 Sandeep Sheth
Sandeep Sheth Debashree Mukherjea
Debashree Mukherjea Leonard P. Rybak
Leonard P. Rybak Vickram Ramkumar
Vickram Ramkumar- 1Department of Pharmacology, Southern Illinois University School of Medicine, Springfield, IL, United States
- 2Department of Surgery (Otolaryngology), Southern Illinois University School of Medicine, Springfield, IL, United States
Evidence of significant hearing loss during the early days of use of cisplatin as a chemotherapeutic agent in cancer patients has stimulated research into the causes and treatment of this side effect. It has generally been accepted that hearing loss is produced by excessive generation of reactive oxygen species (ROS) in cell of the cochlea, which led to the development of various antioxidants as otoprotective agents. Later studies show that ROS could stimulate cochlear inflammation, suggesting the use of anti-inflammatory agents for treatment of hearing loss. In this respect, G-protein coupled receptors, such as adenosine A1 receptor and cannabinoid 2 receptors, have shown efficacy in the treatment of hearing loss in experimental animals by increasing ROS scavenging, suppressing ROS generation, or by decreasing inflammation. Inflammation could be triggered by activation of transient receptor potential vanilloid 1 (TRPV1) channels in the cochlea and possibly other TRP channels. Targeting TRPV1 for knockdown has also been shown to be a useful strategy for ensuring otoprotection. Cisplatin entry into cochlear hair cells is mediated by various transporters, inhibitors of which have been shown to be effective for treating hearing loss. Finally, cisplatin-induced DNA damage and activation of the apoptotic process could be targeted for cisplatin-induced hearing loss. This review focuses on recent development in our understanding of the mechanisms underlying cisplatin-induced hearing loss and provides examples of how drug therapies have been formulated based on these mechanisms.
Cisplatin Ototoxicity
Cisplatin is a widely used and effective drug for the treatment of solid tumors ranging from ovarian, lung, head, and neck to testicular cancer. Dose limiting side effects, such as ototoxicity, neurotoxicity, and nephrotoxicity, are generally encountered with a majority of patients treated with cisplatin-based chemotherapy. Methods to increase diuresis, such as hydration, have been shown to reduce nephrotoxicity. However, to date no effective FDA approved treatment for ototoxicity is available. Cisplatin-induced hearing loss is primarily in the high frequency range. It is bilateral and permanent and severely affects the quality of life for cancer patients. The incidence of cisplatin-induced hearing loss in children ranges from 22 to 77% (Knight et al., 2005; Kushner et al., 2006; Coradini et al., 2007). This range reflects hearing loss observed with the different doses and duration of cisplatin treatment in addition to the different age groups of the patients who were treated. Accordingly, children show greater risk for developing hearing loss following cisplatin treatment than adults (Knight et al., 2005; Kushner et al., 2006; Gurney et al., 2007). Hearing loss is especially difficult for children who are undergoing treatment for brain tumors, such as neuroblastoma. It could affect early speech development and hamper social integration. Development of effective therapies for treating hearing loss is therefore of primary importance.
Several approaches have been undertaken over the past two decades to treat cisplatin ototoxicity. These include the use of localized or systemic administration of antioxidants or drugs which activate endogenous antioxidant systems. Cisplatin induces apoptosis of hair cells through activation of mitochondrial pathway which can be targeted to inhibit cell death. Another approach involves using anti-inflammatory agents which target the pro-inflammatory mechanisms associated with cisplatin treatment in the cochlea. Recent insights into the entry processes of cisplatin in hair cells and other cells in the cochlea should stimulate the development of drugs which specifically block entry of drugs into these cells without affecting cisplatin entry into cancer cells. This report reviews the currently accepted mechanisms underlying cisplatin-induced damage or death to cochlear cells and highlights how these mechanisms could guide the future development of effective otoprotective agents.
Cochlear ROS Generation and Antioxidant Defense System
The normal function of the cochlea requires its high metabolic activity in areas such as the stria vascularis, spiral ligament, and spiral prominence (Salt et al., 1987; Ryan, 1988). This high metabolic activity leads to leakage of electrons from the mitochondrial respiratory chain which can react with oxygen (O2) to form superoxide (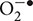 ). Environmental stimuli which increase the metabolic activity of the cochlea, such as loud noise, are expected to increase oxidative stress in the cochlea. The metabolic demand on the cochlea renders it very sensitive to hypoxic events and ischemia-reperfusion injuries (Seidman et al., 1991). Ototoxic drugs, such as cisplatin have been shown to increase the generation of reactive oxygen species (ROS) (Clerici and Yang, 1996; Kopke et al., 1997) by either stimulating enzyme systems linked to this process or by inactivating antioxidant systems (Church et al., 1995; Rybak et al., 1995). Rats injected with cisplatin demonstrate reduced cochlear glutathione (GSH) and antioxidant enzyme activities (Ravi et al., 1995). A primary target of cisplatin for generation of ROS is the NOX3 NADPH oxidase system (Banfi et al., 2004). NOX3 is induced by cisplatin and knockdown of this enzyme by trans-tympanic delivery of siRNA protects against cisplatin-induced ototoxicity (Mukherjea et al., 2010). Since NOX3 is localized primarily to the cochlea, systemic administration of inhibitors of NOX3 could effectively reduce enzyme activity and treat hearing loss. Another active ROS generating system in the cochlea is xanthine oxidase. This enzyme converts hypoxanthine (a metabolite derived from the breakdown of adenosine by adenosine deaminase) to uric acid. Inhibition of this enzyme by allopurinol contributes to reductions in cisplatin-induced ototoxicity and nephrotoxicity when administered with ebselen, a glutathione peroxidase (GSH.Px) mimetic (Lynch et al., 2005).
). Environmental stimuli which increase the metabolic activity of the cochlea, such as loud noise, are expected to increase oxidative stress in the cochlea. The metabolic demand on the cochlea renders it very sensitive to hypoxic events and ischemia-reperfusion injuries (Seidman et al., 1991). Ototoxic drugs, such as cisplatin have been shown to increase the generation of reactive oxygen species (ROS) (Clerici and Yang, 1996; Kopke et al., 1997) by either stimulating enzyme systems linked to this process or by inactivating antioxidant systems (Church et al., 1995; Rybak et al., 1995). Rats injected with cisplatin demonstrate reduced cochlear glutathione (GSH) and antioxidant enzyme activities (Ravi et al., 1995). A primary target of cisplatin for generation of ROS is the NOX3 NADPH oxidase system (Banfi et al., 2004). NOX3 is induced by cisplatin and knockdown of this enzyme by trans-tympanic delivery of siRNA protects against cisplatin-induced ototoxicity (Mukherjea et al., 2010). Since NOX3 is localized primarily to the cochlea, systemic administration of inhibitors of NOX3 could effectively reduce enzyme activity and treat hearing loss. Another active ROS generating system in the cochlea is xanthine oxidase. This enzyme converts hypoxanthine (a metabolite derived from the breakdown of adenosine by adenosine deaminase) to uric acid. Inhibition of this enzyme by allopurinol contributes to reductions in cisplatin-induced ototoxicity and nephrotoxicity when administered with ebselen, a glutathione peroxidase (GSH.Px) mimetic (Lynch et al., 2005).
The cochlea possesses an efficient antioxidant defense system. This includes antioxidants such as vitamin C, vitamin E, and low molecular weight thiols, such as GSH (Kopke et al., 1999). Studies in the guinea pig cochlea show that the highest levels of GSH are present in the basal and intermediate cells of the stria vascularis and in cells of the spiral ligament (Usami et al., 1996). This distribution matches well with the distribution of glutathione S-transferase (el Barbary et al., 1993), an enzyme which conjugates and detoxifies xenobiotics (such as cisplatin). Increased levels of glutathione S-transferase in cancer cells can aid in the inactivation of cisplatin and contribute to resistance to cisplatin in the cochlea. In addition, the cochlea expresses several antioxidant enzymes, which include superoxide dismutase (SOD), GSH.Px, and catalase (CAT). SOD catalyzes the conversion of to H2O2 and O2 while CAT converts H2O2 to O2 and H2O. GSH.Px reduces H2O2 and possibly other peroxides. The enzyme GSH.Px also catalyzes the conversion of reduced GSH to its oxidized form (GSSG), in the process of detoxifying H2O2. In addition, glutathione reductase (GR) is important for the defense against ROS by aiding in the regeneration of GSH from GSSG (Figure 1). Two forms of SOD are expressed in the cochlea. A Cu/Zn isoform of SOD is localized in the cytosol, while a Mn-regulated isoform (Mn-SOD) is localized to the mitochondria (Yao and Rarey, 1996). Mn-SOD is localized to metabolically active sites in the cochlea such as stria vascularis, spiral ligament, spiral prominence, spiral limbus, and organ of Corti (Lai et al., 1996). Activities of SOD were higher in the subcellular fractions of stria vascularis and spiral ligament compared to the organ of Corti (Pierson and Gray, 1982; Yao and Rarey, 1996). High activities of other antioxidant enzymes were also observed in the lateral wall tissues compared to the rest of the cochlea, suggesting their roles in mitigating oxidative stress in these regions (Pierson and Gray, 1982). In the absence of this ROS detoxification system, ROS can produce cellular damage by lipid peroxidation, with increased levels of the lipid peroxide, malondialdehyde, and 4-hydroxynonenal.
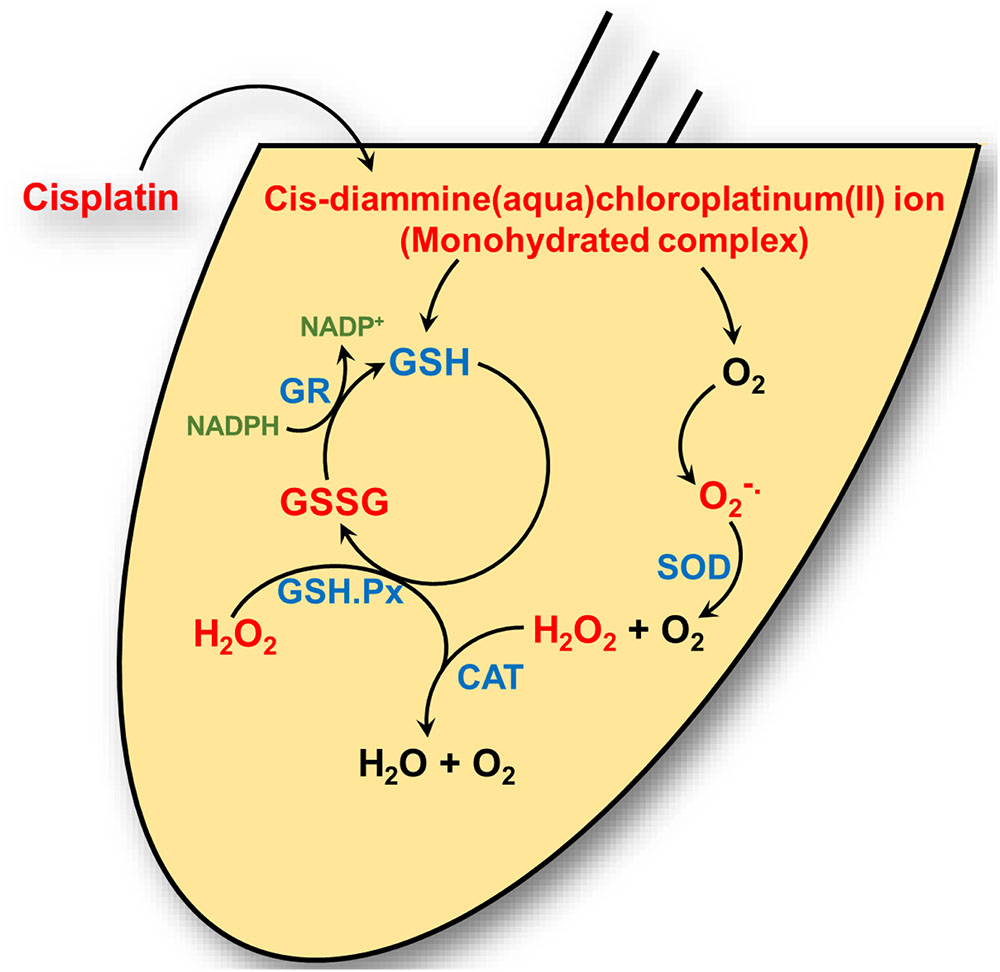
FIGURE 1. Cisplatin’s interaction with the cochlear antioxidant defense system. Cisplatin is converted to a cis-diammine(aqua)chloroplatinum(II) (a monohydrate cisplatin complex) upon entering the cell cytoplasm. These reactive platinum species can react with molecular oxygen (O2) to generate superoxide () which is detoxified by superoxide dismutase (SOD) to hydrogen peroxide (H2O2) and oxygen. Hydrogen peroxide is further detoxified by catalase to water (H2O) and oxygen. Cisplatin reactive intermediates readily bind to and oxidizes the antioxidant reduced glutathione (GSH) to oxidized glutathione (GSSG). Glutathione peroxidase (GSH.Px) consumes GSH to produce GSSG in the process of converting H2O2 into H2O. Glutathione reductase (GR) reduces GSSG to GSH by using the reduced from of nicotinamide adenine dinucleotide phosphate (NADP+), NAPDH, as cofactor.
Increased ROS Generation Contributes to Cisplatin-Induced Hearing Loss
Exposure of explants to ROS induces bleb formation and changes in length of outer hair cells which were attenuated by co-administration of the antioxidant, deferoxamine, or antioxidant enzymes (Clerici et al., 1995). Infusion of ROS into the guinea pig ear led to increased compound action potential (CAP) threshold amplitudes, which were reduced by co-administration of antioxidant or antioxidant enzymes (Clerici and Yang, 1996). Furthermore, it was observed that cochleae obtained from cisplatin-treated animals showed depletion of GSH and reduced activities of antioxidant enzymes, such as SOD, CAT, GSH.Px, and GR along with increased evidence of lipid peroxidation (Rybak et al., 2000). This reduction in antioxidant capacity in the cochlea could result from: (1) covalent binding of cisplatin to sulfhydryl groups within the antioxidant enzymes, causing enzyme inactivation; (2) loss of metal cofactors, such as copper and selenium, which are vital for activity of SOD and GSH.Px; (3) increased ROS which could exhaust antioxidant enzymes; and/or (4) depletion of cochlear antioxidant enzyme cofactors, such as GSH and NADPH, which are essential for GSH.Px and GR activities, respectively (for review see Rybak et al., 2000). The increased oxidative stress within the cochlea could increase lipid peroxidation of membranes, inactivate essential cellular enzymes and membrane transporters, and disrupt ion channel function. The ultimate effect of increased ROS generation is to promote apoptotic and necrotic cell death. These data suggest that ROS plays a critical role in cisplatin-induced ototoxicity and that its inhibition could ameliorate hearing loss.
As discussed below, one important result of increased oxidative stress is induction of inflammatory processes in the cochlea. The importance of managing ROS in mediating otoprotection is underscored by the observation that polymorphisms of glutathione S-transferase gene increase susceptibility to cisplatin hearing loss (Oldenburg et al., 2007). In this report, it was shown that patients inheriting the 105Ile/105Ile-GSTP1 or 105Val/105Ile-GSTP1 alleles had greater hearing loss than those inheriting the 105Val/105Val-GSTP1 alleles (Oldenburg et al., 2007).
Targeting Oxidative Stress for Treating Cisplatin-Induced Hearing Loss
The cochlea is endowed with a number of distinct cellular mechanisms which could contribute to otoprotection. These include various endogenous antioxidant enzymes and antioxidants (detailed above), heat shock proteins, kidney injury molecule-1, anti-apoptotic proteins, transcription factors such as nuclear factor erythroid 2-related factor 2 (Nrf2) and signal transducer and activator of transcription (STAT) proteins, and a number of hormone and G-protein-coupled receptors (Ford et al., 1997; Oh et al., 2000; Mukherjea et al., 2006; So et al., 2006; Borse et al., 2017). Therefore, it is very surprising that these mechanisms are not able to effectively protect against cisplatin ototoxicity. However, it is possible that over the course of cisplatin treatment in animal models and humans that these protective systems become overwhelmed and are no longer able to manage the toxicity. It is believed that exogenously administered antioxidants or other drugs can boost the local protective mechanism to ensure otoprotection.
Early studies were directed at examining the effects of drugs which reduce oxidative stress for treating hearing loss. Studies performed in animals indicated that antioxidants protected against cisplatin-induced hearing loss. Many of these antioxidants are thiol compounds which have high affinities for platinum. This forms the basis for their protection against cisplatin toxicity but also could account for the antitumor interference with cisplatin. Studies showed that N-acetyl cysteine (NAC) protected against cisplatin-ototoxicity in rats (Dickey et al., 2008) and guinea pigs (Choe et al., 2004). Sodium thiosulfate (STS) was also effective against cisplatin-induced hearing loss (Otto et al., 1988). However, systemic administration of this drug led to formation of cisplatin–STS complex, which reduced the levels of cisplatin in circulation needed for effective antitumor therapy (Wimmer et al., 2004). Thus, local application of STS into the cochlea would be required to provide effective otoprotection (Wimmer et al., 2004), as was shown previously (Wang et al., 2003). Several studies have shown that D-methionine, another sulfur-containing compound, protects against cisplatin-induced hearing loss when administered by both the systemic and local routes (Campbell et al., 1996; Korver et al., 2002). At the cellular level, the otoprotection mediated by this compound was associated with its ability to increase the activity of intrinsic antioxidant enzymes (Campbell et al., 2003). Other antioxidants which show promise against cisplatin-induced hearing loss include ebselen, lipoic acid, diethyldithiocarbamate, and 4-methylthiobenzoic acid (Rybak et al., 1999). Similarly, high doses of amifostine provided otoprotection in hamsters but its use was associated with neurotoxicity (Church et al., 2004).
Contrary to the pre-clinical findings, studies performed in humans showed that STS protected against cisplatin-induced nephrotoxicity (Goel et al., 1989) but was ineffective against cisplatin-induced hearing loss (Zuur et al., 2007). Amifostine was also ineffective as an otoprotectant in patients with metastatic melanoma (Ekborn et al., 2002) and in children suffering from neuroblastoma or germ cell tumors and were on a chemotherapeutic regimen including cisplatin (Marina et al., 2005; Sastry and Kellie, 2005). However, later studies showed that higher doses of amifostine were able to provide significant otoprotection (Fouladi et al., 2008).
Several G-protein-coupled receptors have been characterized in the cochlea, activation of which confer protection against cisplatin-induced hearing loss. Immunolabeling studies show distribution of the A1 adenosine receptors (A1ARs) in the stria vascularis, spiral ganglion cells, and organ of Corti (Vlajkovic et al., 2009). In the organ of Corti, the greatest distribution of A1AR is in the inner hair cells, Deiter’s cells and lower levels in the outer hair cells. Expression of A1AR was also observed in mouse organ of Corti-derived cell lines including the UB/OC-1 cells (Kaur et al., 2016) and HEI-OC1 cells (unpublished data). Localized application of A1AR agonist resulted in an increase in the activities of the antioxidative enzymes GSH.Px and SOD (Ford et al., 1997). Furthermore, A1AR agonist also reduced the cisplatin-mediated increase in malondialdehyde levels in the cochlea resulting in protection against cisplatin-induced hair cell damage and hearing loss (Whitworth et al., 2004). No significant benefits was observed following activation of the A2aAR and A3AR (Whitworth et al., 2004) which are also distributed in the cochlea (Vlajkovic et al., 2009). Systemic administration of the adenosine amine congener (ADAC) was shown to protect against cisplatin ototoxicity, presumably by activating the A1AR (Gunewardene et al., 2013). Recent data support an anti-inflammatory role of A1AR activation in the cochlea mediated by suppression of the NOX3 isoform of NADPH oxidase and suppression of STAT1-mediated inflammatory pathway (Kaur et al., 2016). STAT1 activation plays an integral role in cisplatin ototoxicity, as inhibition or knockdown of this transcription factor reduced cisplatin-induced hearing loss (Schmitt et al., 2009; Kaur et al., 2011; Mukherjea et al., 2011). A recent study from our laboratory has further documented an essential role of STAT1 in mediating cisplatin-induced hearing loss, as inhibition of this factor by a green tea extract and a known STAT1 inhibitor, epigallocatechin-3-gallate (EGCG), provided otoprotection (Borse et al., 2017). In a rodent model, EGCG protected against cisplatin-induced hearing loss without compromising cisplatin antitumor efficacy (Borse et al., 2017) (see model depicted in Figure 2). In vitro studies performed in HEI-OC1 cells demonstrate that cannabinoid 2 receptor (CB2) agonists reduce cisplatin-induced cell killing (Jeong et al., 2007). CB2 are also expressed in the stria vascularis, inner hair cells and spiral ganglion cells of the cochlea from adult albino rats (Martin-Saldana et al., 2016). Recent studies from our laboratories support an otoprotective role of CB2 activation in the cochlea, which is mediated at least in part, through inhibition of STAT1 (Ghosh et al., 2016; unpublished data). Thus, the protective action of CB2 could share a similar mechanism as observed by A1AR, namely inhibition of STAT1.
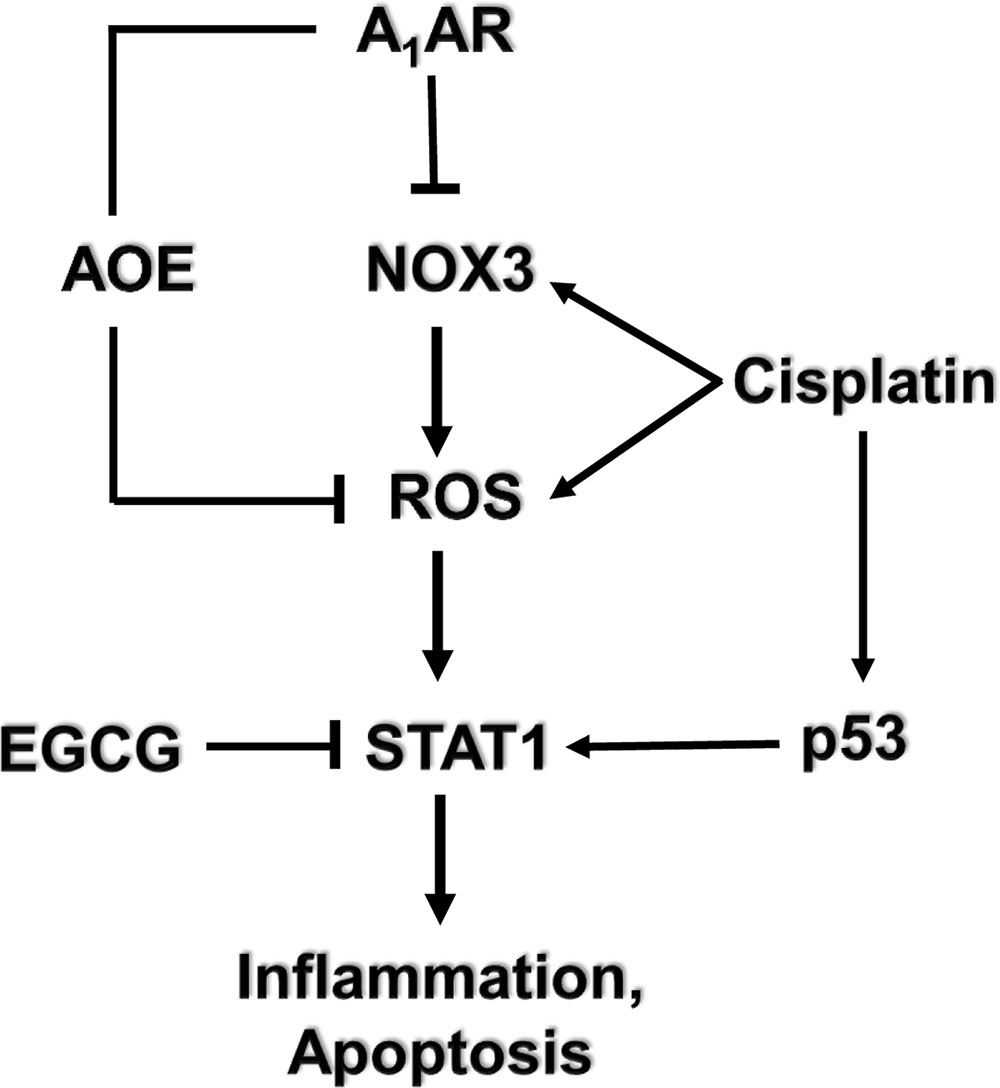
FIGURE 2. Mechanism of cisplatin-induced hearing loss and A1 adenosine receptor (A1AR)-dependent otoprotection. Cisplatin mediates NOX3 activation and reactive oxygen species (ROS) generation. The generation of ROS promotes signal transducer and activator of transcription 1 (STAT1) activation which stimulates the inflammatory process. Activated STAT1 association with active p53 promotes the apoptosis of cochlear cells. The otoprotective effects of A1AR activation is mediated by reducing oxidative stress in the cochlea by activating antioxidant enzymes (AOE) and/or by suppressing the induction of NOX3. EGCG, a known inhibitor of STAT1, has been shown to protect against cisplatin-induced hearing loss.
Additional studies from our laboratory implicated transient receptor potential vanilloid 1 (TRPV1) channels in cisplatin-mediated ototoxicity (Mukherjea et al., 2008). In a rat model, we showed knockdown of these channels by trans-tympanic administration of short interfering (si) RNA protected against cisplatin-induced hearing loss and damage to the outer hair cells (Mukherjea et al., 2008) (Figure 3). Protection was likely mediated by reducing the expression of a downstream target of TRPV1, such as NOX3, activation of which promotes ROS generation and STAT1 activation, as indicated above. STAT1 can promote both the inflammatory and pro-apoptotic actions of cisplatin in the cochlea (Kaur et al., 2011). This study suggests that inhibition of TRPV1 or NOX3 could serve as useful approaches for reducing cisplatin ototoxicity.
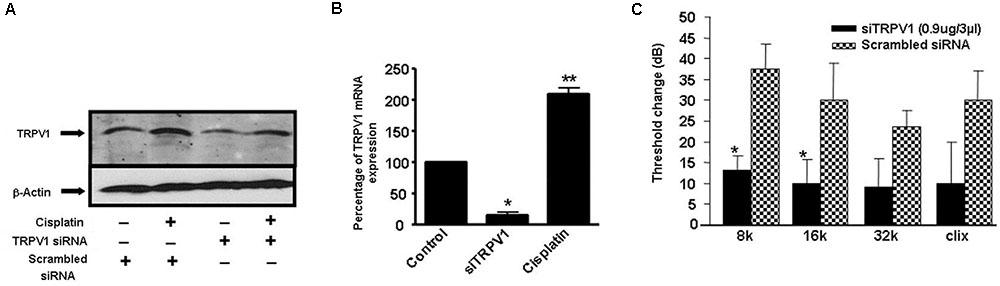
FIGURE 3. Round window administration of siRNA against TRPV1 protects against cisplatin-induced ototoxicity in rats. (A) Round window application of TRPV1 siRNA reduced both, basal and cisplatin-stimulated TRPV1 protein levels in the cochlea, assessed 24 h following cisplatin administration. (B) TRPV1 siRNA suppressed TRPV1 expression in the rat cochlea. (C) Functional studies show that TRPV1 siRNA (0.9 μg) administered by round window application protected against cisplatin-induced elevations in hearing thresholds at all frequencies tested and for click stimuli. Cisplatin (13 mg/kg i.p.) was administered 48 h following siRNA or a scrambled siRNA sequence and post-treatment ABRs were determined after an additional 72 h period. ∗p < 0.05 versus scrambled siRNA-treated cochleae and ∗∗p < 0.05 versus TRPV1 siRNA (n = 5). This figure was adapted from Mukherjea et al. (2008), with permission.
Characterization of cisplatin-induced cell death in HEI-OC1 cells showed induction of apoptosis by increased lipid peroxidation and altered mitochondrial permeability transition. It was shown that the calcium-channel blocker, flunarizine, attenuated cisplatin-induced cell death (So et al., 2006). The mechanism underlying the otoprotective action of flunarizine appears to involve activation of Nrf2 and increased expression of hemeoxygenase-1 (HO-1) (So et al., 2006). Flunarizine also exhibited an anti-inflammatory role, as evidenced from its ability to inhibit the ERK1/2 MAP kinase-nuclear factor (NF)-κB-dependent pathway (So et al., 2008).
Mitochondrial Targets of Cisplatin-Induced Ototoxicity
Bcl-2 Family
The Bcl-2 family of proteins consists of members that form the mitochondrial apoptotic pathway and function as regulators of cell death and cell survival. Among its members, Bcl-2 and Bcl-xL promote cell survival, whereas Bax, Bak, Bcl-XS, Bid, Bad, and Bim induce apoptosis (Siddiqui et al., 2015). The balance between the pro-apoptotic and anti-apoptotic proteins is crucial for the well-being of the cell. However, cellular damage caused by noxious stimuli can tilt this balance in favor of apoptosis. This process is initiated when pro-apoptotic protein such as Bax and Bid translocate from the cytoplasm to the mitochondria. This triggers a sequence of events leading to the permeabilization of the outer mitochondrial membrane, which results in the loss of mitochondrial membrane potential, generation of ROS, and release of cytochrome c from mitochondria into the cytoplasm (Siddiqui et al., 2015).
Several studies have implicated the mitochondrial pathways in the apoptosis of auditory cells after cisplatin treatment. Mongolian gerbils administered cisplatin showed deterioration in the responses of both distortion product otoacoustic emissions (DPOAE) and the endocochlear potential as compared with age-matched controls (Alam et al., 2000). The cisplatin-induced hearing loss was correlated with increased levels of Bax and decreased expression of Bcl-2 in the cells of organ of Corti, spiral ganglion and the lateral wall, as determined by immunohistochemistry. Moreover, cisplatin significantly elevated the ratio of Bax to Bcl-2, an important indicator of apoptosis, in the three representative regions in all turns of the cochlea. Similarly, cochlear hair cells from cisplatin-treated guinea pigs demonstrated apoptosis which was linked to the activation and redistribution of cytosolic Bax and the release of cytochrome c from the mitochondria (Wang et al., 2004). In another study conducted in HEI/OC1 cells, cisplatin-induced apoptosis through mitochondrial pathway which involved truncation of Bid, mitochondrial translocation of Bax, and release of cytochrome c (Devarajan et al., 2002). Recently, cisplatin treatment in UB/OC-1 cells significantly increased expression of Bax, but reduced the levels of Bcl-xL (Borse et al., 2017). Treatment with EGCG, a known inhibitor of STAT1, reversed these cisplatin-mediated effects on the expression of Bax and Bcl-xL (Borse et al., 2017) and protected against hearing loss (Figure 4). These data indicate that cisplatin induces apoptosis through activation of mitochondrial pathway and that inhibiting elements of this pathway could alleviate hearing loss.
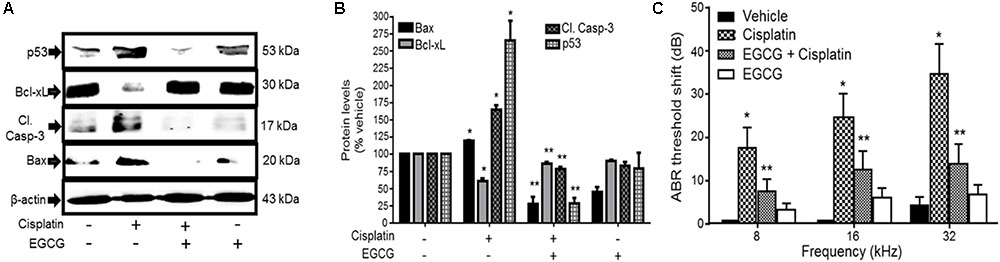
FIGURE 4. Epigallocatechin-3-gallate (EGCG) inhibited cisplatin-induced apoptosis and hearing loss. (A) UB/OC-1 cells pretreated with either vehicle or EGCG (100 μM) followed by cisplatin (20 μM) for 24 h were analyzed for pro-apoptotic proteins, such as p53, cleaved caspase-3 and Bax and anti-apoptotic protein, Bcl-xL. The expression of pro-apoptotic proteins was substantially reduced by EGCG while the reductions in Bcl-xL were attenuated. (B) The bar graph represents results from A after normalization with β-actin bands and are presented as the mean ± SEM. ∗p < 0.05 versus vehicle and ∗∗p < 0.05 versus vehicle + cisplatin (n = 4). (C) Pre-treatment ABR thresholds were recorded in Wistar rats, which were then treated with oral EGCG (100 mg/kg body weight). Cisplatin (11 mg/kg) was administered intraperitoneally 24 h later and animals were continued on daily oral EGCG treatments for additional 3 days. Post-treatment ABRs were performed on day 4. Daily oral administration of EGCG protected from cisplatin-induced ABR threshold shifts at all frequencies tested. ∗p < 0.05 versus vehicle and ∗∗p < 0.05 versus vehicle + cisplatin (n = 4). This figure was adapted from Borse et al. (2017), with permission.
Caspases
Stress signals from the mitochondria also regulate the cleavage of pro-caspase-9 into its active form, caspase-9 (Hengartner, 2000). Activated caspase-9 can then cleave and activate the downstream effector caspase, caspase-3, resulting in apoptotic destruction of the cell. Caspase-3 is also activated by caspase-8, which is an initiator caspase activated by plasma membrane death receptors (Salvesen and Dixit, 1997). Cisplatin induces apoptosis of auditory hair cells and cochlear cell lines via activation of initiator caspase-9 and its effector caspase-3. Cisplatin-induced activation of caspase-9 and caspase-3 was also seen in HEI/OC1 (Devarajan et al., 2002; Chung et al., 2008) cells and UB/OC1 cells (Borse et al., 2017). Wang et al. (2004) reported the activation of caspase-9 and caspase-3, but not caspase-8, in cochlear hair cells of pigmented guinea pigs treated with cisplatin. Furthermore, intracochlear perfusion of specific inhibitors of caspase-9 and caspase-3 protected against cisplatin-induced hair cell death and hearing loss in these animals (Wang et al., 2004). In another study, treatment of neonatal rat organ of Corti explants with caspase-1- and caspase-3-specific inhibitors and a general caspase inhibitor protected more than 80% of the auditory hair cells from cisplatin-induced apoptosis (Liu et al., 1998). Unlike other caspases, caspase-1 is not linked with the induction of apoptosis in cells. Caspase-1 is studied for its role in initiating inflammatory immune responses through formation of inflammasomes. Activation of caspase-1 was recently reported to induce hearing loss after cytomegalovirus (CMV) infection in the inner ear through upregulation of its downstream inflammatory factors, such as interleukin-1β and interleukin-18 (Shi et al., 2015). Therefore, it is possible that caspase-1-specific inhibitor may protect from cisplatin ototoxicity by inhibiting cisplatin-induced inflammation in the cochlea.
Several mechanisms underlying cisplatin-induced caspase activation has been proposed. For example, cisplatin-induced apoptosis and caspase-3 activation in HEI/OC1 cells was reduced after treatment with NF-κB inhibitors, Bay 11-7085 and SN-50 (Chung et al., 2008). This finding indicates that NF-κB is activated by cisplatin and plays a pro-apoptotic role in HEI/OC1 cell death. Cisplatin-induced apoptosis and activation of caspase-3, -8, and -9 were also inhibited by activation of CB2 receptors (Jeong et al., 2007), which are present in the cochlear cells and upregulated by cisplatin (Martin-Saldana et al., 2016). Cisplatin-mediated expression of cleaved-caspase-3 in UB/CO1 cells was inhibited by treatment with EGCG through inhibition of STAT1 (Borse et al., 2017). Taken together, loss of auditory cells as a result of cisplatin-induced apoptosis involves activation of caspases as a final common pathway activated by different upstream signaling pathways described above. Inhibition of one or more of these signaling pathways could effectively rescue hair cell loss and restore hearing.
p53
The general mechanism of action of cisplatin in tumor cells is that it forms crosslinks with the purine bases of the DNA, causing DNA damage. The overwhelming DNA damage caused by formation of DNA adducts interferes with the DNA replication and repair mechanisms, which subsequently induces apoptosis (Dasari and Tchounwou, 2014). The apoptotic signal is initiated by activation (through phosphorylation) of p53, a tumor suppressor gene and an important mediator of DNA damage-induced apoptosis. Accumulating evidence suggest that in response to stress signal, a fraction of activated p53 rapidly translocates to the mitochondria, where it interacts with the various pro- and anti-apoptotic members of Bcl-2 family to either activate or inhibit them, respectively (Vaseva and Moll, 2009). In the mitochondria, p53 also induces loss of mitochondrial membrane potential, cytochrome c release and caspase-3 activation, triggering apoptosis (Marchenko et al., 2000). These findings were consistent with those in p53-deficient cells, where Bax translocation, cytochrome c release, and caspase-3 activation were downregulated, confirming that p53 acts upstream of mitochondrial apoptotic regulators (Morris et al., 2001). Interestingly, overexpression of anti-apoptotic Bcl-2 or Bcl-xL prevented stress signal-mediated mitochondrial accumulation of p53 and apoptosis, suggesting a feedback loop between p53 and mitochondrial apoptotic regulators (Marchenko et al., 2000).
The role of p53 in the regulation of cisplatin-induced apoptosis in the auditory system has been investigated. Application of the p53 inhibitor, pifithrin-α, to cochlear organotypic cultures exposed to cisplatin attenuated hair cell damage (Zhang et al., 2003; Benkafadar et al., 2017). The protection was associated with reduction in the expression of p53, caspase-1, and caspase-3 (Zhang et al., 2003). The validity of these in vitro results was tested in hair cells derived from p53-deficient mice, which exhibited resistance to cisplatin-induced apoptosis (Benkafadar et al., 2017) and reduced caspase-3 activation (Cheng et al., 2005a,b). Although systemic inhibition of p53 protects against cisplatin-induced ototoxicity, this treatment strategy would interfere with the anti-cancer efficacy of cisplatin, restricting its use for treatment in humans. To overcome this problem, Benkafadar et al. (2017) demonstrated that intra-tympanic application of pifithrin-α protects auditory function without compromising the chemotherapeutic efficacy of systemically administered cisplatin. They further showed that systemic administration of pifithrin-α even sensitizes TP53-mutant tumors to cisplatin. These results illustrate the role of p53 as an important regulator of cisplatin-induced apoptosis in auditory hair cells.
Cisplatin Targets DNA in the Cochlea
Previous studies have shown that platinated DNA accumulates in the nuclei of outer hair cells, supporting cells, marginal cells of the stria vascularis and cells in the spiral ligament (van Ruijven et al., 2005). Other studies have also shown DNA adduct in spiral ganglion neurons following cisplatin administration. The cochlear cells express DNA repair enzymes which can reduce the level of DNA adduct formed over time. These repair enzymes, known as nucleotide excision repair (NER) enzymes, are classified as transcriptional-coupled repair (TCR) enzymes or global DNA repair (GDR) enzymes which differ in their targets. Defective TCR function is observed in Cockayne syndrome which is characterized by progressive hearing loss. Outer hair cells from Cockayne syndrome group A (Csa-/-) and group B (Csb-/-) mice were hypersensitive to cisplatin. In contrast, Xpc-/- mice which were deficient in global genome repair enzymes showed normal sensitivity to cisplatin (Rainey et al., 2016). This study implicates DNA damage as one mechanism of cisplatin-induced loss of outer hair cells and hearing loss and suggests that TCR plays a primary role in protecting hair cells from cisplatin-induced hearing loss. The effects of eight single-nucleotide polymorphisms in excision repair cross-complementing group 1, 2, 4, and 5 (ERCC1, ERCC2, ERCC4, and ERCC5) genes and xeroderma pigmentosum complementary group C and A (XPC and XPA) genes were assessed in patients with osteosarcoma who were treated with cisplatin. These studies showed that polymorphisms in DNA repair gene, XPC, was associated with increased cisplatin-induced ototoxicity in cancer patients. Increased ototoxicity was associated with the CC genotype of XPC Lys939Gln (Caronia et al., 2009).
Uptake of Cisplatin Into the Cochlea
One of the major entry ports for cisplatin in the cochlea is the mammalian copper transport 1 (Ctr1). Ctr1 is highly expressed in the cochlea where it is localized to outer hair cells, inner hair cells, stria vascularis, and spiral ganglion neurons and contributes to drug entry and cell apoptosis. Decreasing cisplatin entry by intra-tympanic administration of copper sulfate, a substrate of Ctr1, protects against hearing loss induced by cisplatin (More et al., 2010). Cisplatin entry into cochlear cells is also mediated by organic cation transporter (OCT). Three isoforms of this protein exists, OCT1-3, which are present mainly in the kidneys and liver. Expression of OCT2 has also been detected in the organ of Corti and stria vascularis (Ciarimboli et al., 2010). Inhibition of these transporters with cimetidine protects against cisplatin-induced nephrotoxicity and ototoxicity. OCT knockout mice exhibit reduced toxicity to cisplatin. Single-nucleotide polymorphism in OCT-2 gene protects against ototoxicity in children (Lanvers-Kaminsky et al., 2015). Several studies have reported that the entry of aminoglycosides into cochlear hair cells is mediated by mechanotransducer (MET) channels (Gale et al., 2001; Marcotti et al., 2005; Dai et al., 2006; Wang and Steyger, 2009; Alharazneh et al., 2011). In fact, changes in the structure of aminoglycosides which limits their entry through MET channels are less ototoxic (Huth et al., 2015). Cisplatin has also shown to block MET channels in chick cochlear hair cells (Kimitsuki et al., 1993). Recent studies have indicated that cisplatin-induced damage to hair cells in the zebrafish is dependent on functional MET channels (Thomas et al., 2013). These investigators showed that inhibition of mechanotransduction channels by quinine or EGTA protected against cisplatin-induced hair cell death. Furthermore, these investigators showed that zebrafish mutants which lacked mechanotransduction channels were also resistant to cisplatin-induced hair cell death. These studies suggest that mechanotransduction channels are a major contributor to the entry of cisplatin into hair cells, at least in the zebrafish. Interestingly, chemical inhibition of Ctr1 and OCT-2 did not provide significant protection against killing of lateral line hair cells in zebrafish (Thomas et al., 2013). Preliminary data from our lab indicate a similar pathway of entry of transplatin, the inactive isomer of cisplatin, which protects against cisplatin ototoxicity (Dhukhwa et al., 2017). These studies suggest that MET channels could serve as an additional entry ports for cisplatin into cochlear hair cells. Several other ion channels might also contribute to aminoglycoside uptake into hair cells. In addition to TRPV1, several TRP channels including TRPV4, TRPA1, TRPC3, and TRPML3 are expressed in the cochlea (Cuajungco et al., 2007; Asai et al., 2010). These channels have been shown to allow entry of aminoglycoside into kidney cells (Myrdal and Steyger, 2005). The potential role of these channels in mediating the entry of aminoglycosides and cisplatin into hair cells has not yet been established.
Megalin is a low-density lipoprotein which is highly expressed in the kidney and in the stria vascularis of the cochlea which accumulate high levels of platinum DNA adducts. Patients who showed cisplatin-induced hearing impairment demonstrate a higher frequency of megalin gene polymorphism compared to those with no hearing loss after cisplatin therapy. These findings implicate megalin gene polymorphisms in susceptibility to cisplatin ototoxicity (Riedemann et al., 2008). In the kidneys, megalin has been shown to bind β2-microglobulin, cytochrome c, retinal binding proteins, and polybasic antibiotics (such as gentamicin). These investigators show that the accumulation of cisplatin in the renal proximal tubules is mediated by binding of cisplatin-metallothionein complex to megalin (Klassen et al., 2004). The expression of megalin in the stria vascularis might similarly allow the accumulation of cisplatin into the stria vascularis.
Conclusion
Studies described above have identified a number of different mechanisms which mediated cisplatin ototoxicity and provide the basis for rational drug design to treat this debilitating condition. While most of the drugs which target these mechanisms have shown promise, efficacy studies are still in the experimental animal stage (see Table 1 for list of drugs/drug targets). These studies need to be extended in human clinical trials for final validation. The routes of drug administration would be a major issue, as systemic administration of these drugs could compromise cisplatin chemotherapeutic efficacy. In this regard, the use of a number of cisplatin transporters (namely Ctr1 and OCT2) and antioxidants would have the greatest likelihood of interfering with cisplatin antitumor efficacy (as discussed above). Localized delivery of these drugs into the cochlea would eliminate potential systemic toxicities but would require an additional minor surgical procedure to accomplish this goal. This would allow the use of a majority of the agents listed in this table for protection against hearing loss. Several of these drugs are currently in clinical use or in clinical trials for other indications. These include TNF-α antagonists which are used for the treatment of chronic inflammatory diseases (Aggarwal et al., 2012) and EGCG which is in various clinical trials for the treatment of cancer (Singh et al., 2011). These agents which are effective systemically could serve as initial candidates for treating cisplatin-induced hearing loss and other forms of hearing loss. Other drugs which could be advanced quickly into clinical use are MET channel blockers, such as bulky aminoglycoside antibiotics, which demonstrate low potential for ototoxicity (Huth et al., 2015). Continued research in this area would uncover new mechanisms underlying cisplatin-induced hearing loss and validate novel targets and drugs to treat this condition.
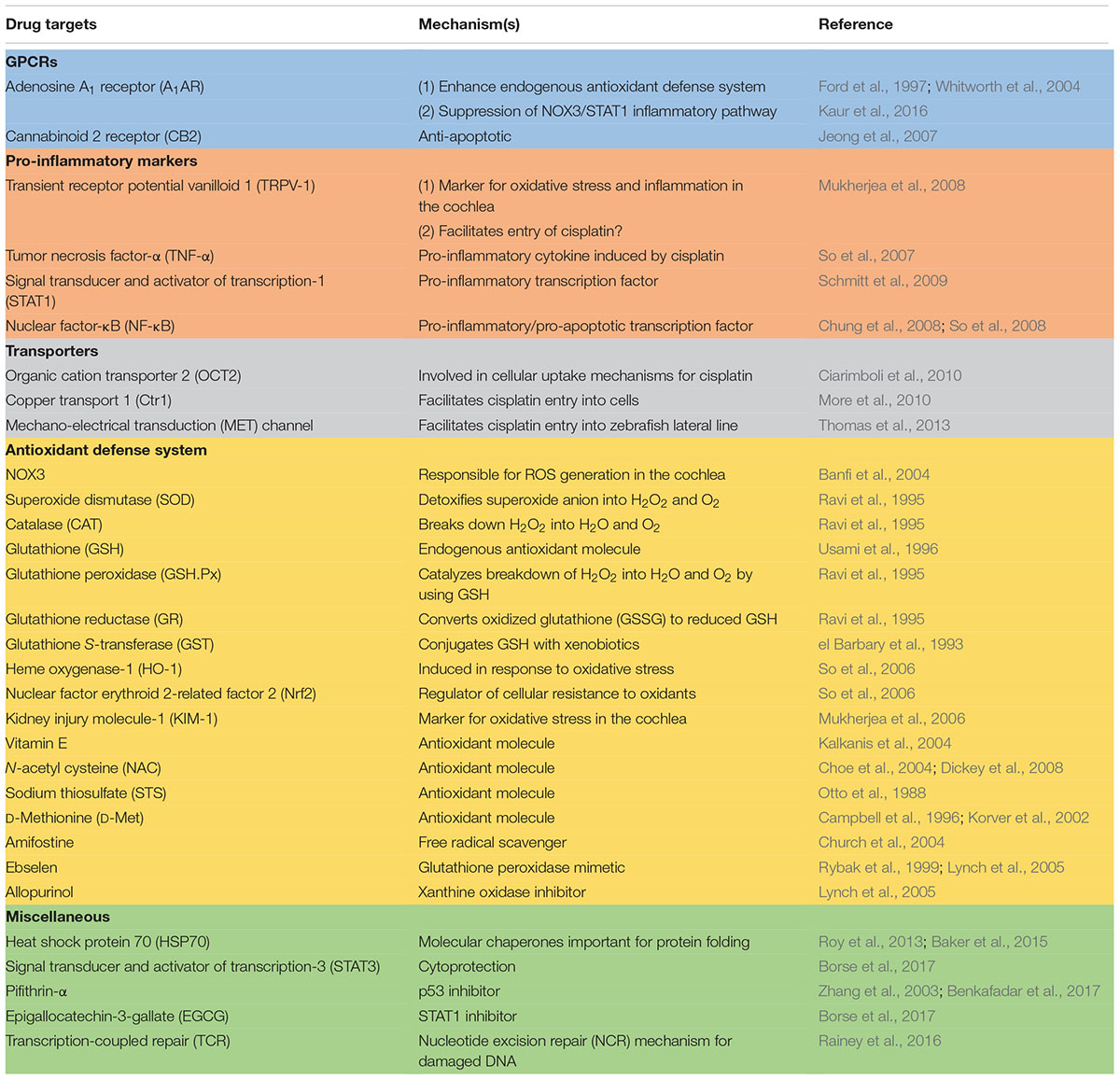
TABLE 1. Potential drug targets for treatment of cisplatin ototoxicity.
Author Contributions
SS and VR conceived and together outlined this review. SS and VR wrote the manuscript. DM and LR critiqued and revised the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to acknowledge the NIH grant support (NCI RO1 CA166907, NIDCD RO1-DC 002396 and RO3 DC011621) for work described in the review which were performed in the authors laboratories.
References
Aggarwal, B. B., Gupta, S. C., and Kim, J. H. (2012). Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey. Blood 119, 651–665. doi: 10.1182/blood-2011-04-325225
Alam, S. A., Ikeda, K., Oshima, T., Suzuki, M., Kawase, T., Kikuchi, T., et al. (2000). Cisplatin-induced apoptotic cell death in Mongolian gerbil cochlea. Hear. Res. 141, 28–38. doi: 10.1016/S0378-5955(99)00211-7
Alharazneh, A., Luk, L., Huth, M., Monfared, A., Steyger, P. S., Cheng, A. G., et al. (2011). Functional hair cell mechanotransducer channels are required for aminoglycoside ototoxicity. PLOS ONE 6:e22347. doi: 10.1371/journal.pone.0022347
Asai, Y., Holt, J. R., and Geleoc, G. S. (2010). A quantitative analysis of the spatiotemporal pattern of transient receptor potential gene expression in the developing mouse cochlea. J. Assoc. Res. Otolaryngol. 11, 27–37. doi: 10.1007/s10162-009-0193-8
Baker, T. G., Roy, S., Brandon, C. S., Kramarenko, I. K., Francis, S. P., Taleb, M., et al. (2015). Heat shock protein-mediated protection against Cisplatin-induced hair cell death. J. Assoc. Res. Otolaryngol. 16, 67–80. doi: 10.1007/s10162-014-0491-7
Banfi, B., Malgrange, B., Knisz, J., Steger, K., Dubois-Dauphin, M., and Krause, K. H. (2004). NOX3, a superoxide-generating NADPH oxidase of the inner ear. J. Biol. Chem. 279, 46065–46072. doi: 10.1074/jbc.M403046200
Benkafadar, N., Menardo, J., Bourien, J., Nouvian, R., Francois, F., Decaudin, D., et al. (2017). Reversible p53 inhibition prevents cisplatin ototoxicity without blocking chemotherapeutic efficacy. EMBO Mol. Med. 9, 7–26. doi: 10.15252/emmm.201606230
Borse, V., Al Aameri, R. F. H., Sheehan, K., Sheth, S., Kaur, T., Mukherjea, D., et al. (2017). Epigallocatechin-3-gallate, a prototypic chemopreventative agent for protection against cisplatin-based ototoxicity. Cell Death Dis. 8:e2921. doi: 10.1038/cddis.2017.314
Campbell, K. C., Meech, R. P., Rybak, L. P., and Hughes, L. F. (2003). The effect of D-methionine on cochlear oxidative state with and without cisplatin administration: mechanisms of otoprotection. J. Am. Acad. Audiol. 14, 144–156.
Campbell, K. C., Rybak, L. P., Meech, R. P., and Hughes, L. (1996). D-methionine provides excellent protection from cisplatin ototoxicity in the rat. Hear. Res. 102, 90–98. doi: 10.1016/S0378-5955(96)00152-9
Caronia, D., Patino-Garcia, A., Milne, R. L., Zalacain-Diez, M., Pita, G., Alonso, M. R., et al. (2009). Common variations in ERCC2 are associated with response to cisplatin chemotherapy and clinical outcome in osteosarcoma patients. Pharmacogenomics J. 9, 347–353. doi: 10.1038/tpj.2009.19
Cheng, A. G., Cunningham, L. L., and Rubel, E. W. (2005a). Mechanisms of hair cell death and protection. Curr. Opin. Otolaryngol. Head Neck Surg. 13, 343–348.
Cheng, A. G., Morrison, R. S., and Rubel, E. W. (2005b). p53 Mediates Cisplatin-Induced Hair Cell Death In Vitro. Mt Royal, NJ: Association for Research in Otolaryngology, 211.
Choe, W. T., Chinosornvatana, N., and Chang, K. W. (2004). Prevention of cisplatin ototoxicity using transtympanic N-acetylcysteine and lactate. Otol. Neurotol. 25, 910–915. doi: 10.1097/00129492-200411000-00009
Chung, W. H., Boo, S. H., Chung, M. K., Lee, H. S., Cho, Y. S., and Hong, S. H. (2008). Proapoptotic effects of NF-kappaB on cisplatin-induced cell death in auditory cell line. Acta Otolaryngol. 128, 1063–1070. doi: 10.1080/00016480701881811
Church, M. W., Blakley, B. W., Burgio, D. L., and Gupta, A. K. (2004). WR-2721 (Amifostine) ameliorates cisplatin-induced hearing loss but causes neurotoxicity in hamsters: dose-dependent effects. J. Assoc. Res. Otolaryngol. 5, 227–237. doi: 10.1007/s10162-004-4011-z
Church, M. W., Kaltenbach, J. A., Blakley, B. W., and Burgio, D. L. (1995). The comparative effects of sodium thiosulfate, diethyldithiocarbamate, fosfomycin and WR-2721 on ameliorating cisplatin-induced ototoxicity. Hear. Res. 86, 195–203. doi: 10.1016/0378-5955(95)00066-D
Ciarimboli, G., Deuster, D., Knief, A., Sperling, M., Holtkamp, M., Edemir, B., et al. (2010). Organic cation transporter 2 mediates cisplatin-induced oto- and nephrotoxicity and is a target for protective interventions. Am. J. Pathol. 176, 1169–1180. doi: 10.2353/ajpath.2010.090610
Clerici, W. J., DiMartino, D. L., and Prasad, M. R. (1995). Direct effects of reactive oxygen species on cochlear outer hair cell shape in vitro. Hear. Res. 84, 30–40. doi: 10.1016/0378-5955(95)00010-2
Clerici, W. J., and Yang, L. (1996). Direct effects of intraperilymphatic reactive oxygen species generation on cochlear function. Hear. Res. 101, 14–22. doi: 10.1016/S0378-5955(96)00126-8
Coradini, P. P., Cigana, L., Selistre, S. G., Rosito, L. S., and Brunetto, A. L. (2007). Ototoxicity from cisplatin therapy in childhood cancer. J. Pediatr. Hematol. Oncol. 29, 355–360. doi: 10.1097/MPH.0b013e318059c220
Cuajungco, M. P., Grimm, C., and Heller, S. (2007). TRP channels as candidates for hearing and balance abnormalities in vertebrates. Biochim. Biophys. Acta 1772, 1022–1027. doi: 10.1016/j.bbadis.2007.01.002
Dai, C. F., Mangiardi, D., Cotanche, D. A., and Steyger, P. S. (2006). Uptake of fluorescent gentamicin by vertebrate sensory cells in vivo. Hear. Res. 213, 64–78. doi: 10.1016/j.heares.2005.11.011
Dasari, S., and Tchounwou, P. B. (2014). Cisplatin in cancer therapy: molecular mechanisms of action. Eur. J. Pharmacol. 740, 364–378. doi: 10.1016/j.ejphar.2014.07.025
Devarajan, P., Savoca, M., Castaneda, M. P., Park, M. S., Esteban-Cruciani, N., Kalinec, G., et al. (2002). Cisplatin-induced apoptosis in auditory cells: role of death receptor and mitochondrial pathways. Hear. Res. 174, 45–54. doi: 10.1016/S0378-5955(02)00634-2
Dhukhwa, A., Mukherjea, D., Ghosh, S., Sheehan, K., Borse, V., Sheth, S., et al. (2017). Transplatin, A Novel Treatment for Cisplatin Ototoxicity. Mt Royal, NJ: Association for Research in Otolaryngology, 402
Dickey, D. T., Muldoon, L. L., Doolittle, N. D., Peterson, D. R., Kraemer, D. F., and Neuwelt, E. A. (2008). Effect of N-acetylcysteine route of administration on chemoprotection against cisplatin-induced toxicity in rat models. Cancer Chemother. Pharmacol. 62, 235–241. doi: 10.1007/s00280-007-0597-2
Ekborn, A., Laurell, G., Johnstrom, P., Wallin, I., Eksborg, S., and Ehrsson, H. (2002). D-Methionine and cisplatin ototoxicity in the guinea pig: D-methionine influences cisplatin pharmacokinetics. Hear. Res. 165, 53–61. doi: 10.1016/S0378-5955(02)00277-0
el Barbary, A., Altschuler, R. A., and Schacht, J. (1993). Glutathione S-transferases in the organ of Corti of the rat: enzymatic activity, subunit composition and immunohistochemical localization. Hear. Res. 71, 80–90. doi: 10.1016/0378-5955(93)90023-T
Ford, M. S., Maggirwar, S. B., Rybak, L. P., Whitworth, C., and Ramkumar, V. (1997). Expression and function of adenosine receptors in the chinchilla cochlea. Hear. Res. 105, 130–140. doi: 10.1016/S0378-5955(96)00204-3
Fouladi, M., Chintagumpala, M., Ashley, D., Kellie, S., Gururangan, S., Hassall, T., et al. (2008). Amifostine protects against cisplatin-induced ototoxicity in children with average-risk medulloblastoma. J. Clin. Oncol. 26, 3749–3755. doi: 10.1200/JCO.2007.14.3974
Gale, J. E., Marcotti, W., Kennedy, H. J., Kros, C. J., and Richardson, G. P. (2001). FM1-43 dye behaves as a permeant blocker of the hair-cell mechanotransducer channel. J. Neurosci. 21, 7013–7025.
Ghosh, S., Mukherjea, D., Sheehan, K., Tupal, S., Borse, V., Dhukhwa, A., et al. (2016). Expression, Regulation and Function of Cannabinoid Receptor 2 in the Cochlea. Mt Royal, NJ: Association for Research in Otolaryngology, 52.
Goel, R., Cleary, S. M., Horton, C., Kirmani, S., Abramson, I., Kelly, C., et al. (1989). Effect of sodium thiosulfate on the pharmacokinetics and toxicity of cisplatin. J. Natl. Cancer Inst. 81, 1552–1560. doi: 10.1093/jnci/81.20.1552
Gunewardene, N., Guo, C. X., Wong, A. C. Y., Thorne, P. R., and Vlajkovic, S. M. (2013). Adenosine amine congener ameliorates cisplatin-induced hearing loss. World J. Otorhinolaryngol. 3, 100–107. doi: 10.5319/wjo.v3.i3.100
Gurney, J. G., Tersak, J. M., Ness, K. K., Landier, W., Matthay, K. K., Schmidt, M. L., et al. (2007). Hearing loss, quality of life, and academic problems in long-term neuroblastoma survivors: a report from the Children’s Oncology Group. Pediatrics 120, e1229–e1236. doi: 10.1542/peds.2007-0178
Huth, M. E., Han, K. H., Sotoudeh, K., Hsieh, Y. J., Effertz, T., Vu, A. A., et al. (2015). Designer aminoglycosides prevent cochlear hair cell loss and hearing loss. J. Clin. Invest. 125, 583–592. doi: 10.1172/JCI77424
Jeong, H. J., Kim, S. J., Moon, P. D., Kim, N. H., Kim, J. S., Park, R. K., et al. (2007). Antiapoptotic mechanism of cannabinoid receptor 2 agonist on cisplatin-induced apoptosis in the HEI-OC1 auditory cell line. J. Neurosci. Res. 85, 896–905. doi: 10.1002/jnr.21168
Kalkanis, J. G., Whitworth, C., and Rybak, L. P. (2004). Vitamin E reduces cisplatin ototoxicity. Laryngoscope 114, 538–542. doi: 10.1097/00005537-200403000-00028
Kaur, T., Borse, V., Sheth, S., Sheehan, K., Ghosh, S., Tupal, S., et al. (2016). Adenosine A1 receptor protects against cisplatin ototoxicity by suppressing the NOX3/STAT1 inflammatory pathway in the cochlea. J. Neurosci. 36, 3962–3977. doi: 10.1523/JNEUROSCI.3111-15.2016
Kaur, T., Mukherjea, D., Sheehan, K., Jajoo, S., Rybak, L. P., and Ramkumar, V. (2011). Short interfering RNA against STAT1 attenuates cisplatin-induced ototoxicity in the rat by suppressing inflammation. Cell Death Dis. 2:e180. doi: 10.1038/cddis.2011.63
Kimitsuki, T., Nakagawa, T., Hisashi, K., Komune, S., and Komiyama, S. (1993). Cisplatin blocks mechano-electric transducer current in chick cochlear hair cells. Hear. Res. 71, 64–68. doi: 10.1016/0378-5955(93)90021-R
Klassen, R. B., Crenshaw, K., Kozyraki, R., Verroust, P. J., Tio, L., Atrian, S., et al. (2004). Megalin mediates renal uptake of heavy metal metallothionein complexes. Am. J. Physiol. Renal Physiol. 287, F393–F403. doi: 10.1152/ajprenal.00233.2003
Knight, K. R., Kraemer, D. F., and Neuwelt, E. A. (2005). Ototoxicity in children receiving platinum chemotherapy: underestimating a commonly occurring toxicity that may influence academic and social development. J. Clin. Oncol. 23, 8588–8596. doi: 10.1200/JCO.2004.00.5355
Kopke, R., Allen, K. A., Henderson, D., Hoffer, M., Frenz, D., and Van de Water, T. (1999). A radical demise. toxins and trauma share common pathways in hair cell death. Ann. N. Y. Acad. Sci. 884, 171–191. doi: 10.1111/j.1749-6632.1999.tb08641.x
Kopke, R. D., Liu, W., Gabaizadeh, R., Jacono, A., Feghali, J., Spray, D., et al. (1997). Use of organotypic cultures of Corti’s organ to study the protective effects of antioxidant molecules on cisplatin-induced damage of auditory hair cells. Am. J. Otol. 18, 559–571.
Korver, K. D., Rybak, L. P., Whitworth, C., and Campbell, K. M. (2002). Round window application of D-methionine provides complete cisplatin otoprotection. Otolaryngol. Head Neck Surg. 126, 683–689. doi: 10.1067/mhn.2002.125299
Kushner, B. H., Budnick, A., Kramer, K., Modak, S., and Cheung, N. K. (2006). Ototoxicity from high-dose use of platinum compounds in patients with neuroblastoma. Cancer 107, 417–422. doi: 10.1002/cncr.22004
Lai, M. T., Ohmichi, T., Egusa, K., Okada, S., and Masuda, Y. (1996). Immunohistochemical localization of manganese superoxide dismutase in the rat cochlea. Eur. Arch. Otorhinolaryngol. 253, 273–277. doi: 10.1007/BF00171142
Lanvers-Kaminsky, C., Sprowl, J. A., Malath, I., Deuster, D., Eveslage, M., Schlatter, E., et al. (2015). Human OCT2 variant c.808G > T confers protection effect against cisplatin-induced ototoxicity. Pharmacogenomics 16, 323–332. doi: 10.2217/pgs.14.182
Liu, W., Staecker, H., Stupak, H., Malgrange, B., Lefebvre, P., and Van De Water, T. R. (1998). Caspase inhibitors prevent cisplatin-induced apoptosis of auditory sensory cells. Neuroreport 9, 2609–2614. doi: 10.1097/00001756-199808030-00034
Lynch, E. D., Gu, R., Pierce, C., and Kil, J. (2005). Reduction of acute cisplatin ototoxicity and nephrotoxicity in rats by oral administration of allopurinol and ebselen. Hear. Res. 201, 81–89. doi: 10.1016/j.heares.2004.08.002
Marchenko, N. D., Zaika, A., and Moll, U. M. (2000). Death signal-induced localization of p53 protein to mitochondria. A potential role in apoptotic signaling. J. Biol. Chem. 275, 16202–16212. doi: 10.1074/jbc.275.21.16202
Marcotti, W., van Netten, S. M., and Kros, C. J. (2005). The aminoglycoside antibiotic dihydrostreptomycin rapidly enters mouse outer hair cells through the mechano-electrical transducer channels. J. Physiol. 567(Pt 2), 505–521. doi: 10.1113/jphysiol.2005.085951
Marina, N., Chang, K. W., Malogolowkin, M., London, W. B., Frazier, A. L., Womer, R. B., et al. (2005). Amifostine does not protect against the ototoxicity of high-dose cisplatin combined with etoposide and bleomycin in pediatric germ-cell tumors: a Children’s Oncology Group study. Cancer 104, 841–847. doi: 10.1002/cncr.21218
Martin-Saldana, S., Trinidad, A., Ramil, E., Sanchez-Lopez, A. J., Coronado, M. J., Martinez-Martinez, E., et al. (2016). Spontaneous cannabinoid receptor 2 (CB2) expression in the cochlea of adult albino rat and its up-regulation after cisplatin treatment. PLOS ONE 11:e0161954. doi: 10.1371/journal.pone.0161954
Meyers, J. R., MacDonald, R. B., Duggan, A., Lenzi, D., Standaert, D. G., Corwin, J. T., et al. (2003). Lighting up the senses: FM1-43 loading of sensory cells through nonselective ion channels. J. Neurosci. 23, 4054–4065.
More, S. S., Akil, O., Ianculescu, A. G., Geier, E. G., Lustig, L. R., and Giacomini, K. M. (2010). Role of the copper transporter, CTR1, in platinum-induced ototoxicity. J. Neurosci. 30, 9500–9509. doi: 10.1523/JNEUROSCI.1544-10.2010
Morris, E. J., Keramaris, E., Rideout, H. J., Slack, R. S., Dyson, N. J., Stefanis, L., et al. (2001). Cyclin-dependent kinases and P53 pathways are activated independently and mediate Bax activation in neurons after DNA damage. J. Neurosci. 21, 5017–5026.
Mukherjea, D., Jajoo, S., Kaur, T., Sheehan, K. E., Ramkumar, V., and Rybak, L. P. (2010). Transtympanic administration of short interfering (si)RNA for the NOX3 isoform of NADPH oxidase protects against cisplatin-induced hearing loss in the rat. Antioxid. Redox Signal. 13, 589–598. doi: 10.1089/ars.2010.3110
Mukherjea, D., Jajoo, S., Sheehan, K., Kaur, T., Sheth, S., Bunch, J., et al. (2011). NOX3 NADPH oxidase couples transient receptor potential vanilloid 1 to signal transducer and activator of transcription 1-mediated inflammation and hearing loss. Antioxid. Redox Signal. 14, 999–1010. doi: 10.1089/ars.2010.3497
Mukherjea, D., Jajoo, S., Whitworth, C., Bunch, J. R., Turner, J. G., Rybak, L. P., et al. (2008). Short interfering RNA against transient receptor potential vanilloid 1 attenuates cisplatin-induced hearing loss in the rat. J. Neurosci. 28, 13056–13065. doi: 10.1523/JNEUROSCI.1307-08.2008
Mukherjea, D., Whitworth, C. A., Nandish, S., Dunaway, G. A., Rybak, L. P., and Ramkumar, V. (2006). Expression of the kidney injury molecule 1 in the rat cochlea and induction by cisplatin. Neuroscience 139, 733–740. doi: 10.1016/j.neuroscience.2005.12.044
Myrdal, S. E., and Steyger, P. S. (2005). TRPV1 regulators mediate gentamicin penetration of cultured kidney cells. Hear. Res. 204, 170–182. doi: 10.1016/j.heares.2005.02.005
Oh, S. H., Yu, W. S., Song, B. H., Lim, D., Koo, J. W., Chang, S. O., et al. (2000). Expression of heat shock protein 72 in rat cochlea with cisplatin-induced acute ototoxicity. Acta Otolaryngol. 120, 146–150. doi: 10.1080/000164800750000793
Oldenburg, J., Kraggerud, S. M., Cvancarova, M., Lothe, R. A., and Fossa, S. D. (2007). Cisplatin-induced long-term hearing impairment is associated with specific glutathione s-transferase genotypes in testicular cancer survivors. J. Clin. Oncol. 25, 708–714. doi: 10.1200/JCO.2006.08.9599
Otto, W. C., Brown, R. D., Gage-White, L., Kupetz, S., Anniko, M., Penny, J. E., et al. (1988). Effects of cisplatin and thiosulfate upon auditory brainstem responses of guinea pigs. Hear. Res. 35, 79–85. doi: 10.1016/0378-5955(88)90042-1
Pierson, M. G., and Gray, B. H. (1982). Superoxide dismutase activity in the cochlea. Hear. Res. 6, 141–151. doi: 10.1016/0378-5955(82)90050-8
Rainey, R. N., Ng, S. Y., Llamas, J., van der Horst, G. T., and Segil, N. (2016). Mutations in cockayne syndrome-associated genes (Csa and Csb) predispose to cisplatin-induced hearing loss in mice. J. Neurosci. 36, 4758–4770. doi: 10.1523/JNEUROSCI.3890-15.2016
Ravi, R., Somani, S. M., and Rybak, L. P. (1995). Mechanism of cisplatin ototoxicity: antioxidant system. Pharmacol. Toxicol. 76, 386–394. doi: 10.1111/j.1600-0773.1995.tb00167.x
Riedemann, L., Lanvers, C., Deuster, D., Peters, U., Boos, J., Jurgens, H., et al. (2008). Megalin genetic polymorphisms and individual sensitivity to the ototoxic effect of cisplatin. Pharmacogenomics J. 8, 23–28. doi: 10.1038/sj.tpj.6500455
Roy, S., Ryals, M. M., Van den Bruele, A. B., Fitzgerald, T. S., and Cunningham, L. L. (2013). Sound preconditioning therapy inhibits ototoxic hearing loss in mice. J. Clin. Invest. 123, 4945–4949. doi: 10.1172/JCI71353
Ryan, A. F. (1988). “Circulation of the inner ear. II. The relationship between metabolism and blood flow in the cochlea,” in Physiology of the Ear, eds A. F. Jahn and J. Santos-Sacchi (New York, NY: Raven Press), 317–325.
Rybak, L. P., Husain, K., Morris, C., Whitworth, C., and Somani, S. (2000). Effect of protective agents against cisplatin ototoxicity. Am. J. Otol. 21, 513–520.
Rybak, L. P., Ravi, R., and Somani, S. M. (1995). Mechanism of protection by diethyldithiocarbamate against cisplatin ototoxicity: antioxidant system. Fundam. Appl. Toxicol. 26, 293–300. doi: 10.1006/faat.1995.1100
Rybak, L. P., Whitworth, C., and Somani, S. (1999). Application of antioxidants and other agents to prevent cisplatin ototoxicity. Laryngoscope 109, 1740–1744. doi: 10.1097/00005537-199911000-00003
Salt, A. N., Melichar, I., and Thalmann, R. (1987). Mechanisms of endocochlear potential generation by stria vascularis. Laryngoscope 97(8 Pt 1), 984–991.
Salvesen, G. S., and Dixit, V. M. (1997). Caspases: intracellular signaling by proteolysis. Cell 91, 443–446. doi: 10.1016/S0092-8674(00)80430-4
Sastry, J., and Kellie, S. J. (2005). Severe neurotoxicity, ototoxicity and nephrotoxicity following high-dose cisplatin and amifostine. Pediatr. Hematol. Oncol. 22, 441–445. doi: 10.1080/08880010590964381
Schmitt, N. C., Rubel, E. W., and Nathanson, N. M. (2009). Cisplatin-induced hair cell death requires STAT1 and is attenuated by epigallocatechin gallate. J. Neurosci. 29, 3843–3851. doi: 10.1523/JNEUROSCI.5842-08.2009
Seidman, M. D., Quirk, W. S., Nuttall, A. L., and Schweitzer, V. G. (1991). The protective effects of allopurinol and superoxide dismutase-polyethylene glycol on ischemic and reperfusion-induced cochlear damage. Otolaryngol. Head Neck Surg. 105, 457–463. doi: 10.1177/019459989110500318
Shi, X., Dong, Y., Li, Y., Zhao, Z., Li, H., Qiu, S., et al. (2015). Inflammasome activation in mouse inner ear in response to MCMV induced hearing loss. J. Otol. 10, 143–149. doi: 10.1016/j.joto.2015.12.001
Siddiqui, W. A., Ahad, A., and Ahsan, H. (2015). The mystery of BCL2 family: Bcl-2 proteins and apoptosis: an update. Arch. Toxicol. 89, 289–317. doi: 10.1007/s00204-014-1448-7
Singh, B. N., Shankar, S., and Srivastava, R. K. (2011). Green tea catechin, epigallocatechin-3-gallate (EGCG): mechanisms, perspectives and clinical applications. Biochem. Pharmacol. 82, 1807–1821. doi: 10.1016/j.bcp.2011.07.093
So, H., Kim, H., Kim, Y., Kim, E., Pae, H. O., Chung, H. T., et al. (2008). Evidence that cisplatin-induced auditory damage is attenuated by downregulation of pro-inflammatory cytokines via Nrf2/HO-1. J. Assoc. Res. Otolaryngol. 9, 290–306. doi: 10.1007/s10162-008-0126-y
So, H., Kim, H., Lee, J. H., Park, C., Kim, Y., Kim, E., et al. (2007). Cisplatin cytotoxicity of auditory cells requires secretions of proinflammatory cytokines via activation of ERK and NF-kappaB. J. Assoc. Res. Otolaryngol. 8, 338–355. doi: 10.1007/s10162-007-0084-9
So, H. S., Kim, H. J., Lee, J. H., Lee, J. H., Park, S. Y., Park, C., et al. (2006). Flunarizine induces Nrf2-mediated transcriptional activation of heme oxygenase-1 in protection of auditory cells from cisplatin. Cell Death Differ. 13, 1763–1775. doi: 10.1038/sj.cdd.4401863
Thomas, A. J., Hailey, D. W., Stawicki, T. M., Wu, P., Coffin, A. B., Rubel, E. W., et al. (2013). Functional mechanotransduction is required for cisplatin-induced hair cell death in the zebrafish lateral line. J. Neurosci. 33, 4405–4414. doi: 10.1523/JNEUROSCI.3940-12.2013
Usami, S., Hjelle, O. P., and Ottersen, O. P. (1996). Differential cellular distribution of glutathione–an endogenous antioxidant–in the guinea pig inner ear. Brain Res. 743, 337–340. doi: 10.1016/S0006-8993(96)01090-6
van Ruijven, M. W., de Groot, J. C., Hendriksen, F., and Smoorenburg, G. F. (2005). Immunohistochemical detection of platinated DNA in the cochlea of cisplatin-treated guinea pigs. Hear. Res. 203, 112–121. doi: 10.1016/j.heares.2004.12.007
Vaseva, A. V., and Moll, U. M. (2009). The mitochondrial p53 pathway. Biochim. Biophys. Acta 1787, 414–420. doi: 10.1016/j.bbabio.2008.10.005
Vlajkovic, S. M., Housley, G. D., and Thorne, P. R. (2009). Adenosine and the auditory system. Curr. Neuropharmacol. 7, 246–256. doi: 10.2174/157015909789152155
Wang, J., Ladrech, S., Pujol, R., Brabet, P., Van De Water, T. R., and Puel, J. L. (2004). Caspase inhibitors, but not c-Jun NH2-terminal kinase inhibitor treatment, prevent cisplatin-induced hearing loss. Cancer Res. 64, 9217–9224. doi: 10.1158/0008-5472.CAN-04-1581
Wang, J., Lloyd Faulconbridge, R. V., Fetoni, A., Guitton, M. J., Pujol, R., and Puel, J. L. (2003). Local application of sodium thiosulfate prevents cisplatin-induced hearing loss in the guinea pig. Neuropharmacology 45, 380–393. doi: 10.1016/S0028-3908(03)00194-1
Wang, Q., and Steyger, P. S. (2009). Trafficking of systemic fluorescent gentamicin into the cochlea and hair cells. J. Assoc. Res. Otolaryngol. 10, 205–219. doi: 10.1007/s10162-009-0160-4
Whitworth, C. A., Ramkumar, V., Jones, B., Tsukasaki, N., and Rybak, L. P. (2004). Protection against cisplatin ototoxicity by adenosine agonists. Biochem. Pharmacol. 67, 1801–1807. doi: 10.1016/j.bcp.2004.01.010
Wimmer, C., Mees, K., Stumpf, P., Welsch, U., Reichel, O., and Suckfull, M. (2004). Round window application of D-methionine, sodium thiosulfate, brain-derived neurotrophic factor, and fibroblast growth factor-2 in cisplatin-induced ototoxicity. Otol. Neurotol. 25, 33–40. doi: 10.1097/00129492-200401000-00007
Yao, X., and Rarey, K. E. (1996). Detection and regulation of Cu/Zn-SOD and Mn-SOD in rat cochlear tissues. Hear. Res. 96, 199–203. doi: 10.1016/0378-5955(96)00050-0
Zhang, M., Liu, W., Ding, D., and Salvi, R. (2003). Pifithrin-alpha suppresses p53 and protects cochlear and vestibular hair cells from cisplatin-induced apoptosis. Neuroscience 120, 191–205. doi: 10.1016/S0306-4522(03)00286-0
Zuur, C. L., Simis, Y. J., Lansdaal, P. E., Hart, A. A., Rasch, C. R., Schornagel, J. H., et al. (2007). Risk factors of ototoxicity after cisplatin-based chemo-irradiation in patients with locally advanced head-and-neck cancer: a multivariate analysis. Int. J. Radiat. Oncol. Biol. Phys. 68, 1320–1325. doi: 10.1016/j.ijrobp.2007.01.042
Keywords: cisplatin, ototoxicity, otoprotection, oxidative stress, apoptosis, antioxidants, anti-inflammatory agents
Citation: Sheth S, Mukherjea D, Rybak LP and Ramkumar V (2017) Mechanisms of Cisplatin-Induced Ototoxicity and Otoprotection. Front. Cell. Neurosci. 11:338. doi: 10.3389/fncel.2017.00338
Received: 31 August 2017; Accepted: 12 October 2017;
Published: 27 October 2017.
Edited by:
Lisa Cunningham, National Institutes of Health (NIH), United StatesReviewed by:
Allison B. Coffin, Washington State University, United StatesAndy Groves, Baylor College of Medicine, United States
Evangelia Tserga, Karolinska Institute (KI), Sweden
Copyright © 2017 Sheth, Mukherjea, Rybak and Ramkumar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vickram Ramkumar, dnJhbWt1bWFyQHNpdW1lZC5lZHU=