 Li Zhou1,2Di Chen3Xu-Ming Huang2Fei Long3Hua Cai3Wen-Xia Yao3Zhong-Cheng Chen4Zhi-Jian Liao5Zhe-Zhi Deng1Sha Tan1Yi-Long Shan1Wei Cai1Yu-Ge Wang1Ri-Hong Yang6Nan Jiang7*Tao Peng3*Ming-Fan Hong8*
Li Zhou1,2Di Chen3Xu-Ming Huang2Fei Long3Hua Cai3Wen-Xia Yao3Zhong-Cheng Chen4Zhi-Jian Liao5Zhe-Zhi Deng1Sha Tan1Yi-Long Shan1Wei Cai1Yu-Ge Wang1Ri-Hong Yang6Nan Jiang7*Tao Peng3*Ming-Fan Hong8* Zheng-Qi Lu1*
Zheng-Qi Lu1*- 1Department of Neurology, The Third Affiliated Hospital of Sun Yat-sen University, Guangzhou, China
- 2Department of Rehabilitation, The First Affiliated Hospital of Clinical Medicine of Guangdong Pharmaceutical University, Guangzhou, China
- 3Laboratory of Viral Immunology, State Key Laboratory of Respiratory Disease, Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences, Sino-French Hoffmann Institute of Immunology, Guangzhou Medical University, Guangzhou, China
- 4Department of Laboratory, Third Affiliated Hospital of Sun Yat-sen University, Guangzhou, China
- 5Institute of Hematology, Guangzhou, China
- 6Department of Pathology, Third Affiliated Hospital of Sun Yat-sen University, Guangzhou, China
- 7Department of Hepatic Surgery, Third Affiliated Hospital of Sun Yat-sen University, Guangzhou, China
- 8Department of Neurology, The First Affiliated Hospital of Clinical Medicine of Guangdong Pharmaceutical University, Guangzhou, China
β-Amyloid protein (Aβ) is thought to cause neuronal loss in Alzheimer’s disease (AD). Aβ treatment promotes the re-activation of a mitotic cycle and induces rapid apoptotic death of neurons. However, the signaling pathways mediating cell-cycle activation during neuron apoptosis have not been determined. We find that Wnt5a acts as a mediator of cortical neuron survival, and Aβ42 promotes cortical neuron apoptosis by downregulating the expression of Wnt5a. Cell-cycle activation is mediated by the reduced inhibitory effect of Wnt5a in Aβ42 treated cortical neurons. Furthermore, Wnt5a signals through the non-canonical Wnt/Ca2+ pathway to suppress cyclin D1 expression and negatively regulate neuronal cell-cycle activation in a cell-autonomous manner. Together, aberrant downregulation of Wnt5a signaling is a crucial step during Aβ42 induced cortical neuron apoptosis and might contribute to AD-related neurodegeneration.
Introduction
Alzheimer’s disease (AD) is a chronic neurodegenerative disease that induces by progressive neuron loss (Davies and Maloney, 1976; Whitehouse et al., 1982; Gómez-Isla et al., 1996). The key events of AD pathogenesis are the production and deposition of amyloid-beta peptides (Aβ; Hardy and Selkoe, 2002; Murphy and LeVine, 2010). Aβ induces DNA synthesis and promotes the re-entry of cell-cycle in cultured cortical neurons and in mouse models of AD (Kruman et al., 2004; Varvel et al., 2008). Aberrant cell-cycle activation is causally related to neuronal apoptosis and might be a root cause of several neurodegenerative disorders (Liu and Greene, 2001; Yang et al., 2001, 2003; Becker and Bonni, 2004; Herrup and Yang, 2007). A series of the proteins report to be involved in AD pathology is cyclin-dependent kinases (CDKs) and their regulators (such as CDK1, CDK4, CDK5, p16, Cyclin B1, Cyclin D1 and Cyclin E; Arendt et al., 1996; McShea et al., 1997; Vincent et al., 1997; Hoozemans et al., 2002; Shukla et al., 2012). Meanwhile, previous studies demonstrated that Aβ peptides will lead to re-enter various phases of the cell cycle in adult neurons. Aβ induces the cleavage of CDK5 regulatory subunit p35 to p25 (Patrick et al., 1999), promotes the expression of Cyclin D1 and B1 (Majd et al., 2008), increases the activity of CDK1, CDK4 and CDK5 (Milton, 2002; Biswas et al., 2007; Lopes et al., 2010). However, the signaling pathways mediating CDKs activation during Aβ induced neuron apoptosis have not been determined.
Recently, more and more evidence suggest a strong relationship between a dysregulation of the Wnt signaling pathway activity and AD (De Ferrari et al., 2014; Riise et al., 2015). Many components of Wnt signaling are altered in AD, such as Dkk1 and β-catenin. Dkk1, which inhibits Wnt signaling by binding to the LRPs and preventing Wnts from forming a complex with Fz and LRPs (Zorn, 2001), is increased in brains of both familial/early-onset AD and sporadic/late-onset AD patients (De Ferrari and Moon, 2006; Boonen et al., 2009). DKK1 is also upregulated in cultured cortical neurons treated with Aβ (Caricasole et al., 2004). β-catenin is reduced in brains of AD patients carrying presenilin-1-inherited mutations (Zhang et al., 1998) and in brains of late-onset AD patients (De Ferrari et al., 2007).
Moreover, Wnt pathway plays an important role in the regulation of cell cycle (Davidson and Niehrs, 2010). Wnt1 blocks the differentiation and enhance the proliferation of PC12 cells by activating cyclin D1 (Issack and Ziff, 1998), the Cyclin D1 is also a direct target gene of the Wnt/β-catenin/LEF-1 pathway through a LEF-1 binding site in the Cyclin D1 promoter (Shtutman et al., 1999), Wnt pathway promotes oral squamous-cell carcinoma cell proliferation by negatively control the expression of the human homolog of the Drosophila headcase (HECA) and reduces the interaction of HECA with CDK2, CDK9, Cyclin A and Cyclin K (Dowejko et al., 2012) and so on. Based on the importance of cell-cycle reactivation in Aβ induced neuronal injury and the essential role of Wnt pathway in activation of cell cycle, we speculate that Aβ may activate the cell-cycle pathway by Wnt family members.
The purpose of this study was to investigate the role of Wnt family members in Aβ42 induced cortical neuron damage. This study is the first to show Aβ42 induced non-canonical Wnt5a/Ca2+ pathway dysfunction and cell-cycle re-action in cortical neurons. Meaningfully, we find Wnt5a can protect Aβ42 treated cortical neurons by alleviating the expression of Cyclin D1. These observations illuminate that aberrant downregulation of Wnt5a signaling is a crucial pathological step that contributes to AD-related neurodegeneration.
Materials and Methods
Reagents
Amyloid β peptide (1–42; Human Aβ42; Abcam plc., #ab120301), Hoechst 33258 (Sigma-Aldrich, #B2883), recombinant mouse Wnt5a protein (Bio-Techne China Co. Ltd. R&D Systems #645-WN), Roscovitine (ROS; Selleck.cn, #S1153), KN62 (Selleck.cn, #S7422), Calmodulin Kinase IINtide, Myristoylated (Ntide; Merck Millipore, #208921) and Z-VAD-FMK (Selleck.cn, #S7023) were added to the media at the indicated concentrations and time points. For transient gene transfection, DIC5 cortical neurons were transfected using the calcium phosphate transfection method described previously (Kingston et al., 2003). Lipofectamine 3000 (Thermo Fisher Scientific Inc.) was used for transient gene or siRNA transfection of HEK293 cells.
The following primary antibodies were used: Anti-Wnt7a antibody (Abcam plc., #ab100792), Anti-Wnt5a antibody (Abcam plc., #ab72583), Anti-beta Tubulin antibody (Abcam plc., #ab6046), Anti-Cyclin D1 antibody (Abcam plc., #ab16663), Anti-E2F1 antibody (Abcam plc., #ab94888), phospho-Rb (Ser795) antibody (Cell Signaling Technology, Inc., #9301), Anti-β-Catenin antibody (Abcam plc., #ab32572), Anti-Histone H1 antibody (Abcam plc., #ab71594), Anti-phospho-CaMKII alpha (Thr286)/beta (Thr287; Abcam plc., #ab32678), Flag (Sigma-Aldrich, #F1804).
Cell Culture
Primary cortical neurons of either sex were established from cortices dissected from newborn C57/BL6 mice (<24 h) as described previously (Sciarretta and Minichiello, 2010). Animals were purchased from the Experimental Animal Center of Sun Yat-sen University. Ethical approval was obtained from the Animal Ethics Committee of Sun Yat-sen University. Briefly, the whole cerebral cortex was isolated and cells were dissociated in a trypsin solution (2.5 mg/mL in Hank’s buffer salt solution) for 10 min at 37°C. The cortex cell suspension was centrifuged and re-suspended, seeded at a density of 1.5 × 106 cells/ml in plates pre-coated with poly-d-lysine (Sigma, St. Louis, MO, USA), and grown in Neurobasal™-A medium containing 2% B27 supplement (Thermo Fisher Scientific Inc., Waltham, MA, USA), L-glutamine (0.25 mM, Invitrogen), GlutaMax-I (0.25 mM, Thermo Fisher Scientific Inc.), 1% penicillin (100 U/ml, Invitrogen), 1% streptomycin (100 μg/ml, Invitrogen) in a humidified incubator with 5% CO2 at 37°C. HEK293 cells were obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA) and were cultured at 37°C in 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen) supplemented with 1% penicillin (100 U/ml, Invitrogen), 1% streptomycin (100 μg/ml, Invitrogen), L-glutamine (292 μg/ml, Invitrogen) and 10% fetal bovine serum (FBS; hyClone Laboratories).
Pathological Studies
Human cortical tissues obtained from surgical resection of patients with IV grade glioblastoma (glioblastoma adjacent normal cortical tissues) were fixed in 4% paraformaldehyde overnight at 4°C. Before sampling, routine processing, and paraffin embedding were performed. Written informed consent had been obtained from the family members of patients before the surgery. The human tissue study was approved by the Ethics Committee of the Third Affiliated Hospital of Sun Yat-sen University.
Animals and Drug Treatment
Male mice (8–12 week-old from Experimental Animal Center of Sun Yat-sen University, Guangzhou, China), maintained at an ambient temperature of 22–24°C under a 12:12 h light:dark cycle, were used in this experiment. Animals were divided into three groups (n = 10 each group): (1) sham-operated plus physiological saline treatment; (2) Human Aβ42 (3 nmol/10 μl) i.c.v. (intracerebroventricularly) injection plus physiological saline treatment; and (3) Human Aβ42 (3 nmol/10 μl) plus recombinant mouse Wnt5a protein treatment (6 nmol/10 μl) i.c.v. injection. To obtain the aggregated form of Aβ42, the peptide solution was placed in an incubator at 37°C for 72 h. Seven days after injection, mice were perfused transcardially with 4% paraformaldehyde in phosphate buffered saline (PBS). The brains were post fixed for 24 h and were embedded in paraffin wax. Serial coronal sections (5 μm thickness) were cut from various sections of the brain.
Surgery
All experimental procedures were carried out in accordance with the guidelines of the Animal Care and Use Committee of Sun Yat-sen University. The mice weighing 20–24 g were anesthetized with sodium pentobarbital (Sigma-Aldrich Co., at a dose of 70 mg/kg) and placed in a stereotaxic apparatus (Stoelting, USA). According to the atlas of Paxinos, Franklin, and Franklin (Paxinos and Franklin’s the Mouse Brain in Stereotaxic Coordinates, Fourth Edition), 8 mm 26-gauge stainless-steel guide cannulas, closed by stylets, were implanted over the lateral ventricle (0.5 mm posterior to bregma, 1.0 mm lateral to midline, 2.0 mm ventral to skull surface). Body temperature of the mice was maintained at 37°C. The injection lasted 5 min and the needle with the syringe was left in place for 2 min after the injection for the completion of drug infusion. After surgery, mice were housed individually. All experiments were carried out between 9:00 a.m. and 6:00 p.m.
Plasmids
The overexpression plasmids of GFP, Wnt5a (mouse) and Cyclin D1 (mouse) were incorporated into the pcDNA-Flag expression vector.
Quantitative Polymerase Chain Reaction (Q-PCR)
Total RNA was extracted and isolated from cells using TRIzol reagent (Invitrogen) as described previously (Peirson and Butler, 2007). First strand cDNA was synthesized from 1 μg of mRNA using Superscript III reverse transcriptase (Invitrogen) and oligo (dT) as primers. Q-PCR was performed in triplicate on an ABI Prism 7000 sequence detection system using an ABI SYBR Green PCR mixture as described by the manufacturer. PCR cycling conditions were as follows: initial denaturation at 95°C for 5–10 min followed by 40 cycles of 95°C for 30 s 1 min of annealing, and 1 min of extension at 72°C. The annealing temperature was adapted for the specific primer set used. Fluorescence data were collected during the annealing stage of amplification and specificity of the amplification was verified by melting curve analysis. Cycle threshold (Ct) values were calculated using identical threshold values for all experiments. β-actin was used as a control and for normalization. Relative RNA expression was calculated using the formula ratio = 2 (Ctref−Cttarget). Data shown represent the mean and SE of three separate experiments. The following primer pairs were used: wnt1 forward (5′-ATG AAC CTT CAC AAC AAC GAG-3′) and reverse (5′-GGT TGC TGC CTC GGT TG-3′); wnt2 forward (5′-CTG GCT CTG GCT CCC TCT G-3′) and reverse (5′-GGA ACT GGT GTT GGC ACT CTG-3′); wnt2b forward (5′-CGT TCG TCT ATG CTA TCT CGT CAG-3′) and reverse (5′-ACA CCG TAA TGG ATG TTG TCA CTA C-3′); wnt3 forward (5′-CAA GCA CAA CAA TGA AGC AGG C-3′) and reverse (5′-TCG GGA CTC ACG GTG TTT CTC-3′); wnt3a forward (5′-CAC CAC CGT CAG CAA CAG CC-3′) and reverse (5′-AGG AGC GTG TCA CTG CGA AAG-3′); wnt4 forward (5′-GAG AAG TGT GGC TGT GAC CGG-3′) and reverse (5′-ATG TTG TCC GAG CAT CCT GAC C-3′); wnt5a forward (5′-CTC CTT CGC CCA GGT TGT TAT AG-3′) and reverse (5′-TGT CTT CGC ACC TTC TCC AAT G-3′); wnt5b forward (5′-ATG CCC GAG AGC GTG AGA AG-3′) and reverse (5′-ACA TTT GCA GGC GAC ATC AGC-3′); wnt6 forward (5′-TGC CCG AGG CGC AAG ACT G-3′) and reverse (5′-ATT GCA AAC ACG AAA GCT GTC TCT C-3′); wnt7a forward (5′-ATC TCC GGA TCG GTG ACT TC-3′) and reverse (5′-AGG CCT GGG ATC TTG TTA CAG-3′); wnt7b forward (5′-TCT CTG CTT TGG CGT CCT CTA C-3′) and reverse (5′-GCC AGG CCA GGA ATC TTG TTG-3′); wnt8a forward (5′-ACG GTG GAA TTG TCC TGA GCA TG-3′) and reverse (5′-GAT GGC AGC AGA GCG GAT GG-3′); wnt8b forward (5′-TTG GGA CCG TTG GAA TTG CC-3′) and reverse (5′-AGT CAT CAC AGC CAC AGT TGT C-3′); wnt9a forward (5′-GCA GCA AGT TTG TCA AGG AGT TCC-3′) and reverse (5′-GCA GGA GCC AGA CAC ACC ATG-3′); wnt9b forward (5′-AAG TAC AGC ACC AAG TTC CTC AGC-3′) and reverse (5′-GAA CAG CAC AGG AGC CTG ACA C-3′); wnt10a forward (5′-CCT GTT CTT CCT ACT GCT GCT GG-3′) and reverse (5′-CGA TCT GGA TGC CCT GGA TAG C-3′); wnt10b forward (5′-TTC TCT CGG GAT TTC TTG GAT TC-3′) and reverse (5′-TGC ACT TCC GCT TCA GGT TTT C-3′); wnt11 forward (5′-CTG AAT CAG ACG CAA CAC TGT AAA C-3′) and reverse (5′-CTC TCT CCA GGT CAA GCA GGT AG-3′); wnt16 forward (5′-AGT AGC GGC ACC AAG GAG AC-3′) and reverse (5′-GAA ACT TTC TGC TGA ACC ACA TGC-3′); β-actin forward (5′-CGT CTT CCC CTC CAT CG-3′) and reverse (5′-CTC GTT AAT GTC ACG CAC-3′).
Immunoblot Analysis
Equal amounts of protein (40–50 mg) were size-fractionated using 6%–15% SDS-PAGE gradient gels. The resolved proteins were electrophoretically transferred onto polyvinylidene difluoride membranes and analyzed by immunoblotting using an ECL chemiluminescence reagent and XAR film (Kodak, XBT-1) according to the manufacturer’s protocol. Primary antibodies were used at optimized dilutions along with the appropriate HRP-conjugated secondary antibodies. The data were collected from at least three independent experiments.
Immunofluorescence
Brainstem neurons were grown on 13 mm round glass coverslips and processed according to the immunofluorescence protocol as described previously (Xie et al., 2011). Cells were placed on ice, washed twice with PBS and fixed with 0.37% PFA in PBS for 10 min followed by permeabilization with 0.1% Triton X-100 in TBS and blocking in 3% donkey serum. Cells were then washed twice in PBS and treated by cold blocking buffer for 1 h. After sequential treatment with NH4Cl (50 mM in 20 mM glycine) for 10 min, the indicated antibody (1:200 in bovine serum albumin) was added and incubated overnight at 4°C. After an additional incubation for 1 h at room temperature with Hoechst 33258 and fluorescein isothiocyanate-conjugated secondary antibody (Invitrogen; 1:400 in bovine serum albumin), the slides were mounted in anti-fading solution (Permafluor, Beckman Coulter, Krefeld, Germany) and stored at 4°C, followed by confocal laser-scanning microscopy. A score of 0–3 was assigned to describe the level of Wnt5a protein based on the red fluorescence area in the cytoplasm of neurons (grade 0, <5%; grade 1, 5%–33%; grade 2, 33%–66%; and grade 3, >66%).
Subcellular Fractionation
Approximately 107 cells were harvested into 10 ml of isotonic fractionation buffer (250 mM sucrose, 0.5 mM EDTA, 20 mM Hepes, and 500 μM Na3VO4 at pH 7.2) supplemented with protease inhibitor cocktail complete (Roche Molecular Biochemicals) and centrifuged at 900 g for 5 min. The pellet was then resuspended in 200 μl fractionation buffer, homogenized with a ball-bearing homogenizer and centrifuged at 900 g for 5 min to enrich the nuclei. The post-nuclear supernatant was centrifuged at 20,000 g for 15 min to collect the heavy membrane fraction enriched in mitochondria, the supernatant was as cytoplasm without mitochondrion.
siRNA Interference
The target sequences for Wnt5a-specific siRNAs were 5′-GAC CUG GUC UAC AUC GAC CTT-3′ and 5′-AGU GCA AUG UCU UCC AAG UTT-3′; Cyclin D1 specific siRNAs were 5′-CCA AUA GGU GUA GGA AAU AGC GCT G-3′ and 5′-AAC ACC AGC TCC TGT GCT GCG-3′ all of which and the negative control siRNA (no silencing small RNA fragment) were synthesized by GenChem Co. (Shanghai, China).
Cell Survival Assays
Cells were stained with Hoechst 33258 (5 μg/ml) to visualize nuclear morphology. Apoptosis was quantified by scoring the number of cells with pyknotic nuclei (chromatin condensation or nuclear fragmentation) relative to the total number of Hoechst 33258-positive cells in the same visual field. Cells were counted in an unbiased manner (at least 1000 cells for each group) and were scored blindly without previous knowledge of their treatment.
CaMKII Activity Assay
Homogenates of treated cortical neurons were quantified and used to detect CaMKII activity, using the SignaTECT® Calcium/Calmodulin-Dependent Protein Kinase Assay System (Promega, #V8161) as described by the manufacturer. In brief, the assay quantifies [γ-32P]-labeling of a proprietary CaMKII-specific, biotinylated peptide substrate that is based on the T286 autophosphorylation site, followed by streptavidin capture; this recognition sequence is conserved in mouse. Controls include calmodulin and substrate omission. The substrate is specific for CaMKII and does not detect CaMKIV, PKC or calcineurin (Hanson et al., 1989; Goueli et al., 1995). Samples were assayed in triplicate and protein concentration was determined using the bicinchoninic acid (BCA) and Western blotting method. Results were expressed as relative CaMKII activity.
Tissue Immunohistochemistry
Human cortical tissues obtained from surgical resection of patients with IV grade glioblastoma (glioblastoma adjacent normal cortical tissues) or mouse brains were fixed in 4% paraformaldehyde in 0.1 M phosphate buffer pH 7.6 overnight at 4°C. Dehydration of tissue was through a series of 80%, 95% (v/v) ethanol 1 h each followed by 100% ethanol overnight. Two 100% (v/v) xylene washes were done for 1 h each and then 1 h in 60°C Paraplast Plus (Tyco/Healthcare). After a change of Paraplast Plus, tissue was incubated in a 60°C vacuum oven for 2 h prior to placing in molds to cool and solidify. Sections, 5 μm thick, were cut and mounted. Sections were deparaffinized by drying on superfrost plus slides (Fisher), heating at 56°C overnight, and then washing through mixed xylenes, 100% ethanol, 95% ethanol, ddH2O. Slides were immersed in 10 mm citrate buffer, pH 6.0, dry heated for 10 min each to unmask antigen sites, and then cooled and washed in PBS. Endogenous peroxidase activity was inhibited by rinsing the slides in 3% hydrogen peroxide for 5 min. Non-specific binding was blocked by 5 min incubation with the Super Block Solution (ScyTek Laboratories). After washing in PBS, sections were incubated for 30 min at room temperature with indicated antibodies. Sections were washed extensively with PBS and subsequently treated with the Ultra Tek Anti-Polyvalent kit (ScyTek Laboratories). Finally, sections were treated with 3,3′-diaminobenzidine as chromogenic and mounted.
Statistical Analysis
All experiments were repeated at least three times using independent culture preparations. All measurements were performed blindly. The significance of difference between means was analyzed by the ANOVA and post hoc Bonferroni/Dunn tests (for multiple comparisons) and by the Student’s t test (for single comparisons). The results are represented as means ± SEM. Statistical significance was determined by value of p < 0.05 or p < 0.01 for all analyses.
Results
Aβ42 Promotes the Decrease of Wnt5a Expression
In order to explore the role of Wnt in Aβ42 induced neuronal cell death, primary cultured mouse cortical neurons were used in this study (Figure 1A). In the current study, after 5 days culture in vitro, cortical neurons were treated with Aβ42. We found Aβ caused a concentration-dependent increase of cortical neuron apoptosis (Figure 1B). A significant of cortical neuron apoptosis was observed at concentrations from 0.5 μM to 4 μM (Figure 1B), these results were consistent with previous studies.
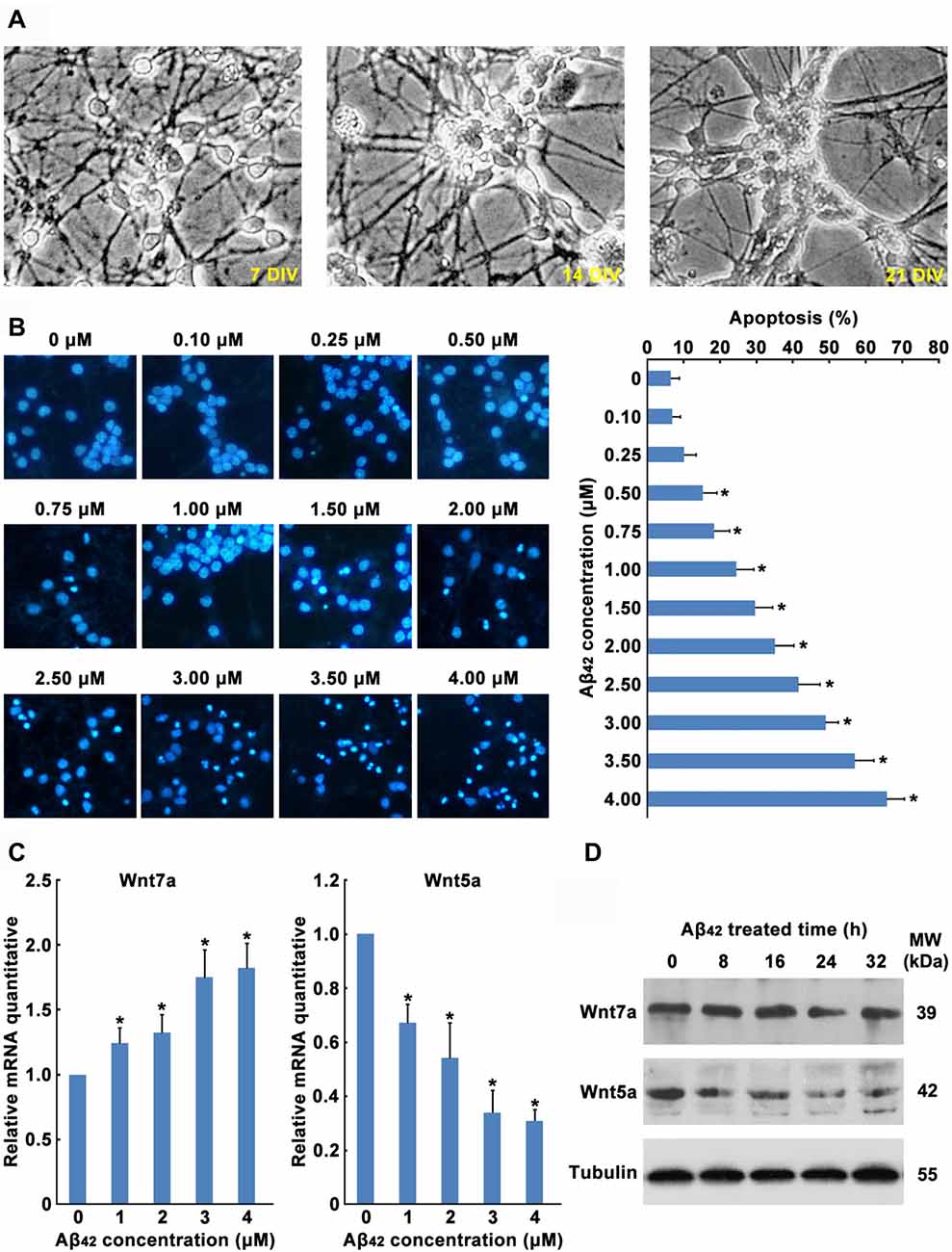
Figure 1. Aβ42 promotes the decrease of Wnt5a expression. (A) Cortical neurons cultured in vitro for 7, 14 and 21 days. (B) DIV7 cortical neurons were treated with Aβ42 at indicated concentrations for 24 h. Then neurons were stained for nucleus (Hoechst 33258, Blue). Apoptosis was determined by the percentage of cells that were had pyknotic nuclei. (C) DIV7 cortical neurons were treated with Aβ42 at indicated concentrations for 8 h, total RNA was extracted and analyzed by Q-PCR. (D) DIV7 cortical neurons were treated with 3 μM Aβ42 at indicated times, total protein was extracted and analyzed by Western blotting. DIV, days in vitro; MW, molecular weight; kDa, kilodalton. All data in this figure represent the means ± SEM of three independent experiments. *P < 0.05.
Earlier reports have shown that Aβ42 may promote neuronal apoptosis by regulating the expression of Wnt family members (Bozyczko-Coyne et al., 2001; Majd et al., 2008). To study which Wnt isoforms take part in accommodation of Aβ42 induced cytotoxicity, the mRNA expression levels of all 19 Wnt gene family members in cortical neurons were analyzed by RT-PCR. As illustrated in Table 1, most Wnt members were expressed only at very low levels in both control and Aβ42 treated group, Aβ42 mainly altered the expression of Wnt5a, Wnt7a, Wnt9b and Wnt11. We next confirmed the mRNA and protein expression levels of these genes. We found the mRNA and protein expression levels of Wnt9b and Wnt11 were undetectable (data not shown); the mRNA expression level of Wnt7a was upregulated after Aβ42 treatment, but the protein level of Wnt7a was not changed (Figures 1C,D); Aβ42 significantly suppressed the expression level of Wnt5a (Figures 1C,D). These results indicate that Aβ42 suppresses the expression of Wnt5a on both mRNA and protein levels.
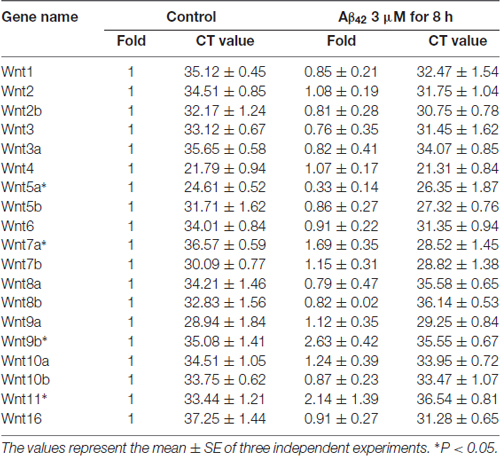
Table 1. Wnts mRNA levels after Aβ42 treatment.
Aβ42 Inhibited the Expression of Wnt5a in Vivo
We next determined the expression level of Wnt5a protein in human brains. Unfortunately, we were unable to obtain postmortem brains derived from AD patients. As an alternative, we collected the adjacent normal brain tissues of malignant glioma from 32- to 86-year-old patients. Immunohistochemical staining of the adjacent normal brain tissues of malignant glioma revealed that Wnt5a levels were consistently expressed in the neurons, this result was consistent with previous study (Howng et al., 2002); however, the Wnt5a levels were decreased with the increase of age (Figure 2A). These results indicate a role of Wnt5a in the aging of human brain.
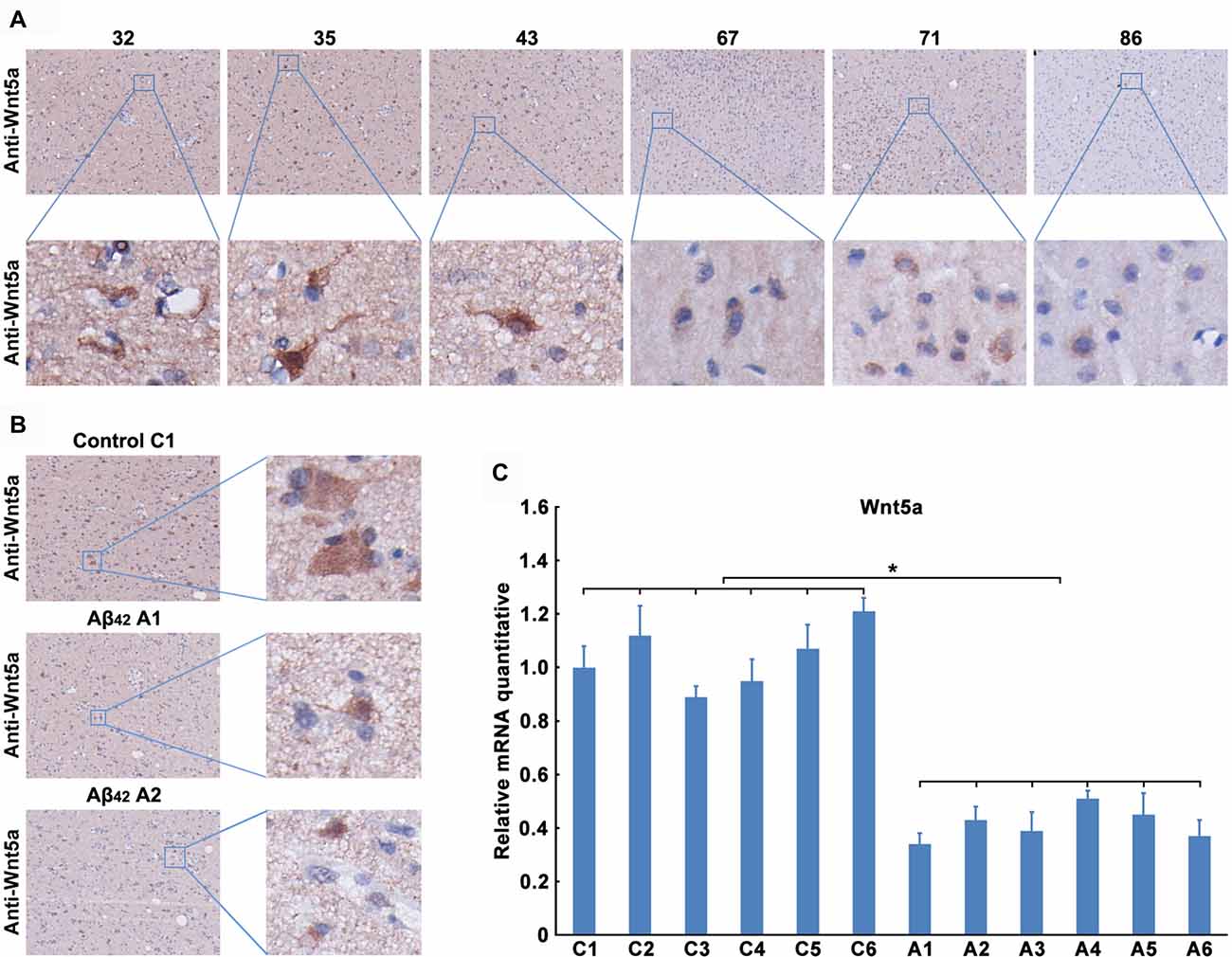
Figure 2. Aβ42 inhibited the expression of Wnt5a in vivo. (A) Wnt5a immunoreactivity was primarily detected in the adjacent normal brain tissues of malignant glioma from 32, 35, 43, 67, 71 and 86 years old patients (magnification of upper panel: ×40; magnification of lower panel: ×400). (B,C) A solution of Aβ42 (3 nmol/10 μl) or Aβ42 (3 nmol/10 μl) combined with recombinant Wnt5a protein (6 nmol/10 μl) was injected into the lateral ventricles of mice. Seven days after treatment, Wnt5a immunoreactivity was primarily detected in mouse brain tissues (B, control C1: control group mouse 1; Aβ42 A1: Aβ42 treated group mouse 1; Aβ42 A2: Aβ42 treated group mouse 2; magnification of left panel: ×40; magnification of right panel: ×400), total RNA was extracted and analyzed by Q-PCR (C, C1–C6: control group mouse 1–6; A1–A6: Aβ42 treated group mouse 1–6). All data in this figure represent the means ± SEM of three independent experiments. *P < 0.05.
To examine the effect of Aβ42 on the expression of Wnt5a in vivo, a solution of Aβ42 was injected into the lateral ventricles of mice. Seven days after the injection, tissues from the cerebral cortex were collected to assess the expression of Wnt5a. We found that the protein level of Wnt5a was decreased in cortical neurons after Aβ42-treatment (Figure 2B). This observation was further supported by quantitative PCR analysis (Figure 2C). Therefore, Aβ42 suppresses the expression of Wnt5a on both mRNA and protein levels in vivo.
Wnt5a Downregulation Contributes to Aβ42 Induced Cortical Neuron Apoptosis
To investigate the function of Wnt5a in cortical neuron survival and apoptosis, two siRNAs were designed to reduce mouse Wnt5a protein levels. These siRNAs completely abolished the expression of a transfected mouse Flag-Wnt5a construct in HEK293 cells (Figure 3A). In cortical neurons, Wnt5a siRNAs were co-transfected with the marker plasmid pCMV-GFP, immunofluorescence analysis using anti-Wnt5a antibody revealed that the expression of Wnt5a was abolished by the two Wnt5a siRNAs (Figure 3B), demonstrating the efficacy of the siRNAs in cortical neurons. Furthermore, siRNAs specifically targeted against Wnt5a induced cortical neuron apoptosis, and the Wnt5a siRNAs induced apoptosis was inhibited by Caspase inhibitor Z-VAD (Figure 3C). These results indicate that Wnt5a is required for the survival of cortical neuron.
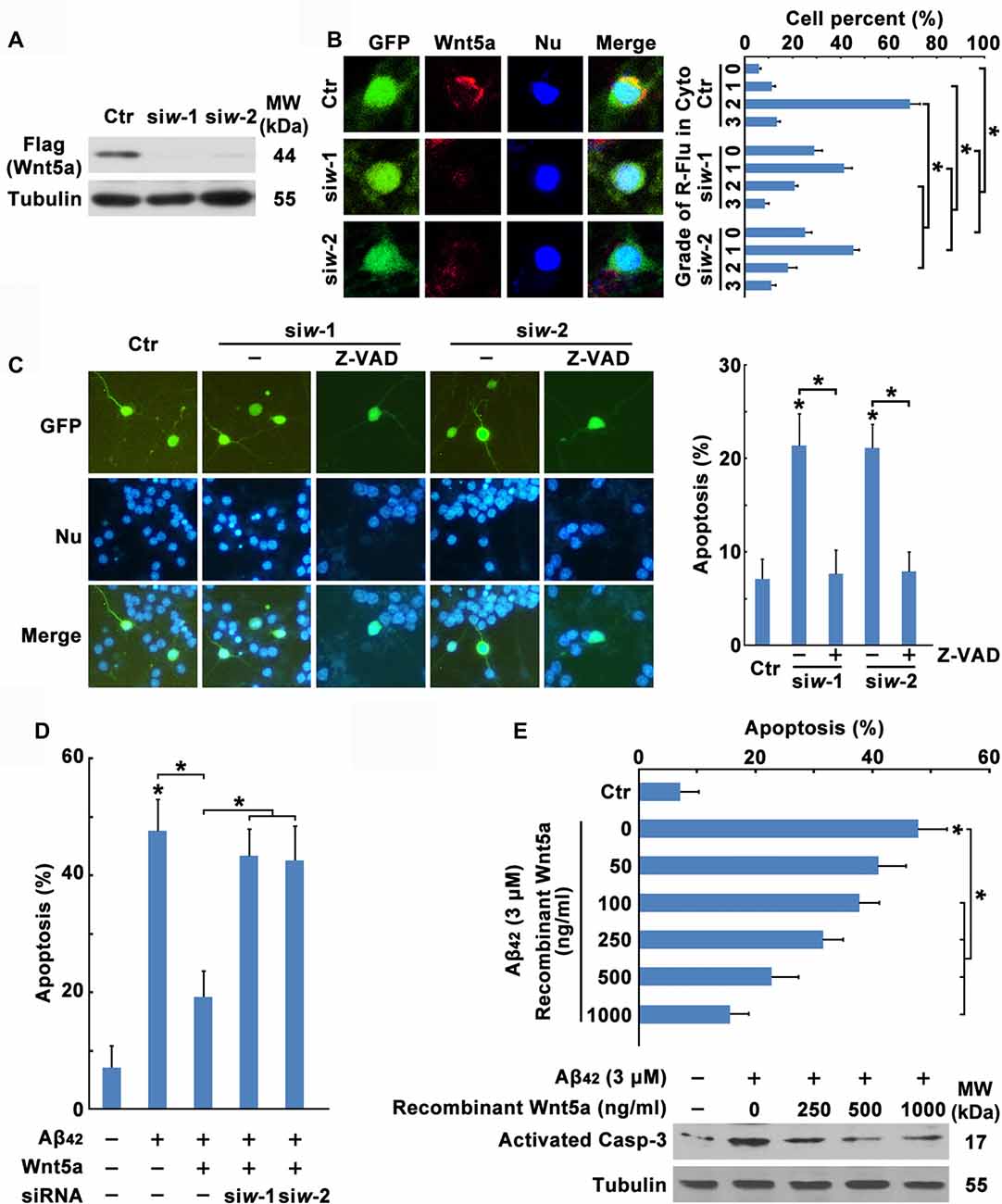
Figure 3. Wnt5a downregulation contributes to Aβ42 induced cortical neuron apoptosis. (A) HEK239 cells were transfected with Flag-Wnt5a and Wnt5a siRNAs for 12 h, then cells were subjected to immunoblot detection. (B) DIV6 cortical neurons were transfected with GFP plasmids and Wnt5a siRNAs for 24 h. Immunofluorescence detection of Wnt5a of neurons appearing in Red, then neurons were stained for nucleus (Blue; left panel). The efficiency of Wnt5a siRNAs were estimated by a score of 0–3 based on the red fluorescence area in the cytoplasm of neurons (grade 0, <5%; grade 1, 5%–33%; grade 2, 33%–66%; and grade 3, >66%; right panel). (C) DIV6 cortical neurons were transfected with GFP plasmids and Wnt5a siRNAs for 12 h following 50 μM Z-VAD treatment for another 12 h, and then neurons were stained for nucleus (Blue). Apoptosis was determined by the percentage of GFP positive cells that were had pyknotic nuclei. (D) DIV6 cortical neurons were transfected with GFP plasmids, Wnt5a plasmids and Wnt5a siRNAs for 24 h, and then neurons were stained for nucleus (Blue). Apoptosis was determined by the percentage of GFP positive cells that were had pyknotic nuclei. (E) DIV6 cortical neurons were treated with 3 μM Aβ42 with or without recombinant Wnt5a protein at indicated concentrations for 24 h, and then neurons were stained for nucleus (Blue). Apoptosis was determined by the percentage of cells that were had pyknotic nuclei (upper panel); or cells were subjected to immunoblot detection (lower panel). MW, molecular weight; kDa, kilodalton. All data in this figure represent the means ± SEM of three independent experiments. *P < 0.05.
To further confirm the effect of Wnt5a on cortical neuron survival, transfection of neurons with wild-type Wnt5a also significantly increased neuron survival during Aβ42 treatment, and the effect induced by Wnt5a overexpression was abolished by Wnt5a siRNAs (Figure 3D). Moreover, we added various concentration (0, 50, 100, 250, 500, 1000 ng/ml) of recombinant Wnt5a protein to the culture medium of Aβ42 treated neurons. The suppressive effect of recombinant Wnt5a protein on Aβ42 induced neuronal apoptosis was dose-dependent (Figure 3E). Taken together, Wnt5a acts as a mediator of cortical neuron survival, and Aβ42 promotes cortical neuron apoptosis by downregulating the expression of Wnt5a.
Wnt5a Inhibits the Expression of Cyclin D1
Neurons in culture exposed to Aβ42 could re-enter the cell cycle, cross the G1-S-phase transition and begin de novo DNA synthesis before apoptotic death occurs (Caricasole et al., 2003; Majd et al., 2008). And cell-cycle activation is causally related to neuronal death (Busser et al., 1998; Giovanni et al., 1999; Liu and Greene, 2001; Yang et al., 2003; Becker and Bonni, 2004). Meanwhile, Wnt pathway is the key pathway in activation of cell cycle (Davidson and Niehrs, 2010). Interestingly, Wnt5a can act as a cell proliferation inhibitor in many cancer cells and mouse B cells (Liang et al., 2003; Kremenevskaja et al., 2005; Bitler et al., 2011), suggesting a cell-cycle suppressor role for Wnt5a in some cell types. We speculate that Aβ may activate the cell-cycle pathway by reducing the expression of Wnt5a.
To determine the effect of Aβ42 in inducing cortical neuron to re-enter the cell cycle, neurons were treated with Aβ42 combined with or without CDKs inhibitor ROS (Bach et al., 2005), results showed that Aβ42 induced neuronal apoptosis was significantly decreased by CDKs inhibitor (Figure 4A). Furthermore, CDKs inhibitor also inhibited Wnt5a siRNAs induced cortical neuron apoptosis (Figure 4B). These findings demonstrate that Aβ42 could promote neurons re-enter the cell cycle by reducing the expression of Wnt5a.
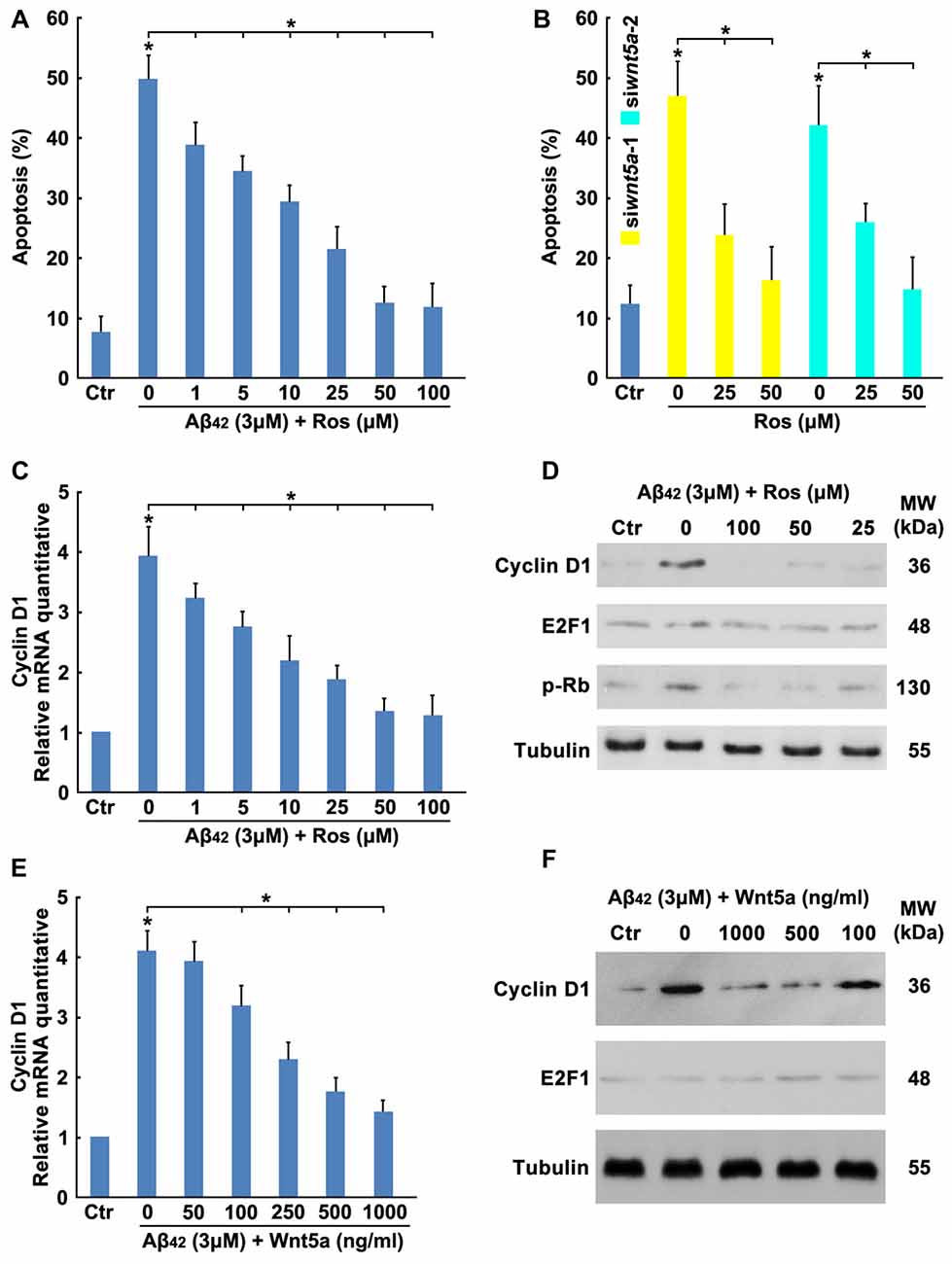
Figure 4. Wnt5a inhibits the expression of Cyclin D1. (A) DIV6 cortical neurons were treated with 3 μM Aβ42 with or without Roscovitine (ROS) at indicated concentrations for 24 h, then apoptosis was determined. (B) DIV6 cortical neurons were treated with Wnt5a siRNAs and ROS at indicated concentrations for 24 h, then apoptosis was determined. (C) DIV6 cortical neurons were treated with 3 μM Aβ42 with or without ROS at indicated concentrations for 12 h, total RNA was extracted and analyzed by Q-PCR. (D) DIV6 cortical neurons were treated with 3 μM Aβ42 with or without ROS at indicated concentrations for 16 h, total protein was extracted and analyzed by Western blotting. (E) DIV6 cortical neurons were treated with 3 μM Aβ42 with or without recombinant Wnt5a protein at indicated concentrations for 12 h, total RNA was extracted and analyzed by Q-PCR. (F) DIV6 cortical neurons were treated with 3 μM Aβ42 with or without recombinant Wnt5a protein at indicated concentrations for 16 h, total protein was extracted and analyzed by Western blotting. MW, molecular weight; kDa, kilodalton. All data in this figure represent the means ± SEM of three independent experiments. *P < 0.05.
Aberrations in cell cycle control are always controlled by cell cycle regulators, such as cyclins, Myc and E2F1 (van den Heuvel, 2005). To study which cell cycle regulators mediated the Aβ42 activated cell cycle, the mRNA expression levels of cyclins, Myc and E2F1 in cortical neurons were analyzed by RT-PCR. As illustrated in Table 2, most cyclin members and Myc were expressed only at very low levels in both control and treatment group, Aβ42 mainly altered the expression of Cyclin D1 and E2F1. We next confirmed that Aβ42 significantly increased the expression level of Cyclin D1, and the upregulation of Cyclin D1 was eliminated by CDKs inhibitor ROS (inhibited the CDK mediated phosphorylation of Rb Ser795; Figures 4C,D). Accordingly, neurons treated with recombinant Wnt5a protein also abolished Aβ42 induced Cyclin D1 expression (Figures 4E,F). However, the protein level of E2F1 was not changed after Aβ42, CDKs inhibitor and Wnt5a treatment (Figures 4C,D). Taken together, these results suggest that Wnt5a prevents cell cycle activation by inhibiting the expression of Cyclin D1 in cortical neurons.
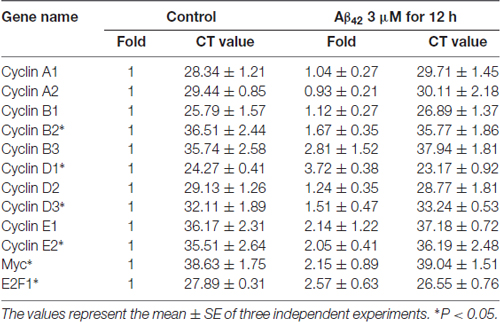
Table 2. Cell cycle regulators mRNA levels after Aβ42 treatment.
Wnt5a Inhibits the Expression of Cyclin D1 in Vivo
We next confirmed the expression level of Cyclin D1 protein in the adjacent normal brain tissues of malignant glioma from 32- to 86-year-old patients. As observed in previous studies (Hoozemans et al., 2002), trace amounts of Cyclin D1 was detected in the brain cortex of 32, 35 and 43 years old people, but the protein levels were upregulated with the increase of age (Figure 5A). These results also indicated a relationship between the neuronal aging and Cyclin D1.
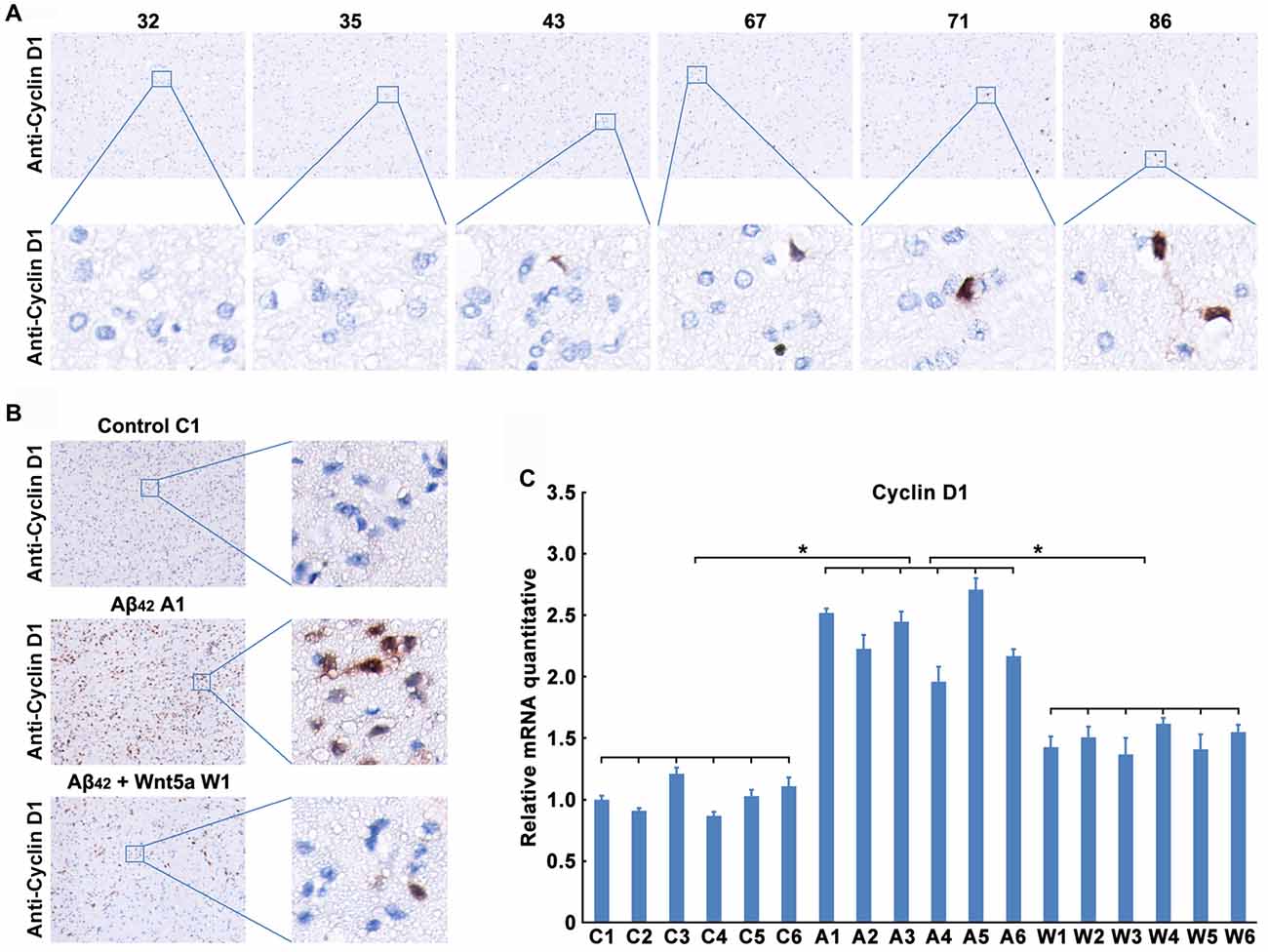
Figure 5. Wnt5a inhibits the expression of Cyclin D1 in vivo. (A) Cyclin D1 immunoreactivity was primarily detected in the adjacent normal brain tissues of malignant glioma from 32, 35, 43, 67, 71 and 86 years old patients (magnification of upper panel: ×40; magnification of lower panel: ×400). (B,C) A solution of Aβ42 (3 nmol/10 μl) or Aβ42 (3 nmol/10 μl) combined with recombinant Wnt5a protein (6 nmol/10 μl) was injected into the lateral ventricles of mice. Seven days after treatment, Cyclin D1 immunoreactivity was primarily detected in mouse brain tissues (B, control C1: control group mouse 1; Aβ42 A1: Aβ42 treated group mouse 1; Aβ42 + Wnt5a W1: Aβ42 and Wnt5a treated group mouse 1; magnification of left panel: ×40; magnification of right panel: ×400), total RNA was extracted and analyzed by Q-PCR (C, C1–C6: control group mouse 1–6; A1–A6: Aβ42 treated group mouse 1–6; W1–W6: Aβ42 and Wnt5a treated group mouse 1–6). All data in this figure represent the means ± SEM of three independent experiments. *P < 0.05.
To examine the effect of Wnt5a on Cyclin D1 in vivo, a solution of Aβ42 or Aβ42 combined with recombinant Wnt5a protein was injected into the lateral ventricles of mice. Seven days after the injection, tissues from the cerebral cortex were collected to assess the expression of Cyclin D1. We found that the protein level of Cyclin D1 was increased in cortical neurons after Aβ42-treatment, and the increase of Cyclin D1 was inhibited by recombinant Wnt5a protein (Figure 5B). This observation was further supported by quantitative PCR analysis (Figure 5C). These in vivo results support the conclusion that Wnt5a could prevent Aβ42 induced Cyclin D1 expression.
Wnt5a/CaMKII Pathway Down-Regulates the Expression of Cyclin D1
The mechanism of Wnt5a-mediated inhibition of the Cyclin D1expression is unclear. Wnt5a has been proposed to activate both the canonical Wnt/β-catenin pathway (He et al., 1997; Toyofuku et al., 2000) and Wnt/Ca2+ pathway (Liang et al., 2003). Activation of the Wnt/Ca2+ pathway has been suggested to block the Wnt/β-catenin signaling cascade (Torres et al., 1996; Kühl et al., 2001; Ishitani et al., 2003). To dissect the mechanistic pathway utilized by Wnt5a in regulating cortical neuron cell-cycle activation, we analyzed β-catenin levels in Aβ42 treated and untreated neurons. No difference was detected in β-catenin levels in whole-cell or in nuclear extracts between Aβ42 treated and untreated neurons, suggesting that Wnt5a was neither activating nor inhibiting the canonical Wnt/β-catenin pathway in cortical neuron (Figures 6A,B).
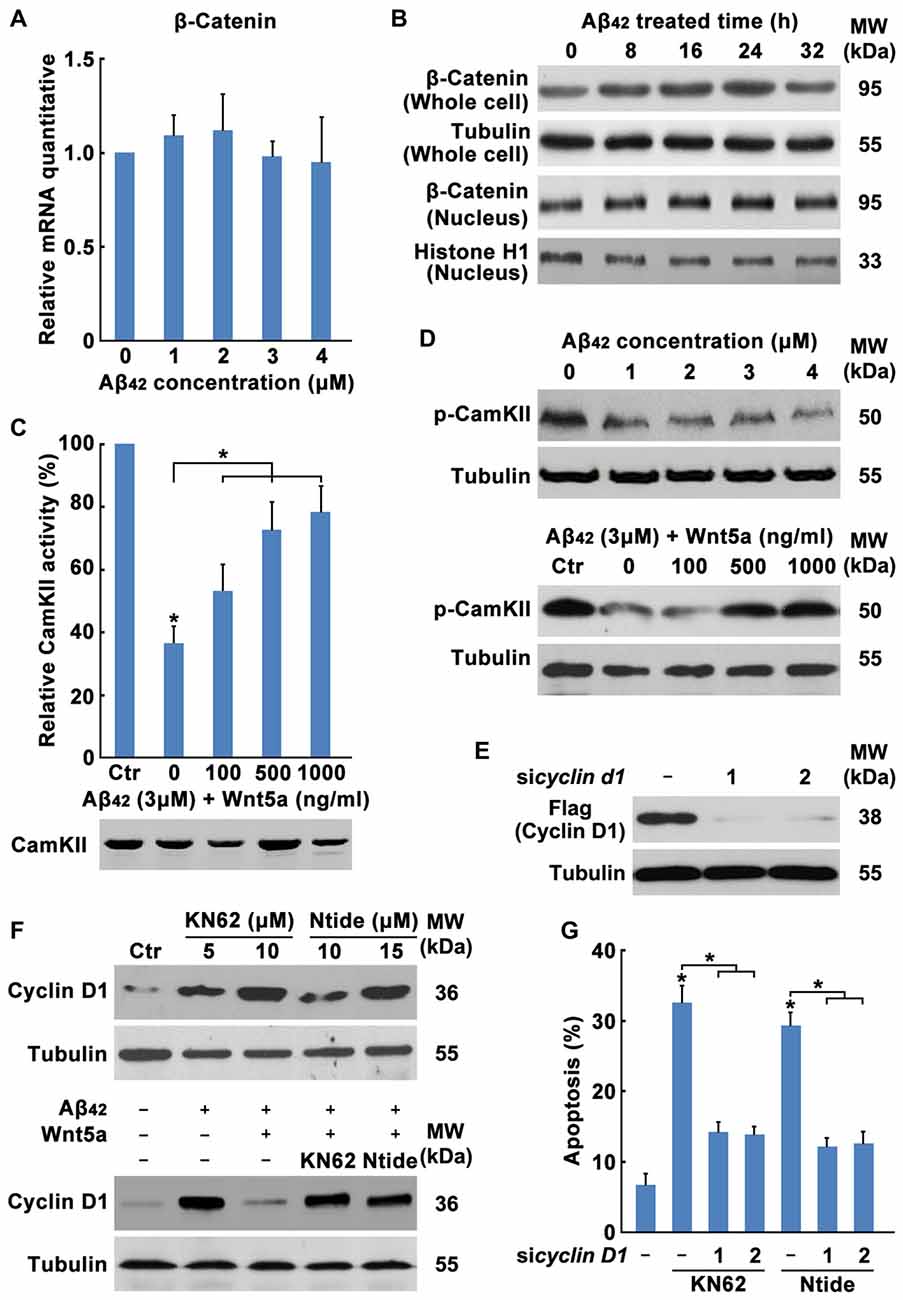
Figure 6. Wnt5a/CaMKII pathway down-regulates the expression of Cyclin D1. (A) DIV7 cortical neurons were treated with Aβ42 at indicated concentrations for 12 h, total RNA was extracted and analyzed by Q-PCR. (B) DIV7 cortical neurons were treated with 3 μM Aβ42 at indicated times, total protein or nuclear protein was extracted and analyzed by Western blotting. (C) DIV6 cortical neurons were treated with 3 μM Aβ42 with or without recombinant Wnt5a protein at indicated concentrations for 12 h. CamKII activity was determined using SignaTECT® Calcium/Calmodulin-Dependent Protein Kinase Assay System (Upper panel). The input total CamKII was analyzed by Western blotting (Lower panel). (D) DIV7 cortical neurons were treated with Aβ42 at indicated concentrations (Upper panel) or treated with 3 μM Aβ42 with/without recombinant Wnt5a protein at indicated concentrations (Lower panel) for 16 h, total protein was extracted and analyzed by Western blotting. (E) DIV7 cortical neurons were treated with KN62 or Calmodulin Kinase II Ntide, Myristoylated (Ntide) at indicated concentrations for 16 h (Left panel), or DIV7 cortical neurons were treated with 3 μM Aβ42 with/without 500 ng/ml recombinant Wnt5a protein or 10 μM KN62 or 15 μM Ntide as indicated for 16 h (Right panel), total protein was extracted and analyzed by Western blotting. (F) HEK239 cells were transfected with Flag-Cyclin D1 and Cyclin D1 siRNAs for 12 h, then cells were subjected to immunoblot detection. (G) DIV7 cortical neurons were treated with 10 μM KN62 or 15 μM Ntide and transfected with or without Cyclin D1 siRNAs for 24 h (Left panel), then apoptosis was determined. MW, molecular weight; kDa, kilodalton. All data in this figure represent the means ± SEM of three independent experiments. *P < 0.05.
Therefore, we examined the Wnt/Ca2+ pathway. CamKII activity assays showed that Aβ42 treated cells possess 63.51% less activity than Aβ42 untreated cells and the decrease of CamKII activity was recovered by Wnt5a recombinant protein stimulation (Figure 6C), even though Western blotting analysis revealed equal amounts of total CamKII in both samples (Figure 6C). Furthermore, we examined the level of phospho-CaMKII alpha (Thr286)/beta (Thr287) by Western blotting. CaMKII phosphorylation was decreased at as early as 0.5 h after Aβ42 treatment and was also recovered by Wnt5a recombinant protein stimulation (Figure 6D). These results indicate that Wnt5a is signaling via the Wnt/Ca2+ pathway to activate CamKII.
To further confirm the ability of Wnt/Ca2+/CaMKII pathway to downregulate Cyclin D1 and inhibit cell-cycle activation, KN62 [a pan-inhibitor of CaMKs (Tokumitsu et al., 1990)] and CaMKII-Ntide [an endogenous selective inhibitor of CaMKII (Chang et al., 1998)], was used to treat cortical neurons. Western blotting analysis revealed that these two inhibitors elevated Cyclin D1 expression of both Aβ42 untreated and Aβ42 plus Wnt5a recombinant protein treated neurons (Figure 6E). Furthermore, we found CamKII inhibitors were sufficient to cause cortical neuron apoptosis in a Cyclin D1 dependent way (Figures 6F,G). Collectively, Wnt5a/CaMKII pathway down-regulates the expression of Cyclin D1.
Wnt5a Promotes Cortical Neuron Survival by Inhibiting Cyclin D1 Expression
Previous studies have established that overexpression of Cyclin D1 may lead to neuronal apoptosis (Kranenburg et al., 1996). To assess whether Cyclin Dl overexpression would induce this phenomenon, two siRNAs were designed to reduce mouse Cyclin D1 protein levels (Figure 6F). siRNAs specifically targeted against Cyclin D1 inhibited Aβ42 induced cortical neuron apoptosis (Figure 7A). Furthermore, we confirmed that overexpression of Cyclin D1 could lead to cortical neuron apoptosis, and Cyclin D1 induced apoptosis was also abolished by Cyclin D1 siRNAs (Figure 7B). These results indicate that Cyclin D1 is required for Aβ42 induced cortical neuron apoptosis.
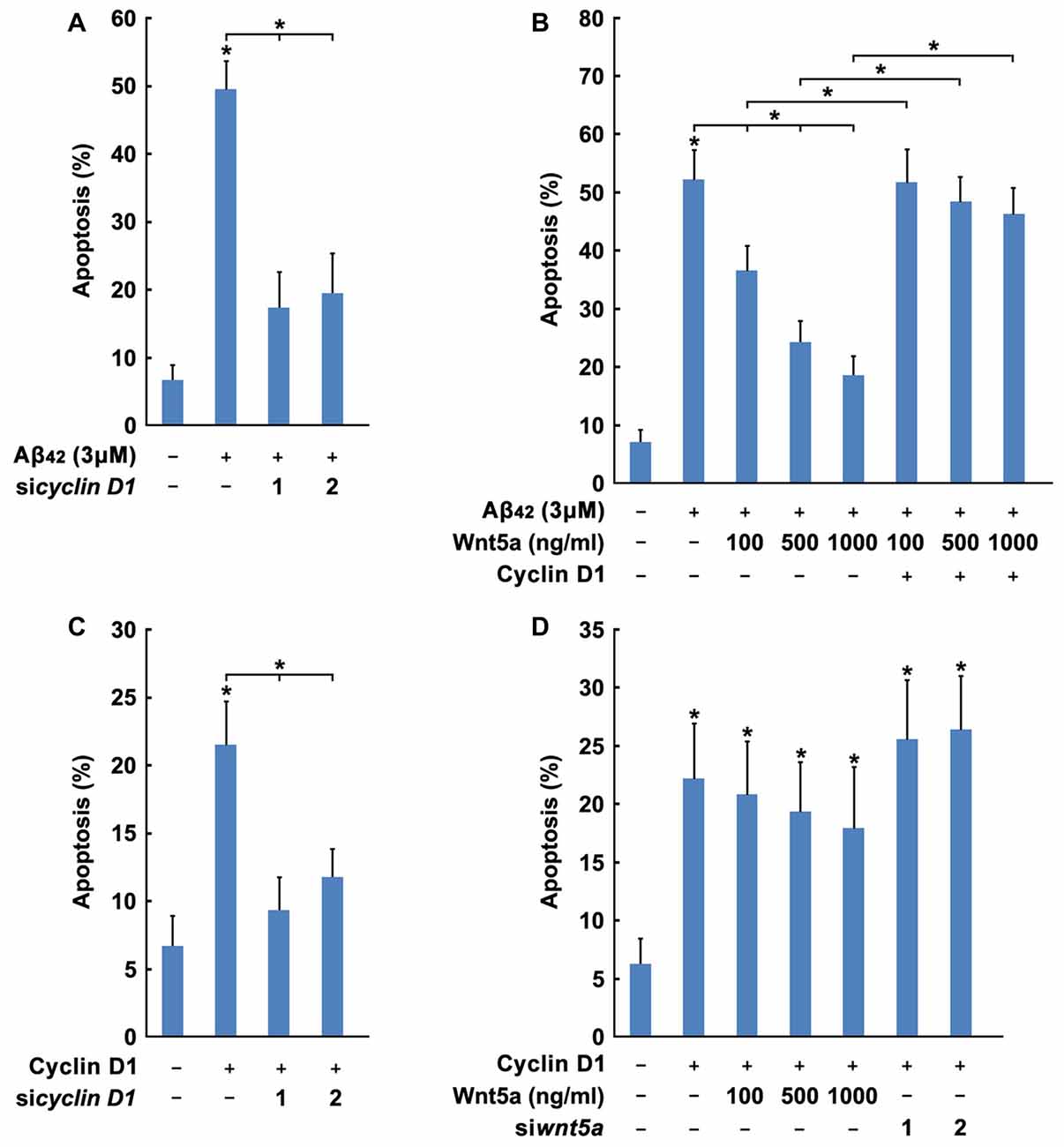
Figure 7. Wnt5a promotes cortical neuron survival by inhibiting Cyclin D1 expression. (A) DIV6 cortical neurons were transfected with GFP plasmids and Cyclin D1 siRNAs for 12 h, and treated with 3 μM Aβ42 for another 12 h. Apoptosis was determined by the percentage of GFP positive cells that were had pyknotic nuclei. (B) DIV6 cortical neurons were transfected with GFP plasmids, Flag-Cyclin D1 plasmids and Cyclin D1 siRNAs for 24 h. Apoptosis was determined by the percentage of GFP positive cells that were had pyknotic nuclei. (C) DIV6 cortical neurons were transfected with or without GFP plasmids, Flag-Cyclin D1 plasmids or control plasmids for 12 h, then treated with 3 μM Aβ42 with/without recombinant Wnt5a protein at indicated concentrations for 24 h. Apoptosis was determined by the percentage of GFP positive cells that were had pyknotic nuclei. (D) DIV6 cortical neurons were transfected with or without GFP plasmids, Flag-Cyclin D1 plasmids or Cyclin D1 siRNAs for 12 h, then treated with with/without recombinant Wnt5a protein at indicated concentrations for 24 h. Apoptosis was determined by the percentage of GFP positive cells that were had pyknotic nuclei. MW, molecular weight; kDa, kilodalton. All data in this figure represent the means ± SEM of three independent experiments. *P < 0.05.
Based on the obvious importance of Wnt5a and Cyclin D1 in cortical neuron survival and apoptosis, we hypothesized that Wnt5a might mediate the cortical neuron survival by inhibiting Cyclin D1 expression. Manipulations of Wnt5a and Cyclin D1, using knockdown, overexpression or recombinant protein approaches, were combined to investigate their functional relationship. The Wnt5a recombinant protein significantly blocked cortical neuron apoptosis in Aβ media (Figure 7C). In contrast, overexpression of wild-type Cyclin D1 removed the protection conferred by Wnt5a recombinant protein (Figure 7C). Overexpression of wild-type Cyclin D1 induced apoptosis could not prevent by adding Wnt5a recombinant protein, and the knockdown of endogenous Wnt5a also could not enhance the apoptosis rate (Figure 7D). These findings demonstrated that Cyclin D1 lies downstream of the Wnt5a-dependent pro-survival signaling cascade.
Discussion
Wnt signaling has a key role in the nervous system (Salinas, 2012; Inestrosa and Varela-Nallar, 2014; Lambert et al., 2016). On the one hand, Wnt signaling plays critical roles in several physiological cellular processes of neurons, including regulate the differentiation and migration of neural progenitor cells, participate in the formation of neuronal circuits, playing roles in dendrite and axon development, dendritic spine formation (Inestrosa and Arenas, 2010; Budnik and Salinas, 2011; Salinas, 2012). On the other hand, the Wnt pathway modulate the survival of mature neurons and aberrant signaling by Wnt pathways is linked to a range of neurodegenerative diseases (Inestrosa and Varela-Nallar, 2014; Purro et al., 2014; Libro et al., 2016; Zhang et al., 2016). Interestingly, previous studies have reported that stimulation of Wnt signaling by exogenous Wnts could protect cultured neurons against Aβ-mediated neurotoxicity (Cerpa et al., 2010; Silva-Alvarez et al., 2013). Exogenous Wnt5a was able to modulate the increase in the anti-apoptotic protein on the outer mitochondrial membrane of hippocampal neurons (Cerpa et al., 2010), suggesting that Wnt5a may protects neurons from the apoptotic effect induced by Aβ oligomers. Wnt5a also plays a pivotal role in the maintenance of normal postsynaptic integrity (Cerpa et al., 2010), and its activation may be of therapeutic interest in patients with AD. However, there is only one factor Dickkopf 1 (Dkk1), a neurodegenerative factor that serves as an antagonist of the canonical Wnt signaling pathway, has been report to inhibit Wnt activity and induct of neuronal cell death (Caricasole et al., 2004; Scali et al., 2006), the direct role of endogenous Wnt family members play in neurodegenerative diseases is still unknown. For the first time, we report that Wnt5a signals through the noncanonical Wnt/Ca2+ pathway to suppress cyclin D1 expression and negatively regulate neuronal cell death by inhibitiing cell-cycle activation.
Cell-cycle activation is causally related to Aβ induced neuronal death (Caricasole et al., 2003; Varvel et al., 2008). In this study, we find endogenous Wnt5a increase the survival of cortical neurons by inhibiting the cell-cycle activation and cell proliferation, and the downregulation of Wnt5a contributes to Aβ toxicity in neuron cultures. Many studies have also reported that administration of exogenous Wnt5a prevented Aβ-induced synaptotoxicity and promoted neuronal survival (Varvel et al., 2008; Varela-Nallar et al., 2012; Silva-Alvarez et al., 2013; Godoy et al., 2014). Recently, Wnt5a has been showed to up-regulated in mouse brains prior to AD phenotypes and in Aβ treated cortical neurons (Li et al., 2011). We think the difference between our and previous studies (Li et al., 2011) may induced by different experimental method. We used specific siRNAs to knockdown the expression of Wnt5a; but Li et al. (2011) used an rabbit-anti-Wnt5a antibody (from Abcam) to perform immunohistochemistry experiment and detect endogenous AD mouse brain Wnt5a, they also used this antibody to suppress Wnt5a signaling. Interestingly, we have tested the same antibody by Western blotting, we found this antibody coud interact with many nonspecific proteins (data not shown), and this Wnt5a antibody may induce non-specific results during the experiment. In fact, Wnt5a has been shown to play different roles in the regulation of neuronal cell-cycle activation and following cell proliferation during different neural cell development stage (Inestrosa and Arenas, 2010). Wnt5a can promote the proliferation of cultured neuronal progenitor cells isolated from postnatal and adult mouse SvZ (subventricular zone of the lateral ventricles in the forebrain), but reduce dopaminergic progenitor proliferation and neurogenesis to promote ventral midbrain morphogenesis in loss-of-function experiments in vivo (Inestrosa and Arenas, 2010), indicating Wnt5a may play as an inhibitor of cell-cycle activation in mature neurons.
Development studies and cell differentiation studies using various model systems have revealed that Wnt5a can signal through both Wnt/β-catenin pathway and Wnt/Ca2+ pathway to regulate cell adhesion, mobility, proliferation, and differentiation (He et al., 1997; Sheldahl et al., 1999; Toyofuku et al., 2000; Kühl et al., 2001). To determine which pathway mediates Wnt5a signaling in mature cortical neurons, we first found expression of Myc or E2F1 was unchanged, expression of cyclin D1 was decreased after Aβ treatment. Whole-cell and nuclear β-catenin level remained unchanged in the presence or absence of Aβ, while activity of CamKII was decreased in Wnt5a null or Aβ treated cells. These results indicate that Wnt5a signals via the Wnt/Ca2+ pathway to inhibit cyclin D1 expression. Because β-catenin levels and Myc expression levels were unchanged in the presence or absence of Wnt5a, it is unlikely that Wnt5a inhibits the Wnt/β-catenin signaling cascade in these cells. However, the mechanism that how Wnt5a signaling activates the Ca2+ pathway to inhibit cyclin D1 expression need further study.
In this study, we encountered some confusing phenomenon. Due to the low transfection efficiency of cortical neurons (<1%), we have to validate the effectiveness of siRNA in HEK293 cells (transfection efficiency >95%). We show that siRNAs against Wnt5a results in a complete loss of expression in HEK293 cells. However, these Wnt5a siRNAs only induces apoptosis in about 20% of transfected cortical neurons. We think there are two reasons causing this result: (1) in HEK293 cells, Wnt5a siRNAs and Flag-Wnt5a expression plasmids were transfected into cells at the same time, the expression of exogenous Flag-Wnt5a was inhibited at the very beginning. Therefore, a complete inhibitory effect has seen in HEK293 cells; and (2) in neurons, both the mRNA and protein levels of endogenous Wnt5a are high. Thus, the Wnt5a siRNAs can only partially reduce the level of endogenous Wnt5a in the transfected neurons. To evaluate the exact effect of Wnt5a siRNAs, a score of 0–3 was assigned to describe the levels of Wnt5a protein based on the red fluorescence area in the cytoplasm of neurons (grade 0, <5%; grade 1, 5%–33%; grade 2, 33%–66%; and grade 3, >66%; Figue 3B). After Wnt5a siRNAs treatment, the ratio of cortical neurons in grade 0 increased from 6% to 25%–30%, and this is consistent with the percent of cortiacl neuron apoptosis induced by Cyclin D1 siRNAs.
Conclusion
Our studies reveal that Wnt5a increases neuronal survival by negative regulating cell-cycle activation in vitro and in vivo. This conclusion is supported in part by previous experiments in which adding exogenous Wnt5a in hippocampal neurons and cortical neurons. The downregulation of Wnt5a signaling might be an important contributor of AD-related neurodegeneration.
Author Contributions
LZ conceived of the study, and participated in its design and helped to draft the manuscript; LZ also carried out the cell culture, Q-PCR, pathological studies, animal surgery, immunoblot analysis, CaMKII activity assay and cell survival assays. DC and Y-GW carried out the cell culture, animal surgery and plasmid construct. X-MH and FL carried out the Q-PCR, animal surgery and immunofluorescence. HC, ST, Y-LS and WC carried out the subcellular fractionation. W-XY carried out the tissue immunohistochemistry. Z-CC carried out the siRNA interference. Z-JL, Z-ZD and WC carried out the immunoblot analysis. R-HY carried out the pathological studies and tissue immunohistochemistry. TP, NJ and M-FH conceived of the study and participated in its design. Z-QL conceived of the study, and participated in its design and coordination and helped to draft the manuscript; Z-QL also carried out the pathological studies and animal surgery. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 81271328); the Science and Technology Planning Project of Guangdong Province, China (Grant No. 2012B060300013 and 2011B031800255); the Science and Technology Program of Guangzhou, China (Grant No. 2011Y2-00017-1).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Arendt, T., Rödel, L., Gartner, U., and Holzer, M. (1996). Expression of the cyclin-dependent kinase inhibitor p16 in Alzheimer’s disease. Neuroreport 7, 3047–3049. doi: 10.1097/00001756-199611250-00050
Bach, S., Knockaert, M., Reinhardt, J., Lozach, O., Schmitt, S., Baratte, B., et al. (2005). Roscovitine targets, protein kinases and pyridoxal kinase. J. Biol. Chem. 280, 31208–31219. doi: 10.1074/jbc.m500806200
Becker, E. B., and Bonni, A. (2004). Cell cycle regulation of neuronal apoptosis in development and disease. Prog. Neurobiol. 72, 1–25. doi: 10.1016/j.pneurobio.2003.12.005
Biswas, S. C., Shi, Y., Vonsattel, J.-P. G., Leung, C. L., Troy, C. M., and Greene, L. A. (2007). Bim is elevated in Alzheimer’s disease neurons and is required for β-amyloid-induced neuronal apoptosis. J. Neurosci. 27, 893–900. doi: 10.1523/jneurosci.3524-06.2007
Bitler, B. G., Nicodemus, J. P., Li, H., Cai, Q., Wu, H., Hua, X., et al. (2011). Wnt5a suppresses epithelial ovarian cancer by promoting cellular senescence. Cancer Res. 71, 6184–6194. doi: 10.1158/0008-5472.can-11-1341
Boonen, R. A., van Tijn, P., and Zivkovic, D. (2009). Wnt signaling in Alzheimer’s disease: up or down, that is the question. Ageing Res. Rev. 8, 71–82. doi: 10.1016/j.arr.2008.11.003
Bozyczko-Coyne, D., O’kane, T. M., Wu, Z.-L., Dobrzanski, P., Murthy, S., Vaught, J. L., et al. (2001). CEP-1347/KT-7515, an inhibitor of SAPK/JNK pathway activation, promotes survival and blocks multiple events associated with Aβ-induced cortical neuron apoptosis. J. Neurochem. 77, 849–863. doi: 10.1046/j.1471-4159.2001.00294.x
Budnik, V., and Salinas, P. C. (2011). Wnt signaling during synaptic development and plasticity. Curr. Opin. Neurobiol. 21, 151–159. doi: 10.1016/j.conb.2010.12.002
Busser, J., Geldmacher, D. S., and Herrup, K. (1998). Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer’s disease brain. J. Neurosci. 18, 2801–2807.
Caricasole, A., Copani, A., Caraci, F., Aronica, E., Rozemuller, A. J., Caruso, A., et al. (2004). Induction of Dickkopf-1, a negative modulator of the Wnt pathway, is associated with neuronal degeneration in Alzheimer’s brain. J. Neurosci. 24, 6021–6027. doi: 10.1523/jneurosci.1381-04.2004
Caricasole, A., Copani, A., Caruso, A., Caraci, F., Iacovelli, L., Sortino, M. A., et al. (2003). The Wnt pathway, cell-cycle activation and β-amyloid: novel therapeutic strategies in Alzheimer’s disease? Trends Pharmacol. Sci. 24, 233–238. doi: 10.1016/s0165-6147(03)00100-7
Cerpa, W., Farías, G. G., Godoy, J. A., Fuenzalida, M., Bonansco, C., and Inestrosa, N. C. (2010). Wnt-5a occludes Aβ oligomer-induced depression of glutamatergic transmission in hippocampal neurons. Mol. Neurodegener. 5:3. doi: 10.1186/1750-1326-5-3
Chang, B. H., Mukherji, S., and Soderling, T. R. (1998). Characterization of a calmodulin kinase II inhibitor protein in brain. Proc. Natl. Acad. Sci. U S A 95, 10890–10895. doi: 10.1073/pnas.95.18.10890
Davidson, G., and Niehrs, C. (2010). Emerging links between CDK cell cycle regulators and Wnt signaling. Trends Cell Biol. 20, 453–460. doi: 10.1016/j.tcb.2010.05.002
Davies, P., and Maloney, A. J. (1976). Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 2:1403. doi: 10.1016/s0140-6736(76)91936-x
De Ferrari, G. V., Avila, M. E., Medina, M. A., Perez-Palma, E., Bustos, B. I., and Alarcon, M. A. (2014). Wnt/β-catenin signaling in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 13, 745–754. doi: 10.2174/1871527312666131223113900
De Ferrari, G. V., and Moon, R. T. (2006). The ups and downs of Wnt signaling in prevalent neurological disorders. Oncogene 25, 7545–7553. doi: 10.1038/sj.onc.1210064
De Ferrari, G. V., Papassotiropoulos, A., Biechele, T., Wavrant De-Vrieze, F., Avila, M. E., Major, M. B., et al. (2007). Common genetic variation within the low-density lipoprotein receptor-related protein 6 and late-onset Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 104, 9434–9439. doi: 10.1073/pnas.0603523104
Dowejko, A., Bauer, R., Bauer, K., Müller-Richter, U. D., and Reichert, T. E. (2012). The human HECA interacts with cyclins and CDKs to antagonize Wnt-mediated proliferation and chemoresistance of head and neck cancer cells. Exp. Cell Res. 318, 489–499. doi: 10.1016/j.yexcr.2011.11.004
Giovanni, A., Wirtz-Brugger, F., Keramaris, E., Slack, R., and Park, D. S. (1999). Involvement of cell cycle elements, cyclin-dependent kinases, pRb and E2F × DP, in B-amyloid-induced neuronal death. J. Biol. Chem. 274, 19011–19016. doi: 10.1074/jbc.274.27.19011
Godoy, J. A., Arrázola, M. S., Ordenes, D., Silva-Alvarez, C., Braidy, N., and Inestrosa, N. C. (2014). Wnt-5a ligand modulates mitochondrial fission-fusion in rat hippocampal neurons. J. Biol. Chem. 289, 36179–36193. doi: 10.1074/jbc.m114.557009
Gómez-Isla, T., Price, J. L., Mckeel, D. W. Jr., Morris, J. C., Growdon, J. H., and Hyman, B. T. (1996). Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J. Neurosci. 16, 4491–4500.
Goueli, B. S., Hsiao, K., Tereba, A., and Goueli, S. A. (1995). A novel and simple method to assay the activity of individual protein kinases in a crude tissue extract. Anal. Biochem. 225, 10–17. doi: 10.1006/abio.1995.1100
Hanson, P. I., Kapiloff, M. S., Lou, L. L., Rosenfeld, M. G., and Schulman, H. (1989). Expression of a multifunctional Ca2+/calmodulin-dependent protein kinase and mutational analysis of its autoregulation. Neuron 3, 59–70. doi: 10.1016/0896-6273(89)90115-3
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
He, X., Saint-Jeannet, J.-P., Wang, Y., Nathans, J., Dawid, I., and Varmus, H. (1997). A member of the Frizzled protein family mediating axis induction by Wnt-5A. Science 275, 1652–1654. doi: 10.1126/science.275.5306.1652
Herrup, K., and Yang, Y. (2007). Cell cycle regulation in the postmitotic neuron: oxymoron or new biology? Nat. Rev. Neurosci. 8, 368–378. doi: 10.1038/nrn2124
Hoozemans, J. J., Brückner, M. K., Rozemuller, A. J., Veerhuis, R., Eikelenboom, P., and Arendt, T. (2002). Cyclin D1 and cyclin E are co-localized with cyclo-oxygenase 2 (COX-2) in pyramidal neurons in Alzheimer disease temporal cortex. J. Neuropathol. Exp. Neurol. 61, 678–688. doi: 10.1093/jnen/61.8.678
Howng, S. L., Wu, C. H., Cheng, T. S., Sy, W. D., Lin, P. C., Wang, C., et al. (2002). Differential expression of Wnt genes, β-catenin and E-cadherin in human brain tumors. Cancer Lett. 183, 95–101. doi: 10.1016/s0304-3835(02)00085-x
Inestrosa, N. C., and Arenas, E. (2010). Emerging roles of Wnts in the adult nervous system. Nat. Rev. Neurosci. 11, 77–86. doi: 10.1038/nrn2755
Inestrosa, N. C., and Varela-Nallar, L. (2014). Wnt signaling in the nervous system and in Alzheimer’s disease. J. Mol. Cell Biol. 6, 64–74. doi: 10.1093/jmcb/mjt051
Ishitani, T., Kishida, S., Hyodo-Miura, J., Ueno, N., Yasuda, J., Waterman, M., et al. (2003). The TAK1-NLK mitogen-activated protein kinase cascade functions in the Wnt-5a/Ca2+ pathway to antagonize Wnt/beta-catenin signaling. Mol. Cell. Biol. 23, 131–139. doi: 10.1128/mcb.23.1.131-139.2003
Issack, P. S., and Ziff, E. B. (1998). Altered expression of helix-loop-helix transcriptional regulators and cyclin D1 in Wnt-1-transformed PC12 cells. Cell Growth Differ. 9, 837–845.
Kingston, R. E., Chen, C. A., and Rose, J. K. (2003). Calcium phosphate transfection. Curr. Protoc. Mol. Biol. Chapter9: Unit 9.1. doi: 10.1002/0471142727.mb0901s63
Kranenburg, O., van der Eb, A. J., and Zantema, A. (1996). Cyclin D1 is an essential mediator of apoptotic neuronal cell death. EMBO J. 15, 46–54.
Kremenevskaja, N., von Wasielewski, R., Rao, A. S., Schöfl, C., Andersson, T., and Brabant, G. (2005). Wnt-5a has tumor suppressor activity in thyroid carcinoma. Oncogene 24, 2144–2154. doi: 10.1038/sj.onc.1208370
Kruman, I. I., Wersto, R. P., Cardozo-Pelaez, F., Smilenov, L., Chan, S. L., Chrest, F. J., et al. (2004). Cell cycle activation linked to neuronal cell death initiated by DNA damage. Neuron 41, 549–561. doi: 10.1016/s0896-6273(04)00017-0
Kühl, M., Geis, K., Sheldahl, L. C., Pukrop, T., Moon, R. T., and Wedlich, D. (2001). Antagonistic regulation of convergent extension movements in xenopus by Wnt/β-catenin and Wnt/Ca2+ signaling. Mech. Dev. 106, 61–76. doi: 10.1016/s0925-4773(01)00416-6
Lambert, C., Cisternas, P., and Inestrosa, N. C. (2016). Role of Wnt signaling in central nervous system injury. Mol. Neurobiol. 53, 2297–2311. doi: 10.1007/s12035-015-9138-x
Li, B., Zhong, L., Yang, X., Andersson, T., Huang, M., and Tang, S. J. (2011). WNT5A signaling contributes to Aβ-induced neuroinflammation and neurotoxicity. PLoS One 6:e22920. doi: 10.1371/journal.pone.0022920
Liang, H., Chen, Q., Coles, A. H., Anderson, S. J., Pihan, G., Bradley, A., et al. (2003). Wnt5a inhibits B cell proliferation and functions as a tumor suppressor in hematopoietic tissue. Cancer Cell 4, 349–360. doi: 10.1016/s1535-6108(03)00268-x
Libro, R., Bramanti, P., and Mazzon, E. (2016). The role of the Wnt canonical signaling in neurodegenerative diseases. Life Sci. 158, 78–88. doi: 10.1016/j.lfs.2016.06.024
Liu, D. X., and Greene, L. A. (2001). Neuronal apoptosis at the G1/S cell cycle checkpoint. Cell Tissue Res. 305, 217–228. doi: 10.1007/s004410100396
Lopes, J. P., Oliveira, C. R., and Agostinho, P. (2010). Neurodegeneration in an Aβ-induced model of Alzheimer’s disease: the role of Cdk5. Aging Cell 9, 64–77. doi: 10.1111/j.1474-9726.2009.00536.x
Majd, S., Zarifkar, A., Rastegar, K., and Takhshid, M. A. (2008). Different fibrillar Aβ 1–42 concentrations induce adult hippocampal neurons to reenter various phases of the cell cycle. Brain Res. 1218, 224–229. doi: 10.1016/j.brainres.2008.04.050
McShea, A., Harris, P. L., Webster, K. R., Wahl, A. F., and Smith, M. A. (1997). Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer’s disease. Am. J. Pathol. 150, 1933–1939.
Milton, N. G. (2002). The amyloid-β peptide binds to cyclin B1 and increases human cyclin-dependent kinase-1 activity. Neurosci. Lett. 322, 131–133. doi: 10.1016/s0304-3940(02)00081-2
Murphy, M. P., and LeVine, H. III. (2010). Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimers. Dis. 19, 311–323. doi: 10.3233/JAD-2010-1221
Patrick, G. N., Zukerberg, L., Nikolic, M., de la Monte, S., Dikkes, P., and Tsai, L. H. (1999). Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 402, 615–622. doi: 10.1038/45159
Peirson, S. N., and Butler, J. N. (2007). Quantitative polymerase chain reaction. Methods Mol. Biol. 362, 349–362. doi: 10.1007/978-1-59745-257-1_25
Purro, S. A., Galli, S., and Salinas, P. C. (2014). Dysfunction of Wnt signaling and synaptic disassembly in neurodegenerative diseases. J. Mol. Cell Biol. 6, 75–80. doi: 10.1093/jmcb/mjt049
Riise, J., Plath, N., Pakkenberg, B., and Parachikova, A. (2015). Aberrant Wnt signaling pathway in medial temporal lobe structures of Alzheimer’s disease. J. Neural Transm. (Vienna) 122, 1303–1318. doi: 10.1007/s00702-015-1375-7
Salinas, P. C. (2012). Wnt signaling in the vertebrate central nervous system: from axon guidance to synaptic function. Cold Spring Harb. Perspect. Biol. 4:a008003. doi: 10.1101/cshperspect.a008003
Scali, C., Caraci, F., Gianfriddo, M., Diodato, E., Roncarati, R., Pollio, G., et al. (2006). Inhibition of Wnt signaling, modulation of Tau phosphorylation and induction of neuronal cell death by DKK1. Neurobiol. Dis. 24, 254–265. doi: 10.1016/j.nbd.2006.06.016
Sciarretta, C., and Minichiello, L. (2010). The preparation of primary cortical neuron cultures and a practical application using immunofluorescent cytochemistry. Methods Mol. Biol. 633, 221–231. doi: 10.1007/978-1-59745-019-5_16
Sheldahl, L. C., Park, M., Malbon, C. C., and Moon, R. T. (1999). Protein kinase C is differentially stimulated by Wnt and frizzled homologs in a G-protein-dependent manner. Curr. Biol. 9, 695–698. doi: 10.1016/s0960-9822(99)80310-8
Shtutman, M., Zhurinsky, J., Simcha, I., Albanese, C., D’amico, M., Pestell, R., et al. (1999). The cyclin D1 gene is a target of the β-catenin/LEF-1 pathway. Proc. Natl. Acad. Sci. U S A 96, 5522–5527. doi: 10.1073/pnas.96.10.5522
Shukla, V., Skuntz, S., and Pant, H. C. (2012). Deregulated Cdk5 activity is involved in inducing Alzheimer’s disease. Arch. Med. Res. 43, 655–662. doi: 10.1016/j.arcmed.2012.10.015
Silva-Alvarez, C., Arrázola, M. S., Godoy, J. A., Ordenes, D., and Inestrosa, N. C. (2013). Canonical Wnt signaling protects hippocampal neurons from Aβ oligomers: role of non-canonical Wnt-5a/Ca2+ in mitochondrial dynamics. Front. Cell. Neurosci. 7:97. doi: 10.3389/fncel.2013.00097
Tokumitsu, H., Chijiwa, T., Hagiwara, M., Mizutani, A., Terasawa, M., and Hidaka, H. (1990). KN-62, 1-[N,O-bis(5-isoquinolinesulfonyl)-N-methyl-L-tyrosyl]-4-phenylpiperazi ne, a specific inhibitor of Ca2+/calmodulin-dependent protein kinase II. J. Biol. Chem. 265, 4315–4320.
Torres, M. A., Yang-Snyder, J. A., Purcell, S. M., Demarais, A. A., Mcgrew, L. L., and Moon, R. T. (1996). Activities of the Wnt-1 class of secreted signaling factors are antagonized by the Wnt-5A class and by a dominant negative cadherin in early xenopus development. J. Cell Biol. 133, 1123–1137. doi: 10.1083/jcb.133.5.1123
Toyofuku, T., Hong, Z., Kuzuya, T., Tada, M., and Hori, M. (2000). Wnt/frizzled-2 signaling induces aggregation and adhesion among cardiac myocytes by increased cadherin-β-catenin complex. J. Cell Biol. 150, 225–241. doi: 10.1083/jcb.150.1.225
van den Heuvel, S. (2005). Cell-Cycle Regulation. WormBook doi: 10.1895/wormbook.1.28.1 [Epub ahead of Print].
Varela-Nallar, L., Parodi, J., Farias, G. G., and Inestrosa, N. C. (2012). Wnt-5a is a synaptogenic factor with neuroprotective properties against Aβ toxicity. Neurodegener. Dis. 10, 23–26. doi: 10.1159/000333360
Varvel, N. H., Bhaskar, K., Patil, A. R., Pimplikar, S. W., Herrup, K., and Lamb, B. T. (2008). Aβ oligomers induce neuronal cell cycle events in Alzheimer’s disease. J. Neurosci. 28, 10786–10793. doi: 10.1523/JNEUROSCI.2441-08.2008
Vincent, I., Jicha, G., Rosado, M., and Dickson, D. W. (1997). Aberrant expression of mitotic cdc2/cyclin B1 kinase in degenerating neurons of Alzheimer’s disease brain. J. Neurosci. 17, 3588–3598.
Whitehouse, P. J., Price, D. L., Struble, R. G., Clark, A. W., Coyle, J. T., and Delon, M. R. (1982). Alzheimer’s disease and senile dementia: loss of neurons in the basal forebrain. Science 215, 1237–1239. doi: 10.1126/science.7058341
Xie, B., Wang, C., Zheng, Z., Song, B., Ma, C., Thiel, G., et al. (2011). Egr-1 transactivates Bim gene expression to promote neuronal apoptosis. J. Neurosci. 31, 5032–5044. doi: 10.1523/jneurosci.5504-10.2011
Yang, Y., Geldmacher, D. S., and Herrup, K. (2001). DNA replication precedes neuronal cell death in Alzheimer’s disease. J. Neurosci. 21, 2661–2668.
Yang, Y., Mufson, E. J., and Herrup, K. (2003). Neuronal cell death is preceded by cell cycle events at all stages of Alzheimer’s disease. J. Neurosci. 23, 2557–2563.
Zhang, L., Deng, J., Pan, Q., Zhan, Y., Fan, J. B., Zhang, K., et al. (2016). Targeted methylation sequencing reveals dysregulated Wnt signaling in Parkinson disease. J. Genet. Genomics 43, 587–592. doi: 10.1016/j.jgg.2016.05.002
Zhang, Z., Hartmann, H., Do, V. M., Abramowski, D., Sturchler-Pierrat, C., Staufenbiel, M., et al. (1998). Destabilization of beta-catenin by mutations in presenilin-1 potentiates neuronal apoptosis. Nature 395, 698–702. doi: 10.1038/27208
Keywords: Wnt5a, β-Amyloid protein, Alzheimer’s disease, cortical neuron, apoptosis, cell-cycle activation, Cyclin D1
Citation: Zhou L, Chen D, Huang X-M, Long F, Cai H, Yao W-X, Chen Z-C, Liao Z-J, Deng Z-Z, Tan S, Shan Y-L, Cai W, Wang Y-G, Yang R-H, Jiang N, Peng T, Hong M-F and Lu Z-Q (2017) Wnt5a Promotes Cortical Neuron Survival by Inhibiting Cell-Cycle Activation. Front. Cell. Neurosci. 11:281. doi: 10.3389/fncel.2017.00281
Received: 06 May 2017; Accepted: 30 August 2017;
Published: 29 September 2017.
Edited by:
Lavinia Alberi, SICHH, SwitzerlandReviewed by:
Swananda Marathe, Indian Institute of Science, IndiaAlessandro Rufini, University of Leicester, United Kingdom
Copyright © 2017 Zhou, Chen, Huang, Long, Cai, Yao, Chen, Liao, Deng, Tan, Shan, Cai, Wang, Yang, Jiang, Peng, Hong and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nan Jiang, bmFuamlhbmdfc3lzdUAxNjMuY29t
Tao Peng, cHRfZ3ptZWRpY2FsdW5AMTYzLmNvbQ==
Ming-Fan Hong, cWlueWFuZzE5ODcwMkBzaW5hLmNvbQ==
Zheng-Qi Lu, emhlbmdxaV9sdTIwMTJAc2luYS5jb20=