., Raveney, B. J., Klemann, A. K., Ozawa, T., von Hörsten, S., Shudo, K., et al. (2009). Synthetic retinoid AM80 inhibits Th17 cells and ameliorates experimental autoimmune encephalomyelitis. Am. J. Pathol. 174, 2234–2245. doi: 10.2353/ajpath.2009.081084
Korn, T., Bettelli, E., Oukka, M., and Kuchroo, V. K. (2009). IL-17 and Th17 cells. Annu. Rev. Immunol. 27, 485–517. doi: 10.1146/annurev.immunol.021908.132710
Korn, T., and Kallies, A. (2017). T cell responses in the central nervous system. Nat. Rev. Immunol. 17, 179–194. doi: 10.1038/nri.2016.144
Krol, J., Loedige, I., and Filipowicz, W. (2010). The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 11, 597–610. doi: 10.1038/nrg2843
Lai, Y.-Y., Shen, F., Cai, W.-S., Chen, J.-W., Feng, J.-H., Cao, J., et al. (2016). MiR-384 regulated IRS1 expression and suppressed cell proliferation of human hepatocellular carcinoma. Tumour Biol. 37, 14165–14171. doi: 10.1007/s13277-016-5233-5
Li, J., Wan, Y., Guo, Q., Zou, L., Zhang, J., Fang, Y., et al. (2010). Altered microRNA expression profile with miR-146a upregulation in CD4+ T cells from patients with rheumatoid arthritis. Arthritis Res. Ther. 12:R81. doi: 10.1186/ar3006
Liu, L., and Duff, K. (2008). A technique for serial collection of cerebrospinal fluid from the cisterna magna in mouse. J. Vis. Exp. 21:960. doi: 10.3791/960
Liu, X., Ren, S., Qu, X., Ge, C., Cheng, K., and Zhao, R. C. (2015). Mesenchymal stem cells inhibit Th17 cells differentiation via IFN-γ-mediated SOCS3 activation. Immunol. Res. 61, 219–229. doi: 10.1007/s12026-014-8612-2
Liu, C. G., Wang, J.-L., Li, L., and Wang, P.-C. (2014). MicroRNA-384 regulates both amyloid precursor protein and β-secretase expression and is a potential biomarker for Alzheimer’s disease. Int. J. Mol. Med. 34, 160–166. doi: 10.3892/ijmm.2014.1780
Liu, Y., You, C., Zhang, Z., Zhang, J., and Yan, H. (2015). Roles of Treg/Th17 cell imbalance and neuronal damage in the visual dysfunction observed in experimental autoimmune optic neuritis chronologically. Neuromolecular Med. 17, 391–403. doi: 10.1007/s12017-015-8368-4
Lochner, M., Wang, Z., and Sparwasser, T. (2015). The special relationship in the development and function of T helper 17 and regulatory T cells. Prog. Mol. Biol. Transl. Sci. 136, 99–129. doi: 10.1016/bs.pmbts.2015.07.013
Murugaiyan, G., Beynon, V., Mittal, A., Joller, N., and Weiner, H. L. (2011). Silencing microRNA-155 ameliorates experimental autoimmune encephalomyelitis. J. Immunol. 187, 2213–2221. doi: 10.4049/jimmunol.1003952
Murugaiyan, G., da Cunha, A. P., Ajay, A. K., Joller, N., Garo, L. P., Kumaradevan, S., et al. (2015). MicroRNA-21 promotes Th17 differentiation and mediates experimental autoimmune encephalomyelitis. J. Clin. Invest. 125, 1069–1080. doi: 10.1172/JCI74347
Mycko, M. P., Cichalewska, M., Machlanska, A., Cwiklinska, H., Mariasiewicz, M., and Selmaj, K. W. (2012). MicroRNA-301a regulation of a T-helper 17 immune response controls autoimmune demyelination. Proc. Natl. Acad. Sci. U S A 109, E1248–E1257. doi: 10.1073/pnas.1114325109
O’Connell, R. M., Kahn, D., Gibson, W. S., Round, J. L., Scholz, R. L., Chaudhuri, A. A., et al. (2010). MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity 33, 607–619. doi: 10.1016/j.immuni.2010.09.009
Ogata, K., Sumida, K., Miyata, K., Kushida, M., Kuwamura, M., and Yamate, J. (2015). Circulating miR-9* and miR-384-5p as potential indicators for trimethyltin-induced neurotoxicity. Toxicol. Pathol. 43, 198–208. doi: 10.1177/0192623314530533
Qu, X., Liu, X., Cheng, K., Yang, R., and Zhao, R. C. (2012). Mesenchymal stem cells inhibit Th17 cell differentiation by IL-10 secretion. Exp. Hematol. 40, 761–770. doi: 10.1016/j.exphem.2012.05.006
Rostami, A., and Ciric, B. (2013). Role of Th17 cells in the pathogenesis of CNS inflammatory demyelination. J. Neurol. Sci. 333, 76–87. doi: 10.1016/j.jns.2013.03.002
Singh, R. P., Massachi, I., Manickavel, S., Singh, S., Rao, N. P., Hasan, S., et al. (2013). The role of miRNA in inflammation and autoimmunity. Autoimmun. Rev. 12, 1160–1165. doi: 10.1016/j.autrev.2013.07.003
Tzartos, J. S., Friese, M. A., Craner, M. J., Palace, J., Newcombe, J., Esiri, M. M., et al. (2008). Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am. J. Pathol. 172, 146–155. doi: 10.2353/ajpath.2008.070690
Venkatesha, S. H., Dudics, S., Weingartner, E., So, E. C., Pedra, J., and Moudgil, K. D. (2015). Altered Th17/Treg balance and dysregulated IL-1β response influence susceptibility/resistance to experimental autoimmune arthritis. Int. J. Immunopathol. Pharmacol. 28, 318–328. doi: 10.1177/0394632015595757
Wu, D., Wang, J., and Zhang, M. (2016). Altered Th17/Treg ratio in nasal polyps with distinct cytokine profile: association with patterns of inflammation and mucosal remodeling. Medicine (Baltimore) 95:e2998. doi: 10.1097/MD.0000000000002998
Yu, Z.-X., Ji, M.-S., Yan, J., Cai, Y., Liu, J., Yang, H.-F., et al. (2015). The ratio of Th17/Treg cells as a risk indicator in early acute respiratory distress syndrome. Crit. Care 19:82. doi: 10.1186/s13054-015-0811-2
Zhernakova, A., Withoff, S., and Wijmenga, C. (2013). Clinical implications of shared genetics and pathogenesis in autoimmune diseases. Nat. Rev. Endocrinol. 9, 646–659. doi: 10.1038/nrendo.2013.161
Zhu, L., Chen, H., Liu, M., Yuan, Y., Wang, Z., Chen, Y., et al. (2016). Treg/Th17 cell imbalance and IL-6 profile in patients with unexplained recurrent spontaneous abortion. Reprod. Sci. doi: 10.1177/1933719116670517 [Epub ahead of print].
Zhu, S., Pan, W., Song, X., Liu, Y., Shao, X., Tang, Y., et al. (2012). The microRNA miR-23b suppresses IL-17-associated autoimmune inflammation by targeting TAB2, TAB3 and IKK-α. Nat. Med. 18, 1077–1086. doi: 10.1038/nm.2815
Keywords: miR-384, experimental autoimmune encephalomyelitis, Th17/Treg, SOCS3, multiple sclerosis
Citation: Qu X, Han J, Zhang Y, Wang Y, Zhou J, Fan H and Yao R (2017) MiR-384 Regulates the Th17/Treg Ratio during Experimental Autoimmune Encephalomyelitis Pathogenesis. Front. Cell. Neurosci. 11:88. doi: 10.3389/fncel.2017.00088
Received: 07 November 2016; Accepted: 14 March 2017;
Published: 28 March 2017.
Edited by:
Antonio Gambardella, Magna Græcia University, ItalyReviewed by:
Elsa Fabbretti, University of Nova Gorica, SloveniaAndrew Chan, Ruhr University Bochum, Germany
Copyright © 2017 Qu, Han, Zhang, Wang, Zhou, Fan and Yao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ruiqin Yao, d2VueGlfeWFvQDE2My5jb20=
† These authors have contributed equally to this work.