 Farr Niere
Farr Niere Kimberly F. Raab-Graham
Kimberly F. Raab-Graham- Department of Physiology and Pharmacology, Wake Forest School of Medicine, Winston-Salem, NC, United States
The mammalian/mechanistic target of rapamycin complex 1 (mTORC1) serves as a regulator of mRNA translation. Recent studies suggest that mTORC1 may also serve as a local, voltage sensor in the postsynaptic region of neurons. Considering biochemical, bioinformatics and imaging data, we hypothesize that the activity state of mTORC1 dynamically regulates local membrane potential by promoting and repressing protein synthesis of select mRNAs. Our hypothesis suggests that mTORC1 uses positive and negative feedback pathways, in a branch-specific manner, to maintain neuronal excitability within an optimal range. In some dendritic branches, mTORC1 activity oscillates between the “On” and “Off” states. We define this as negative feedback. In contrast, positive feedback is defined as the pathway that leads to a prolonged depolarized or hyperpolarized resting membrane potential, whereby mTORC1 activity is constitutively on or off, respectively. We propose that inactivation of mTORC1 increases the expression of voltage-gated potassium alpha (Kv1.1 and 1.2) and beta (Kvβ2) subunits, ensuring that the membrane resets to its resting membrane potential after experiencing increased synaptic activity. In turn, reduced mTORC1 activity increases the protein expression of syntaxin-1A and promotes the surface expression of the ionotropic glutamate receptor N-methyl-D-aspartate (NMDA)-type subunit 1 (GluN1) that facilitates increased calcium entry to turn mTORC1 back on. Under conditions such as learning and memory, mTORC1 activity is required to be high for longer periods of time. Thus, the arm of the pathway that promotes syntaxin-1A and Kv1 protein synthesis will be repressed. Moreover, dendritic branches that have low mTORC1 activity with increased Kv expression would balance dendrites with constitutively high mTORC1 activity, allowing for the neuron to maintain its overall activity level within an ideal operating range. Finally, such a model suggests that recruitment of more positive feedback dendritic branches within a neuron is likely to lead to neurodegenerative disorders.
Introduction
The mammalian/mechanistic target of rapamycin (mTOR) is a ubiquitous serine/threonine kinase that is involved in many cellular processes (Hay and Sonenberg, 2004; Zoncu et al., 2011; Laplante and Sabatini, 2012). mTOR forms complexes with two distinct sets of proteins to give rise to mTORC1 and mTORC2, for complex 1 and 2 respectively. mTORC1 is well-characterized for its important roles in nutrient sensing and mRNA translation regulation (Hay and Sonenberg, 2004; Hoeffer and Klann, 2010; Santini and Klann, 2011; Switon et al., 2017). Historically, phosphorylation of mTORC1, which is thought to activate or turn on the protein, is required to signal to its downstream targets to promote mRNA translation or protein synthesis. Recent work, however, is redefining this view by demonstrating that mTORC1 activity is equally important in repressing protein synthesis of select mRNAs (Raab-Graham et al., 2006; Auerbach et al., 2011; Sosanya et al., 2013; Niere et al., 2016). The initial discovery that mTORC1 activity represses the local, dendritic mRNA translation of the voltage-gated potassium channel subunit, Kv1.1 was pivotal in reexamining mTORC1’s function. In hindsight, the discovery was foretelling mTORC1’s function in membrane excitability. Our subsequent study revealed that acute reduction of mTORC1 activity by rapamycin in vivo preferentially alters the expression of proteins involved in ion homeostasis and regulation of the membrane potential (Raab-Graham et al., 2006; Niere et al., 2016). Unlike mTORC1, mTORC2 is insensitive to acute rapamycin treatment (Sarbassov et al., 2006; Lamming et al., 2012; Saxton and Sabatini, 2017). For the purpose of this hypothesis article, we primarily consider mTORC1 regulation of local protein synthesis as an underlying mechanism that alters membrane potential within individual dendrites. To the best of our knowledge, the role of mTORC1 in regulating local, dendritic membrane potential is unexplored. Deciphering the mechanism by which mTORC1 controls the cell’s membrane potential is fundamental, since neurological disorders with aberrant mTORC1 activity present abnormal electrochemical membrane properties (Poolos and Johnston, 2012; Schmunk and Gargus, 2013).
Local Protein Synthesis in Dendrites
Local mRNA translation is a spatiotemporal mechanism that ensures rapid protein synthesis within a limited region (Steward and Schuman, 2001). Numerous mRNAs have already been identified in neuronal processes; and because of their location, they are poised to be immediately translated in response to activity (Raab-Graham et al., 2006; Cajigas et al., 2012; Niere et al., 2012). In the dendrites, for example, the activity-regulated cytoskeleton-associated (Arc) protein, which facilitates the removal of α-amino-3-hydroxy-5-methyl-isoxazolepropionic acid (AMPA)-selective ionotropic glutamate receptors (GluA), is rapidly synthesized—within 5 min—upon activating group I metabotropic glutamate receptors (mGluRs), resulting in long-term synaptic depression (LTD; Park et al., 2008; Waung et al., 2008; Niere et al., 2012). Additionally, the dendrite-rich region of hippocampal CA1 stratum radiatum contains several mRNAs that code for ion channels, bringing to mind that other ion channel proteins, like Kv1.1, can be locally synthesized upon the right cues, (Table 1; Raab-Graham et al., 2006; Cajigas et al., 2012). The compartmentalization of these mRNAs, away from the soma or axons, suggests that their translation will only alter dendritic membrane properties in a site-specific manner. While the necessary elements (e.g., mRNAs, translation factors and location) to regulate dendritic membrane potential actively are readily available, it still remains unknown whether a general mechanism exists that can coordinate the expression of ion channels, receptors and their associated proteins to change the membrane potential dynamically.
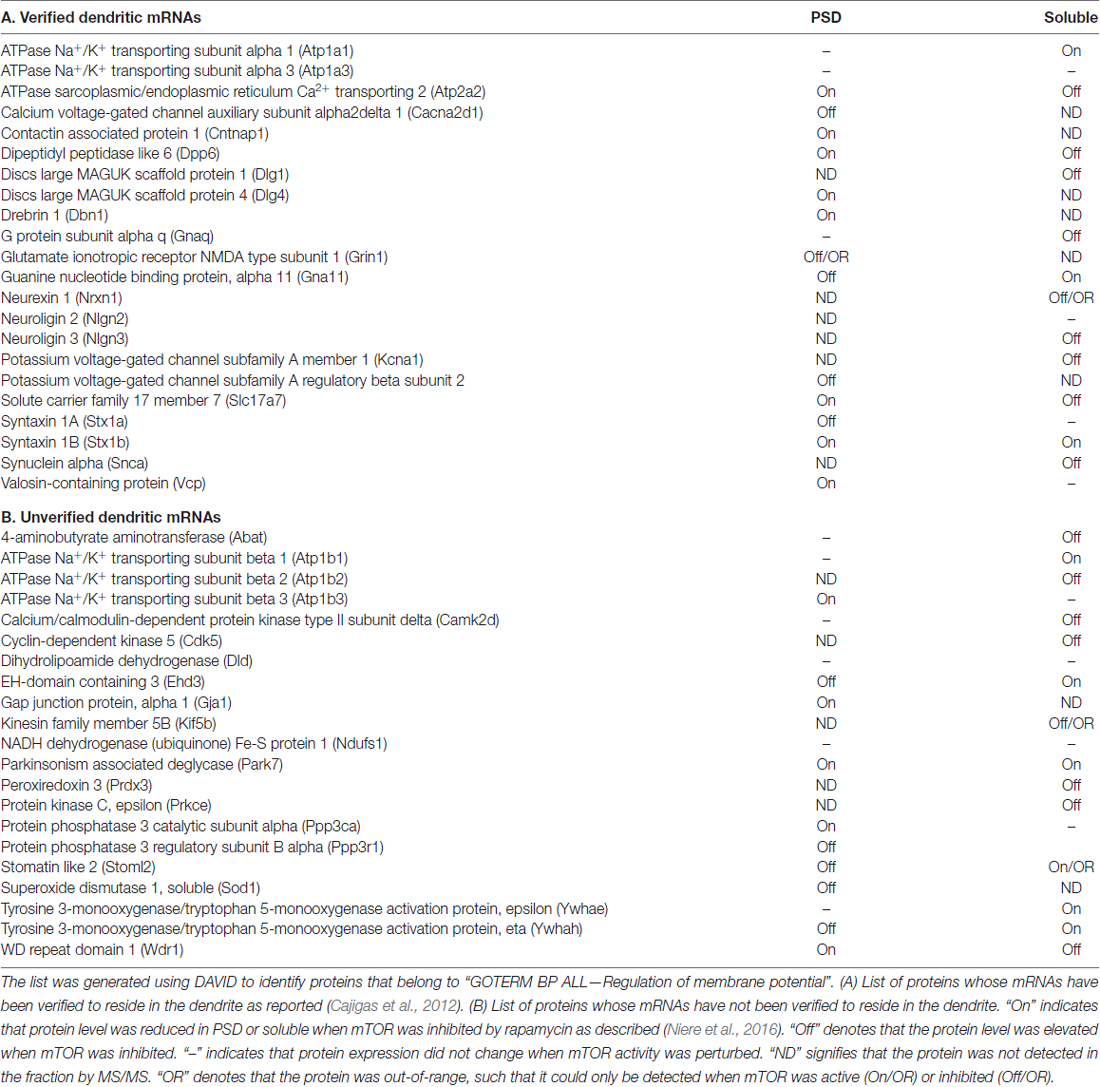
Table 1. List of membrane potential-associated proteins identified by MS/MS in the PSD and soluble fractions of cortices from rats injected with rapamycin or DMSO.
mTORC1-Regulated Protein Synthesis and Plasticity
Engagement of receptor-mediated signaling such as glutamate, brain-derived neurotrophic factor (BDNF), and γ-amino butyric acid (GABA) receptors under distinct neuronal conditions alters membrane potential and involves mTORC1 (Hou and Klann, 2004; Takei et al., 2004; Inamura et al., 2005; Bateup et al., 2011; Weston et al., 2012, 2014; Workman et al., 2013, 2015). Our recent finding that acute inhibition of mTORC1 activity disrupts the expression of proteins that are involved in ion homeostasis and membrane potential at the synapse supports the close relationship of mTORC1 signaling in maintaining a normal electrochemical gradient of the postsynaptic membrane (Niere et al., 2016). Using the unbiased approach of tandem mass spectrometry (MS/MS) to identify changes in protein composition at the postsynaptic site, we found proteins whose functions affect the membrane potential (Table 1). Several of the mRNAs that encode these proteins curiously reside in the dendrites, suggesting that membrane potential-associated proteins can be locally translated with the right cues (Raab-Graham et al., 2006; Cajigas et al., 2012). We also identified proteins whose levels increased (mTORC1-Off), decreased (mTORC1-On) or remained consistent (mTORC1-independent) with mTORC1 inhibition (Niere et al., 2016).
Dynamic Expression of Dendritic Voltage-Gated Potassium Channel
A family of ion channels that significantly impact the membrane potential is made up of voltage-gated potassium channels (Kv). (Magee and Johnston, 2005; Metz et al., 2007; Pongs, 2008; Remy et al., 2010). While we have a good grasp on the mechanics of Kv channels and their influence on the electrical properties of the cell membrane, little is known about posttranscriptional mechanisms that allow them to respond accordingly to changes in their extracellular environment. The members of the subfamily of Kv1 channels are of particular interest, as their presence or absence can profoundly affect the resting membrane potential and the generation of action potentials (Rho et al., 1999; Brew et al., 2003; Gittelman and Tempel, 2006; Kirchheim et al., 2013; Sosanya et al., 2015a). Kv1 channels form octomers that consist of four pore-forming α subunits (Kvα1.X) and four cytoplasmic β (Kvβ) subunits (Trimmer, 1998; Gutman et al., 2005). The α subunit Kv1.1 is profoundly vital as majority of disease-associated mutations in Kv1 are in the KCNA gene, which codes for Kv1.1 (Ovsepian et al., 2016). Because of the inherent fluid structure of the membrane, ion channels are inserted into and removed from the membrane (Hoffman et al., 1997; Yuste, 1997). The proper surface expression of Kv1.1-containing channels depends on the co-assembly of Kv1.1 with other Kvα1-type (e.g., Kv1.2, Kv1.3 or Kv1.4) channels to mask the strong endoplasmic reticulum (ER) retention signal of Kv1.1 (Vacher et al., 2007). In the dendrites, inhibition of mTORC1 promotes new synthesis of Kv1.1 protein and its expression on the membrane (Raab-Graham et al., 2006). With the identification of Kv1.1 and in anticipation of identifying other “Kv1.1-like” proteins whose levels are negatively regulated by mTORC1 activity, we have designated Kv1.1 and “Kv1.1-like” proteins as “mTORC1-Off”, since they require mTORC1 activity to be turned off or inhibited to undergo mRNA translation. Conversely, “mTORC1-On” proteins require turning on or activation of mTORC1 to be synthesized.
Dysregulated Protein Synthesis Underlies Neurological Disorders
Healthy cells require mTORC1 activity to be within a dynamic range, since neurons in static mTORC1 states (i.e., over- or underactive) are linked to several neurological disorders (Pei and Hugon, 2008; Hoeffer and Klann, 2010; Ma et al., 2010; Bové et al., 2011; Jernigan et al., 2011; Ricciardi et al., 2011; Santini and Klann, 2011; Costa-Mattioli and Monteggia, 2013; Switon et al., 2017). Disorders with excessive protein synthesis are generally associated with overactive mTORC1, while reduced protein production with underactive mTORC1. But in light of the discovery that mRNA translation occurs both when mTORC1 is turned on or off, a state of fixed mTORC1 activity can conceivably promote and repress the protein synthesis of dendritically localized mRNAs to pathological levels. A prime example of a neurological disorder with hyperactive mTORC1 is temporal lobe epilepsy (TLE; Buckmaster et al., 2009; Zeng et al., 2009; Pun et al., 2012; Sha et al., 2012; Wong, 2014; Sosanya et al., 2015a). It has been suggested that overactive mTORC1 in epilepsy promotes excessive protein synthesis that engenders neuronal hyperexcitability. Equally as important, overactive mTORC1 represses several ion channels that reduces excitability. This arm of active mTORC1 also supports epilepsy by giving rise to aberrant synchronous activity of neurons and neuronal circuits (Raab-Graham et al., 2006; Graef and Godwin, 2010; Poolos and Johnston, 2012; Brewster et al., 2013; Sosanya et al., 2013, 2015a; Niere et al., 2016). The imbalance caused by increased synthesis of mTORC1-On proteins and decreased production of mTORC1-Off proteins thrusts the neuronal activity outside the optimal operating range, consequently bringing forth devastating neurological disorders.
mTORC1 as a Local Voltage Sensor
mTORC1 controls mRNA translation of ion channels, ionotropic receptors and their associated proteins (Gong et al., 2006; Raab-Graham et al., 2006; Antion et al., 2008; Bateup et al., 2011, 2013; Huang et al., 2012; Weston et al., 2012; Sosanya et al., 2013, 2015a). In light of these findings and our recent discoveries, we hypothesize that mTORC1 serves as a local voltage sensor by coordinating the levels of postsynaptic proteins that regulate the membrane’s electrical property. We propose below that mTORC1 employs negative and positive feedback mechanisms to modify the electrical features of the dendritic membrane.
Materials and Methods
Sample Preparation
Sprague Dawley rats and C57BL/6 mice (4–6 weeks old) were used. Rapamycin was administered intraperitoneally at 10 mg/kg into rats and 1 mg/kg into mice. As control, animals received an equal volume and concentration of DMSO, which was the vehicle for rapamycin. Synaptoneurosomes were prepared from cortices by size filtration (Workman et al., 2013). The postsynaptic density fraction was isolated as described (Niere et al., 2016). Briefly, synaptoneurosomes were subjected to Triton X-100 solubilization. The PSD and soluble fractions constituted the Triton X-100 insoluble and soluble portions, respectively.
Western Blots
Total lysates, synaptoneurosomes and PSD were solubilized in RIPA buffer before running SDS-PAGE. Protein concentrations were measured using BCA protein assay. Equal amount of protein from each sample was loaded and run in SDS-PAGE gel. Proteins were immunoblotted with primary antibodies against phospho-mTOR Ser2448 (1:2000; Cell Signaling), mTOR (1:5000; LifeTechnologies), Kv1.1 (1:1000 NeuroMab), Kv1.1 (1:1000; LifeSpan BioSciences), Kv1.2 (NeuroMab), Kvβ2 (NeuroMab), actin (1:10,000; Sigma), tubulin (1:50,000; Abcam). Immunoblots were washed and incubated with the appropriate fluorescence- (LiCor) or HRP-conjugated (ThermoFisher Scientific) secondary antibodies (1:5000). Protein expression was quantified by densitometric analysis using ImageJ (National Institutes of Health) software. For the PSD fraction, total protein as measured by densitometry of Ponceau-S staining was used to normalize Kv1.1 and Kv1.2 protein expression.
Immunoprecipitation
Rat cortices were homogenized in tris-buffered saline (50 mM). Membrane fraction was sedimented at 14,000 RPM for 20 min at 4°C. Isolated membranes were solubilized (20 mM HEPES, pH 7.2 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, protease inhibitor, 0.5% SDS) to extract membrane proteins. Preclearing and immunoprecipitation were performed with Protein A/G-agarose suspension (Roche) using the manufacturer’s directions. 4 μg of rabbit-anti-Kv1.1 (LifeSpan Biosciences) or rabbit IgG (control, Santa Cruz Biotechnologies) were used to precipitate the Kv1.1 protein complex from the pre-cleared samples (1 mg of protein). Laemmli sample buffer (1×, BioRad) was used to dissociate the antibody-antigen complex from the agarose beads, after which the supernatant was run in SDS-polyacrylamide gel by electrophoresis. To probe for Kv1.2 protein, mouse-anti-Kv1.2 (NeuroMab) was used. Immunoblots were visualized with the appropriate fluorescence-conjugated secondary antibodies (1:5000; LiCor).
Immunofluorescence
Primary hippocampal rat neurons were prepared from postnatal rats (day 0–2) and fixed in PFA at room temperature as described (Niere et al., 2012; Workman et al., 2013). The following primary antibodies were used: rabbit-anti-Kv1.1 (1:200; LifeSpan Biosciences), mouse-anti-Kv1.2 (1:200; NeuroMab), and chicken-anti-MAP2 (1:2000; Aves Labs). Appropriate secondary antibodies—AlexaFluor-488, 555 and 647 (Invitrogen)—were used to visualize the proteins of interest. Kv1.1, Kv1.2 and MAP2 protein expression were quantified in secondary dendrites that were 50 μm in length and at least 20 μm away from the soma as described (Niere et al., 2012, 2016). A Pearson’s correlation coefficient (PCC) was determined to measure Kv1.1–1.2 colocalization in the dendrites as described (Niere et al., 2016). Z-stacks of seven planes at 1 μm/plane were acquired for each dendrite. Only the plane that had the brightest MAP2 staining was analyzed for colocalization. The ImageJ (NIH) plugin JACoP was used to determine the PCC for each dendrite. The same threshold paradigm was used across all conditions using the threshold function of JACoP.
Biochemical and Bioinformatics Analyses of Neuronal Proteome Expose An mTORC1-Mediated Regulation of Local, Postsynaptic Excitability
We have previously shown that stimulation of the ionotropic glutamate receptor N-methyl-D-aspartate (NMDA)-type (GluN or NMDAR) led to downstream activation of mTORC1 that can be blocked by (2 R)-amino-5-phosphonovaleric acid (AP5)—NMDAR antagonist—and rapamycin (Raab-Graham et al., 2006; Sosanya et al., 2013, 2015a). Recently, our MS/MS data suggest that mTORC1 regulates the expression of three proteins that can cause dynamic changes in membrane excitability: syntaxin 1A (Stx1A), syntaxin 1B (Stx1B), and Kv1.1. These proteins work together to engage negative and positive feedback pathways that control mTORC1 activity (Figure 1). The negative feedback back loop has two arms to turn mTORC1 on or off. Turning off mTORC1 activity promotes the expression of Stx1A, an mTORC1-Off protein. Stx1A serves to switch mTORC1 activity from an “Off” to an “On” state by increasing the insertion of NMDARs to the extracellular membrane. With the increase in mTORC1 activity, the mTORC1-On protein, Stx1B, is then synthesized. Stx1B causes the endocytosis of NMDARs, thus promoting the transition of mTORC1 from the “On” to “Off” state (Figure 1). Notably, when mTORC1 is on, then the translation of the positive feedback mRNAs (i.e., Kv1.1, Kv1.2, Kvβ2) are repressed. In contrast, reduction of mTORC1 activity also enhances Kv1.1, Kv1.2, and Kvβ2 protein levels that work to sustain the mTORC1-Off state. Whether a dendritic branch utilizes positive or negative feedback is likely to depend on the presence or absence of Stx1A and/or Stx1B within a dendritic branch (Figure 1).
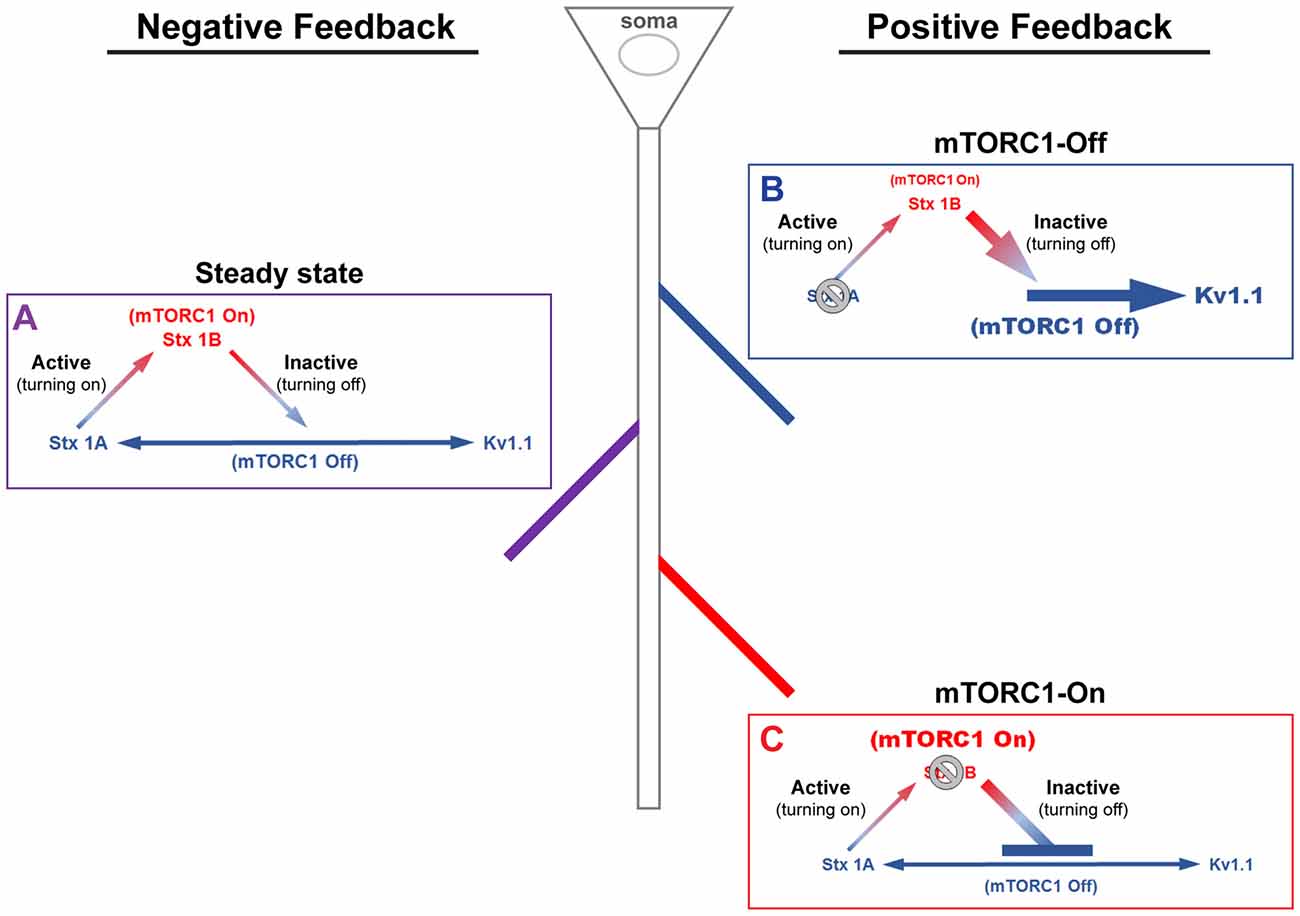
Figure 1. A model of mammalian/mechanistic target of rapamycin complex 1 (mTORC1)-mediated regulation of postsynaptic membrane excitability. mTORC1 regulates membrane excitability by coordinating the expression of mTORC1-On and Off proteins. (A) At steady-state, mTORC1 dynamically switches between On and Off forms in response to neuronal activity. Turning on mTORC1 (Active; ascending arrow) increases the level of syntaxin 1B (Stx1B), an mTORC1-On protein that promotes endocytosis of N-methyl-D-aspartate (NMDA) receptors (NMDARs). Removal of NMDARs reduces membrane excitability and turns off mTORC1 activity (Inactive; descending arrow). Turning off mTORC1 increases the expression of mTORC1-Off proteins, syntaxin 1A (Stx1A; bottom, left arrow) and voltage-gated potassium channel (Kv1.1, bottom, right arrow). Stx1A shuttles NMDARs to the membrane surface, which turns on mTORC1 and increases membrane excitability. Kv1.1, on the other hand, dampens synaptic input. The number of activated NMDARs at the surface, which turns on mTORC1, acts as a signal to stop Kv1.1 mRNA translation. (B) In the absence of Stx1A (or Stx1a mRNA), a positive feedback mechanism could be triggered, whereby mTORC1 remains turned off. The inability to reinsert NMDARs would further lower the membrane potential and support Kv1.1 protein synthesis, reducing the threshold for synaptic activation. (C) The absence of Stx1B (or Stx1b mRNA) could initiate another positive feedback mechanism, such that mTORC1 activity is constitutively on and the membrane remains potentiated. In postsynaptic regions that lack Stx1B, suppression of NMDAR exocytosis would cause mTORC1 to stay active and lower Kv1.1 expression. This state may be another mechanism that supports long-term potentiation.
mTORC1-Mediated Positive Feedback Regulation of Local, Dendritic Membrane Potential Through Kv1.1 Protein Synthesis
Potassium channels are likened to “shock absorbers” as they dampen dendritic membrane depolarization that can arise from calcium entry (Hoffman et al., 1997; Yuste, 1997; Yuan and Chen, 2006). Inhibiting the activity of mTORC1, interestingly, elevates Kv1.1 protein expression only in the dendrites (Raab-Graham et al., 2006; Sosanya et al., 2013, 2015a). While we have determined the molecular mechanism that describes local Kv1.1 protein synthesis, the functional significance of increased Kv1.1 protein when mTORC1 activity is reduced is not fully understood (Sosanya et al., 2013, 2015a). We propose that the mTORC1-Off-dependent translation of Kv1.1 serves to ensure that the membrane resets to a normal resting potential, thereby maintaining neuronal excitability to be within an optimal operating range.
Increased Kv1.2 and Kvβ2 Protein Levels in mTORC1-Off State May Lead to Increased Postsynaptic Surface Expression of Kv1.1
Kv1.1 is an obligate heteromultimeric channel in mammalian neurons (Manganas and Trimmer, 2000). mTORC1 inhibition promotes total and surface expression of dendritic Kv1.1 that are open at rest (Hopkins et al., 1994; Smart et al., 1998; Brew et al., 2003; Raab-Graham et al., 2006; Sosanya et al., 2013, 2015a). In rapamycin, which reduces mTORC1 activity globally, elevated expression of Kv1.1 increases the threshold for action potential firing (Sosanya et al., 2015a). However, the proteins that assemble with dendritic Kv1.1 to facilitate its surface expression are unknown. Two promising candidate proteins identified by our MS/MS that can facilitate the surface expression of Kv1.1 are Kv1.2 and β2 (Manganas and Trimmer, 2000). Thus, we set out to determine if Kv1.2 or β2 subunits are synthesized and co-assemble with Kv1.1 when mTORC1 activity is reduced.
We first compared Kv1.2 and β2 expression of cortical total membrane fractions isolated from rats that received an intraperitoneal (i.p.,) injection of carrier (DMSO) or the mTORC1 inhibitor rapamycin. To ensure that rapamycin reduced mTOR activity we quantified the phosphorylated or active form of mTOR normalized to total mTOR by Western blot analysis. Rapamycin reduced mTOR activity by ~30% (Figure 2A). Notably, Kv1.1, 1.2 and β2 protein levels increased by ~87%, 20% and 40%, respectively, with mTORC1 inhibition (Figures 2B–D).
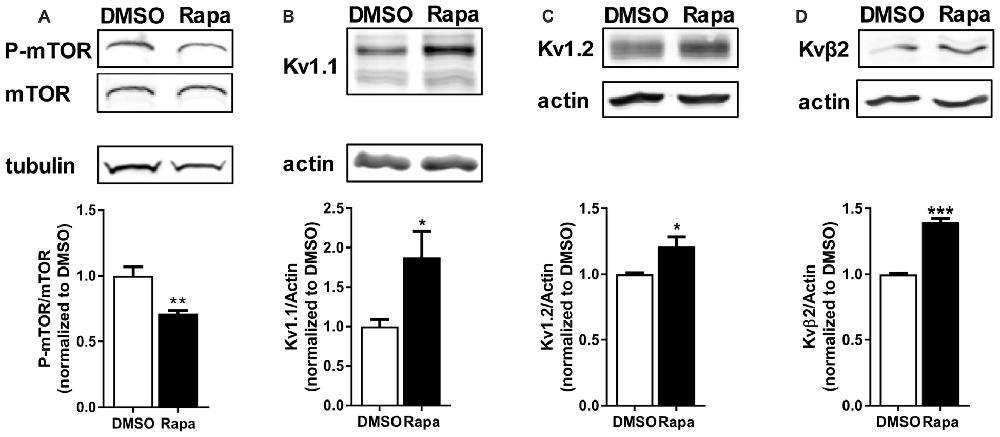
Figure 2. Reduction of mTOR activity increased protein levels of potassium channels and their associated protein. Intraperitoneal (i.p.,) administration of rapamycin (Rapa; 1 mg/kg; 1 h), an mTOR inhibitor, in mice (A) reduced mTOR activity (DMSO = 1.00 ± 0.07; Rapa = 0.71 ± 0.02), but increased (B) Kv1.1 (DMSO = 1.00 ± 0.09; Rapa = 1.88 ± 0.33), (C) Kv1.2 (DMSO = 1.00 ± 0.01; Rapa = 1.21 ± 0.07), and (D) Kvβ2 (DMSO = 1.00 ± 0.01; Rapa = 1.40 ± 0.03) as measured by densitometry analysis. P-mTOR and mTOR denote phosphorylated mTOR and total mTOR, respectively. Representative images and quantification (average ± SEM) are shown. N = 4 (DMSO) and 3 (Rapa) animals. Statistics: Student’s t-test; *p < 0.05, **p < 0.01, ***p < 0.001.
Because mTORC1 inhibition led to a small but significant increase in Kv1.2 expression, we predicted that the number of Kv1.2 subunits that associate with new Kv1.1 channel protein might increase. To test this hypothesis, we immunoprecipitated Kv1.1 from cortical membranes isolated from mice treated with carrier (DMSO) or rapamycin and Western blotted for Kv1.2. Indeed, the amount of surface-expressed Kv1.2 (~90 kD) that co-immunoprecipitated with Kv1.1 in rapamycin-treated samples increased by ~72% (Figure 3; Zhu et al., 2003). These data altogether suggest that more Kv1.1 subunits co-assemble with Kv1.2 with mTORC1 inhibition.
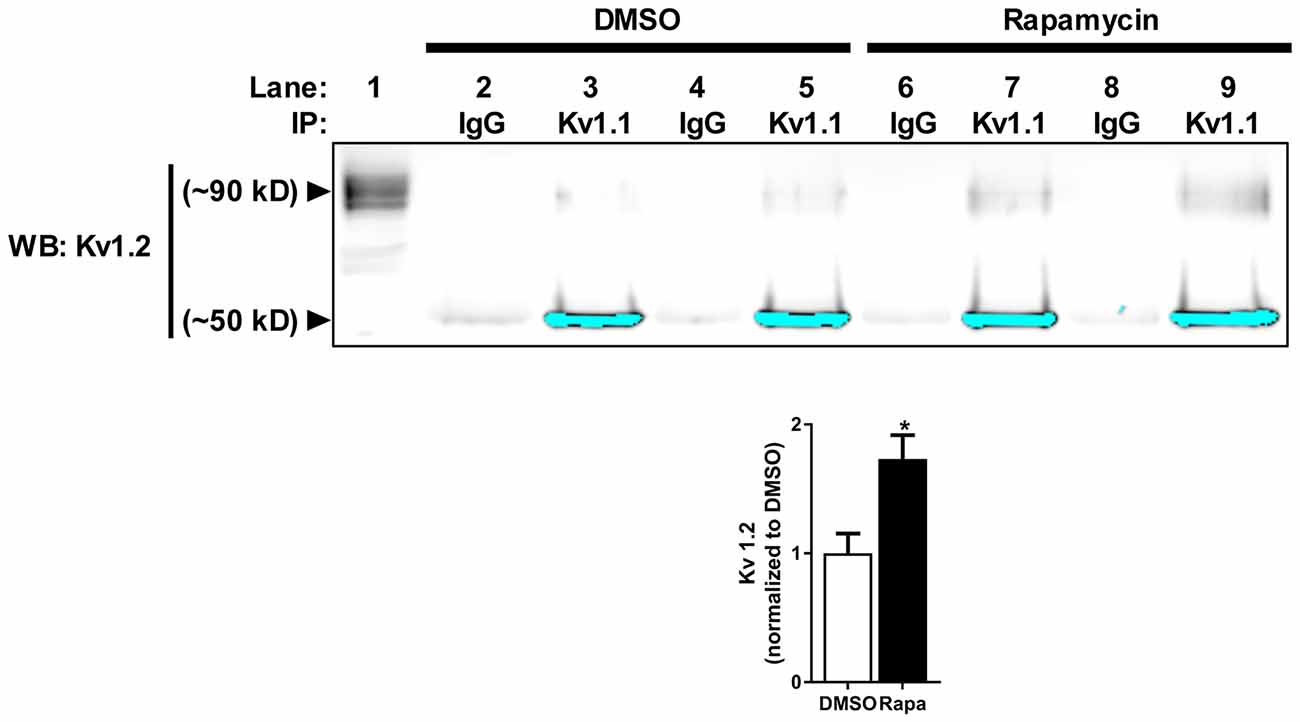
Figure 3. Reduction of mTOR activity increases Kv1.1–1.2 association in vivo. Kv1.2 subunit co-immunoprecipitates with Kv1.1. The high molecular weight (~90 kD) Kv1.2 subunit assembles more with Kv1.1 in mice treated with rapamycin (1.73 ± 0.19) compared to DMSO (1.00 ± 0.15) as measured by densitomery analysis. IP denotes the antibody used to immunoprecipitate Kv1.1 from samples (1 mg/mL) in lanes 2–9. IgG serves as antibody control for immunoprecipiration (lanes 2, 4, 6 and 8). Kv1.2 is enriched in samples that contain Kv1.1 antibody-conjugated beads (lanes 3, 5, 7 and 9) compared to IgG-conjugated beads. The signals at ~50 kD are the low molecular Kv1.2 species and have been saturated to visualize Kv1.2 at ~90 kD. The high molecular weight is the extracellular fraction, and the low molecular is the intracellular fraction of Kv1.2 (Zhu et al., 2003). Lane 1 is the input lane, which is 1% (10 μL) of the total volume that was used to immunoprecipitate Kv1.1. Representative images and quantification (average ± SEM) are shown. N = 4 animals per condition. Statistics: Student’s t-test; *p < 0.05.
To determine if the increase in Kv1.1–1.2 heteromultimeric channels with mTORC1 inhibition takes place in the dendrites, we performed imaging experiments that double-labeled Kv1.1 and Kv1.2 in cultured hippocampal neurons. As mentioned above, we previously showed that NMDAR stimulation led to downstream mTOR activation (Raab-Graham et al., 2006; Sosanya et al., 2013, 2015a). We initiated these studies by first determining whether Kv1.1 and Kv1.2 levels increased in the dendrites of neurons that were treated with the NMDAR inhibitor AP5 relative to carrier (H2O; Figures 4A–B). Indeed, both Kv1.1 and Kv1.2 expression significantly increased in the dendrites of hippocampal neurons with mTORC1 inhibition. Microtubule-associated protein (MAP2), a dendritic marker, remained constant between treatments (Figure 4B). Consistent with our Western blot results, blocking NMDAR/mTOR signaling increased the dendritic expression of Kv1.1 and Kv1.2 (Figures 3, 4). We also examined if we could detect increased colocalization between Kv1.1 and Kv1.2 in the dendrites, since our co-immunoprecipitation (coIP) data indicated elevated Kv1.1–1.2 association when mTORC1 activity is attenuated (Figure 3). Corroborating our coIP data, we detected more Kv1.1–1.2 colocalization in the dendrites treated with AP5 by Pearson’s correlation analysis (CTL = 0.10 ± 0.02; AP5 = 0.18 ± 0.02; Figure 4B).
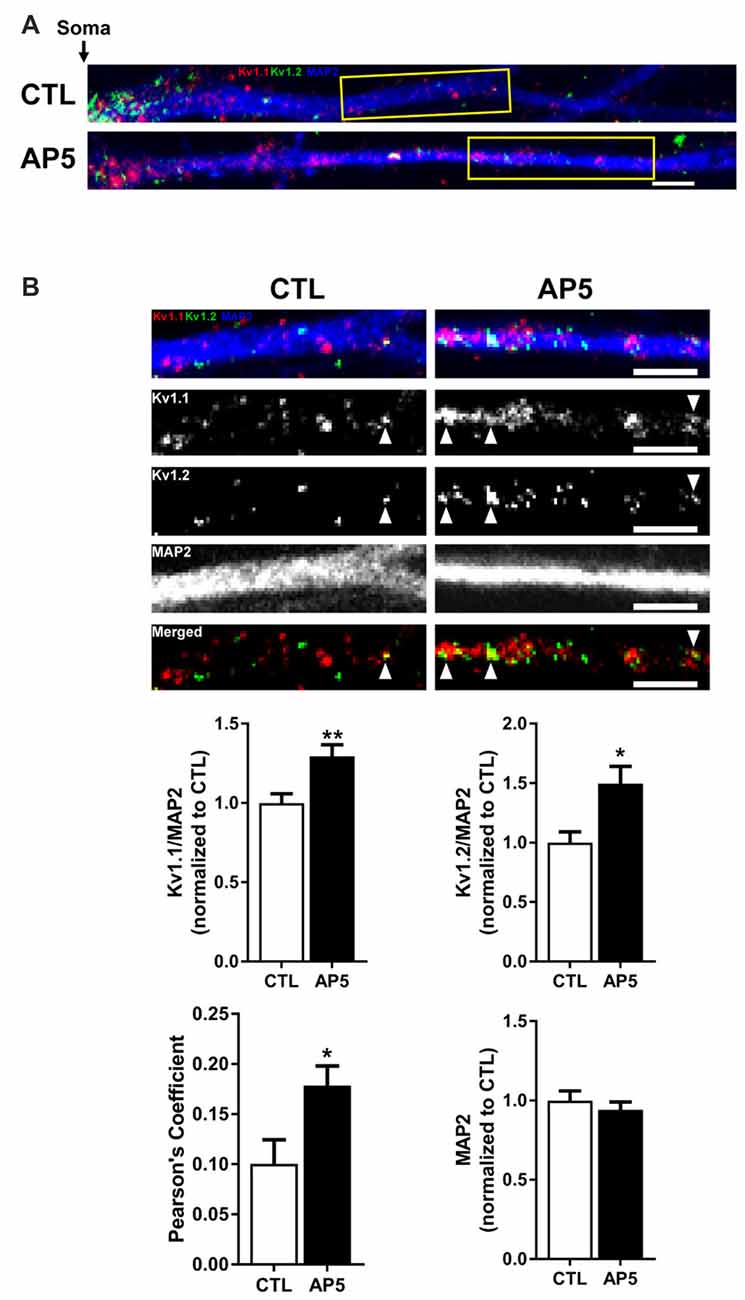
Figure 4. NMDA receptor blockade by AP5 elevates Kv1.1 and Kv1.2 protein in dendrites. (A) Representative dendrites starting from the edge of the soma (indicated by the black arrow) of dissociated hippocampal neurons treated with (top) water (CTL; control) or (bottom) AP5, an NMDA receptor blocker. The dendrites were immunostained with antibodies against Kv1.1 (red), Kv1.2 (green) and the dendritic marker MAP2 (blue). Dendrites outlined in yellow are magnified in (B). (B, Top) Kv1.1, Kv1.2 and MAP2 expression were measured in distal dendrites (at least 20 μm from the soma). White arrowhead indicates colocalized Kv1.1 and Kv1.2 signals. The bottom panel of dendrites show the merged Kv1.1 and Kv1.2 immunostaining. Colocalized Kv1.1 and Kv1.2 signals appear yellow and are quantified by Pearson’s Coefficient. (Below) Quantification (average ± SEM) of fluorescent signal normalized by MAP2 signal indicating more Kv1.1 and Kv1.2 in NMDA receptor blocker AP5 compared to control (CTL). Kv1.1 (CTL = 1.00 ± 0.06, N = 24 dendrites; AP5 = 1.29 ± 0.07, N = 30 dendrites). Kv1.2 (CTL = 1.00 ± 0.09, N = 11 dendrites; AP5 = 1.50 ± 0.15, N = 16 dendrites). MAP2 (CTL = 1.00 ± 0.06; AP5 = 0.94 ± 0.05). NMDA receptor blockade increases colocalization of Kv1.1 and Kv1.2 proteins (CTL = 0.10 ± 0.02, N = 18 dendrites; AP5 = 0.18 ± 0.02, N = 21 dendrites) Statistics: Student’s t-test; *p < 0.05, **p < 0.01.
While we observed elevated levels of Kv1.1 and Kv1.2 proteins at the dendrites, we were curious whether these changes extended to the PSD. We utilized a biochemical approach that isolates neuronal subcellular fractions using filtration and detergent solubility to examine Kv1.1 and Kv1.2 expression in a PSD-enriched fraction. It is well established that resident postsynaptic density (PSD) proteins are not soluble in the detergent Triton X-100 (Fiszer and Robertis, 1967). Thus, we reasoned that we could determine Kv1.1 and 1.2 by biochemically isolating the lysate (total), Triton X-soluble (dendrites and axons), and Triton X-insoluble (PSD) as outlined in Figure 2 of Niere et al. (2016), (Fiszer and Robertis, 1967; Cohen et al., 1977; Rao and Steward, 1991; Villasana et al., 2006). Summarily, we first assessed the purity of fractionated samples by Western blotting for well-characterized resident proteins: postsynaptic density protein of 95 kD (PSD95, PSD marker) and synapsin 1 (presynaptic marker). The PSD (P) fraction was enriched for PSD95, while little synapsin was detected. The soluble (S) fraction, which mainly contains dendrites and axons, was enriched in synapsin and devoid of PSD95. To ensure that mTORC1 activity was inhibited by rapamycin in the PSD, we probed for the phosphorylated or active form of ribosomal S6 protein (P-S6), the downstream marker of mTORC1, and normalized P-S6 over total S6 protein. As expected, rapamycin reduced P-S6 signal by ~88% (Niere et al., 2016). Western blot analysis for Kv1.1 and Kv1.2 in these fractions indicated that Kv1.1 increased in both the PSD and the soluble fractions, while Kv1.2 only increased in the PSD with rapamycin (Figure 5). Control proteins, PSD95 and tubulin, did not significantly change between treatments. These data suggest that Kv1.1–1.2 heteromultimers are likely to reside in the PSD when mTORC1 activity is low.
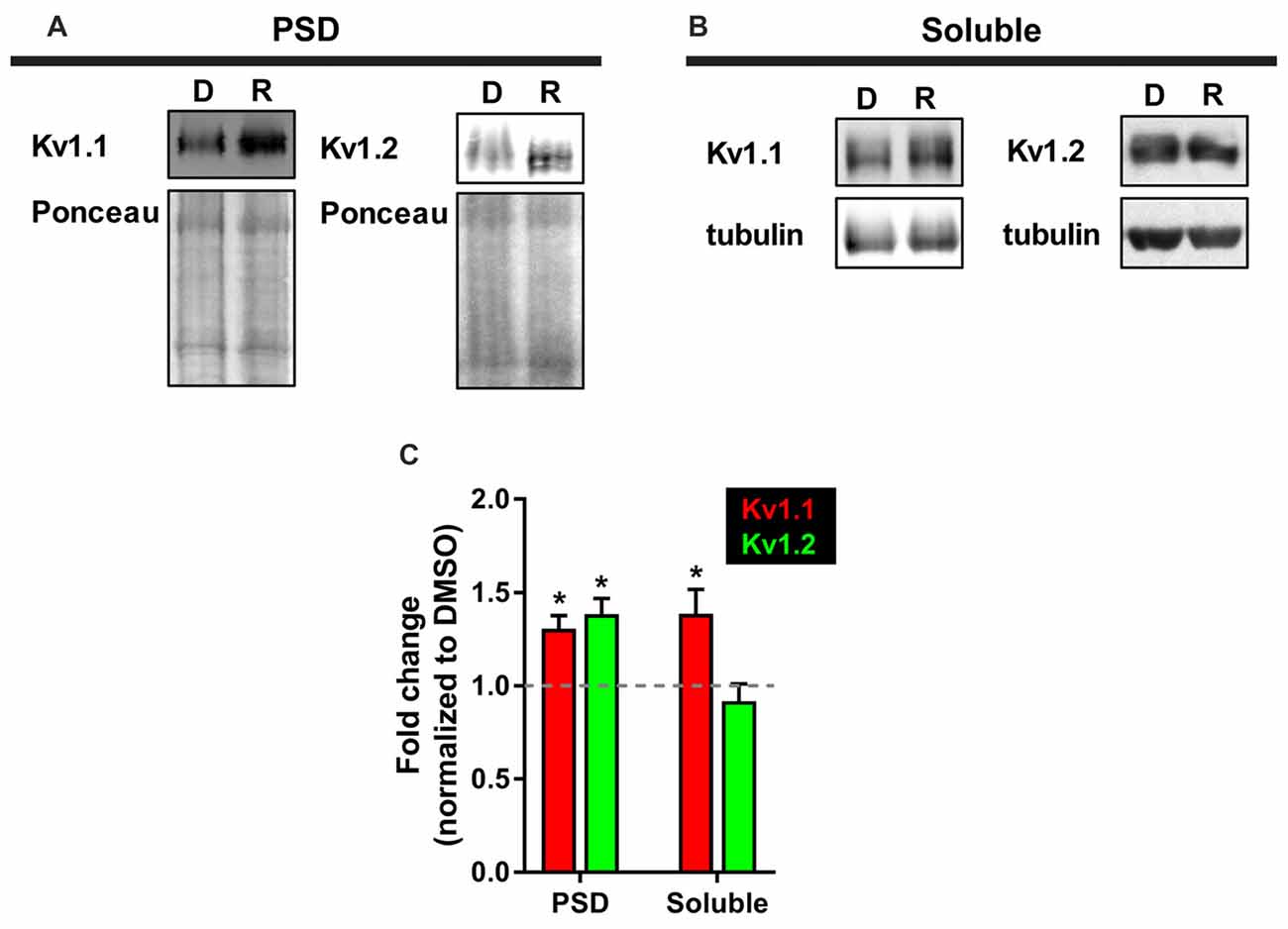
Figure 5. More Kv1.1 and Kv1.2 cofractionate in the PSD with mTORC1 inhibition in vivo. (A) Representative Western blots depicting increased levels of Kv1.1 and Kv1.2 in the PSD fraction (Triton X-100 insoluble) of synaptoneurosomes with mTORC1 inhibition by rapamycin (R). DMSO (D) served as control. Total protein as measured by densitometry of Ponceau-S staining was used to normalize Kv1.1 and Kv1.2 protein expression. (B) In the soluble fraction (Triton X-100 soluble) of synaptoneurosomes, only Kv1.1 protein was increased with rapamycin. Tubulin was used to normalize Kv1.1 and Kv1.2 protein levels. (C) Quantification (average ± SEM) is expressed as densitometric ratio between protein values in rapamycin-treated and DMSO-treated rats. Kv1.1 (PSD = 1.31 ± 0.07; Soluble = 1.39 ± 0.13). Kv1.2 (PSD = 1.39 ± 0.08; Soluble = 0.92 ± 0.09). N = 3 animals per condition. Statistics: Student’s t-test; *p < 0.05.
Leucine-Rich Glioma Inactivated-1 (Lgi1) Protein and Kv1.1 May Sustain mTORC1-Off State and Reduced Membrane Excitability Postsynaptically
Our neuronal proteome analysis of different cellular compartments detected three proteins known to interact with Kv1.1 channels that are regulated by mTORC1 activity. Of particular interest is the highly prominent epilepsy-associated protein Lgi1 (Anderson, 2010; Yokoi et al., 2012; Kegel et al., 2013; Ohkawa et al., 2013). PSD (P) and soluble (S) fractions composed of axons and dendrites—were derived from synaptoneurosomes and subjected to Triton X-100 solubilization. Rapamycin increased the expression of Lgi1 in the Triton X-100 insoluble, PSD (P) fraction, while reducing it in the Triton X-100 soluble (S) fraction—(P = 129%, S = 65% of control; Niere et al., 2016). Interestingly, Lgi1 null mice exhibit neuronal hyperexcitability and spontaneous seizures and fail to live beyond 25 days (Chabrol et al., 2010; Fukata et al., 2010; Yu et al., 2010). Lgi1 is viewed as an antiepileptogenic protein as it converts Kv1.1 channels from A-type channels that inactivate quickly to a delayed rectifier by preventing Kv1.1–Kvβ1 interaction (Chernova et al., 1998; Fukata et al., 2006, 2010; Schulte et al., 2006). Thus, the increased postsynaptic expression of Lgi1 could further enhance the conductance of Kv1.1 in the PSD and in turn keep mTORC1 in a dephosphorylated state. In addition, the reduced expression in the soluble fraction suggests that Kv1.1 expression in the dendrites and/or axons may associate with Kvβ1 and quickly inactivate, similar to A-type channels.
Bidirectional Regulation of Cyclin-Dependent Kinase 5 (Cdk 5) May Support mTORC1-Off-Dependent Reduction of Postsynaptic Membrane Excitability
We found that Cdk5 was differentially regulated in soluble and PSD fractions (Table 1). mTORC1 inhibition increased Cdk5 protein expression in the soluble. Phosphorylation of Kvβ2 by Cdk2/5 had been shown to disrupt Kvβ2’s binding to EB1, thus releasing the channel complex from microtubules and allowing Kv1.1-complex to traffic to the plasma membrane (Gu et al., 2006; Vacher and Trimmer, 2011). The increased level of Cdk5, therefore, may facilitate the local insertion of Kv1.1-containing channels in the dendrites when mTORC1 is inhibited. In the PSD, however, we did not detect Cdk5 (Table 1) but found Kv1.1 and Kv1.2 (Figure 5). These findings suggest that while Cdk5 may be essential for the trafficking of Kv1.1 channels to the dendritic surface, this function of Cdk5 may not be critical in the PSD. The absence of microtubules in spines also support the notion that Cdk5 is not necessary to traffic the Kv1.1 complex to the membrane of the PSD compartment. The masking of the ER retention signal of Kv1.1 by associating with Kv1.2 in the PSD may be sufficient to traffic the Kv1.1 complex to the membrane surface (Manganas and Trimmer, 2000; Tiffany et al., 2000).
Potential Physiological Impact of Compartmentalized mTORC1 Activity
These data collectively suggest that mTORC1 may determine the subcellular localization of Kv1.1-associated proteins and Kv1.1-containing channels. Localized Kv1.1 complex, in turn, will dictate the electrical properties of the compartment and whether multiple compartments (i.e., spines and/or dendritic branches) can interact. For example, potassium channels have been suggested to serve as gate keepers, electrically isolating neuronal compartments (Metz et al., 2007; Harnett et al., 2013). When mTORC1 activity is low in the PSD, the expression of Kv1.1, 1.2, and Lgi1 (sustained potassium current) is likely to temper synaptic stimulation by preventing the opening of voltage-dependent calcium channels and thus confining the synaptic signal to the stimulated spine itself (Harnett et al., 2012). In a situation where mTORC1 activity is high in the spine but low in the associated dendritic branch, reduced Lgi1 would suggest that Kv1.1 could associate with Kvβ1 (transient or A-type channel). Kv1.1-β1 would quickly inactivate upon depolarization allowing for the interaction of nearby stimulated spines (Harnett et al., 2013). In this case, dendritic branches (daughter branches) are likely to remain uncoupled from their parent dendrite. When mTORC1 activity is high throughout the dendritic branch, then Kv expression is repressed. As a consequence, synaptic stimulation will generate local calcium and/or sodium spikes that invade into the parent dendrite (Golding et al., 1999; Frick et al., 2003; Losonczy et al., 2008; Makara et al., 2009). mTORC1 activity, therefore, serves as a local voltage sensor, with increased activity reducing barriers (Kv channels) between compartments allowing for dendritic integration.
mTORC1-Mediated Negative Feedback Regulation of The Postsynaptic Membrane Excitability Through The Differential Expression of Stx1A, Stx1B, and GluN1 Proteins
A family of proteins that stood out in our MS/MS analysis of the proteome that localize with the PSD was syntaxin 1 (Niere et al., 2016). We discovered that mTORC1 regulated syntaxin-1 protein levels and that the two isoforms, Stx1A and Stx1B, differentially responded to mTORC1 activity level (Table 1). Attenuating mTORC1 activity with the mTOR specific inhibitor rapamycin (10 mg/kg, 1 h) increased Stx1A expression (~54%) but reduced Stx1B (~24%; Niere et al., 2016). Therefore, we classified Stx1A as mTORC1-Off and Stx1B as mTORC1-On. Syntaxin-1 is well-characterized as a presynaptic protein that regulates neurotransmitter release; however, its function postsynaptically remains unclear (Bennett et al., 1992, 1993; Rizo and Südhof, 2002, 2012; Rizo and Xu, 2015). Contrary to earlier assumptions, recent studies now reveal that presynaptic syntaxin-1A and 1B perform distinct roles (Mishima et al., 2014; Schubert et al., 2014; Vlaskamp et al., 2016). In light of these findings, we propose that syntaxin-1A and syntaxin-1B perform different roles in mediating mTORC1-dependent regulation of postsynaptic membrane potential as described below.
Another mTORC1-regulated protein that caught our attention was the ionotropic glutamate receptor NMDA-type subunit 1 (GluN1). GluN1 is the obligate subunit for functional NMDARs (Cull-Candy and Leszkiewicz, 2004; Paoletti, 2011). Interestingly, GluN1 was dramatically elevated with reduced mTORC1 activity, such that we could only detect it in the PSD fraction by MS/MS after administering rapamycin as reported (Niere et al., 2016). GluN1, hence, is an mTORC1-Off protein. Because the mRNAs encoding GluN1 and syntaxins 1A and 1B are in the dendrites, we hypothesize that they are postsynaptically translated in response to changes in mTORC1 activity (Cajigas et al., 2012). Namely, Stx1B is synthesized when mTORC1 is turned on and GluN1 and Stx1A when mTORC1 is turned off.
In this section, we consider our data in the context of what others have demonstrated regarding syntaxins 1A, 1B and NR1. We propose that these proteins maintain the oscillation of mTORC1 between “On” and “Off” states that determines the excitability of the postsynaptic membrane (Figure 1). We are calling this pathway the negative feedback loop. The “active” direction of the negative feedback loop begins when mTORC1 is turned off and ends when mTORC1 is turned on. In this process, repression of mTORC1 activity increases the protein levels of Stx1A and GluN1 in the PSD and resultantly turns mTORC1 on. The “inactive” direction of the negative feedback loop toggles mTORC1 from “On” to “Off” state. In the negative path, Stx1B mediates the switch in mTORC1 activity.
mTORC1-Dependent Expression of Stx1A and GluN1 May Increase Postsynaptic Membrane Excitability
A recent study demonstrated that postsynaptic Stx1A preferentially associates with NMDARs over α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs; Hussain et al., 2016). Earlier studies implicated syntaxins 3 and 4 but not syntaxin-1 in AMPAR exocytosis during LTP (Kennedy et al., 2010; Jurado et al., 2013; Jurado, 2014). In light of these recent findings, we predict that in states where mTORC1 activity is attenuated, the elevated levels of NR1 and syntaxin-1 serve to increase the surface expression of NMDARs at the postsynaptic membrane. Increased NMDAR density can contribute to postsynaptic excitability through their interaction with other voltage-sensitive conductances (e.g., voltage-gated calcium channels and calcium-activated potassium channels; Poolos and Kocsis, 1990; Pongrácz et al., 1992; Schiller et al., 2000; Schiller and Schiller, 2001; Stocker, 2004; Antic et al., 2010; Faber, 2010; Shah et al., 2010). Therefore, turning down mTORC1 activity acts as a signal to increase dendritic membrane excitability.
Completing the active-negative feedback pathway, increased NMDAR surface expression can escalate mTOR activation through calcium-mediated signaling cascade (Tang and Schuman, 2002; Gong et al., 2006; Hoeffer and Klann, 2009). This, in turn, will increase the expression of mTORC1-On proteins, such as Stx1B but decrease the levels of NR1 and Stx1A (Figure 1). We predict that turning on mTORC1 elevates Stx1B postsynaptically and leads to degradation of NMDARs. Consequently, removal of NMDARs will turn off mTORC1. NMDAR activation can lead to its internalization (Vissel et al., 2001; Barria and Malinow, 2002; Nong et al., 2003). Several mechanisms control NMDAR internalization, recycling and degradation (Roche et al., 2001; Prybylowski et al., 2002; Scott et al., 2004). The NMDAR subunits GluN1 and GluN2 contain endocytic sorting domains (Scott et al., 2004). GluN2B contains two motifs and thus GluN2B can be directed either to the recycling or late endosome/lysosomal pathway. GluN1, however, solely contains the motif that directs the receptor to the lysosomal or degradation path. The degradative path of GluN1 is highly interesting as our MS/MS data only detects an increase in GluN1 when mTORC1 activity is attenuated (Table 1, Niere et al., 2016). As a requisite subunit for functional NMDARs, mTORC1-dependent upregulation of NR1 may be critical in maintaining membrane excitability (Cull-Candy and Leszkiewicz, 2004; Paoletti, 2011).
mTORC1-Dependent Expression of Stx1B May Reduce Postsynaptic Membrane Excitability
Syntaxin-11 has recently been implicated in late endosome-lysosome fusion (Offenhäuser et al., 2011; van der Sluijs et al., 2013). Curiously, syntaxin-11 protein and mRNA are essentially absent in the brain (Valdez et al., 1999). A comparison of different syntaxins reveals that syntaxin-1B and syntaxin-11 have extensive homology (Tang et al., 1998). Thus, we suspect that in the brain, syntaxin-1B may be the functional homolog to syntaxin-11 by tethering NR1 to the lysosomes for degradation when mTORC1 is active.
By sensing calcium levels mediated by NMDARs, mTORC1 accordingly tunes the electrical property of the postsynaptic membrane through a negative feedback mechanism that relies on the protein expression of syntaxins 1A and 1B and NR1 (Table 1 and Figure 1). mTORC1 increases membrane excitability by synchronously increasing Stx1A and decreasing Stx1B to escalate surface expression of NMDARs through elevated NR1 protein levels. However, as mTORC1 gets turned on by increasing calcium concentration through NMDAR entry, mTORC1 simultaneously reduces Stx1A and elevates Stx1B protein levels to inhibit NMDAR trafficking to the membrane. This response can lead to a reduction in mTORC1 activity.
Local Voltage Sensing of mTORC1 Is Vital for Normal Postsynaptic Function and Requires Dynamic Expression of mTORC1-On and off Proteins
In consideration of our data and published observations, we have attempted to synthesize a mechanism by which mTORC1 can detect changes in local, postsynaptic membrane potential. This proposed mechanism implicates mTORC1 as a voltage sensor that can promote protein synthesis of specific receptors, ion channels, and associated proteins that can keep mTORC1 activity in the optimal range. Interestingly, protein syntheses of several ionotropic receptors, ion channels, and their associated proteins are dependent on mTOR activity (Tang et al., 2002; Cammalleri et al., 2003; Schratt et al., 2004; Raab-Graham et al., 2006; Liao et al., 2007; Gobert et al., 2008; Meyuhas and Kahan, 2015).
Our biochemical, bioinformatics and imaging data have identified molecules that can mediate mTORC1-dependent regulation of the postsynaptic membrane. Emerging data support a role for clustered synaptic plasticity both in vivo and in vitro (Govindarajan et al., 2006; Losonczy and Magee, 2006; Losonczy et al., 2008; Kleindienst et al., 2011; Makino and Malinow, 2011). However, to test this hypothesis, electrophysiological experiments and/or imaging experiments that evaluate the necessity of Stx1A and Stx1B to shuttle GluN receptors to and from the postsynaptic membrane are critical. The relationship between acute changes in local, postsynaptic mTORC1 activity and surface membrane expression of GluN should also be tested. Work by Makino and Malinow (2011) elegantly demonstrate fluorescent tagged GluA1 receptors enriched in spines that are clustered together upon sensory experience. Perhaps a similar strategy can be utilized for GluN1.
A-type potassium channels have been implicated as a critical factor in determining dendritic branch strength (Losonczy et al., 2008; Makara et al., 2009). The molecular identity underlying the A-type current, however, has been suggested to be Kv4.2. Interestingly, Kv4.2 is an mTOR-Off protein (Lee et al., 2011). In light of our data, Kv1.1 could also serve as a gatekeeper of branch strength and compartmentalization (Figures 2–5; Sosanya et al., 2015b). Determining the contribution of Kv1.1 to branch strength, however, is more difficult due to its presence in both axons and dendrites when mTOR is off. Genetic tools that block or increase branch-specific dendritic targeting of Kv1.1 are necessary to adequately test our hypothesis.
How can positive and negative feedback pathways that control mTORC1 activity take place in a single neuron? One way this is achieved is by localizing different populations of mRNAs-RNA binding protein (RNP) complex to specific dendritic branches. For example, we have recently shown that the RNA binding protein HuD favors 1 daughter branch over the other (Sosanya et al., 2015b). Our model suggests that the choice between translating Stx1A and/or Stx1B mRNA dictates whether a dendrite will utilize a negative or positive feedback pathway. Future studies should include the identification of the RNA binding factors that regulate Stx1A and/or Stx1B mRNA translation to determine if they are expressed in unique dendritic branches similar to HuD.
Many mTOR-related diseases present mTORC1 activity that is outside the ideal physiological range. The recruitment of a positive feedback may underlie mTORC1-related diseases. For example, excessive repression of the inactive arm would drive the hyperactivity of mTOR and the repression of Kv1.1, 1.2, β2 and Lgi1 (Figure 1C). This is likely to be the case in animal models of diseases that exhibit neurological disorders that are common in Alzheimer’s disease, autism spectrum disorder, and tuberous sclerosis complex, all disease where patients are reported to suffer from seizures (Swiech et al., 2008; Bateup et al., 2011, 2013; Bové et al., 2011; Hays et al., 2011; Ronesi et al., 2012; Contractor et al., 2015; Tamagnini et al., 2015a,b; Oh et al., 2016). In contrast, extended use of the inactive arm would induce constitutive expression of Kv1.1, 1.2, β2, and Lgi1 that could lead to dangerously low mTOR activity and diseases with hypoactive mTOR (e.g., Parkinson’s disease, Rett’s syndrome, and major depressive disorder; Karege et al., 2007; Swiech et al., 2008; Taneja et al., 2009; Bové et al., 2011; Jernigan et al., 2011; Ricciardi et al., 2011; Chandran et al., 2013; Figure 1B). Considering the presence of mTORC1 in the postsynaptic region and its ability to regulate the levels of ion channels, ionotropic receptors, and their associated proteins that determine conductance and trafficking, it is germane to define the link between dysregulated mTORC1 activity and abnormal membrane excitability. While it may be that the proposed positive and negative feedback mechanisms governing mTORC1-dependent regulation of membrane potential may be simple as presented, consideration and refinement of this hypothesis is an important step that will have broad implications not only in neurological disorders but in other fields that investigate excitable membranes.
Ethics Statement
This study was carried out in accordance with the recommendations of National Institutes of Health’s Guide for the Care and Use of Laboratory Animals. The protocol was approved by the University of Texas Institutional Animal Care and Use Committee.
Author Contributions
FN and KFR-G designed the experiments and wrote the article. FN performed the experiments and analyzed the data.
Funding
This work was supported by National Science Foundation (NSF) IOS-1355158 (KFR-G), NSF PRFB DBI-1306528 (FN), National Institutes of Health (NH)-National Institute on Alcohol Abuse and Alcoholism (NIAA) pilot grant provided by the Integrated Neuroscience Initiative on Alcoholism (KFR-G), US Department of Defense USAMRMC Award W81XWH-14-10061 (KFR-G), NIH-National Institute on Aging (NIA) pilot grant provided by Wake Forest University Health Sciences Alzheimer’s Disease Core Center P30AG049638 (KFR-G), and NIH-National Institute on Drug Abuse (NIDA) pilot grant provided by Wake Forest Health Sciences Center for the Neurobiology of Addiction Treatment (CNAT) P50DA06634 (KFR-G).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Anderson, M. P. (2010). Arrested glutamatergic synapse development in human partial epilepsy. Epilepsy Curr. 10, 153–158. doi: 10.1111/j.1535-7511.2010.01386.x
Antic, S. D., Zhou, W. L., Moore, A. R., Short, S. M., and Ikonomu, K. D. (2010). The decade of the dendritic NMDA spike. J. Neurosci. Res. 88, 2991–3001. doi: 10.1002/jnr.22444
Antion, M. D., Hou, L., Wong, H., Hoeffer, C. A., and Klann, E. (2008). mGluR-dependent long-term depression is associated with increased phosphorylation of S6 and synthesis of elongation factor 1A but remains expressed in S6K-deficient mice. Mol. Cell. Biol. 28, 2996–3007. doi: 10.1128/MCB.00201-08
Auerbach, B. D., Osterweil, E. K., and Bear, M. F. (2011). Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature 480, 63–68. doi: 10.1038/nature10658
Barria, A., and Malinow, R. (2002). Subunit-specific NMDA receptor trafficking to synapses. Neuron 35, 345–353. doi: 10.1016/s0896-6273(02)00776-6
Bateup, H. S., Johnson, C. A., Denefrio, C. L., Saulnier, J. L., Kornacker, K., and Sabatini, B. L. (2013). Excitatory/inhibitory synaptic imbalance leads to hippocampal hyperexcitability in mouse models of tuberous sclerosis. Neuron 78, 510–522. doi: 10.1016/j.neuron.2013.03.017
Bateup, H. S., Takasaki, K. T., Saulnier, J. L., Denefrio, C. L., and Sabatini, B. L. (2011). Loss of Tsc1 in vivo impairs hippocampal mGluR-LTD and increases excitatory synaptic function. J. Neurosci. 31, 8862–8869. doi: 10.1523/JNEUROSCI.1617-11.2011
Bennett, M. K., Calakos, N., and Scheller, R. H. (1992). Syntaxin: a synaptic protein implicated in docking of synaptic vesicles at presynaptic active zones. Science 257, 255–259. doi: 10.1126/science.1321498
Bennett, M. K., García-Arrarás, J. E., Elferink, L. A., Peterson, K., Fleming, A. M., Hazuka, C. D., et al. (1993). The syntaxin family of vesicular transport receptors. Cell 74, 863–873. doi: 10.1016/0092-8674(93)90466-4
Bové, J., Martínez-Vicente, M., and Vila, M. (2011). Fighting neurodegeneration with rapamycin: mechanistic insights. Nat. Rev. Neurosci. 12, 437–452. doi: 10.1038/nrn3068
Brew, H. M., Hallows, J. L., and Tempel, B. L. (2003). Hyperexcitability and reduced low threshold potassium currents in auditory neurons of mice lacking the channel subunit Kv1.1. J. Physiol. 548, 1–20. doi: 10.1113/jphysiol.2002.035568
Brewster, A. L., Lugo, J. N., Patil, V. V., Lee, W. L., Qian, Y., Vanegas, F., et al. (2013). Rapamycin reverses status epilepticus-induced memory deficits and dendritic damage. PLoS One 8:e57808. doi: 10.1371/journal.pone.0057808
Buckmaster, P. S., Ingram, E. A., and Wen, X. (2009). Inhibition of the mammalian target of rapamycin signaling pathway suppresses dentate granule cell axon sprouting in a rodent model of temporal lobe epilepsy. J. Neurosci. 29, 8259–8269. doi: 10.1523/JNEUROSCI.4179-08.2009
Cajigas, I. J., Tushev, G., Will, T. J., tom Dieck, S., Fuerst, N., and Schuman, E. M. (2012). The local transcriptome in the synaptic neuropil revealed by deep sequencing and high-resolution imaging. Neuron 74, 453–466. doi: 10.1016/j.neuron.2012.02.036
Cammalleri, M., Lütjens, R., Berton, F., King, A. R., Simpson, C., Francesconi, W., et al. (2003). Time-restricted role for dendritic activation of the mTOR-p70S6K pathway in the induction of late-phase long-term potentiation in the CA1. Proc. Natl. Acad. Sci. U S A 100, 14368–14373. doi: 10.1073/pnas.2336098100
Chabrol, E., Navarro, V., Provenzano, G., Cohen, I., Dinocourt, C., Rivaud-Péchoux, S., et al. (2010). Electroclinical characterization of epileptic seizures in leucine-rich, glioma-inactivated 1-deficient mice. Brain 133, 2749–2762. doi: 10.1093/brain/awq171
Chandran, A., Iyo, A. H., Jernigan, C. S., Legutko, B., Austin, M. C., and Karolewicz, B. (2013). Reduced phosphorylation of the mTOR signaling pathway components in the amygdala of rats exposed to chronic stress. Prog. Neuropsychopharmacol. Biol. Psychiatry 40, 240–245. doi: 10.1016/j.pnpbp.2012.08.001
Chernova, O. B., Somerville, R. P., and Cowell, J. K. (1998). A novel gene, LGI1, from 10q24 is rearranged and downregulated in malignant brain tumors. Oncogene 17, 2873–2881. doi: 10.1038/sj.onc.1202481
Cohen, R. S., Blomberg, F., Berzins, K., and Siekevitz, P. (1977). The structure of postsynaptic densities isolated from dog cerebral cortex. I. Overall morphology and protein composition. J. Cell Biol. 74, 181–203. doi: 10.1083/jcb.74.1.181
Contractor, A., Klyachko, V. A., and Portera-Cailliau, C. (2015). Altered neuronal and circuit excitability in fragile X syndrome. Neuron 87, 699–715. doi: 10.1016/j.neuron.2015.06.017
Costa-Mattioli, M., and Monteggia, L. M. (2013). mTOR complexes in neurodevelopmental and neuropsychiatric disorders. Nat. Neurosci. 16, 1537–1543. doi: 10.1038/nn.3546
Cull-Candy, S. G., and Leszkiewicz, D. N. (2004). Role of distinct NMDA receptor subtypes at central synapses. Sci. STKE 2004:re16. doi: 10.1126/stke.2552004re16
Faber, E. S. (2010). Functional interplay between NMDA receptors, SK channels and voltage-gated Ca2+ channels regulates synaptic excitability in the medial prefrontal cortex. J. Physiol. 588, 1281–1292. doi: 10.1113/jphysiol.2009.185645
Fiszer, S., and Robertis, E. D. (1967). Action of triton X-100 on ultrastructure and membrane-bound- enzymes of isolated nerve endings from rat brain. Brain Res. 5, 31–44. doi: 10.1016/0006-8993(67)90217-x
Frick, A., Magee, J., Koester, H. J., Migliore, M., and Johnston, D. (2003). Normalization of Ca2+ signals by small oblique dendrites of CA1 pyramidal neurons. J. Neurosci. 23, 3243–3250.
Fukata, Y., Adesnik, H., Iwanaga, T., Bredt, D. S., Nicoll, R. A., and Fukata, M. (2006). Epilepsy-related ligand/receptor complex LGI1 and ADAM22 regulate synaptic transmission. Science 313, 1792–1795. doi: 10.1126/science.1129947
Fukata, Y., Lovero, K. L., Iwanaga, T., Watanabe, A., Yokoi, N., Tabuchi, K., et al. (2010). Disruption of LGI1-linked synaptic complex causes abnormal synaptic transmission and epilepsy. Proc. Natl. Acad. Sci. U S A 107, 3799–3804. doi: 10.1073/pnas.0914537107
Gittelman, J. X., and Tempel, B. L. (2006). Kv1.1-containing channels are critical for temporal precision during spike initiation. J. Neurophysiol. 96, 1203–1214. doi: 10.1152/jn.00092.2005
Gobert, D., Topolnik, L., Azzi, M., Huang, L., Badeaux, F., Desgroseillers, L., et al. (2008). Forskolin induction of late-LTP and up-regulation of 5′ TOP mRNAs translation via mTOR, ERK, and PI3K in hippocampal pyramidal cells. J. Neurochem. 106, 1160–1174. doi: 10.1111/j.1471-4159.2008.05470.x
Golding, N. L., Jung, H. Y., Mickus, T., and Spruston, N. (1999). Dendritic calcium spike initiation and repolarization are controlled by distinct potassium channel subtypes in CA1 pyramidal neurons. J. Neurosci. 19, 8789–8798.
Gong, R., Park, C. S., Abbassi, N. R., and Tang, S. J. (2006). Roles of glutamate receptors and the mammalian target of rapamycin (mTOR) signaling pathway in activity-dependent dendritic protein synthesis in hippocampal neurons. J. Biol. Chem. 281, 18802–18815. doi: 10.1074/jbc.m512524200
Govindarajan, A., Kelleher, R. J., and Tonegawa, S. (2006). A clustered plasticity model of long-term memory engrams. Nat. Rev. Neurosci. 7, 575–583. doi: 10.1038/nrn1937
Graef, J. D., and Godwin, D. W. (2010). Intrinsic plasticity in acquired epilepsy: too much of a good thing? Neuroscientist 16, 487–495. doi: 10.1177/1073858409358776
Gu, C., Zhou, W., Puthenveedu, M. A., Xu, M., Jan, Y. N., and Jan, L. Y. (2006). The microtubule plus-end tracking protein EB1 is required for Kv1 voltage-gated K+ channel axonal targeting. Neuron 52, 803–816. doi: 10.1016/j.neuron.2006.10.022
Gutman, G. A., Chandy, K. G., Grissmer, S., Lazdunski, M., McKinnon, D., Pardo, L. A., et al. (2005). International union of pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol. Rev. 57, 473–508. doi: 10.1124/pr.57.4.10
Harnett, M. T., Makara, J. K., Spruston, N., Kath, W. L., and Magee, J. C. (2012). Synaptic amplification by dendritic spines enhances input cooperativity. Nature 491, 599–602. doi: 10.1038/nature11554
Harnett, M. T., Xu, N. L., Magee, J. C., and Williams, S. R. (2013). Potassium channels control the interaction between active dendritic integration compartments in layer 5 cortical pyramidal neurons. Neuron 79, 516–529. doi: 10.1016/j.neuron.2013.06.005
Hay, N., and Sonenberg, N. (2004). Upstream and downstream of mTOR. Genes Dev. 18, 1926–1945. doi: 10.1101/gad.1212704
Hays, S. A., Huber, K. M., and Gibson, J. R. (2011). Altered neocortical rhythmic activity states in Fmr1 KO mice are due to enhanced mGluR5 signaling and involve changes in excitatory circuitry. J. Neurosci. 31, 14223–14234. doi: 10.1523/JNEUROSCI.3157-11.2011
Hoeffer, C. A., and Klann, E. (2009). “NMDA receptors and translational control,” in Biology of The NMDA Receptor, ed. A. M. Van Dongen (Boca Raton, FL: CRC Press).
Hoeffer, C. A., and Klann, E. (2010). mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 33, 67–75. doi: 10.1016/j.tins.2009.11.003
Hoffman, D. A., Magee, J. C., Colbert, C. M., and Johnston, D. (1997). K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature 387, 869–875. doi: 10.1038/43119
Hopkins, W. F., Allen, M. L., Houamed, K. M., and Tempel, B. L. (1994). Properties of voltage-gated K+ currents expressed in Xenopus oocytes by mKv1.1, mKv1.2 and their heteromultimers as revealed by mutagenesis of the dendrotoxin-binding site in mKv1.1. Pflugers Arch. 428, 382–390. doi: 10.1007/bf00724522
Hou, L., and Klann, E. (2004). Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. J. Neurosci. 24, 6352–6361. doi: 10.1523/JNEUROSCI.0995-04.2004
Huang, X., McMahon, J., Yang, J., Shin, D., and Huang, Y. (2012). Rapamycin down-regulates KCC2 expression and increases seizure susceptibility to convulsants in immature rats. Neuroscience 219, 33–47. doi: 10.1016/j.neuroscience.2012.05.003
Hussain, S., Ringsevjen, H., Egbenya, D. L., Skjervold, T. L., and Davanger, S. (2016). SNARE protein syntaxin-1 colocalizes closely with NMDA receptor subunit NR2B in postsynaptic spines in the hippocampus. Front. Mol. Neurosci. 9:10. doi: 10.3389/fnmol.2016.00010
Inamura, N., Nawa, H., and Takei, N. (2005). Enhancement of translation elongation in neurons by brain-derived neurotrophic factor: implications for mammalian target of rapamycin signaling. J. Neurochem. 95, 1438–1445. doi: 10.1111/j.1471-4159.2005.03466.x
Jernigan, C. S., Goswami, D. B., Austin, M. C., Iyo, A. H., Chandran, A., Stockmeier, C. A., et al. (2011). The mTOR signaling pathway in the prefrontal cortex is compromised in major depressive disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 35, 1774–1779. doi: 10.1016/j.pnpbp.2011.05.010
Jurado, S. (2014). The dendritic SNARE fusion machinery involved in AMPARs insertion during long-term potentiation. Front. Cell. Neurosci. 8:407. doi: 10.3389/fncel.2014.00407
Jurado, S., Goswami, D., Zhang, Y., Molina, A. J., Sudhof, T. C., and Malenka, R. C. (2013). LTP requires a unique postsynaptic SNARE fusion machinery. Neuron 77, 542–558. doi: 10.1016/j.neuron.2012.11.029
Karege, F., Perroud, N., Burkhardt, S., Schwald, M., Ballmann, E., La Harpe, R., et al. (2007). Alteration in kinase activity but not in protein levels of protein kinase B and glycogen synthase kinase-3β in ventral prefrontal cortex of depressed suicide victims. Biol. Psychiatry 61, 240–245. doi: 10.1016/j.biopsych.2006.04.036
Kegel, L., Aunin, E., Meijer, D., and Bermingham, J. R. (2013). LGI proteins in the nervous system. ASN Neuro 5, 167–181. doi: 10.1042/AN20120095
Kennedy, M. J., Davison, I. G., Robinson, C. G., and Ehlers, M. D. (2010). Syntaxin-4 defines a domain for activity-dependent exocytosis in dendritic spines. Cell 141, 524–535. doi: 10.1016/j.cell.2010.02.042
Kirchheim, F., Tinnes, S., Haas, C. A., Stegen, M., and Wolfart, J. (2013). Regulation of action potential delays via voltage-gated potassium Kv1.1 channels in dentate granule cells during hippocampal epilepsy. Front. Cell. Neurosci. 7:248. doi: 10.3389/fncel.2013.00248
Kleindienst, T., Winnubst, J., Roth-Alpermann, C., Bonhoeffer, T., and Lohmann, C. (2011). Activity-dependent clustering of functional synaptic inputs on developing hippocampal dendrites. Neuron 72, 1012–1024. doi: 10.1016/j.neuron.2011.10.015
Lamming, D. W., Ye, L., Katajisto, P., Goncalves, M. D., Saitoh, M., Stevens, D. M., et al. (2012). Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 335, 1638–1643. doi: 10.1126/science.1215135
Laplante, M., and Sabatini, D. M. (2012). mTOR signaling in growth control and disease. Cell 149, 274–293. doi: 10.1016/j.cell.2012.03.017
Lee, H. Y., Ge, W.-P., Huang, W., He, Y., Wang, G. X., Rowson-Baldwin, A., et al. (2011). Bidirectional regulation of dendritic voltage-gated potassium channels by the fragile X mental retardation protein. Neuron 72, 630–642. doi: 10.1016/j.neuron.2011.09.033
Liao, L., Pilotte, J., Xu, T., Wong, C. C., Edelman, G. M., Vanderklish, P., et al. (2007). BDNF induces widespread changes in synaptic protein content and up-regulates components of the translation machinery: an analysis using high-throughput proteomics. J. Proteome Res. 6, 1059–1071. doi: 10.1021/pr060358f
Losonczy, A., and Magee, J. C. (2006). Integrative properties of radial oblique dendrites in hippocampal CA1 pyramidal neurons. Neuron 50, 291–307. doi: 10.1016/j.neuron.2006.03.016
Losonczy, A., Makara, J. K., and Magee, J. C. (2008). Compartmentalized dendritic plasticity and input feature storage in neurons. Nature 452, 436–441. doi: 10.1038/nature06725
Ma, T., Hoeffer, C. A., Capetillo-Zarate, E., Yu, F., Wong, H., Lin, M. T., et al. (2010). Dysregulation of the mTOR pathway mediates impairment of synaptic plasticity in a mouse model of Alzheimer’s disease. PLoS One 5:e12845. doi: 10.1371/journal.pone.0012845
Magee, J. C., and Johnston, D. (2005). Plasticity of dendritic function. Curr. Opin. Neurobiol. 15, 334–342. doi: 10.1016/j.conb.2005.05.013
Makara, J. K., Losonczy, A., Wen, Q., and Magee, J. C. (2009). Experience-dependent compartmentalized dendritic plasticity in rat hippocampal CA1 pyramidal neurons. Nat. Neurosci. 12, 1485–1487. doi: 10.1038/nn.2428
Makino, H., and Malinow, R. (2011). Compartmentalized versus global synaptic plasticity on dendrites controlled by experience. Neuron 72, 1001–1011. doi: 10.1016/j.neuron.2011.09.036
Manganas, L. N., and Trimmer, J. S. (2000). Subunit composition determines Kv1 potassium channel surface expression. J. Biol. Chem. 275, 29685–29693. doi: 10.1074/jbc.m005010200
Metz, A. E., Spruston, N., and Martina, M. (2007). Dendritic D-type potassium currents inhibit the spike afterdepolarization in rat hippocampal CA1 pyramidal neurons. J. Physiol. 581, 175–187. doi: 10.1113/jphysiol.2006.127068
Meyuhas, O., and Kahan, T. (2015). The race to decipher the top secrets of TOP mRNAs. Biochim. Biophys. Acta 1849, 801–811. doi: 10.1016/j.bbagrm.2014.08.015
Mishima, T., Fujiwara, T., Sanada, M., Kofuji, T., Kanai-Azuma, M., and Akagawa, K. (2014). Syntaxin 1B, but not syntaxin 1A, is necessary for the regulation of synaptic vesicle exocytosis and of the readily releasable pool at central synapses. PLoS One 9:e90004. doi: 10.1371/journal.pone.0090004
Niere, F., Namjoshi, S., Song, E., Dilly, G. A., Schoenhard, G., Zemelman, B. V., et al. (2016). Analysis of proteins that rapidly change upon mechanistic/mammalian target of rapamycin complex 1 (mTORC1) repression identifies Parkinson protein 7 (PARK7) as a novel protein aberrantly expressed in tuberous sclerosis complex (TSC). Mol. Cell. Proteomics 15, 426–444. doi: 10.1074/mcp.M115.055079
Niere, F., Wilkerson, J. R., and Huber, K. M. (2012). Evidence for a fragile X mental retardation protein-mediated translational switch in metabotropic glutamate receptor-triggered Arc translation and long-term depression. J. Neurosci. 32, 5924–5936. doi: 10.1523/JNEUROSCI.4650-11.2012
Nong, Y., Huang, Y. Q., Ju, W., Kalia, L. V., Ahmadian, G., Wang, Y. T., et al. (2003). Glycine binding primes NMDA receptor internalization. Nature 422, 302–307. doi: 10.1038/nature01497
Offenhäuser, C., Lei, N., Roy, S., Collins, B. M., Stow, J. L., and Murray, R. Z. (2011). Syntaxin 11 binds Vti1b and regulates late endosome to lysosome fusion in macrophages. Traffic 12, 762–773. doi: 10.1111/j.1600-0854.2011.01189.x
Oh, M. M., Simkin, D., and Disterhoft, J. F. (2016). Intrinsic hippocampal excitability changes of opposite signs and different origins in CA1 and CA3 pyramidal neurons underlie aging-related cognitive deficits. Front. Syst. Neurosci. 10:52. doi: 10.3389/fnsys.2016.00052
Ohkawa, T., Fukata, Y., Yamasaki, M., Miyazaki, T., Yokoi, N., Takashima, H., et al. (2013). Autoantibodies to epilepsy-related LGI1 in limbic encephalitis neutralize LGI1-ADAM22 interaction and reduce synaptic AMPA receptors. J. Neurosci. 33, 18161–18174. doi: 10.1523/JNEUROSCI.3506-13.2013
Ovsepian, S. V., LeBerre, M., Steuber, V., O’Leary, V. B., Leibold, C., and Oliver Dolly, J. (2016). Distinctive role of Kv1.1 subunit in the biology and functions of low threshold K+ channels with implications for neurological disease. Pharmacol. Ther. 159, 93–101. doi: 10.1016/j.pharmthera.2016.01.005
Paoletti, P. (2011). Molecular basis of NMDA receptor functional diversity. Eur. J. Neurosci. 33, 1351–1365. doi: 10.1111/j.1460-9568.2011.07628.x
Park, S., Park, J. M., Kim, S., Kim, J. A., Shepherd, J. D., Smith-Hicks, C. L., et al. (2008). Elongation factor 2 and fragile X mental retardation protein control the dynamic translation of Arc/Arg3.1 essential for mGluR-LTD. Neuron 59, 70–83. doi: 10.1016/j.neuron.2008.05.023
Pei, J. J., and Hugon, J. (2008). mTOR-dependent signaling in Alzheimer’s disease. J. Cell. Mol. Med. 12, 2525–2532. doi: 10.1111/j.1582-4934.2008.00509.x
Pongrácz, F., Poolos, N. P., Kocsis, J. D., and Shepherd, G. M. (1992). A model of NMDA receptor-mediated activity in dendrites of hippocampal CA1 pyramidal neurons. J. Neurophysiol. 68, 2248–2259.
Pongs, O. (2008). Regulation of excitability by potassium channels. Results Probl. Cell Differ. 44, 145–161. doi: 10.1007/400_2007_032
Poolos, N. P., and Johnston, D. (2012). Dendritic ion channelopathy in acquired epilepsy. Epilepsia 53, 32–40. doi: 10.1111/epi.12033
Poolos, N. P., and Kocsis, J. D. (1990). Dendritic action potentials activated by NMDA receptor-mediated EPSPs in CA1 hippocampal pyramidal cells. Brain Res. 524, 342–346. doi: 10.1016/0006-8993(90)90714-m
Prybylowski, K., Fu, Z., Losi, G., Hawkins, L. M., Luo, J., Chang, K., et al. (2002). Relationship between availability of NMDA receptor subunits and their expression at the synapse. J. Neurosci. 22, 8902–8910.
Pun, R. Y., Rolle, I. J., Lasarge, C. L., Hosford, B. E., Rosen, J. M., Uhl, J. D., et al. (2012). Excessive activation of mTOR in postnatally generated granule cells is sufficient to cause epilepsy. Neuron 75, 1022–1034. doi: 10.1016/j.neuron.2012.08.002
Raab-Graham, K. F., Haddick, P. C., Jan, Y. N., and Jan, L. Y. (2006). Activity- and mTOR-dependent suppression of Kv1.1 channel mRNA translation in dendrites. Science 314, 144–148. doi: 10.1126/science.1131693
Rao, A., and Steward, O. (1991). Evidence that protein constituents of postsynaptic membrane specializations are locally synthesized: analysis of proteins synthesized within synaptosomes. J. Neurosci. 11, 2881–2895.
Remy, S., Beck, H., and Yaari, Y. (2010). Plasticity of voltage-gated ion channels in pyramidal cell dendrites. Curr. Opin. Neurobiol. 20, 503–509. doi: 10.1016/j.conb.2010.06.006
Rho, J. M., Szot, P., Tempel, B. L., and Schwartzkroin, P. A. (1999). Developmental seizure susceptibility of Kv1.1 potassium channel knockout mice. Dev. Neurosci. 21, 320–327. doi: 10.1159/000017381
Ricciardi, S., Boggio, E. M., Grosso, S., Lonetti, G., Forlani, G., Stefanelli, G., et al. (2011). Reduced AKT/mTOR signaling and protein synthesis dysregulation in a Rett syndrome animal model. Hum. Mol. Genet. 20, 1182–1196. doi: 10.1093/hmg/ddq563
Rizo, J., and Südhof, T. C. (2002). Snares and Munc18 in synaptic vesicle fusion. Nat. Rev. Neurosci. 3, 641–653. doi: 10.1038/nrn898
Rizo, J., and Südhof, T. C. (2012). The membrane fusion enigma: SNAREs, Sec1/Munc18 proteins and their accomplices—guilty as charged? Annu. Rev. Cell Dev. Biol. 28, 279–308. doi: 10.1146/annurev-cellbio-101011-155818
Rizo, J., and Xu, J. (2015). The synaptic vesicle release machinery. Annu. Rev. Biophys. 44, 339–367. doi: 10.1146/annurev-biophys-060414-034057
Roche, K. W., Standley, S., McCallum, J., Dune Ly, C., Ehlers, M. D., and Wenthold, R. J. (2001). Molecular determinants of NMDA receptor internalization. Nat. Neurosci. 4, 794–802. doi: 10.1038/90498
Ronesi, J. A., Collins, K. A., Hays, S. A., Tsai, N. P., Guo, W., Birnbaum, S. G., et al. (2012). Disrupted Homer scaffolds mediate abnormal mGluR5 function in a mouse model of fragile X syndrome. Nat. Neurosci. 15, 431–440. doi: 10.1038/nn.3033
Santini, E., and Klann, E. (2011). Dysregulated mTORC1-dependent translational control: from brain disorders to psychoactive drugs. Front. Behav. Neurosci. 5:76. doi: 10.3389/fnbeh.2011.00076
Sarbassov, D. D., Ali, S. M., Sengupta, S., Sheen, J. H., Hsu, P. P., Bagley, A. F., et al. (2006). Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 22, 159–168. doi: 10.1016/j.molcel.2006.03.029
Saxton, R. A., and Sabatini, D. M. (2017). mTOR signaling in growth, metabolism, and disease. Cell 168, 960–976. doi: 10.1016/j.cell.2017.03.035
Schiller, J., Major, G., Koester, H. J., and Schiller, Y. (2000). NMDA spikes in basal dendrites of cortical pyramidal neurons. Nature 404, 285–289. doi: 10.1038/35005094
Schiller, J., and Schiller, Y. (2001). NMDA receptor-mediated dendritic spikes and coincident signal amplification. Curr. Opin. Neurobiol. 11, 343–348. doi: 10.1016/s0959-4388(00)00217-8
Schmunk, G., and Gargus, J. J. (2013). Channelopathy pathogenesis in autism spectrum disorders. Front. Genet. 4:222. doi: 10.3389/fgene.2013.00222
Schratt, G. M., Nigh, E. A., Chen, W. G., Hu, L., and Greenberg, M. E. (2004). BDNF regulates the translation of a select group of mRNAs by a mammalian target of rapamycin-phosphatidylinositol 3-kinase-dependent pathway during neuronal development. J. Neurosci. 24, 7366–7377. doi: 10.1523/JNEUROSCI.1739-04.2004
Schubert, J., Siekierska, A., Langlois, M., May, P., Huneau, C., Becker, F., et al. (2014). Mutations in STX1B, encoding a presynaptic protein, cause fever-associated epilepsy syndromes. Nat. Genet. 46, 1327–1332. doi: 10.1038/ng.3130
Schulte, U., Thumfart, J. O., Klöcker, N., Sailer, C. A., Bildl, W., Biniossek, M., et al. (2006). The epilepsy-linked Lgi1 protein assembles into presynaptic Kv1 channels and inhibits inactivation by Kvβ1. Neuron 49, 697–706. doi: 10.1016/j.neuron.2006.01.033
Scott, D. B., Michailidis, I., Mu, Y., Logothetis, D., and Ehlers, M. D. (2004). Endocytosis and degradative sorting of NMDA receptors by conserved membrane-proximal signals. J. Neurosci. 24, 7096–7109. doi: 10.1523/JNEUROSCI.0780-04.2004
Sha, L. Z., Xing, X. L., Zhang, D., Yao, Y., Dou, W. C., Jin, L. R., et al. (2012). Mapping the spatio-temporal pattern of the mammalian target of rapamycin (mTOR) activation in temporal lobe epilepsy. PLoS One 7:e39152. doi: 10.1371/journal.pone.0039152
Shah, M. M., Hammond, R. S., and Hoffman, D. A. (2010). Dendritic ion channel trafficking and plasticity. Trends Neurosci. 33, 307–316. doi: 10.1016/j.tins.2010.03.002
Smart, S. L., Lopantsev, V., Zhang, C. L., Robbins, C. A., Wang, H., Chiu, S. Y., et al. (1998). Deletion of the Kv1.1 potassium channel causes epilepsy in mice. Neuron 20, 809–819. doi: 10.1016/s0896-6273(00)81018-1
Sosanya, N. M., Brager, D. H., Wolfe, S., Niere, F., and Raab-Graham, K. F. (2015a). Rapamycin reveals an mTOR-independent repression of Kv1.1 expression during epileptogenesis. Neurobiol. Dis. 73, 96–105. doi: 10.1016/j.nbd.2014.09.011
Sosanya, N. M., Cacheaux, L. P., Workman, E. R., Niere, F., Perrone-Bizzozero, N. I., and Raab-Graham, K. F. (2015b). Mammalian target of rapamycin (mTOR) tagging promotes dendritic branch variability through the capture of Ca2+/calmodulin-dependent protein kinase II α (CaMKIIα) mRNAs by the RNA-binding protein HuD. J. Biol. Chem. 290, 16357–16371. doi: 10.1074/jbc.M114.599399
Sosanya, N. M., Huang, P. P., Cacheaux, L. P., Chen, C. J., Nguyen, K., Perrone-Bizzozero, N. I., et al. (2013). Degradation of high affinity HuD targets releases Kv1.1 mRNA from miR-129 repression by mTORC1. J. Cell Biol. 202, 53–69. doi: 10.1083/jcb.201212089
Steward, O., and Schuman, E. M. (2001). Protein synthesis at synaptic sites on dendrites. Annu. Rev. Neurosci. 24, 299–325. doi: 10.1146/annurev.neuro.24.1.299
Stocker, M. (2004). Ca2+-activated K+ channels: molecular determinants and function of the SK family. Nat. Rev. Neurosci. 5, 758–770. doi: 10.1038/nrn1516
Swiech, L., Perycz, M., Malik, A., and Jaworski, J. (2008). Role of mTOR in physiology and pathology of the nervous system. Biochim. Biophys. Acta 1784, 116–132. doi: 10.1016/j.bbapap.2007.08.015
Switon, K., Kotulska, K., Janusz-Kaminska, A., Zmorzynska, J., and Jaworski, J. (2017). Molecular neurobiology of mTOR. Neuroscience 341, 112–153. doi: 10.1016/j.neuroscience.2016.11.017
Takei, N., Inamura, N., Kawamura, M., Namba, H., Hara, K., Yonezawa, K., et al. (2004). Brain-derived neurotrophic factor induces mammalian target of rapamycin-dependent local activation of translation machinery and protein synthesis in neuronal dendrites. J. Neurosci. 24, 9760–9769. doi: 10.1523/JNEUROSCI.1427-04.2004
Tamagnini, F., Novelia, J., Kerrigan, T. L., Brown, J. T., Tsaneva-Atanasova, K., and Randall, A. D. (2015a). Altered intrinsic excitability of hippocampal CA1 pyramidal neurons in aged PDAPP mice. Front. Cell. Neurosci. 9:372. doi: 10.3389/fncel.2015.00372
Tamagnini, F., Scullion, S., Brown, J. T., and Randall, A. D. (2015b). Intrinsic excitability changes induced by acute treatment of hippocampal CA1 pyramidal neurons with exogenous amyloid β peptide. Hippocampus 25, 786–797. doi: 10.1002/hipo.22403
Taneja, P., Ogier, M., Brooks-Harris, G., Schmid, D. A., Katz, D. M., and Nelson, S. B. (2009). Pathophysiology of locus ceruleus neurons in a mouse model of Rett syndrome. J. Neurosci. 29, 12187–12195. doi: 10.1523/JNEUROSCI.3156-09.2009
Tang, B. L., Low, D. Y., and Hong, W. (1998). Syntaxin 11: a member of the syntaxin family without a carboxyl terminal transmembrane domain. Biochem. Biophys. Res. Commun. 245, 627–632. doi: 10.1006/bbrc.1998.8490
Tang, S. J., Reis, G., Kang, H., Gingras, A. C., Sonenberg, N., and Schuman, E. M. (2002). A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc. Natl. Acad. Sci. U S A 99, 467–472. doi: 10.1073/pnas.012605299
Tang, S. J., and Schuman, E. M. (2002). Protein synthesis in the dendrite. Philos. Trans. R. Soc. Lond. B Biol. Sci. 357, 521–529. doi: 10.1098/rstb.2001.0887
Tiffany, A. M., Manganas, L. N., Kim, E., Hsueh, Y. P., Sheng, M., and Trimmer, J. S. (2000). PSD-95 and SAP97 exhibit distinct mechanisms for regulating K+ channel surface expression and clustering. J. Cell Biol. 148, 147–158. doi: 10.1083/jcb.148.1.147
Trimmer, J. S. (1998). Regulation of ion channel expression by cytoplasmic subunits. Curr. Opin. Neurobiol. 8, 370–374. doi: 10.1016/s0959-4388(98)80063-9
Vacher, H., Mohapatra, D. P., Misonou, H., and Trimmer, J. S. (2007). Regulation of Kv1 channel trafficking by the mamba snake neurotoxin dendrotoxin K. FASEB J. 21, 906–914. doi: 10.1096/fj.06-7229com
Vacher, H., and Trimmer, J. S. (2011). Diverse roles for auxiliary subunits in phosphorylation-dependent regulation of mammalian brain voltage-gated potassium channels. Pflugers Arch. 462, 631–643. doi: 10.1007/s00424-011-1004-8
Valdez, A. C., Cabaniols, J. P., Brown, M. J., and Roche, P. A. (1999). Syntaxin 11 is associated with SNAP-23 on late endosomes and the trans-Golgi network. J. Cell Sci. 112, 845–854. doi: 10.1007/978-3-211-76310-0_24
van der Sluijs, P., Zibouche, M., and van Kerkhof, P. (2013). Late steps in secretory lysosome exocytosis in cytotoxic lymphocytes. Front. Immunol. 4:359. doi: 10.3389/fimmu.2013.00359
Villasana, L. E., Klann, E., and Tejada-Simon, M. V. (2006). Rapid isolation of synaptoneurosomes and postsynaptic densities from adult mouse hippocampus. J. Neurosci. Methods 158, 30–36. doi: 10.1016/j.jneumeth.2006.05.008
Vissel, B., Krupp, J. J., Heinemann, S. F., and Westbrook, G. L. (2001). A use-dependent tyrosine dephosphorylation of NMDA receptors is independent of ion flux. Nat. Neurosci. 4, 587–596. doi: 10.1038/88404
Vlaskamp, D. R., Rump, P., Callenbach, P. M., Vos, Y. J., Sikkema-Raddatz, B., van Ravenswaaij-Arts, C. M., et al. (2016). Haploinsufficiency of the STX1B gene is associated with myoclonic astatic epilepsy. Eur. J. Paediatr. Neurol. 20, 489–492. doi: 10.1016/j.ejpn.2015.12.014
Waung, M. W., Pfeiffer, B. E., Nosyreva, E. D., Ronesi, J. A., and Huber, K. M. (2008). Rapid translation of Arc/Arg3.1 selectively mediates mGluR-dependent LTD through persistent increases in AMPAR endocytosis rate. Neuron 59, 84–97. doi: 10.1016/j.neuron.2008.05.014
Weston, M. C., Chen, H., and Swann, J. W. (2012). Multiple roles for mammalian target of rapamycin signaling in both glutamatergic and GABAergic synaptic transmission. J. Neurosci. 32, 11441–11452. doi: 10.1523/JNEUROSCI.1283-12.2012
Weston, M. C., Chen, H., and Swann, J. W. (2014). Loss of mTOR repressors Tsc1 or Pten has divergent effects on excitatory and inhibitory synaptic transmission in single hippocampal neuron cultures. Front. Mol. Neurosci. 7:1. doi: 10.3389/fnmol.2014.00001
Wong, M. (2014). mTOR strikes again: mTORC1 activation causes epilepsy independent of overt pathological changes. Epilepsy Curr. 14, 41–43. doi: 10.5698/1535-7597-14.1.41
Workman, E. R., Haddick, P. C., Bush, K., Dilly, G. A., Niere, F., Zemelman, B. V., et al. (2015). Rapid antidepressants stimulate the decoupling of GABAB receptors from GIRK/Kir3 channels through increased protein stability of 14–3-3η. Mol. Psychiatry 20, 298–310. doi: 10.1038/mp.2014.165
Workman, E. R., Niere, F., and Raab-Graham, K. F. (2013). mTORC1-dependent protein synthesis underlying rapid antidepressant effect requires GABABR signaling. Neuropharmacology 73, 192–203. doi: 10.1016/j.neuropharm.2013.05.037
Yokoi, N., Fukata, M., and Fukata, Y. (2012). Synaptic plasticity regulated by protein-protein interactions and posttranslational modifications. Int. Rev. Cell Mol. Biol. 297, 1–43. doi: 10.1016/B978-0-12-394308-8.00001-7
Yu, Y. E., Wen, L., Silva, J., Li, Z., Head, K., Sossey-Alaoui, K., et al. (2010). Lgi1 null mutant mice exhibit myoclonic seizures and CA1 neuronal hyperexcitability. Hum. Mol. Genet. 19, 1702–1711. doi: 10.1093/hmg/ddq047
Yuan, L. L., and Chen, X. (2006). Diversity of potassium channels in neuronal dendrites. Prog. Neurobiol. 78, 374–389. doi: 10.1016/j.pneurobio.2006.03.003
Yuste, R. (1997). Potassium channels. Dendritic shock absorbers. Nature 387:851, 853. doi: 10.1038/43063
Zeng, L. H., Rensing, N. R., and Wong, M. (2009). The mammalian target of rapamycin signaling pathway mediates epileptogenesis in a model of temporal lobe epilepsy. J. Neurosci. 29, 6964–6972. doi: 10.1523/JNEUROSCI.0066-09.2009
Zhu, J., Watanabe, I., Poholek, A., Koss, M., Gomez, B., Yan, C., et al. (2003). Allowed N-glycosylation sites on the Kv1.2 potassium channel S1–S2 linker: implications for linker secondary structure and the glycosylation effect on channel function. Biochem. J. 375, 769–775. doi: 10.1042/bj20030517
Keywords: mTOR, syntaxin, glutamate receptors, ion channels, neurological disorders
Citation: Niere F and Raab-Graham KF (2017) mTORC1 Is a Local, Postsynaptic Voltage Sensor Regulated by Positive and Negative Feedback Pathways. Front. Cell. Neurosci. 11:152. doi: 10.3389/fncel.2017.00152
Received: 31 January 2017; Accepted: 09 May 2017;
Published: 30 May 2017.
Edited by:
Daniel Johnston, University of Texas at Austin, United StatesReviewed by:
Charles Albert Hoeffer, University of Colorado Boulder, United StatesDax Hoffman, National Institutes of Health, United States
Copyright © 2017 Niere and Raab-Graham. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kimberly F. Raab-Graham, a3JhYWJncmFAd2FrZWhlYWx0aC5lZHU=