 Soledad Bárez-López
Soledad Bárez-López Ana Guadaño-Ferraz
Ana Guadaño-Ferraz- 1Department of Endocrine and Nervous System Pathophysiology, Instituto de Investigaciones Biomédicas Alberto Sols, Consejo Superior de Investigaciones Científicas (CSIC), Universidad Autónoma de Madrid (UAM), Madrid, Spain
- 2Center for Biomedical Research on Rare Diseases (Ciberer), Instituto de Salud Carlos III, Madrid, Spain
Thyroid hormones (THs) play an essential role in the development of all vertebrates; in particular adequate TH content is crucial for proper neurodevelopment. TH availability and action in the brain are precisely regulated by several mechanisms, including the secretion of THs by the thyroid gland, the transport of THs to the brain and neural cells, THs activation and inactivation by the metabolic enzymes deiodinases and, in the fetus, transplacental passage of maternal THs. Although these mechanisms have been extensively studied in rats, in the last decade, models of genetically modified mice have been more frequently used to understand the role of the main proteins involved in TH signaling in health and disease. Despite this, there is little knowledge about the mechanisms underlying THs availability in the mouse brain. This mini-review article gathers information from findings in rats, and the latest findings in mice regarding the ontogeny of TH action and the sources of THs to the brain, with special focus on neurodevelopmental stages. Unraveling TH economy and action in the mouse brain may help to better understand the physiology and pathophysiology of TH signaling in brain and may contribute to addressing the neurological alterations due to hypo and hyperthyroidism and TH resistance syndromes.
Introduction
Thyroid hormones (THs) are essential for the correct development of vertebrates controlling cell growth and metabolism in all tissues. The developing CNS is particularly sensitive to impairments in TH signaling. It is well known that TH deficit leads to numerous alterations the severity of which depends on the timing and the cause of the hormonal deficit (Morreale de Escobar et al., 2004; Zoeller and Rovet, 2004). While the wide range of clinical manifestations of adult onset hypothyroidism are usually reversed with an appropriate treatment, THs deficit during development can lead to irreversible neurological alterations unless treated with a timely initiation of hormonal replacement therapy (Morreale de Escobar et al., 2000), highlighting the importance of both appropriate levels of THs and timing of action.
THs are synthesized in the thyroid gland. Out of the total hormonal secretion of the thyroid gland approximately 93% is as thyroxine or 3,5,3′,5′-tetraiodo-L-thyronine (T4) and 7% as 3,5,3′-triodo-L-thyronine (T3) which is the active nuclear form (Bernal, 2015). The main pathway of THs action is at the genomic level by regulating gene expression through the binding of T3 to nuclear TH receptors (TRs) which function as ligand-modulated transcription factors (Cheng et al., 2010). In the brain deiodinases (D2 and D3) play an essential role in maintaining appropriate levels of T3. D2 catalyzes the conversion of T4 into the genomically active T3 while D3 catalyzes the deiodination of T4 and T3 into 3,3′5′-triiodo-L-thyronine (rT3) and 3,3′-diiodo-Lthyronine (T2) respectively, inactivating TH action (Bianco and Kim, 2006). Another crucial step for TH action is the transport of THs across the brain barriers and the plasma membrane of target cells by TH membrane transporter proteins. Among the many TH transporters that have been shown to mediate TH transport, the monocarboxylate transporter 8 (MCT8) presents the highest specificity for THs. Mutations in this transporter lead to the Allan-Herndon-Dudley syndrome with severe endocrine and neurological alterations (Visser, 2007; Fu et al., 2013; Bernal, 2015). In view of this, several regulatory mechanisms fine-tune the brain content of T4 and T3. These include TH secretion by the thyroid gland, TH transport to the brain and neural cells, activity of deiodinases, and in the fetus, transplacental passage of maternal THs (Obregon et al., 1984; Morreale de Escobar et al., 1990).
Most of the knowledge regarding TH action in the developing brain has arisen from studies in rats. However, due to the advantages of using genetic engineering approaches to generate genetically modified animals in mice over rat, the mouse has become the most widely used model to study thyroid physiology and TH action in brain. The use of genetically modified mice is providing new insights into thyroid physiological and pathological events. Despite this, many events underlying TH availability in the brain during mouse fetal development still need to be unraveled in order to fully understand TH economy in the developing brain. The aim of this review is to give an up-to-date insight into the evidence that has led to the understanding of TH availability and action in the developing brain of both rats and mice especially during embryonic stages.
Ontogeny of Thyroid Hormone Action
In the rat, tissues from whole embryos are provided with small amounts of T4 and T3 from at least embryonic day 11 (E11; Obregon et al., 1984), well before the onset of fetal thyroid gland function, which starts after E17. As the fetal thyroid gland starts functioning, the proportion of fetal THs increases so at term around 17.5% of the fetal pool of T4 (Morreale de Escobar et al., 1990) and 47% of the fetal pool of T3 are of maternal origin (Grijota-Martínez et al., 2011). In the mouse, the contribution of maternal T4 at term is larger than in rats as 60% of the total TH content in peripheral tissues derive directly from the mother (Bárez-López et al., 2017b).
The few studies reporting T3 and T4 content in the developing rat brain have been restricted to periods coinciding with or following the onset of fetal thyroid function at E17–E21 (Morreale de Escobar et al., 1985; Ruiz De Oña et al., 1988; Figure 1). In mice, T4 content in brain has been detected from at least E16 (Dong et al., 2015) which is just before the onset of the fetal thyroid gland that in mice starts around E16.5 (Fernández et al., 2015), and T3 has been detected from at least E18 (Ferrara et al., 2013; Bárez-López et al., 2017b).
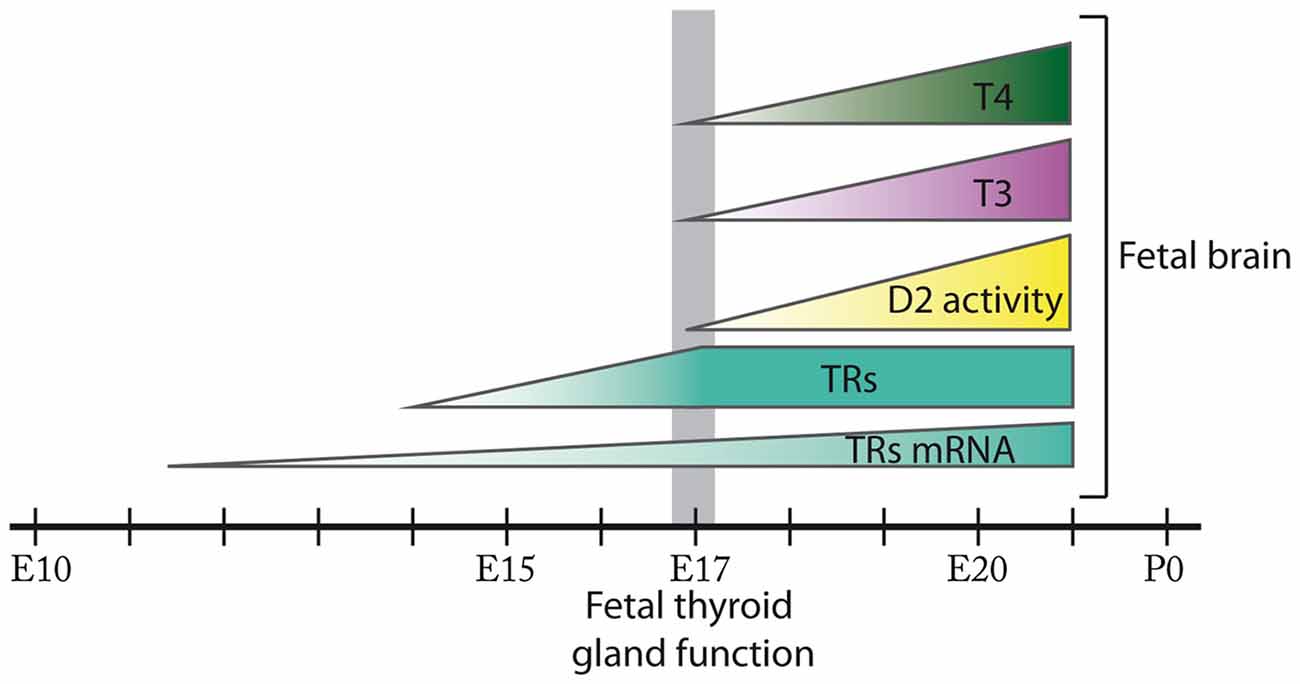
Figure 1. Ontogenesis of thyroid hormone action during fetal brain development in the rat. In the rat brain, thyroid hormone receptors (TRs) mRNA expression is detected from E11.5 and TRs from E14. D2 activity in addition to 3,5,3′,5′-tetraiodo-L-thyronine (T4) and 3,5,3′-triodo-L-thyronine (T3) content are present in the rat fetal brain from at least E17 when the onset of fetal thyroid function takes place. D2 activity, T4 and T3 content progressively increase in the rat fetal brain from E17 until E21. The size of the figures does not represent relative abundance. Most of these events remain to be fully elucidated in the mouse.
In the rat, mRNA for TRs has been detected from E11.5 at the neural tube and from E12.5 in certain areas of the prosencephalon, mesencephalon and the rhombencephalon (Bradley et al., 1992). In addition, T3 binding assays have revealed the presence of TRs in brain from E14, preceding the development of thyroid function, and their concentration has been shown to increase progressively until E17 (Perez-Castillo et al., 1985). Furthermore, at least from E21 there is a T3 receptor occupancy of 38% (Ferreiro et al., 1990; Figure 1). To our knowledge there are no studies describing the expression of TRs in the fetal mouse brain.
In the rat, deiodinase activity assays have detected D2 in the brain from as early as E17 with increasing activity until the day prior to birth (Ruiz De Oña et al., 1991; Figure 1). In the mouse, Dio2 mRNA expression has been detected in the brain from at least E15 (Bárez-López et al., 2017b), suggesting that maternal THs are necessary in the fetal brain before the maturation of the fetal thyroid gland. Interestingly, D2 and D3 are developmentally regulated in the brain (Bates et al., 1999; Escámez et al., 1999; Guadaño-Ferraz et al., 1999) as during the rat early postnatal development the maturational patterns of D2 and D3 activities reveal a progressive increase in D2 activity while D3 activity follows the opposite pattern (Kaplan and Yaskoski, 1981).
Regarding TH transporters there are no studies describing their spatiotemporal expression pattern during embryonic development in neither rats nor mice. There is a very complete study describing in detail the expression pattern of the main TH transporters in the mouse brain (Mct8, organic anion transporter family member 1c1 (Oatp1c1), L-type amino acid transporter 1, L-type amino acid transporter 2 and monocarboxylate transporter 10) from postnatal day 6 (P6) and this study revealed a precise mRNA expression pattern for each of the transporters which changed with age (Müller and Heuer, 2014). Other physiological studies have demonstrated that Mct8-deficient mice present a state of brain hyperthyroidism from E18 until P5 and that the combined inactivation of Mct8 and Lat2 suppresses this hyperthyroidism (Ferrara et al., 2013; Núñez et al., 2014) suggesting a crucial role for TH transporters in brain TH homeostasis. The dynamic expression of TH transporters in the brain as well as the functional implications of their deficiency highlights the relevance of TH transporters activity during fetal neurodevelopment and the consequent need to increase our understanding of their spatiotemporal expression pattern and function.
Sources of Thyroid Hormones to The Rodent Brain
The main source of T3 in the brain is provided by the local conversion of T4 into T3 by D2 as, in the adult rat brain, 80% of nuclear bound T3 is formed locally from T4 (Crantz et al., 1982). Further studies in adult hypothyroid rats revealed that constant infusion of T3 at relatively low doses was enough to normalize T3 levels in peripheral tissues while much higher doses were necessary for the cerebral cortex (CCx) to reach normal values. On the other hand, when T4 was administered, T3 values were normalized in the CCx at lower doses than those necessary to restore T3 levels in other peripheral tissues. Furthermore, T3 content in the CCx remained within the physiological range regardless of the increase in the dose of the T4 administered (Escobar-Morreale et al., 1999). These studies revealed that T4 is the primary source of T3 in the brain and that local conversion of T4 into T3 is tightly regulated avoiding excess of T3. A previous study had proposed a close metabolic coupling between glial cells and neurons by which T4 would be taken up from the blood and the cerebrospinal fluid by astrocytes and tanycytes, where it would be deiodinated into T3 by D2 activity, and would finally be released for utilization by neurons (Guadaño-Ferraz et al., 1997). Years later, studies with transgenic mice validated and completed this model for T3 availability to neural cells (Ceballos et al., 2009; Freitas et al., 2010; Morte et al., 2010a). Currently, the model states that brain T3 has a double origin: a fraction is available directly from the circulation, and another is produced locally from T4 in the astrocytes by D2. To enter the brain, circulating T4 and T3 need to cross the brain barriers through specific TH transporters. Based on the transporters’ location the current model in rodents indicates that: (i) T3 and T4 cross the blood-brain barrier (BBB) through Mct8 (Dumitrescu et al., 2006; Trajkovic et al., 2007) into the extracellular fluid where they can directly reach the neural cells in the proximity of the blood vessels; and that (ii) T4, but not T3, crosses the BBB through the Oatp1c1 (Mayerl et al., 2014) directly into the astrocytes through their end-feet in contact with the blood vessels, and astrocytes produce additional T3 by D2 activity that can be then transported to the neurons (reviewed in Morte and Bernal, 2014).
For the postnatal and adult brain, studies in transgenic mice suggest that each of these two routes can contribute to at least 50% of the total pool of brain T3. D2-deficient mice present around a 50% decrease in the content of T3 in the CCx at P15 (Galton et al., 2007) and 40% decrease at 3 months of age (Bárez-López et al., 2014). Furthermore, Mct8-deficient mice also present a 50% reduction in the content of T3 in the CCx (Dumitrescu et al., 2006; Trajkovic et al., 2007). Together these data suggest that in the mice postnatal brain around 50% of the total brain T3 derives directly from T3 uptake from the circulation and another 50% is locally generated in the astrocytes from T4.
In contrast, studies in rats demonstrated that during fetal development the brain depends almost entirely on the T3 locally generated by D2. A pioneer study demonstrated that infusion of T4 to hypothyroid pregnant rats was sufficient to increase T3 content in the fetal brain to similar values of control fetuses while T3 concentrations increased very little in plasma and lung (Morreale de Escobar et al., 1988). Subsequent studies evidenced that the high doses of T3 administered to hypothyroid pregnant rats were able to cross the placenta but failed to normalize T3 content in the fetal brain and to induce the expression of T3-dependent genes. In contrast, physiological doses of T4 could cross both the placenta and the fetal brain barriers, normalizing fetal brain T3 concentrations by T4 deiodination and increasing neuronal gene expression (Calvo et al., 1990; Grijota-Martínez et al., 2011). The role of D2 activity in this process is further supported by D2 activity ontogeny studies that revealed significant D2 activity in rat fetal brain which highly increased prior to birth and was inversely regulated by increasing doses of T4 (Ruiz De Oña et al., 1988). The reason why the fetal brain is not permeable to T3 is not known and cannot be explained by lack of the Mct8 transporter as this is present at the BBB during fetal development (Grijota-Martínez et al., 2011). Likewise for the mouse, recent studies have demonstrated that maternal T4 is an essential source of TH in the developing mouse fetal brain and that T3 content is regulated through the conversion of T4 into T3 by D2 activity (Bárez-López et al., 2017b). However, the contribution of maternal T3 to the total fetal brain T3 content in mice still remains to be elucidated.
It still remains unclear whether maternal THs are necessary before the onset of the fetal thyroid gland. Some studies in rats indicated that maternal hypothyroidism leads to defects in the proliferation of some neuronal precursors that is usually completed by E12 (Narayanan and Narayanan, 1985), and to defects in the migration of some proliferating cells that normally finishes at E16–E17 (Lucio et al., 1997). In mice the treatment of pregnant dams with goitrogens from E13 to E16 led to a decrease in the T4 content in the fetal CCx and had an effect in the expression of a few T3-dependent genes (Dong et al., 2015). These findings might indicate that maternal THs are necessary before the fetal thyroid gland starts functioning, however, these defects could arise from maternal hypothyroidism side effects or from a direct effect of the goitrogens used to induce hypothyroidism. However one study in rats evidenced that maternal hypothyroxinemia derived from low iodine intake results in abnormal migration of neurons that is usually complete before the fetal thyroid function, strongly suggesting that maternal THs are necessary before the onset of the fetal thyroid gland (Lavado-Autric et al., 2003).
Another essential aspect that needs to be clarified is the contribution of maternal THs to the fetal brain under fetal euthyroid conditions. Studies in rats revealed that fetuses coming from thyroidectomized pregnant dams presented only a small decrease in the brain content of T4, with no changes in T3 content at perinatal stages (Berbel et al., 2010; Grijota-Martínez et al., 2011) and unaffected expression of T3-dependent genes (Morte et al., 2010b; Grijota-Martínez et al., 2011). Likewise, in another study, brain D2 activity of fetuses coming from thyroidectomized pregnant rats was not different from those coming from control dams (Ruiz De Oña et al., 1988). These findings in rats can be attributed to the progressive activity of the fetal thyroid gland indicating that fetal THs are the main regulators of TH-dependent fetal gene expression. Nevertheless, these results do not rule out a specific contribution from the mother. As the contribution of the fetal thyroid gland seems to be larger in rats than in mice (Bárez-López et al., 2017b), it should not be assumed that mice will follow the same pattern as rats. The difficulty of performing successful thyroidectomies in mice might hinder the assessment of the role of maternal THs in mice fetal brain under euthyroid fetal conditions.
Brain Barriers and Thyroid Hormones
The passage of substances from the blood to the brain is regulated by the BBB and the blood-cerebrospinal fluid barrier (BCSFB). The BBB is formed by endothelial cells within CNS microvessels while the BCSFB is formed by a single epithelial cell layer in the choroid plexus and multiple epithelial cell layers in the arachnoid membrane (Johanson et al., 2011). These barriers tightly control the influx and efflux of molecules and ions at the blood-brain interface to meet neural cell needs and to protect the brain from toxins and pathogens (Obermeier et al., 2013). The severe alterations present in MCT8-deficient patients (Fu et al., 2013) revealed the relevance of TH transporters and the importance of TH transport across brain barriers (Ceballos et al., 2009; Iwayama et al., 2016; Vatine et al., 2017). Studies in the rat indicated that the contribution of the choroid plexus to the total brain TH content is about 18% (Chanoine et al., 1992) and, since the BBB surface area is much greater than that of the choroid plexus, the BBB has been considered the major pathway for TH entry into the brain (Dratman et al., 1991).
However, what is the situation in the fetal brain during the development of the BBB (Obermeier et al., 2013) and before the maturation of astrocytes (Daneman et al., 2010)? In a recent study we detected Dio2 mRNA in the mouse brain in the meninges, the ependymal layer of the lateral ventricles and the choroid plexus at E15 and E18 (Bárez-López et al., 2017b). Dio2 expression in the meninges, the ependymal layer of the lateral ventricles and the choroid plexus diminished by P3 and has not been detected in these structures in the adult brain neither in mice (Bárez-López et al., 2017a) nor rats (Wittmann et al., 2014). This indicates an important role of D2 at the BCSFB during fetal stages, especially since it has been suggested that the BCSFB could have a more important role during early stages of brain growth and development as the choroid plexus may have a greater transport capacity (Keep and Jones, 1990). Moreover, the volume of the lateral ventricles is larger at these stages in comparison to the rest of the brain (O’Rahilly and Müller, 2008). This suggests that the T4 transported through the choroid plexus could be deiodinated into T3 by D2 activity at the BCSFB and from the cerebrospinal fluid the T3 could reach the brain cells and exert and action at the genomic level (Figure 2). Furthermore, the generation of T3 by D2 at the meninges could be controlling the synthesis of retinoid acid (Gil-Ibáñez et al., 2014) regulating cortical neurogenesis (Siegenthaler et al., 2009). It has also been demonstrated that D2 deficiency during perinatal neurodevelopment leads to a state of brain hypothyroidism with reduced expression of T3-dependent genes (Bárez-López et al., 2017b). Altogether the current data indicate that conversion of T4 into T3 by D2 activity might be the only source of T3 during mouse brain development.
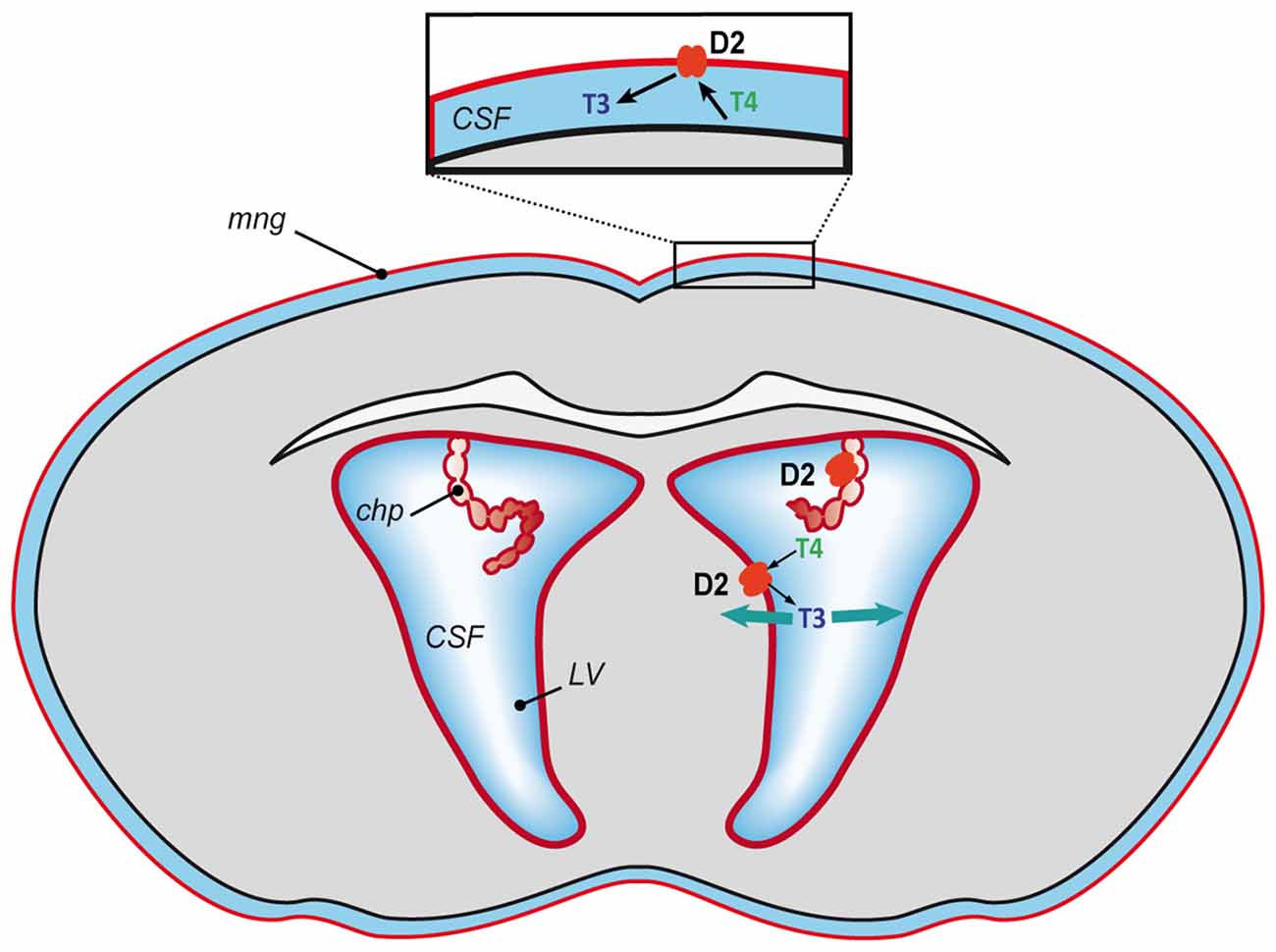
Figure 2. Proposed model of T3 availability to the mouse developing brain. T4 mostly of maternal origin reaches the brain, gets transported into the cerebrospinal fluid and converted into T3 by D2 (in red) at the blood-cerebrospinal fluid barrier (meninges and choroid plexus) and at the lateral ventricles. From the cerebrospinal fluid the T3 could spread throughout the brain and access the neural cells to exert its action. CFS, cerebrospinal fluid; chp, choroid plexus; LV, lateral ventricles and mng, meninges.
Conclusion
Due to the crucial role of TH action during brain development and to consequences of TH deficit, it is essential to understand the physiological mechanisms that underlie T4 and T3 availability to the developing brain in order to obtain insight into pathological conditions. This will permit the development of appropriate treatments and establish timings to prevent, restore or at least palliate possible impairments, for example for MCT8-deficient patients. Studies in animal models have provided a great deal of information about the events underlying TH action during brain development, however, there are still many aspects that need to be elucidated. Among all the unresolved matters both in rat and mouse, a better understanding of the contribution of maternal THs to the fetal brain under euthyroid conditions, of the role of TH transporters during brain development and of the contribution of the BCSFB to TH brain availability would facilitate potential clinical applications in the future.
Author Contributions
SB-L and AG-F have contributed in the drafting of the article and have revised it critically for important intellectual content and for the final approval of the submitted version. SB-L has designed the figures.
Funding
This work was supported by the Spanish Ministry of Economy and Competitiveness (grant number SAF2014-54919-R). SB-L is a recipient of a contract from the Sherman Foundation (grant number OTR02211). The cost of this publication has been paid in part by FEDER funds.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Juan Bernal for critical reading of the manuscript and helpful discussions, Ewan McDonald for editorial help and Javier Pérez for helping with the artwork.
Abbreviations
BBB, blood brain barrier; BCSFB, blood-cerebrospinal fluid barrier; D2, deiodinase type-2; D3, deiodinases type-3; E, embryonic day; MCT8, monocarboxylate transporter 8; Oatp1c1, organic anion transporter family member 1C1; P, postnatal day; T3, 3,5,3′-triodo-L-thyronine; T4, 3,5,3′,5′-tetraiodo-L-thyronine; THs, thyroid hormones; TRs, thyroid hormone receptors.
References
Bárez-López, S., Bosch-García, D., Gómez-Andrés, D., Pulido-Valdeolivas, I., Montero-Pedrazuela, A., Obregon, M. J., et al. (2014). Abnormal motor phenotype at adult stages in mice lacking type 2 deiodinase. PLoS One 9:e103857. doi: 10.1371/journal.pone.0103857
Bárez-López, S., Montero-Pedrazuela, A., Bosch-García, D., Venero, C., and Guadaño-Ferraz, A. (2017a). Increased anxiety and fear memory in adult mice lacking type 2 deiodinase. Psychoneuroendocrinology 84, 51–60. doi: 10.1016/j.psyneuen.2017.06.013
Bárez-López, S., Obregon, M. J., Bernal, J., and Guadaño-Ferraz, A. (2017b). Thyroid hormone economy in the perinatal mouse brain: implications for cerebral cortex development. Cereb. Cortex 12, 1–11. doi: 10.1093/cercor/bhx088
Bates, J. M., St Germain, D. L., and Galton, V. A. (1999). Expression profiles of the three iodothyronine deiodinases, D1, D2, and D3, in the developing rat. Endocrinology 140, 844–851. doi: 10.1210/en.140.2.844
Berbel, P., Navarro, D., Ausó, E., Varea, E., Rodríguez, A. E., Ballesta, J. J., et al. (2010). Role of late maternal thyroid hormones in cerebral cortex development: an experimental model for human prematurity. Cereb. Cortex 20, 1462–1475. doi: 10.1093/cercor/bhp212
Bernal, J. (2015). “Thyroid hormones in brain development and function,” in Endotext. eds L. J. De Groot, G. Chrousos, K. Dungan, K. R. Feingold, A. Grossman, J. M. Hershman, et al. (South Dartmouth, MA: MDText.com, Inc.). Available online at: http://www.thyroidmanager.org/
Bianco, A. C., and Kim, B. W. (2006). Deiodinases: implications of the local control of thyroid hormone action. J. Clin. Invest. 116, 2571–2579. doi: 10.1172/JCI29812
Bradley, D. J., Towle, H. C., and Young, W. S. III (1992). Spatial and temporal expression of alpha- and beta-thyroid hormone receptor mRNAs, including the beta 2-subtype, in the developing mammalian nervous system. J. Neurosci. 12, 2288–2302.
Calvo, R., Obregon, M. J., Ruiz de Oña, C., Escobar del Rey, F., and Morreale de Escobar, G. (1990). Congenital hypothyroidism, as studied in rats. Crucial role of maternal thyroxine but not of 3,5,3′-triiodothyronine in the protection of the fetal brain. J. Clin. Invest. 86, 889–899. doi: 10.1172/jci114790
Ceballos, A., Belinchon, M. M., Sanchez-Mendoza, E., Grijota-Martinez, C., Dumitrescu, A. M., Refetoff, S., et al. (2009). Importance of monocarboxylate transporter 8 for the blood-brain barrier-dependent availability of 3,5,3′-triiodo-L-thyronine. Endocrinology 150, 2491–2496. doi: 10.1210/en.2008-1616
Chanoine, J. P., Alex, S., Fang, S. L., Stone, S., Leonard, J. L., Körhle, J., et al. (1992). Role of transthyretin in the transport of thyroxine from the blood to the choroid plexus, the cerebrospinal fluid and the brain. Endocrinology 130, 933–938. doi: 10.1210/en.130.2.933
Cheng, S.-Y., Leonard, J. L., and Davis, P. J. (2010). Molecular aspects of thyroid hormone actions. Endocr. Rev. 31, 139–170. doi: 10.1210/er.2009-0007
Crantz, F. R., Silva, J. E., and Larsen, P. R. (1982). An analysis of the sources and quantity of 3,5,3′-triiodothyronine specifically bound to nuclear receptors in rat cerebral cortex and cerebellum. Endocrinology 110, 367–375. doi: 10.1210/endo-110-2-367
Daneman, R., Zhou, L., Kebede, A. A., and Barres, B. A. (2010). Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 468, 562–566. doi: 10.1038/nature09513
Dong, H., You, S. H., Williams, A., Wade, M. G., Yauk, C. L., and Thomas Zoeller, R. (2015). Transient maternal hypothyroxinemia potentiates the transcriptional response to exogenous thyroid hormone in the fetal cerebral cortex before the onset of fetal thyroid function: a messenger and microRNA profiling study. Cereb. Cortex 25, 1735–1745. doi: 10.1093/cercor/bht364
Dratman, M. B., Crutchfield, F. L., and Schoenhoff, M. B. (1991). Transport of iodothyronines from bloodstream to brain: contributions by blood:brain and choroid plexus:cerebrospinal fluid barriers. Brain Res. 554, 229–236. doi: 10.1016/0006-8993(91)90194-z
Dumitrescu, A. M., Liao, X. H., Weiss, R. E., Millen, K., and Refetoff, S. (2006). Tissue-specific thyroid hormone deprivation and excess in monocarboxylate transporter (mct) 8-deficient mice. Endocrinology 147, 4036–4043. doi: 10.1210/en.2006-0390
Escámez, M. J., Guadaño-Ferraz, A., Cuadrado, A., and Bernal, J. (1999). Type 3 iodothyronine deiodinase is selectively expressed in areas related to sexual differentiation in the newborn rat brain. Endocrinology 140, 5443–5446. doi: 10.1210/en.140.11.5443
Escobar-Morreale, H. F., Obregon, M. J., Escobar del Rey, F., and Morreale de Escobar, G. (1999). Tissue-specific patterns of changes in 3,5,3′-triiodo-L-thyronine concentrations in thyroidectomized rats infused with increasing doses of the hormone. Which are the regulatory mechanisms? Biochimie 81, 453–462. doi: 10.1016/s0300-9084(99)80095-9
Fernández, L. P., López-Márquez, A., and Santisteban, P. (2015). Thyroid transcription factors in development, differentiation and disease. Nat. Rev. Endocrinol. 11, 29–42. doi: 10.1038/nrendo.2014.186
Ferrara, A. M., Liao, X. H., Gil-Ibáñez, P., Marcinkowski, T., Bernal, J., Weiss, R. E., et al. (2013). Changes in thyroid status during perinatal development of MCT8-deficient male mice. Endocrinology 154, 2533–2541. doi: 10.1210/en.2012-2031
Ferreiro, B., Pastor, R., and Bernal, J. (1990). T3 receptor occupancy and T3 levels in plasma and cytosol during rat brain development. Acta Endocrinol. (Copenh) 123, 95–99. doi: 10.1530/acta.0.1230095
Freitas, B. C., Gereben, B., Castillo, M., Kallo, I., Zeold, A., Egri, P., et al. (2010). Paracrine signaling by glial cell-derived triiodothyronine activates neuronal gene expression in the rodent brain and human cells. J. Clin. Invest. 120, 2206–2217. doi: 10.1172/JCI41977
Fu, J., Refetoff, S., and Dumitrescu, A. M. (2013). Inherited defects of thyroid hormone-cell-membrane transport: review of recent findings. Curr. Opin. Endocrinol. Diabetes Obes. 20, 434–440. doi: 10.1097/01.med.0000432531.03233.ad
Galton, V. A., Wood, E. T., St. Germain, E. A., Withrow, C. A., Aldrich, G., St Germain, G. M., et al. (2007). Thyroid hormone homeostasis and action in the type 2 deiodinase-deficient rodent brain during development. Endocrinology 148, 3080–3088. doi: 10.1210/en.2006-1727
Gil-Ibáñez, P., Bernal, J., and Morte, B. (2014). Thyroid hormone regulation of gene expression in primary cerebrocortical cells: role of thyroid hormone receptor subtypes and interactions with retinoic acid and glucocorticoids. PLoS One 9:e91692. doi: 10.1371/journal.pone.0091692
Grijota-Martínez, C., Díez, D., Morreale de Escobar, G., Bernal, J., and Morte, B. (2011). Lack of action of exogenously administered T3 on the fetal rat brain despite expression of the monocarboxylate transporter 8. Endocrinology 152, 1713–1721. doi: 10.1210/en.2010-1014
Guadaño-Ferraz, A., Escámez, M. J., Rausell, E., and Bernal, J. (1999). Expression of type 2 iodothyronine deiodinase in hypothyroid rat brain indicates an important role of thyroid hormone in the development of specific primary sensory systems. J. Neurosci. 19, 3430–3439.
Guadaño-Ferraz, A., Obregón, M. J., St. Germain, D. L., and Bernal, J. (1997). The type 2 iodothyronine deiodinase is expressed primarily in glial cells in the neonatal rat brain. Proc. Natl. Acad. Sci. U S A 94, 10391–10396. doi: 10.1073/pnas.94.19.10391
Iwayama, H., Liao, X. H., Braun, L., Bárez-López, S., Kaspar, B., Weiss, R. E., et al. (2016). Adeno associated virus 9-based gene therapy delivers a functional monocarboxylate transporter 8, improving thyroid hormone availability to the brain of Mct8-deficient mice. Thyroid 26, 1311–1319. doi: 10.1089/thy.2016.0060
Johanson, C. E., Stopa, E. G., and McMillan, P. N. (2011). The blood-cerebrospinal fluid barrier: structure and functional significance. Methods Mol. Biol. 686, 101–131. doi: 10.1007/978-1-60761-938-3_4
Kaplan, M. M., and Yaskoski, K. A. (1981). Maturational patterns of iodothyronine phenolic and tyrosyl ring deiodinase activities in rat cerebrum, cerebellum, and hypothalamus. J. Clin. Invest. 67, 1208–1214. doi: 10.1172/jci110136
Keep, R. F., and Jones, H. C. (1990). A morphometric study on the development of the lateral ventricle choroid plexus, choroid plexus capillaries and ventricular ependyma in the rat. Dev. Brain Res. 56, 47–53. doi: 10.1016/0165-3806(90)90163-s
Lavado-Autric, R., Ausó, E., García-Velasco, J. V., Arufe Mdel, C., Escobar del Rey, F., Berbel, P., et al. (2003). Early maternal hypothyroxinemia alters histogenesis and cerebral cortex cytoarchitecture of the progeny. J. Clin. Invest. 111, 1073–1082. doi: 10.1172/jci16262
Lucio, R. A., García, J. V., Ramón Cerezo, J., Pacheco, P., Innocenti, G. M., and Berbel, P. (1997). The development of auditory callosal connections in normal and hypothyroid rats. Cereb. Cortex 7, 303–316. doi: 10.1093/cercor/7.4.303
Mayerl, S., Müller, J., Bauer, R., Richert, S., Kassmann, C. M., Darras, V. M., et al. (2014). Transporters MCT8 and OATP1C1 maintain murine brain thyroid hormone homeostasis. J. Clin. Invest. 124, 1987–1999. doi: 10.1172/jci70324
Morreale de Escobar, G., Calvo, R., Obregon, M. J., and Escobar Del Rey, F. (1990). Contribution of maternal thyroxine to fetal thyroxine pools in normal rats near term. Endocrinology 126, 2765–2777. doi: 10.1210/endo-126-5-2765
Morreale de Escobar, G., Obregon, M. J., and del Rey, F. E. (2004). Maternal thyroid hormones early in pregnancy and fetal brain development. Best Pract. Res. Clin. Endocrinol. Metab. 18, 225–248. doi: 10.1016/j.beem.2004.03.012
Morreale de Escobar, G., Obregón, M. J., and Escobar del Rey, F. (2000). Is neuropsychological development related to maternal hypothyroidism or to maternal hypothyroxinemia? J. Clin. Endocrinol. Metab. 85, 3975–3987. doi: 10.1210/jcem.85.11.6961
Morreale de Escobar, G., Obregon, M. J., Ruiz de Oña, C., and Escobar del Rey, F. (1988). Transfer of thyroxine from the mother to the rat fetus near term: effects on brain 3,5,3′-triiodothyronine deficiency. Endocrinology 122, 1521–1531. doi: 10.1210/endo-122-4-1521
Morreale de Escobar, G., Pastor, R., Obregon, M. J., and Escobar del Rey, F. (1985). Effects of maternal hypothyroidism on the weight and thyroid hormone content of rat embryonic tissues, before and after onset of fetal thyroid function. Endocrinology 117, 1890–1900. doi: 10.1210/endo-117-5-1890
Morte, B., and Bernal, J. (2014). Thyroid hormone action: astrocyte-neuron communication. Front. Endocrinol. (Lausanne) 5:82. doi: 10.3389/fendo.2014.00082
Morte, B., Ceballos, A., Diez, D., Grijota-Martínez, C., Dumitrescu, A. M., Di Cosmo, C., et al. (2010a). Thyroid hormone-regulated mouse cerebral cortex genes are differentially dependent on the source of the hormone: a study in monocarboxylate transporter-8- and deiodinase-2-deficient mice. Endocrinology 151, 2381–2387. doi: 10.1210/en.2009-0944
Morte, B., Díez, D., Ausó, E., Belinchón, M. M., Gil-Ibáñez, P., Grijota-Martínez, C., et al. (2010b). Thyroid hormone regulation of gene expression in the developing rat fetal cerebral cortex: prominent role of the Ca2+/calmodulin-dependent protein kinase IV pathway. Endocrinology 151, 810–820. doi: 10.1210/en.2009-0958
Müller, J., and Heuer, H. (2014). Expression pattern of thyroid hormone transporters in the postnatal mouse brain. Front. Endocrinol. (Lausanne) 5:92. doi: 10.3389/fendo.2014.00092
Narayanan, C. H., and Narayanan, Y. (1985). Cell formation in the motor nucleus and mesencephalic nucleus of the trigeminal nerve of rats made hypothyroid by propylthiouracil. Exp. Brain Res. 59, 257–266. doi: 10.1007/BF00230905
Núñez, B., Martínez De Mena, R., Obregon, M. J., Font-Llitjos, M., Nunes, V., Palacín, M., et al. (2014). Cerebral cortex hyperthyroidism of newborn mct8-deficient mice transiently suppressed by lat2 inactivation. PLoS One 9:e96915. doi: 10.1371/journal.pone.0096915
Obermeier, B., Daneman, R., and Ransohoff, R. M. (2013). Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 19, 1584–1596. doi: 10.1038/nm.3407
Obregon, M. J., Mallol, J., Pastor, R., Morreale De Escobar, G., and Escobar Del Rey, F. (1984). L-thyroxine and 3,5,3′-triiodo-L-thyronine in rat embryos before onset of fetal thyroid function. Endocrinology 114, 305–307. doi: 10.1210/endo-114-1-305
O’Rahilly, R., and Müller, F. (2008). Significant features in the early prenatal development of the human brain. Ann. Anat. 190, 105–118. doi: 10.1016/j.aanat.2008.01.001
Perez-Castillo, A., Bernal, J., Ferreiro, B., and Pans, T. (1985). The early ontogenesis of thyroid hormone receptor in the rat fetus. Endocrinology 117, 2457–2461. doi: 10.1210/endo-117-6-2457
Ruiz De Oña, C., Morreale De Escobar, G., Calvo, R., Escobar Del Rey, F., and Obregón, M. J. (1991). Thyroid hormones and 5′-deiodinase in the rat fetus late in gestation: effects of maternal hypothyroidism. Endocrinology 128, 422–432. doi: 10.1210/endo-128-1-422
Ruiz De Oña, C., Obregón, M. J., Escobar Del Rey, F., and Morreale De Escobar, G. (1988). Developmental changes in rat brain 5′-deiodinase and thyroid hormones during the fetal period: the effects of fetal hypothyroidism and maternal thyroid hormones. Pediatr. Res. 24, 588–594. doi: 10.1203/00006450-198811000-00010
Siegenthaler, J. A., Ashique, A. M., Zarbalis, K., Patterson, K. P., Hecht, J. H., Kane, M. A., et al. (2009). Retinoic acid from the meninges regulates cortical neuron generation. Cell 139, 597–609. doi: 10.1016/j.cell.2009.10.004
Trajkovic, M., Visser, T. J., Mittag, J., Horn, S., Lukas, J., Darras, V. M., et al. (2007). Abnormal thyroid hormone metabolism in mice lacking the monocarboxylate transporter 8. J. Clin. Invest. 117, 627–635. doi: 10.1172/JCI28253
Vatine, G. D., Al-Ahmad, A., Barriga, B. K., Svendsen, S., Salim, A., Garcia, L., et al. (2017). Modeling psychomotor retardation using iPSCs from MCT8-deficient patients indicates a prominent role for the blood-brain barrier. Cell Stem Cell 20, 831.e5–843.e5. doi: 10.1016/j.stem.2017.04.002
Wittmann, G., Harney, J. W., Singru, P. S., Nouriel, S. S., Reed Larsen, P., and Lechan, R. M. (2014). Inflammation-inducible type 2 deiodinase expression in the leptomeninges, choroid plexus and at brain blood vessels in male rodents. Endocrinology 155, 2009–2019. doi: 10.1210/en.2013-2154
Keywords: thyroid hormones, brain, rodents, type 2 deiodinase, MCT8, neurodevelopment, brain barriers
Citation: Bárez-López S and Guadaño-Ferraz A (2017) Thyroid Hormone Availability and Action during Brain Development in Rodents. Front. Cell. Neurosci. 11:240. doi: 10.3389/fncel.2017.00240
Received: 14 June 2017; Accepted: 31 July 2017;
Published: 14 August 2017.
Edited by:
Davide Cervia, Università degli Studi della Tuscia, ItalyReviewed by:
Gabor Wittmann, Tufts Medical Center, United StatesMario Valentino, University of Malta, Malta
Copyright © 2017 Bárez-López and Guadaño-Ferraz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Soledad Bárez-López, c2JhcmV6QGlpYi51YW0uZXM=