 Iya Prytkova1,2
Iya Prytkova1,2 Kristen J. Brennand1,2,3,4,5*
Kristen J. Brennand1,2,3,4,5*- 1Department of Neuroscience, Icahn School of Medicine at Mount Sinai, New York, NY, United States
- 2Friedman Brain Institute, Icahn School of Medicine at Mount Sinai, New York, NY, United States
- 3Department of Genetics and Genomics, Icahn School of Medicine at Mount Sinai, New York, NY, United States
- 4Icahn Institute of Genomics and Multiscale Biology, Icahn School of Medicine at Mount Sinai, New York, NY, United States
- 5Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY, United States
Excitatory dopaminergic neurons, inhibitory GABAergic neurons, microglia, and oligodendrocytes have all been implicated in schizophrenia (SZ) network pathology. Still, SZ has been a difficult disorder to study, not only because of the limitations of animal models in capturing the complexity of the human mind, but also because it is greatly polygenic, with high rates of variability across the population. The advent of patient-derived pluripotent stem cells and induced neural and glial cultures has brought hope for modeling the molecular dysfunction underlying SZ pathology in a patient-specific manner. Here I review the successes of the patient-specific induced cultures in generating different cell types for the study of SZ, with special emphasis on the utility of co-culture techniques, both two- and three-dimensional, for modeling network dysfunction in disease.
Introduction
Schizophrenia (SZ) is a common debilitating psychiatric disorder which affects approximately 1% of the world population and accounts for a significant socio-economic burden (Rössler et al., 2005). The disease is characterized by the presence of positive symptoms, such as hallucinations, delusions, and paranoia, and negative symptoms, such as social withdrawal and flattened affect; SZ is also marked by cognitive deficits such as disorganized thoughts and difficulty concentrating (Barnhill, 2013). The symptoms may appear in a variety of constellations. Additionally, individuals with SZ have a high risk of suicide, high prevalence of substance abuse, and a high rate of homelessness (Rössler et al., 2005). Despite being recognized as early as 1896 (Rössler et al., 2005), disease etiology has remained elusive. The rate of heritability has been estimated to be around 80%, but the concordance rate among monozygotic twins is around 50% (Cardno et al., 1999), indicating that genetic contribution alone is insufficient to fully account for disease risk. Moreover, genome wide associated studies (GWAS) by the Schizophrenia Working Group of the Psychiatric Genomics Consortium (2014) have identified over one hundred loci associated with the disease, consistent with the clinical heterogeneity of the disorder.
Network Dysfunction in Schizophrenia
Overwhelming genetic evidence now demonstrates that SZ is a complex genetic disorder (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014) reflecting cumulative risk from more than 100 rare and common variants. Although the genetic and symptomatic heterogeneity of SZ implicates a wide variety of neurotransmitter, neuroimmune, neuroanatomical, and neurodevelopmental processes (reviewed xx), the precise cell type(s) responsible for the cell autonomous defects underlying the earliest disease processes remain unresolved. Historically, the effectiveness of D2-receptor antagonists as anti-psychotics shaped much of the thinking surrounding the neurobiological mechanisms of SZ (Delay et al., 1952; Carlsson et al., 1957); while successful in some cases at alleviating the positive and, to a lesser extent (Kirkpatrick et al., 2001), negative symptoms of SZ, these drugs produce a plethora of side effects which lead to high rates of discontinuation (Lieberman et al., 2005). Glutamatergic signaling via n-methyl-D-aspartate receptor (NMDA-R) has also been repeatedly linked to disordered cognition and sensory processing (for review see Kantrowitz and Javitt, 2012); NMDA-R antagonists, such as phencyclidine and ketamine, mimic some cognitive and behavioral symptoms of SZ (Kehrer et al., 2008). Another possibility is that the loss of NMDA receptors on GABAergic interneurons leads to hyper-activation of cortical forebrain neurons (Olney et al., 1999), thus leading to an overall excitation/inhibition imbalance (Homayoun and Moghaddam, 2007). Since glutamatergic signaling on GABAergic interneurons has an important regulatory function on the activity of dopaminergic neurons, it seems reasonable that genetic variants impacting any (or all) of these cell types could lead to convergent elements of symptomology across genetically heterogeneous patients.
There is a strong neurodevelopmental component to SZ risk (Weinberger, 1995). Most cases of SZ are diagnosed in adolescence or young adulthood (Lewis and Lieberman, 2000), although there are rare cases of childhood onset (Asarnow, 1994). During these stages of development, extensive synaptic pruning and overall loss of gray matter occurs (Huttenlocher and Dabholkar, 1997). Neuroimaging studies have shown a loss of frontal and hippocampal volume in SZ patients; post-mortem tissue analyses have further identified decreased spine density and synaptic connectivity (Glantz and Lewis, 2000; McGlashan and Hoffman, 2000; Faludi and Mirnics, 2011). Microglia and oligodendrocytes have also been implicated in SZ pathology, due to their roles in synaptic pruning (Paolicelli et al., 2011) and cell-to-cell communication (Tkachev et al., 2003), respectively.
The limitations of animal models in re-capitulating human development, the heterogeneity of this complex genetic disorder, and the variable response to pharmacological therapies underscore the necessity for modeling SZ in human cells. Neurons and glia derived from human induced pluripotent stem cells (hiPSCs) offer an opportunity to investigate the complex cellular and molecular interactions involved in SZ. Despite the fairly recent advent of hiPSCs in Takahashi et al. (2007), numerous advances have facilitated improved reprogramming of patient somatic cells into neurons, either directly or via a pluripotent stem cell intermediate. Below I review the available protocols for generating various neuronal cultures, with a focus on the application of each method to modeling cellular dysfunction in SZ. Finally, I address current limitations and future strategies to improve upon this approach.
Patient hiPscs-Derived Brain Cells for Modeling Schizophrenia
Cellular and molecular dysfunction in SZ has traditionally been studied by examining post-mortem patient brain tissue or through animal models. Post-mortem tissue studies are limited by degradation of key biomolecules (i.e., RNA, DNA, proteins, epigenetic marks) due to delays between death and sample processing (Ferrer et al., 2008). On the other hand, animal models carry the benefit of in vivo manipulations at different developmental stages and isogenic controls. However, animal models of SZ rely either on targeted lesions, administration of psychotropic drugs such as phencyclidine, or on manipulating a single gene associated with the disorder (Brennand and Gage, 2011; Flores et al., 2016). These approaches have limited etiological relevance, given the highly polygenic nature of SZ discussed above. By contrast, patient hiPSC-derived neurons and glia offer a unique opportunity to investigate the full genetic landscape contributing to SZ while monitoring neural development (Table 1).
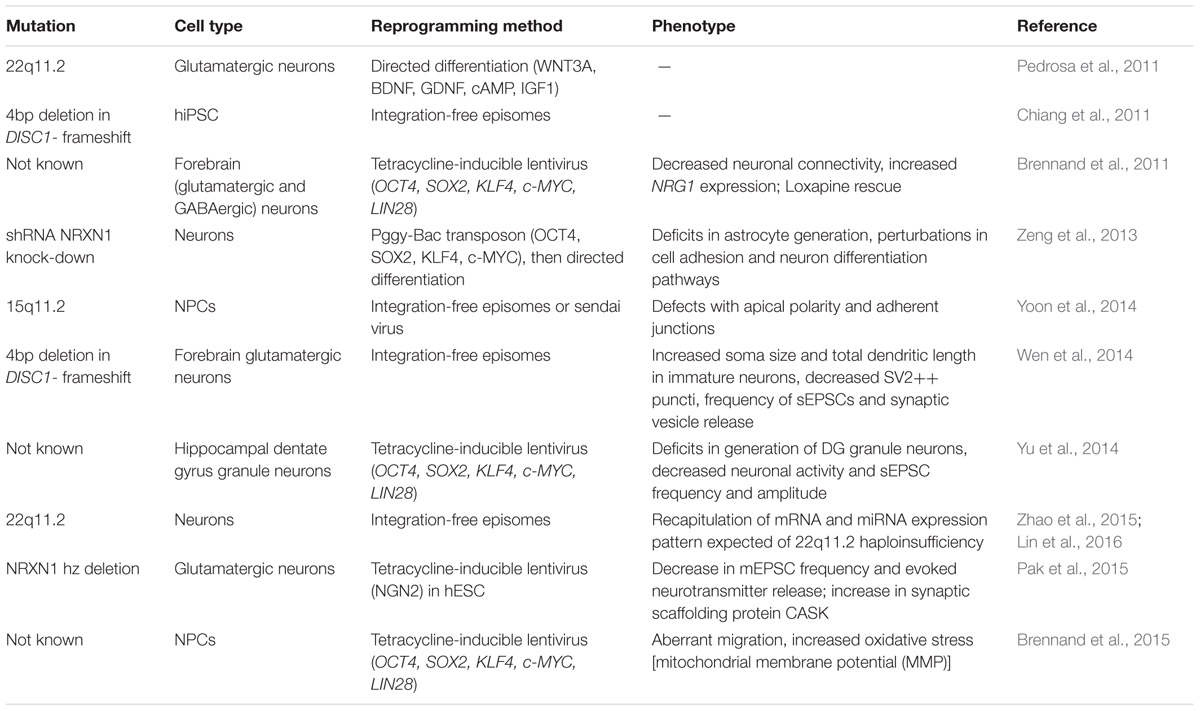
TABLE 1. Recent human induced pluripotent stem cell (hiPSC)-based studies of schizophrenia.
Neural Cultures
Several groups have generated heterogeneous neuronal populations from a variety patient derived hiPSCs, focusing either on genetically defined (Chiang et al., 2011; Pedrosa et al., 2011; Wen et al., 2014) or unknown genetic (Brennand et al., 2011, 2015) patient cohorts. Chiang et al. (2011) were the first to report hiPSCs lines from three SZ patients harboring DISC1 mutations, which have been long implicated in SZ and other neuropsychiatric disorders, including autism and major depression. Although this mutation is known to occur in only two rare family pedigrees, the DISC1 protein is thought to bind many proteins involved in neuronal development and synapse formation (Duan et al., 2007; Porteous et al., 2014), potentially providing broader insights into disease mechanisms. Wen et al. (2014) advanced this work by generating fore-brain like neurons from SZ patients with a DISC1 mutations as well as isogenic hiPSC lines, correcting mutations in one of the patients and introducing frameshift deletions in DISC1 into control hiPSCs. The corrected isogenic lines showed an improvement in synaptic functionality, as evidenced by the increase in synaptic markers (co-localization of SYN1 and PSD95) and increased amplitude and frequency of spontaneous synaptic currents, while the mutated isogenic lines showed a deficit in synaptic activity. The researchers then performed RNA-seq to gain understanding of the affected molecular pathways, and, for the first time, showed differential expression in DISC1-interacting proteins. Moreover, they identified differential regulation of several genes coding for presynaptic proteins (including neurexin 1, synaptophysin, and synaptoporin) and transporters (including MEF2C), implicating mutant DISC1 as a hub of transcriptional regulation (Wen et al., 2014).
Several copy number variations (CNVs) have been associated with increased SZ risk, including 22q11.2, 15q11.2, and NRXN1 loci (for review see St Clair, 2009). HiPSC studies of 22q11.2 deletions, found in approximately 1% of SZ cases (Bassett and Chow, 2008), have identified differential gene expression in pathways regulating cell cycle and development (Lin et al., 2016) as well as impaired neurite outgrowth and cellular migration (Toyoshima et al., 2016). Investigation of patient hiPSCs with a 15q11.2 microdeletion revealed deficits in apical polarity and adherens junctions due to CYFIP1 haploinsufficiency; genetic association analyses on these hiPSCs uncovered an epistatic interaction between a WAVE signaling mediator (involved in cytoskeleton development) and CYFIP1 (Yoon et al., 2014). Finally, NRXN1 expression was reduced in human embryonic stem cells (hESCs) by heterozygous deletion (Pak et al., 2015) and hiPSCs by shRNA (Zeng et al., 2013). Heterozygous mutant neurexins resulted in impaired neurotransmitter release and elevation of CASK (synaptic scaffolding protein) (Pak et al., 2015), and half reduction in NRXN1 expression led to changes in gene expression of cell adhesion and neuron differentiation pathways (Zeng et al., 2013). These studies highlight the utility of in vitro studies for manipulating human genes in human cells and generating human isogenic controls.
By contrast, Brennand et al. (2011) took a more generalized approach and generated hiPSC-derived neurons from four SZ patients who lacked a unifying genotype, but were selected on the basis of the likelihood of a genetic component to their disease (as supported by relatives affected by psychiatric disease or early age of onset). The group generated a mix of excitatory, inhibitory, and dopaminergic neurons, evidenced by the presence of VGLUT1, GAD67, and tyrosine hydroxylase, respectively. These patient-derived neurons formed fewer processes, exhibited decreased neuronal connectivity (assayed by trans-neuronal spread of rabies virus) and decreased spine density (Brennand et al., 2011). Most strikingly, many of the gene expression pathways perturbed in SZ hiPSC neurons were also detected in immature neural progenitor cells, indicating that these pathways were altered even before the establishment of post-mitotic neurons (Brennand et al., 2015). Overall, these findings mimic the decreased neuronal connectivity and synaptic function shown in post-mortem tissue studies (16, 33–35).
Co-culture Systems
The above investigations demonstrate the utility of patient hiPSC-derived neurons for modeling cellular interactions, gaining insight in the impact of a given mutation on said interactions, and elucidating the molecular mechanisms responsible. Most importantly, this is achieved using human cells, thus providing much more translational value than animal models. Despite these advantages, a number of issues surround induced neural protocols (see Engel et al., 2016), especially in the context of modeling synaptogenesis and network dysfunction. First, gene expression studies indicate hiPSC neurons best resemble fetal brain tissue (Brennand et al., 2015). Second, hiPSC neurons require extensive cultivation periods of up to 7 months (Maroof et al., 2013; Nicholas et al., 2013; Suzuki and Vanderhaeghen, 2015). Moreover, pure neuronal cultures are limited in their ability to accurately re-capitulate network functionality, since glial cells play an important role in regulating neuronal activity and establishing myelination.
Several groups have developed astrocyte-neuron co-culture systems, using both rodent and human astrocytes with human iPSC-derived neurons, with promising results for maturation and network activity (Tang et al., 2013; Muratore et al., 2014; Odawara et al., 2014; Kuijlaars et al., 2016). Indeed, the notion that astrocytes enhance synapse maturation is well established from rodent cell cultures (Araque and Perea, 2004). Odawara et al. (2014) utilized multiple electrode array (MEA) plates to co-culture rat astrocytes with hiPSC-derived neurons and found increased electrophysiological activity after 3 weeks of plating, with neurons continuing to increase activity for more than 3 months after plating. Muratore et al. (2014) performed a direct comparison of several neuronal differentiation protocols and demonstrated a significant increase in vGLUT1 expression by day 40 in neurons cultured on mouse astrocytes, pointing to accelerated maturation of excitatory neurons. Kuijlaars et al. (2016) were the first to utilize astrocyte co-culture with solely human cells in 2016. Using commercially available fetal astrocytes, the group observed synchronized calcium oscillations in hiPSC-derived neurons 3–4 weeks after final plating, indicating sustained network activity (Kuijlaars et al., 2016).
Although astrocyte co-cultures have yet to be utilized to model neuropsychiatric disease, these studies are very promising, particularly the human-only system employed by Kuijlaars et al. (2016). Human neuron-astrocyte co-culture systems may be useful not only for improved neuronal maturation but also for elucidating astrocyte contribution to the disease state, especially taking into consideration the role of astrocytes in modulating synaptic connection and signaling (Goudriaan et al., 2014). Post-mortem tissue studies have shown decreases in astrocyte density in certain regions (Williams et al., 2013) and increases in astrocyte markers and neuroinflammation in a subset of patients (Catts et al., 2014). Co-culturing astrocytes and neurons derived from the same patient may more closely mimic the cellular and molecular dynamics of the individual disease state. Additionally, co-culturing patient astrocytes with healthy control neurons and healthy control astrocytes may help elucidate the contribution of each cell type to SZ etiology.
Oligodendrocyte dysfunction and myelin deficits have also been heavily implicated in SZ by genome association and post-mortem studies (Burns et al., 2003; Davis et al., 2003; Tkachev et al., 2003; Takahashi et al., 2011; Goudriaan et al., 2014; Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014). Specifically, volume reduction in myelin observed in SZ patients is consistent with deficits in neuronal communication and synaptic plasticity thought to underlie SZ pathology (reviewed by Takahashi et al., 2011). A hiPSC model of patient oligodendrocytes and healthy neurons would allow to investigate the precise consequences of patient-specific variation in oligodendrocyte genes on myelin formation, interactions with neurons, and synaptic plasticity. Additionally, given the well-established role of myelin in neuronal maturation (Hasegawa et al., 1992), co-culture with oligodendrocytes will likely improve the immature state of current neural hiPSC cultures. Although to date no co-culture system of hiPSC-derived oligodendrocytes and neurons has been reported, oligodendrocytes have been successfully generated from hiPSCs (Livesey et al., 2016; Ehrlich et al., 2017). Most excitingly, Ehrlich et al. (2017) demonstrated successful integration of hiPSC-derived oligodendrocytes into mouse brains, giving hope that this myelination can be reiterated in vitro with human cells.
Microglia mediate synaptic pruning during adolescence (Paolicelli et al., 2011) (although see Sekar et al., 2016 for neuronal contribution to synaptic pruning in SZ) and respond to immune threats by producing inflammatory cytokines (Kreutzberg, 1996); accordingly, abnormal pruning (Feinberg, 1982) and neuroinflammation (Ashdown et al., 2005; Monji et al., 2013) are thought to contribute to the etiology of SZ. Studying these functions in a hiPSC system would allow scientists to mix and match patient and control microglia and neurons, in order to determine the driving force behind abnormal cellular connectivity and function. Critically, microglia-like cells have been successfully generated from hiPSCs by several groups in the last year (Muffat et al., 2016; Abud et al., 2017; Pandya et al., 2017). These cells resemble fetal and adult human microglia in their gene expression patterns and show phagocytic activity in vitro and in vivo. A co-culture application for modeling synaptic pruning would benefit from more mature, myelinated neurons; however, hiPSC microglia show potential for modeling inflammatory activation early in development in a co-culture system.
Modeling Regional Specificity and Circuitry
Although several protocols bias hiPSC differentiation into region-specific neurons, these methodologies fall far short of generating defined circuitry. Typically, hiPSCs are first transformed into neural progenitor cells (NPCs) through dual SMAD inhibition, which involves antagonism of bone morphogenic protein (BMP) and transforming growth factor beta (TGF-β) (Chambers et al., 2009). Such directed differentiation into NPCs (in contrast to allowing hiPSC colonies to differentiate in neural medium as in the studies by Brennand et al. (2011) and Pedrosa et al. (2011) significantly reduces variability in the subsequently derived neurons. The NPCs are then subjected to treatment with growth factors found in distinct regions of the developing brain. Thus, scientists have generated neurons specific to the medial and caudal ganglionic eminences (sites of GABAergic neuron development) (Ahn et al., 2016), cerebral cortex (Shi et al., 2012), and the dentate gyrus (Yu et al., 2014). Although directed-differentiation protocols are thought to more closely mimic in vivo development, the protracted timeline poses a major disadvantage. Viral-based induction strategies have been utilized to overexpress exogenous transcription factors to produce excitatory (Ho et al., 2016) and cortical inhibitory neurons (Colasante et al., 2015); however, these strategies carry the caveat of additional artificiality and may fail to affect all of the transcription factors necessary to induce regional specificity (see Chanda et al., 2013 and Pak et al., 2015 for evidence on same phenotype across different cell culture methods).
Even with regional specificity and micro-circuit modeling, two-dimensional cultures fail to recapitulate the three-dimensional organization of the brain and circuits therein. The ongoing development of three dimensional neural culture (alternatively called brain organoids or cortical spheroids) shows significant promise for modeling network activity and circuit organization. Paşca et al. (2015) derived cortical spheres containing non-reactive astrocytes and functional cortical neurons with laminated organization. Qian et al. (2016) designed a miniature bioreactor system to ensure proper circulation of nutrients in media and produced region-specific organoids that mimicked forebrain, midbrain, and hypothalamus expressing appropriate markers and resembling respective cytoarchitecture. Most recently, Birey et al. (2017) generated and fused together pallium- and subpallium-resembling organoids to model development and migration of inhibitory neurons. After 3 weeks of contact between the pallium and subpallium (ventral forebrain) organoids, inhibitory neurons from the subpallium sphere migrated into the pallium sphere, yielding functional excitatory and inhibitory post-synaptic connections. Creation of such chimeric organoids is an exciting prospect for modeling developmental cellular migration and connectivity in a 3D system.
Although organoids have yet to be applied to SZ, their potential for modeling neural developmental and electrophysiological anomalies is obvious. Much work remains, however, to adequately model circuit development and contributions of non-neural cells to SZ pathology. Currently, brain organoids contain only relatively immature neurons and non-reactive astrocytes, with oligodendrocytes and microglia still lacking. Potential injection of hiPSC-derived oligodendrocytes and microglia (discussed in the section “Co-culture System”) has promise for incorporation into organoid tissue, and the potential to recapitulate exciting in vivo findings that SZ patient hiPSC-derived glial cells function differently when injected into a mouse brain (Windrem et al., 2017), but differences in growing conditions and developmental requirements still pose major challenges in vitro.
While there is solid evidence that the neuropathologies of SZ often precede symptom onset, there are also clear adolescent neurobiological developmental processes (GABAergic maturation, excitatory synaptic pruning, microglia- and astrocyte-mediated neuroinflammation) and environmental exposures (cannabis abuse, stress) that can accelerate or exacerbate genetic risk (well-reviewed van Os et al., 2010). Moving forward, there is an urgent need to add further complexity to our hiPSC-based models in order to model these additional processes. Such efforts will involve not just improving the complexity of cell types, regional patterning and circuit formation, but also the restoration of critical features of “age” and “maturity” to hiPSC-derived cultures. While reprogramming of fibroblasts into hiPSCs erases age-related epigenetic marks and gene expression profiles, these can be at least partially maintained by inducing patient somatic cells directly into neurons (Mertens et al., 2015; Huh et al., 2016). Moreover, some environmental insults may be relatively easy to introduce, such as maternal infection/stress during development (Boksa, 2008), cannabis use (Henquet et al., 2005) or hypoxia during delivery (Cannon et al., 2002), while others such as trauma from childhood abuse and neglect (Read et al., 2005) may only be vaguely approximated by stress-related molecules such as corticosteroids or inflammatory cytokines.
Concluding Remarks
Despite being in a nascent stage of methodological development, hiPSC-derived neurons show promise for modeling molecular and cellular deficits of SZ. Because of the complex etiology and great genetic variability of the disorder, a patient-specific approach may be illuminating from a mechanistic perspective as well as for drug screening. Already, hiPSC-derived neurons have recapitulated deficits in synaptic maturation and connectivity. Furthermore, advances in generating organoids show promise for modeling network activity in three dimensions, thus better recapitulating brain development and circuitry.
Of course, many limitations accompany the use of tissue culture to model psychiatric disease. Aside from being limited to molecular and cellular phenotypes with no recourse for assessing behavioral consequences, current methods produce immature neurons, limiting studies to the developmental predisposition of disease rather than the actual disease state. Nonetheless, modeling predisposition may uncover targets for preventative therapeutic interventions for patients with a high familial risk of psychiatric disease.
Given the developmental underpinnings and genetic heterogeneity of SZ, hiPSC cultures should complement animal models. Overall, subtype specific hiPSC-derived neuronal and glial (astrocyte, oligodendrocyte, and microglia) culture methods are rapidly evolving. Improved neuronal maturation, combined with novel three dimensional cultures, present an exciting opportunity to investigate circuit function and connectivity in patient-specific cells.
Author Contributions
IP gathered references and wrote the manuscript. KB revised the manuscript and provided additional references.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
KB is a New York Stem Cell Foundation – Robertson Investigator. The Brennand Laboratory is supported by the Brain and Behavior Research Foundation, Brain Research Foundation, NIH grants R01 MH101454 and R01 MH106056, and the New York Stem Cell Foundation. Special thanks to Patricia Casaccia and Anne Schaefer for feedback on this manuscript.
References
Abud, E. M., Ramirez, R. N., Martinez, E. S., Healy, L. M., Nguyen, C. H. H., Newman, S. A., et al. (2017). iPSC-derived human microglia-like cells to study neurological diseases. Neuron 94, 278.e9–293.e9. doi: 10.1016/j.neuron.2017.03.042
Ahn, S., Kim, T.-G., Kim, K.-S., and Chung, S. (2016). Differentiation of human pluripotent stem cells into Medial Ganglionic Eminence vs. Caudal Ganglionic Eminence cells. Methods 101, 103–112. doi: 10.1016/j.ymeth.2015.09.009
Araque, A., and Perea, G. (2004). Glial modulation of synaptic transmission in culture. Glia 47, 241–248. doi: 10.1002/glia.20026
Asarnow, J. R. (1994). Childhood-onset schizophrenia. J. Child Psychol. Psychiatry 35, 1345–1371. doi: 10.1111/j.1469-7610.1994.tb01280.x
Ashdown, H., Dumont, Y., Ng, M., Poole, S., Boksa, P., and Luheshi, G. N. (2005). The role of cytokines in mediating effects of prenatal infection on the fetus: implications for schizophrenia. Mol. Psychiatry 11, 47–55. doi: 10.1038/sj.mp.4001748
Barnhill, J. W. (2013). “Schizophrenia spectrum and other psychotic disorders,” in DSM-5 Clinical Cases. DSM Library, ed. American Psychiatric Association (Lake St. Louis, MO: American Psychiatric Publishing). doi: 10.1176/appi.books.9781585624836.jb02
Bassett, A. S., and Chow, E. W. (2008). Schizophrenia and 22q11.2 deletion syndrome. Curr. Psychiatry Rep. 10, 148–157. doi: 10.1007/s11920-008-0026-1
Birey, F., Andersen, J., Makinson, C. D., Islam, S., Wei, W., Huber, N., et al. (2017). Assembly of functionally integrated human forebrain spheroids. Nature 545, 54–59. doi: 10.1038/nature22330
Boksa, P. (2008). Maternal infection during pregnancy and schizophrenia. J. Psychiatry Neurosci. 33, 183–185.
Brennand, K., Savas, J. N., Kim, Y., Tran, N., Simone, A., Hashimoto-Torii, K., et al. (2015). Phenotypic differences in hiPSC NPCs derived from patients with schizophrenia. Mol. Psychiatry 20, 361–368. doi: 10.1038/mp.2014.22
Brennand, K., Simone, A., Jou, J., Gelboin-Burkhart, C., Tran, N., Sangar, S., et al. (2011). Modeling schizophrenia using hiPSC neurons. Nature 473, 221–225. doi: 10.1038/nature09915
Brennand, K. J., and Gage, F. H. (2011). Concise review: the promise of hiPSC-based studies of schizophrenia. Stem Cells 29, 1915–1922. doi: 10.1002/stem.762
Burns, J., Job, D., Bastin, M. E., Whalley, H., Macgillivray, T., Johnstone, E. C., et al. (2003). Structural disconnectivity in schizophrenia: a diffusion tensor magnetic resonance imaging study. Br. J. Psychiatry 182, 439–443. doi: 10.1192/bjp.182.5.439
Cannon, T. D., van Erp, T. G., Rosso, I. M., Huttunen, M., Lönnqvist, J., Pirkola, T., et al. (2002). Fetal hypoxia and structural brain abnormalities in schizophrenic patients, their siblings, and controls. Arch. Gen. Psychiatry 59, 35–41. doi: 10.1001/archpsyc.59.1.35
Cardno, A. G., Marshall, E. J., Coid, B., Macdonald, A. M., Ribchester, T. R., Davies, N. J., et al. (1999). Heritability estimates for psychotic disorders: the Maudsley twin psychosis series. Arch. Gen. Psychiatry 56, 162–168. doi: 10.1001/archpsyc.56.2.162
Carlsson, A., Lindqvist, M., and Magnusson, T. (1957). 3,4-Dihydroxyphenylalanine and 5-hydroxytryptophan as reserpine antagonists. Nature 180, 1200. doi: 10.1038/1801200a0
Catts, V. S., Wong, J., Fillman, S. G., Fung, S. J., and Shannon Weickert, C. (2014). Increased expression of astrocyte markers in schizophrenia: association with neuroinflammation. Aust. N. Z. J. Psychiatry 48, 722–734. doi: 10.1177/0004867414531078
Chambers, S. M., Fasano, C. A., Papapetrou, E. P., Tomishima, M., Sadelain, M., and Studer, L. (2009). Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 27, 275–280. doi: 10.1038/nbt.1529
Chanda, S., Marro, S., Wernig, M., and Südhof, T. C. (2013). Neurons generated by direct conversion of fibroblasts reproduce synaptic phenotype caused by autism-associated neuroligin-3 mutation. Proc. Natl. Acad. Sci. U.S.A. 110, 16622–16627. doi: 10.1073/pnas.1316240110
Chiang, C.-H., Su, Y., Wen, Z., Yoritomo, N., Ross, C. A., Margolis, R. L., et al. (2011). Integration-free induced pluripotent stem cells derived from schizophrenia patients with a DISC1 mutation. Mol. Psychiatry 16, 358–360. doi: 10.1038/mp.2011.13
Colasante, G., Lignani, G., Rubio, A., Medrihan, L., Yekhlef, L., Sessa, A., et al. (2015). Rapid conversion of fibroblasts into functional forebrain GABAergic interneurons by direct genetic reprogramming. Cell Stem Cell 17, 719–734. doi: 10.1016/j.stem.2015.09.002
Davis, K. L., Stewart, D. G., Friedman, J. I., Buchsbaum, M., Harvey, P. D., Hof, P. R., et al. (2003). White matter changes in schizophrenia: evidence for myelin-related dysfunction. Arch. Gen. Psychiatry 60, 443–456. doi: 10.1001/archpsyc.60.5.443
Delay, J., Deniker, P., and Harl, J. M. (1952). Therapeutic use in psychiatry of phenothiazine of central elective action (4560 RP). Ann. Med. Psychol. 110 2, 112–117.
Duan, X., Chang, J. H., Ge, S., Faulkner, R. L., Kim, J. Y., Kitabatake, Y., et al. (2007). Disrupted-in-schizophrenia 1 regulates integration of newly generated neurons in the adult brain. Cell 130, 1146–1158. doi: 10.1016/j.cell.2007.07.010
Ehrlich, M., Mozafari, S., Glatza, M., Starost, L., Velychko, S., Hallmann, A.-L., et al. (2017). Rapid and efficient generation of oligodendrocytes from human induced pluripotent stem cells using transcription factors. Proc. Natl. Acad. Sci. U.S.A. 114, E2243–E2252. doi: 10.1073/pnas.1614412114
Engel, M., Do-Ha, D., Muñoz, S. S., and Ooi, L. (2016). Common pitfalls of stem cell differentiation: a guide to improving protocols for neurodegenerative disease models and research. Cell. Mol. Life Sci. 73, 3693–3709. doi: 10.1007/s00018-016-2265-3
Faludi, G., and Mirnics, K. (2011). Synaptic changes in the brain of subjects with schizophrenia. Int. J. Dev. Neurosci. 29, 305–309. doi: 10.1016/j.ijdevneu.2011.02.013
Feinberg, I. (1982). Schizophrenia: caused by a fault in programmed synaptic elimination during adolescence? J. Psychiatr. Res. 17, 319–334. doi: 10.1016/0022-3956(82)90038-3
Ferrer, I., Martinez, A., Boluda, S., Parchi, P., and Barrachina, M. (2008). Brain banks: benefits, limitations and cautions concerning the use of post-mortem brain tissue for molecular studies. Cell Tissue Bank. 9, 181–194. doi: 10.1007/s10561-008-9077-0
Flores, G., Morales-Medina, J. C., and Diaz, A. (2016). Neuronal and brain morphological changes in animal models of schizophrenia. Behav. Brain Res. 301, 190–203. doi: 10.1016/j.bbr.2015.12.034
Glantz, L. A., and Lewis, D. A. (2000). Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch. Gen. Psychiatry 57, 65–73. doi: 10.1001/archpsyc.57.1.65
Goudriaan, A., de Leeuw, C., Ripke, S., Hultman, C. M., Sklar, P., Sullivan, P. F., et al. (2014). Specific glial functions contribute to schizophrenia susceptibility. Schizophr. Bull. 40, 925–935. doi: 10.1093/schbul/sbt109
Hasegawa, M., Houdou, S., Mito, T., Takashima, S., Asanuma, K., and Ohno, T. (1992). Development of myelination in the human fetal and infant cerebrum: a myelin basic protein immunohistochemical study. Brain Dev. 14, 1–6. doi: 10.1016/S0387-7604(12)80271-3
Henquet, C., Murray, R., Linszen, D., and van Os, J. (2005). The environment and schizophrenia: the role of cannabis use. Schizophr. Bull. 31, 608–612. doi: 10.1093/schbul/sbi027
Ho, S.-M., Hartley, B. J., Tcw, J., Beaumont, M., Stafford, K., Slesinger, P. A., et al. (2016). Rapid Ngn2-induction of excitatory neurons from hiPSC-derived neural progenitor cells. Methods 101, 113–124. doi: 10.1016/j.ymeth.2015.11.019
Homayoun, H., and Moghaddam, B. (2007). NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J. Neurosci. 27, 11496–11500. doi: 10.1523/JNEUROSCI.2213-07.2007
Huh, C. J., Zhang, B., Victor, M. B., Dahiya, S., Batista, L. F., Horvath, S., et al. (2016). Maintenance of age in human neurons generated by microRNA-based neuronal conversion of fibroblasts. eLife 5:e18648. doi: 10.7554/eLife.18648
Huttenlocher, P. R., and Dabholkar, A. S. (1997). Regional differences in synaptogenesis in human cerebral cortex. J. Comp. Neurol. 387, 167–178. doi: 10.1002/(SICI)1096-9861(19971020)387:2<167::AID-CNE1>3.0.CO;2-Z
Kantrowitz, J., and Javitt, D. C. (2012). Glutamatergic transmission in schizophrenia: from basic research to clinical practice. Curr. Opin. Psychiatry 25, 96–102. doi: 10.1097/YCO.0b013e32835035b2
Kehrer, C., Maziashvili, N., Dugladze, T., and Gloveli, T. (2008). Altered excitatory-inhibitory balance in the NMDA-hypofunction model of schizophrenia. Front. Mol. Neurosci. 1:6. doi: 10.3389/neuro.02.006.2008
Kirkpatrick, B., Buchanan, R. W., Ross, D. E., and Carpenter, W. T. (2001). A separate disease within the syndrome of schizophrenia. Arch. Gen. Psychiatry 58, 165–171. doi: 10.1001/archpsyc.58.2.165
Kreutzberg, G. W. (1996). Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 19, 312–318. doi: 10.1016/0166-2236(96)10049-7
Kuijlaars, J., Oyelami, T., Diels, A., Rohrbacher, J., Versweyveld, S., Meneghello, G., et al. (2016). Sustained synchronized neuronal network activity in a human astrocyte co-culture system. Sci. Rep. 6:36529. doi: 10.1038/srep36529
Lewis, D. A., and Lieberman, J. A. (2000). Catching up on schizophrenia: natural history and neurobiology. Neuron 28, 325–334. doi: 10.1016/S0896-6273(00)00111-2
Lieberman, J. A., Scott Stroup, T., McEvoy, J. P., Swartz, M. S., Rosenheck, R. A., Perkins, D. O., et al. (2005). Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. New Engl. J. Med. 353, 1209–1223. doi: 10.1056/NEJMoa051688
Lin, M., Pedrosa, E., Hrabovsky, A., Chen, J., Puliafito, B. R., Gilbert, S. R., et al. (2016). Integrative transcriptome network analysis of iPSC-derived neurons from schizophrenia and schizoaffective disorder patients with 22q11.2 deletion. BMC Syst. Biol. 10:105. doi: 10.1186/s12918-016-0366-0
Livesey, M. R., Magnani, D., Cleary, E. M., Vasistha, N. A., James, O. T., Selvaraj, B. T., et al. (2016). Maturation and electrophysiological properties of human pluripotent stem cell-derived oligodendrocytes. Stem Cells 34, 1040–1053. doi: 10.1002/stem.2273
Maroof, A. M., Keros, S., Tyson, J. A., Ying, S.-W., Ganat, Y. M., Merkle, F. T., et al. (2013). Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell 12, 559–572. doi: 10.1016/j.stem.2013.04.008
McGlashan, T. H., and Hoffman, R. E. (2000). Schizophrenia as a disorder of developmentally reduced synaptic connectivity. Arch. Gen. Psychiatry 57, 637–648. doi: 10.1001/archpsyc.57.7.637
Mertens, J., Paquola, A. C., Ku, M., Hatch, E., Böhnke, L., Ladjevardi, S., et al. (2015). Directly reprogrammed human neurons retain aging-associated transcriptomic signatures and reveal age-related nucleocytoplasmic defects. Cell Stem Cell 17, 705–718. doi: 10.1016/j.stem.2015.09.001
Monji, A., Kato, T. A., Mizoguchi, Y., Horikawa, H., Seki, Y., Kasai, M., et al. (2013). Neuroinflammation in schizophrenia especially focused on the role of microglia. Prog. NeuroPsychopharmacol. Biol. Psychiatry 42, 115–121. doi: 10.1016/j.pnpbp.2011.12.002
Muffat, J., Li, Y., Yuan, B., Mitalipova, M., Omer, A., Corcoran, S., et al. (2016). Efficient Derivation of microglia-like cells from human pluripotent stem cells. Nat. Med. 22, 1358–1367. doi: 10.1038/nm.4189
Muratore, C. R., Srikanth, P., Callahan, D. G., and Young-Pearse, T. L. (2014). Comparison and optimization of hiPSC forebrain cortical differentiation protocols. PLOS ONE 9:e105807. doi: 10.1371/journal.pone.0105807
Nicholas, C. R., Chen, J., Tang, Y., Southwell, D. G., Chalmers, N., Vogt, D., et al. (2013). Functional maturation of hPSC-derived forebrain interneurons requires an extended timeline and mimics human neural development. Cell Stem Cell 12, 573–586. doi: 10.1016/j.stem.2013.04.005
Odawara, A., Saitoh, Y., Alhebshi, A. H., Gotoh, M., and Suzuki, I. (2014). Long-term electrophysiological activity and pharmacological response of a human induced pluripotent stem cell-derived neuron and astrocyte co-culture. Biochem. Biophys. Res. Commun. 443, 1176–1181. doi: 10.1016/j.bbrc.2013.12.142
Olney, J. W., Newcomer, J. W., and Farber, N. B. (1999). NMDA receptor hypofunction model of schizophrenia. J. Psychiatr. Res. 33, 523–533. doi: 10.1016/S0022-3956(99)00029-1
Pak, C., Danko, T., Zhang, Y., Aoto, J., Anderson, A., Maxeiner, S., et al. (2015). Human neuropsychiatric disease modeling using conditional deletion reveals synaptic transmission defects caused by heterozygous mutations in NRXN1. Cell Stem Cell 17, 316–328. doi: 10.1016/j.stem.2015.07.017
Pandya, H., Shen, M. J., Ichikawa, D. M., Sedlock, A. B., Choi, Y., Johnson, K. R., et al. (2017). Differentiation of human and murine induced pluripotent stem cells to microglia-like cells. Nat. Neurosci. 20, 753–759. doi: 10.1038/nn.4534
Paolicelli, R. C., Bolasco, G., Pagani, F., Maggi, L., Scianni, M., Panzanelli, P., et al. (2011). Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458. doi: 10.1126/science.1202529
Paşca, A. M., Sloan, S. A., Clarke, L. E., Tian, Y., Makinson, C. D., Huber, N., et al. (2015). Functional cortical neurons and astrocytes from human pluripotent stem cells in 3d culture. Nat. Methods 12, 671–678. doi: 10.1038/nmeth.3415
Pedrosa, E., Sandler, V., Shah, A., Carroll, R., Chang, C., Rockowitz, S., et al. (2011). Development of patient-specific neurons in schizophrenia using induced pluripotent stem cells. J. Neurogenet. 25, 88–103. doi: 10.3109/01677063.2011.597908
Porteous, D. J., Thomson, P. A., Millar, J. K., Evans, K. L., Hennah, W., Soares, D. C., et al. (2014). DISC1 as a genetic risk factor for schizophrenia and related major mental illness: response to Sullivan. Mol. Psychiatry 19, 141–143. doi: 10.1038/mp.2013.160
Qian, X., Nguyen, H. N., Song, M. M., Hadiono, C., Ogden, S. C., Hammack, C., et al. (2016). Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell 165, 1238–1254. doi: 10.1016/j.cell.2016.04.032
Read, J., van Os, J., Morrison, A. P., and Ross, C. A. (2005). Childhood trauma, psychosis and schizophrenia: a literature review with theoretical and clinical implications. Acta Psychiatr. Scand. 112, 330–350. doi: 10.1111/j.1600-0447.2005.00634.x
Rössler, W., Salize, H. J., van Os, J., and Riecher-Rössler, A. (2005). Size of burden of schizophrenia and psychotic disorders. Eur. Neuropsychopharmacol. 15, 399–409. doi: 10.1016/j.euroneuro.2005.04.009
Schizophrenia Working Group of the Psychiatric Genomics Consortium (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427. doi: 10.1038/nature13595
Sekar, A., Bialas, A. R., de Rivera, H., Davis, A., Hammond, T. R., Kamitaki, N., et al. (2016). Schizophrenia risk from complex variation of complement component 4. Nature 530, 177–183. doi: 10.1038/nature16549
Shi, Y., Kirwan, P., and Livesey, F. J. (2012). Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nat. Protoc. 7, 1836–1846. doi: 10.1038/nprot.2012.116
St Clair, D. (2009). Copy number variation and schizophrenia. Schizophr. Bull. 35, 9–12. doi: 10.1093/schbul/sbn147
Suzuki, I. K., and Vanderhaeghen, P. (2015). Is this a brain which i see before me? Modeling human neural development with pluripotent stem cells. Development 142, 3138–3150. doi: 10.1242/dev.120568
Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K., et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872. doi: 10.1016/j.cell.2007.11.019
Takahashi, N., Sakurai, T., Davis, K. L., and Buxbaum, J. D. (2011). Linking oligodendrocyte and myelin dysfunction to neurocircuitry abnormalities in schizophrenia. Prog. Neurobiol. 93, 13–24. doi: 10.1016/j.pneurobio.2010.09.004
Tang, X., Zhou, L., Wagner, A. M., Marchetto, M. C. N., Muotri, A. R., Gage, F. H., et al. (2013). Astroglial cells regulate the developmental timeline of human neurons differentiated from induced pluripotent stem cells. Stem Cell Res. 11, 743–757. doi: 10.1016/j.scr.2013.05.002
Tkachev, D., Mimmack, M. L., Ryan, M. M., Wayland, M., Freeman, T., Jones, P. B., et al. (2003). Oligodendrocyte dysfunction in schizophrenia and bipolar disorder. Lancet 362, 798–805. doi: 10.1016/S0140-6736(03)14289-4
Toyoshima, M., Akamatsu, W., Okada, Y., Ohnishi, T., Balan, S., Hisano, Y., et al. (2016). Analysis of induced pluripotent stem cells carrying 22q11.2 deletion. Transl. Psychiatry 6, e934. doi: 10.1038/tp.2016.206
van Os, J., Kenis, G., and Rutten, B. (2010). The environment and schizophrenia. Nature 468, 203–212. doi: 10.1038/nature09563
Weinberger, D. R. (1995). From neuropathology to neurodevelopment. Lancet 346, 552–557. doi: 10.1016/S0140-6736(95)91386-6
Wen, Z., Nguyen, H. N., Guo, Z., Lalli, M. A., Wang, X., Su, Y., et al. (2014). Synaptic dysregulation in a human iPS cell model of mental disorders. Nature 515, 414–418. doi: 10.1038/nature13716
Williams, M. R., Hampton, T., Pearce, R. K. B., Hirsch, S. R., Ansorge, O., Thom, M., et al. (2013). Astrocyte decrease in the subgenual cingulate and callosal genu in schizophrenia. Eur. Arch. Psychiatry Clin. Neurosci. 263, 41–52. doi: 10.1007/s00406-012-0328-5
Windrem, M. S., Osipovitch, M., Liu, Z., Bates, J., Chandler-Militello, D., Zou, L., et al. (2017). Human iPSC glial mouse chimeras reveal glial contributions to schizophrenia. Cell Stem Cell 21, 195.e6–208.e6. doi: 10.1016/j.stem.2017.06.012
Yoon, K.-J., Nguyen, H. N., Ursini, G., Zhang, F., Kim, N.-S., Wen, Z., et al. (2014). Modeling a genetic risk for schizophrenia in iPSCs and mice reveals neural stem cell deficits associated with adherens junctions and polarity. Cell Stem Cell 15, 79–91. doi: 10.1016/j.stem.2014.05.003
Yu, D. X., Di Giorgio, F. P., Yao, J., Marchetto, M. C., Brennand, K., Wright, R., et al. (2014). Modeling hippocampal neurogenesis using human pluripotent stem cells. Stem Cell Rep. 2, 295–310. doi: 10.1016/j.stemcr.2014.01.009
Zeng, L., Zhang, P., Shi, L., Yamamoto, V., Lu, W., and Wang, K. (2013). Functional impacts of NRXN1 knockdown on neurodevelopment in stem cell models. PLOS ONE 8:e59685. doi: 10.1371/journal.pone.0059685
Zhao, D., Lin, M., Chen, J., Pedrosa, E., Hrabovsky, A., Fourcade, H. M., et al. (2015). MicroRNA profiling of neurons generated using induced pluripotent stem cells derived from patients with schizophrenia and schizoaffective disorder, and 22q11.2 del. PLOS ONE 10:e0132387. doi: 10.1371/journal.pone.0132387
Keywords: human induced pluripotent stem cells, induced neurons, co-culture, organoids, schizophrenia
Citation: Prytkova I and Brennand KJ (2017) Prospects for Modeling Abnormal Neuronal Function in Schizophrenia Using Human Induced Pluripotent Stem Cells. Front. Cell. Neurosci. 11:360. doi: 10.3389/fncel.2017.00360
Received: 22 August 2017; Accepted: 03 November 2017;
Published: 23 November 2017.
Edited by:
Chao Deng, University of Wollongong, AustraliaReviewed by:
Tomiki Sumiyoshi, National Center of Neurology and Psychiatry, JapanStewart Alan Anderson, University of Pennsylvania, United States
Yuriko Iwakura, Niigata University, Japan
Copyright © 2017 Prytkova and Brennand. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kristen J. Brennand, a3Jpc3Rlbi5icmVubmFuZEBtc3NtLmVkdQ==