 Stefano Gambardella1*
Stefano Gambardella1* Rosangela Ferese1
Rosangela Ferese1 Francesca Biagioni1
Francesca Biagioni1 Carla L. Busceti1
Carla L. Busceti1 Rosa Campopiano1Anna M. P. Griguoli1
Rosa Campopiano1Anna M. P. Griguoli1 Fiona Limanaqi2
Fiona Limanaqi2 Giuseppe Novelli1,3Marianna Storto1
Giuseppe Novelli1,3Marianna Storto1 Francesco Fornai1,2
Francesco Fornai1,2- 1IRCCS Neuromed, Pozzilli, Italy
- 2Department of Translational Research and New Technologies in Medicine and Surgery, University of Pisa, Pisa, Italy
- 3Department of Biomedicine and Prevention, School of Medicine, University of Rome Tor Vergata, Rome, Italy
The functional anatomy of the reticular formation (RF) encompasses a constellation of brain regions which are reciprocally connected to sub-serve a variety of functions. Recent evidence indicates that neuronal degeneration within one of these regions spreads synaptically along brainstem circuitries. This is exemplified by the recruitment of various brainstem reticular nuclei in specific Parkinson’s disease (PD) phenotypes, and by retrospective analysis of lethargic post-encephalitic parkinsonism. In fact, the spreading to various monoamine reticular nuclei can be associated with occurrence of specific motor and non-motor symptoms (NMS). This led to re-consider PD as a brainstem monoamine disorder (BMD). This definition surpasses the anatomy of meso-striatal motor control to include a variety of non-motor domains. This concept clearly emerges from the quite specific clinical-anatomical correlation which can be drawn in specific paradigms of PD genotypes. Therefore, this review article focuses on the genetics and neuroanatomy of three PD genotypes/phenotypes which can be selected as prototype paradigms for a differential recruitment of the RF leading to differential occurrence of NMS: (i) Parkin-PD, where NMS are rarely reported; (ii) LRRK2-PD and slight SNC point mutations, where the prevalence of NMS resembles idiopathic PD; (iii) Severe SNCA point mutations and multiplications, where NMS are highly represented.
Introduction
In the process of re-defining Parkinson’s disease (PD) a task force is working on various symptoms which, despite being unrelated to the extra-pyramidal motor system, are now considered as fundamental features of PD (Poewe, 2008; Fornai and Ruggieri, 2013; Marras and Chaudhuri, 2016; Wei et al., 2016). Although most cardinal symptoms of PD involve the extra-pyramidal motor system, an in depth knowledge of PD patients led to describe a variety of non-motor alterations as well as pyramidal motor dysfunctions which are presently under intense scrutiny (Li et al., 2010; Fornai and Ruggieri, 2013; Fornai et al., 2013; Natale et al., 2013; Xu et al., 2015; Zou et al., 2016). In fact, the occurrence of these symptoms may be helpful to discern between various PD genotypes/phenotypes, while it provides new vistas on the variety of brainstem reticular nuclei, which may or may not, be recruited during the disease course (Chao et al., 2015). Despite being missed out for a long time or being considered as an unexpected complication of a pure extra-pyramidal motor disorder, non-motor symptoms (NMS) are now a fundamental feature of PD. At the same time NMS provides an unusual perspective to elucidate the anatomical network sub-serving a brainstem monoamine disorder (BMD) which is the anatomical core of PD. In PD, NMS are often present during the disease course (Chaudhuri et al., 2006; Li et al., 2010; Bonnet et al., 2012; Chege and McColl, 2014; Bastide et al., 2015; Gao et al., 2015). In fact, brain areas, which control extra-pyramidal motor systems concomitantly, affect other neurological and psychiatric domains. This is evident when examining several reticular brainstem nuclei, whose functions affect both extra-pyramidal motor circuitries and a variety of non-motor domains during the neuropathology of sporadic PD (S-PD; Fornai and Ruggieri, 2013). Within these sub-cortical regions a variety of activities beyond extra-pyramidal motor control take place, which explains why non-motor alterations should be expected to occur rather than being unusual in most PD patients. In a recent article, we emphasized such a concept focusing on the involvement of a number of brainstem monoamine nuclei (Fornai and Ruggieri, 2013). In this analysis, we proposed the definition of BMD as more balanced to define the neuroanatomy of PD. In fact, a constellation of nuclei belonging to the brainstem reticular formation (BRF), are affected at various disease/stage severity. There is an appreciable site-specificity, which connects neuroanatomy with the onset of NMS and this works as a general model to build a clinical anatomical correlation within various PD syndromes. Within this context, progress in neurogenetics provided a powerful tool to improve PD nosography since specific gene/protein alterations connect quite specifically with clusters of both motor and NMS, which in turn, are related to a damage in quite selective brain areas. This configures PD as a spectrum of brainstem disorders which ranges between the occurrence of solely extra-pyramidal motor symptoms, up to the coexistence of pyramidal, extra-pyramidal symptoms along with NMS. Remarkably, a deep insight in the genetics of PD, along with the progressive awareness of NMS in PD, provided two key elements, which fostered the need to re-define PD itself. This is listed in the form of three paradigms shown in Figure 1 as PD due to mutations in three different loci (Parkin; LRRK2; SNCA). Similarly, Figure 2 shows the anatomy of the BRF and highlights those reticular nuclei which are often recruited in each paradigm of PD (Parkin; LRRK2; SNCA). When combining Figures 1, 2, one may get a general correlation between PD genotype, the occurrence of specific NMS, and the recruitment of specific brainstem reticular nuclei. For instance, Parkin disease represents an almost pure extra-pyramidal motor disorder. This is well described by Doherty et al. (2013) showing the paucity of symptoms (Figure 1) and affected brain areas (Figure 2), limited to the substantia nigra pars compacta (SNpc) dorsal tier, locus coeruleus (LC) and dorsal motor nucleus of the vagus. Conversely, the specific point mutation p.G51D in the SNCA gene (Lesage et al., 2013) or multiplications of SNCA gene itself leads to a plethora of non-motor alterations (Figure 1). In this latter SNCA-dependent syndrome, motor alterations surpass the extra-pyramidal circuitry featuring also a pyramidal disorder with fatal prognosis (Mutez et al., 2011; Chen et al., 2015; Kiely et al., 2015). The p.G51D mutation additionally shifts the disease towards multiple system atrophy (MSA). The great amount of NMS shown in Figure 1 is consistent with massive recruitment of brainstem nuclei shown in Figure 2. Roughly, in the middle of these extremes we describe most LRRK2-associated PD, which features extra-pyramidal motor symptoms along with some NMS (Zimprich et al., 2004). This resembles most SNCA point mutations but p.G51D, as well as S-PD patients (Figure 1).
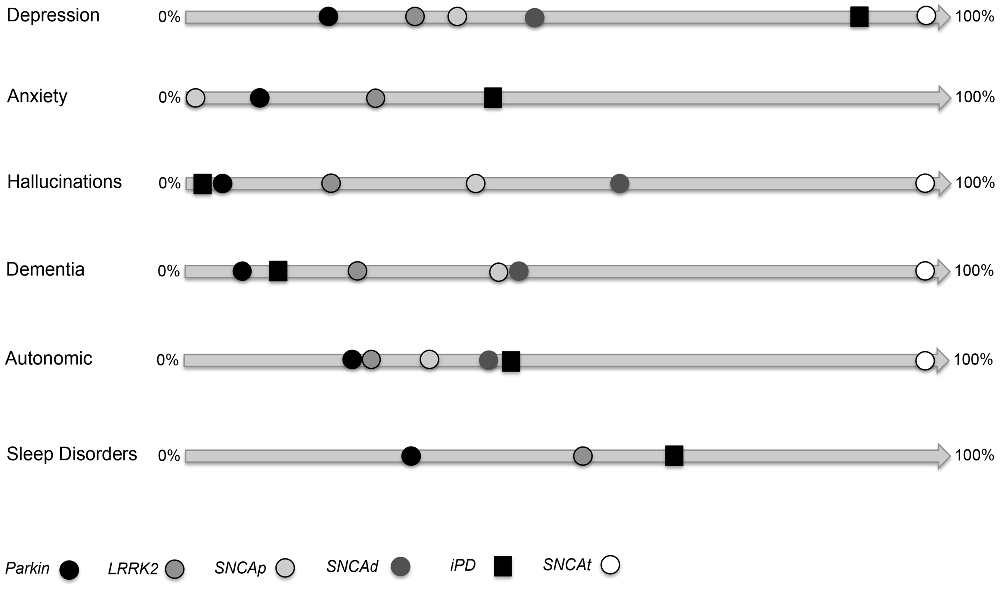
Figure 1. Non-motor symptoms (NMS) and paradigms of genetic Parkinson’s disease (PD). This image shows the frequencies of each NMS (depression, anxiety, hallucination, dementia, autonomic dysfunction, sleep disorders) for Parkinsonism related to Parkin, LRRK2, SNCA point mutation (SNCAp), SNCA duplication (SNCAd), SNCA triplication or homozygote duplication (SNCAt). Image adapted from tables reported in Chaudhuri et al. (2015).
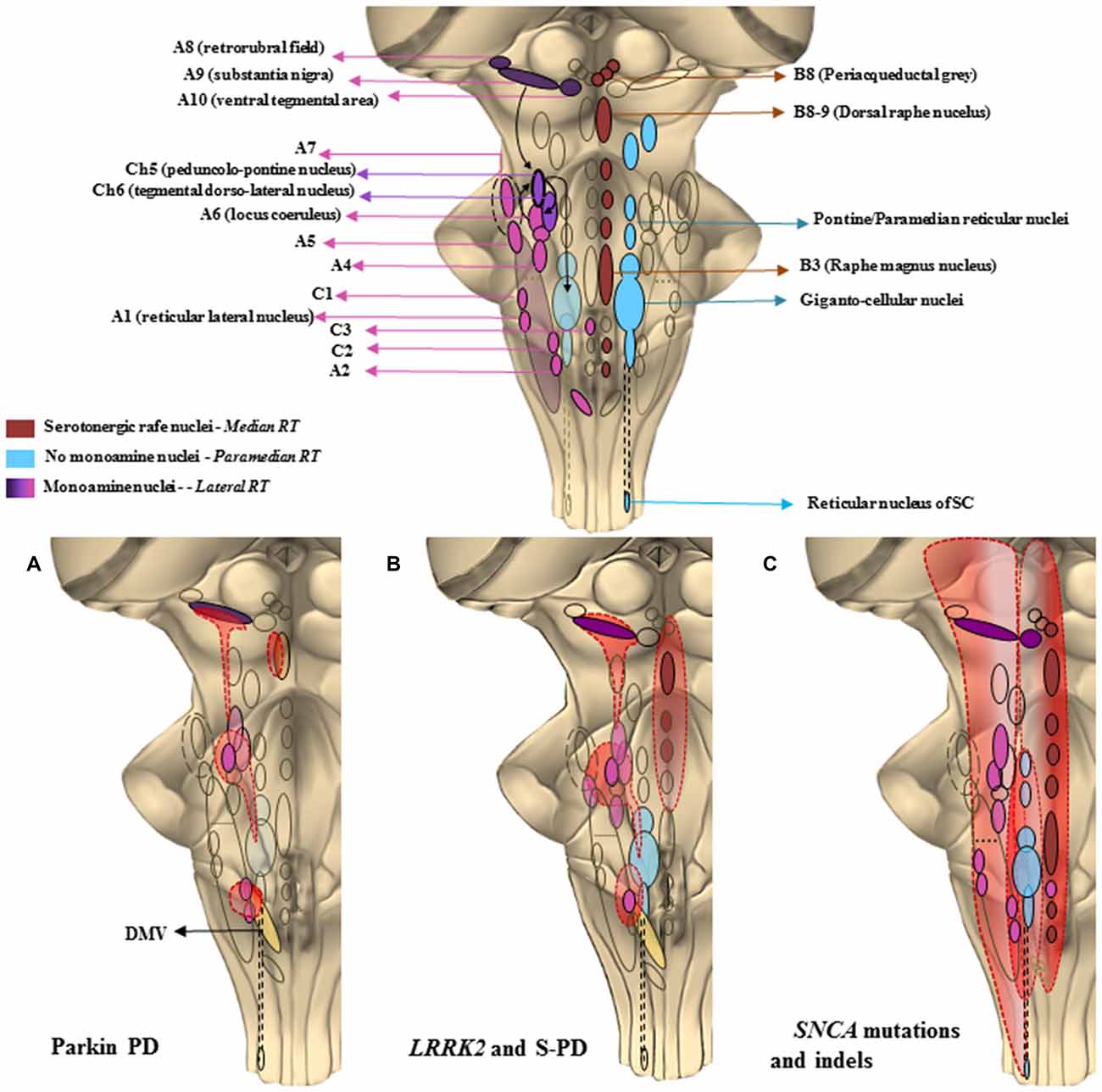
Figure 2. The chemical neuroanatomy of the brainstem reticular formation (BRF) and its involvement in specific PD paradigms. This cartoon offers a schematic description of those brainstem areas properly belonging to the reticular formation (RF), which may be the key in PD pathology, as well as their selective recruitment in the three PD paradigms taken into account. In the upper part is shown the constellation of the RF nuclei following a neurotransmitter chemical classification. The isodendritic morphology of the neurons composing the RF nuclei, configures them as crucial stations of both afferent and efferent projections descending and projecting up to the cortex and spinal cord (SC). This network of overlapping connections is involved in a plenty of either extrapyramidal motor and non-motor functions. The major monoamine containing areas, mainly localized in the lateral RF except from C3, are the noradrenergic (A1–A7) adrenergic (C1–C3) dopaminergic (A8–A10) and cholinergic (Ch5–Ch6) nuclei. These are crucial for respiratory activity and for regulating blood pressure and heart rate, micturition, sweat, sleep-wakink cycle as well as descending motor control. Serotonergic nuclei are found in the median RF raphe nuclei, mainly in the B3, B8 and B9 areas. They control vegetative functions such as mood, sleep and sexual behavior, depression and pain. The medial RF, found between the median and the lateral column, is a region lacking monoamine nuclei, but whose giganto-cellular and paramedianpontine nuclei act as a station for fibers connecting with monoamine regions such as A6 (LC) and Ch6. They are involved in voluntary movement regulation, as well as in optical, acoustic and olfactory control due to their connections respectively, to the spinal cord and to the main cranial nerves’ nuclei. In the second part of the figure, it is shown how a progressive and selective anatomic recruitment of such nuclei may be phenotypically and genotypically related to specific PD subtypes. (A) Parkin PD—Impairment of ventral SNpc (A9), mild impairment of LC (A6) leading to solely motor symptoms. A minimal alteration of the dorsal raphe nucleus (B8–9) may lead to apathia or anhedonia. DMV is affected as well, which may lead to a partial impairment of the overlapping C2/A2 area controlling the parasympathetic outflow. (B) LRRK2 and slighter SNCA point mutations—Featuring the typical PD motor symptoms, along with the presence of sleep disorders, depression and dementia which are here related with a more extended involvement of monoamine nuclei of the lateral RF as well as of the median raphe nuclei. (C) Severe SNCA point mutations and large gene rearrangements—They present as a predominance of non-motor autonomic, psychotic and cognitive symptoms relying on the massive involvement of rostral and caudal areas of the RF extending above and beyond the brainstem.
The Case of Parkin Disease
The Parkin gene (OMIM 602544), codes for a protein called Parkin, a E3 ubiquitin-ligase, which tags altered proteins by ubiquitin chains (Hristova et al., 2009; Yoshii et al., 2011). The loss of function of parkin activity leads to a restricted pathology of the mesencephalic RF (SNpc; Takahashi et al., 1994), which is affected only in the ventral tier (Doherty et al., 2013). This is quite odd in PD since we are now aware that in most PD cases the pontine nucleus of LC (A6) is massively affected and represents a hallmark for pathological diagnosis (Braak et al., 2000; Dickson, 2012). Mutations in Parkin produce autosomic recessive (AR) parkinsonism with early onset. Alterations are spread over the entire gene and include deletions and duplications of one or more exons in more than 50% of cases (Matsumine et al., 1997; Kitada et al., 1998; Abbas et al., 1999; Labandeira-Garcia et al., 2011). Motor symptoms of PD patients with two Parkin mutations present with classic parkinsonism, slow disease progression and more symmetrical onset, with fewer NMS than classic PD (Lesage and Brice, 2009; Lohmann et al., 2009; Kasten et al., 2010), supporting the concept of limited pathology (Kägi et al., 2010). In line with this, the frequency of dementia in Parkin mutations matches the frequency of the general population above the age of 65 (Khan et al., 2003; Macedo et al., 2009; Xu et al., 2012). Even psychiatric symptoms are almost absent (Lohmann et al., 2009; Alcalay et al., 2014). These observations may be unexpected since the occurrence of psychiatric disorders PD patients is connected with dopamine (DA)-induced motor fluctuations. Indeed, these patients undergo DA-dependent motor fluctuations early in their life, nonetheless these motor fluctuations do not contaminate the psychiatric domain. This evidence is critical to understand the neurobiology of DA-dependent psychiatric disorders. The occurrence of fluctuations in response to DA replacement therapy is concomitant with a massive loss of DA axons (Lohmann et al., 2009). Thus, the DA replacement therapy generates peaks and valleys of extracellular DA concentrations, which trigger a non-canonical transduction pathway within post-synaptic neurons (Gerfen, 2006; Biagioni et al., 2009). When the DA axons are present, the DA substitution therapy does not generate fluctuations. In fact, the excess of DA concentrations is quickly buffered by the powerful uptake from surrounding DA terminals via the DA transporter (DAT). In PARK2 patients, the loss of DA selectively occurs in the dorsal striatum which produces motor fluctuations only consistent with the loss of ventral tier of the SNpc. In contrast, the meso-limbic DA rising from the reticular nucleus of ventral tegmental area (VTA) pathways is spared preserving DA fluctuations in the ventral striatum and brain areas, which are key for psychopathology. Other psychiatric symptoms such as depression, panic, and anxiety are absent as well, which is likely to depend on sparing 5-hydroxytryptamine (5-HT) and the NA neurons of the RF (Figure 2; Fornai et al., 2013). In fact, depressive symptoms in PD as a whole syndrome, or a few items such as apathy or anhedonia, are likely to rely on degeneration of reticular 5-HT dorsal raphe nucleus and/or NA LC nucleus (Politis and Loane, 2011). Interestingly, although PINK1 mutation carriers are clinically indistinguishable from Parkin mutation carriers, frequency of depression is higher in PINK1 carriers, calling for more detailed anatomical comparisons between these two diseases (Schneider and Klein, 2010; Ricciardi et al., 2014; Chaudhuri et al., 2015). Autonomic dysfunction often relies on medullary A1, A2 and C1 and C2 noradrenergic and adrenergic neurons, respectively, which provide the descending fibers to preganglionic sympathetic neurons (Figure 2). The absence of disease spreading caudally may explain the preservation of blood pressure, micturition, sweating and other autonomic functions in PARK2 patients (Palma and Kaufmann, 2014). This is further explained by the lack of involvement of post-ganglionic ortho-sympathetic neurons, which otherwise degenerate in most PD cases. The lack of upstream disease progression and preservation of cholinergic and noradrenergic ascending reticular pathways as shown in Figure 2 may explain why cognitive dysfunction and sleep disorders are absent in these patients. As reported for PINK1 and DJ-1, these PD patients show a largely preserved sense of smell, adding evidence to the observations that autosomal dominant forms of monogenic parkinsonism exhibit more severe olfactory impairment than the recessive ones (Yoritaka et al., 2011). Only in a few cases slight psychiatric symptoms may occur in Parkin PD patients (Cooney and Stacy, 2016; Lynch and Fujioka, 2016).
LRRK2 Mutations
Mutations of the LRRK2 gene (leucine-rich repeat kinase 2, OMIM 609007) is considered the most common genetic cause of late-onset, autosomal-dominant (AD) familial PD (PARK8; Paisán-Ruíz et al., 2004). The gene codes for a member of the leucine-rich repeat kinase family, widely expressed in isocortex, striatum, cerebellum, BRF and hippocampus (Sweet et al., 2015).
Several LRRK2 mutations have been reported so far. These genotypes differently affect the amount of catecholamine nuclei of the BRF, and produce various NMS patterns (Vitte et al., 2010). LRRK2 overlaps with alpha-synuclein pathology in the BRF, where deposition of LRRK2 appears to anticipate alpha-synuclein pathology (Alegre-Abarrategui et al., 2008). This makes the involvement of brainstem reticular nuclei in LRRK2 PD closely resembling point SNCA mutations and idiopathic PD.
In fact, clinical data support that in LRKK2 patients the prevalence of NMS is similar to idiopatic PD (i-PD; Estanga et al., 2014; Gaig et al., 2014).
Depression is quite common and can affect up to 40% of individuals with LRRK2 mutations (Chaudhuri et al., 2006; Langston, 2006; Schrag and Schott, 2006; Shanker et al., 2011; Gaig et al., 2014; Pont-Sunyer et al., 2015). Similarly, REM sleep behavioral disorder (RBD) is quite frequent (Trinh et al., 2014). This suggests a recruitment of ascending reticular nuclei extending beyond the SNpc. In fact, just like idiopathic PD, LRRK2 involves the substantia nigra and LC (Vitte et al., 2010).
The occurrence of dementia in 17% of LRKK2 patients is lower than i-PD (Aarsland and Kurz, 2010). This appears to rely on altered synaptic activity within hippocampus. Supporting this concept, transgenic mice expressing p.G2019S LRRK2 mutations show altered long-term depression (LTD) with potential impairment of learning and memory as shown in PD patients carrying LRRK2 mutations (Shanker et al., 2011; Sweet et al., 2015). Olfactory functions is preserved in PD patients carrying the LRRK2 mutations which is confirmed in preclinical models where mice expressing p.R1441C LRKK2 mutations exhibit normal olfactory function (Tsika et al., 2014). These latter findings may be in contradiction with the classic Braak staging of PD. Nonetheless, the brainstem progression involves monoamine containing nuclei of the RF and starts from the dorsal nucleus of the vagus and nucleus solitarius (Kingsbury et al., 2010). According to Figure 2 this area corresponds roughly to the A2/C2 catecholamine region of the medulla. The cortical pathology is independent from the recruitment of the brainstem and so would be the olfactory allocortex including the olfactory bulb.
Slight and Severe SNCA Point Mutations and Multiplications
The SNCA gene (alpha-synuclein, OMIM 163890) was the first gene to be associated with familial parkinsonism (Houlden and Singleton, 2012). This gene codes for the protein alpha-synuclein which is altered in both familial PD (F-PD) and S-PD (Oczkowska et al., 2013). Both point mutations (PARK1) and gene multiplications (PARK4), have been reported to produce PD with different age at onset, penetrance and clinical motor and non-motor features (Singleton et al., 2003; Puschmann et al., 2009; Kiely et al., 2013).
SNCA Multiplications
Triplication carriers have disease onset about 10 years earlier and a more rapid disease course than duplication carriers, who overall closely resemble i-PD patients, while higher clinical variability has been reported for different point mutations (Miller et al., 2004; Ferese et al., 2015). NMS are constantly present in all SNCA patients. Remarkably, those patients carrying four copies of SNCA undergo a severe phenotype of PD that leads to death in a few years from diagnosis. These patients possess early onset sleep disorders, autonomic dysfunction, and psychotic episodes, with massive involvement of the BRF (Figure 2). This is evidenced by a massive deposition of alpha- synuclein oligomers in the BRF (Roberts et al., 2015). These oligomers are made up of prion-like partially proteinase-K resistant alpha-synuclein just like oligomers in the brainstem of PD patients. These patients show rapid cognitive and motor deterioration which surpasses the extrapyramidal circuitry affecting the corticospinal pathway as evidenced by a pyramidal syndrome which adds on the extrapyramidal movement disorder which is the main cause of death in these patients (Singleton et al., 2003; Ferese et al., 2015). These latter syndromes indicate that neuronal degeneration recruits brain areas above the brainstem level extending well beyond the classic neuropathology described by Tretiakoff in 1919 (Parent and Parent, 2010).
SNCA Duplication
SNCA duplications lead to various NMS (dysautonomia, RBD, hallucinations, depression; Konno et al., 2016) albeit with lower prevalence compared with carriers of four SNCA copies, but still higher compared with i-PD. SNCA duplication shows an incomplete penetrance in some carriers and, interestingly, NMS are reported in both healthy people and PD patients carrying this kind of mutation. Such carriers are reported to show an impairment of reward learning, suggesting that a copy number variation of the SNCA gene may be associated with NMS (especially selective learning impairment) more than producing classic motor parkinsonism (Kéri et al., 2010). In contrast, olfactory dysfunction and RBD are observed only in symptomatic carriers (Nishioka et al., 2009).
SNCA Point Mutations
High clinical variability has been reported in SNCA point mutations, where NMS are less frequent if compared with SNCA multiplications, and dementia is also less frequent when compared with i-PD patients (Aarsland et al., 2005; Aarsland and Kurz, 2010). NMS features are variably associated with different point mutations. p.A53T and p.A30P are reported to correlate with dementia (Puschmann et al., 2009), p.E46K with dementia and visual hallucinations (Zarranz et al., 2004), and p.G209A with olfactory dysfunction and RBD. In G209A carriers, prominent motor decline and deterioration of autonomic and cognitive function occur.
Remarkably, the point mutation p.G51D leads to a rapidly evolving syndrome. In this case, despite no gene multiplication occurs, there is a widespread neuropathology that, similarly to patients carrying four SNCA copies, leads to the involvement of the pyramidal tract. Even in this case, the cortico-spinal degeneration is the main cause of death with a clinical course and neuropathology which surpasses PD and features MSA, where a plethora of NMS and massive areas in the brainstem are affected to extend rostrally to the prosencephalon and caudally to the spinal cord (Lesage et al., 2013; Kiely et al., 2015).
Conclusions
The occurrence of both motor NMS in most cases of PD is now well established. This is related to the engagement of a constellation of brain areas mostly located in the core of the BRF. The severity and variety of motor symptoms and mostly the various occurrence of NMS produce different PD syndromes, which appear more and more as a variety of diseases. This is substantiated by the number of genetic alterations, which may produce PD, and it is confirmed by the variety brain nuclei which may be involved. The present manuscript suggests a clinical anatomical correlation based on different genetic alterations which produce PD. These genetic conditions were used as paradigms for a clear-cut separation of different PD syndromes. The various involvement of brainstem reticular nuclei in these three paradigms was discussed.
Evidence was provided that the onset of an almost pure extrapyramidal syndrome due to a Parkin mutation was related to a quite unusual pathology mostly confined to the ventral tier of the SNpc. On the other hand, the multiplications of the SNCA genes and the p.G51D SNCA point mutation were related to the most severe phenotype in which a plethora of NMS (psychotic and mood disorder, sleep disorder, cognitive alterations, autonomic dysfunctions) were associated with a massive involvement of several nuclei of the BRF. Remarkably the most severe condition (point mutation p.G51D is definitely considered as MSA rather than PD). In the middle of these paradigms, we described the clinical anatomical correlation in the case of most LRRK2 mutation and SNCA point mutation, which resembles S-PD. An interesting correlation was evident between specific NMS and specific brainstem reticular nuclei. This is quite remarkable for anxiety, mood and sleep disorders which associate with serotonergic neurons and lateral reticular noradrenergic nuclei. Similarly, the occurrence of autonomic dysfunctions witnesses for the involvement of the bulbar noradrenergic and adrenergic areas (A1/A2 and C1/C2, respectively). The impairment of oculo-motor activities suggests the impairment in the paramedianpontine RF, while the presence of DA-dependent psychotic symptoms relates with the involvement of the mesencephalic VTA of the RF which projects to limbic and iso-cortical regions. In the most severe phenotypes, the disease surpasses the definition of BMD and configures as a MSA where the early involvement of the brainstem is expected to spread quickly to distant brain regions also including the motor cortex and spinal cord. The spreading of symptoms in the disease course is likely to rely on the prion-like properties of key proteins such as alpha-synuclein. The variety of brain areas is not totally unexpected in the light of the remarkable branching and collateralization of the iso-dendritic neurons forming the core of the BRF which represents a powerful stream to drive physiological and altered synaptic activity.
Author Contributions
SG: coordination and writing the review. RF: pubmed research and state of the art about genetic of parkin. FB: pubmed research and state of the art about genetic of LRRK2. CLB: pubmed research and state of the art about genetic of SNCA. RC: participated in critically revising the article. AMPG: participated in drafting the article. FL: pubmed research and state of the art about genetic of SNCA duplication and triplication. GN: participated in critically revising the article for important intellectual content. MS: participated in drafting the article. FF: coordinating and writing the article. Participated in drafting the article. Participated in critically revising the article for important intellectual content.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aarsland, D., and Kurz, M. W. (2010). The epidemiology of dementia associated with Parkinson’s disease. Brain Pathol. 20, 633–639. doi: 10.1111/j.1750-3639.2009.00369.x
Aarsland, D., Zaccai, J., and Brayne, C. (2005). A systematic review of prevalence studies of dementia in Parkinson’s disease. Mov. Disord. 20, 1255–1263. doi: 10.1002/mds.20527
Abbas, N., Lücking, C. B., Ricard, S., Dürr, A., Bonifati, V., De Michele, G., et al. (1999). A wide variety of mutations in the parkin gene are responsible for autosomal recessive parkinsonism in Europe. French Parkinson’s disease genetics study group and the european consortium on genetic susceptibility in Parkinson’s disease. Hum. Mol. Genet. 8, 567–574. doi: 10.1093/hmg/8.4.567
Alcalay, R. N., Caccappolo, E., Mejia-Santana, H., Tang, M. X., Rosado, L., Orbe Reilly, M., et al. (2014). Cognitive and motor function in long-duration PARKIN-associated Parkinson disease. JAMA Neurol. 71, 62–67. doi: 10.1001/jamaneurol.2013.4498
Alegre-Abarrategui, J., Ansorge, O., Esiri, M., and Wade-Martins, R. (2008). LRRK2 is a component of granular α-synuclein pathology in the brainstem of Parkinson’s disease. Neuropathol. Appl. Neurobiol. 34, 272–283. doi: 10.1111/j.1365-2990.2007.00888.x
Bastide, M. F., Meissner, W. G., Picconi, B., Fasano, S., Fernagut, P. O., Feyder, M., et al. (2015). Pathophysiology of L-dopa-induced motor and non-motor complications in Parkinson’s disease. Prog. Neurobiol. 132, 96–168. doi: 10.1016/j.pneurobio.2015.07.002
Biagioni, F., Pellegrini, A., Ruggieri, S., Murri, L., Paparelli, A., and Fornai, F. (2009). Behavioural sensitisation during dopamine replacement therapy in Parkinson’s disease is reminiscent of the addicted brain. Curr. Top. Med. Chem. 9, 894–902. doi: 10.2174/156802609789378245
Bonnet, A. M., Jutras, M. F., Czernecki, V., Corvol, J. C., and Vidailhet, M. (2012). Nonmotor symptoms in Parkinson’s disease in 2012: relevant clinical aspects. Parkinsons Dis. 2012:198316. doi: 10.1155/2012/198316
Braak, H., Rüb, U., Sandmann-Keil, D., Gai, W. P., de Vos, R. A., Jansen Steur, E. N., et al. (2000). Parkinson’s disease: affection of brain stem nuclei controlling premotor and motor neurons of the somatomotor system. Acta Neuropathol. 99, 489–495. doi: 10.1007/s004010051150
Chao, Y. X., Chew, L. M., Deng, X., and Tan, E. K. (2015). Nonmotor symptoms in sporadic versus familial forms of Parkinson’s disease. Neurodegener. Dis. Manag. 5, 147–153. doi: 10.2217/nmt.14.57
Chaudhuri, K. R., Healy, D. G., Schapira, A. H., and National Institute for Clinical Excellence. (2006). Non-motor symptoms of Parkinson’s disease: diagnosis and management. Lancet Neurol. 5, 235–245. doi: 10.1016/S1474-4422(06)70373-8
Chaudhuri, R., Tolosa, E., Schapira, A. H. V., and Poewe, W. (2015). Non-Motor Symptoms of Parkinson’s Disease. 2nd Edn. Oxford: Oxford University Press.
Chege, P. M., and McColl, G. (2014). Caenorhabditis elegans: a model to investigate oxidative stress and metal dyshomeostasis in Parkinson’s disease. Front. Aging Neurosci. 89:2014. doi: 10.3389/fnagi.2014.00089
Chen, D., Wei, X., Zou, J., Wang, R., Liu, X., Xu, X., et al. (2015). Contra-directional expression of serum homocysteine and uric acid as important biomarkers of multiple system atrophy severity: a cross-sectional study. Front. Cell. Neurosci. 9:247. doi: 10.3389/fncel.2015.00247
Cooney, J. W., and Stacy, M. (2016). Neuropsychiatric issues in Parkinson’s disease. Curr. Neurol. Neurosci. Rep. 16:49. doi: 10.1007/s11910-016-0647-45
Dickson, D. W. (2012). Parkinson’s disease and parkinsonism: neuropathology. Cold Spring Harb. Perspect. Med. 2:a009258. doi: 10.1101/cshperspect.a009258
Doherty, K. M., Silveira-Moriyama, L., Parkkinen, L., Healy, D. G., Farrell, M., Mencacci, N. E., et al. (2013). Parkin disease: a clinicopathologic entity? JAMA Neurol. 70, 571–579. doi: 10.1001/jamaneurol.2013.172
Estanga, A., Rodriguez-Oroz, M. C., Ruiz-Martinez, J., Barandiaran, M., Gorostidi, A., Bergareche, A., et al. (2014). Cognitive dysfunction in Parkinson’s disease related to the R1441G mutation in LRRK2. Parkinsonism Relat. Disord. 10, 1097–1100. doi: 10.1016/j.parkreldis.2014.07.005
Ferese, R., Modugno, N., Campopiano, R., Santilli, M., Zampatti, S., Giardina, E., et al. (2015). Four copies of SNCA responsible for autosomal dominant Parkinson’s disease in two italian siblings. Parkinsons Dis. 2015:546462. doi: 10.1155/2015/546462
Fornai, F., Frati, A., Gesi, M., Fulceri, F., Paparelli, S., Falleni, A., et al. (2013). Neurobiology and neuroanatomy of psychiatric symptoms in parkinsonism. Arch. Ital. Biol. 151, 179–191.
Fornai, F., and Ruggieri, S. (2013). Re-defining Parkinson’s disease. Arch. Ital. Biol. 151, 137–142.
Gaig, C., Vilas, D., Infante, J., Sierra, M., García-Gorostiaga, I., Buongiorno, M., et al. (2014). Nonmotor symptoms in LRRK2 G2019S associated Parkinson’s disease. PLoS One 9:108982. doi: 10.1371/journal.pone.0108982
Gao, H., Wei, X., Liao, J., Wang, R., Xu, J., Liu, X., et al. (2015). Lower bone mineral density in patients with Parkinson’s disease: a cross-sectional study from chinese mainland. Front. Aging Neurosci. 7:203. doi: 10.3389/fnagi.2015.00203
Gerfen, C. R. (2006). Indirect-pathway neurons lose their spines in Parkinson disease. Nat. Neurosci. 9, 157–158. doi: 10.1038/nn0206-157
Houlden, H., and Singleton, A. B. (2012). The genetics and neuropathology of Parkinson’s disease. Acta Neuropathol. 124, 325–338. doi: 10.1007/s00401-012-1013-5
Hristova, V. A., Beasley, S. A., Rylett, R. J., and Shaw, G. S. (2009). Identification of a novel Zn2+- binding domain in the autosomal recessive juvenile Parkinson-related E3 ligase parkin. J. Biol. Chem. 284, 14978–14986. doi: 10.1074/jbc.M808700200
Kägi, G., Klein, C., Wood, N. W., Schneider, S. A., Pramstaller, P. P., Tadic, V., et al. (2010). Nonmotor symptoms in Parkin gene-related parkinsonism. Mov. Disord. 25, 1279–1284. doi: 10.1002/mds.22897
Kasten, M., Kertelge, L., Brüggemann, N., van der Vegt, J., Schmidt, A., Tadic, V., et al. (2010). Nonmotor symptoms in genetic Parkinson disease. Arch. Neurol. 67, 670–676. doi: 10.1001/archneurol.67.6.670
Kéri, S., Moustafa, A. A., Myers, C. E., Benedek, G., and Gluck, M. A. (2010). α-Synuclein gene duplication impairs reward learning. Proc. Natl. Acad. Sci. U S A 107, 15992–15994. doi: 10.1073/pnas.1006068107
Khan, N. L., Graham, E., Critchley, P., Schrag, A. E., Wood, N. W., Lees, A. J., et al. (2003). Parkin disease: a phenotypic study of a large case series. Brain 126, 1279–1292. doi: 10.1093/brain/awg142
Kiely, A. P., Asi, Y. T., Kara, E., Limousin, P., Ling, H., Lewis, P., et al. (2013). α-Synucleinopathy associated with G51D SNCA mutation:a link between Parkinson’s disease and multiple system atrophy? Acta Neuropathol. 125, 753–769. doi: 10.1007/s00401-013-1096-7
Kiely, A. P., Ling, H., Asi, Y. T., Kara, E., Proukakis, C., Schapira, A. H., et al. (2015). Distinct clinical and neuropathological features of G51D SNCA mutation cases compared with SNCA duplication and H50Q mutation. Mol. Neurodegener. 10:41. doi: 10.1186/s13024-015-0038-3
Kingsbury, A. E., Bandopadhyay, R., Silveira-Moriyama, L., Ayling, H., Kallis, C., Sterlacci, W., et al. (2010). Brain stem pathology in Parkinson’s disease: an evaluation of the Braak staging model. Mov. Disord. 25, 2508–2515. doi: 10.1002/mds.23305
Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S., et al. (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608. doi: 10.1038/33416
Konno, T., Ross, O. A., Puschmann, A., Dickson, D. W., and Wszolek, Z. K. (2016). Autosomal dominant Parkinson’s disease caused by SNCA duplications. Parkinsonism Relat. Disord. 22, S1–S6. doi: 10.1016/j.parkreldis.2015.09.007
Labandeira-Garcia, J. L., Rodriguez-Pallares, J., Villar-Cheda, B., Rodríguez-Perez, A. I., Garrido-Gil, P., and Guerra, M. J. (2011). Aging, angiotensin system and dopaminergic degeneration in the substantia nigra. Aging Dis. 2, 257–274.
Langston, J. W. (2006). The Parkinson’s complex: parkinsonism is just the tip of the iceberg. Ann. Neurol. 59, 591–596. doi: 10.1002/ana.20834
Lesage, S., Anheim, M., Letournel, F., Bousset, L., Honoré, A., Rozas, N., et al. (2013). G51D α-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol. 73, 459–471. doi: 10.1002/ana.23894
Lesage, S., and Brice, A. (2009). Parkinson’s disease: from monogenic forms to genetic susceptibility factors. Hum. Mol. Genet. 18, 48–59. doi: 10.1093/hmg/ddp012
Li, H., Zhang, M., Chen, L., Zhang, J., Pei, Z., Hu, A., et al. (2010). Nonmotor symptoms are independently associated with impaired health-related quality of life in Chinesepatients with Parkinson’s disease. Mov. Disord. 25, 2740–2746. doi: 10.1002/mds.23368
Lohmann, E., Thobois, S., Lesage, S., Broussolle, E., du Montcel, S. T., Ribeiro, M. J., et al. (2009). A multidisciplinary study of patients with early-onset PD with and without parkin mutations. Neurology 72, 110–116. doi: 10.1212/01.WNL.0000327098.86861.d4
Lynch, T., and Fujioka, S. (2016). Impulsive and compulsive parkin disease. Neurology 24, 1426–1427. doi: 10.1212/WNL.0000000000003188
Macedo, M. G., Verbaan, D., Fang, Y., van Rooden, S. M., Visser, M., Anar, B., et al. (2009). Genotypic and phenotypic characteristics of Dutch patients with early onset Parkinson’s disease. Mov. Disord. 242, 196–203. doi: 10.1002/mds.22287
Marras, C., and Chaudhuri, K. R. (2016). Nonmotor features of Parkinson’s disease subtypes. Mov. Disord. 31, 1095–1102. doi: 10.1002/mds.26510
Matsumine, H., Saito, M., Shimoda-Matsubayashi, S., Tanaka, H., Ishikawa, A., Nakagawa-Hattori, Y., et al. (1997). Localization of a gene for an autosomal recessive form of juvenile Parkinsonism to chromosome 6q25.2–27. Am. J. Hum. Genet. 60, 588–596.
Miller, D. W., Hague, S. M., Clarimon, J., Baptista, M., Gwinn-Hardy, K., Cookson, M. R., et al. (2004). α-synuclein in blood and brain from familial Parkinson disease with SNCA locus triplication. Neurology 62, 1835–1838. doi: 10.1212/01.WNL.0000127517.33208.f4
Mutez, E., Leprêtre, F., Le Rhun, E., Larvor, L., Duflot, A., Mouroux, V., et al. (2011). SNCA locus duplication carriers: from genetics to Parkinson disease phenotypes. Hum. Mutat. 32, E2079–E2090. doi: 10.1002/humu.21459
Natale, G., Biagioni, F., Vivacqua, G., D’Este, L., Fumagalli, L., and Fornai, F. (2013). The neurobiology of dysautonomia in Parkinson’s disease. Arch. Ital. Biol. 151, 203–218.
Nishioka, K., Ross, O. A., Ishii, K., Kachergus, J. M., Ishiwata, K., Kitagawa, M., et al. (2009). Expanding the clinical phenotype of SNCA duplication carriers. Mov. Disord. 24, 1811–1819. doi: 10.1002/mds.22682
Oczkowska, A., Kozubski, W., Lianeri, M., and Dorszewska, J. (2013). Mutations in PRKN and SNCA genes important for the progress of Parkinson’s disease. Curr. Genomics 14, 502–517. doi: 10.2174/1389202914666131210205839
Paisán-Ruíz, C., Jain, S., Evans, E. W., Gilks, W. P., Simón, J., van der Brug, M., et al. (2004). Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 18, 595–600. doi: 10.1016/j.neuron.2004.10.023
Palma, J.-A., and Kaufmann, H. (2014). Autonomic disorders predicting Parkinson’s disease. Parkinsonism Relat. Disord. 20, S94–S98. doi: 10.1016/s1353-8020(13)70024-5
Parent, M., and Parent, A. (2010). Substantia nigra and Parkinson’s disease: a brief history of their long and intimate relationship. Can. J. Neurol. Sci. 37, 313–319. doi: 10.1017/s0317167100010209
Poewe, W. (2008). Non-motor symptoms in Parkinson’s disease. Eur. J. Neurol. 15, 14–20. doi: 10.1111/j.1468-1331.2008.02056.x
Politis, M., and Loane, C. (2011). Serotonergic dysfunction in Parkinson’s disease and its relevance to disability. ScientificWorldJournal 11, 1726–1734. doi: 10.1100/2011/172893
Pont-Sunyer, C., Iranzo, A., Gaig, C., Fernández-Arcos, A., Vilas, D., and Valldeoriola, F. (2015). Sleep disorders in parkinsonian and nonparkinsonian LRRK2 mutation carriers. PLoS One 10:e0132368. doi: 10.1371/journal.pone.0132368
Puschmann, A., Ross, O. A., Vilariño-Güell, C., Lincoln, S. J., Kachergus, J. M., and Cobb, S. A. (2009). A swedish family with de novo α-synuclein A53T mutation: evidence for early cortical dysfunction. Parkinsonism Relat. Disord. 15, 627–632. doi: 10.1016/j.parkreldis.2009.06.007
Ricciardi, L., Petrucci, S., Guidubaldi, A., Ialongo, T., Serra, L., Ferraris, A., et al. (2014). Phenotypic variability of PINK1 expression: 12 years’ clinical follow-up of two Italian families. Mov. Disord. 29, 1561–1566. doi: 10.1002/mds.25994
Roberts, R. F., Wade-Martins, R., and Alegre-Abarrategui, J. (2015). Direct visualization of α-synuclein oligomers reveals previously undetected pathology in Parkinson’s disease brain. Brain 138, 1642–1657. doi: 10.1093/brain/awv040
Schneider, S. A., and Klein, C. (2010). PINK1 Type of Young-Onset Parkinson Disease. GeneReviews®[Internet]. Seattle, WA: University of Washington.
Schrag, A., and Schott, J. M. (2006). Epidemiological, clinical, and genetic characteristics of early-onset parkinsonism. Lancet Neurol. 5, 355–363. doi: 10.1016/S1474-4422(06)70411-2
Shanker, V., Groves, M., Heiman, G., Palmese, C., Saunders-Pullman, R., Ozelius, L., et al. (2011). Mood and cognition in leucine-rich repeat kinase 2 G2019S Parkinson’s disease. Mov. Disord. 26, 1875–1880. doi: 10.1002/mds.23746
Singleton, A. B., Farrer, M., Johnson, J., Singleton, A., Hague, S., Kachergus, J., et al. (2003). α-Synuclein locus triplication causes Parkinson’s disease. Science 302:841. doi: 10.1126/science.1090278
Sweet, E. S., Saunier-Rebori, B., Yue, Z., and Blitzer, R. D. (2015). The Parkinson’s disease-associated mutation LRRK2–G2019S impairs synaptic plasticity in mouse hippocampus. J. Neurosci. 35, 11190–11195. doi: 10.1523/JNEUROSCI.0040-15.2015
Takahashi, H., Ohama, E., Suzuki, S., Horikawa, Y., Ishikawa, A., Morita, T., et al. (1994). Familial juvenile parkinsonism: clinical and pathologic study in a family. Neurology 44, 437–441. doi: 10.1212/wnl.44.3_part_1.437
Trinh, J., Amouri, R., Duda, J. E., Morley, J. F., Read, M., Donald, A., et al. (2014). Comparative study of Parkinson’s disease and leucine-rich repeat kinase 2 p.G2019S parkinsonism. Neurobiol. Aging 35, 1125–1131. doi: 10.1016/j.neurobiolaging.2013.11.015
Tsika, E., Kannan, M., Foo, C. S., Dikeman, D., Glauser, L., Gellhaar, S., et al. (2014). Conditional expression of Parkinson’s disease-related R1441C LRRK2 in midbrain dopaminergic neurons of mice causes nuclear abnormalities without neurodegeneration. Neurobiol. Dis. 71, 345–358. doi: 10.1016/j.nbd.2014.08.027
Vitte, J., Traver, S., Maués De Paula, A., Lesage, S., Rovelli, G., Corti, O., et al. (2010). Leucine-rich repeat kinase 2 is associated with the endoplasmic reticulum in dopaminergic neurons and accumulates in the core of Lewy bodies in Parkinson disease. J. Neuropathol. Exp. Neurol. 69, 959–972. doi: 10.1097/nen.0b013e3181efc01c
Wei, X., Yan, R., Chen, Z., Weng, R., Liu, X., Gao, H., et al. (2016). Combined diffusion tensor imaging and arterial spin labeling as markers of early Parkinson’s disease. Sci. Rep. 6:33762. doi: 10.1038/srep33762
Xu, Y., Wei, X., Liu, X., Liao, J., Lin, J., Zhu, C., et al. (2015). Low cerebral glucose metabolism: a potential predictor for the severity of vascular parkinsonism and Parkinson’s disease. Aging Dis. 6, 426–436. doi: 10.14336/AD.2015.0204
Xu, Y., Yan, J., Zhou, P., Li, J., Gao, H., Xia, Y., et al. (2012). Neurotransmitter receptors and cognitive dysfunction in Alzheimer’s disease and Parkinson’s disease. Prog. Neurobiol. 97, 1–13. doi: 10.1016/j.pneurobio.2012.02.002
Yoritaka, A., Shimo, Y., Shimo, Y., Inoue, Y., Yoshino, H., and Hattori, N. (2011). Nonmotor symptoms in patients with PARK2 mutations. Parkinsons Dis. 2011:473640. doi: 10.4061/2011/473640
Yoshii, S. R., Kishi, C., Ishihara, N., and Mizushima, N. (2011). Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J. Biol. Chem. 286, 19630–19640. doi: 10.1074/jbc.M110.209338
Zarranz, J. J., Alegre, J., Gómez-Esteban, J. C., Lezcano, E., Ros, R., Ampuero, I., et al. (2004). The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 55, 164–173. doi: 10.1002/ana.10795
Zimprich, A., Biskup, S., Leitner, P., Lichtner, P., Farrer, M., Lincoln, S., et al. (2004). Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44, 601–607. doi: 10.1016/j.neuron.2004.11.005
Keywords: Parkinson’s disease, non-motor symptoms, pyramidal syndrome, genetic Parkinsonism, genotype-phenotype correlation
Citation: Gambardella S, Ferese R, Biagioni F, Busceti CL, Campopiano R, Griguoli AMP, Limanaqi F, Novelli G, Storto M and Fornai F (2017) The Monoamine Brainstem Reticular Formation as a Paradigm for Re-Defining Various Phenotypes of Parkinson’s Disease Owing Genetic and Anatomical Specificity. Front. Cell. Neurosci. 11:102. doi: 10.3389/fncel.2017.00102
Received: 16 December 2016; Accepted: 27 March 2017;
Published: 18 April 2017.
Edited by:
Pier Giorgio Mastroberardino, Erasmus University Rotterdam, NetherlandsReviewed by:
Gonzalo Alvarez-Bolado, Heidelberg University, GermanyDennis Qing Wang, Third Affiliated Hospital of Sun Yat-sen University, China
Copyright © 2017 Gambardella, Ferese, Biagioni, Busceti, Campopiano, Griguoli, Limanaqi, Novelli, Storto and Fornai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefano Gambardella, c3RlZmFuby5nYW1iYXJkZWxsYUBuZXVyb21lZC5pdA==