 Michal Vascak
Michal Vascak Jianli Sun
Jianli Sun Matthew Baer
Matthew Baer Kimberle M. Jacobs
Kimberle M. Jacobs John T. Povlishock
John T. Povlishock- Department of Anatomy and Neurobiology, Virginia Commonwealth University School of Medicine, Richmond, VA, United States
The axon initial segment (AIS) is the site of action potential (AP) initiation, thus a crucial regulator of neuronal activity. In excitatory pyramidal neurons, the high density of voltage-gated sodium channels (NaV1.6) at the distal AIS regulates AP initiation. A surrogate AIS marker, ankyrin-G (ankG) is a structural protein regulating neuronal functional via clustering voltage-gated ion channels. In neuronal circuits, changes in presynaptic input can alter postsynaptic output via AIS structural-functional plasticity. Recently, we showed experimental mild traumatic brain injury (mTBI) evokes neocortical circuit disruption via diffuse axonal injury (DAI) of excitatory and inhibitory neuronal systems. A key finding was that mTBI-induced neocortical electrophysiological changes involved non-DAI/ intact excitatory pyramidal neurons consistent with AIS-specific alterations. In the current study we employed Thy1-yellow fluorescent protein (YFP)-H mice to test if mTBI induces AIS structural and/or functional plasticity within intact pyramidal neurons 2 days after mTBI. We used confocal microscopy to assess intact YFP+ pyramidal neurons in layer 5 of primary somatosensory barrel field (S1BF), whose axons were continuous from the soma of origin to the subcortical white matter (SCWM). YFP+ axonal traces were superimposed on ankG and NaV1.6 immunofluorescent profiles to determine AIS position and length. We found that while mTBI had no effect on ankG start position, the length significantly decreased from the distal end, consistent with the site of AP initiation at the AIS. However, NaV1.6 structure did not change after mTBI, suggesting uncoupling from ankG. Parallel quantitative analysis of presynaptic inhibitory terminals along the postsynaptic perisomatic domain of these same intact YFP+ excitatory pyramidal neurons revealed a significant decrease in GABAergic bouton density. Also within this non-DAI population, patch-clamp recordings of intact YFP+ pyramidal neurons showed AP acceleration decreased 2 days post-mTBI, consistent with AIS functional plasticity. Simulations of realistic pyramidal neuron computational models using experimentally determined AIS lengths showed a subtle decrease is NaV1.6 density is sufficient to attenuate AP acceleration. Collectively, these findings highlight the complexity of mTBI-induced neocortical circuit disruption, involving changes in extrinsic/presynaptic inhibitory perisomatic input interfaced with intrinsic/postsynaptic intact excitatory neuron AIS output.
Introduction
Traumatic brain injury (TBI) constitutes a major global healthcare problem that extracts a devastating personal and societal toll (Langlois et al., 2006). Historically, TBI research focused on severe head injuries associated with high-speed motor vehicle incidents. In the last decade, the incidence of severe TBI decreased dramatically (Thurman et al., 1999; Coronado et al., 2011), with milder forms of TBI elicited by contact sports and blast waves during combat now predominating (Borg et al., 2004; Cassidy et al., 2004; Styrke et al., 2007; Hoge et al., 2008). Mild TBI (mTBI) is frequently termed the “silent epidemic” because of its relatively subtle nature typically occurring without any evidence of macroscopic brain damage such as contusion or hematoma formation (Alexander, 1995; Povlishock and Katz, 2005). However, recent findings in patients with mTBI linking acute cognitive network dysfunction with potential life-long morbidity have captured the attention of media and raised public concern (Mannix et al., 2016). Despite the focus of research shifting to mTBI (CDC, 2003), its cellular and physiological substrates remain controversial (Shaw, 2002; Büki and Povlishock, 2006; Cohen et al., 2007; Andriessen et al., 2010; Johnson et al., 2013).
Using advanced structural neuroimaging of patients with TBI ranging from mild-to-severe, multiple groups have demonstrated that diffuse axonal injury (DAI) occurs within various subcortical white matter (SCWM) tracts and callosal domains (Salmond et al., 2006; Bazarian et al., 2007; Mayer et al., 2010; Kinnunen et al., 2011). These findings led to the inference that white matter DAI causes cortical network dysfunction that underlies acute and/or chronic cognitive impairment (Bonnelle et al., 2011; Sharp et al., 2011, 2014; Johnson et al., 2013). However, other groups using the same imaging tools have not confirmed this finding in well-controlled studies of patients with mTBI and clinically significant cognitive dysfunction (Zhang et al., 2010; Mac Donald et al., 2011; Ilvesmäki et al., 2014; Wäljas et al., 2014). In fact, some reports suggest the potential for primary neocortical gray involvement (Newcombe et al., 2011; Bouix et al., 2013; Ling et al., 2013), wherein local information processing relies on balanced excitatory and inhibitory unitary neuronal activity to support function of large-scale distributed neurocognitive networks (Mountcastle, 1979; Mesulam, 1990; Douglas et al., 1995; Varela et al., 2001; Hasenstaub et al., 2005; Raichle, 2010). Several multi-modal studies using functional MRI and electrophysiological approaches have reported local neocortical network dysfunction following mTBI, supporting this premise (Mayer et al., 2011; Sponheim et al., 2011; Tremblay et al., 2011; Bashir et al., 2012; Huang et al., 2017; Palacios et al., 2017).
Although these issues are extremely difficult to rigorously assess in humans, our laboratory critically evaluated the potential for mTBI-induced neocortical change in a well-controlled animal model. Specifically, following experimental mTBI using transgenic mice, we observed primary neocortical damage involving scattered DAI within a subset of layer 5 pyramidal neurons expressing a yellow variant of green fluorescent protein (YFP) labeling the soma as well as their dendrites and axons (Greer et al., 2011). Perhaps even more biologically significant was our parallel observation that the non-DAI (intact) YFP+ neuronal population showed altered electrophysiological properties without any evidence of overt structural damage (Greer et al., 2012; Hånell et al., 2015a; Sun and Jacobs, 2016). Over time, these same intact YFP+ pyramidal neurons became hyperexcitable, entirely consistent with some form of mTBI-induced circuit disruption within local neocortical networks (Zhang and Raichle, 2010; Wolf and Koch, 2016).
At present, the neurobiological basis for this hyperexcitability in this intact neuronal population remains unknown. While several different mechanisms can alter neuronal excitability, our observations are most consistent with structural-functional changes within the axon initial segment (AIS), the site of action potential (AP) generation (Stuart et al., 1997; Palmer and Stuart, 2006). The AIS is a critical subdomain regulating neuronal excitability by integrating synaptic input to determine whether to fire an AP, thereby acting as a gatekeeper of neuronal output (Bender and Trussell, 2012; Kole and Stuart, 2012). Specifically, the high density of voltage-gated sodium channels (NaV) at the distal AIS sets the threshold for AP generation (Kole et al., 2008; Hu et al., 2009; Popovic et al., 2011). Ankyrin-G (ankG), a widely used AIS marker, is the master structural organizer that clusters ion channels via linkage to the subaxolemmal cytoskeleton (Zhou et al., 1998; Jenkins and Bennett, 2001; Yang et al., 2007). In sensory circuits, presynaptic input fine-tunes AIS structure (Kuba, 2012; Gutzmann et al., 2014; Kuba et al., 2014) altering neuronal excitability (Kuba et al., 2010). Because pyramidal neuron AIS function is strongly regulated by perisomatic GABAergic synaptic transmission from parvalbumin (PV)-expressing interneurons (Cobb et al., 1995; Pouille and Scanziani, 2001; Freund, 2003; Klausberger et al., 2003; Klausberger and Somogyi, 2008; Cardin et al., 2009; Atallah et al., 2012; Pouille et al., 2013; Lazarus et al., 2015; Wefelmeyer et al., 2015), concomitant involvement of these inhibitory inputs could potentially induce changes in AIS structure-function after mTBI (Buffington and Rasband, 2011; Baalman et al., 2013).
To address these AIS-related issues, the current report builds upon our previous studies that utilized YFP-expressing transgenic mice in a well-characterized model of mTBI to examine the potential for changes in layer 5 pyramidal neuron AIS and its associated perisomatic domain, both of which are critical for regulating neuronal excitability. Specifically, to assess AIS structural plasticity, immunohistochemical approaches targeting ankG and NaV1.6 distribution were quantitatively assessed in the early stage post-mTBI using confocal microscopy. These assessments of AIS structural plasticity in non-DAI/intact layer 5 pyramidal neurons were also accompanied by parallel morphological studies examining presynaptic GABAergic bouton density. Lastly, these imaging studies were interfaced with patch-clamp electrophysiology and computational modeling of layer 5 pyramidal neurons to establish a functional match with the AIS structural plasticity posited above. Using these multifaceted approaches, we show for the first time that mTBI evokes structural plasticity at the distal AIS, occurring independent of detectable changes in sodium channel distribution, in a specific subset of neocortical neurons with intact axons. These same events within intact neurons were also recognized to occur in concert with PV+ GABAergic terminal loss at their perisomatic domain. Lastly, the observed intrinsic/postsynaptic (AIS) and extrinsic/presynaptic (perisomatic) structural plasticity within intact neurons were also accompanied by a reduction in AP acceleration, likely reflecting a change in NaV1.6 density. Collectively, these findings reshape our understanding of mTBI and have major implications as we continue to better understand the brain’s response to milder injury in terms of the functionality of its intrinsic networks within neocortex.
Materials and Methods
Animal maintenance and experimental protocols were approved by the Virginia Commonwealth University Institutional Animal Care and Use Committee and complied with principles in the National Research Council Guide for the Care and Use of Laboratory Animals: 8th Edition.
Breeding and Genotyping of YFP-H Transgenic Mice
The Thy1-YFP-H line [B6Cg-TgN(Thy1-YFP-H)2Jrs, stock number 003782] was obtained from Jackson Laboratories (Bar Harbor, ME) and maintained as heterozygotes upon a C57BL/6J background. Inheritance of the fluorescent transgene in these was determined from an ear punch taken at weaning (~21 days). The tissue from the ear punch was mounted on a glass slide and examined using a FITC filter on an Olympus DP71 digital camera (Olympus, Center Valley, PA, USA) where YFP expression could easily be identified in mice that inherited the transgene (YFP-H mice). Transgene expression in these mice is under the control of the neuronal specific Thy1 promoter, resulting in YFP expression within the neocortex that is primarily restricted to layer 5 pyramidal neurons (Feng et al., 2000).
Experimental Design
This study used a total of 18 young adult (age: 6–10 weeks; weight: 20–24 g) male mice randomly assigned1 to either sham-injury (control) or mTBI (experimental) groups. Based on our previous work showing mTBI consistently generates DAI within primary somatosensory barrel field (S1BF), we assessed layer 5 intact YFP+ pyramidal neurons for AIS structural-functional changes at 2 days postinjury within this well-characterized region of neocortical gray matter (Greer et al., 2011, 2013; Hånell et al., 2015b). For confocal microscopy studies, a total of 11 mice (age 8–10 weeks) were surgically prepared for sham-injury (n = 5) or mTBI (n = 6) and perfused for immunohistochemical labeling of tissue sections. To identify the AIS we fluorescently labeled ankG, the master scaffolding protein that is widely used as a surrogate marker for AIS position and length (Hedstrom et al., 2008; Grubb and Burrone, 2010; Gutzmann et al., 2014). Additionally, we targeted NaV1.6, which sets the threshold for AP generation (Kole et al., 2008), to structurally assess the membrane channels underpinning AIS function/output. In addition to intrinsic/postsynaptic AIS structural plasticity within intact YFP+ pyramidal neurons, we assessed their perisomatic domain for extrinsic/presynaptic changes in GABAergic bouton density. To probe for any AIS functional plasticity correlated with potential structural changes we recorded intrinsic electrophysiological data from intact YFP+ pyramidal neurons in layer 5 of S1BF within ex vivo slice preparations from a total of 7 mice (age 6–8 weeks) at 2 days following sham-injury (n = 3) or mTBI (n = 4). Lastly, we determined the functional consequences of any potential AIS structural changes using a realistic computational model of a layer 5 pyramidal neuron based on experimentally derived properties (Hallermann et al., 2012).
Surgical Preparation and Central Fluid Percussion Injury
To model mTBI we used midline central fluid percussion injury (cFPI) first described by Dixon and associates using rats (Dixon et al., 1987), which our lab modified for mice as described previously (Greer et al., 2011). Briefly, anesthetized mice were surgically prepared for cFPI induction by installing a hub surrounding a craniectomy centered on the superior sagittal suture, midway between bregma and lambda. Intraoperative rectal temperature was maintained at 37 ± 0.2°C using a thermostatically controlled heating pad (Harvard Apparatus). Additionally, heart rate beats per minute (BPM), respiratory rate (RPM) and arterial blood oxygenation (SpO2) were monitored using a thigh-clamp pulse oximeter sensor (MouseOx; STARR Life Sciences) to ensure maintenance of physiological homeostasis. After a post-operative recovery (~1.5 h), mice were re-anesthetized and then connected to the fluid percussion apparatus (Custom Design and Fabrication, Virginia Commonwealth University) forming a closed mechanical system. Releasing the pendulum, striking the piston in the fluid-filled cylinder generated a mild pressure wave (~12 ms) that was delivered onto the intact dura. This action simulates human brain inertial loading during trauma-induced rapid acceleration-deceleration causing a mild diffuse brain injury (Dixon et al., 1987). The mean ± standard error of the mean (SEM) of the peak amplitude of the pressure wave (1.6 ± 0.03 atmospheres, n = 10 mice) was measured by a transducer and displayed on an oscilloscope (Tektronix TDS 210). For sham-injury, an identical procedure was used with the exception of the pendulum’s release. Mice were disconnected from the apparatus immediately postinjury and visually monitored while removing the hub, suturing the incision and checking reflexes. None of the mice showed signs of seizure or apnea. Severity of injury (peak amplitude) and duration of loss of righting reflex, a rodent behavioral surrogate of loss of consciousness, were recorded for each animal (Grimm et al., 2015). We determined the degree of mTBI by comparing loss of righting reflex duration with shams (Morehead et al., 1994). After recovering from loss of righting reflex, animals were transferred to a warmed cage to maintain normothermia and monitored before returned to the vivarium.
Perfusion and Tissue Processing
Mice received a lethal dose of sodium pentobarbital (1.6 mg/g IP) 2 days postinjury. After loss of pain reflexes mice were transcardially perfused, first with heparinized (10 units/ml) saline for 1 min then 4% paraformaldehyde in Millonig’s buffer pH 7.4 for 20 min. Brains were dissected and then sectioned coronally at 60 μm using a vibratome (Leica VT1000S). Sections directly below the craniectomy (bregma level −0.6 to −2.4 mm) were collected in 24-well plates filled with Millonig’s buffer pH 7.4. To quantitatively assess for intrinsic AIS and extrinsic/synaptic perisomatic structural plasticity, for each animal, we labeled ankG, NaV1.6 and double-labeled glutamate decarboxylase-67 (GAD67) with PV in sections taken from a randomly selected and two adjacent wells, respectively, containing caudal S1BF (bregma level −1.5 to −2.0). This was done because of the consistency with which cFPI generates DAI within this well characterized area of neocortex, which was also the rational for choosing this region-of-interest (ROI) in our previously published electrophysiological studies (Greer et al., 2012; Hånell et al., 2015a; Sun and Jacobs, 2016).
Immunohistochemistry
Free-floating sections were rinsed with phosphate buffered saline (PBS). Heat-induced epitope retrieval was performed by incubating sections in 10 mM sodium citrate buffer pH 8.5 for 10 min in an 80°C water bath (Jiao et al., 1999). After cooling to room temperature sections were rinsed with PBS then incubated for 1 h at room temperature with 10% normal goat serum, 2% fish skin gelatin and 0.5% Triton X-100 in PBS. To mask any potential endogenous mouse immunoglobulin, the blocking buffer was supplemented with Mouse-on-Mouse reagent (Vector Laboratories, MKB-2213). Then, sections were rinsed with 1% normal goat serum, 1% fish skin gelatin, and 0.5% Triton X-100 in PBS (working buffer). Primary antibody solutions were prepared by dilution with working buffer and the sections were incubated overnight at 4°C with agitation. Specifically, we used monoclonal antibodies against ankG (1:500; mouse IgG2a, clone N106/36; NeuroMab), NaV1.6 (1:200; mouse IgG1, clone K87A/10; NeuroMab), PV (1:2000; mouse IgG1; Swant, PV235) and GAD67 (1:1000; mouse IgG2a, clone 1G10.2; Millipore, MAB5406). For qualitative double-labeled overview images, we used a polyclonal antibody against NaV1.6 (1:200; Alomone). The following day, sections were rinsed with working buffer and incubated with isotype-specific goat-derived secondary antibodies conjugated to Alexa Fluor 568 for quantitative analyses and a combination of Alexa Fluor 568 and 633 for qualitative colocalization (1:500: ThermoFisher Scientific) for 2 h at room temperature. After final rinses using working buffer then PBS, sections were mounted on glass slides and cover-slipped using non-hardening Vectashield with DAPI (Vector Laboratories, H-1200) to prevent shrinkage and preserve morphology of tissue.
The AIS quantitative analysis on the YFP+ background used separate single-labeled sections opposed to double-labeled single sections in an effort to match the fluorescent quantum yield and stoichiometry via monoclonal primary antibodies to immunolabel ankG and NaV1.6, which were both visualized using Alexa Fluor 568 (Lichtman and Conchello, 2005). In the same vein, mouse immunoglobulin isotype-specific secondary antibodies were used in all studies, which also optimized the signal-to-noise ratio (Manning et al., 2012). Parallel control studies were conducted to ensure both primary and secondary antibody fidelity (Lorincz and Nusser, 2008b). In all cases, primary antibody omission abolished immunoreactivity. Secondary antibody specificity evaluated via cross-reactivity showed no signal between all possible primary × secondary host and/or isotype combinations.
Confocal Microscopy
Image acquisition was performed using a laser-scanning confocal microscope (LSM 710, Carl Zeiss). Since sham vs. mTBI groups could be readily differentiated based on YFP+ profiles, we used DAPI visualized under epifluorescence to center the stage over the ROI. In this way, the investigator was blinded from the experimental/dependent variable channel, thus adhering to stereological principles including random sampling. Using a 10× objective (low-power) the field-of-view (FOV) was centered over the S1BF region along the dorsolateral edge of the hippocampus. We used continuous laser scanning to guide rotation of the FOV until it was orthogonal to the underlying SCWM. Images were acquired with optimal Nyquist sampling using Plan-Apochromat 10×/0.45 NA (XY = 0.41 μm/pixel; Z = 5.8 μm), 20×/0.8 NA (XY = 0.152 μm/pixel; Z = 0.94 μm), 40×/1.4 (XY = 0.094 μm/pixel; Z = 0.52 μm) and 63×/1.2 NA (XY = 0.088 μm/pixel) oil immersion objective lenses. All multichannel images were acquired using sequential scanning at the lowest possible laser power to avoid crosstalk (488 Argon, 561 DPSS and 633 HeNe). All images for quantitative analysis were acquired using identical laser settings per channel across samples. Gain and offset were adjusted for optimal signal range. The pinhole was set to 1.0 Airy unit for the red channel (i.e., ankG, NaV1.6 and GAD67). To maintain identical optical slice thickness with respect to the red channel, in multichannel z-stack images the green (YFP) and far-red (PV) the pinhole ranged from 0.8 to 1.2 Airy units.
Intact YFP+ Pyramidal Neuron Sampling
Within the 10× overview images we measured ever YFP+ pyramidal neuron that met our inclusion criteria. Specifically, intact neurons were defined by YFP+ axons that were continuous from the soma of origin to the SCWM interface. YFP+ that were transected during tissue processing were excluded in both sham and mTBI samples. Since AIS structure and composition vary even within a particular neuronal subtype (Kuba, 2012; King et al., 2014), we set a priori conditions to control for any potential confounds. Specifically, layer 5 is subdivided into layer 5a and 5b, which are populated by multiple subtypes of pyramidal neurons with different morphological and physiological properties, as well as projections to different anatomical regions of the brain (Chagnac-Amitai et al., 1990; Schubert et al., 2006; Hattox and Nelson, 2007). To control for potential confounding of layer 5 pyramidal neurons based on neocortical depth, for each AIS sample we also measured the distance from the axo-somatic vertex to where YFP+ profiles entered SCWM, indicated by the sharp change in trajectory. Additionally, we excluded pyramidal neurons with axons emerging from basal dendrites because of their unique intrinsic and synaptic properties (Thome et al., 2014; Hamada et al., 2016). Overall, the number of intact YFP+ pyramidal neurons meeting our inclusion criteria ranged from 5 to 14 per section. This data set met the recommended minimum of five measurements per animal to obtain a robust and unbiased estimates of variance using multilevel modeling described below (Walsh, 1947; Maas and Hox, 2004; Galbraith et al., 2010). Further, the Wilcoxon test showed sample sizes between sham and mTBI groups were similar for both ankG (sham: n = 53 AIS from 5 mice; mTBI: n = 54 AIS from 6 mice; X2 = 0.079, p = 0.7782) and NaV1.6 (sham: n = 54 AIS from 5 mice; mTBI: n = 44 AIS from 6 mice; X2 = 3.36, p = 0.0666) analyses of AIS structure. These samples sizes allowed us to detect a 1.5 μm (5%–10%) change in AIS end position with 80% power (G*Power; Franz Faul, Kiel University) based on mean ± SD sham ankG end position (25.5 ± 2.94 μm; n = 50 AIS from 3 mice) determined from pilot experiments. For perisomatic GABAergic bouton density, sample sizes between sham (n = 48 somas from 5 mice) and mTBI (n = 50 somas from 5 mice; one section lost during processing) groups were also similar (X2 = 0.1, p = 0.7518; Wilcoxon test).
Quantification of AIS
To capture ankG and NaV1.6 fluorescent profiles along YFP+ intact pyramidal neurons, a 40× objective at 3× zoom (760 × 760 pixels) in z-stacks (mean 4.5 μm, range 2.7–5.3 μm). Because of the diffuse nature of YFP+ pathology following mTBI, these constrained images allowed tracing the YFP+ axon through and past the AIS region in a blinded fashion. Further, we set a limit of 10 optical slices to minimize error caused by tilted AIS orientation in the z-plane. AIS measurements were performed using a previously described method (Grubb and Burrone, 2010; Evans et al., 2013). Specifically, z-stacks were collapsed into a single maximum intensity projections that were imported into MATLAB software (MathWorks) for analyses using custom-written functions (Matthew Grubb and Thomas Watkins, King’s College London, UK; freely available at www.mathworks.com/matlabcentral/fileexchange/28181-ais-quantification). While visualizing only the YFP channel, an axonal profile was traced starting at the edge of the soma, continuing distally along the axon, through and past the region of the AIS. Immunofluorescent profiles of ankG/NaV1.6 were then superimposed on traces of YFP+ axons. At each pixel along this profile, fluorescent intensity values were averaged over a 3 × 3 pixel square centered on the pixel of interest. Averaged profile values were then smoothed using a 40-point (~5 mm) sliding mean and normalized between 1 (maximum smoothed fluorescence) and 0 (minimum smoothed fluorescence). With respect to the soma edge, delineated at the axo-somatic vertex, ankG and NaV1.6 proximal (start) and distal (end) positions were obtained at points where the profiles reached thresholds of 0.33 and 0.5 relative to maximum fluorescence, respectively. These optimal threshold values were determined empirically and varying ± 0.2 did not change the overall pattern of results.
Quantification of Perisomatic Bouton Density
The total number of GAD67+ and GAD67+/PV+ puncta were quantified along the perisomatic domain of YFP+ intact pyramidal neurons within layer 5 of S1BF. Single optical slices (0.9 μm thick) were captured using a 63× objective at 3× zoom (512 × 512 pixels, 45 μm × 45 μm; resolution = 0.088 μm/pixel) using sequential scanning and a single laser intensity for each channel for all samples. Confocal images were imported to Fiji (a distribution of ImageJ) then processed and analyzed using custom written macros for automated analyses. YFP images were converted into binary images to segment the pyramidal neuron profile, which was then dilated and eroded to generate a 2 μm thick band approximating the perimeter/perisomatic domain. Quantification of puncta within this band was performed using both gray scale and segmented/binary images. Specifically, perisomatic GAD67+ puncta in gray scale (8-bit) images were quantified using the “Find Maxima” function. To quantify perisomatic GAD67+/PV+ puncta, profiles in each channel were segmented from background subtracted 8-bit images by converting into binary using a minimum gray-value threshold. Varying the threshold ±10 gray-values did not change the overall pattern of results. GAD67+/PV+ colocalized populations were isolated using the “Image Calculator” function. The “Particle Analysis” function with appropriate size and shape exclusion filters was used to determine the total number of objects per unit area (FOV). GAD67+ (PV+) puncta within this band were quantified and normalized by dividing by the area of the band to determine perisomatic bouton density, summarized as puncta per 100 μm2.
Computational Modeling
Simulations in Neuron v7.1 used a previously published realistic model of neocortical layer 5 pyramidal neuron AP initiation (Hallermann et al., 2012). The model was based on Neurolucida reconstructions using ion channel properties determined from experimental recordings. Simulations were executed using default setting with the exception of AIS ion channel densities and length modifications based on our experimental observations. Specifically, the default AIS length (63 μm) in this model was ~60% longer than our experimental measurements. To compare the effect of AIS length using our experimentally observed values (~24–26 μm), the peak NaV1.6 density at the distal AIS was increased proportionally from the default value of 7000–11,200 pS μm−2. Importantly, this control NaV1.6 density remained within the realistic estimates reported by Hallermann et al. (2012). Default current-clamp parameters were used to evoke an AP within 5 ms of injection. Overall, simulations using our adjusted AIS parameters resulted in AP waveforms that were qualitatively similar to those produced using default settings and also our electrophysiological recordings. This allowed us to investigate the kinetics (i.e., acceleration; described below) of the AP at the AIS in isolation from the AP at the soma. Specifically, we compared the effect of changing AIS length using experimentally determined values (i.e., sham vs. mTBI) and NaV1.6 density on AP acceleration. Note, in these layer 5 pyramidal neuron simulations, AIS length was modulated at the distal position with respect to the soma of origin. Additionally, changing length or peak NaV1.6 density at the distal AIS did not change the distribution ratio of ion channels.
Electrophysiology
In addition to the above, a separate cohort of sham-injured (n = 3) and mTBI (n = 4) mice were anesthetized with isoflurane and decapitated for quick brain removal 2 days postinjury. The brains were immediately chilled in ice-cold oxygenated sucrose-modified artificial cerebral spinal fluid (aCSF) slicing solution (mM: 2.5 KCl, 10 MgSO4, 0.5 CaCl2, 1.25 NaH2PO4, 234 sucrose, 11 glucose and 26 NaHCO3). Using a vibratome (VT 1200, Leica Microsystems) brains were coronally sectioned at 300 μm and then incubated for 30–45 min at 34°C in an oxygenated aCSF (mM: 126 NaCl, 3.5 KCl, 1 MgSO4, 1.2 CaCl2, 1.25 NaH2PO4, 10 glucose and 26 NaHCO3). Thereafter, slices remained at room temperature until placed in the recording chamber maintained at 32 ± 0.5°C.
As previously described (Sun and Jacobs, 2016), whole-cell patch-clamp recordings were performed under infrared Dodt contrast microscopy (Zeiss AxioExaminer). A 60× water-immersion objective was used to visually identify YFP+ layer 5 pyramidal neurons of S1BF with axons descending from the soma into the white matter (intact) or ending with an axonal swelling (DAI), deep to the surface of the slice to avoid those transected by the vibratome. We have shown previously that these morphologies are easily identified in the living slice for YFP+ layer 5 pyramidal neurons (Greer et al., 2012). Consistent with our structural assessments, only these intact YFP+ pyramidal neurons were recorded in both sham and mTBI slices. Additionally, all layer 5 YFP+ samples had an apical dendrite, a characteristic morphological feature of pyramidal neurons. The slices were continuously perfused with aCSF solution saturated with 95% O2 and 5% CO2. Patch electrodes (final resistances, 2–4 MΩ) were pulled from borosilicate glass (World Precision Instruments) on a horizontal Flaming-Brown microelectrode puller (Model P-97, Sutter Instruments). The intracellular solution contained (in mM): 130 K-gluconate, 10 Hepes, 11 EGTA, 2.0 MgCl2, 2.0 CaCl2, 4 Na-ATP and 0.2 Na-GTP. Electrode capacitance was electronically compensated. Data were acquired and digitized at 200 kHz using a MultiClamp 700B amplifier and Digidata 1440A with pClamp software, respectively (Molecular Devices). Whole-cell patch-clamp was approached under voltage-clamp mode at −65 mV. After 2–5 min stabilization, APs were recorded in current-clamp mode to obtain intrinsic property measurements. Pipette Capacitance Neutralization was set at 9.6 pF, Bessel Filter at bypass, auto Bridge Balance, and Gain at one. APs were evoked with 10 depolarizing steps (30 ms) beginning with a 100 pA step, and increased by 10 pA every 5 s while neurons were maintained at −60 mV. Access resistance was continuously monitored and rechecked after each recording. If the series resistance increased by 20% at any time, the recording was terminated. Previously we showed that there was no significant difference between the results for naïve and sham animals (Greer et al., 2012). Despite this similarity, for added rigor the control group contained only age- and survival time-matched sham-injured animals. Additionally, passive membrane properties (mean ± standard deviation; SD) compared using a t test showed no statistical differences in capacitance (pF: sham = 196 ± 46.5, mTBI = 176 ± 28.4; t25 = −1.35, p = 0.1882), input resistance (MΩ: sham = 67 ± 21.5, mTBI = 79 ± 25.1; t25 = 1.28, p = 0.2111), access resistance (MΩ: sham = 9.2 ± 3.21, mTBI = 9.11 ± 3.06; t25= −0.07, p = 0.9409), or resting membrane potential (mV: sham = −69.8 ± 3.03, mTBI = −70.7 ± 5.79; t25 = −0.49, p = 0.6298) between sham (n = 13 cells from 3 mice) and mTBI (n = 14 cells from 4 mice) groups.
In this report, we use a newly implemented protocol for intrinsic property analysis that allows us to measure the first AP on each sweep (5 sweeps per cell). To assess AP kinetics, we applied three successive seven-point boxcar filters to the membrane voltage, and then calculated the first and then second derivative, which yielded two peaks. Specifically, the first and second peak in the plot of the second derivative of the membrane voltage corresponded to AP acceleration at the AIS and soma, respectively (Khaliq and Raman, 2006; Meeks and Mennerick, 2007). All recorded cells from sham and mTBI slices resulted in two peaks, and their amplitudes were measured for each of five APs per cell. For statistical analyses, the first AP on each of the five sweeps was nested within each cell. There were two cells in sham slices where only four sweeps were recorded. In total, we recorded and analyzed 63 AP from sham and 70 AP from mTBI mice. The number of recorded cells per animal, (range = 2–6) was similar (X2 = 0.5283, p = 0.4673) between sham (n = 3 mice) and mTBI (n = 4 mice). For statistical analysis using multilevel modeling described below, each neuron had and additional nested set of five APs.
Statistics
Statistical analysis of data sets was performed using JMP Pro version 12.2.0 (a distribution of SAS). Data sets were assessed for normality using quantile (QQ) plots and the Shapiro-Wilk test. For all analyses, the variance in data between sham and mTBI groups was not significantly different (p > 0.05, Brown-Forsythe test). Physiology data and loss of righting reflex were statistically analyzed using unpaired t tests. Statistical analyses (described below) tested for covariance of distance from SCWM with AIS measurements. If covariance was significant, AIS measurements were leveraged with respect to distance from SCWM. Non-leveraged vs. leveraged AIS measurements were statistically assessed for significant difference using the Wilcoxon signed-rank test. For this test, the statistical unit corresponded to the average difference (non-leveraged−leveraged) in AIS measurements per animal.
To determine statistically significant differences among intact YFP+ pyramidal neuron outcome variables (distance from SCWM, AIS structure, perisomatic bouton density and AIS function) between sham-injury and mTBI groups, we employed multilevel modeling (Nieuwenhuis et al., 2011; Button et al., 2013; Aarts et al., 2014). For statistical analysis, we constructed a multilevel model using an experimental condition at the animal level (sham-injury or mTBI), where the outcome variable (e.g., AIS length) consists of multiple measurements per animal (i.e., nested data). In other words, each animal had nested data consisting of a series of measurements. Because some variability is expected in the average outcome measure as a function of each animal, the data from each animal are not independent (Nieuwenhuis et al., 2011). Thus, failure to account for such dependent data can inflate the Type I error rate (α) above the standard nominal value of 0.05 (Snijders and Bosker, 1993). Multilevel modeling accounts for this within-animal dependence on the outcome measure (e.g., AIS length) and thus preserves the true Type I error rate for the statistical test (Walsh, 1947; Galbraith et al., 2010), which we have set to α = 0.05.
Statistical analysis using multilevel modeling performs linear regressions in a sequential fashion (Aarts et al., 2014). In addition to accounting for intra-animal variability, this statistical model also allowed us to test whether distance from SCWM was a covariate of AIS length, and if there were any interactions with sham vs. mTBI groups. To conduct multilevel modeling, first an F test was used to determine if the multilevel model accounted for the variability in the data set. If the F test yielded p < 0.05, we inferred that the model fit the data and continued our analysis. We then evaluated the adjusted coefficient of determination (R2), which describes how much of the variability in the data is accounted for in the multilevel model (e.g., the proportion of AIS length that is a function of mTBI and distance to SCWM). In predictive statistics, the R2 can be used to assess the effect size of a multilevel linear regression model, where 0.01, 0.09 and 0.25 are defined as small, medium and large effects, respectively (Aarts et al., 2014). In next step, a two-tailed t test in linear regression terms was used to determine whether each specific parameter (e.g., sham vs. mTBI and distance to SCWM) had a significant effect on the outcome measure (e.g., AIS length), with the threshold for significance set at α < 0.05 adjusted for multiple comparisons. When analyzing only two experimental groups (e.g., sham vs. mTBI), a generally accepted index for reporting a standardized effect size is Cohen’s d, which equals the difference in the means of the two groups in units of SD. Cohen defined 0.2, 0.5 and 0.8 as small, medium and large effects, respectively (Cohen, 1988).
For ankG multilevel modeling, intact YFP+ pyramidal neuron summary statistics (t ratio, degrees of freedom, and p-value) and outcome measures (mean ± SEM) were calculated from nested data sets (e.g., N = 53 AIS total, 10–11 AIS nested per animal, n = 5 mice). To summarize multilevel modeling statistics, we report the number of animals per group (n) and total number of measurements per animal (N), the degrees of freedom (denoted as the subscript in the t ratio, and the p-value). All statistical data are summarized using mean ± SEM, unless otherwise noted. Descriptive data are summarized with mean and 95% confidence intervals (CI). All statistical tests were two-tailed and considered significantly different for p < 0.05.
Results
Intraoperative physiology was normal and consistent with previous reports on mice under isoflurane anesthesia (Cesarovic et al., 2010; Ewald et al., 2011; Hånell et al., 2015b). Specifically, the arterial oxygen saturation (sham = 97.8 ± 0.1%; mTBI = 97.7 ± 0.2%), heart rate (sham = 513 ± 12 BPM; mTBI = 505 ± 14 BPM) and RPM (sham = 63 ± 6 RPM; mTBI = 74 ± 6 BRPM) were similar between sham (n = 8 mice) and mTBI (n = 10 mice) groups (SpO2: t12.8 = 0.73, p = 0.4770; BPM: t16.0 = 0.42, p = 0.6811; RPM: t15.4 = −1.23, p = 0.2251; t test). Consistent with our previous reports (Greer et al., 2011; Hånell et al., 2015b), the loss of righting reflex duration in mTBI mice (4.7 ± 0.24 min; n = 10) was significantly greater (t11.4 = 7.13, p < 0.0001; t test) than shams (1.2 ± 0.42 min; n = 8). Additionally, mTBI mice for confocal (4.7 ± 0.39 min; n = 6) and electrophysiological (4.6 ± 0.25 min; n = 4) studies had similar loss of righting reflex (t7.8 = −0.35, p = 0.7337; t test). Importantly, signs of hypoxia/apnea were not observed during surgical preparation or postinjury. Overall, these physiological assessments did not show any evidence of confounding mechanisms that play a role in secondary insults.
Macroscopically, both sham and mTBI brain tissue appeared normal without evidence of surgically induced lesions (Figures 1A–E), as previously reported. Post-mTBI tissue sections revealed no macroscopic change (Figure 2) consistent with the mild and diffuse nature of cFPI. Importantly, the dorsal neocortex underneath the craniectomy site did not show evidence of focal contusion, cavitation, or overt subarachnoid hemorrhage induced by the fluid pressure wave (Figure 2B). Overall, the brain parenchyma was devoid of overt hemorrhage, the ventricular system maintained a regular contour, with no evidence of trauma-related ventricular enlargement. Taken together these data support our premise that cFPI in YFP-H mice is a reproducible model of mTBI.
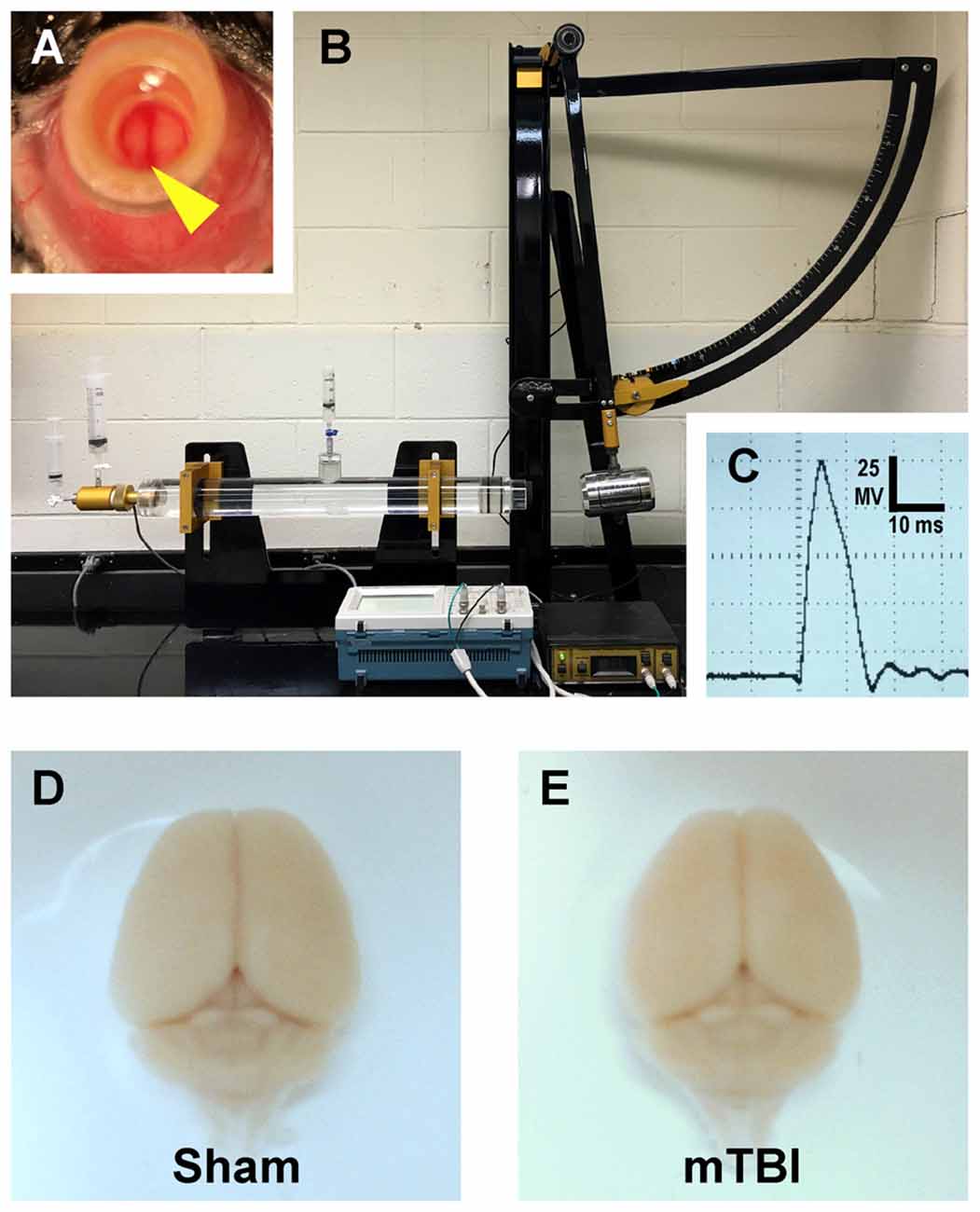
Figure 1. Mouse model of mild traumatic brain injury (mTBI) via midline central fluid percussion injury (cFPI). (A) Dorsal view showing craniectomy site with the dura intact. The superior sagittal sinus (yellow arrowhead) can be seen through the saline-filled hub. (B) Fluid percussion injury device with the pendulum in the cocked position. Severity of injury is determined by arc height. After releasing the trigger the pendulum strikes a piston in a cylinder reservoir generating a fluid pressure wave. The mouse cranium is attached to the cylinder at the opposite end via the installed hub, forming a closed mechanical system. (C) A representative fluid pressure wave that is transmitted directly onto the intact dura. (D,E) Representative images of dissected brains perfused 2 days after cFPI-induced mTBI. Sham (D) and mTBI (E) brains show no evidence of surgically-induced damage. After cFPI, there is no evidence of mass lesions and/or contusion. There is no focal damage at the craniectomy site where the pressure wave entered the cranium.
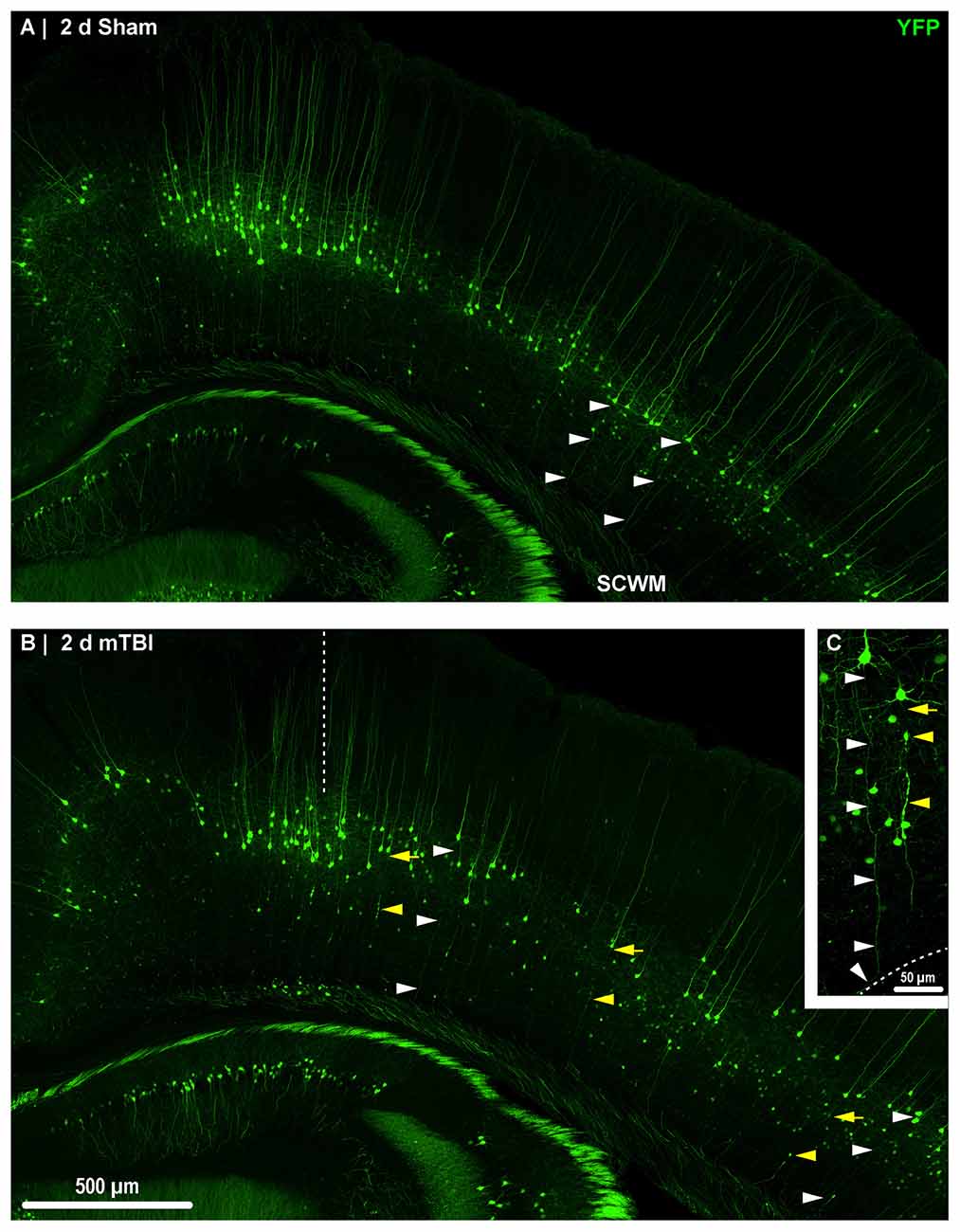
Figure 2. Yellow fluorescent protein (YFP) expression in neocortical layer 5 pyramidal neurons. Representative images of sections from sham (A) and mTBI (B) brains shown in Figure 1. Thy1 driven YFP expression results in restricted labeling of a discrete subset of layer 5 pyramidal neurons. (A–C) The selective YFP expression allows identification of individual axons that be traced from their soma of origin to the subcortical white matter (SCWM; white arrowheads). (B) Two days after mTBI, evidence of diffuse axonal injury (DAI) indicated by YFP+ axonal swellings is seen dispersed across the dorsolateral neocortex (yellow arrowheads). The craniectomy boundary is depicted by the vertical dashed line. No evidence of focal injury is observed under the craniectomy site. (C) A representative intact YFP+ pyramidal neuron juxtaposed by an injured axon. The intact axonal profile can be continuously traced from the soma of origin to where it enters the SCWM (dashed line), indicated by the acute change in trajectory.
YFP+ Pyramidal Neuron DAI
Consistent with previous work from our laboratory and others that utilized the YFP-H strain (Feng et al., 2000; Greer et al., 2011; Antón-Fernández et al., 2015; Hånell et al., 2015b), we observed YFP+ pyramidal neurons in sham (Figures 3A,D) and mTBI (Figures 4A,D) mice primarily within layer 5 neocortex (Figure 2). Following sham-injury, we did not observe any YFP+ axonal swellings or morphological irregularities indicative of axonal damage within neocortex or underlying SCWM (Figure 2A). Descending axons from YFP+ pyramidal neurons in shams demonstrated a continuous trail from their soma of origin to the SCWM interface. Following cFPI, all tissue sections processed for YFP visualization and parallel immunohistochemical analyses revealed a pattern of microscopic change consistent with that routinely described in rat and mouse cFPI models. Despite the overall preservation of brain parenchymal integrity typical of a mild diffuse TBI in both humans and rodents, the fluid pressure wave consistently evoked YFP+ axonal swellings indicating DAI. At 2 days post-mTBI, comparable to previous reports, we observed pathologic YFP+ axonal profiles in continuity with their somas of origin (Figures 2B, 4A) and their detached axonal segments distributed throughout layer 5/6 of the dorsolateral neocortex corresponding to S1BF (Figure 2B). Underscoring the diffuse nature of mild cFPI, the distribution of YFP+ pyramidal neurons proximal and distal axonal swellings were interspersed among numerous YFP+ profiles showing no morphological evidence of either primary axonal injury or retrograde neuronal involvement. Similar to shams, we readily identified these non-DAI/intact YFP+ pyramidal neurons via tracing YFP+ axons continuously from the edge of soma of origin to the SCWM interface (Figure 2C). Importantly, we observed consistent YFP+ pyramidal neuron axonal injury within the same neocortical regions of all mTBI mice, indicating a generalized diffuse response to cFPI-induced mTBI in mice rather than an isolated focal event.
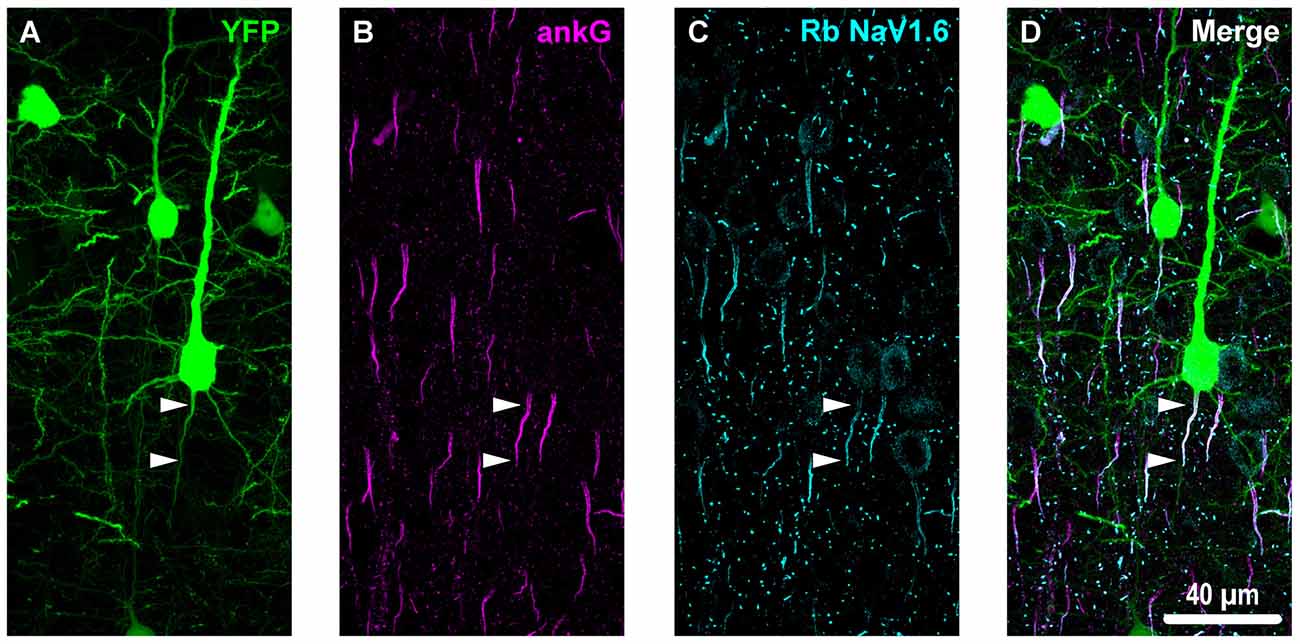
Figure 3. Overview of ankyrin-G (ankG) and voltagegated sodium channels (NaV1.6) immunofluorescent profiles in YFP mice. (A) Neocortical layer 5 YFP+ pyramidal neurons (green). (B) AnkG immunoreactivity visualized using Alexa Fluor 568 (magenta) shows characteristic tapered morphology. (C) NaV1.6 immunolabeled with a polyclonal rabbit (Rb) antibody visualized using Alexa Fluor 633 (cyan) reveals shorter profiles corresponding to the distal axon initial segment (AIS). Punctate profiles indicate Nodes of Ranvier. (D) Composite image clearly demonstrating the utility of ankG as an AIS marker. The proximal axon of the YFP+ pyramidal neuron in the center of the field-of-view (FOV) colocalizes with ankG and NaV1.6, which is expressed at the distal AIS. Several ankG+ profiles highlight NaV1.6 expression at the distal AIS.
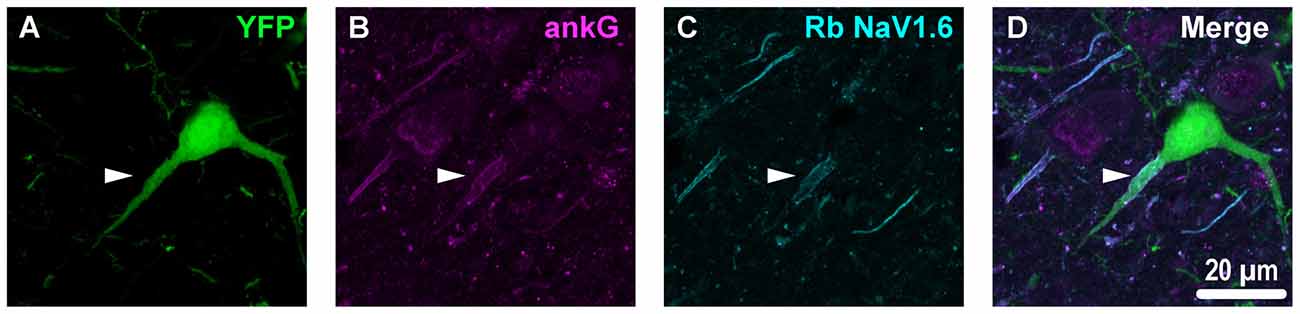
Figure 4. YFP+ pyramidal neuron axonal injury attenuates ankG and NaV1.6 expression at the AIS. (A–C) Representative images of YFP (A) ankG (B) and NaV1.6 (C). (A) Evidence of DAI occurring at the AIS of a YFP+ pyramidal neuron (green) is indicated by a severely swollen proximal segment (arrowhead). (B,C) AnkG (magenta) and NaV1.6 (cyan) expression persists, but is substantially attenuated with abnormal morphology after mTBI-induced AIS injury. (D) Composite image showing ankG and NaV1.6 within the swollen AIS of a YFP+ pyramidal neuron.
Intact YFP+ Pyramidal Neuron Responses
AIS Structural Plasticity
To estimate AIS position and length (Figure 6), we quantified immunofluorescent profiles of ankG, a key structural protein underpinning AIS function (Grubb and Burrone, 2010). Consistent with previous reports from our laboratory and others (Greer et al., 2013; Gutzmann et al., 2014; Clark et al., 2016), ankG labeling throughout the neocortex was restricted to the AIS and nodes of Ranvier (Figure 3B). Importantly, ankG labeling of intact YFP+ pyramidal neurons clearly delineated the AIS in sections from both sham and mTBI mice (Figures 6B,E). For ankG multilevel modeling, intact YFP+ pyramidal neuron summary of outcome measures (mean ± SEM) and statistics were calculated from nested data sets (sham: N = 53 AIS total, 10–11 AIS nested per animal, n = 5 mice; mTBI: N = 54 AIS total, 5–13 AIS nested per animal, n = 6 mice). The sampling distribution of intact YFP+ pyramidal neurons with respect to distance from the SCWM interface (Figure 5) was symmetrical between groups (sham = 372 ± 14 μm, mTBI = 368 ± 14 μm; t8.0 = 0.18, p = 0.8585). Within this same data set (Figure 6), the ankG start position with respect to soma of origin was similar between groups (sham = 2.1 ± 0.16 μm, mTBI = 2.2 ± 0.16 μm; t6.41 = −0.50, p = 0.6342). In contrast, we observed a statistically significant decrease by 6.8% (−1.8 ± 0.59) in ankG end position (sham = 26.6 ± 0.42 μm, mTBI = 24.8 ± 0.42 μm; t7.4 = −3.08, p = 0.0167). Overall, this resulted in ankG length significantly decreasing by 7.8% (−1.9 ± 0.58 μm) from the distal end (sham = 24.4 ± 0.41 μm, mTBI = 22.5 ± 0.41 μm; t7.7 = −3.30, p = 0.0114; R2 = 0.16). Because layer 5 pyramidal neurons are heterogeneous (Chagnac-Amitai et al., 1990; Hattox and Nelson, 2007; Le Bé et al., 2007), varying structurally and functionally with cortical depth (e.g., layer 5a vs. 5b), we controlled for this potential confound in analyzing plasticity (Jacob et al., 2012) by measuring the AIS distance from SCWM. Multilevel analysis (N = 107 AIS total) revealed that distance from SCWM is a significant covariate (dSCWM) of both ankG end position (t88.0 = 4.07, p < 0.0001) and overall length (t79.0 = 4.52, p < 0.0001). Specifically, ankG end position (dSCWM = 0.019 ± 0.005) and length (dSCWM = 0.020 ± 0.004) are directly proportional to distance from SCWM. The equal slopes of the best-fit lines for ankG length as a function of distance from SCWM (Figure 6I) indicated that there was no interaction with experimental groups (end position: t86.0 = −0.82, p = 0.3120; length: t80.3 = 0.69, p = 0.5512). Consistent with the symmetrical sample distribution intact YFP+ pyramidal neurons in layer 5 S1BF (Figure 5), the statistical model yielded similar values after leveraging, with respect to distance from SCWM, ankG end position (sham = 26.6 ± 0.36 μm, mTBI = 24.9 ± 0.39 μm) and length (sham = 24.4 ± 0.33 μm, mTBI = 22.6 ± 0.35 μm). Additionally, the leveraged ankG length after mTBI was significantly shorter by 7.4% (−1.8 ± 0.48 μm) compared to shams (t7.4 = −3.74, p = 0.0066; R2 = 0.28). Notably, accounting for this covariate resulted in 20% decrease in SEM (0.41–0.33) and a 75% increase in the R2 (0.16–0.28). Hence, while this reduction in ankG length is relatively modest, multilevel modeling revealed that mTBI accounted for almost 30% of the variability between data sets, and the effect size was robust (Cohen’s d = 0.76). Further, ankG was shortened from the distal AIS microdomain that contains the “trigger zone” for AP firing, where a highly dense distribution of NaV1.6 sets the threshold for AP initiation (Kole et al., 2008).
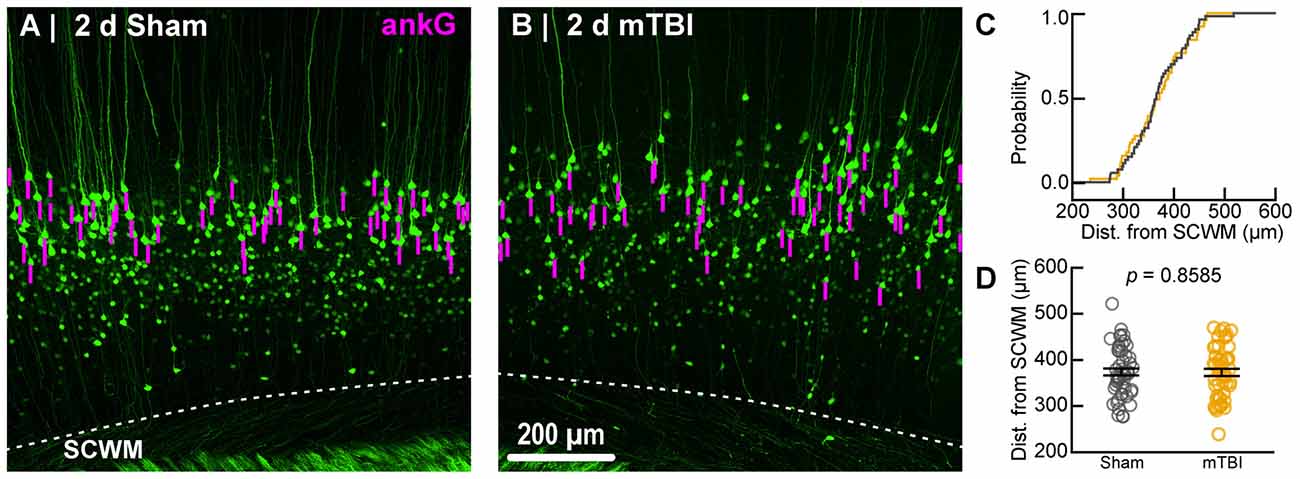
Figure 5. Sampling distribution of intact YFP+ pyramidal neurons for ankG quantitative analysis. (A,B) Superimposed maximum intensity projections of 10× overview images for sham (A) and mTBI (B). The intact YFP+ pyramidal neuron sampling distribution (magenta bars) is symmetric with respect to the SCWM. (C) Cumulative frequency distribution plot shows a high degree of overlap between sham (gray) and mTBI (gold). (D) Quantitative analysis confirmed no sampling difference. Statistics: multilevel analysis with animals as a random variable. Data summarized with means and standard error of the mean (SEM) error bars.
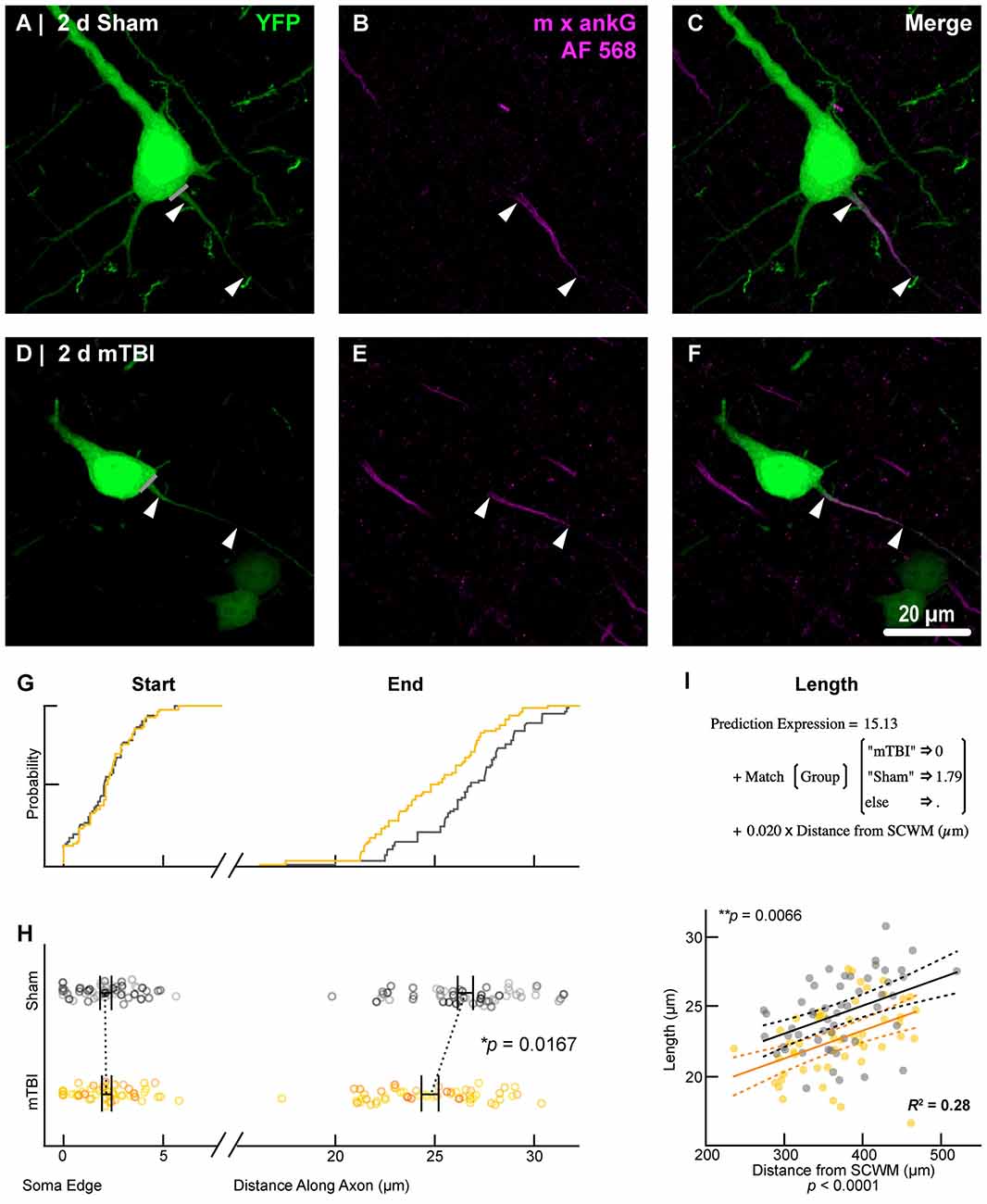
Figure 6. AnkG length decreases from the distal AIS after 2 days post-mTBI. Representative images of sham (A–C) and mTBI (D–F) of YFP (A,D; green) and ankG (B,E; magenta). (C,F) Composite image showing YFP+ axon colocalization with ankG, demarcating the AIS. YFP traces were taken from the line drawn across the ado-somatic vertex (A,D), through and past the region of the AIS (C,F). (G–I) Quantitative analysis of ankG profiles of sham (gray) and mTBI (gold). (G) Cumulative frequency distribution plots showing overlap in start position but a proximal shift in mTBI end position. (H) Multilevel analysis of start and end positions with animals as a random variable depicted by different shades. Data summarized with means and SEM error bars. Note that there is no change in ankG start position; however, there was a significant decrease in the end position. This resulted in an overall decrease in length from the distal AIS. (I) Multilevel analysis of ankG length with animals as a random variable and distance from SCWM as a covariate. Plot of ankG length as a function of distance from SCWM (solid lines) with 95% confidence intervals (CI; dashed lines) for sham and mTBI. Note that distance from SCWM is a significant covariate of ankG length, but there is no interaction with experimental groups indicated seen as a parallel shift in lines.
To estimate the distal AIS “trigger zone” position and length (Figure 8), we quantified immunofluorescent profiles of NaV1.6. Consistent with previous reports from other labs (Lorincz and Nusser, 2008a; King et al., 2014), NaV1.6 labeling throughout the neocortex was restricted to the distal AIS and nodes of Ranvier (Figure 3C). Importantly, NaV1.6 labeling of intact YFP+ pyramidal neurons clearly delineated the distal AIS in sections from both sham and mTBI mice (Figures 8B,E). For NaV1.6 multilevel modeling, intact YFP+ pyramidal neuron summary of outcome measures (mean ± SEM) and statistics were calculated from nested data sets (sham: N = 54 AIS total, 7–14 AIS nested per animal, n = 5 mice; mTBI: N = 44 AIS total, 5–9 AIS nested per animal, n = 6 mice). The sampling distribution of intact YFP+ pyramidal neurons with respect to distance from the SCWM interface (Figure 7) was not statistical different between groups (sham = 339 ± 16 μm, mTBI = 384 ± 15 μm; t9.55 = 1.99, p = 0.0761). Within this same data set (Figure 8), the NaV1.6 start position with respect to soma of origin was similar between groups (sham = 11.0 ± 0.28 μm, mTBI = 11.0 ± 0.32 μm; t5.3 = 0.12, p = 0.8708). Unlike our ankG findings at the distal AIS, we did not observe a statistically significant difference in NaV1.6 end position (sham = 26.0 ± 0.61 μm, mTBI = 27.0 ± 0.59 μm; t9.7 = 1.20, p = 0.2876) or overall length (sham = 15.0 ± 0.60 μm, mTBI = 16.0 ± 0.60 μm; t9.9 = 1.24, p = 0.2448).
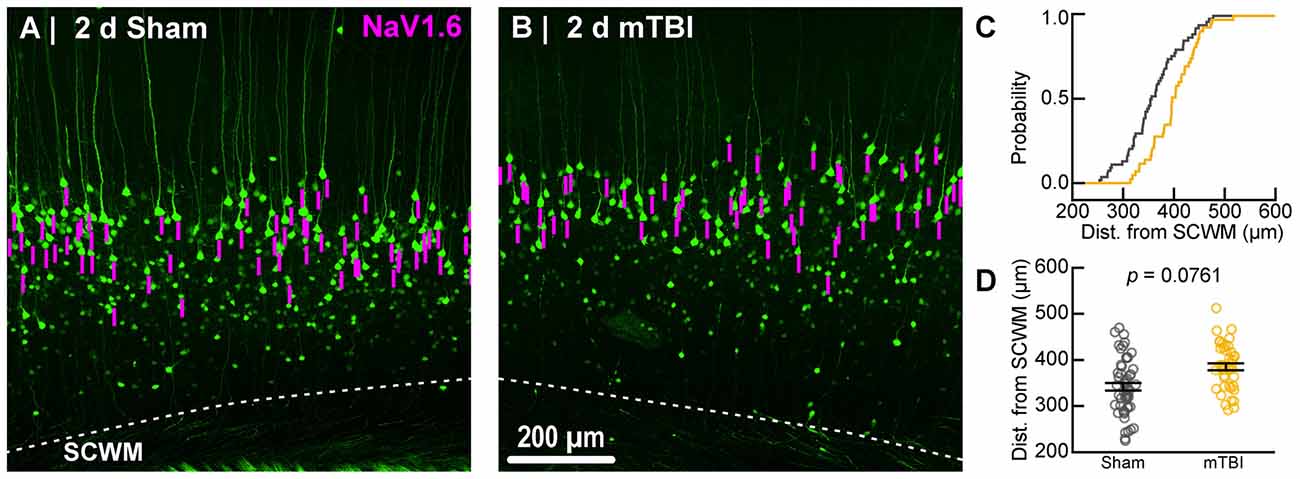
Figure 7. Sampling distribution of intact YFP+ pyramidal neurons for NaV1.6 quantitative analysis. (A,B) Superimposed maximum intensity projections of 10× overview images for sham (A) and mTBI (B). The intact YFP+ pyramidal neuron sampling distribution (magenta bars) with respect to the SCWM appeared slightly asymmetric, with mTBI having several samples located more superficially (upper right aspect). (C) Cumulative frequency distribution plot showing mTBI (gold) samples are shifted toward increased distance from SCWM compared to sham (gray). (D) Quantitative analysis confirmed revealed there was no statistically significant difference in sampling. Statistics: multilevel analysis with animals as a random variable. Data summarized with means and SEM error bars.
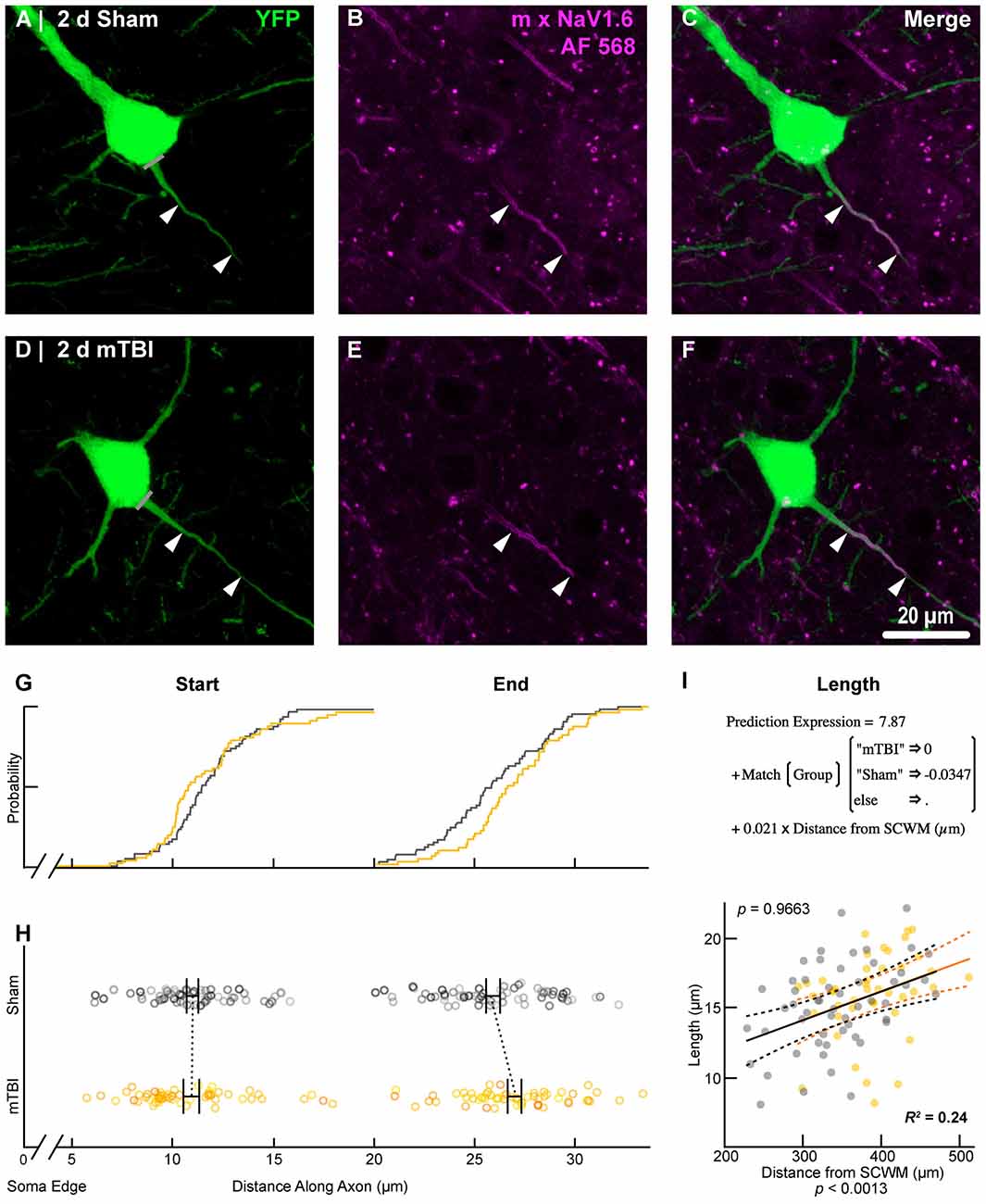
Figure 8. NaV1.6 geometry does not change after mTBI. Representative images of sham (A–C) and mTBI (D–F) of YFP (A,D; green) and NaV1.6 (B,E; magenta). (C,F) Composite image showing YFP+ axon colocalization with NaV1.6, demarcating the distal AIS. (G–I) Quantitative analysis of NaV1.6 profiles of sham (gray) and mTBI (gold). (G) Cumulative frequency distribution plots showing overlap in start position but a distal shift in mTBI end position. (H) Multilevel analysis of start and end positions with animals as a random variable depicted by different shades. Data summarized with means and SEM error bars. Note that there was no change in NaV1.6 start or end position. (I) Multilevel analysis of NaV1.6 length with animals as a random variable and distance from SCWM as a covariate. Plot of NaV1.6 length as a function of distance from SCWM (solid lines) with 95% CI (dashed lines) for sham and mTBI. Distance from SCWM is a significant covariate of NaV1.6 length. This accounted for the overlap in sham and mTBI plots compared to the shift seen in the uncorrected distribution of the end position (G).
In line with our ankG findings (Figure 6I), multilevel analysis also revealed that distance from SCWM is a significant covariate of both NaV1.6 end position (t71.7 = 3.80, p = 0.0003; dSCWM = 0.020 ± 0.005) and NaV1.6 length (t59.9 = 3.38, p = 0.0013; dSCWM = 0.021 ± 0.006; Figure 8I). Notably, the coefficient of distance from SCWM (dSCWM) was remarkably similar between ankG and NaV1.6 for both end position and length (average = 0.02, 95% CI: 0.018–0.021; n = 4). This observations provided further evidence supporting that AIS length is directly proportional to distance from SCWM, wherein upper vs. lower pyramidal neurons have longer and shorter AIS, respectively. Specifically, the statistical model predicts AIS length from distal end increases ~2 μm for every 100 μm from SCWM (Figures 6I, 8I). Considering layer 5 S1BF is ~300 μm thick (Figures 5, 7), this corresponds to ~6 μm difference in AIS length from the distal end between upper and lower pyramidal neuron populations. While the sampling distributions with respect to the distance from SCWM interface between sham and mTBI were not significantly different (as described above), in light of this covariate’s magnitude, we compared the means of non-leveraged vs. leveraged with respect to distance from SCWM of NaV1.6 end position and length using the Wilcoxon signed-rank test. For sham data (n = 5 mice), including the distance from SCWM covariate in our multilevel model did result in a statistically significant difference between the means NaV1.6 end position (leveraged = 26.1 ± 0.52 μm; X2 = −2.5, p = 0.6250) or length (leveraged = 15.4 ± 0.55 μm; X2 = −6.5, p = 0.1250). In contrast, for mTBI data (n = 6 mice) the distance from SCWM covariate in the multilevel model yielded significantly decreased mean values for both NaV1.6 end position (leveraged = 26.2 ± 0.52 μm; X2 = 10.5, p = 0.0313) and length (leveraged = 15.4 ± 0.56 μm; X2 = 10.5, p = 0.0313). This difference in NaV1.6 end position (−0.70 ± 0.38) and length (−0.58 ± 0.42) from the distal AIS is consistent with our model’s predicted effect of a −45 μm difference between sham (339 μm) and mTBI (384 μm) mean distance from SCWM (e.g., 0.02 × −45 μm = −0.9 μm; Figures 7C,D).
Collectively, these data suggest disparate responses between ankG and NaV1.6 at the distal AIS 2 days after mTBI. Restructuring our multilevel model by changing the experimental condition to AIS component allowed us to directly assess for differences between ankG and NaV1.6 end position. As described above, we accounted for nested measurements per animal. Additionally, in this multilevel model we also accounted for the dependency resulting from sampling two sections per animal for single labeling of ankG and NaV1.6. To determine if there was a differential response at the distal AIS, we compared the leveraged means of ankG and NaV1.6 end positions in both sham and mTBI data sets. In shams (n = 5 mice), the leveraged end position for ankG (26.3 ± 0.55 μm; N = 53 AIS, 10–11 AIS nested per animal) and NaV1.6 (26.0 ± 0.55 μm; N = 54 AIS, 7–14 AIS nested per animal) were similar (t8.2 = 0.47, p = 0.6522; N = 10 sections, 2 per animal). Conversely, after mTBI (n = 6 mice) the leveraged end position for ankG (25.0 ± 0.34 μm; N = 54 AIS, 5–13 AIS nested per animal, 54 AIS total) was less distal than NaV1.6 (26.5 ± 0.37 μm; N = 44 AIS, 5–9 AIS nested per animal). This difference of 5.6% (–1.48 ± 0.50) between ankG and NaV1.6 end positions was statistically significant (t8.6 = −2.94, p = 0.0172; N = 12 sections, 2 per animal). Overall, the multilevel model accounted for a large proportion of the variability between data sets (R2 = 0.23) and the effect size was substantial (Cohen’s d = 0.61). Taken together, these data show that ankG structural plasticity is uncoupled from NaV1.6 at 2 days post-mTBI. Further, these data provide compelling evidence for S1BF layer 5 pyramidal neuron AIS length as a function of distance from the SCWM. Lastly, these findings reveal that AIS length both after mTBI and as a function of distance from the SCWM is modulated at the site of AP initiation that is regulated by NaV1.6.
Perisomatic GABAergic Bouton Density
In concert with intrinsic/postsynaptic AIS structural plasticity, we observed a loss of GABAergic inputs along the perisomatic domain of intact YFP+ pyramidal neurons in layer 5 S1BF, revealing evidence for extrinsic/presynaptic change 2 days after mTBI (Figure 9). Immunolabeling GAD67, the GABA-synthesizing enzyme enriched in PV+ presynaptic boutons (Fish et al., 2011) allowed us to conduct a high precision quantitative analysis of the major source of inhibitory input at the perisomatic domain, which strongly regulates AIS activity. We quantified perisomatic GABAergic bouton density of only intact YFP+ pyramidal neurons in S1BF using the same sampling method for AIS studies (Figures 5, 7). For perisomatic GABAergic bouton density (puncta/100 μm2) multilevel modeling, intact YFP+ pyramidal neuron summary of outcome measures (mean ± SEM) and statistics were calculated from nested data sets (sham: N = 48 somas total, 5–13 somas nested per animal, n = 5 mice; mTBI: N = 50 somas total, 5–14 somas nested per animal, n = 5 mice). Using two different quantitative methods, we observed a decrease in perisomatic GABAergic bouton density in both GAD67+ (Figures 9B,F,J) and GAD67+/PV+ (Figures 9C,G,K,L) immunoreactive profiles 2 days after mTBI. Specifically, the density of GAD67+ profiles in gray-scale images between sham (13.0 ± 0.57 puncta/100 μm2) and mTBI (10.9 ± 0.57 puncta/100 μm2) were significantly different; t7.8 = −2.61, p = 0.0321). Similarly, the density of GAD67+/PV+ profiles in binary images between sham (12.0 ± 0.55 puncta/100 μm2) and mTBI (8.7 ± 0.56 puncta/100 μm2) were also significantly different; t5.4 = −4.04, p = 0.0086). The magnitudes of change were comparable between gray and binary quantitative methods, with intact YFP+ pyramidal neuron perisomatic GABAergic bouton density decreasing by 16% and 24%, respectively. Additionally, the effect sizes were robust for both methods (Cohen’s d: gray-scale = 0.71; binary = 0.99). Lastly, analysis of the cumulative distribution plot of the size of PV+/GAD67+ puncta (segmented particles in binary images) revealed that the greatest decrease occurred in particles that were the average size (~1 μm2) of GABAergic puncta (Figure 9L). Taken together, these data provide substantial evidence for diffuse presynaptic GABAergic terminal loss 2 days after mTBI.
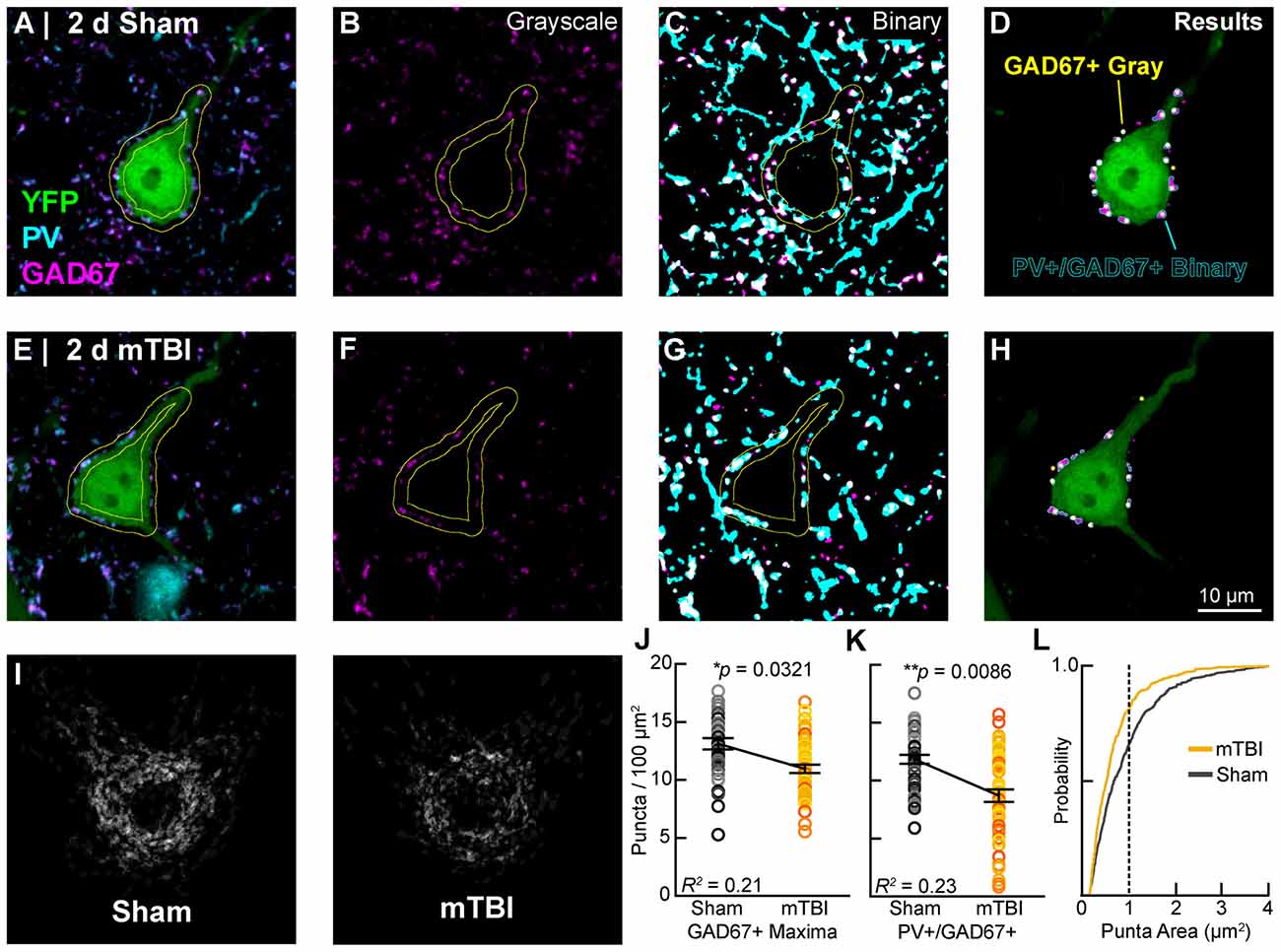
Figure 9. Intact YFP+ pyramidal neuron perisomatic GABAergic bouton density decreases 2 days post-mTBI. Representative images of sham (A–D) and mTBI (E–H) glutamate decarboxylase-67 (GAD67+; B,F; magenta) and parvalbumin (PV+; A,C,E,G; cyan) punch along the perimeter of YFP+ pyramidal neurons (A,E,D,H; green). Perisomatic bouton density was quantified in 8-bit gray scale images of GAD67 expression (B,F,J) and also binary images of colocalized PV+/GAD67+ puncta (C,G,I,K,L). (D,H) Outcome of automated quantification using gray scale and binary images was comparable. Perisomatic GAD67 expression (magenta) and result of gray scale analysis (yellow dots) largely overlapped with results of PV+/GAD67+ binary image analysis (cyan outlines). (I) A qualitative decrease in perisomatic GABAergic bouton density is seen in superimposed images of binary PV+/GAD67+ profiles. (J,K) Multilevel analysis with animals as a random variable depicted by different shades for sham (gray) and mTBI (gold/orange). Data summarized with means and SEM error bars. GAD67+ maxima in gray-scale images and PV+/GAD67 puncta in binary images were both significantly reduced in mTBI vs. sham. The absolute values as well as relative decrease were similar between gray-scale and binary image analyses. (L) Cumulative frequency distribution of PV+/GAD67+ puncta area. Vertical line at 1 μm2 corresponds to average size of a GABAergic presynaptic bouton. This shift in the mTBI plot shows that PV+/GAD67+ puncta approximating the average area of a GABAergic bouton accounting for the majority of the loss.
AIS Functional Plasticity
Our findings of presynaptic input loss in concert with postsynaptic AIS remodeling have been associated with alterations of neuronal excitability in sensory circuits reported by others (Kuba et al., 2010, 2014). Appreciating the known intrinsic properties of the AIS and the mechanisms that modulate its structure and function (Bender and Trussell, 2012), we followed our morphological studies with electrophysiological assessments at 2 days post-mTBI. Specifically, we employed whole-cell patch-clamp analysis of intact YFP+ pyramidal neurons in layer 5 S1BF to compare intrinsic AIS functional properties between sham (n = 13 cells from 3 mice; 4–5 AP nested per cell, N = 63 AP total) and mTBI (n = 14 cells from 4 mice; 5 AP nested per cell, N = 70 AP total) groups using multilevel modeling. Similar to shams, current injection evoked an AP in all patched intact YFP+ pyramidal neurons after mTBI, consistent with the diffuse and subtle nature of mTBI pathophysiology. While AP waveforms were comparable between sham and mTBI (Figures 10A,B), AP decay tau significantly decreased by 79% after mTBI (p = 0.0014; Table 1). Additionally, we found that AP threshold was ~3 mV more depolarized after mTBI (−46.2 ± 1.0 mV) compared to shams (−49.6 ± 1.0 mV). This difference in AP threshold was statistical significant (t25 = 2.41, p = 0.0236; Cohen’s d = 0.95), and consistent with altered AIS intrinsic function. While AP initiation occurs at the AIS, after spiking the signal travels both anterograde down the axon, and retrograde toward the soma. Therefore, AP recorded by patching the cell body reflects a combination of AP generation at the AIS and back-propagation in the soma (Hu et al., 2009). To further dissect intrinsic AIS function from patch-recordings at the cell body, we calculated the second derivative of membrane voltage (i.e., AP acceleration), which showed two peaks (Figure 10C). In the plot of the second derivative, the amplitude of the first and second peaks corresponded to AP acceleration at the AIS and soma, respectively (Khaliq and Raman, 2006; Meeks and Mennerick, 2007). Interestingly, multilevel modeling revealed AP acceleration is significantly reduced at the AIS (t25 = −2.57, p = 0.0164), but not the soma (t25 = 0.58, p = 0.5701; Figures 10C,D). Specifically, AP acceleration at the AIS decreased by 21% after mTBI (4104 ± 296 mV/μs2) compared to shams (5203 ± 308 mV/μs2), and the effect size was large (Cohen’s d = 1.01). AP threshold in pyramidal neurons is set by NaV1.6 channels localized at the distal AIS (Kole et al., 2008). These data showing AP threshold depolarization and attenuation of AP acceleration specifically at the AIS are consistent with alteration of NaV1.6 membrane distribution after mTBI. Together, our morphological and electrophysiological studies provide significant evidence of intact YFP+ pyramidal neuron AIS structural and functional plasticity after mTBI.
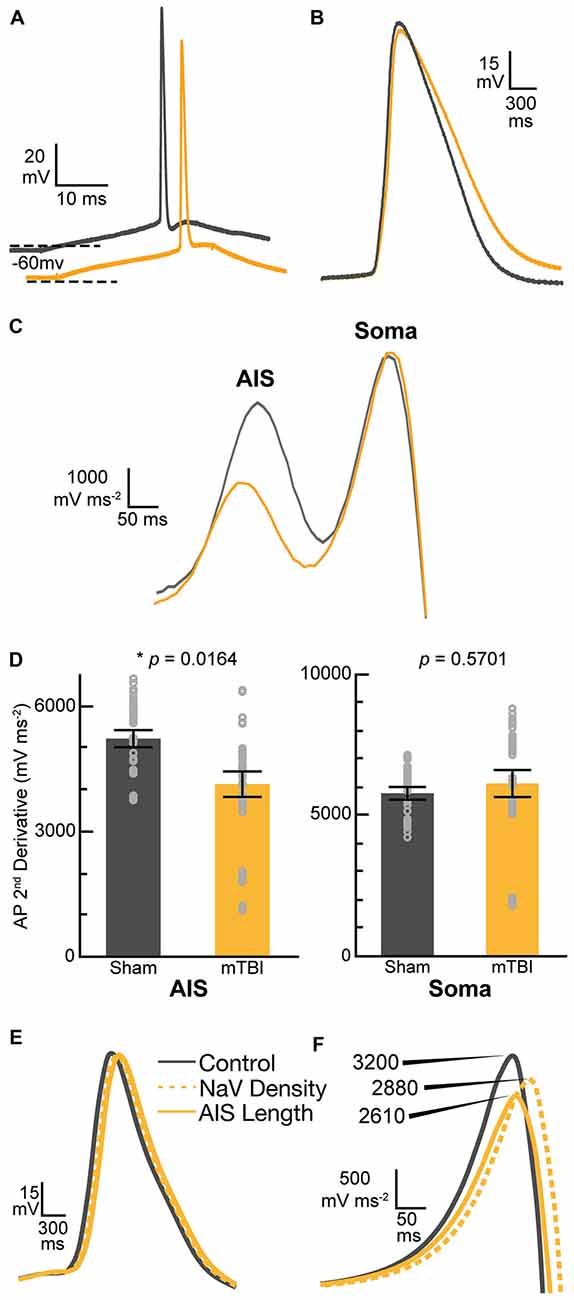
Figure 10. Action potential (AP) acceleration at the AIS is reduced 2 days post-mTBI. (A–D) Whole-cell patch-clamp analysis of intact YFP+ pyramidal neurons of layer 5 primary somatosensory barrel field (S1BF) in sham (gray) and mTBI (gold) ex vivo live slices. (A) Representative traces of AP recorded at somas. The mTBI trace is offset for better comparison. Dashed lines indicate holding potential. (B) Enlarged traces from (A) showing AP waveform. Note slightly longer AP decay after mTBI. (C) Plot of second derivative of the membrane voltage. The first and second peak amplitudes were used to compare AP acceleration at the AIS and soma, respectively. (D) Multilevel analysis of the second derivative of membrane voltage (AP acceleration) with animals as a random variable and accounting for the five recorded AP nested within each cell. Data summarized with means and 95% CI error bars. The peak amplitude at the AIS was significantly reduced, reflecting attenuated AP acceleration. In contrast, there was no change in the AP acceleration at the soma. (E,F) Computational modeling of a layer 5 pyramidal neuron. Evoked AP waveforms in simulations (E) using experimentally observed AIS lengths were similar to AP recorded from intact YFP+ pyramidal neurons (A). Overall, changing AIS length or NaV density at the distal AIS by 7% did not have an overt effect on AP waveform. (F) The plots of computationally derived second derivatives of membrane voltage were also comparable to experimental observations (B). Modeling data showed that a 7% decrease in AIS length or NaV1.6 density at the distal AIS both decrease peak amplitude of the second derivative plot, consistent with attenuated AP acceleration.
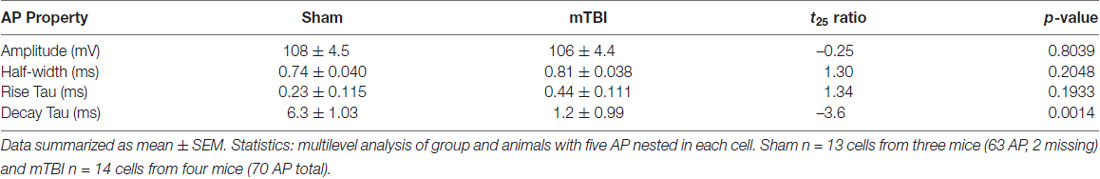
Table 1. Intrinsic action potential (AP) properties of intact yellow fluorescent protein (YFP+) pyramidal neurons 2 days postinjury.
Layer 5 Pyramidal Neuron Modeling
While AIS excitability varies with axial geometry such as length (Kuba et al., 2010), voltage-gated ion channel density is also a critical determinant of intrinsic AP properties (Kole et al., 2008). To investigate the functional consequences of AIS structural plasticity and explore the effect varying NaV1.6 density we used an anatomically and physiologically accurate computational model of a layer 5 pyramidal neuron (Hallermann et al., 2012). This computational model allowed us to probe AP dynamics specifically at the AIS. The AP waveforms in the model (Figure 10E) were qualitatively similar to those observed experimentally (Figure 10B). Comparing the effect of AIS length differences based on our experimental observations of ankG immunofluorescent profiles, we found that shortening the length by 7% resulted in a 19% decrease in the peak of the second derivative of membrane voltage, reflecting attenuated AP acceleration (Figure 10F). To help elucidate potential functional consequences of ankG and NaV1.6 uncoupling we evaluated the effect of a proportionate decrease in NaV density at the distal AIS. Without changing AIS length, a 7% decrease in NaV density at the distal AIS also attenuated AP acceleration by 10%. Thus, AP kinetics at the AIS depends on both geometry and density of functional components. Collectively, these morphological and computational findings suggest that even subtle AIS changes may alter AP spike dynamics among intact pyramidal neurons after mTBI.
Discussion
This study provides compelling evidence of intact pyramidal neuron AIS structural and functional plasticity after clinically relevant mTBI in mice. Consistent with human mTBI, midline cFPI reproducibly evoked DAI without mass lesions, neocortical contusion, and/or overt cell death (Mittl et al., 1994; Saatman et al., 2008; Andriessen et al., 2010; Bigler and Maxwell, 2012; Yuh et al., 2013; Shultz et al., 2016). Exploiting restricted YFP expression in a discrete subset of layer 5 pyramidal neurons and an established AIS marker (ankG), we confirmed subcellular structural plasticity within the non-DAI/intact population. In this mTBI-mediated response, the loss of presynaptic GABAergic boutons surrounding the perisomatic domain of intact pyramidal neurons suggested compromised AIS inhibition mediated by PV+ interneuron input. Consistent with AIS structural change, patch-clamp recordings of intact YFP+ layer 5 pyramidal neurons revealed attenuated AP acceleration after mTBI. This finding was further supported by simulations using a realistic layer 5 pyramidal neuron model that showed even a subtle decrease in AIS length or NaV1.6 density attenuates AP acceleration. Taken together, these findings demonstrate significant structural and functional changes of both intrinsic and extrinsic/synaptic components that are major regulators of AIS activity. The conclusions of this study depart from current thought on mTBI, which has been based primarily on the role of structural disconnection of white matter tracts (Sharp and Ham, 2011; Sharp et al., 2014). Collectively, the novel subcellular and neurophysiologic substrates identified in this study significantly extend our knowledge of neocortical circuit dysfunction following mTBI.
cFPI: Clinical Relevance and Utility for Evaluating Neocortical Circuit Dysfunction
Several features of our cFPI model underscore its clinical relevance and utility for evaluating both DAI and intact neuronal populations in neocortical circuits after mTBI. We used loss of righting reflex in mice as a surrogate for loss of consciousness in humans to assess injury severity, which had a similar duration ranging 3–5 min (Shaw, 2002; Malec et al., 2007; Schrader et al., 2009). While this was our only behavioral assessment, we monitored arterial oxygen saturation, heart rate, RPM and core body temperature in all mice used for structural and functional studies. Specifically, all mice were in physiological homeostasis. Importantly, evaluating these parameters help distinguish our work from most other investigators who do not perform routine physiologic monitoring to exclude secondary insults, which could severely confound the assessment of network disruption via diffuse structural and functional changes.
Capitalizing on restricted neuronal labeling with YFP including the total axonal length, readily traceable within the neocortical gray, allowed the unequivocal identification of mTBI-induced DAI vs. non-DAI/intact populations in both fixed tissue sections and live slices. Specifically, we readily identified intact pyramidal neurons by following their YFP+ axons from the soma of origin to the SCWM interface. Previously, we reported that mTBI induces DAI primarily within the AIS of the YFP+ layer 5 pyramidal neurons (Greer et al., 2013). Additionally, in a former publication we showed that the most commonly used DAI marker (β-amyloid precursor protein) did not colocalize with YFP axons in either the SCWM or corpus callosum (Hånell et al., 2015b). Taken together, these findings strongly support the premise that virtually all YFP+ pyramidal neuron DAI occurs within the neocortical gray. Based on this premise, intact YFP+ pyramidal neurons vs. those undergoing DAI can be reliably identified by examining their axonal segments spanning neocortical layers 5/6 to the SCWM interface.
Pyramidal neurons are the major excitatory component in cortical networks (Fairen et al., 1984; DeFelipe and Fariñas, 1992; Somogyi et al., 1998). Previously we showed that intact YFP+ pyramidal neurons intrinsic and extrinsic/synaptic properties were altered post-mTBI and developed over time (Greer et al., 2012; Hånell et al., 2015a; Sun and Jacobs, 2016). Initially after mTBI, the intrinsic properties of intact pyramidal neurons paralleled that found in the DAI population, suggesting that ion and/or neurotransmitter imbalance had an indiscriminant overriding effect on currents (Greer et al., 2012). However, by 2 days post-mTBI a functional divergence was identified wherein the intact YFP+ pyramidal neuron population showed distinct electrophysiological changes implicating AIS structural-functional alteration. These changes occurred in parallel with overall increased network excitability (Hånell et al., 2015a), implicating loss of inhibition (Isaacson and Scanziani, 2011; Yizhar et al., 2011; Lazarus et al., 2015). Collectively, these previous findings by our laboratory formed the basis of the current study focusing on the effect of mTBI on intrinsic structure and function of the AIS as well as the extrinsic/synaptic GABAergic inputs that control its output (Cobb et al., 1995; Pouille and Scanziani, 2001; Freund, 2003; Klausberger et al., 2003; Klausberger and Somogyi, 2008; Cardin et al., 2009; Atallah et al., 2012; Pouille et al., 2013; Lazarus et al., 2015; Wefelmeyer et al., 2015).
Building upon our previous studies in YFP-H mice, we succeeded in conducting a highly detailed analysis within this specific subset of neocortical layer 5 intact pyramidal neurons showing no evidence of DAI. This was a crucial component of our study since the AIS ion channel distribution differs across neuronal subtypes (Lorincz and Nusser, 2008a), and even within a neuronal subtype (Hu et al., 2009). Additionally, as seen shown in the current study, there is substantial variability in position and length. Further, because DAI affects only a small fraction of neurons within a pyramidal neuron population (Greer et al., 2011) that represent 80% of all neocortical neurons (DeFelipe and Fariñas, 1992), quantification of such heterogeneous population (Molnár and Cheung, 2006) remains an important confounder. Our previous reports underscore this issue, showing that the neocortical pyramidal neuron AIS has a predilection for DAI (Greer et al., 2013). In the current study we show DAI occurring within the AIS severely disrupts ankG and NaV1.6 expression (Figure 4). Hence, even a 5% burden of DAI (Greer et al., 2011) causing AIS disruption would substantially contribute to data variability.
Layer 5 Pyramidal Neuron AIS “Tuning”
To the best of our knowledge, this is the first study to account for these issues by examining only intact, Thy1-expressing, layer 5 pyramidal neurons. The important distinction in the current report is that we detected a decrease in AIS length within 2 days after cFPI-induced mTBI specifically within the layer 5 pyramidal neuron populations with no overt structural axonal abnormalities and/or evidence of DAI. For structural studies, we relied on immunofluorescent labeling of ankG and NaV1.6 as surrogate markers of the AIS and associated “trigger zone” at the distal end, respectively (Grubb et al., 2011; Kole and Stuart, 2012). An unexpected finding in the current study that followed from our a priori control parameters was that distance from SCWM was a significant covariate of AIS length (Figure 6I). Specifically, intact YFP+ pyramidal neurons AIS length from the distal end varied within the ~300 μm span of layer 5 neocortex. Importantly, this pattern was observed in both ankG and NaV1.6 assessments, which yielded virtually the same coefficient of 0.02 in each of the statistical models. This observed variance in AIS structure is consistent with known properties of layer 5 pyramidal neurons which are subdivided into layer 5a and 5b and populated by multiple subtypes characterized by different morphological and electrophysiological features (Chagnac-Amitai et al., 1990; Schubert et al., 2001; Hattox and Nelson, 2007). Additionally, the novel findings in this study are consistent with the “fine-tuning” of the AIS previously described in visual (Gutzmann et al., 2014), as well as auditory circuits (Kuba et al., 2010; Kuba, 2012). Specifically, Gutzmann et al. (2014) showed AIS length from the distal end varies between layer 2/3 and layer 5, representing functionally distinct regions within a neocortical column (Mountcastle, 1979; Somogyi et al., 1998; DeFelipe et al., 2003). Building on this previous work showing inter-laminar differences, in the current report we demonstrate intra-laminar AIS variability, consistent with the presence of layer 5 pyramidal neuron subpopulations (Molnár and Cheung, 2006). While AIS varying as a function of neocortical depth was observed in both 2 days sham-injury and mTBI mice, the lack of interaction between distance from SCWM and experimental groups supports that this finding reflects a novel AIS property with important basic science implications beyond the scope of our injury paradigm.
Uncoupling of ankG and NaV1.6 after mTBI
The current report highlights the importance of measuring both AIS proximal and distal positions with respect to soma of origin. Since structural remodeling can occur at the start and/or end of the AIS (Grubb and Burrone, 2010; Kuba et al., 2010; Evans et al., 2013, 2015), knowledge of AIS position with respect to soma of origin is an important metric. This issue becomes even more critical when factoring proximal and distal AIS subdomains are populated by different ion channel subtypes (Lorincz and Nusser, 2008a; Hu et al., 2009). A key morphological finding in this current study is that while there was no change in ankG position with respect to the soma of origin, the length significantly decreased from the distal end (Figure 6H). This finding adds to the repertoire of studies demonstrating AIS length modulation with altered neuronal activity (Evans et al., 2015), sensory circuit development (Kuba et al., 2010; Gutzmann et al., 2014), models of injury (Baalman et al., 2013; Hinman et al., 2013), and also congenital or acquired diseases (Kaphzan et al., 2011; Hamada and Kole, 2015; Clark et al., 2016).
Surprisingly, we did not observe parallel changes in NaV1.6 length at the distal AIS, the site of AP initiation (Stuart et al., 1997; Palmer and Stuart, 2006; Kole et al., 2008; Popovic et al., 2011). To our knowledge, this is the first report that demonstrates ankG remodeling uncoupled from changes in NaV1.6 geometric distribution. Hence, this observation raises the question whether other AIS components may stabilize NaV1.6 in the absence of ankG. For example, the cell adhesion molecule neurofascin-186, which is known to stabilize the AIS (Ratcliffe et al., 2001; Hedstrom et al., 2007; Zonta et al., 2011; Kriebel et al., 2012). Alternatively, a decrease in ankG length could elicit a subtle decrease NaV1.6 density (Hedstrom et al., 2008; Gasser et al., 2012) in the absence of length change. In such a case, a decrease in ankG length from the distal end resulting in NaV1.6 redistribution may be a potential mechanism underlying AIS functional changes discussed below. On the other hand, NaV1.6 activity and/or association with ankG may be modulated biochemically (Garrido et al., 2003; Bréchet et al., 2008; Hund et al., 2010; Evans et al., 2015). Notably, is the potential involvement of calcineurin, a phosphatase that has been implicated in both AIS plasticity (Evans et al., 2013, 2015) and mTBI axonal pathophysiology (Singleton et al., 2001; Marmarou and Povlishock, 2006; Reeves et al., 2007; Campbell et al., 2012). Ultimately, in addition to this studies limitation of a single time point, our morphological analyses were confined to ankG and NaV1.6. Thus, future studies are needed with more time points that target other AIS structural proteins as well as different voltage-gate ion channels modulating AP dynamics.
Evidence for AIS Functional Plasticity
Following our structural studies with electrophysiological assessments revealed further evidence for intact YFP+ pyramidal neuron AIS plasticity after mTBI. While AP waveform was not overtly different after mTBI, we did find a significant change in the decay tau. Interestingly, this suggested potential involvement of potassium channels within the AIS, which have been shown to redistribute after sensory deprivation (Kuba et al., 2015) and modulate AP waveform (Kole et al., 2007). Additionally, we observed that the AP threshold was more depolarized after mTBI. Since NaV1.6 channels set AP threshold (Kole et al., 2008), this provided indirect evidence for intrinsic AIS functional plasticity localized at the distal AIS. Notwithstanding the maintenance of NaV1.6 geometry after mTBI, patch-clamp recordings show AP acceleration is attenuated at the AIS but not the soma. Together, these observations lend further support to the supposition that subtler AIS ion channel redistribution and/or post-translation modification may be operant in the absence of morphological changes. Computer simulations using a realistic model of a layer 5 pyramidal neuron supported this premise. Specifically, decreasing either AIS length by experimentally observed values or NaV1.6 density by the same proportion also decreased AP acceleration, consistent with the data reported by Hallermann et al. (2012) in their characterization of this layer 5 pyramidal neuron model. All together, these data reflect slower NaV1.6 activation specifically at the AIS. Based on these collective findings, we now posit that at 2 days post-mTBI the AIS is in a state of flux, wherein the initial ankG structural remodeling precedes the NaV1.6 geometric redistribution that leads to length reduction.
Diffuse Loss of Perisomatic GABAergic Terminals
Neocortical balance of excitation-inhibition is regulated by PV+ interneurons that are highly interconnected with local pyramidal neurons (Gulyás et al., 1999; Pouille and Scanziani, 2001; Isaacson and Scanziani, 2011; Packer and Yuste, 2011; Hu et al., 2014; Scholl et al., 2015). The proximity of PV+ terminals to the AIS allows for strong inhibitory control over AIS output (Cobb et al., 1995; Klausberger et al., 2003; Freund and Katona, 2007; Atallah et al., 2012; Pouille et al., 2013; Lazarus et al., 2015; Wefelmeyer et al., 2015). Previously, it has been demonstrated that fulminant deafferentation of sensory input can induce AIS structural plasticity associated with altered neuronal excitability (Kuba et al., 2010). In light of the known features of AIS structure-function and that mTBI-induced abnormal network activity is associated with imbalanced excitation-inhibition, we also evaluated extrinsic/synaptic structural changes in concert with our AIS investigations. In the current study, quantitative analysis of layer 5 intact YFP+ pyramidal neuron perisomatic GAD67+ and PV+/GAD67+ puncta revealed a significant decrease in density. While determining the actual functional consequences of this structural finding is beyond the scope of the current study, this parallel observation to our AIS findings provides additional evidence that local neocortical disruption is a consistent feature of mTBI. In future studies, it would be important to determine if such loss of presynaptic GABAergic input elicits postsynaptic AIS shortening to decrease pyramidal neuron excitability to restore network homeostasis after mTBI.
While the mechanism underlying PV+ interneuron terminal loss was not critically addressed in the current study, recently we showed for the first time that locally projecting neocortical GABAergic interneurons undergo DAI (Vascak et al., 2017). Specifically, we showed that the majority of GABAergic DAI is represented by the PV+ interneuron subclass. Importantly, PV+ interneuron DAI manifested shortly after mTBI and occurred primarily in the perisomatic/AIS domain, prior to the initial branch point of their expansive axonal arbor. Since PV+ interneurons target hundreds of postsynaptic pyramidal neurons (Woodruff et al., 2010; Packer and Yuste, 2011; Taniguchi et al., 2013; Scholl et al., 2015; Takács et al., 2015), disconnection prior to the initial branch point results in widespread, diffuse inhibitory terminal loss. In our recent study, widespread PV+ axonal debris fields within 1 day post-mTBI indicated diffuse terminal degeneration. Consistent with PV+ interneuron DAI and anterograde deafferentation of postsynaptic target sites, in an earlier report from our laboratory we showed ultrastructural evidence of diffuse terminal degeneration along the perisomatic domain of healthy pyramidal neurons within layer 5 neocortex within 2 days post-mTBI (Singleton et al., 2002). Together, these findings suggest that the observed loss of PV+ terminals along the perisomatic domain of intact YFP+ pyramidal neurons is likely due to DAI-induced deafferentation of target postsynaptic sites.
Conclusion
The AIS is a highly complex and dynamic domain (Hallermann et al., 2012; King et al., 2014; Evans et al., 2015). Intrinsically linked structure-function relationships between scaffolding proteins and ion channels are a crucial for AIS activity (Zhou et al., 1998; Jenkins and Bennett, 2001; Yang et al., 2007). In this way, the neocortical pyramidal neuron AIS is a major determinant of network function and homeostatic plasticity (Grubb et al., 2011; Bender and Trussell, 2012; Kole and Stuart, 2012). This premise has been evaluated in several studies showing AIS disruption associated with multiple disease states involving an imbalance of network function (Kaphzan et al., 2011; Hinman et al., 2013; Hamada and Kole, 2015; Clark et al., 2016). The findings of this study broaden the cerebral landscape affected by mTBI, emphasizing the role of postsynaptic functional disruption of intact neurons. Based on the current data, we posit that AIS plasticity may constitute a compensatory response to mTBI. However, future studies are needed to determine whether AIS structural-functional plasticity is adaptive or maladaptive, which has important implications for restoring excitatory-inhibitory balance in neocortical circuits disrupted by mTBI.
Author Contributions
MV, KMJ and JTP contributed to the design of the experiments. MV performed all surgeries and confocal imaging. JS collected all electrophysiological data. Both MV and MB performed statistical data analyses. MV, JS, KMJ and JTP contributed to writing of the manuscript.
Funding
This work was funded by National Institutes of Health (NIH) Grant NS077675.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Carol Davis, Susan Walker and Jesse Sims for their invaluable technical assistance, Scott Henderson and Frances White for sharing their expertise in confocal microscopy, Jeff Dupree, Anders Hanell, Audrey Lafrenaye, Vishal Patel and Chris Jurgens for their scientific insights.
Abbreviations
aCSF, artificial cerebral spinal fluid; AIS, axon initial segment; ankG, ankyrin-G; AP, action potential(s); BPM, beats per minute; cFPI, central fluid percussion injury; CI, confidence interval; DAI, diffuse axonal injury; dSCWM, coefficient of distance from subcortical white mater; FOV, field-of-view; GAD67, glutamate decarboxylase (67 kDa); Kv1, voltage-gated potassium channel; mTBI, mild traumatic brain injury; NaV, voltage-gated sodium channel; PBS, phosphate buffered saline; PV, parvalbumin; ROI, region-of-interest; RPM, respirations per minute; S1BF, primary somatosensory barrel field; SCWM, subcortical white matter; SD, standard deviation; SEM, standard error of the mean; TBI, traumatic brain injury; YFP, yellow fluorescent protein.
Footnotes
References
Aarts, E., Verhage, M., Veenvliet, J. V., Dolan, C. V., and van der Sluis, S. (2014). A solution to dependency: using multilevel analysis to accommodate nested data. Nat. Neurosci. 17, 491–496. doi: 10.1038/nn.3648
Alexander, M. P. (1995). Mild traumatic brain injury: pathophysiology, natural history, and clinical management. Neurology 45, 1253–1260. doi: 10.1212/WNL.45.7.1253
Andriessen, T. M. J. C., Jacobs, B., and Vos, P. E. (2010). Clinical characteristics and pathophysiological mechanisms of focal and diffuse traumatic brain injury. J. Cell. Mol. Med. 14, 2381–2392. doi: 10.1111/j.1582-4934.2010.01164.x
Antón-Fernández, A., Rubio-Garrido, P., DeFelipe, J., and Muñoz, A. (2015). Selective presence of a giant saccular organelle in the axon initial segment of a subpopulation of layer V pyramidal neurons. Brain Struct. Funct. 220, 869–884. doi: 10.1007/s00429-013-0689-1
Atallah, B. V., Bruns, W., Carandini, M., and Scanziani, M. (2012). Parvalbumin-expressing interneurons linearly transform cortical responses to visual stimuli. Neuron 73, 159–170. doi: 10.1016/j.neuron.2011.12.013
Baalman, K. L., Cotton, R. J., Rasband, S. N., and Rasband, M. N. (2013). Blast wave exposure impairs memory and decreases axon initial segment length. J. Neurotrauma 30, 741–751. doi: 10.1089/neu.2012.2478
Bashir, S., Vernet, M., Yoo, W.-K., Mizrahi, I., Theoret, H., and Pascual-Leone, A. (2012). Changes in cortical plasticity after mild traumatic brain injury. Restor. Neurol. Neurosci. 30, 277–282. doi: 10.3233/RNN-2012-110207
Bazarian, J. J., Zhong, J., Blyth, B., Zhu, T., Kavcic, V., and Peterson, D. (2007). Diffusion tensor imaging detects clinically important axonal damage after mild traumatic brain injury: a pilot study. J. Neurotrauma 24, 1447–1459. doi: 10.1089/neu.2007.0241
Bender, K. J., and Trussell, L. O. (2012). The physiology of the axon initial segment. Annu. Rev. Neurosci. 35, 249–265. doi: 10.1146/annurev-neuro-062111-150339
Bigler, E. D., and Maxwell, W. L. (2012). Neuropathology of mild traumatic brain injury: relationship to neuroimaging findings. Brain Imaging Behav. 6, 108–136. doi: 10.1007/s11682-011-9145-0
Bonnelle, V., Leech, R., Kinnunen, K. M., Ham, T. E., Beckmann, C. F., De Boissezon, X., et al. (2011). Default mode network connectivity predicts sustained attention deficits after traumatic brain injury. J. Neurosci. 31, 13442–13451. doi: 10.1523/JNEUROSCI.1163-11.2011
Borg, J., Holm, L., Cassidy, J. D., Peloso, P. M., Carroll, L. J., von Holst, H., et al. (2004). Diagnostic procedures in mild traumatic brain injury: results of the WHO collaborating centre task force on mild traumatic brain injury. J. Rehabil. Med. 36, 61–75. doi: 10.1080/16501960410023822
Bouix, S., Pasternak, O., Rathi, Y., Pelavin, P. E., Zafonte, R., and Shenton, M. E. (2013). Increased gray matter diffusion anisotropy in patients with persistent post-concussive symptoms following mild traumatic brain injury. PLoS One 8:e66205. doi: 10.1371/journal.pone.0066205
Bréchet, A., Fache, M. P., Brachet, A., Ferracci, G., Baude, A., Irondelle, M., et al. (2008). Protein kinase CK2 contributes to the organization of sodium channels in axonal membranes by regulating their interactions with ankyrin G. J. Cell Biol. 183, 1101–1114. doi: 10.1083/jcb.200805169
Buffington, S. A., and Rasband, M. N. (2011). The axon initial segment in nervous system disease and injury. Eur. J. Neurosci. 34, 1609–1619. doi: 10.1111/j.1460-9568.2011.07875.x
Büki, A., and Povlishock, J. T. (2006). All roads lead to disconnection?—Traumatic axonal injury revisited. Acta Neurochir. (Wien) 148, 181–193; discussion 193–194. doi: 10.1007/s00701-005-0674-4
Button, K. S., Ioannidis, J. P. A., Mokrysz, C., Nosek, B. A., Flint, J., Robinson, E. S. J., et al. (2013). Power failure: why small sample size undermines the reliability of neuroscience. Nat. Rev. Neurosci. 14, 365–376. doi: 10.1038/nrn3475
Campbell, J. N., Register, D., and Churn, S. B. (2012). Traumatic brain injury causes an FK506-sensitive loss and an overgrowth of dendritic spines in rat forebrain. J. Neurotrauma 29, 201–217. doi: 10.1089/neu.2011.1761
Cardin, J. A., Carlén, M., Meletis, K., Knoblich, U., Zhang, F., Deisseroth, K., et al. (2009). Driving fast-spiking cells induces γ rhythm and controls sensory responses. Nature 459, 663–667. doi: 10.1038/nature08002
Cassidy, J. D., Carroll, L. J., Peloso, P. M., Borg, J., von Holst, H., Holm, L., et al. (2004). Incidence, risk factors and prevention of mild traumatic brain injury: results of the WHO collaborating centre task force on mild traumatic brain injury. J. Rehabil. Med. 36, 28–60. doi: 10.1080/16501960410023732
CDC. (2003). Report to Congress on Mild Traumatic Brain Injury in the United States: Steps to Prevent a Serious Public Health Problem. Atlanta, GA: Centers for Disease Control and Prevention.
Cesarovic, N., Nicholls, F., Rettich, A., Kronen, P., Hässig, M., Jirkof, P., et al. (2010). Isoflurane and sevoflurane provide equally effective anaesthesia in laboratory mice. Lab. Anim. 44, 329–336. doi: 10.1258/la.2010.009085
Chagnac-Amitai, Y., Luhmann, H. J., and Prince, D. A. (1990). Burst generating and regular spiking layer 5 pyramidal neurons of rat neocortex have different morphological features. J. Comp. Neurol. 296, 598–613. doi: 10.1002/cne.902960407
Clark, K. C., Josephson, A., Benusa, S. D., Hartley, R. K., Baer, M., Thummala, S., et al. (2016). Compromised axon initial segment integrity in EAE is preceded by microglial reactivity and contact. Glia 64, 1190–1209. doi: 10.1002/glia.22991
Cobb, S. R., Buhl, E. H., Halasy, K., Paulsen, O., and Somogyi, P. (1995). Synchronization of neuronal activity in hippocampus by individual GABAergic interneurons. Nature 378, 75–78. doi: 10.1038/378075a0
Cohen, A. S., Pfister, B. J., Schwarzbach, E., Grady, M. S., Goforth, P. B., and Satin, L. S. (2007). Injury-induced alterations in CNS electrophysiology. Prog. Brain Res. 161, 143–169. doi: 10.1016/s0079-6123(06)61010-8
Coronado, V. G., Xu, L., Basavaraju, S. V., McGuire, L. C., Wald, M. M., Faul, M. D., et al. (2011). Surveillance for traumatic brain injury-related deaths—United States, 1997–2007. MMWR Surveill. Summ. 60, 1–32.
DeFelipe, J., Alonso-Nanclares, L., and Arellano, J. I. (2003). Microstructure of the neocortex: comparative aspects. J. Neurocytol. 31, 299–316. doi: 10.1023/A:1024130211265
DeFelipe, J., and Fariñas, I. (1992). The pyramidal neuron of the cerebral cortex: morphological and chemical characteristics of the synaptic inputs. Prog. Neurobiol. 39, 563–607. doi: 10.1016/0301-0082(92)90015-7
Dixon, C. E., Lyeth, B. G., Povlishock, J. T., Findling, R. L., Hamm, R. J., Marmarou, A., et al. (1987). A fluid percussion model of experimental brain injury in the rat. J. Neurosurg. 67, 110–119. doi: 10.3171/jns.1987.67.1.0110
Douglas, R. J., Koch, C., Mahowald, M., Martin, K. A., and Suarez, H. H. (1995). Recurrent excitation in neocortical circuits. Science 269, 981–985. doi: 10.1126/science.7638624
Evans, M. D., Dumitrescu, A. S., Kruijssen, D. L. H., Taylor, S. E., and Grubb, M. S. (2015). Rapid modulation of axon initial segment length influences repetitive spike firing. Cell Rep. 13, 1233–1245. doi: 10.1016/j.celrep.2015.09.066
Evans, M. D., Sammons, R. P., Lebron, S., Dumitrescu, A. S., Watkins, T. B. K., Uebele, V. N., et al. (2013). Calcineurin signaling mediates activity-dependent relocation of the axon initial segment. J. Neurosci. 33, 6950–6963. doi: 10.1523/JNEUROSCI.0277-13.2013
Ewald, A. J., Werb, Z., and Egeblad, M. (2011). Monitoring of vital signs for long-term survival of mice under anesthesia. Cold Spring Harb. Protoc. 2011:pdb.prot5563. doi: 10.1101/pdb.prot5563
Fairen, A., DeFelipe, J., and Regidor, J. (1984). “Nonpyramidal neurons: general account,” in Cellular Components of the Cerebral Cortex, eds A. Peters and E. G. Jones (New York, NY: Plenum Press), 201–253.
Feng, G., Mellor, R. H., Bernstein, M., Keller-Peck, C., Nguyen, Q. T., Wallace, M., et al. (2000). Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron 28, 41–51. doi: 10.1016/s0896-6273(00)00084-2
Fish, K. N., Sweet, R. A., and Lewis, D. A. (2011). Differential distribution of proteins regulating GABA synthesis and reuptake in axon boutons of subpopulations of cortical interneurons. Cereb. Cortex 21, 2450–2460. doi: 10.1093/cercor/bhr007
Freund, T. F. (2003). Interneuron Diversity series: rhythm and mood in perisomatic inhibition. Trends Neurosci. 26, 489–495. doi: 10.1016/s0166-2236(03)00227-3
Freund, T. F., and Katona, I. (2007). Perisomatic inhibition. Neuron 56, 33–42. doi: 10.1016/j.neuron.2007.09.012
Galbraith, S., Daniel, J. A., and Vissel, B. (2010). A study of clustered data and approaches to its analysis. J. Neurosci. 30, 10601–10608. doi: 10.1523/JNEUROSCI.0362-10.2010
Garrido, J. J., Giraud, P., Carlier, E., Fernandes, F., Moussif, A., Fache, M.-P., et al. (2003). A targeting motif involved in sodium channel clustering at the axonal initial segment. Science 300, 2091–2094. doi: 10.1126/science.1085167
Gasser, A., Ho, T. S.-Y., Cheng, X., Chang, K.-J., Waxman, S. G., Rasband, M. N., et al. (2012). An ankyrinG-binding motif is necessary and sufficient for targeting Nav1.6 sodium channels to axon initial segments and nodes of Ranvier. J. Neurosci. 32, 7232–7243. doi: 10.1523/JNEUROSCI.5434-11.2012
Greer, J. E., Hånell, A., McGinn, M. J., and Povlishock, J. T. (2013). Mild traumatic brain injury in the mouse induces axotomy primarily within the axon initial segment. Acta Neuropathol. 126, 59–74. doi: 10.1007/s00401-013-1119-4
Greer, J. E., McGinn, M. J., and Povlishock, J. T. (2011). Diffuse traumatic axonal injury in the mouse induces atrophy, c-Jun activation, and axonal outgrowth in the axotomized neuronal population. J. Neurosci. 31, 5089–5105. doi: 10.1523/JNEUROSCI.5103-10.2011
Greer, J. E., Povlishock, J. T., and Jacobs, K. M. (2012). Electrophysiological abnormalities in both axotomized and nonaxotomized pyramidal neurons following mild traumatic brain injury. J. Neurosci. 32, 6682–6687. doi: 10.1523/JNEUROSCI.0881-12.2012
Grimm, K., Lamont, L., Tranquilli, W., Greene, S., and Roberston, S. (2015). Veterinary Anesthesia and Analgesia. 5th Edn. Ames, IA: John Wiley and Sons.
Grubb, M. S., and Burrone, J. (2010). Activity-dependent relocation of the axon initial segment fine-tunes neuronal excitability. Nature 465, 1070–1074. doi: 10.1038/nature09160
Grubb, M. S., Shu, Y., Kuba, H., Rasband, M. N., Wimmer, V. C., and Bender, K. J. (2011). Short- and long-term plasticity at the axon initial segment. J. Neurosci. 31, 16049–16055. doi: 10.1523/JNEUROSCI.4064-11.2011
Gulyás, A. I., Megías, M., Emri, Z., and Freund, T. F. (1999). Total number and ratio of excitatory and inhibitory synapses converging onto single interneurons of different types in the CA1 area of the rat hippocampus. J. Neurosci. 19, 10082–10097.
Gutzmann, A., Ergül, N., Grossmann, R., Schultz, C., Wahle, P., and Engelhardt, M. (2014). A period of structural plasticity at the axon initial segment in developing visual cortex. Front. Neuroanat. 8:11. doi: 10.3389/fnana.2014.00011
Hallermann, S., de Kock, C. P. J., Stuart, G. J., and Kole, M. H. P. (2012). State and location dependence of action potential metabolic cost in cortical pyramidal neurons. Nat. Neurosci. 15, 1007–1014. doi: 10.1038/nn.3132
Hamada, M. S., Goethals, S., de Vries, S. I., Brette, R., and Kole, M. H. P. (2016). Covariation of axon initial segment location and dendritic tree normalizes the somatic action potential. Proc. Natl. Acad. Sci. U S A 113, 14841–14846. doi: 10.1073/pnas.1607548113
Hamada, M. S., and Kole, M. H. P. (2015). Myelin loss and axonal ion channel adaptations associated with gray matter neuronal hyperexcitability. J. Neurosci. 35, 7272–7286. doi: 10.1523/JNEUROSCI.4747-14.2015
Hånell, A., Greer, J. E., and Jacobs, K. M. (2015a). Increased network excitability due to altered synaptic inputs to neocortical layer V intact and axotomized pyramidal neurons after mild traumatic brain injury. J. Neurotrauma 32, 1590–1598. doi: 10.1089/neu.2014.3592
Hånell, A., Greer, J. E., McGinn, M. J., and Povlishock, J. T. (2015b). Traumatic brain injury-induced axonal phenotypes react differently to treatment. Acta Neuropathol. 129, 317–332. doi: 10.1007/s00401-014-1376-x
Hasenstaub, A., Shu, Y., Haider, B., Kraushaar, U., Duque, A., and McCormick, D. A. (2005). Inhibitory postsynaptic potentials carry synchronized frequency information in active cortical networks. Neuron 47, 423–435. doi: 10.1016/j.neuron.2005.06.016
Hattox, A. M., and Nelson, S. B. (2007). Layer V neurons in mouse cortex projecting to different targets have distinct physiological properties. J. Neurophysiol. 98, 3330–3340. doi: 10.1152/jn.00397.2007
Hedstrom, K. L., Ogawa, Y., and Rasband, M. N. (2008). AnkyrinG is required for maintenance of the axon initial segment and neuronal polarity. J. Cell Biol. 183, 635–640. doi: 10.1083/jcb.200806112
Hedstrom, K. L., Xu, X., Ogawa, Y., Frischknecht, R., Seidenbecher, C. I., Shrager, P., et al. (2007). Neurofascin assembles a specialized extracellular matrix at the axon initial segment. J. Cell Biol. 178, 875–886. doi: 10.1083/jcb.200705119
Hinman, J. D., Rasband, M. N., and Carmichael, S. T. (2013). Remodeling of the axon initial segment after focal cortical and white matter stroke. Stroke 44, 182–189. doi: 10.1161/STROKEAHA.112.668749
Hoge, C. W., McGurk, D., Thomas, J. L., Cox, A. L., Engel, C. C., and Castro, C. A. (2008). Mild traumatic brain injury in U.S. Soldiers returning from Iraq. N. Engl. J. Med. 358, 453–463. doi: 10.1056/NEJMoa072972
Hu, H., Gan, J., and Jonas, P. (2014). Interneurons. Fast-spiking, parvalbumin+ GABAergic interneurons: from cellular design to microcircuit function. Science 345:1255263. doi: 10.1126/science.1255263
Hu, W., Tian, C., Li, T., Yang, M., Hou, H., and Shu, Y. (2009). Distinct contributions of Nav1.6 and Nav1.2 in action potential initiation and backpropagation. Nat. Neurosci. 12, 996–1002. doi: 10.1038/nn.2359
Huang, M.-X., Harrington, D. L., Robb Swan, A., Angeles Quinto, A., Nichols, S., Drake, A., et al. (2017). Resting-state magnetoencephalography reveals different patterns of aberrant functional connectivity in combat-related mild traumatic brain injury. J. Neurotrauma 34, 1412–1426. doi: 10.1089/neu.2016.4581
Hund, T. J., Koval, O. M., Li, J., Wright, P. J., Qian, L., Snyder, J. S., et al. (2010). A βIV-spectrin/CaMKII signaling complex is essential for membrane excitability in mice. J. Clin. Invest. 120, 3508–3519. doi: 10.1172/JCI43621
Ilvesmäki, T., Luoto, T. M., Hakulinen, U., Brander, A., Ryymin, P., Eskola, H., et al. (2014). Acute mild traumatic brain injury is not associated with white matter change on diffusion tensor imaging. Brain 137, 1876–1882. doi: 10.1093/brain/awu095
Isaacson, J. S., and Scanziani, M. (2011). How inhibition shapes cortical activity. Neuron 72, 231–243. doi: 10.1016/j.neuron.2011.09.027
Jacob, V., Petreanu, L., Wright, N., Svoboda, K., and Fox, K. (2012). Regular spiking and intrinsic bursting pyramidal cells show orthogonal forms of experience-dependent plasticity in layer V of barrel cortex. Neuron 73, 391–404. doi: 10.1016/j.neuron.2011.11.034
Jenkins, S. M., and Bennett, V. (2001). Ankyrin-G coordinates assembly of the spectrin-based membrane skeleton, voltage-gated sodium channels, and L1 CAMs at Purkinje neuron initial segments. J. Cell Biol. 155, 739–746. doi: 10.1083/jcb.200109026
Jiao, Y., Sun, Z., Lee, T., Fusco, F. R., Kimble, T. D., Meade, C. A., et al. (1999). A simple and sensitive antigen retrieval method for free-floating and slide-mounted tissue sections. J. Neurosci. Methods 93, 149–162. doi: 10.1016/s0165-0270(99)00142-9
Johnson, V. E., Stewart, W., and Smith, D. H. (2013). Axonal pathology in traumatic brain injury. Exp. Neurol. 246, 35–43. doi: 10.1016/j.expneurol.2012.01.013
Kaphzan, H., Buffington, S. A., Jung, J. I., Rasband, M. N., and Klann, E. (2011). Alterations in intrinsic membrane properties and the axon initial segment in a mouse model of Angelman syndrome. J. Neurosci. 31, 17637–17648. doi: 10.1523/JNEUROSCI.4162-11.2011
Khaliq, Z. M., and Raman, I. M. (2006). Relative contributions of axonal and somatic Na channels to action potential initiation in cerebellar Purkinje neurons. J. Neurosci. 26, 1935–1944. doi: 10.1523/JNEUROSCI.4664-05.2006
King, A. N., Manning, C. F., and Trimmer, J. S. (2014). A unique ion channel clustering domain on the axon initial segment of mammalian neurons. J. Comp. Neurol. 522, 2594–2608. doi: 10.1002/cne.23551
Kinnunen, K. M., Greenwood, R., Powell, J. H., Leech, R., Hawkins, P. C., Bonnelle, V., et al. (2011). White matter damage and cognitive impairment after traumatic brain injury. Brain 134, 449–463. doi: 10.1093/brain/awq347
Klausberger, T., and Somogyi, P. (2008). Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science 321, 53–57. doi: 10.1126/science.1149381
Klausberger, T., Magill, P. J., Márton, L. F., Roberts, J. D. B., Cobden, P. M., Buzsáki, G., et al. (2003). Brain-state- and cell-type-specific firing of hippocampal interneurons in vivo. Nature 421, 844–848. doi: 10.1038/nature01374
Kole, M. H. P., Ilschner, S. U., Kampa, B. M., Williams, S. R., Ruben, P. C., and Stuart, G. J. (2008). Action potential generation requires a high sodium channel density in the axon initial segment. Nat. Neurosci. 11, 178–186. doi: 10.1038/nn2040
Kole, M. H. P., Letzkus, J. J., and Stuart, G. J. (2007). Axon initial segment Kv1 channels control axonal action potential waveform and synaptic efficacy. Neuron 55, 633–647. doi: 10.1016/j.neuron.2007.07.031
Kole, M. H. P., and Stuart, G. J. (2012). Signal processing in the axon initial segment. Neuron 73, 235–247. doi: 10.1016/j.neuron.2012.01.007
Kriebel, M., Wuchter, J., Trinks, S., and Volkmer, H. (2012). Neurofascin: a switch between neuronal plasticity and stability. Int. J. Biochem. Cell Biol. 44, 694–697. doi: 10.1016/j.biocel.2012.01.012
Kuba, H. (2012). Structural tuning and plasticity of the axon initial segment in auditory neurons. J. Physiol. 590, 5571–5579. doi: 10.1113/jphysiol.2012.237305
Kuba, H., Adachi, R., and Ohmori, H. (2014). Activity-dependent and activity-independent development of the axon initial segment. J. Neurosci. 34, 3443–3453. doi: 10.1523/JNEUROSCI.4357-13.2014
Kuba, H., Oichi, Y., and Ohmori, H. (2010). Presynaptic activity regulates Na+ channel distribution at the axon initial segment. Nature 465, 1075–1078. doi: 10.1038/nature09087
Kuba, H., Yamada, R., Ishiguro, G., and Adachi, R. (2015). Redistribution of Kv1 and Kv7 enhances neuronal excitability during structural axon initial segment plasticity. Nat. Commun. 6:8815. doi: 10.1038/ncomms9815
Langlois, J. A., Rutland-Brown, W., and Wald, M. M. (2006). The epidemiology and impact of traumatic brain injury: a brief overview. J. Head Trauma Rehabil. 21, 375–378. doi: 10.1097/00001199-200609000-00001
Lazarus, M. S., Krishnan, K., and Huang, Z. J. (2015). GAD67 deficiency in parvalbumin interneurons produces deficits in inhibitory transmission and network disinhibition in mouse prefrontal cortex. Cereb. Cortex 25, 1290–1296. doi: 10.1093/cercor/bht322
Le Bé, J.-V., Silberberg, G., Wang, Y., and Markram, H. (2007). Morphological, electrophysiological, and synaptic properties of corticocallosal pyramidal cells in the neonatal rat neocortex. Cereb. Cortex 17, 2204–2213. doi: 10.1093/cercor/bhl127
Lichtman, J. W., and Conchello, J.-A. (2005). Fluorescence microscopy. Nat. Methods 2, 910–919. doi: 10.1038/nmeth817
Ling, J. M., Klimaj, S., Toulouse, T., and Mayer, A. R. (2013). A prospective study of gray matter abnormalities in mild traumatic brain injury. Neurology 81, 2121–2127. doi: 10.1212/01.WNL.0000437302.36064.b1
Lorincz, A., and Nusser, Z. (2008a). Cell-type-dependent molecular composition of the axon initial segment. J. Neurosci. 28, 14329–14340. doi: 10.1523/JNEUROSCI.4833-08.2008
Lorincz, A., and Nusser, Z. (2008b). Specificity of immunoreactions: the importance of testing specificity in each method. J. Neurosci. 28, 9083–9086. doi: 10.1523/JNEUROSCI.2494-08.2008
Maas, C. J. M., and Hox, J. J. (2004). Robustness issues in multilevel regression analysis. Stat. Neerl. 58, 127–137. doi: 10.1046/j.0039-0402.2003.00252.x
Mac Donald, C. L., Johnson, A. M., Cooper, D., Nelson, E. C., Werner, N. J., Shimony, J. S., et al. (2011). Detection of blast-related traumatic brain injury in U.S. military personnel. N. Engl. J. Med. 364, 2091–2100. doi: 10.1056/NEJMoa1008069
Malec, J. F., Brown, A. W., Leibson, C. L., Flaada, J. T., Mandrekar, J. N., Diehl, N. N., et al. (2007). The mayo classification system for traumatic brain injury severity. J. Neurotrauma 24, 1417–1424. doi: 10.1089/neu.2006.0245
Manning, C. F., Bundros, A. M., and Trimmer, J. S. (2012). Benefits and pitfalls of secondary antibodies: why choosing the right secondary is of primary importance. PLoS One 7:e38313. doi: 10.1371/journal.pone.0038313
Mannix, R., Meehan, W. P. III, and Pascual-Leone, A. (2016). Sports-related concussions—media, science and policy. Nat. Rev. Neurol. 12, 486–490. doi: 10.1038/nrneurol.2016.99
Marmarou, C. R., and Povlishock, J. T. (2006). Administration of the immunophilin ligand FK506 differentially attenuates neurofilament compaction and impaired axonal transport in injured axons following diffuse traumatic brain injury. Exp. Neurol. 197, 353–362. doi: 10.1016/j.expneurol.2005.10.003
Mayer, A. R., Ling, J., Mannell, M. V., Gasparovic, C., Phillips, J. P., Doezema, D., et al. (2010). A prospective diffusion tensor imaging study in mild traumatic brain injury. Neurology 74, 643–650. doi: 10.1212/WNL.0b013e3181d0ccdd
Mayer, A. R., Mannell, M. V., Ling, J., Gasparovic, C., and Yeo, R. A. (2011). Functional connectivity in mild traumatic brain injury. Hum. Brain Mapp. 32, 1825–1835. doi: 10.1002/hbm.21151
Meeks, J. P., and Mennerick, S. (2007). Action potential initiation and propagation in CA3 pyramidal axons. J. Neurophysiol. 97, 3460–3472. doi: 10.1152/jn.01288.2006
Mesulam, M. M. (1990). Large-scale neurocognitive networks and distributed processing for attention, language, and memory. Ann. Neurol. 28, 597–613. doi: 10.1002/ana.410280502
Mittl, R. L., Grossman, R. I., Hiehle, J. F., Hurst, R. W., Kauder, D. R., Gennarelli, T. A., et al. (1994). Prevalence of MR evidence of diffuse axonal injury in patients with mild head injury and normal head CT findings. Am. J. Neuroradiol. 15, 1583–1589.
Molnár, Z., and Cheung, A. F. P. (2006). Towards the classification of subpopulations of layer V pyramidal projection neurons. Neurosci. Res. 55, 105–115. doi: 10.1016/j.neures.2006.02.008
Morehead, M., Bartus, R. T., Dean, R. L., Miotke, J. A., Murphy, S., Sall, J., et al. (1994). Histopathologic consequences of moderate concussion in an animal model: correlations with duration of unconsciousness. J. Neurotrauma 11, 657–667. doi: 10.1089/neu.1994.11.657
Mountcastle, V. (1979). “An organizing principle for cerebral function: the unit module and the distributed system,” in The Neurosciences, Fourth Study Program, eds F. Schmidt and F. Worden (Cambridge, MA: MIT Press), 21–42.
Newcombe, V. F. J., Outtrim, J. G., Chatfield, D. A., Manktelow, A., Hutchinson, P. J., Coles, J. P., et al. (2011). Parcellating the neuroanatomical basis of impaired decision-making in traumatic brain injury. Brain 134, 759–768. doi: 10.1093/brain/awq388
Nieuwenhuis, S., Forstmann, B. U., and Wagenmakers, E.-J. (2011). Erroneous analyses of interactions in neuroscience: a problem of significance. Nat. Neurosci. 14, 1105–1107. doi: 10.1038/nn.2886
Packer, A. M., and Yuste, R. (2011). Dense, unspecific connectivity of neocortical parvalbumin-positive interneurons: a canonical microcircuit for inhibition? J. Neurosci. 31, 13260–13271. doi: 10.1523/JNEUROSCI.3131-11.2011
Palacios, E. M., Yuh, E. L., Chang, Y.-S., Yue, J. K., Schnyer, D. M., Okonkwo, D. O., et al. (2017). Resting-state functional connectivity alterations associated with six-month outcomes in mild traumatic brain injury. J. Neurotrauma 34, 1546–1557. doi: 10.1089/neu.2016.4752
Palmer, L. M., and Stuart, G. J. (2006). Site of action potential initiation in layer 5 pyramidal neurons. J. Neurosci. 26, 1854–1863. doi: 10.1523/JNEUROSCI.4812-05.2006
Popovic, M. A., Foust, A. J., McCormick, D. A., and Zecevic, D. (2011). The spatio-temporal characteristics of action potential initiation in layer 5 pyramidal neurons: a voltage imaging study. J. Physiol. 589, 4167–4187. doi: 10.1113/jphysiol.2011.209015
Pouille, F., and Scanziani, M. (2001). Enforcement of temporal fidelity in pyramidal cells by somatic feed-forward inhibition. Science 293, 1159–1163. doi: 10.1126/science.1060342
Pouille, F., Watkinson, O., Scanziani, M., and Trevelyan, A. J. (2013). The contribution of synaptic location to inhibitory gain control in pyramidal cells. Physiol. Rep. 1:e00067. doi: 10.1002/phy2.67
Povlishock, J. T., and Katz, D. I. (2005). Update of neuropathology and neurological recovery after traumatic brain injury. J. Head Trauma Rehabil. 20, 76–94. doi: 10.1097/00001199-200501000-00008
Raichle, M. E. (2010). Two views of brain function. Trends Cogn. Sci. 14, 180–190. doi: 10.1016/j.tics.2010.01.008
Ratcliffe, C. F., Westenbroek, R. E., Curtis, R., and Catterall, W. A. (2001). Sodium channel β-1 and β-3 subunits associate with neurofascin through their extracellular immunoglobulin-like domain. J. Cell Biol. 154, 427–434. doi: 10.1083/jcb.200102086
Reeves, T. M., Phillips, L. L., Lee, N. N., and Povlishock, J. T. (2007). Preferential neuroprotective effect of tacrolimus (FK506) on unmyelinated axons following traumatic brain injury. Brain Res. 1154, 225–236. doi: 10.1016/j.brainres.2007.04.002
Saatman, K. E., Duhaime, A.-C., Bullock, R., Maas, A. I. R., Valadka, A., and Manley, G. T. (2008). Classification of traumatic brain injury for targeted therapies. J. Neurotrauma 25, 719–738. doi: 10.1089/neu.2008.0586
Salmond, C. H., Menon, D. K., Chatfield, D. A., Williams, G. B., Pena, A., Sahakian, B. J., et al. (2006). Diffusion tensor imaging in chronic head injury survivors: correlations with learning and memory indices. Neuroimage 29, 117–124. doi: 10.1016/j.neuroimage.2005.07.012
Scholl, B., Pattadkal, J. J., Dilly, G. A., Priebe, N. J., and Zemelman, B. V. (2015). Local integration accounts for weak selectivity of mouse neocortical parvalbumin interneurons. Neuron 87, 424–436. doi: 10.1016/j.neuron.2015.06.030
Schrader, H., Mickeviciene, D., Gleizniene, R., Jakstiene, S., Surkiene, D., Stovner, L. J., et al. (2009). Magnetic resonance imaging after most common form of concussion. BMC Med. Imaging 9:11. doi: 10.1186/1471-2342-9-11
Schubert, D., Kötter, R., Luhmann, H. J., and Staiger, J. F. (2006). Morphology, electrophysiology and functional input connectivity of pyramidal neurons characterizes a genuine layer va in the primary somatosensory cortex. Cereb. Cortex 16, 223–236. doi: 10.1093/cercor/bhi100
Schubert, D., Staiger, J. F., Cho, N., Kötter, R., Zilles, K., and Luhmann, H. J. (2001). Layer-specific intracolumnar and transcolumnar functional connectivity of layer V pyramidal cells in rat barrel cortex. J. Neurosci. 21, 3580–3592.
Sharp, D. J., Beckmann, C. F., Greenwood, R., Kinnunen, K. M., Bonnelle, V., De Boissezon, X., et al. (2011). Default mode network functional and structural connectivity after traumatic brain injury. Brain 134, 2233–2247. doi: 10.1093/brain/awr175
Sharp, D. J., and Ham, T. E. (2011). Investigating white matter injury after mild traumatic brain injury. Curr. Opin. Neurol. 24, 558–563. doi: 10.1097/WCO.0b013e32834cd523
Sharp, D. J., Scott, G., and Leech, R. (2014). Network dysfunction after traumatic brain injury. Nat. Rev. Neurol. 10, 156–166. doi: 10.1038/nrneurol.2014.15
Shaw, N. A. (2002). The neurophysiology of concussion. Prog. Neurobiol. 67, 281–344. doi: 10.1016/s0301-0082(02)00018-7
Shultz, S. R., McDonald, S. J., Vonder Haar, C., Meconi, A., Vink, R., van Donkelaar, P., et al. (2016). The potential for animal models to provide insight into mild traumatic brain injury: translational challenges and strategies. Neurosci. Biobehav. Rev. doi: 10.1016/j.neubiorev.2016.09.014 [Epub ahead of print].
Singleton, R. H., Stone, J. R., Okonkwo, D. O., Pellicane, A. J., and Povlishock, J. T. (2001). The immunophilin ligand FK506 attenuates axonal injury in an impact-acceleration model of traumatic brain injury. J. Neurotrauma 18, 607–614. doi: 10.1089/089771501750291846
Singleton, R. H., Zhu, J., Stone, J. R., and Povlishock, J. T. (2002). Traumatically induced axotomy adjacent to the soma does not result in acute neuronal death. J. Neurosci. 22, 791–802.
Snijders, T. A. B., and Bosker, R. J. (1993). Standard errors and sample sizes for two-level research. J. Educ. Stat. 18, 237–259. doi: 10.3102/10769986018003237
Somogyi, P., Tamás, G., Lujan, R., and Buhl, E. H. (1998). Salient features of synaptic organisation in the cerebral cortex. Brain Res. Rev. 26, 113–135.
Sponheim, S. R., McGuire, K. A., Kang, S. S., Davenport, N. D., Aviyente, S., Bernat, E. M., et al. (2011). Evidence of disrupted functional connectivity in the brain after combat-related blast injury. Neuroimage 54, S21–S29. doi: 10.1016/j.neuroimage.2010.09.007
Stuart, G., Schiller, J., and Sakmann, B. (1997). Action potential initiation and propagation in rat neocortical pyramidal neurons. J. Physiol. 505, 617–632. doi: 10.1111/j.1469-7793.1997.617ba.x
Styrke, J., Stålnacke, B.-M., Sojka, P., and Björnstig, U. (2007). Traumatic brain injuries in a well-defined population: epidemiological aspects and severity. J. Neurotrauma 24, 1425–1436. doi: 10.1089/neu.2007.0266
Sun, J., and Jacobs, K. M. (2016). Knockout of cyclophilin-d provides partial amelioration of intrinsic and synaptic properties altered by mild traumatic brain injury. Front. Syst. Neurosci. 10:63. doi: 10.3389/fnsys.2016.00063
Takács, V. T., Szonyi, A., Freund, T. F., Nyiri, G., and Gulyás, A. I. (2015). Quantitative ultrastructural analysis of basket and axo-axonic cell terminals in the mouse hippocampus. Brain Struct. Funct. 220, 919–940. doi: 10.1007/s00429-013-0692-6
Taniguchi, H., Lu, J., and Huang, Z. J. (2013). The spatial and temporal origin of chandelier cells in mouse neocortex. Science 339, 70–74. doi: 10.1126/science.1227622
Thome, C., Kelly, T., Yanez, A., Schultz, C., Engelhardt, M., Cambridge, S. B., et al. (2014). Axon-carrying dendrites convey privileged synaptic input in hippocampal neurons. Neuron 83, 1418–1430. doi: 10.1016/j.neuron.2014.08.013
Thurman, D. J., Alverson, C., Dunn, K. A., Guerrero, J., and Sniezek, J. E. (1999). Traumatic brain injury in the united states: a public health perspective. J. Head Trauma Rehabil. 14, 602–615. doi: 10.1097/00001199-199912000-00009
Tremblay, S., de Beaumont, L., Lassonde, M., and Théoret, H. (2011). Evidence for the specificity of intracortical inhibitory dysfunction in asymptomatic concussed athletes. J. Neurotrauma 28, 493–502. doi: 10.1089/neu.2010.1615
Varela, F., Lachaux, J. P., Rodriguez, E., and Martinerie, J. (2001). The brainweb: phase synchronization and large-scale integration. Nat. Rev. Neurosci. 2, 229–239. doi: 10.1038/35067550
Vascak, M., Jin, X., Jacobs, K. M., and Povlishock, J. T. (2017). Mild traumatic brain injury induces structural and functional disconnection of local neocortical inhibitory networks via parvalbumin interneuron diffuse axonal injury. Cereb. Cortex 4, 1–20. doi: 10.1093/cercor/bhx058
Wäljas, M., Lange, R. T., Hakulinen, U., Huhtala, H., Dastidar, P., Hartikainen, K., et al. (2014). Biopsychosocial outcome after uncomplicated mild traumatic brain injury. J. Neurotrauma 31, 108–124. doi: 10.1089/neu.2013.2941
Walsh, J. E. (1947). Concerning the effect of intraclass correlation on certain significance tests. Ann. Math. Stat. 18, 88–96. doi: 10.1214/aoms/1177730495
Wefelmeyer, W., Cattaert, D., and Burrone, J. (2015). Activity-dependent mismatch between axo-axonic synapses and the axon initial segment controls neuronal output. Proc. Natl. Acad. Sci. U S A 112, 9757–9762. doi: 10.1073/pnas.1502902112
Wolf, J. A., and Koch, P. F. (2016). Disruption of network synchrony and cognitive dysfunction after traumatic brain injury. Front. Syst. Neurosci. 10:43. doi: 10.3389/fnsys.2016.00043
Woodruff, A. R., Anderson, S. A., and Yuste, R. (2010). The enigmatic function of chandelier cells. Front. Neurosci. 4:201. doi: 10.3389/fnins.2010.00201
Yang, Y., Ogawa, Y., Hedstrom, K. L., and Rasband, M. N. (2007). βIV spectrin is recruited to axon initial segments and nodes of Ranvier by ankyrinG. J. Cell Biol. 176, 509–519. doi: 10.1083/jcb.200610128
Yizhar, O., Fenno, L. E., Prigge, M., Schneider, F., Davidson, T. J., O’Shea, D. J., et al. (2011). Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature 477, 171–178. doi: 10.1038/nature10360
Yuh, E. L., Mukherjee, P., Lingsma, H. F., Yue, J. K., Ferguson, A. R., Gordon, W. A., et al. (2013). Magnetic resonance imaging improves 3-month outcome prediction in mild traumatic brain injury. Ann. Neurol. 73, 224–235. doi: 10.1002/ana.23783
Zhang, K., Johnson, B., Pennell, D., Ray, W., Sebastianelli, W., and Slobounov, S. (2010). Are functional deficits in concussed individuals consistent with white matter structural alterations: combined FMRI and DTI study. Exp. Brain Res. 204, 57–70. doi: 10.1007/s00221-010-2294-3
Zhang, D., and Raichle, M. E. (2010). Disease and the brain’s dark energy. Nat. Rev. Neurol. 6, 15–28. doi: 10.1038/nrneurol.2009.198
Zhou, D., Lambert, S., Malen, P. L., Carpenter, S., Boland, L. M., and Bennett, V. (1998). AnkyrinG is required for clustering of voltage-gated Na channels at axon initial segments and for normal action potential firing. J. Cell Biol. 143, 1295–1304. doi: 10.1083/jcb.143.5.1295
Keywords: mild traumatic brain injury, axon initial segment, GABAergic synapse, plasticity, action potential initiation, layer 5 pyramidal neurons, transgenic mouse models, excitation-inhibition balance
Citation: Vascak M, Sun J, Baer M, Jacobs KM and Povlishock JT (2017) Mild Traumatic Brain Injury Evokes Pyramidal Neuron Axon Initial Segment Plasticity and Diffuse Presynaptic Inhibitory Terminal Loss. Front. Cell. Neurosci. 11:157. doi: 10.3389/fncel.2017.00157
Received: 21 February 2017; Accepted: 17 May 2017;
Published: 06 June 2017.
Edited by:
Maren Engelhardt, Heidelberg University, GermanyReviewed by:
Matthew N. Rasband, Baylor College of Medicine, United StatesLisa Mapelli, University of Pavia, Italy
Copyright © 2017 Vascak, Sun, Baer, Jacobs and Povlishock. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John T. Povlishock, am9obi5wb3ZsaXNob2NrQHZjdWhlYWx0aC5vcmc=