 Sean B. Christensen1
Sean B. Christensen1 Arik J. Hone1
Arik J. Hone1 Isabelle Roux2†Julie Kniazeff3Jean-Philippe Pin3Grégory Upert4
Isabelle Roux2†Julie Kniazeff3Jean-Philippe Pin3Grégory Upert4 Denis Servent4
Denis Servent4 Elisabeth Glowatzki2,5
Elisabeth Glowatzki2,5 J. Michael McIntosh1,6,7*
J. Michael McIntosh1,6,7*- 1Department of Biology, University of Utah, Salt Lake City, UT, United States
- 2Department of Otolaryngology, Head and Neck Surgery, The Center for Hearing and Balance and the Center for Sensory Biology, The Johns Hopkins University School of Medicine, Baltimore, MD, United States
- 3IGF, Centre National de la Recherche Scientifique, Institut National de la Santé et de la Recherche Médicale, Université Montpellier, Montpellier, France
- 4Service d'Ingénierie Moléculaire des Protéines, CEA, Université Paris-Saclay, Gif-sur-Yvette, France
- 5Department of Neuroscience, The Johns Hopkins University School of Medicine, Baltimore, MD, United States
- 6George E. Whalen Veterans Affairs Medical Center, Salt Lake City, UT, United States
- 7Department of Psychiatry, University of Utah, Salt Lake City, UT, United States
Transcripts for α9 and α10 nicotinic acetylcholine receptor (nAChR) subunits are found in diverse tissues. The function of α9α10 nAChRs is best known in mechanosensory cochlear hair cells, but elsewhere their roles are less well-understood. α9α10 nAChRs have been implicated as analgesic targets and α-conotoxins that block α9α10 nAChRs produce analgesia. However, some of these peptides show large potency differences between species. Additionally several studies have indicated that these conotoxins may also activate GABAB receptors (GABABRs). To further address these issues, we cloned the cDNAs of mouse α9 and α10 nAChR subunits. When heterologously expressed in Xenopus oocytes, the resulting α9α10 nAChRs had the expected pharmacology of being activated by acetylcholine and choline but not by nicotine. A conotoxin analog, RgIA4, potently, and selectively blocked mouse α9α10 nAChRs with low nanomolar affinity indicating that RgIA4 may be effectively used to study murine α9α10 nAChR function. Previous reports indicated that RgIA4 attenuates chemotherapy-induced cold allodynia. Here we demonstrate that RgIA4 analgesic effects following oxaliplatin treatment are sustained for 21 days after last RgIA4 administration indicating that RgIA4 may provide enduring protection against nerve damage. RgIA4 lacks activity at GABAB receptors; a bioluminescence resonance energy transfer assay was used to demonstrate that two other analgesic α-conotoxins, Vc1.1 and AuIB, also do not activate GABABRs expressed in HEK cells. Together these findings further support the targeting of α9α10 nAChRs in the treatment of pain.
Introduction
Neuropathic pain, caused by lesion or disease of the somatosensory system, is estimated to afflict 7–10% of the population (van Hecke et al., 2014). Common causes include traumatic nerve injury, metabolic disorders such as diabetes, viral infection, including herpes or HIV, and nerve injury induced by cancer chemotherapy. Although a variety of treatment options are available, a significant fraction of patients are treatment refractory leaving millions to endure long lasting pain states (Costigan et al., 2009; Colloca et al., 2017). In addition, polypharmacy to control pain can lead to significant side effect burden (Colloca et al., 2017).
Opioids, inspired by opium isolated from the poppy Papaver somniferum, are one of the most powerful available pain-reducing medications. Unfortunately, chronic neuropathic pain is not well-treated by opioids. In addition, opioid use is plagued by effect tolerance, addiction, and unintentional overdose. Over 12 million people in the U.S. are estimated to have abused opioids (Brady et al., 2016). Thus, non-opioid alternatives are a high priority.
Classical neuronal nicotinic acetylcholine receptors are ligand-gated ion channels assembled in pentamers composed of various α or α/β subunit combinations. Agonists of nAChRs have been intensively investigated as potential analgesics. The tobacco plant toxin nicotine is a nAChR agonist that has long been known to have antinociceptive properties; however dosage requirements and side effects from non-selective activity on multiple nAChR subtypes preclude practical use as an analgesic. Considerable effort has been expended to develop selective agonists of CNS α4β2 nAChRs and more recently α7 nAChR agonists and positive allosteric modulators as potential therapeutic agents (Umana et al., 2013).
The α9α10 subtype of nAChR has also been proposed as a novel analgesic target (Vincler et al., 2006; Vincler and McIntosh, 2007; McIntosh et al., 2009; Del Bufalo et al., 2014; Mohammadi and Christie, 2014). Interestingly, rather than CNS targeted agonists, peripherally acting α9α10 antagonists have shown promise (Vincler et al., 2006; Holtman et al., 2011; Zheng et al., 2011; Wala et al., 2012; Luo et al., 2015).
A particularly rich source of nAChR antagonist are marine mollusks of the genus Conus (Azam and McIntosh, 2009; Lebbe et al., 2014; Dutertre et al., 2017). Cone snails are carnivores that immobilize their prey by injecting them with a complex mixture of toxins. There are ~700 species of Conus. Of the species examined thus far, the great majority have documented components that are structurally similar to peptides known to block nAChRs. Thus, Conus may have thousands of novel nAChR targeted peptides. A very valuable online database, ConoServer, has documented reported Conus sequences (Kaas et al., 2012).
Three Conus peptides, that act on α9α10 nAChRs, Vc1.1, RgIA, and GeXIVA and analogs thereof have shown analgesic activity in several models of neuropathic pain (Satkunanathan et al., 2005; Vincler et al., 2006; Clark et al., 2010; Carstens et al., 2011; Di Cesare Mannelli et al., 2014; Luo et al., 2015; Castro et al., 2016; Pacini et al., 2016; Romero et al., 2017). Vc1.1 reached phase II human clinical trials, but was withdrawn in part, because it was discovered that Vc1.1 was much less potent at the human vs. rat α9α10 nAChR (Metabolic, 2007). Thus, species analysis is particularly important for this subgroup of α-conotoxins.
To our knowledge there are no previous reports of the properties of heterologously expressed mouse α9α10 nAChRs. In this report, nAChR subunits α9 and α10 were cloned and we examined the subtype selectivity of a newly developed antagonist α-conotoxin RgIA4 for mouse nAChRs. Further, we demonstrate the long acting analgesic effects of this peptide in a mouse model of chemotherapy induced neuropathy.
Materials and Methods
Animals
Animals for RNA extraction: 4 day old C57BL/6J mice (stock number 000664, Jackson Laboratory, Bar Harbor, ME, USA) were euthanized, and their inner ears were quickly removed from the temporal bones. All experimental procedures involving animals were approved by the Johns Hopkins University Animal Care and Use Committee. Animals for oxaliplatin experiments: Wild-type mice were CBA/CaJ (Jackson Labs, Bar Harbor ME). Germline α9 knockout (KO) mice (Abazeed et al., 2013) originally on a 129Sv/Ev and CBA/CaJ background were crossed using an accelerated backcrossing program (Jackson Labs) until 99.5% identity with wild-type CBA/CaJ mice was achieved. Mice were then further backcrossed with wild-type CBA/CaJ mice an additional three generations. Experiments involving animals have been reported according to ARRIVE guidelines (Kilkenny et al., 2010). All efforts were made to minimize animal suffering and to reduce the number of animals used. All experimental procedures were in accordance with the National Institutes of Health guidelines for the care and use of laboratory animals and were performed under approved protocols at the University of Utah.
RNA Extractions and Reverse Transcription-Polymerase Chain Reactions (RT-PCR)
Mouse cochlear neuroepithelia were isolated in RNAlater stabilization solution (ThermoFisher Scientific, Waltham, MA, USA) and quickly frozen. Total RNA was extracted using Trizol reagent (Invitrogen, ThermoFisher Scientific). Reverse transcriptase reactions were performed using Superscript III (ThermoFisher Scientific) with random primers p(dN)6 (Roche, Sigma-Aldrich, St Louis, MO, USA) and polymerase chain reaction (PCR) using the high fidelity TaKaRa LA Taq DNA Polymerase (TaKaRa Bio, Mountain View, CA, USA).
Primers 5′-cgACTAGTgttgggaaaggATGaaccggccccatccc-3′ and 5′-cgCTCGAGctaatctgctcttgctatgatcaagacgg-3′ were used to amplify Chrna9 coding sequence, and primers 5′-cgACTAGTccagcagggcctgttgctttacatctcc-3′ and 5′-cgCTCGAGttacagggcttgcaccagtaccaggaggc-3′, Chrna10 coding sequence. In both cases, these primers were designed to add SpeI and XhoI restriction sites 5′ and 3′ respectively of the coding sequences and to amplify part of the 5′UTR.
The 1,467 and 1,423 bp fragments amplified were isolated and purified after agarose gel separation and inserted into pGEMT vector (pGEMT Vector System, Promega). Sequences of the clones obtained were compared to the direct sequences of pooled independent PCR amplifications using cochlear neuroepithelia RT-PCR as template, to reference sequences (NM_001081104, NM_001081424), and to C57BL/6J genomic sequences using BLAT—UCSC Genome Browser (University of California—Santa Cruz, CA, USA) (GRCm38/mm10).
The sequence alignments presented in Figure 1 were obtained using Clustal Omega (Sievers et al., 2011) (MegAlign Pro, DNASTAR Lasergene, Madison, WI, USA). The following sequences were used as reference: Mus musculus (NP_001074573, NP_001074893), Rattus norvegicus (NP_075219, NP_072161), Homo sapiens (NP_060051, NP_065135) for α9 and α10 subunits, respectively.
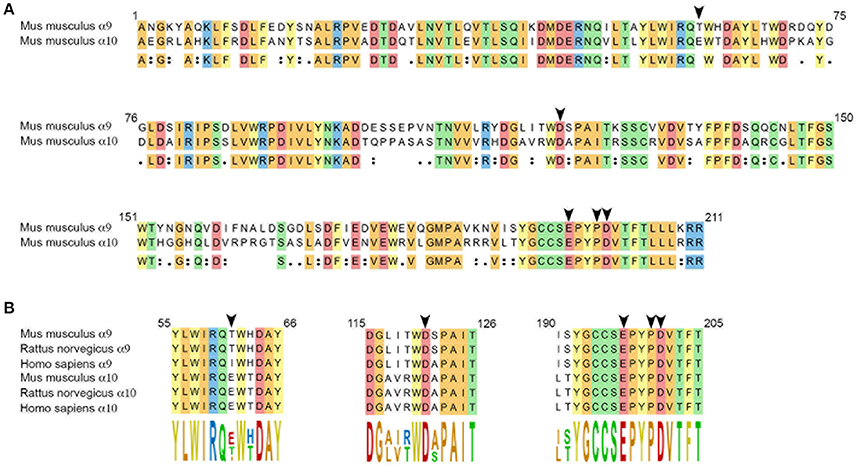
Figure 1. (A) Amino acid sequence alignment of the extracellular domains of mouse α9 and α10 subunits. Numbering complies with the numbering of the α9 nAChR subunit presented in the crystal structure of the human α9 extracellular domain (Zouridakis et al., 2014). Arrow heads point at residues thought to interact with α-CT× RgIA4: T61 and D121 (α9) and E197, P200, and D201 (α10), based on comparison with the binding of rat α9 and α10 subunits to α-conotoxin RgIA (Perez et al., 2009; Azam et al., 2015). Conserved residues are highlighted, colors were chosen depending on the chemical properties of the residues. (B) Sequence alignments of the α-conotoxin RgIA–interacting domains of mouse, rat and human α9, and α10 nAChR subunits.
Plasmid Constructs and Preparation
Chrna9 and Chrna10 cDNAs were further subcloned into a modified pSGEM vector (originally developed by Dr. Michael Hollmann, Ruhr University, Bochum, Germany), using SpeI and XhoI restriction sites. This vector allows the expression of a transcript including the 5'UTR of the β-globin of Xenopus laevis, the coding sequence of the nAChR subunit preceded by its native 5'UTR, and followed by the 3'UTR of the β-globin of X. laevis. It was optimized to increase the expression (translational efficiency) of the nAChR subunit in oocytes (Filchakova and McIntosh, 2013).
Plasmid DNA were isolated from bacteria using Maxiprep Qiagen plasmid kits (Qiagen, Hilden, Germany). The sequences of the expression plasmids were confirmed by automated DNA sequencing in the Johns Hopkins University School of Medicine DNA sequencing Core facility.
Agonist Concentration Response Analysis
X. laevis oocytes were injected with 25 ng of each cRNA encoding α9 and α10 subunits as previously described (Cartier et al., 1996) and incubated for 48 h at 17°C in ND96 prior to use. Two-electrode voltage clamp electrophysiology was conducted to assess for functional expression of α9α10 nAChRs as previously described (Azam et al., 2008). The oocyte membranes were clamped at a holding potential of −70 mV and stimulated with 1 s pulses of 1 mM ACh once every 60 s. After a steady-state response baseline was observed, the oocytes were then stimulated with ascending concentrations of ACh, choline, or nicotine. Each oocyte was stimulated first with ACh followed by choline then nicotine. The maximal response value for activation by ACh was determined using the Hill equation: Y = Bottom + (Top-Bottom)/(1+10∧((LogEC50−X)*Hill slope)). The responses to all concentrations of ACh, choline, and nicotine were then normalized to this value and calculated as a % response.
Antagonist Response Analysis
Xenopus laevis oocytes were injected with 15–30 ng of cRNA (equal amounts for each nAChR subunit) and incubated for 48 h at 17°C in ND96 prior to use. The mouse α7 subunit was co-injected with an equal amount of ric3 to increase expression. The mouse α9 and α10 subunits were made as a part of this study;all other mouse clones were kindly provided by Dr. Jerry Stitzel (University of Colorado, Boulder, CO, USA). Two-electrode voltage clamp electrophysiology was used with oocyte membranes clamped at a holding potential of –70 mV. Oocytes were stimulated with 1 s pulses of ACh every 60 s at the following concentrations: for adult and fetal muscle 10 μM was used; for mouse α7, 200 μM; and for all other subunit combinations 100 μM ACh was used. After a steady baseline of ACh pulses was achieved using ND96, the solution was switched to ND96 containing various toxin concentrations and ACh pulses were observed for toxin response. All dose response curves, including IC50 and hillslope data, were calculated using non-linear regression (curve fit) sigmoidal dose response (variable slope), (GraphPad Prism, San Diego, CA, USA).
Oxaliplatin-Induced Cold Allodynia
Oxaliplatin (MedChem Express, Monmouth Junction, NJ) was dissolved at 0.6 μg/μl in 0.9% NaCl. RgIA4 was dissolved at 0.01 μg/μl in 0.9% NaCl. Mice were injected i.p. daily with oxaliplatin (3.5 mg/kg) or 0.9% saline (vehicle). Additionally, mice were injected s.c. with RgIA4 (40 μg/kg) or vehicle. Treatment weeks included injection on Thurs, Fri, and Mon–Wed. On Thurs, 24 h after last injection, mice were assessed for cold allodynia. Injections stopped on day 21, with assessment occurring on day 22 and subsequent testing occurring once a week for three additional weeks. Testing was conducted using a cold plate test chamber (IITC, Inc Life Science, Woodland, CA). Animals were allowed to acclimate in the chamber at room temperature (23°C) for 5 min. Temperature was then lowered at a rate of 10°C per minute. The testing was stopped when the animal lifted both forepaws and shaking or licking occurred. Alternating lifting of forepaws was not scored. Throughout the study period, experimenters were blinded as to the identity of the injected compounds.
Competition Binding Assay
A competition binding assay on intact HEK293 cells was performed using the Tag-lite™ technology (Cisbio bioassays) (Zwier et al., 2010). HEK293 cells were transfected with plasmids encoding the rat GABAB1 subunit fused to a Snap tag at the N-terminus and the rat GABAB2 subunit and seeded in 96-well black-walled plates at a density of 100,000 cells per well. Twenty-four hours after transfection, the cells were labeled with 300 nM SNAP-Lumi4Tb (Cisbio bioassays, Codolet, France) for 1 h at 37°C in Tag-lite buffer. After extensive washing with Tag-lite buffer, the cells were incubated with the indicated α-conotoxins. Non-fluorescent ligands (GABA or CGP54626) or buffer together with 10 nM fluorescent CGP54626-Red (Cisbio bioassays, Codolet, France) were incubated for 3 h at 4°C prior to signal detection. The fluorescence was collected at 620 and 665 nm using a Pherastar plate reader (BMG Labtech, Ortenberg, Germany), 50 ms after laser excitation at 337 nm. The FRET signal was then calculated as the ratio (signal at 665 nm)/(signal at 620 nm) × 104 and normalized to specific binding. Non-specific binding was determined in the presence of a high concentration of unlabeled CGP54626 (1 μM).
Bioluminescence Resonance Energy Transfer Assay
Bioluminescence Resonance Energy Transfer (BRET) assay was performed on HEK293 cells by recording heterotrimeric G protein dissociation following receptor activation as previously described (Gales et al., 2006). Briefly, HEK293 cells were transfected with plasmids encoding the rat receptor subunits (GABAB1a and GABAB2) and the G protein heterotrimer fused to BRET chromophores [Gαi1-Rluc or GαoA−Rluc, Gβ2 and Gγ2-Venus (Comps-Agrar et al., 2011)]. Cells were seeded in a 96-well white plates at a density of 100,000 cells per well. Twenty-four hours after transfection, the cells were washed with phosphate buffered saline prior to Coelenterazine h substrate (5 μM) and GABA (1 mM) or α-conotoxin (1 μM) addition. The BRET signal was recorded over time using the Mithras LB 940 plate reader (Berthold Biotechnologies, Bad Wildbad, Germany) that allows the sequential integration of light signals detected with two filter settings (Rluc filter, 485 ± 20 nm and Venus/YPF filter, 530 ± 25 nm). The BRET signal is determined as the ratio between Venus and Rluc emissions.
Results
Heterologously Expressed Mouse α9 and α10 Subunits Form Functional nAChRs
In the developing mouse cochlea, an ACh response and cholinergic efferent activity with pharmacology consistent with activation of α9 and α10 nAChR subunits, can be detected in cochlear inner hair cells at P4 (Roux et al., 2016). We cloned the cDNAs of α9 and α10 subunits expressed in P4 C57BL/6J mouse cochlear neuroepithelium (see Materials and Methods). At the protein level, α9 isolated from mouse showed 97% identity with rat, and 91% identity with human homologs. α10 showed 98% identity with rat and 92% identity with human homologs. Sequences of the extracellular domains of mouse α9 and α10 are shown in Figure 1. Residues previously determined to be critical for α-conotoxin RgIA binding (Azam et al., 2015) are indicated by arrowheads in Figure 1B. Note the complete conservation of these residues between rat and mouse sequences.
Each cDNA insert was subcloned into the pSGEM Xenopus oocyte expression vector and used for cRNA preparation. The cRNAs were then injected into oocytes to assess whether mouse α9 and α10 subunits could assemble into functional receptors. The oocytes were subjected to two-electrode voltage clamp electrophysiology and assessed for functional responses to nicotinic agonists (Figure 2). Under these conditions, the oocytes responded robustly to 1 mM ACh as evidenced by current amplitudes that were often in excess of 10 μA. Choline also evoked currents in these oocytes and behaved as a partial agonist relative to ACh. Nic on the other hand failed to evoke currents at concentration from 100 nM to 10 mM. This pharmacological profile is similar to that reported for heterologously expressed human and rat α9-containing nAChRs (Elgoyhen et al., 1994, 2001; Verbitsky et al., 2000; Sgard et al., 2002).
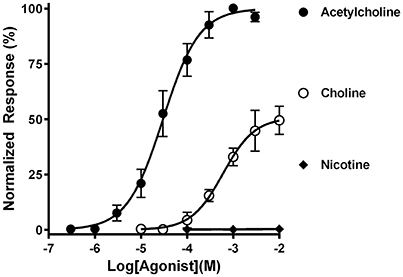
Figure 2. Functional characterization of mouse α9α10 nAChRs heterologously expressed in Xenopus laevis oocytes. Oocytes expressing mouse α9α10 nAChRs were subjected to two-electrode voltage clamp electrophysiology as described in Materials and Methods. The EC50for activation of α9α10 nAChRs by ACh was 30.1 (23.5–38.7) μM and the Hill slope was 1.1 (0.8–1.4) (n = 5). The EC50 for activation by choline was 601 (311–1160) μM; Hill slope 1.3 (0.5–2.1) maximal efficacy was 50 ± 5% relative to ACh (n = 4). Nicotine failed to evoke currents in all oocytes tested (0.4 ± 0.2%) (n = 4). The values in parenthesis denote the 95% confidence interval.
RgIA4 Potently and Selectively Blocks Mouse α9α10 nAChRs
α-conotoxins that block the α9α10 nAChR were previously shown to vary widely in their potency for rat vs. human nAChRs, with these peptides being orders of magnitude less potent on the human α9α10 nAChR. We therefore assessed the potency of RgIA4 on mouse α9α10 nAChRs. RgIA4 potently blocked the ACh response of the α9α10 nAChR subtype with an IC50 of 1.2 nM (Figure 3). α-conotoxin [V11L;V16D]ArIB is a potent antagonist of homomeric α7 nAChRs (Whiteaker et al., 2007). Other antagonists of α7 nAChRs, including methyllycaconitine (MLA) and α-bungartoxin also potently block α9α10 nAChRs. We therefore assessed the activity of α-conotoxin [V11L;V16D]ArIB on mouse α9α10 nAChRs. In contrast to RgIA4, [V11L;V16D]ArIB (10 μM) blocked only 12 ± 8.9% of 100 μM ACh-induced current. Thus, RgIA4 is > 10,000-fold more potent than α-conotoxin [V11L;V16D]ArIB on mouse α9α10 nAChRs.
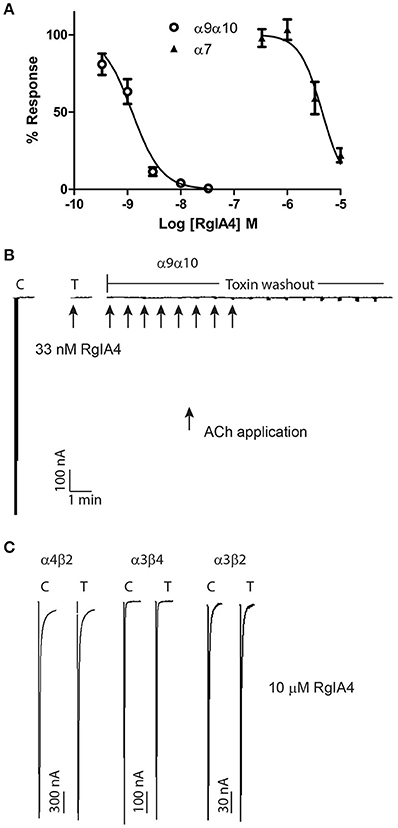
Figure 3. RgIA4 selectively blocks mouse α9α10 nAChRs. nAChRs were expressed in Xenopus laevis oocytes. (A) The concentration response curves for α9α10 and α7 are shown. The IC50 for α9α10 was 1.2 (0.95–1.6) nM with a Hill slope of 1.6 (0.91–2.2). The IC50 for α7 was 4.5 (3.1–6.4) μM with a Hill slope of 2.0 (0.84–3.2). The values in parenthesis denote the 95% confidence intervals; n = 3–7 for each condition. (B) Recovery from toxin block of α9α10 is relatively slow. Example trace of block by 33 nM RgIA4. Recovery after 15 min toxin washout was ~2%. Arrows indicate application of 10 μM ACh. (C) At all other indicated nAChR subtypes, RgIA4 (10 μM) blocked less than 50% of the ACh-evoked response; the percent of the ACh-response for each subtype was α1β1δϵ, 95.0 ± 3.8; α1β1δγ, 86.2 ± 3.8; α3β2, 106 ± 2.9; α3β4, 105 ± 0.7; α4β2, 102 ± 3.3; α4β4, 74.3 ± 3.0. n = 3–4 for each condition; ± is S.E.M. Example traces for α4β2, α3β4, and α3β2 are shown. Results are summarized in Table 1.
The activity of RgIA4 was further assessed on additional mouse nAChR subtypes including the fetal and adult muscle subtypes α1β1δγ, α1β1δϵ, and neuronal subtypes α2β2, α3β2, α3β4, α2β4, α4β2, α4β4, and α7 as described in Materials and Methods. For each of the α/β subunit heteromers, 10 μM peptide blocked less than 50% of current (Table 1). The IC50 for α7 was 4.5 μM (95% confidence interval 3.1–6.4 μM). Block of the α9α10 nAChR by a low concentration (33 nM) RgIA4 abolished ACh-induced current. Recovery from block following peptide washout was very slow (~2% at 15 min, Figure 3). In contrast, recovery from block of α7 was rapid. After two min of washout of 5 μM RgIA4 (the approximate IC50 concentration), the ACh current recovered to 101.6 ± 6.9% of pre-compound application baseline (n = 3, data not shown).

Table 1. RgIA4 IC50 values for various nAChR subtypes.
RgIA4 Provides Long Lasting Protection against Chemotherapy Induced Cold Allodynia
Chemotherapy induced neuropathy is a common and dose limiting side effect of cancer treatment. Pain from relatively mild cold temperatures (cold allodynia) is particularly problematic following oxaliplatin treatment. There are no FDA approved medications to prevent this complication. RgIA4 was previously shown to prevent cold allodynia in oxaliplatin treated rats and mice. Effects lasted up to 72 h post-treatment (Romero et al., 2017). An unanswered question is whether the RgIA4 prevention of cold allodynia represented temporary delay in development of the oxaliplatin-induced side effect or was indicative of more long lasting effects. Here we evaluated the effects of 3 weeks of treatment with RgIA4 followed by a further 3 weeks of observation. Four groups of mice were treated with either saline + saline, oxaliplatin + saline, saline + RgIA4 or oxaliplatin + RgIA4. This treatment regimen continued for 3 weeks after which all treatments were stopped. Mice were assessed at weekly intervals during and after treatment for the presence of cold allodynia. Experimenters were blinded to treatment conditions. Oxaliplatin produced cold allodynia, an effect that significantly differed from control beginning by week three and continuing for 2 weeks post-oxaliplatin treatment. Oxaliplatin/RgIA4 treated animal differed significantly from oxaliplatin/saline treated animals as measured at weeks 2–5. RgIA4 treated animals did not differ from saline treated controls at any tested time point (Figure 4).
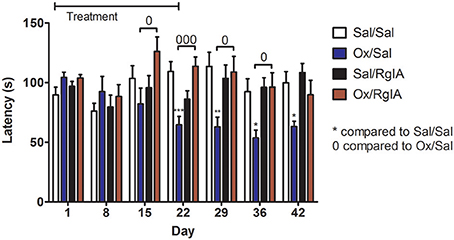
Figure 4. RgIA4 prevents oxaliplatin-induced cold allodynia. Mice were injected once per day with oxaliplatin (3.5 mg/kg i.p) or 0.9% saline (vehicle) for control animals. In addition, mice were injected once per day with RgIA4 (40 μg/kg, s.c.) or vehicle. During the treatment period, mice were injected for 2 days, followed by a 2 day break, followed by injection for another 3 days. Twenty-four hours after last injection, mice were assessed for cold allodynia using a cold-plate as described in Materials and Methods. This injection schedule was repeated for two additional weeks. Treatments were stopped on day 21; cold-plate assessment was performed 24 h later on day 22; subsequent testings were performed once a week for three additional weeks. Time to respond to decreasing temperature was measured and indicated on the y axis as the mean ± SEM (n = 8) for each experimental group. Experimenters were blinded to treatment conditions. Data was analyzed using a one-way ANOVA with Dunnett's Multiple Comparison Test, P-values were indicated as: *P < 0.05, **P < 0.01, and ***P < 0.001 for significant difference from vehicle/vehicle control. And 0P < 0.05, 000P < 0.001, and indicating significant difference compared with oxaliplatin/vehicle mice.
Binding and Functional Activity of Structurally Related Conotoxins on GABAB Receptors
RgIA4 was chosen for the present study because it potently binds α9α10 nAChRs but not multiple other tested targets including opioid receptors or GABAB receptors (GABABR) (Romero et al., 2017). In addition to RgIA4, other structurally related nAChR targeted α-conotoxins are analgesic in several models of neuropathic pain. Among these α-conotoxins, the α9α10 antagonist Vc1.1 has been repeatedly shown to be analgesic. Several studies have shown that Vc1.1 blocks N-type calcium channels in dorsal root ganglion, an effect blocked by GABAB antagonists. In addition, GABAB antagonists prevented analgesic effects of Vc1.1, strongly implicating GABAB receptors as contributing to the therapeutic effects (Callaghan et al., 2008; Callaghan and Adams, 2010; Klimis et al., 2011; Huynh et al., 2015; Castro et al., 2016). However, other studies have failed to confirm GABAB agonist activity (McIntosh et al., 2009; Napier et al., 2012; Wright et al., 2015), prompting debate as to whether the analgesic actions of Vc1.1 and related peptides occur via block of α9α10 nAChRs or stimulation of GABABR (Adams et al., 2012; Mohammadi and Christie, 2015). Separately, AuIB was shown to be analgesic, to stimulate GABABRs but not to block α9α10 nAChRs (Klimis et al., 2011). To further examine the structure, function relationship of conotoxins acting on GABABRs we first conducted a fluorescent ligand competition binding assay using GABABRs expressed in HEK293 cells. Lumi4-Tb-labeled GABABRs were incubated with the fluorescent ligand CGP54626-Red, together with test compounds as described in Material and Methods. Test compounds were assessed for their ability to modulate the homogeneous time resolved fluorescence resonance energy transfer (HTR-FRET) signal between Lumi4-TB and the red fluorescent acceptor. Vc1.1, ImI (which shares the first 8 amino acids of Vc1.1) (Johnson et al., 1995), AuIB, and the closely related AuIA (Luo et al., 1998) were examined (see Table 2 for peptide sequences). No displacement CGP54626-Red by any of the conotoxins was observed (Figure 5A), indicating that the toxins do not bind to the orthosteric GABAB binding site. We next assessed for agonist activity of conotoxins at G-protein-coupled GABABRs using a bioluminescence resonance energy transfer (BRET) assay. Agonist binding to GABABR activates heterotrimeric G proteins. HEK cells expressing GABABR coupled to BRET chomophore labeled G-proteins (Gαi1/αoA-Rluc, Gβ2, and Gγ2-Venus) were utilized to monitor interaction between Gαi or Gαo and Gγ2. The association between Gαi/o-Rluc and Gγ2-Venus at basal state produces a high FRET signal. GABAB binding to the GABABR induces G-protein activation and movement of the Gγ2 subunit away from the Gαi/o subunit leading to a decrease in BRET. GABA robustly decreased BRET, consistent with agonist activity whereas the conotoxins had no effect (Figure 5B).
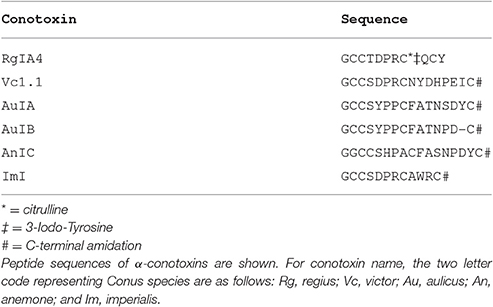
Table 2. Amino acid sequence of selected α-conotoxins.
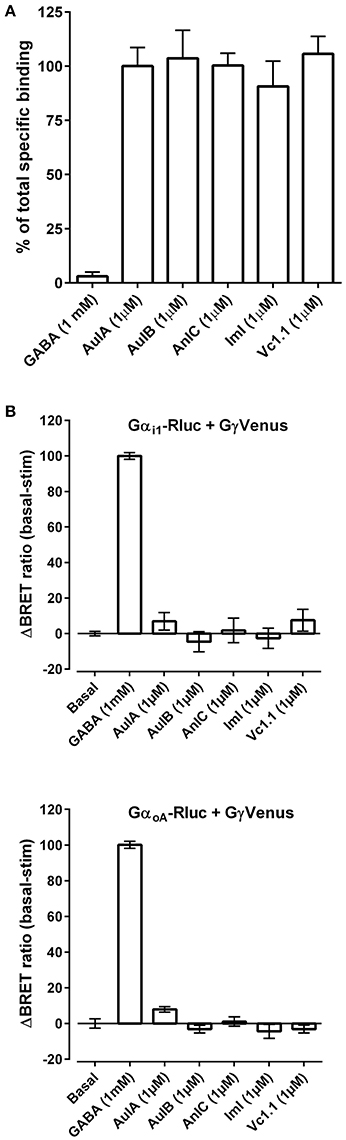
Figure 5. (A) α-Conotoxins do not displace the orthosteric GABABR antagonist CGP54626-Red. GABA, CGP54626, and the indicated α-Ctxs (at 1 μM) were assessed for the ability to displace CGP54626-Red binding from GABABR heterologously expressed in HEK293 cells as described in Material and Methods. Three replicates were obtained for each value and the experiment was repeated three times. (B) α-Conotoxins do not activate GABABR. The indicated α-conotoxins were tested at 1 μM for their ability to activate GABABR heterolgously expressed together with fluorescently tagged G protein subunits in HEK293 cells. At resting state, the G-protein subunits are in close proximity resulting in a strong basal bioluminescence resonance energy transfer (BRET) signal. Activation of GABABR was assessed by monitoring dissociation of Gαi1-Rluc (top) or GαoA-Rluc (bottom) from β2γ2-Venus as measured by the change in BRET. The results after 10 min of toxin incubation are expressed as a percentage of GABA-induced BRET change from basal fluorescence levels. Three replicates were obtained for each value and the experiment was repeated three times.
Discussion
α-Conotoxins are small, disulfide rich peptides found in the venom of cone snails. These peptides are typically 12–20 amino acids in length. Some have high subtype selectivity for mammalian nAChRs enabling the dissection of functional roles of these subtypes (Azam and McIntosh, 2009; Lebbe et al., 2014; Dutertre et al., 2017). α-Conotoxin RgIA potently blocks α9α10 vs. other nAChRs subtypes in rat. However, an amino acid difference in the (–) face of the α9 subunit in human vs. rat renders low potency for the α9α10 nAChR (Azam and McIntosh, 2012; Azam et al., 2015). To overcome this species difference, we developed a peptide analog of RgIA, known as RgIA4, wherein 5 of the 13 amino acids were modified. These modifications led to high potency and selectivity for the human α9α10 nAChR (Romero et al., 2017). In this report we have assessed the potency and selectivity of RgIA4 for mouse nAChRs. To do so, we cloned mouse α9 and α10 subunits and placed each of these in a high expression vector suitable for expression in Xenopus oocytes.
Application of acetylcholine on mouse α9α10 nAChRs expressed in oocytes induced robust currents. Choline, although sometimes referred to as an α7 selective agonist, also potently activated α9α10 nAChRs consistent with what has been reported for rat and human α9α10 nAChRs (Elgoyhen et al., 1994, 2001; Verbitsky et al., 2000; Sgard et al., 2002). The α9α10 nAChR, as expressed in oocytes and natively expressed in cochlear hair cells, is unique among nAChR subtypes in that it is not activated by nicotine (Elgoyhen et al., 2001). Likewise, nicotine did not activate Xenopus oocyte-expressed mouse α9α10 nAChRs. Recently, however, nicotine has been shown to act as an agonist of α9* nAChRs in immune cell monocytes (Hecker et al., 2015; Richter et al., 2016; Backhaus et al., 2017). These nAChRs do not appear to function as canonical ion channels (no ACh- or nicotine-induced ion currents are observed in patch clamp experiments). In these particular cells, agonist stimulation results in alteration of cytokine release. The full subunit composition and structure of immune cell α9* nAChRs is unknown. In some instances, these immune cell nAChRs may be composed of a combination of α7, α9, and α10 subunits (Hecker et al., 2015; Backhaus et al., 2017).
The α7, α9, and α10 subunits are closely related subunits and are the only nAChRs thus far demonstrated to function without a β subunit partner. Interestingly all three subunits may provide the principal or (+) face of the agonist binding interface (Boffi et al., 2017). The parent peptide, RgIA binds to the α10/α9 subunit interface (Azam and McIntosh, 2012; Azam et al., 2015). RgIA4 potently blocked mouse α9α10 nAChRs and the block was selective for this subtype. Block was only slowly reversed upon peptide washout, a characteristic that may allow for synthesis of useful fluorescently or radioactively tagged derivatives. The next most potent block by RgIA4 was that of α7 homomers, but here the IC50 was 3,700-fold lower than that of α9α10 and recovery occurred following 2 min of peptide washout. Thus, as for rat and human, RgIA4 represents a novel probe for dissecting function of mouse α9α10 nAChRs vs. α7 nAChRs. Conversely, α-conotoxin [V11L;V16D]ArIB (Whiteaker et al., 2007) potently blocks mouse α7 nAChRs but not α9α10 nAChRs indicating that RgIA4 and [V11L;V16D]ArIB may be used as complimentary reagents.
Increasing lifespan is associated with rising cancer incidence. Cancer chemotherapy is increasingly lifesaving, but associated with significant side effect burden. Commonly used chemotherapeutic agents, including platinum-based drugs, taxanes and thalidomide analogs, are neurotoxic. Neuropathy is one of the most common chemotherapy induced side effects (Banach et al., 2017). The severity of acute peripheral neuropathy is a significant risk factor for severe chronic neuropathy (Hershman et al., 2014). Oxaliplatin is used as a first-line treatment for colorectal cancer. However, oxaliplatin-induced neuropathy can limit both the dose and duration of therapy (Avan et al., 2015). RgIA4 was recently shown to prevent the development of oxaliplatin-induced cold allodynia, an effect which lasted up to 72 h after last injection of peptide (Romero et al., 2017). Here we further examined the potential duration of the analgesic effect of RgIA4. Co-administration of RgIA4 with oxaliplatin prevented the development of cold allodynia. After cessation of oxaliplatin, cold allodynia continued in control mice, but was not observed at any measured time point in mice treated with RgIA4. Thus, RgIA4 treatment prevented the development of oxaliplatin-induced cold allodynia and had prolonged efficacy (total experiment length was 6 weeks). These prolonged effects suggests that similar agents might be administered, along with oxaliplatin, during cancer chemotherapy as a method to provide potentially disease modifying effects with respect to development of neuropathy.
Several conotoxins or derivatives have now been reported to have analgesic effects. Some of these toxins block α9α10 nAChRs as expressed in oocytes, yet others, such as α-conotoxin AuIB do not (Klimis et al., 2011). These findings have called into question whether the analgesic effects involve an α9α10 nAChR dependent pathway. In addition, several reports have indicated that conotoxins, including Vc1.1, analogs of Vc1.1, RgIA and AuIB block N-type calcium channels in DRG neurons, an effect that is prevented by GABABR antagonists (Callaghan et al., 2008; Callaghan and Adams, 2010; Klimis et al., 2011; Cuny et al., 2012; van Lierop et al., 2013; Berecki et al., 2014; Huynh et al., 2015; Castro et al., 2016). This has led to an alternative hypothesis that these conotoxins exert their analgesic effect by stimulating G-protein coupled GABABR (Adams et al., 2012; Adams and Berecki, 2013; Sadeghi et al., 2017). In support of this, post-translationally modified analogs of Vc1.1 that have α9α10 activity but lack GABABR activity also lack analgesic activity (Nevin et al., 2007). However, several laboratories have failed to observe conotoxin-evoked GABABR responses; these assays have utilized DRG neurons, spinal cord slices and Xenopus oocyte-expressed GABABR (Xiao et al., 2009; Napier et al., 2012; Huynh et al., 2015; Wright et al., 2015).
RgIA4 was previously shown to not stimulate GABABR as expressed in DRG or immortalized cell lines (Romero et al., 2017). To further evaluate the potential structure function relationship of other conotoxins on GABABR we used a FRET-based binding assay and functional BRET assay. Peptides analyzed included Vc1.1, AuIB, and three other conotoxins with similar amino acid sequences (Table 2). None of these conotoxins prevented the binding of the competitive orthosteric antagonist CGP54626-Red. This is consistent with previous binding studies performed in which Vc1.1 did not compete for binding with [3H]CGP54626 in HEK293T cells (McIntosh et al., 2009) or in DRG neurons (Adams and Berecki, 2013). The latter studies led to the proposal that Vc1.1 acts at an allosteric site to exert its agonist effect (Adams et al., 2012; Adams and Berecki, 2013). However, in the current study, neither Vc1.1 nor any of the examined conotoxins activated the GABABR as measured with a G-protein-based BRET assay. This is surprising given that Vc1.1 activation of GABABRs was previously reported to be dependent on the G-protein signaling cascade; block of N-type calcium channels was eliminated by pertussis toxin, intracellular GDPβS, or an inhibitor of pp60c-src tyrosine kinase (Callaghan et al., 2008).
We are unsure how to reconcile the GABABR mediated effects reported in multiple compelling studies using DRG neurons (Callaghan et al., 2008; Callaghan and Adams, 2010; Klimis et al., 2011; Cuny et al., 2012; van Lierop et al., 2013; Berecki et al., 2014; Huynh et al., 2015; Castro et al., 2016), (but see Wright et al., 2015 that did not replicate principle findings of these studies) and the lack of effects seen in other assays (McIntosh et al., 2009; Napier et al., 2012), now including the GABABR BRET assay. There is known interplay between nicotinic and GABAergic systems. CNS nAChRs modulate GABA release (Radcliffe et al., 1999). In addition, GABAB autoreceptors inhibit ACh-evoked GABA release (McClure-Begley et al., 2014). However, Vc1.1 and RgIA were shown to block N-type calcium channels in cultured neurons from α9 KO mice indicating that block of α9 nAChRs could not account for GABABR effects observed in that preparation (Callaghan and Adams, 2010). Cross-regulation of cholinergic and GABAergic systems may also be indirect. For example, microRNAs can repress multiple targets; miRNA-608 targets both acetylcholinesterase and the Rho GTPase CDC42 that is involved in GABAA synapse formation (Hanin et al., 2014). In the present study, the primary GABAB signaling complex (the GABABR and G-proteins α, β, and γ subunits) was expressed in HEK cells. It is possible that in DRG neurons other additional and unknown protein partners may be involved and necessary for conotoxins to act as an agonist. Regardless, the present findings become an interesting piece of a puzzle that must be reconciled with the GABABR hypothesis.
Acute pain is protective to the animal; yet pain can extend for weeks or years, well after healing of the initial injury, and well beyond pain's adaptive utility. Another intriguing finding that must be explained is how these α-conotoxins produce their long lasting effects. RgIA4, a peptide, is rapidly cleared from plasma with a half-life of < 20 min (Mercado et al., 2016). Plasma levels are undetectable at 4 h. Despite the slow reversal of RgIA4 action, block (or stimulation) of any receptor would not be expected to be present for weeks after last compound administration. How then to explain these long lasting effects? The answer is unknown at the present time, but one possible clue comes from examination of the histopathological effects of Vc1.1 and RgIA administration. Nerve injury produces an influx of T-cells and macrophages into the area of nerve injury (Austin et al., 2012; Ji et al., 2016; Lees et al., 2017). Some of these immune cell responses may help to heal damaged tissue while others may lead to further pathology. Alterations in microglia are also implicated in neuropathic pain states; RgIA was also shown to alter levels of CNS microglia (Di Cesare Mannelli et al., 2014). The interplay between neurons and immune cells are increasingly recognized as important in the pathophysiology of neuropathic pain (Austin et al., 2012; Grace et al., 2014; Ji et al., 2016; Peng et al., 2016; Lees et al., 2017). It is possible that the α-conotoxins modulate subpopulations of immune cells in a fashion that favorably alters the development of a pain state. There is precedent for attenuation of neuroimmune signaling to produce long lasting reversal of neuropathic pain. A single injection of adenosine 2A receptor agonists attenuated chronic constriction injury-induced upregulation of spinal cord microglia and astrocytes and reversed mechanical and thermal hyperalgesia, effects that lasted at least 4 weeks (Loram et al., 2009, 2013). Functional α9α10 nAChRs have recently been reported in monocytes where they influence P2X receptor release of IL-1β (Hecker et al., 2015; Richter et al., 2016; Backhaus et al., 2017). Transcripts for α9 and α10 subunits have been reported in other immune cell subtypes (Hao et al., 2011). The functions of α9α10 receptors in these cells, and those that are affected by nerve injury are unknown.
Analgesic effects of nAChR antagonists are not limited to the α-conotoxins. Small molecule antatonists of α9α10 nAChRs have been shown to have analgesic effects in chronic constriction injury and chemotherapy (vincristine)- induced neuropathy (Holtman et al., 2011; Zheng et al., 2011; Wala et al., 2012) (see Hone et al., 2017 for review). Another α9α10 nAChR antagonist conotoxin, GeXIVA (Luo et al., 2015) structurally unrelated to the α-conotoxins was also shown to produce long lasting analgesic effects (Li et al., 2016). These findings together with those of the present report further implicate α9α10 nAChRs in the treatment of neuropathic pain.
Author Contributions
SC, AH, IR, JK, and GU all performed the experiments. SC, AH, IR, JK, JP, GU, DS, EG, and JM all participated in experimental design and the preparing and writing of the manuscript.
Conflict of Interest Statement
Conotoxins, including some of those referenced in this paper have been patented by the University of Utah with JM listed as an inventor. JM has received funding from Kineta Inc. The other authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by National Institutes of Health (NIH) Grants R01-DC-006476 and R01-DC-012957 (to EG); NIH Grant R03-DC-013374 (to IR); and NIH grants P01 GM48677, R01 GM103801 (to JM). Funding was also from the Agence National de la Recherche (ANR-12-BSV2-0015) and from the Fondation Recherche Médicale (FRM DEQ20130326522 to JK and JP). The FRET and BRET experiments were performed using the ARPEGE platform facility at the Institut de Génomique Fonctionnelle (Montpellier, France).
References
Abazeed, M. E., Adams, D. J., Hurov, K. E., Tamayo, P., Creighton, C. J., Sonkin, D., et al. (2013). Integrative radiogenomic profiling of squamous cell lung cancer. Cancer Res. 73, 6289–6298. doi: 10.1158/0008-5472.CAN-13-1616
Adams, D. J., and Berecki, G. (2013). Mechanisms of conotoxin inhibition of N-type (Ca(v)2.2) calcium channels. Biochim. Biophys. Acta 1828, 1619–1628. doi: 10.1016/j.bbamem.2013.01.019
Adams, D. J., Callaghan, B., and Berecki, G. (2012). Analgesic conotoxins: block and G protein-coupled receptor modulation of N-type (Ca(V) 2.2) calcium channels. Br. J. Pharmacol. 166, 486–500. doi: 10.1111/j.1476-5381.2011.01781.x
Austin, P. J., Kim, C. F., Perera, C. J., Moalem-Taylor, G., and Regulatory, T. (2012). cells attenuate neuropathic pain following peripheral nerve injury and experimental autoimmune neuritis. Pain 153, 1916–1931. doi: 10.1016/j.pain.2012.06.005
Avan, A., Postma, T. J., Ceresa, C., Avan, A., Cavaletti, G., Giovannetti, E., et al. (2015). Platinum-induced neurotoxicity and preventive strategies: past, present, and future. Oncologist 20, 411–432. doi: 10.1634/theoncologist.2014-0044
Azam, L., and McIntosh, J. M. (2009). Alpha-conotoxins as pharmacological probes of nicotinic acetylcholine receptors. Acta Pharmacol. Sin. 30, 771–783. doi: 10.1038/aps.2009.47
Azam, L., and McIntosh, J. M. (2012). Molecular basis for the differential sensitivity of rat and human α9α10 nAChRs to α-conotoxin RgIA. J. Neurochem. 122, 1137–1144. doi: 10.1111/j.1471-4159.2012.07867.x
Azam, L., Papakyriakou, A., Zouridakis, M., Giastas, P., Tzartos, S. J., and McIntosh, J. M. (2015). Molecular interaction of alpha-conotoxin RgIA with the rat α9α10 nicotinic acetylcholine receptor. Mol. Pharmacol. 87, 855–864. doi: 10.1124/mol.114.096511
Azam, L., Yoshikami, D., and McIntosh, J. M. (2008). Amino acid residues that confer high selectivity of the alpha6 nicotinic acetylcholine receptor subunit to alpha-conotoxin MII[S4A,E11A,L15A]. J. Biol. Chem. 283, 11625–11632. doi: 10.1074/jbc.M710288200
Backhaus, S., Zakrzewicz, A., Richter, K., Damm, J., Wilker, S., Fuchs-Moll, G., et al. (2017). Differential contribution of subunit interfaces to α9α10 nicotinic acetylcholine receptor function. J. Lipid Res. 58, 1055–1066. doi: 10.1194/jlr.M071506
Banach, M., Juranek, J. K., and Zygulska, A. L. (2017). Chemotherapy-induced neuropathies-a growing problem for patients and health care providers. Brain Behav. 7:e00558. doi: 10.1002/brb3.558
Berecki, G., McArthur, J. R., Cuny, H., Clark, R. J., and Adams, D. J. (2014). Differential Cav2.1 and Cav2.3 channel inhibition by baclofen and alpha-conotoxin Vc1.1 via GABAB receptor activation. J. Gen. Physiol. 143, 465–479. doi: 10.1085/jgp.201311104
Boffi, J. C., Marcovich, I., Gill-Thind, J. K., Corradi, J., Collins, T., Lipovsek, M. M., et al. (2017). Differential contribution of subunit interfaces to α9α10 nicotinic acetylcholine receptor function. Mol. Pharmacol. 91, 250–262. doi: 10.1124/mol.116.107482
Brady, K. T., McCauley, J. L., and Back, S. E. (2016). Prescription opioid misuse, abuse, and treatment in the united states: an update. Am. J. Psychiatry 173, 18–26. doi: 10.1176/appi.ajp.2015.15020262
Callaghan, B., and Adams, D. J. (2010). Analgesic α-conotoxins Vc1.1 and RgIA inhibit N-type calcium channels in sensory neurons of α9 nicotinic receptor knockout mice. Channels 4, 51–54. doi: 10.4161/chan.4.1.10281
Callaghan, B., Haythornthwaite, A., Berecki, G., Clark, R. J., Craik, D. J., and Adams, D. J. (2008). Analgesic alpha-conotoxins Vc1.1 and Rg1A inhibit N-type calcium channels in rat sensory neurons via GABAB receptor activation. J. Neurosci. 28, 10943–10951. doi: 10.1523/JNEUROSCI.3594-08.2008
Carstens, B. B., Clark, R. J., Daly, N. L., Harvey, P. J., Kaas, Q., and Craik, D. J. (2011). Engineering of conotoxins for the treatment of pain. Curr. Pharm. Des. 17, 4242–4253. doi: 10.2174/138161211798999401
Cartier, G. E., Yoshikami, D., Gray, W. R., Luo, S., Olivera, B. M., and McIntosh, J. M. (1996). A new alpha-conotoxin which targets alpha3beta2 nicotinic acetylcholine receptors. J. Biol. Chem. 271, 7522–7528. doi: 10.1074/jbc.271.13.7522
Castro, J., Harrington, A. M., Garcia-Caraballo, S., Maddern, J., Grundy, L., Zhang, J., et al. (2016). α-Conotoxin Vc1.1 inhibits human dorsal root ganglion neuroexcitability and mouse colonic nociception via GABAB receptors. Gut 66, 1083–1094. doi: 10.1136/gutjnl-2015-310971
Clark, R. J., Jensen, J., Nevin, S. T., Callaghan, B. P., Adams, D. J., and Craik, D. J. (2010). The engineering of an orally active conotoxin for the treatment of neuropathic pain. Angew. Chem. Int. Ed. Engl. 49, 6545–6548. doi: 10.1002/anie.201000620
Colloca, L., Ludman, T., Bouhassira, D., Baron, R., Dickenson, A. H., Yarnitsky, D., et al. (2017). Neuropathic pain. Nat. Rev. Dis. Primers 3:17002. doi: 10.1038/nrdp.2017.2
Comps-Agrar, L., Kniazeff, J., Norskov-Lauritsen, L., Maurel, D., Gassmann, M., Gregor, N., et al. (2011). The oligomeric state sets GABA(B) receptor signalling efficacy. EMBO J. 30, 2336–2349. doi: 10.1038/emboj.2011.143
Costigan, M., Scholz, J., and Woolf, C. J. (2009). Neuropathic pain: a maladaptive response of the nervous system to damage. Annu. Rev. Neurosci. 32, 1–32. doi: 10.1146/annurev.neuro.051508.135531
Cuny, H., de Faoite, A., Huynh, T. G., Yasuda, T., Berecki, G., and Adams, D. J. (2012). Gamma-Aminobutyric acid type B (GABAB) receptor expression is needed for inhibition of N-type (Cav2.2) calcium channels by analgesic alpha-conotoxins. J. Biol. Chem. 287, 23948–23957. doi: 10.1074/jbc.M112.342998
Del Bufalo, A., Cesario, A., Salinaro, G., Fini, M., and Russo, P. (2014). Alpha9 alpha10 nicotinic acetylcholine receptors as target for the treatment of chronic pain. Curr. Pharm. Des. 20, 6042–6047. doi: 10.2174/1381612820666140314150634
Di Cesare Mannelli, L., Cinci, L., Micheli, L., Zanardelli, M., Pacini, A., McIntosh, J. M., et al. (2014). α-conotoxin RgIA protects against the development of nerve injury-induced chronic pain and prevents both neuronal and glial derangement. Pain 155, 1986–1995. doi: 10.1016/j.pain.2014.06.023
Dutertre, S., Nicke, A., and Tsetlin, V. (2017). Nicotinic acetylcholine receptor inhibitors derived from snake and snail venoms. Neuropharmacology doi: 10.1016/j.neuropharm.2017.06.011. [Epub ahead of print].
Elgoyhen, A. B., Johnson, D. S., Boulter, J., Vetter, D. E., and Heinemann, S. (1994). Alpha9: an acetylcholine receptor with novel pharmacological properties expressed in rat cochlear hair cells. Cell 79, 705–715. doi: 10.1016/0092-8674(94)90555-X
Elgoyhen, A. B., Vetter, D. E., Katz, E., Rothlin, C. V., Heinemann, S. F., and Boulter, J. (2001). α10: a determinant of nicotinic cholinergic receptor function in mammalian vestibular and cochlear mechanosensory hair cells. Proc. Natl. Acad. Sci. U.S.A. 98, 3501–3506. doi: 10.1073/pnas.051622798
Filchakova, O., and McIntosh, J. M. (2013). Functional expression of human α9* nicotinic acetylcholine receptors in X. laevis oocytes is dependent on the α9 subunit 5' UTR. PLoS ONE 8:e64655. doi: 10.1371/journal.pone.0064655
Gales, C., Van Durm, J. J., Schaak, S., Pontier, S., Percherancier, Y., Audet, M., et al. (2006). Probing the activation-promoted structural rearrangements in preassembled receptor-G protein complexes. Nat. Struct. Mol. Biol. 13, 778–786. doi: 10.1038/nsmb1134
Grace, P. M., Hutchinson, M. R., Maier, S. F., and Watkins, L. R. (2014). Pathological pain and the neuroimmune interface. Nat. Rev. Immunol. 14, 217–231. doi: 10.1038/nri3621
Hanin, G., Shenhar-Tsarfaty, S., Yayon, N., Yau, Y. H., Bennett, E. R., Sklan, E. H., et al. (2014). Competing targets of microRNA-608 affect anxiety and hypertension. Hum. Mol. Genet. 23, 4569–4580. doi: 10.1093/hmg/ddu170
Hao, J., Simard, A. R., Turner, G. H., Wu, J., Whiteaker, P., Lukas, R. J., et al. (2011). Attenuation of CNS inflammatory responses by nicotine involves α7 and non-α7 nicotinic receptors. Exp. Neurol. 227, 110–119. doi: 10.1016/j.expneurol.2010.09.020
Hecker, A., Kullmar, M., Wilker, S., Richter, K., Zakrzewicz, A., Atanasova, S., et al. (2015). Phosphocholine-modified macromolecules and canonical nicotinic agonists inhibit ATP-Induced IL-1beta Release. J. Immunol. 195, 2325–2334. doi: 10.4049/jimmunol.1400974
Hershman, D. L., Lacchetti, C., Dworkin, R. H., Lavoie Smith, E. M., Bleeker, J., Cavaletti, G., et al. (2014). American Society of Clinical, Prevention and management of chemotherapy-induced peripheral neuropathy in survivors of adult cancers: American Society of Clinical Oncology clinical practice guideline. J. Clin. Oncol. 32, 1941–1967. doi: 10.1200/JCO.2013.54.0914
Holtman, J. R., Dwoskin, L. P., Dowell, C., Wala, E. P., Zhang, Z., Crooks, P. A., et al. (2011). The novel small molecule α9α10 nicotinic acetylcholine receptor antagonist ZZ-204G is analgesic. Eur. J. Pharmacol. 670, 500–508. doi: 10.1016/j.ejphar.2011.08.053
Hone, A. J., Servent, D., and McIntosh, J. M. (2017). α9 nicotinic acetylcholine receptors and the modulation of pain. Br. J. Pharmacol. doi: 10.1111/bph.13931. [Epub ahead of print].
Huynh, T. G., Cuny, H., Slesinger, P. A., and Adams, D. J. (2015). Novel mechanism of voltage-gated N-type (Cav2.2) calcium channel inhibition revealed through α-conotoxin Vc1.1 activation of the GABA(B) receptor. Mol. Pharmacol. 87, 240–250. doi: 10.1124/mol.114.096156
Ji, R. R., Chamessian, A., and Zhang, Y. Q. (2016). Pain regulation by non-neuronal cells and inflammation. Science 354, 572–577. doi: 10.1126/science.aaf8924
Johnson, D. S., Martinez, J., Elgoyhen, A. B., Heinemann, S. F., and McIntosh, J. M. (1995). Alpha-Conotoxin ImI exhibits subtype-specific nicotinic acetylcholine receptor blockade: preferential inhibition of homomeric alpha 7 and alpha 9 receptors. Mol. Pharmacol. 48, 194–199.
Kaas, Q., Yu, R., Jin, A. H., Dutertre, S., and Craik, D. J. (2012). ConoServer: updated content, knowledge, and discovery tools in the conopeptide database. Nucleic Acids Res. 40, D325–D330. doi: 10.1093/nar/gkr886
Kilkenny, C., Browne, W. J., Cuthill, I. C., Emerson, M., and Altman, D. G. (2010). Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. J. Pharmacol. Pharmacother. 1, 94–99. doi: 10.4103/0976-500X.72351
Klimis, H., Adams, D. J., Callaghan, B., Nevin, S., Alewood, P. F., Vaughan, C. W., et al. (2011). A novel mechanism of inhibition of high-voltage activated calcium channels by α-conotoxins contributes to relief of nerve injury-induced neuropathic pain. Pain 152, 259–266. doi: 10.1016/j.pain.2010.09.007
Lebbe, E. K., Peigneur, S., Wijesekara, I., and Tytgat, J. (2014), Conotoxins targeting nicotinic acetylcholine receptors: an overview. Mar. Drugs 12, 2970–3004. doi: 10.3390/md12052970
Lees, J. G., Makker, P. G., Tonkin, R. S., Abdulla, M., Park, S. B., Goldstein, D., et al. (2017). Immune-mediated processes implicated in chemotherapy-induced peripheral neuropathy. Eur. J. Cancer 73, 22–29. doi: 10.1016/j.ejca.2016.12.006
Li, X., Hu, Y., Wu, Y., Huang, Y., Yu, S., Ding, Q., et al. (2016). Anti-hypersensitive effect of intramuscular administration of αO-conotoxin GeXIVA[1,2] and GeXIVA[1,4] in rats of neuropathic pain. Prog. Neuropsychopharmacol. Biol. Psychiatry 66, 112–119. doi: 10.1016/j.pnpbp.2015.12.005
Loram, L. C., Harrison, J. A., Sloane, E. M., Hutchinson, M. R., Sholar, P., Taylor, F. R., et al. (2009). Enduring reversal of neuropathic pain by a single intrathecal injection of adenosine 2A receptor agonists: a novel therapy for neuropathic pain. J. Neurosci. 29, 14015–14025. doi: 10.1523/JNEUROSCI.3447-09.2009
Loram, L. C., Taylor, F. R., Strand, K. A., Harrison, J. A., Rzasalynn, R., Sholar, P., et al. (2013). Intrathecal injection of adenosine 2A receptor agonists reversed neuropathic allodynia through protein kinase (PK)A/PKC signaling. Brain Behav. Immun. 33, 112–122. doi: 10.1016/j.bbi.2013.06.004
Luo, S., Kulak, J. M., Cartier, G. E., Jacobsen, R. B., Yoshikami, D., Olivera, B. M., et al. (1998). Alpha-conotoxin AuIB selectively blocks alpha3 beta4 nicotinic acetylcholine receptors and nicotine-evoked norepinephrine release. J. Neurosci. 18, 8571–8579.
Luo, S., Zhangsun, D., Harvey, P. J., Kaas, Q., Wu, Y., Zhu, X., et al. (2015). Cloning, synthesis, and characterization of αO-conotoxin GeXIVA, a potent alpha9alpha10 nicotinic acetylcholine receptor antagonist. Proc. Natl. Acad. Sci. U.S.A. 112, E4026–E4035. doi: 10.1073/pnas.1503617112
McClure-Begley, T. D., Grady, S. R., Marks, M. J., Collins, A. C., and Stitzel, J. A. (2014). Presynaptic GABAB autoreceptor regulation of nicotinic acetylcholine receptor mediated [3H]-GABA release from mouse synaptosomes. Biochem. Pharmacol. 91, 87–96. doi: 10.1016/j.bcp.2014.06.010
McIntosh, J. M., Absalom, N., Chebib, M., Elgoyhen, A. B., and Vincler, M. (2009). Alpha9 nicotinic acetylcholine receptors and the treatment of pain. Biochem. Pharmacol. 78, 693–702. doi: 10.1016/j.bcp.2009.05.020
Mercado, J., Xu, M., Norton, K., Peckham, D., Posakony, J., Tarcha, E., et al. (2016). “A novel, non-opioid, cone snail peptide-based analgesic as a therapeutic alternative for the treatment of chronic pain,” in 35th Annual Scientific Meeting of the American Pain Society (Austin, TX).
Metabolic (2007). Metabolic, Metabolic Discontinues Clinical Trial Programme for Neuropathic Pain Drug, ACV1. Available online at: http://www.asx.com.au/asxpdf/20070814/pdf/313yjgpf7jl4lg.pdf.
Mohammadi, S. A., and Christie, M. J. (2015). Conotoxin interactions with α9α10-nAChRs: is the α9α10-nicotinic acetylcholine receptor an important therapeutic target for pain management? Toxins 7, 3916–3932. doi: 10.3390/toxins7103916
Mohammadi, S., and Christie, M. J. (2014). α9-nicotinic acetylcholine receptors contribute to the maintenance of chronic mechanical hyperalgesia, but not thermal or mechanical allodynia. Mol. Pain 10:64. doi: 10.1186/1744-8069-10-64
Napier, I. A., Klimis, H., Rycroft, B. K., Jin, A. H., Alewood, P. F., Motin, L., et al. (2012). Intrathecal α-conotoxins Vc1.1, AuIB and MII acting on distinct nicotinic receptor subtypes reverse signs of neuropathic pain. Neuropharmacology 62, 2202–2207. doi: 10.1016/j.neuropharm.2012.01.016
Nevin, S. T., Clark, R. J., Klimis, H., Christie, M. J., Craik, D. J., and Adams, D. J. (2007). Are alpha9alpha10 nicotinic acetylcholine receptors a pain target for alpha-conotoxins? Mol. Pharmacol. 72, 1406–1410. doi: 10.1124/mol.107.040568
Pacini, A., Micheli, L., Maresca, M., Branca, J. J., McIntosh, J. M., Ghelardini, C., et al. (2016). The α9α10 nicotinic receptor antagonist alpha-conotoxin RgIA prevents neuropathic pain induced by oxaliplatin treatment. Exp. Neurol. 282, 37–48. doi: 10.1016/j.expneurol.2016.04.022
Peng, J., Gu, N., Zhou, L., B Eyo, U., Murugan, M., Gan, W. B., et al. (2016). Microglia and monocytes synergistically promote the transition from acute to chronic pain after nerve injury. Nat. Commun. 7:12029. doi: 10.1038/ncomms12029
Perez, E. G., Cassels, B. K., and Zapata-Torres, G. (2009). Molecular modeling of the α9α10 nicotinic acetylcholine receptor subtype. Bioorg. Med. Chem. Lett. 19, 251–254. doi: 10.1016/j.bmcl.2008.10.094
Radcliffe, K. A., Fisher, J. L., Gray, R., and Dani, J. A. (1999). Nicotinic modulation of glutamate and GABA synaptic transmission of hippocampal neurons. Ann. N Y. Acad. Sci. 868, 591–610. doi: 10.1111/j.1749-6632.1999.tb11332.x
Richter, K., Mathes, V., Fronius, M., Althaus, M., Hecker, A., Krasteva-Christ, G., et al. (2016). Phosphocholine - an agonist of metabotropic but not of ionotropic functions of α9-containing nicotinic acetylcholine receptors. Sci. Rep. 6:28660. doi: 10.1038/srep28660
Romero, H. K., Christensen, S. B., Di Cesare Mannelli, L., Gajewiak, J., Ramachandra, R., Elmslie, K. S., et al. (2017). Inhibition of α9α10 nicotinic acetylcholine receptors prevents chemotherapy-induced neuropathic pain. Proc. Natl. Acad. Sci. U.S.A. 114, E1825–E1832. doi: 10.1073/pnas.1621433114
Roux, I., Wu, J. S., McIntosh, J. M., and Glowatzki, E. (2016). Assessment of the expression and role of the α1-nAChR subunit in efferent cholinergic function during the development of the mammalian cochlea. J. Neurophysiol. 116, 479–492. doi: 10.1152/jn.01038.2015
Sadeghi, M., McArthur, J. R., Finol-Urdaneta, R. K., and Adams, D. J. (2017). Analgesic conopeptides targeting G protein-coupled receptors reduce excitability of sensory neurons. Neuropharmacology doi: 10.1016/j.neuropharm.2017.05.020. [Epub ahead of print].
Satkunanathan, N., Livett, B., Gayler, K., Sandall, D., Down, J., and Khalil, Z. (2005). Alpha-conotoxin Vc1.1 alleviates neuropathic pain and accelerates functional recovery of injured neurones. Brain Res. 1059, 149–158. doi: 10.1016/j.brainres.2005.08.009
Sgard, F., Charpantier, E., Bertrand, S., Walker, N., Caput, D., Graham, D., et al. (2002). A novel human nicotinic receptor subunit, alpha10, that confers functionality to the alpha9-subunit. Mol. Pharmacol. 61, 150–159. doi: 10.1124/mol.61.1.150
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7:539. doi: 10.1038/msb.2011.75
Umana, I. C., Daniele, C. A., and McGehee, D. S. (2013). Neuronal nicotinic receptors as analgesic targets: it's a winding road. Biochem. Pharmacol. 86, 1208–1214. doi: 10.1016/j.bcp.2013.08.001
van Hecke, O., Austin, S. K., Khan, R. A., Smith, B. H., and Torrance, N. (2014). Neuropathic pain in the general population: a systematic review of epidemiological studies. Pain 155, 654–662. doi: 10.1016/j.pain.2013.11.013
van Lierop, B. J., Robinson, S. D., Kompella, S. N., Belgi, A., McArthur, J. R., Hung, A., et al. (2013). Dicarba alpha-conotoxin Vc1.1 analogues with differential selectivity for nicotinic acetylcholine and GABAB receptors. ACS Chem. Biol. 8, 1815–1821. doi: 10.1021/cb4002393
Verbitsky, M., Rothlin, C. V., Katz, E., and Elgoyhen, A. B. (2000). Mixed nicotinic-muscarinic properties of the alpha9 nicotinic cholinergic receptor. Neuropharmacology 39, 2515–2524. doi: 10.1016/S0028-3908(00)00124-6
Vincler, M., and McIntosh, J. M. (2007). Targeting the alpha9alpha10 nicotinic acetylcholine receptor to treat severe pain. Expert Opin. Ther. Targets 11, 891–897. doi: 10.1517/14728222.11.7.891
Vincler, M., Wittenauer, S., Parker, R., Ellison, M., Olivera, B. M., and McIntosh, J. M. (2006). Molecular mechanism for analgesia involving specific antagonism of alpha9alpha10 nicotinic acetylcholine receptors. Proc. Natl. Acad. Sci. U.S.A. 103, 17880–17884. doi: 10.1073/pnas.0608715103
Wala, E. P., Crooks, P. A., McIntosh, J. M., and Holtman, J. R. Jr. (2012). Novel small molecule alpha9alpha10 nicotinic receptor antagonist prevents and reverses chemotherapy-evoked neuropathic pain in rats. Anesth. Analg. 115, 713–720. doi: 10.1213/ANE.0b013e31825a3c72
Whiteaker, P., Christensen, S., Yoshikami, D., Dowell, C., Watkins, M., Gulyas, J., et al. (2007). Discovery, synthesis, and structure activity of a highly selective alpha7 nicotinic acetylcholine receptor antagonist. Biochemistry 46, 6628–6638. doi: 10.1021/bi7004202
Wright, A. B., Norimatsu, Y., McIntosh, J. M., and Elmslie, K. S. (2015). Limited efficacy of alpha-conopeptides, Vc1.1 and RgIA, to inhibit sensory neuron CaV current. eNeuro 2:ENEURO.0057-14.2015. doi: 10.1523/ENEURO.0057-14.2015
Xiao, C., Nashmi, R., McKinney, S., Cai, H., McIntosh, J. M., and Lester, H. A. (2009). Chronic nicotine selectively enhances α4β2* nicotinic acetylcholine receptors in the nigrostriatal dopamine pathway. J. Neurosci. 29, 12428–12439. doi: 10.1523/JNEUROSCI.2939-09.2009
Zheng, G., Zhang, Z., Dowell, C., Wala, E., Dwoskin, L. P., Holtman, J. R., et al. (2011). Discovery of non-peptide, small molecule antagonists of α9α10 nicotinic acetylcholine receptors as novel analgesics for the treatment of neuropathic and tonic inflammatory pain. Bioorg. Med. Chem. Lett. 21, 2476–2479. doi: 10.1016/j.bmcl.2011.02.043
Zouridakis, M., Giastas, P., Zarkadas, E., Chroni-Tzartou, D., Bregestovski, P., and Tzartos, S. J. (2014). Crystal structures of free and antagonist-bound states of human α9 nicotinic receptor extracellular domain. Nat. Struct. Mol. Biol. 21, 976–980. doi: 10.1038/nsmb.2900
Keywords: nicotinic, chemotherapy, neuropathic pain, α9α10, conotoxins
Citation: Christensen SB, Hone AJ, Roux I, Kniazeff J, Pin J-P, Upert G, Servent D, Glowatzki E and McIntosh JM (2017) RgIA4 Potently Blocks Mouse α9α10 nAChRs and Provides Long Lasting Protection against Oxaliplatin-Induced Cold Allodynia. Front. Cell. Neurosci. 11:219. doi: 10.3389/fncel.2017.00219
Received: 19 May 2017; Accepted: 06 July 2017;
Published: 21 July 2017.
Edited by:
Barbara Jane Morley, Boys Town National Research Hospital, United StatesReviewed by:
Matilde Cordero-Erausquin, Centre National de la Recherche Scientifique (CNRS), FranceHermona Soreq, Hebrew University of Jerusalem, Israel
Copyright © 2017 Christensen, Hone, Roux, Kniazeff, Pin, Upert, Servent, Glowatzki and McIntosh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: J. Michael McIntosh, bWNpbnRvc2gubWlrZUBnbWFpbC5jb20=
†Present Address: Isabelle Roux, Molecular Biology and Genetics Section, Porter Neuroscience Research Center, National Institute on Deafness and Other Communication Disorders—National Institutes of Health, Bethesda, MD, United States