 Men C. Tan1,2†
Men C. Tan1,2† Jocelyn Widagdo1,2*†
Jocelyn Widagdo1,2*† Yu Q. Chau1,2
Yu Q. Chau1,2 Tianyi Zhu1,2
Tianyi Zhu1,2 Justin J.-L. Wong3,4
Justin J.-L. Wong3,4 Allen Cheung2
Allen Cheung2 Victor Anggono1,2*
Victor Anggono1,2*- 1Clem Jones Centre for Ageing Dementia Research, The University of Queensland, Brisbane, QLD, Australia
- 2Queensland Brain Institute, The University of Queensland, Brisbane, QLD, Australia
- 3Gene and Stem Cell Therapy Program, Centenary Institute, Sydney, NSW, Australia
- 4Sydney Medical School, University of Sydney, Sydney, NSW, Australia
Transcription of new RNA is crucial for maintaining synaptic plasticity, learning and memory. Although the importance of synaptic plasticity-related messenger RNAs (mRNAs) is well established, the role of a large group of long non-coding RNAs (lncRNAs) in long-term potentiation (LTP) is not known. In this study, we demonstrated the expression of a lncRNA cluster, namely maternally expressed gene 3 (Meg3), retrotransposon-like gene 1-anti-sense (Rtl1-AS), Meg8 and Meg9, which is located in the maternally imprinted Dlk1-Dio3 region on mouse chromosome 12qF1, in primary cortical neurons following glycine stimulation in an N-Methyl-D-aspartate receptor (NMDAR)-dependent manner. Importantly, we also validated the expression of Meg3, Meg8 and Meg9 in the hippocampus of mice following cued fear conditioning in vivo. Interestingly, Meg3 is the only lncRNA that is expressed in the nucleus and cytoplasm. Further analysis revealed that Meg3 loss of function blocked the glycine-induced increase of the GluA1 subunit of AMPA receptors on the plasma membrane, a major hallmark of LTP. This aberrant trafficking of AMPA receptors correlated with the dysregulation of the phosphatidylinoside-3-kinase (PI3K)/AKT signaling pathway and the downregulation of the lipid phosphatase and tensin homolog (PTEN). These findings provide the first evidence for a functional role of the lncRNA Meg3 in the intricate regulation of the PTEN/PI3K/AKT signaling cascade during synaptic plasticity in neurons.
Introduction
The ability of neurons to modulate the strength of their connectivity, a process known as synaptic plasticity, has long been postulated as a cellular mechanism of learning and memory (Nicoll, 2017). One of the best-studied forms of such plasticity is long-term potentiation (LTP) which produces a persistent increase in the synaptic strength in neurons, characterized by an increase in the size of dendritic spines and the number of synaptic AMPA-type glutamate receptors (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs)). A prominent theory of LTP and long-term memory holds that new messenger RNAs (mRNAs) must be transcribed and new proteins must be synthesized in order to maintain the long-lasting increase in the magnitude of synaptic response and memory consolidation (Huang et al., 1996; Adams and Dudek, 2005). Initial studies have shown that the inhibition of transcription and translation block the synthesis of new AMPARs and late-phase LTP in acute hippocampal slices (Frey et al., 1993; Nguyen et al., 1994; Nayak et al., 1998). More importantly, they also block the maintenance of LTP and long-term memory retention in vivo (Frey et al., 1996; Kandel, 2001; Alberini, 2009; Radwanska et al., 2011; Hagena and Manahan-Vaughan, 2013). Interestingly, commonly used transcription inhibitors such as actinomycin D prevent not only transcription of new mRNAs, but all RNA polymerase II-dependent transcription, which includes various forms of non-coding RNAs.
Long non-coding RNAs (lncRNAs), broadly classified as being more than 200 nucleotides in size, represent a large proportion of the transcriptome and are widely expressed in the mammalian brain in a cell type- and developmental stage-specific manner (Mercer et al., 2008), but a vast proportion are still functionally uncharacterized. In the brain, a number of lncRNAs have been shown to play prominent roles during neural development and differentiation (Bond et al., 2009; Bernard et al., 2010; Roberts et al., 2014), and they are increasingly associated with human neurological disorders (Briggs et al., 2015). However, the role of lncRNAs in post-mitotic neurons is not known. Mechanistically, lncRNAs are capable of binding DNA, RNA and proteins, allowing them to perform a sensory, guiding or scaffolding function and influence multiple processes, from transcriptional regulation to the modulation of protein activity (Mercer and Mattick, 2013). The versatility and dynamics of lncRNAs are highly suited for the rapid neuronal response to extracellular signals, synaptic plasticity and adaptive behaviors. However, despite some evidence of stimulus-dependent expression of neuronal lncRNAs (Briggs et al., 2015; Maag et al., 2015), there is currently no empirical support for lncRNA function in synaptic plasticity or the regulation of learning and memory.
The Dlk1-Dio3 imprinted region, located on mouse distal chromosome 12qF1 (or human chromosome 14q32), contains a cluster of lncRNA genes that are selectively expressed from the maternally inherited allele, namely maternally expressed gene 3 (Meg3), retrotransposon-like gene 1-anti-sense (Rtl1-AS), Meg8 and Meg9 (da Rocha et al., 2008). Meg3, also known as gene trap locus 2 (Gtl2), is highly expressed in neurons in the forebrain of both developing and adult mice (McLaughlin et al., 2006; Qu et al., 2013). However, genetic deletion resulting in the loss of Meg3 from the maternal allele causes perinatal death in mice (Zhou et al., 2010), precluding further observation of any potential neurological phenotypes. The cellular function of Meg3 in regulating cell death and differentiation has been well studied, and it is thought to play a role as a tumor suppressor as its expression is often markedly downregulated in various types of cancers (Zhou et al., 2012). However, despite being highly expressed in the brain, Meg3 currently has no known function in post-mitotic neurons.
In the present study, we reveal that the expression of all four lncRNAs within the Dlk1-Dio3 imprinted locus is dynamically upregulated in primary cultured neurons following glycine stimulation, a validated method of chemically inducing N-Methyl-D-aspartate receptor (NMDAR)-dependent LTP in vitro (Lu et al., 2001). Importantly, increased expression of these lncRNAs can also be observed in the hippocampus of mice that undergo a fear-conditioned associative learning paradigm. Short hairpin RNA (shRNA)- and antisense oligonucleotide (ASO)-mediated knockdown of Meg3, the transcript of which is present in both the nucleus and cytoplasm, enhances surface AMPAR expression under basal conditions and prevents a further increase in the expression of these receptors on the plasma membrane following glycine stimulation. These effects are associated with an overall activation of the phosphatidylinoside-3-kinase (PI3K)/AKT signaling pathway, which plays a major role in the activity-dependent delivery of AMPARs to the plasma membrane. These results provide insights into the role of Meg3 in modulating the PI3K/AKT/phosphatase and tensin homolog (PTEN) signaling pathway in order to fine-tune the level of surface AMPARs during synaptic plasticity.
Materials and Methods
Animals
Adult male C57BL/6 mice (8–12 weeks old) were used for the fear-conditioning experiments, whereas embryonic day 17 mice were used for primary neuronal cultures. Mice were housed on a 12 h light/dark schedule, and fed ad libitum. This study was carried out in accordance with the recommendations of Australian code for the care and use of animals for scientific purposes by the National Health and Medical Research Council. All testing was conducted during the light phase in red light-illuminated rooms following protocols approved by the University of Queensland Animal Ethics Committee.
Antibodies
Specific antibodies against CREB (cAMP response element binding protein, 86B10), phosphorylated CREB (Ser133; 87G3), ERK1/2 (extracellular signal-regulated kinase 1/2, 137F5), phosphorylated ERK (Thr202/Tyr204; E10), AKT (C67E7), phosphorylated AKT (Thr308; D25E6), phosphorylated AKT (Ser473; D9E), p85 (19H8), phosphorylated p85 (Tyr458; 4288), PTEN (D4.3), β-actin (13E5) and GAPDH (14C10) were purchased from Cell Signaling Technology. The GluA1 (MAB2263) and phosphorylated GluA1 (Ser831; AB5847) antibodies were from Millipore.
Primary Neuronal Cultures
Cortical neurons were prepared and maintained in Neurobasal medium containing 2% B27, 2 mM Glutamax, 50 U/ml penicillin, 50 μg/ml streptomycin, and 1% fetal bovine serum as described previously (Anggono et al., 2013; Widagdo et al., 2015). All reagents were obtained from Invitrogen.
Glycine-Induced Chem-LTP
Chem-LTP was performed on mouse cortical neurons at days in vitro (DIV) 15–17 as previously described, with slight modifications (Hussain et al., 2014). Briefly, neurons were incubated for 1 h in artificial cerebrospinal fluid (ACSF; 125 mM NaCl, 2.5 mM KCl, 1.5 mM CaCl2, 25 mM HEPES, pH 7.4, 33 mM glucose, 1 mM MgCl2, 500 nM tetrodotoxin (Tocris), 20 μM bicuculline (Tocris), 1 μM strychnine (Sigma)) prior to 10-min incubation with 200 μM glycine in magnesium-free ACSF to induce chem-LTP (Figure 1A). For the 20 and 40 min time points, neurons were recovered in ACSF for another 10 or 30 min prior to lysis. To block NMDAR activity, 50 μM D-APV (Tocris) was added to the ACSF 10 min prior to the induction of LTP, and remained present throughout the treatment.
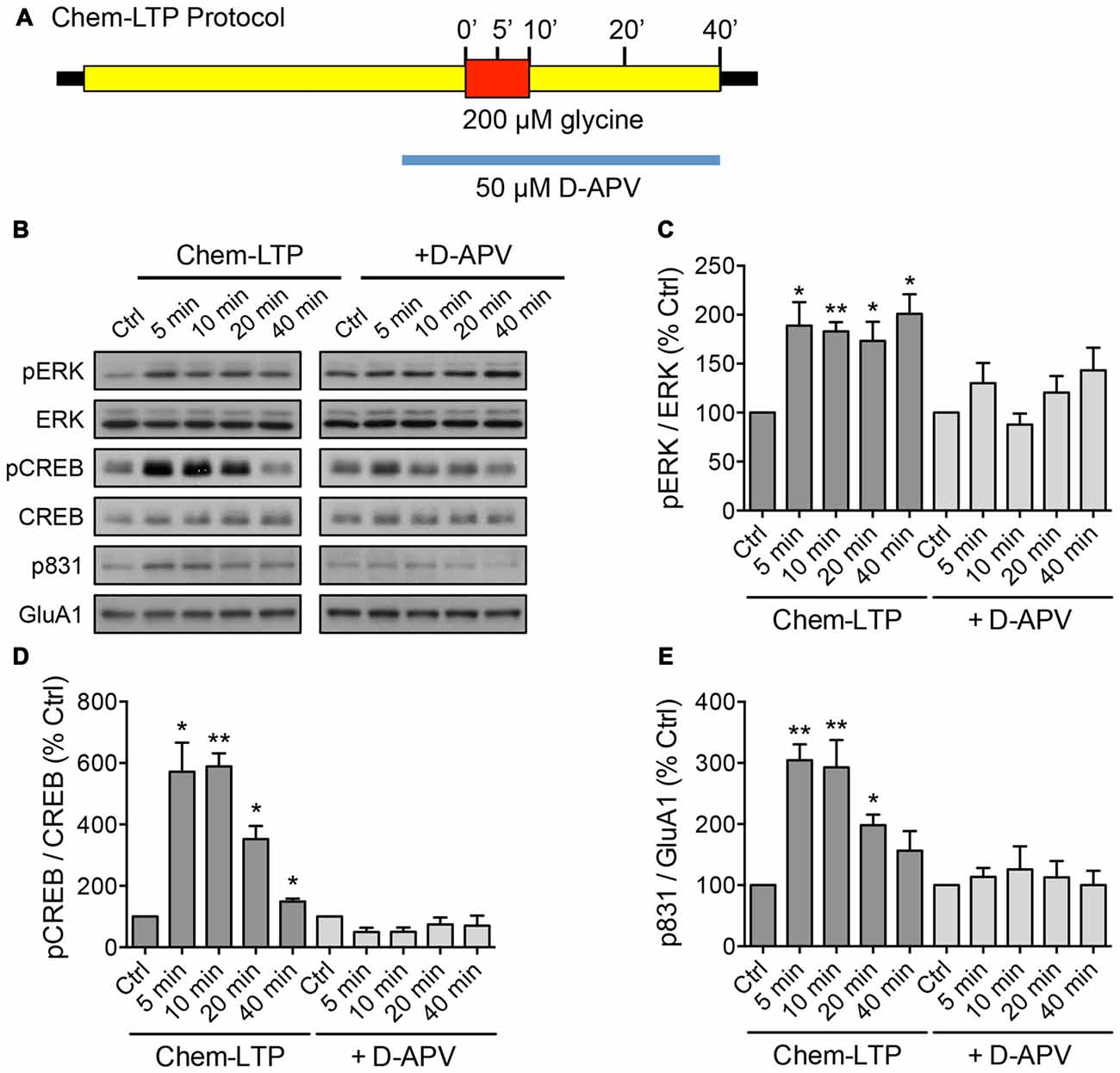
Figure 1. Glycine induces the phosphorylation of extracellular signal-regulated kinase (ERK), cAMP response element binding protein (CREB) and GluA1 in mouse cortical neurons. (A) Schematic diagram of the glycine-induced chem-long-term potentiation (LTP) protocol performed in days in vitro (DIV) 15–17 neurons. The N-Methyl-D-aspartate receptor (NMDAR)-dependent effect was determined by including the NMDAR antagonist, D-APV in the treatment. Protein lysates were collected by direct lysis in SDS sample buffer at different time points as indicated in the diagram. (B–E) Western blot analyses of phosphorylated CREB (Ser133), total CREB, phosphorylated ERK (Thr202/Tyr204), total ERK, phosphorylated GluA1 (Ser831) and total GluA1 levels during chem-LTP in the presence or absence of the NMDAR antagonist, D-APV. Quantitation of pCREB/CREB, pERK/ERK and p831/GluA1 was normalized to the 0 min control in each set. Data represent the mean ± SEM of four independent experiments (n = 4, one-way ANOVA, *P < 0.05, **P < 0.01).
RNA Extraction and Quantitative Real-Time PCR (qPCR)
Total RNA was extracted with the Directzol RNA MiniPrep kit (Zymo Research), including on-column DNAse treatment. For nuclear and cytoplasmic fractions, cortical neurons were lyzed in RLN buffer (50 mM Tris-HCl pH 8.0, 140 mM NaCl, 1.5 mM MgCl2, 0.5% Nonidet P-40, 1000 U/ml RNaseOUT, 1 mM DTT) as previously described (Djebali et al., 2012), with slight modifications. The lysate was spun at 300× g for 3 min at 4°C to pellet the nuclei. RNA from the supernatant (cytoplasm) and the nuclear pellet was extracted using the Directzol kit (Zymo) by adding 6–7 volumes of TRI reagent. For the nuclear pellet, the sample was further passed through a syringe needle. Equal amounts of RNA were reverse transcribed using the QuantiTect Reverse Transcription kit (Qiagen). Quantitative real-time polymerase chain reaction (qPCR) analysis was performed using the QuantiFast SYBR green kit (Qiagen) in a CFX96/384 Touch Real-Time PCR system (Bio-Rad). Gene expression was calculated using the standard comparative Ct method after normalizing to the housekeeping gene, Actb or GapDH. The sequences for the primers used were as follows: Meg3 (Forward: 5′-GGGAGCAGCTATGGATCACC-3′; Reverse: 5′-ATAGCGCCCCCTATTCATGC-3′), Rtl1-AS (Forward: 5′-TAGACCAGGACCTCTCTGCC-3′; Reverse: 5′-GCGGCATGGTTCACAAAACT-3′), Meg8 (Forward: 5′-GCAGAACCACTGGTGGAAGTT-3′; Reverse: 5′-GTGTCCCGTGTCCACAATAG-3′), Meg9 (Forward: 5′-AGGCTATCACCATCCCCCTT-3′; Reverse: 5′-TCCTAGACCTTGCCCGATGA-3′), Actb (Forward: 5′-CGGTTCCGATGCCCTGAGGCTCTT-3′; Reverse: 5′-CGTCACACTTCATGATGGAATTGA-3′), GapDH (Forward: 5′-TGCCCCCATGTTTGTGATG-3′; Reverse: 5′-TGTGGTCATGAGCCCTTCC-3′), Egr1 (Forward: 5′-GCCTTCGCTCACTCCACTAT-3′; Reverse: 5′-CTGGGTTTGATGAGCTGGGA-3′ Gria2 (Forward: 5′-CGAGGGGATATTTTGTGGATGC-3′; Reverse: 5′-ACTGAACCATCCCTACCCGAAA-3′), U6 (Forward: 5′-TCGCTTCGGCAGCACATATAC-3′; Reverse: 5′-TTCACGAATTTGCGTGTCATCC-3′).
Fear Conditioning Paradigm
Two-month old male mice were subjected to a cued fear conditioning paradigm. The “paired” group received five pairings of 2 min 80 dB white noise as the conditioned stimulus (CS), co-terminated with a 1 s 0.7 mA footshock as the unconditioned stimulus (US). The “unpaired” group was exposed to the same CS and US but with randomly applied footshock. The “context” group received the CS only and the “naïve” group comprised home-caged mates that did not undergo training. All animals were returned to their cages prior to being sacrificed.
Lentiviral-Mediated Knockdown of Meg3
Two shRNA sequences targeting mouse Meg3 (sh#1: 5′-ACAAGATGCTTACAGAAATAC-3′; sh#2: 5′-TGAGTGATAGACTACATATAT-3′) were cloned into an FG12 plasmid containing a GFP cassette, and packaged into HEK293 cells as previously described (Widagdo et al., 2016). Neurons were transduced with lentivirus between DIV 8–11 for 6 h, after which they were further incubated for 5 days prior to harvesting or conducting the chem-LTP assay. The efficacy of the transduction was assessed by the GFP expression of neurons, which was typically >90%.
ASO-Mediated Knockdown of Meg3
Neurons were incubated with 1 μM ASO between DIV 12–14 for 48 h prior to harvesting or conducting the chem-LTP assay. The ASOs were 20 nucleotides in length, wherein the central gap segment comprising 10 2′-deoxynucleosides was flanked on the 5′ and 3′ ends by five 2′-O-methoxyethyl-modified nucleosides. All internucleoside linkages were phosphorothioate linkages. The sequences of the ASOs were as follows: control ASO: 5′-CCUUCCCTGAAGGTTCCUCC-3′ (Graham et al., 2007); Meg3 ASOs: a mixture of two Meg3 targeting sequences (ASO-1: 5′-AACAGCAAATGGCACAGGAA-3′, ASO2: 5′-UUAUUTATGGACCTCAGGUG-3′).
Surface Biotinylation Assay
The chemical labeling of surface proteins with biotin was performed as described previously (Anggono et al., 2011). Briefly, neurons were washed twice with ACSF and incubated with 0.5 mg/ml Sulfo-NHS-SS-Biotin (Pierce) for 30 min on ice. Free biotin was quenched by washing the cells twice with ice-cold 50 mM glycine (pH 7.4 in ACSF). Cultures were lyzed and sonicated in RIPA buffer (1% Triton X-100, 0.5% Na-deoxycholate, 0.1% SDS, 2 mM EDTA, 2 mM EGTA, 50 mM NaF, 10 mM Na-pyrophosphate in Tris-buffered saline) and incubated with Neutravidin beads (Pierce) for 3 h at 4°C. Beads were washed three times, eluted with 2× SDS sample buffer, then analyzed by western blotting.
Results
Activity-Dependent Upregulation of lncRNAs within the Dlk1-Dio3 Locus In Vitro and In Vivo
To screen for lncRNAs which may play a role in synaptic plasticity, we first established an in vitro model of chemically induced LTP (chem-LTP) in primary cultured neurons (Figure 1A). Mouse cortical neurons were treated with 200 μM glycine, an NMDAR co-agonist, for 10 min, a protocol that is known to induce NMDAR-dependent insertion of new AMPARs and enhance network activity, resulting in a long-lasting strengthening of excitatory neurotransmission (Lu et al., 2001). This chem-LTP protocol caused a robust and persistent activation of the mitogen-activated protein kinase signaling cascade, as shown by a robust increase in ERK1/2 phosphorylation at Thr-202 and Tyr-204 (Figures 1B,C). In addition, glycine stimulation also led to enhanced phosphorylation of the transcription factor CREB and the GluA1 subunit of AMPARs at Ser-133 and Ser-831, respectively (Figures 1B,D,E), all of which are biochemical hallmarks of LTP (Ehlers, 2003). Glycine-induced increases in ERK, CREB and GluA1 phosphorylation were effectively blocked by D-APV, a selective NMDAR antagonist (Figures 1B–E). Using this chem-LTP paradigm, we profiled the expression levels of well-annotated lncRNAs in mouse primary neurons by qPCR. Unexpectedly, we detected a concurrent upregulation of lncRNAs located within the maternally imprinted Dlk1-Dio3 locus, namely Meg3, Rtl1-AS, Meg8 and Meg9 (Figures 2A–E). In contrast, the mRNA expression of Gria2 was not altered by glycine stimulation (Supplementary Figure S1), suggesting that the response profile for Meg3, Rtl1-AS, Meg8 and Meg9 was indeed target specific. Moreover, the application of D-APV abolished glycine-induced expression of these lncRNAs, suggesting that their rapid induction is dependent on NMDAR activation (Figures 2B–E).
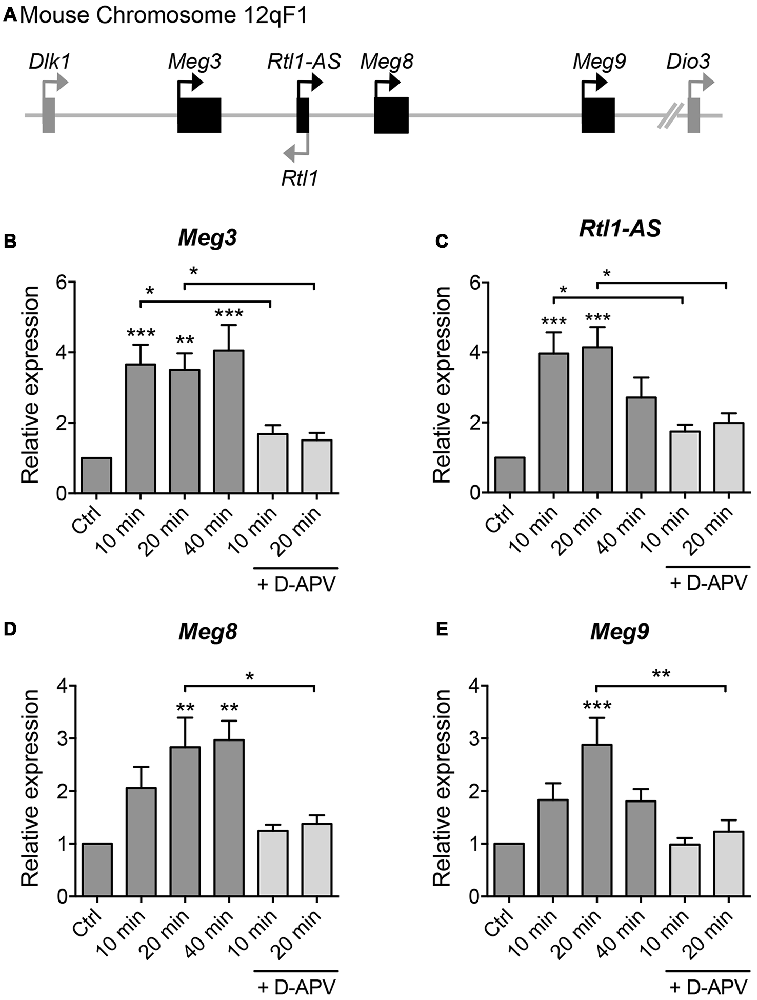
Figure 2. Glycine stimulation induces the expression of long non-coding RNAs (lncRNAs) within the Dlk-Dio3 imprinted locus in an NMDAR-dependent manner. (A) Schematic representation of Dlk1-Dio3 locus in the mouse chromosome 12qF1, which contains a cluster of maternally expressed lncRNAs (black rectangle). (B–E) Quantitative real-time polymerase chain reaction (qPCR) analyses of maternally expressed gene 3 (Meg3), retrotransposon-like gene 1-anti-sense (Rtl1-AS), Meg8 and Meg9 in primary neurons following glycine stimulation, after normalization with Actb. Data represent the mean ± SEM of at least nine independent experiments (n = 9–11, one-way ANOVA, *P < 0.05, **P < 0.01, ***P < 0.001).
Given that LTP plays an important role in learning and memory, we next evaluated the expression of the aforementioned maternally imprinted lncRNAs in response to a learning paradigm in vivo. We performed a cued fear conditioning assay and analyzed the lncRNA expression levels in the hippocampus of mice that: (a) had not been exposed to the training protocol (naïve); (b) were exposed to the cage and tone without receiving foot shocks (context); (c) were subjected to foot shocks that co-terminated with the tone (paired); or (d) received random shocks in the same context (unpaired) (Figure 3A). qPCR analysis revealed that the expression levels of Meg3, Meg8 and Meg9 were significantly upregulated in mice subjected to the paired protocol compared to naïve animals, or those exposed to random shocks or context only (Figures 3B–D). The expression of Rtl1-AS exhibited a trend for upregulation in the paired animals but did not reach statistical significance when compared to the level in naïve mice (Figure 3E). These results suggest that among the Dlk1-Dio3 lncRNAs, the expression levels of at least Meg3, Meg8 and Meg9 are elevated during associative learning in vivo. Interestingly, when examined 24 h post-training, the expression of Meg9 remained high although it did not reach statistical significance, whereas the expression of Meg3 was significantly downregulated (Supplementary Figure S2). Collectively, these data show that the expression of the Dlk1-Dio3 derived lncRNAs is dynamically regulated by neuronal activity both in vitro and in vivo.
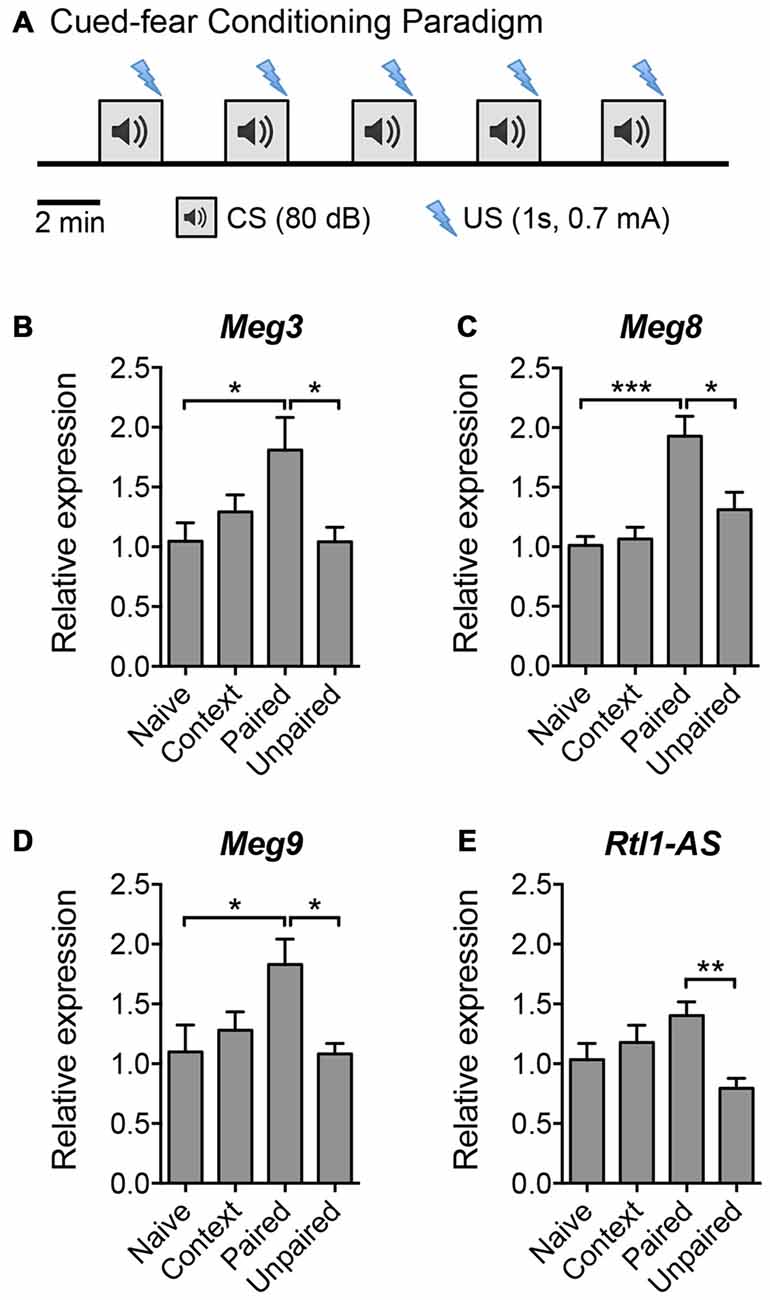
Figure 3. The expression of Dlk1-Dio3 imprinted lncRNAs in the mouse hippocampus following cued fear conditioning. (A) Schematic representation of the cued fear conditioning paradigm consisting of five pairings of the conditioned stimulus (CS, white noise) and the unconditioned stimulus (US, footshock). The US was randomized in the unpaired protocol. Mice were sacrificed 2 h post-training for hippocampal tissue isolation and RNA extraction. (B–E) qPCR analyses of Meg3, Meg8, Meg9 and Rtl1-AS expression after normalization with Actb. Data represent the mean ± SEM (n = 5–6, one-way ANOVA, *P < 0.05, **P < 0.01, ***P < 0.001).
Loss of Meg3 Expression Inhibits Glycine-Induced Increase in Surface GluA1
The majority of lncRNAs reside in the nucleus and associate with chromatin to perform epigenetic-related functions. The Dlk1-Dio3-derived lncRNAs have been shown to interact with the Polycomb Repressive Complex 2 (PRC2) chromatin modifying complex (Zhao et al., 2010; Kaneko et al., 2014). We performed a biochemical nuclear/cytoplasmic fractionation assay and confirmed that all of the Dlk1-Dio3 lncRNAs were highly enriched in the nucleus, with the exception of Meg3, approximately one-third of which was found in the cytoplasmic fraction (Figure 4A). qPCR analysis revealed a proportional increase in the expression of nuclear and cytoplasmic Meg3 in neurons that underwent chem-LTP stimulation (Supplementary Figure S3A). Intrigued by the fact that Meg3 may play a role outside the nucleus, we focused on its neuronal function in subsequent experiments.
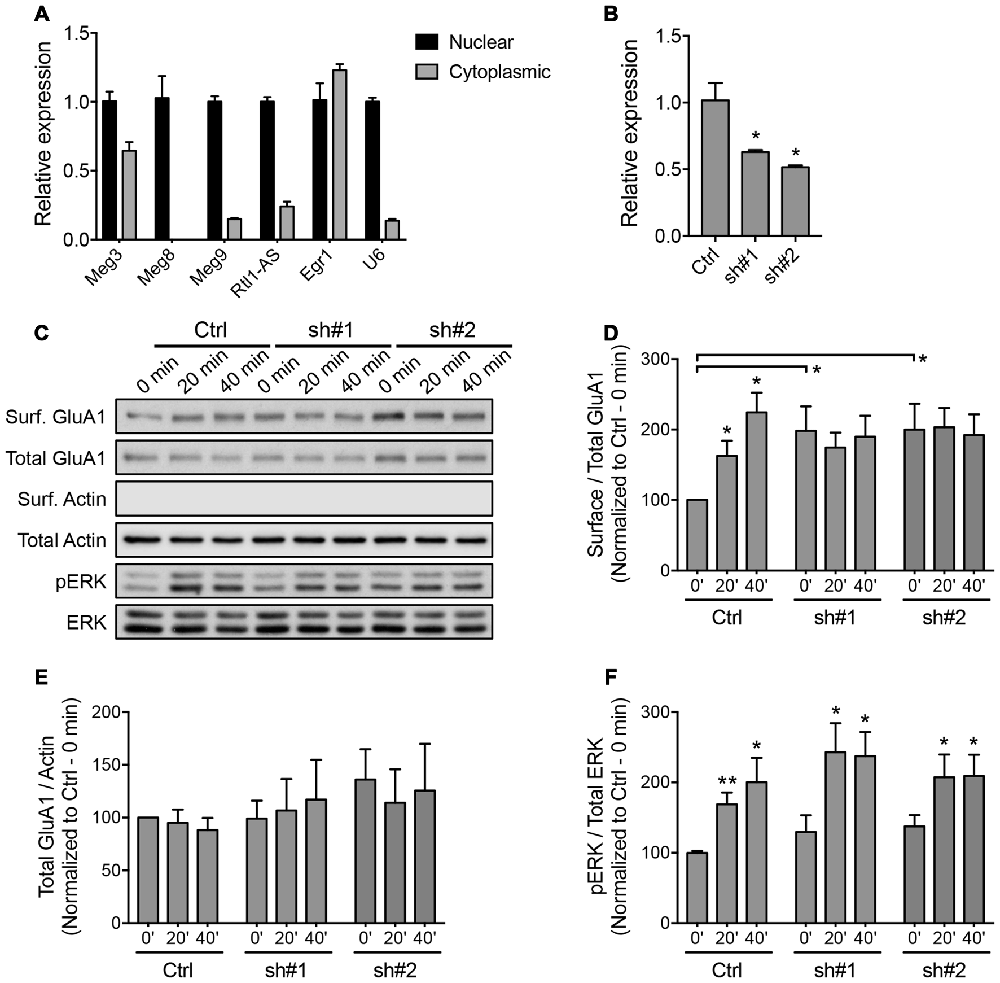
Figure 4. Downregulation of Meg3 disrupts α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) trafficking in primary neurons. (A) qPCR analysis of the Dlk1-Dio3 lncRNAs in the nuclear and cytoplasmic fractions of cortical neurons (n = 3). (B) The efficiency of lentiviral mediated knockdown of Meg3 in cortical neurons using two independent short hairpins RNA (shRNAs) or the FG12 control vector was quantified by qPCR analysis (n = 3, unpaired t-test). (C) Surface biotinylation assay performed in Meg3 knockdown and control neurons following chem-LTP. Representative western blots show the protein levels in the surface and total fractions. The absence of β-actin in the surface fractions was used to validate the biotinylation assay. (D,E) The effects of Meg3 knockdown on the levels of surface and total GluA1 expression were quantified as (D) surface/total receptor ratio and (E) total receptor/β-actin ratios, respectively, and normalized to the 0 min time point of the control cells. Data represent the mean ± SEM of three independent experiments (n = 4–5, two-way ANOVA, *P < 0.05). (F) The effect of Meg3 knockdown on the activation of ERK during chem-LTP was quantified by measuring the ratios of phosphorylated over total ERK (n = 8–9, two-way ANOVA, *P < 0.05, **P < 0.01).
To evaluate the functional importance of Meg3 in primary neurons, we designed two independent shRNAs targeting the exonic region of the Meg3 sequence. Cortical neurons transduced with lentiviral particles expressing these two Meg3 shRNAs exhibited a significant reduction in Meg3 expression compared to neurons that were transduced with control lentiviral particles (Figure 4B). A similar reduction in Meg3 expression was also observed in both the nuclear and cytoplasmic fractions (Supplementary Figure S3B). Using the shRNA-mediated knockdown approach, we analyzed the effect of Meg3 loss of function on chem-LTP by examining the levels of AMPAR expression on the plasma membrane. Surface biotinylation assays revealed a persistent increase in the surface expression of the GluA1 subunit of AMPARs up to 40 min post-chem-LTP induction (Figures 4C,D), consistent with the central dogma that the persistent increase in the number of surface AMPARs supports the induction and maintenance of LTP. Surprisingly, Meg3 knockdown blocked the glycine-induced increase in surface GluA1, which seemed to be occluded due to an elevated level of GluA1 on the plasma membrane under basal conditions (Figures 4C,D). Interestingly, Meg3 knockdown did not have significant effects on the expression of total GluA1 proteins or NMDAR-dependent activation of the ERK signaling pathway (Figures 4C,E,F). Similar observations were made using the ASO-mediated knockdown approach (Supplementary Figure S4). Collectively, these data suggest that the aberrant levels of surface GluA1 are likely due to impaired AMPAR trafficking.
Loss of Meg3 Expression Dysregulates the PTEN/PI3K/AKT Signaling Pathway
We next sought to identify an upstream regulator of AMPAR insertion, which has previously been linked to Meg3 in other cellular systems. Several recent studies have demonstrated a crosstalk between Meg3 and the PI3K/AKT pathway. In macrophages and endothelial cells, the downregulation of Meg3 leads to the activation of PI3K/AKT signaling through an unknown mechanism (Pawar et al., 2016; Qiu et al., 2016). In neurons, the PI3K/AKT pathway plays an important role in neuronal survival, synaptic plasticity, learning and memory (Sanna et al., 2002; Man et al., 2003; Opazo et al., 2003; Horwood et al., 2006). PI3K has previously been shown to regulate AMPAR insertion during LTP, with inhibition of PI3K in neurons blocking LTP (Passafaro et al., 2001; Man et al., 2003). To test whether Meg3 was able to modulate the PI3K/AKT signaling pathway in neurons, we first examined the phosphorylation level of the p85 regulatory subunit of PI3K at Tyr-458 as a proxy of PI3K activity during chem-LTP. In control neurons, glycine increased the level of phosphorylated p85 without altering the total protein expression (Figures 5A–C). In contrast, Meg3 knockdown prevented the chem-LTP-dependent increase in the level of p85 phosphorylation (Figures 5A–C). Interestingly, the level of phosphorylated p85 was significantly higher under basal conditions in Meg3 knockdown neurons than in control cells, which could prevent a further increase during chem-LTP (Figures 5A,B). Next, we analyzed the levels of AKT phosphorylation at Thr-308 and Ser-473, which are direct downstream targets of PI3K. We found that glycine induced a robust increase in the levels of phosphorylated AKT (Figures 5A,D,E) in control neurons. However, these increases were prevented in Meg3 knockdown neurons (Figures 5A,D,E). Interestingly, the level of phospho-Thr-308 was significantly higher under basal conditions in Meg3 knockdown neurons compared to control cells (Figures 5A,D). In contrast, the total AKT level was not affected by the reduction in Meg3 expression (Figure 5F), ruling out translation as the mode of action. Consistent with previous findings, the loss of Meg3 function caused partial overactivation of PI3K/AKT signaling, which correlates well with the higher accumulation of GluA1 on the plasma membrane under basal conditions. To test whether the overactivation of the PI3K/AKT signaling pathway was directly responsible for the increased level of surface GluA1 in Meg3 knockdown neurons, we treated these neurons with two different PI3K inhibitors, wortmannin and LY294002. However, these treatments failed to rescue GluA1 surface expression in Meg3 knockdown neurons (Supplementary Figure S5).
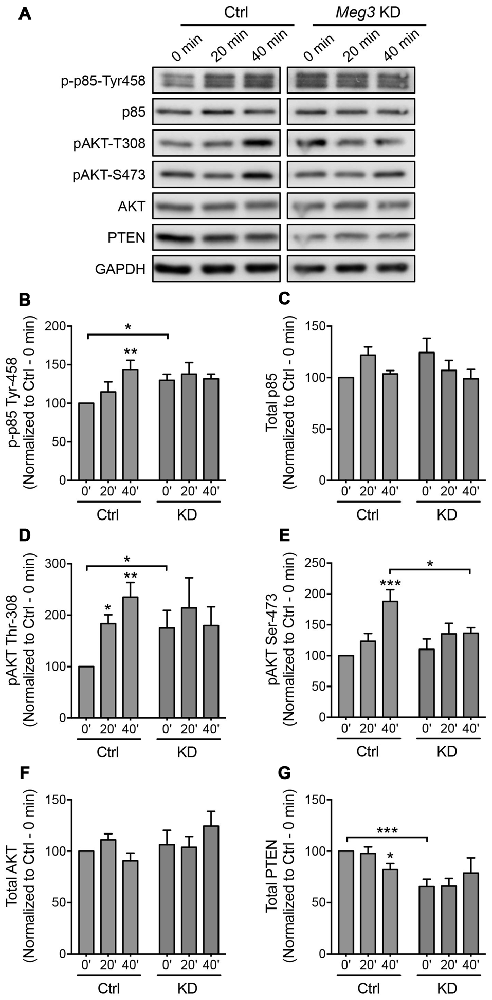
Figure 5. Meg3 knockdown dysregulates phosphatase and tensin homolog (PTEN)/phosphatidylinoside-3-kinase (PI3K)/AKT signaling during chem-LTP. (A) Cortical neurons were transduced with either Meg3 shRNA#2 or FG12 control lentiviral particles for 5 days and subjected to chem-LTP assay. Protein lysates were analyzed by western blotting. (B–G) Activation of the p85 (B) and AKT (D,E) pathways during chem-LTP was measured by quantifying the levels of p85 (Tyr-458) and AKT (Thr-308 and Ser-473) phosphorylation over total protein expression. The relative expression of total p85 (C) AKT (F) and PTEN (G) normalized to GAPDH in control and Meg3 knockdown neurons. Data represent the mean ± SEM of four independent experiments (n = 7, two-way ANOVA, *P < 0.05, **P < 0.01, ***P < 0.001).
The cellular level of phosphatidylinositol (3,4,5)-triphosphate (PIP3) is tightly regulated by the lipid phosphatase, PTEN, which converts PIP3 into phosphatidylinositol (4,5)-bisphosphate (PIP2). This removes the localization of AKT from the plasma membrane, thereby counteracting the PI3K/AKT signaling pathway. Mutations that lead to PTEN loss of function have been associated with hyperactivity of AKT and its downstream target (Lugo et al., 2014). Intrigued by the failure of PI3K inhibitors to restore GluA1 surface expression in Meg3 knockdown neurons, we next examined the level of PTEN protein expression in neurons. We found that the level of PTEN protein was significantly reduced in neurons following glycine stimulation (Figures 5A,G). Interestingly, PTEN expression was significantly downregulated in Meg3 knockdown neurons and was no longer modulated by glycine (Figures 5A,G). In summary, our data demonstrate that Meg3 is a critical regulator of the PTEN/PI3K/AKT signaling pathway and is crucial in modulating the number of GluA1-containing AMPARs on the neuronal plasma membrane.
Discussion
LncRNAs constitute a vast proportion of the mammalian transcriptome and have recently emerged as critical regulators of many cellular and physiological functions. They are highly expressed in the brain and are known to play important roles in synaptogenesis and neuronal differentiation (Briggs et al., 2015). The expression of many lncRNAs is dynamically regulated by neuronal activity, making them prime candidates for controlling neuronal plasticity (Maag et al., 2015). However, there is currently no direct evidence demonstrating the involvement of lncRNAs in synaptic plasticity. In this study, we used the well-established chem-LTP protocol to induce NMDAR-dependent synaptic potentiation, and discovered a rapid induction of lncRNAs embedded within the Dlk1-Dio3 locus in primary cortical neurons. The co-expression of these imprinted lncRNAs was striking, although it has been postulated that Meg3 and the downstream lncRNAs may be transcribed as a large polycistronic unit (~200 kb) prior to further processing in some tissues due to the lack of well-defined promoters for the downstream genes (da Rocha et al., 2008). The binding of CREB to the cAMP response element located in the proximal region of the Meg3 promoter may be responsible for the activation of Meg3 transcription, and potentially that of other lncRNAs within the Dlk1-Dio3 region during glycine stimulation (Zhao et al., 2006). Genetic deletion of the Meg3 promoter impacts not only its own expression, but also that of the other downstream lncRNAs within the Dlk1-Dio3 imprinted region (Zhou et al., 2010). Interestingly, the downstream lncRNAs, Rtl1-AS and Meg9 harbor multiple clusters of miRNAs, many of which have been implicated in the pathophysiology of schizophrenia (Hagan et al., 2009; Gardiner et al., 2012). Although evidence of impaired LTP-like plasticity in patients with schizophrenia has been reported (Hasan et al., 2011; Frantseva et al., 2014), the molecular mechanisms underpinning this process are not well understood.
Using a cued-fear conditioning paradigm, we also found that the expression of a subset of the Dlk1-Dio3 derived lncRNAs, including Meg3, Meg8 and Meg9, was significantly upregulated in the hippocampus 2 h post-training. These data demonstrate that the expression of the Dlk1-Dio3-derived lncRNAs can also be dynamically regulated by associative learning in vivo. Although it is generally accepted that fear conditioning induces LTP (Rogan et al., 1997; DeAndrade et al., 2012), we currently do not have evidence to show the occurrence of NMDAR-dependent LTP in the hippocampi of mice that underwent this behavioral training. Notwithstanding that the learning-induced elevation in the expression levels of these lncRNAs is strictly dependent on the association between tones and shocks, we have not directly demonstrated the requirement of NMDAR activation in this process. Further experiments are required to establish the mechanisms that underlie the learning-induced expression of lncRNAs, as well as to determine their roles in the acquisition and/or recall of fear memory.
One of the major determinants of LTP is the number of AMPARs on the postsynaptic membrane, which is tightly regulated by multiple factors from their synthesis, endosomal trafficking, and endocytosis to the insertion onto the cell surface and their degradation rate (Anggono and Huganir, 2012). Previous studies have shown that certain miRNAs that target the 3′ untranslated region (UTR) of the Gria1 and Gria2 mRNAs, which encode the GluA1 and GluA2 subunits of AMPARs, respectively, are potent epigenetic regulators of LTP (miR-137: Olde Loohuis et al., 2015; miR-124: Gascon et al., 2014; Hou et al., 2015). However, the involvement of lncRNAs in regulating the surface expression of AMPARs in neurons is unknown. In this study, we focused solely on Meg3 because it is expressed not only in the nucleus, but also in the cytoplasm, a feature that is unique among the Dlk1-Dio3 lncRNAs. To determine whether Meg3 plays a role in controlling the surface expression of the GluA1 subunit, we performed loss of function studies using two independent approaches, namely the shRNA- and ASO-mediated knockdown of Meg3. Surprisingly, we discovered that Meg3 knockdown led to a significant increase in surface GluA1 expression concomitant with the overactivation of PI3K/AKT signaling. This pathway is known to play a crucial role in the exocytosis of AMPARs and LTP (Passafaro et al., 2001; Man et al., 2003; Opazo et al., 2003). Interestingly, Meg3 knockdown only caused partial activation of AKT, as shown by an enhanced level of AKT phosphorylation at Thr-308 (a PDK1 site), but not at Ser-473 (an mTORC2 site). In order to test if the overactivation of PI3K was responsible for the basal increase in surface GluA1, we treated Meg3 knockdown neurons with the PI3K inhibitors wortmannin and LY294002 for 1 h. However, the levels of surface GluA1 expression remained high. Given that the total protein level of PTEN is downregulated in Meg3 knockdown neurons, it is plausible that a reciprocal increase in AKT signaling is still sufficient to enhance the expression of surface GluA1, even in the presence of PI3K inhibitors. Our data are consistent with previous studies which have demonstrated the potential role of Meg3 as an endogenous competing RNA for a subset of miRNAs that regulate the level of PTEN in neurons (Peng et al., 2015; Yang et al., 2016). Accurate measurement of the strength and duration of PTEN/PI3K/AKT signaling will need to be investigated in a future study. This will include the determination of the levels of PIP3 in Meg3 knockdown neurons.
The main finding of this study is that the loss of Meg3 function blocks the NMDAR-dependent increase in surface GluA1 in primary cortical neurons. Due to the enhanced level of surface GluA1 in Meg3 knockdown neurons, the activity-dependent increase in surface GluA1 expression seems to be occluded. However, we cannot rule out the possibility that the knockdown of Meg3 leads to both an increase in surface AMPARs and a disturbance in AMPAR forward trafficking during glycine stimulation. In line with the latter hypothesis, we found that Meg3 is indeed required for the full activation of AKT, which is necessary for AMPAR insertion onto the plasma membrane during glycine-induced synaptic potentiation. To further elucidate the role of Meg3 in this activity-dependent AMPAR insertion, further experiments involving Meg3 overexpression and rescue (in Meg3 knockdown neurons) should be performed. The complexity of Meg3 genomic structure (which yields multiple splice variants) and its large size (>10 kb) present challenges that preclude us from conducting these experiments in our current study (Schuster-Gossler et al., 1998; Hagan et al., 2009). Future experiments involving the use of the inducible dCAS9 system to transiently inhibit the transcription of Meg3 would be ideal to decipher the role of Meg3 in activity-dependent synaptic potentiation in neurons.
To reconcile our seemingly counterintuitive findings, in that Meg3 knockdown enhances surface GluA1 and yet its expression is upregulated during chem-LTP, we propose the following working model (Figure 6). We hypothesize that the loss of Meg3 function leads to a partial overactivation of AKT signaling via two distinct mechanisms, namely enhanced PI3K activity and the downregulation of PTEN expression, which subsequently lead to an increase in surface GluA1 expression. Based on emerging evidence in the literature, these effects are likely to be mediated by the loss of Meg3 interactions with a subset of miRNAs that regulate essential components of the PTEN/PI3K/AKT signaling pathway (Peng et al., 2015; Pawar et al., 2016; Qiu et al., 2016; Yang et al., 2016). On the other hand, Meg3 may regulate synaptic plasticity through distinct regulatory mechanisms such as: (a) an epigenetic mechanism via PRC2-dependent chromatin remodeling: (b) a post-transcriptional function through its interaction with miRNAs; or (c) the allosteric modulation of enzymes, including kinases, phosphatases or phospholipids. In the last of these mechanisms, it is plausible that Meg3 could physically bind to the PI3K subunits (Koirala et al., 2017), PIP3 (Lin et al., 2017) or other downstream components of the PTEN/PI3K/AKT signaling pathway during LTP to ensure the correct activation of this pathway to maintain LTP. In summary, this study provides the first evidence demonstrating an involvement of the lncRNA Meg3 in fine-tuning the activity of the PTEN/PI3K/AKT signaling pathway to regulate the optimal number of surface AMPARs in neurons during synaptic potentiation. Further studies are required to delineate the exact mechanisms by which Meg3 and other lncRNAs contribute to synaptic plasticity, as well as the role of lncRNAs in adaptive behaviors.
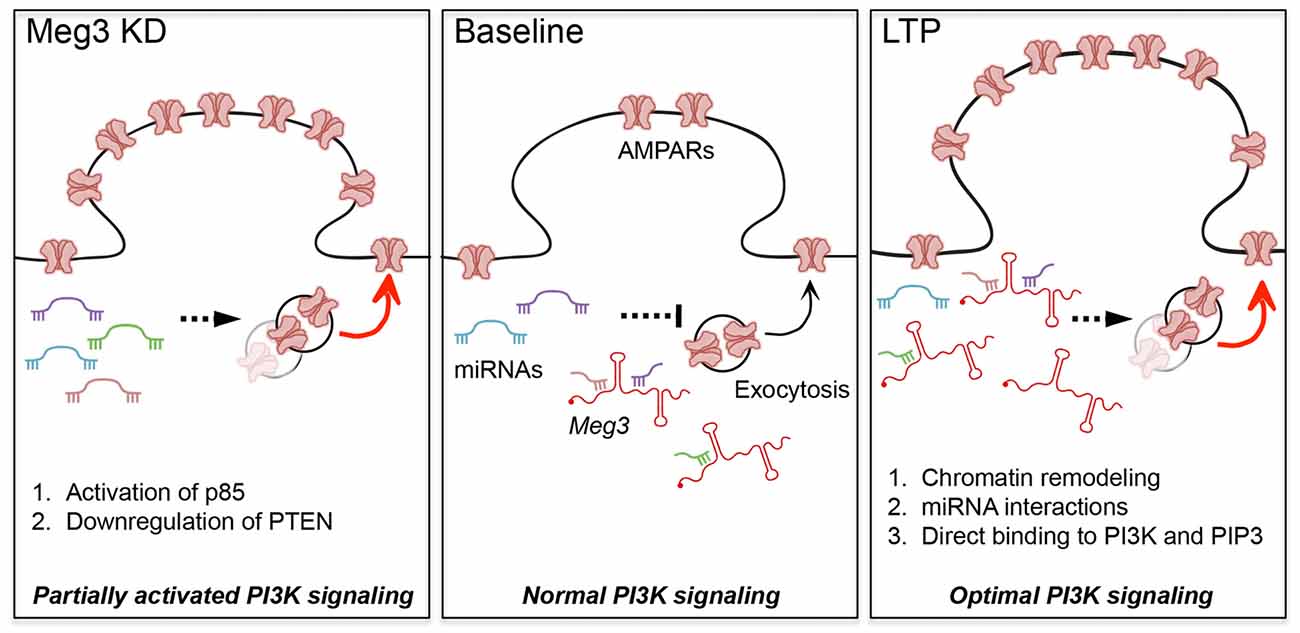
Figure 6. Proposed models for the roles of Meg3 in regulating surface GluA1 expression and activity-dependent synaptic potentiation. Meg3 has been proposed to act as an endogenous competing RNA for a subset of miRNAs that regulate the level of PTEN in neurons. We hypothesize that Meg3 is involved in fine-tuning the activity of the PTEN/PI3K/AKT signaling pathway to regulate the optimal number of surface AMPARs in neurons. (Left panel) Downregulation of Meg3 leads to a partial overactivation of AKT signaling via two distinct mechanisms, namely enhanced PI3K activity and the downregulation of PTEN expression, which subsequently lead to an increase in surface GluA1 expression. One of the underlying mechanisms might involve the loss of Meg3 interactions with a subset of miRNAs that regulate essential components of the PTEN/PI3K/AKT signaling pathway. (Right panel) Our data suggest that Meg3 is required for full activation of AKT and activity-dependent delivery of AMPARs to the plasma membrane during chem-LTP. We propose that Meg3 may regulate synaptic plasticity through distinct regulatory mechanisms such as: (1) polycomb repressive complex 2 (PRC2)-dependent chromatin remodeling; (2) interaction with miRNAs; or (3) the allosteric modulation of PI3K and/or PIP3, ensuring the optimal activation of PI3K signaling to maintain LTP.
Author Contributions
MCT, JW, YQC, TZ and VA performed experiments. JW and VA designed research and wrote the manuscript. VA conceived the project and supervised the research. All authors analyzed the results and approved the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was funded in part by grants from the John T. Reid Charitable Trusts, the University of Queensland (ECR122) and the Clem Jones Centre for Ageing Dementia Research to VA. JW is an Australian Research Council DECRA Fellow (DE170100112). We thank Rowan Tweedale for editing of this manuscript.
Abbreviations
AMPARs, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors; ASO, antisense oligonucleotide; CREB, cAMP response element binding protein; DIV, days in vitro; ERK, extracellular signal-regulated kinase; lncRNA, long non-coding RNA; LTP, long-term potentiation; Meg, maternally expressed gene; NMDA, N-methyl-D-aspartate; PI3K, phosphatidylinoside-3-kinase; PTEN, phosphatase and tensin homolog; Rtl1-AS, retrotransposon-like gene 1-antisense.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fncel.2017.00124/full#supplementary-material
References
Adams, J. P., and Dudek, S. M. (2005). Late-phase long-term potentiation: getting to the nucleus. Nat. Rev. Neurosci. 6, 737–743. doi: 10.1038/nrn1749
Alberini, C. M. (2009). Transcription factors in long-term memory and synaptic plasticity. Physiol. Rev. 89, 121–145. doi: 10.1152/physrev.00017.2008
Anggono, V., Clem, R. L., and Huganir, R. L. (2011). PICK1 loss of function occludes homeostatic synaptic scaling. J. Neurosci. 31, 2188–2196. doi: 10.1523/JNEUROSCI.5633-10.2011
Anggono, V., and Huganir, R. L. (2012). Regulation of AMPA receptor trafficking and synaptic plasticity. Curr. Opin. Neurobiol. 22, 461–469. doi: 10.1016/j.conb.2011.12.006
Anggono, V., Koç-Schmitz, Y., Widagdo, J., Kormann, J., Quan, A., Chen, C.-M., et al. (2013). PICK1 interacts with PACSIN to regulate AMPA receptor internalization and cerebellar long-term depression. Proc. Natl. Acad. Sci. U S A 110, 13976–13981. doi: 10.1073/pnas.1312467110
Bernard, D., Prasanth, K. V., Tripathi, V., Colasse, S., Nakamura, T., Xuan, Z., et al. (2010). A long nuclear-retained non-coding RNA regulates synaptogenesis by modulating gene expression. EMBO J. 29, 3082–3093. doi: 10.1038/emboj.2010.199
Bond, A. M., Vangompel, M. J. W., Sametsky, E. A., Clark, M. F., Savage, J. C., Disterhoft, J. F., et al. (2009). Balanced gene regulation by an embryonic brain ncRNA is critical for adult hippocampal GABA circuitry. Nat. Neurosci. 12, 1020–1027. doi: 10.1038/nn.2371
Briggs, J. A., Wolvetang, E. J., Mattick, J. S., Rinn, J. L., and Barry, G. (2015). Mechanisms of long non-coding RNAs in mammalian nervous system development, plasticity, disease, and evolution. Neuron 88, 861–877. doi: 10.1016/j.neuron.2015.09.045
da Rocha, S. T., Edwards, C. A., Ito, M., Ogata, T., and Ferguson-Smith, A. C. (2008). Genomic imprinting at the mammalian Dlk1-Dio3 domain. Trends Genet. 24, 306–316. doi: 10.1016/j.tig.2008.03.011
DeAndrade, M. P., Zhang, L., Doroodchi, A., Yokoi, F., Cheetham, C. C., Chen, H.-X., et al. (2012). Enhanced hippocampal long-term potentiation and fear memory in Btbd9 mutant mice. PLoS One 7:e35518. doi: 10.1371/journal.pone.0035518
Djebali, S., Davis, C. A., Merkel, A., Dobin, A., Lassmann, T., Mortazavi, A., et al. (2012). Landscape of transcription in human cells. Nature 489, 101–108. doi: 10.1038/nature11233
Ehlers, M. D. (2003). Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat. Neurosci. 6, 231–242. doi: 10.1038/nn1013
Frantseva, M., Cui, J., Farzan, F., Chinta, L. V., Perez Velazquez, J. L., and Daskalakis, Z. J. (2014). Disrupted cortical conductivity in schizophrenia: TMS-EEG study. Cereb. Cortex 24, 211–221. doi: 10.1093/cercor/bhs304
Frey, U., Frey, S., Schollmeier, F., and Krug, M. (1996). Influence of actinomycin D, a RNA synthesis inhibitor, on long-term potentiation in rat hippocampal neurons in vivo and in vitro. J. Physiol. 490, 703–711. doi: 10.1113/jphysiol.1996.sp021179
Frey, U., Huang, Y. Y., and Kandel, E. R. (1993). Effects of cAMP simulate a late stage of LTP in hippocampal CA1 neurons. Science 260, 1661–1664. doi: 10.1126/science.8389057
Gardiner, E., Beveridge, N. J., Wu, J. Q., Carr, V., Scott, R. J., Tooney, P. A., et al. (2012). Imprinted DLK1-DIO3 region of 14q32 defines a schizophrenia-associated miRNA signature in peripheral blood mononuclear cells. Mol. Psychiatry 17, 827–840. doi: 10.1038/mp.2011.78
Gascon, E., Lynch, K., Ruan, H., Almeida, S., Verheyden, J. M., Seeley, W. W., et al. (2014). Alterations in microRNA-124 and AMPA receptors contribute to social behavioral deficits in frontotemporal dementia. Nat. Med. 20, 1444–1451. doi: 10.1038/nm.3717
Graham, M. J., Lemonidis, K. M., Whipple, C. P., Subramaniam, A., Monia, B. P., Crooke, S. T., et al. (2007). Antisense inhibition of proprotein convertase subtilisin/kexin type 9 reduces serum LDL in hyperlipidemic mice. J. Lipid Res. 48, 763–767. doi: 10.1194/jlr.c600025-jlr200
Hagan, J. P., O’Neill, B. L., Stewart, C. L., Kozlov, S. V., and Croce, C. M. (2009). At least ten genes define the imprinted Dlk1-Dio3 cluster on mouse chromosome 12qF1. PLoS One 4:e4352. doi: 10.1371/journal.pone.0004352
Hagena, H., and Manahan-Vaughan, D. (2013). Differentiation in the protein synthesis-dependency of persistent synaptic plasticity in mossy fiber and associational/commissural CA3 synapses in vivo. Front. Integr. Neurosci. 7:10. doi: 10.3389/fnint.2013.00010
Hasan, A., Nitsche, M. A., Rein, B., Schneider-Axmann, T., Guse, B., Gruber, O., et al. (2011). Dysfunctional long-term potentiation-like plasticity in schizophrenia revealed by transcranial direct current stimulation. Behav. Brain Res. 224, 15–22. doi: 10.1016/j.bbr.2011.05.017
Horwood, J. M., Dufour, F., Laroche, S., and Davis, S. (2006). Signalling mechanisms mediated by the phosphoinositide 3-kinase/Akt cascade in synaptic plasticity and memory in the rat. Eur. J. Neurosci. 23, 3375–3384. doi: 10.1111/j.1460-9568.2006.04859.x
Hou, Q., Ruan, H., Gilbert, J., Wang, G., Ma, Q., Yao, W.-D., et al. (2015). MicroRNA miR124 is required for the expression of homeostatic synaptic plasticity. Nat. Commun. 6:10045. doi: 10.1038/ncomms10045
Huang, Y. Y., Nguyen, P. V., Abel, T., and Kandel, E. R. (1996). Long-lasting forms of synaptic potentiation in the mammalian hippocampus. Learn. Mem. 3, 74–85. doi: 10.1101/lm.3.2-3.74
Hussain, N. K., Diering, G. H., Sole, J., Anggono, V., and Huganir, R. L. (2014). Sorting Nexin 27 regulates basal and activity-dependent trafficking of AMPARs. Proc. Natl. Acad. Sci. U S A 111, 11840–11845. doi: 10.1073/pnas.1412415111
Kandel, E. R. (2001). The molecular biology of memory storage: a dialogue between genes and synapses. Science 294, 1030–1038. doi: 10.1126/science.1067020
Kaneko, S., Bonasio, R., Saldaña-Meyer, R., Yoshida, T., Son, J., Nishino, K., et al. (2014). Interactions between JARID2 and noncoding RNAs regulate PRC2 recruitment to chromatin. Mol. Cell 53, 290–300. doi: 10.1016/j.molcel.2013.11.012
Koirala, P., Huang, J., Ho, T.-T., Wu, F., Ding, X., and Mo, Y.-Y. (2017). LncRNA AK023948 is a positive regulator of AKT. Nat. Commun. 8:14422. doi: 10.1038/ncomms14422
Lin, A., Hu, Q., Li, C., Xing, Z., Ma, G., Wang, C., et al. (2017). The LINK-A lncRNA interacts with PtdIns(3,4,5)P3 to hyperactivate AKT and confer resistance to AKT inhibitors. Nat. Cell Biol. 19, 238–251. doi: 10.1038/ncb3473
Lu, W., Man, H., Ju, W., Trimble, W. S., MacDonald, J. F., and Wang, Y. T. (2001). Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron 29, 243–254. doi: 10.1016/s0896-6273(01)00194-5
Lugo, J. N., Smith, G. D., Arbuckle, E. P., White, J., Holley, A. J., Floruta, C. M., et al. (2014). Deletion of PTEN produces autism-like behavioral deficits and alterations in synaptic proteins. Front. Mol. Neurosci. 7:27. doi: 10.3389/fnmol.2014.00027
Maag, J. L. V., Panja, D., Sporild, I., Patil, S., Kaczorowski, D. C., Bramham, C. R., et al. (2015). Dynamic expression of long noncoding RNAs and repeat elements in synaptic plasticity. Front. Neurosci. 9:351. doi: 10.3389/fnins.2015.00351
Man, H.-Y., Wang, Q., Lu, W.-Y., Ju, W., Ahmadian, G., Liu, L., et al. (2003). Activation of PI3-kinase is required for AMPA receptor insertion during LTP of mEPSCs in cultured hippocampal neurons. Neuron 38, 611–624. doi: 10.1016/s0896-6273(03)00228-9
McLaughlin, D., Vidaki, M., Renieri, E., and Karagogeos, D. (2006). Expression pattern of the maternally imprinted gene Gtl2 in the forebrain during embryonic development and adulthood. Gene Expr. Patterns 6, 394–399. doi: 10.1016/j.modgep.2005.09.007
Mercer, T. R., and Mattick, J. S. (2013). Structure and function of long noncoding RNAs in epigenetic regulation. Nat. Struct. and Mol. Biol. 20, 300–307. doi: 10.1038/nsmb.2480
Mercer, T. R., Dinger, M. E., Sunkin, S. M., Mehler, M. F., and Mattick, J. S. (2008). Specific expression of long noncoding RNAs in the mouse brain. Proc. Natl. Acad. Sci. U S A 105, 716–721. doi: 10.1073/pnas.0706729105
Nayak, A., Zastrow, D. J., Lickteig, R., Zahniser, N. R., and Browning, M. D. (1998). Maintenance of late-phase LTP is accompanied by PKA-dependent increase in AMPA receptor synthesis. Nature 394, 680–683. doi: 10.1038/29305
Nguyen, P. V., Abel, T., and Kandel, E. R. (1994). Requirement of a critical period of transcription for induction of a late phase of LTP. Science 265, 1104–1107. doi: 10.1126/science.8066450
Nicoll, R. A. (2017). A brief history of long-term potentiation. Neuron 93, 281–290. doi: 10.1016/j.neuron.2016.12.015
Olde Loohuis, N. F. M., Ba, W., Stoerchel, P. H., Kos, A., Jager, A., Schratt, G., et al. (2015). MicroRNA-137 controls AMPA-receptor-mediated transmission and mGluR-dependent LTD. Cell Rep. 11, 1876–1884. doi: 10.1016/j.celrep.2015.05.040
Opazo, P., Watabe, A. M., Grant, S. G. N., and O’Dell, T. J. (2003). Phosphatidylinositol 3-kinase regulates the induction of long-term potentiation through extracellular signal-related kinase-independent mechanisms. J. Neurosci. 23, 3679–3688.
Passafaro, M., Piëch, V., and Sheng, M. (2001). Subunit-specific temporal and spatial patterns of AMPA receptor exocytosis in hippocampal neurons. Nat. Neurosci. 4, 917–926. doi: 10.1038/nn0901-917
Pawar, K., Hanisch, C., Palma Vera, S. E., Einspanier, R., and Sharbati, S. (2016). Down regulated lncRNA MEG3 eliminates mycobacteria in macrophages via autophagy. Sci. Rep. 6:19416. doi: 10.1038/srep19416
Peng, W., Si, S., Zhang, Q., Li, C., Zhao, F., Wang, F., et al. (2015). Long non-coding RNA MEG3 functions as a competing endogenous RNA to regulate gastric cancer progression. J. Exp. Clin. Cancer Res. 34:79. doi: 10.1186/s13046-015-0197-7
Qiu, G.-Z., Tian, W., Fu, H.-T., Li, C.-P., and Liu, B. (2016). Long noncoding RNA-MEG3 is involved in diabetes mellitus-related microvascular dysfunction. Biochem. Biophys. Res. Commun. 471, 135–141. doi: 10.1016/j.bbrc.2016.01.164
Qu, C., Jiang, T., Li, Y., Wang, X., Cao, H., Xu, H., et al. (2013). Gene expression and IG-DMR hypomethylation of maternally expressed gene 3 in developing corticospinal neurons. Gene Expr. Patterns 13, 51–56. doi: 10.1016/j.gep.2012.11.003
Radwanska, K., Medvedev, N. I., Pereira, G. S., Engmann, O., Thiede, N., Moraes, M. F. D., et al. (2011). Mechanism for long-term memory formation when synaptic strengthening is impaired. Proc. Natl. Acad. Sci. U S A 108, 18471–18475. doi: 10.1073/pnas.1109680108
Roberts, T. C., Morris, K. V., and Wood, M. J. A. (2014). The role of long non-coding RNAs in neurodevelopment, brain function and neurological disease. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369:20130507. doi: 10.1098/rstb.2013.0507
Rogan, M. T., Stäubli, U. V., and LeDoux, J. E. (1997). Fear conditioning induces associative long-term potentiation in the amygdala. Nature 390, 604–607. doi: 10.1038/37601
Sanna, P. P., Cammalleri, M., Berton, F., Simpson, C., Lutjens, R., Bloom, F. E., et al. (2002). Phosphatidylinositol 3-kinase is required for the expression but not for the induction or the maintenance of long-term potentiation in the hippocampal CA1 region. J. Neurosci. 22, 3359–3365.
Schuster-Gossler, K., Bilinski, P., Sado, T., Ferguson-Smith, A., and Gossler, A. (1998). The mouse Gtl2 gene is differentially expressed during embryonic development, encodes multiple alternatively spliced transcripts and may act as an RNA. Dev. Dyn. 212, 214–228. doi: 10.1002/(SICI)1097-0177(199806)212:2<214::AID-AJA6>3.0.CO;2-K
Widagdo, J., Chai, Y. J., Ridder, M. C., Chau, Y. Q., Johnson, R. C., Sah, P., et al. (2015). Activity-dependent ubiquitination of GluA1 and GluA2 regulates AMPA receptor intracellular sorting and degradation. Cell Rep. 10, 783–795. doi: 10.1016/j.celrep.2015.01.015
Widagdo, J., Zhao, Q.-Y., Kempen, M.-J., Tan, M. C., Ratnu, V. S., Wei, W., et al. (2016). Experience-dependent accumulation of N6-Methyladenosine in the prefrontal cortex is associated with memory processes in mice. J. Neurosci. 36, 6771–6777. doi: 10.1523/JNEUROSCI.4053-15.2016
Yang, N.-Q., Luo, X.-J., Zhang, J., Wang, G.-M., and Guo, J.-M. (2016). Crosstalk between Meg3 and miR-1297 regulates growth of testicular germ cell tumor through PTEN/PI3K/AKT pathway. Am. J. Transl. Res. 8, 1091–1099.
Zhao, J., Ohsumi, T. K., Kung, J. T., Ogawa, Y., Grau, D. J., Sarma, K., et al. (2010). Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol. Cell 40, 939–953. doi: 10.1016/j.molcel.2010.12.011
Zhao, J., Zhang, X., Zhou, Y., Ansell, P. J., and Klibanski, A. (2006). Cyclic AMP stimulates MEG3 gene expression in cells through a cAMP-response element (CRE) in the MEG3 proximal promoter region. Int. J. Biochem. Cell Biol. 38, 1808–1820. doi: 10.1016/j.biocel.2006.05.004
Zhou, Y., Cheunsuchon, P., Nakayama, Y., Lawlor, M. W., Zhong, Y., Rice, K. A., et al. (2010). Activation of paternally expressed genes and perinatal death caused by deletion of the Gtl2 gene. Development 137, 2643–2652. doi: 10.1242/dev.045724
Keywords: long non-coding RNA, AMPA receptors, receptor trafficking, maternally expressed gene 3 (Meg3), PTEN/PI3K/AKT, synaptic potentiation, LTP, epigenetics
Citation: Tan MC, Widagdo J, Chau YQ, Zhu T, Wong JJ-L, Cheung A and Anggono V (2017) The Activity-Induced Long Non-Coding RNA Meg3 Modulates AMPA Receptor Surface Expression in Primary Cortical Neurons. Front. Cell. Neurosci. 11:124. doi: 10.3389/fncel.2017.00124
Received: 07 February 2017; Accepted: 12 April 2017;
Published: 03 May 2017.
Edited by:
Hansen Wang, University of Toronto, CanadaReviewed by:
Christine Gall, University of California, Irvine, USASilvia Bassani, Istituto di Neuroscienze (CNR), Italy
Arianna Maffei, Stony Brook University, USA
Alanna Watt, McGill University, Canada
Copyright © 2017 Tan, Widagdo, Chau, Zhu, Wong, Cheung and Anggono. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jocelyn Widagdo, ai53aWRhZ2RvQHVxLmVkdS5hdQ==
Victor Anggono di5hbmdnb25vQHVxLmVkdS5hdQ==
† These authors have contributed equally to this work.