 Paloma González-Sánchez1,2,3
Paloma González-Sánchez1,2,3 Araceli del Arco
Araceli del Arco José A. Esteban
José A. Esteban Jorgina Satrústegui
Jorgina Satrústegui- 1Department of Molecular Biology, Centro de Biología Molecular Severo Ochoa, Consejo Superior de Investigaciones Científicas-Universidad Autónoma de Madrid (CSIC-UAM), Madrid, Spain
- 2Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), Madrid, Spain
- 3Instituto de Investigación Sanitaria Fundación Jiménez Díaz (IIS-FJD), Madrid, Spain
- 4Facultad de Ciencias Ambientales y Bioquímica, Universidad de Castilla la Mancha, Toledo, Spain
- 5Department of Molecular Neurobiology, Centro de Biología Molecular Severo Ochoa, Consejo Superior de Investigaciones Científicas-Universidad Autónoma de Madrid (CSIC-UAM), Madrid, Spain
Store-operated calcium entry (SOCE) is a Calcium (Ca2+) influx pathway activated by depletion of intracellular stores that occurs in eukaryotic cells. In neurons, the presence and functions of SOCE are still in question. Here, we show evidences for the existence of SOCE in primary mouse cortical neurons. Endoplasmic reticulum (ER)-Ca2+ depletion using thapsigargin (Tg) triggered a maintained cytosolic Ca2+ increase, which rapidly returned to basal level in the presence of the SOCE blockers 2-Aminoethoxydiphenyl borate (2-APB) and YM-58483. Neural SOCE is also engaged by activation of metabotropic glutamate receptors (mGluRs) with (S)-3,5-dihydroxyphenylglycine (DHPG) (agonist of group I mGluRs), being an essential mechanism to maintain the mGluR-driven Ca2+ signal. Activation of group I of mGluRs triggers long-term depression (LTD) in many brain regions, but the underlying mechanism and, specifically, the necessity of Ca2+ increase in the postsynaptic neuron is controversial. In primary cortical neurons, we now show that the inhibition of Ca2+ influx through SOCE impaired DHPG-LTD, pointing out a key function of calcium and SOCE in synaptic plasticity.
Introduction
Calcium (Ca2+) is a universal second messenger that regulates numerous cellular processes of all eukaryotic cells, including metabolism, muscle contraction, exocytosis, transcription of numerous genes or programmed cell death (Brini et al., 2013). In neurons, it also plays a critical role in the transmission of synaptic information by controlling neurotransmitter release (Südhof, 2012). In addition, calcium is the intracellular messenger for many forms of activity-dependent synaptic plasticity acting both in the presynaptic and/or postsynaptic neuron (Zucker, 1999; Cavazzini et al., 2005). These roles are performed thanks to abundant and specific ligand- and voltage-gated Ca2+ channels which exert a tight control on Ca2+ dynamics (Grienberger and Konnerth, 2012; Brini et al., 2014). Ca2+ stores also have a relevant role in the regulation of the intracellular Ca2+ concentration (Verkhratsky, 2005).
Ca2+ homeostasis of the endoplasmic reticulum (ER) is controlled by several and well-orchestrated mechanisms (Stutzmann and Mattson, 2011). In mammals, the release of Ca2+ from ER is sensed by the stromal interaction molecule (STIM) proteins, STIM1 and STIM2 (Liou et al., 2005; Roos et al., 2005). After a decrease in ER-Ca2+ levels, STIM proteins oligomerize and migrate to the ER-plasma membrane (PM) junctions, where they interact with Orai calcium channels, Orai1, Orai2 or Orai3 (Feske et al., 2005; Gwack et al., 2007) to allow Ca2+ influx into the cytosol. This mechanism is called store-operated calcium entry (SOCE; Putney, 1986), and it has been extensively studied in non-excitable cells, for which SOCE acts as a principal Ca2+ entry pathway (Parekh and Putney, 2005; Prakriya and Lewis, 2015). Conversely, the relevance of neuronal SOCE is being debated (Lu and Fivaz, 2016). In the last few years, SOCE activity has been observed in hippocampal (Emptage et al., 2001; Baba et al., 2003; Kann et al., 2012; Sun et al., 2014; Samtleben et al., 2015), cortical (Berna-Erro et al., 2009; Klejman et al., 2009; Gruszczynska-Biegala et al., 2011), cerebellar (Baba et al., 2003; Hartmann et al., 2014), sensory (Gemes et al., 2011) and dorsal horn neurons (Xia et al., 2014). Moreover, two independent groups found that STIM1 directly modulates depolarization-induced opening of the voltage-gated Ca2+ channel CaV1.2 (Park et al., 2010; Wang et al., 2010). Neuronal SOCE is thought to perform different roles: the refilling of ER-Ca2+, which is continuously emptying at rest (Samtleben et al., 2015), regulation of neuronal gene expression (Lalonde et al., 2014), and the maturation and maintenance of dendritic spines (Sun et al., 2014; Korkotian et al., 2017). An impairment in SOCE function related to a mislocalization of mitochondria close to Ca2+ entry sites has been shown to occur in Charcot-Marie-Tooth disease related to recessive mutations in ganglioside-induced differentiation associated protein 1 (GDAP1; Pla-Martín et al., 2013; González-Sánchez et al., 2017).
Recently, several studies have suggested a relevant role for ER-Ca2+ stores and SOCE in synaptic plasticity (Bardo et al., 2006; Majewski and Kuznicki, 2015; Moccia et al., 2015; Segal and Korkotian, 2016). SOCE has been shown to control neuronal Ca2+ dynamics during synaptic activity in different neurons (Moccia et al., 2015). A direct link between SOCE and AMPA receptor-dependent Ca2+ signal has been proposed (Gruszczynska-Biegala et al., 2016). A mechanism that links activation of postsynaptic NMDA receptors and L-type Ca2+ channels has been linked to signaling by the ER-Ca2+ sensor STIM1 (Dittmer et al., 2017). STIM1 has also been proposed to be a key regulator of Ca2+ signaling downstream metabotropic glutamate receptor (mGluR) stimulation (Hartmann et al., 2014; Hou et al., 2015).
Activation of group I of mGluRs (mGluRs I) using electrical or pharmacological stimulation ((S)-3,5-dihydroxyphenylglycine (DHPG)) leads to long-term depression (LTD) in many brain regions (Lüscher and Huber, 2010). mGluRs I are canonically coupled to Gαq/11 and the activation of phospholipase Cβ (PLC), inositol triphosphate (IP3) generation, release of Ca2+ from intracellular stores, and protein kinase C (PKC) activation (Abe et al., 1992; Aramori and Nakanishi, 1992). However, depending on the induction protocol and the brain region, there are differences in the involvement of the PLC pathway and postsynaptic Ca2+ rise in mGluR-LTD (Gladding et al., 2009; Lüscher and Huber, 2010).
Although LTD takes place in many brain regions, most studies have focused on hippocampal neurons. The aim of the present study was to investigate the role of SOCE in cortical neuron LTD, specifically upon mGluR stimulation.
Materials and Methods
Animals
Wild-type mice with a mixed C57BL6/Sv129 genetic background were used. Mice were housed in a humidity- and temperature-controlled room on a 12 h light/dark cycle, receiving water and food ad libitum. All the experimental protocols performed in this study were performed in accordance with procedures approved in the Directive 86/609/EEC of the European Union and with approval of the Institutional Ethical Committee of the Center of Molecular Biology Severo Ochoa and Universidad Autonoma de Madrid (CEEA-CBMSO-23/159) and Area de Protection Animal Comunidad de Madrid (PROEX 352/15). All efforts were made to minimize animal suffering.
Primary Neuronal Culture
Cortical neuronal cultures were prepared from E15 to E16 mouse embryos as described earlier (Ramos et al., 2003; Pardo et al., 2006). Neurons were plated at a density of 5 × 105 cells/cm2 on poly-L-lysine and laminin-coated pretreated glass coverslips in Neurobasal medium supplemented with 2% B27, 1% glutamax (all from Gibco Invitrogen, Carlsbad, CA, USA) and 100 mg/ml penicillin–streptomycin. On the fifth day in vitro (DIV) half of the plating medium was removed from each well and replenished with BrainPhys medium (Bardy et al., 2015; Stem cell Technologies, Seattle, WA, USA) supplemented with 2% B27 and 100 mg/ml penicillin–streptomycin. Neurons represented >80% of the total cell population (Ramos et al., 2003; Pardo et al., 2006). The cultures were maintained at 37°C in a humidified atmosphere of 5% CO2. The culture medium was partially replaced every other day. Cultures were used for experimentation between 9 and 11 DIV. BrainPhys was chosen as culture medium for these studies because it has an inorganic salt concentration, glucose level and osmolarity similar to those reported for the brain, and neurons grown in this culture medium show high viability and maintain synaptic network activity during long time in culture (Bardy et al., 2015), i.e., conditions adequate for the study of possible roles of SOCE in synaptic activity. However, we have verified that SOCE activity in cortical neurons was independent of the culture medium, and we have observed a similar and consistent Ca2+ entry after ER-Ca2+ discharge using thapsigargin (Tg) in cortical neurons cultured in both BrainPhys and Neurobasal medium.
Measurement of Cytosolic Ca2+ Signals
Cytosolic calcium imaging with Fura-2 was performed as described by Ruiz et al. (1998). Neurons were plated at 2 × 105 cells/well onto 12 mm round coverslips. Cells were loaded with Fura-2AM by incubation in 2.5 mM D-glucose Ca2+-free HCSS with 5 μM Fura-2AM and 50 μM pluronic F.127 acid (both from Molecular Probes, Invitrogen, Carlsbad, CA, USA), for 30 min at 37 °C, and rinsed with HCSS 2 mM CaCl2, for 30 min. Fluorescence (emission 510 nm) ratio of Ca2+-free (F380) to Ca2+-bound probe (F340) was analyzed using Aquacosmos 2.5 software (Hamamatsu) and Metafluor for Leica developed by Metamorph (Universal Imaging). Regions of interest (ROIs) were selected covering single cells. SOCE analysis was performed using a 2 mM CaCl2 and 1 μM tetrodotoxin (TTX, Tocris Bioscience, Bristol, UK) medium, ER-Ca2+ was depleted using 1 μM of Tg (Alomone Labs, Jerusalem, Israel). Drugs used to study the influence of other neuronal Ca2+ channels were: 10 μM 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), 10 μM MK-801 ((5S,10R)–(+)-5-Methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine hydrogen maleate), 50 μM NiCl2 (all from Sigma-Aldrich, St. Louis, MO, USA) and 5 μM MPEP (2-Methyl-6-(phenylethynyl) pyridine) (Tocris Bioscience, Bristol, UK). mGluR analysis was performed in a 2 mM CaCl2 and 1 μM TTX medium, where 200 μM DHPG (Tocris Bioscience, Bristol, UK) was added in absence or presence of the SOCE blockers 10 μM YM-58483 (Tocris Bioscience, Bristol, UK) or 50 μM 2-Aminoethoxydiphenyl borate (2-APB; Sigma-Aldrich, St. Louis, MO, USA).
Quantitative Real Time PCR (qRT-PCR)
Levels of messenger RNA (mRNA) in neurons were determined by quantitative real time PCR (qRT-PCR). Specific intron-spanning primers for PCR amplification of the SOCE genes: Orai1, 2 and 3, Stim1 and 2, Trpc1, 2, 3, 4, 5, 6 and 7, were designed using Universal Probe Library web1 (Roche, Basel, Switzerland). RNA extraction from 8 to 9 DIV neurons was done with RNeasy mini kit (QIAGEN, Hilden, Germany) following the manufacturer’s instructions. One microgram of total RNA was subjected to first strand cDNA synthesis using random hexamers with Avian Myeloblastosis Virus (AMV) reverse transcriptase (Promega, Madison, WI, USA) and quantified using the spectrophotometer NanoDrop 1000 (ThermoFisher, Waltham, MA, USA).
For qPCR, 2.5 ng of the cDNA synthesized was amplified using the ABI Prism 7900HT real-time PCR System (Applied Biosystems, Foster City, CA, USA). The amplification protocol was: hot start (10 min 95°C) and 40 amplification cycles (15 s 95°C, 1 min 60°C). All reactions were carried out in triplicate. Primer sequences for cDNA analysis by qRT-PCR were as follows (5′-3′): Orai1 forward tacttaagccgcgccaag; Orai1 reverse acttccaccatcgctacca; Orai2 forward cacagacgctagccacgag; Orai2 reverse atgggcacattgagctctg; Orai3 forward cacatctgctctgctgtcg; Orai3 reverse aggcctggtgggtattcat; Stim1 forward cagggactgtactgaagatgaca; Stim1 reverse aggtgattatgccgagtcaag; Stim2 forward gagggcgcagagtgtgag; Stim2 reverse tttagagccatgcggacct; Trpc1 forward tgtggttgtgattgtgctga; Trpc1 reverse tccattctttatcctcatgatttg; Trpc2 forward cccatcgggacctttacc; Trpc2 reverse tcgaaggcggtaggacac; Trpc3 forward ttaattatggtctgggttcttgg; Trpc3 reverse tccacaactgcacgatgtact; Trpc4 forward aaggaagccagaaagcttcg; Trpc4 reverse ccaggttcctcatcacctct; Trpc5 forward ggcgatgcattactctacgc; Trpc5 reverse atcatcagcgtgggaacct; Trpc6 forward tactggtgtgctccttgcag; Trpc6 reverse caaacttcatgaacggtcctc; Trpc7 forward aacgatgaagtcaatgaaggtg; Trpc7 reverse ccagctctcctgtagcctga. We performed an absolute quantification using linearized plasmids containing the cDNA fragments and the standard curve method. For quantification of the PCR products, we used the fluorescent dye SYBR-Green (SYBR® Green, Biorad, Hercules, CA, USA). The parameter analyzed was the quantification cycle (Cq). A standard curve of each amplicon (from 10 to 108 copies) was used and the number of copies of SOCE genes was obtained by extrapolation. The PCR efficiency was calculated for each pair of specific primers and was applied where appropriate.
Western Blot
Cortical neurons were plated at a density of 1 × 106 cells/well on poly-L-lysine and laminin-coated pretreated six wells plates. Cells were collected with a scrapper into a homogenization buffer (250 mM Sucrose, 20 mM Hepes, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, Complete protease inhibitor cocktail mini-EDTA free, (Roche, Basel, Switzerland) and adjusted to pH 7.4). The samples were homogenized by sonication, and aliquots of 30 μg protein (Bradford assay) were separated by SDS-PAGE and transferred to nitrocellulose membranes. Primary antibodies used were α-ORAI1 (1:200) rabbit polyclonal, α-ORAI2 (1:200) rabbit polyclonal, α-STIM2 (1:500) rabbit polyclonal, α-TRPC1 (1:500) rabbit polyclonal, α-TRPC4 (1:500) rabbit polyclonal, (all from Alomone Labs, Jerusalem, Israel) and α−βActin (1:5000) mouse monoclonal (Sigma-Aldrich, St. Louis, MO, USA).
Electrophysiological Recordings and mEPSC Analysis
Voltage-clamp whole-cell recordings were performed in DIV 10–11 cortical neurons. Neurons at 37°C were continuously perfused with a recording medium containing (in mM) 120 NaCl, 0.8 MgCl2, 5.4 KCl, 25 HEPES, 2.5 D-glucose, 2 CaCl2 and supplemented with 1 μM TTX to prevent action potential-evoked responses; pH was adjusted to 7.4. Patch electrodes with resistances between 4–6 MΩ were filled with an internal solution containing (in mM) 115 K gluconate, 20 KCl, 10 HEPES, 2 MgCl2, 4 Na2-ATP, 0.3 Na3-GTP, pH adjusted to 7.3 and osmolarity ~290 mOsm. Neurons were voltage-clamped at −60 mV. Electrophysiological recordings and data acquisition were carried out with a Multiclamp 700B amplifier and Clampfit 10.7 software (Molecular Devices, Sunnyvale, CA, USA). Miniature excitatory postsynaptic currents (mEPSC) were detected and analyzed using the Clampfit EventDetection module. Only events with an amplitude >8 pA, an exponential decay and a monotonic rising phase, which could be clearly discriminated from the background noise, were considered as mEPSCs. mEPSC amplitude and frequency were compared during a 2-min baseline period and in 2-min windows 15 min after 200 μM DHPG application (5 min). Decay time constants (τ) were calculated from single exponential fits of averaged mEPSC traces.
Statistical Analysis
Statistical analyses were performed using STATISTICA software, version 7 (StatSoft). Shapiro-Wilk test was applied to determine the distribution of the data. In normal distributions, one-way or two-way ANOVA test, following of a post hoc Bonferroni test were applied. In non-normal distributions (mEPSC parameters) Wilcoxon matched paired test was applied. Significance was *p < 0.05, **p < 0.01, ***p < 0.001. All data are expressed as the mean ± SEM.
Results
Store-Operated Ca2+ Entry Is Induced in Cortical Neurons after Store Depletion
To assess the presence of SOCE pathway in primary cortical neurons, we depleted ER stores using 1 μM Tg, the sarco/endoplasmatic reticulum Ca2+ ATPase (SERCA) inhibitor, in a medium with 1 μM TTX, in order to prevent neuronal activity. Tg application resulted in a sustained increase in cytosolic calcium (Figure 1A, black circles) that lasted for several minutes. To determine whether Ca2+ entering the cell through SOC channels was contributing to this cytosolic Ca2+ rise, we applied different widely used SOCE blockers along with Tg: 2-APB and YM-58483 (also called BTP2; Xia et al., 2014; Prakriya and Lewis, 2015). YM-58483 is highly specific and has been shown not to interfere with voltage-gated calcium channels (VGCC; Xia et al., 2014), and 2-APB is a commonly used bimodal SOCE modulator which decreases SOCE activity at high concentrations and also inhibits IP3 signaling (Prakriya and Lewis, 2015). In neurons, it is known that ER Ca2+ is continuously being released and replenished and consequently SOCE is active even in resting conditions (Samtleben et al., 2015). To avoid a drop in ER calcium caused by SOCE inhibition, the SOCE blockers were not preincubated but applied together with Tg. In the presence of 10 μM YM-58483 or 50 μM 2-APB, the Tg-induced cytosolic Ca2+ signal was strongly attenuated, with a rapid decrease 1–2 min after onset (Figures 1A–C). This result is consistent with a Ca2+ entry through SOC channel upon depletion of Ca2+ ER stores with Tg.
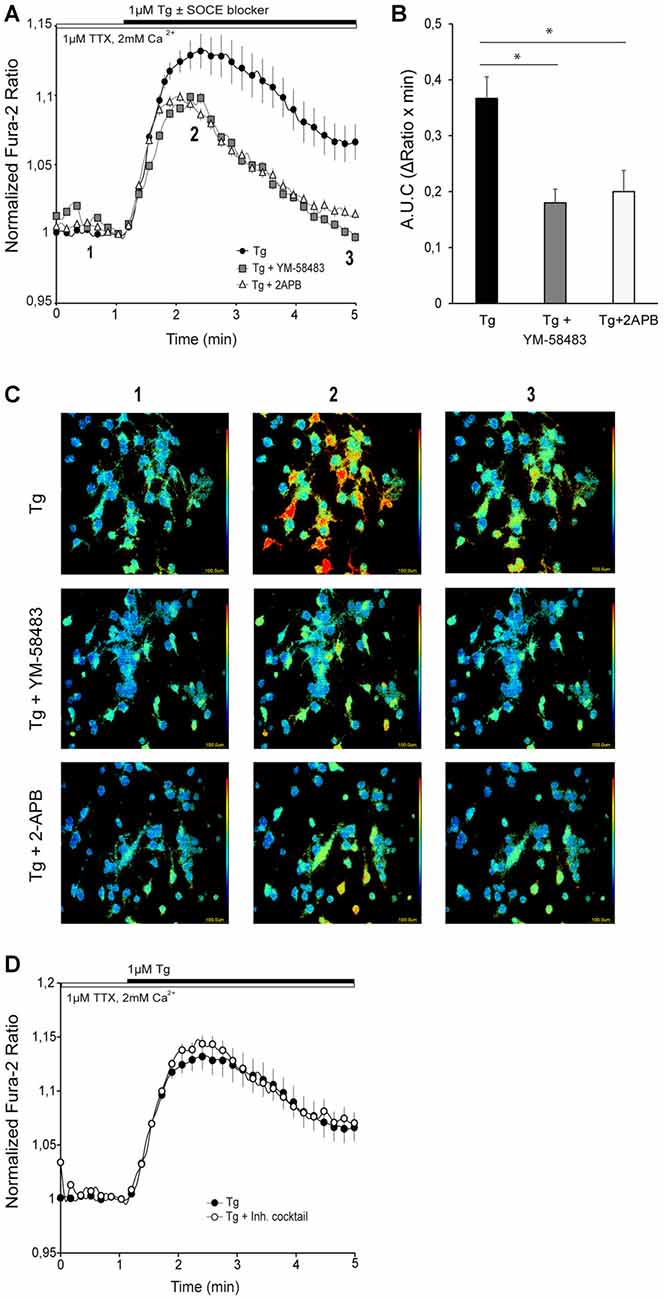
Figure 1. Store-operated calcium entry (SOCE) activity in primary cortical neurons. (A) Fura-2 [Ca2+]i signals in cortical neurons in HCSS medium containing 2 mM CaCl2 and 1 μM tetrodotoxin (TTX), upon addition of 1 μM thapsigargin (Tg) ± 10 μM YM-58483 or 50 μM (2-Aminoethoxydiphenyl borate (2-APB) were indicated. The numbers refer to the frames reported in the (C). (B) Quantification of area under SOCE curve (A.U.C, ΔRatio·min). Data were obtained from three independent experiments (n = 3), and in each one a total of 25–30 cells were analyzed. (C) Representative color-coded Fura-2 [Ca2+]i ratio images from basal situation (1), immediately after Tg ± SOCE blockers addition (2), and the end of the measurement. Pseudo-color ratio images were obtained using Aquacosmos 2.5 software (Hamamatsu). (D) Fura-2 [Ca2+]i signals in cortical neurons in HCSS medium containing 2 mM CaCl2 and 1 μM TTX ± inhibitory cocktail (10 μM CNQX (6-cyano-7-nitroquinoxaline-2,3-dione), 10 μM MK-801, 5 μM MPEP, 50 μM NiCl2), upon addition of 1 μM Tg. Data were obtained from three independent experiments (n = 3), and in each one a total of 25–30 cells were analyzed. All data are normalized to the initial values and are expressed as mean ± SEM. Means were compared using one-way ANOVA, *p < 0.05, post hoc Bonferroni test.
In addition, to test the possibility that neuronal activity contributed to this calcium signal, cytosolic Ca2+ levels were recorded in the presence of an inhibitor cocktail containing 10 μM CNQX (an AMPA/Kainate receptor inhibitor), 10 μM MK-801 (NMDA receptor inhibitor) and 5 μM MPEP (a mGluR inhibitor) to block excitatory action of ionotropic and metabotropic glutamate receptors, 10 μM TTX to block voltage-gated sodium channels, and 50 μM NiCl2 to block voltage-activated T-type calcium channels (the inhibition cocktail was present during the whole experiment). Figure 1D shows that the cytosolic Ca2+ increase triggered by emptying ER-Ca2+ was similar both in the absence and the presence of the inhibitory cocktail. This result contrasts with those recently reported in rat cortical neurons using a different SOCE induction protocol, in which AMPA receptors appeared to contribute to SOC mediated Ca2+ entry (Gruszczynska-Biegala et al., 2016). This difference could arise from the protocols used to measure SOCE activity in neurons. In this study, ER-Ca2+ depletion was triggered in a medium containing physiological calcium concentration, while Gruszczynska-Biegala et al. (2016) employed the Ca2+ addback protocol, in which addition of Ca2+ to the external medium may activate neuronal Ca2+ channels by changing the membrane excitability as described elsewhere (Lu and Fivaz, 2016).
SOC Channel Proteins Are Expressed in Mouse Cortical Neurons
We have studied the different candidate proteins forming SOC channels in other cell types, Orai and Trpc families as calcium channels located in PM and Stim family as calcium sensor in ER-membrane. We first investigated the mRNA levels of the different isoforms of the SOCE components, Orai, Stim and Trpc families, by RT-qPCR in 8–9 DIV cultured cortical neurons from four independent embryos. The profiles obtained for each embryo were quite similar to one another and we found that the isoforms forming the pore channel with highest mRNA levels were Orai1, Orai2, Trpc1 and Trpc4, while Stim1 and Stim2 mRNA levels were similar (Figure 2A). Then, protein levels of the SOCE components with highest mRNA levels were investigated by Western Blot. As positive controls, we used the human neuroblastoma SH-SY5Y cell line, which expresses these proteins (Olianas et al., 2014). We observed a band at 50 kDa with the ORAI1 antibody, a single band at 110 kDa with the TRPC4 antibody and a band at 90 kDa with the STIM2 antibody, but not bands corresponding to ORAI2 and TRPC1 proteins (Figure 2B). These results show that in cultured cortical neurons, ORAI1, STIM2 and TRPC4 were the predominant members of their respective families, in agreement with previous reports in cortical and other neurons of the central nervous system (CNS; Berna-Erro et al., 2009; Steinbeck et al., 2011; Korkotian et al., 2017). STIM1 proteins are also present in mouse cortical neurons (Hou et al., 2015; Guner et al., 2017), but STIM2 has been described as predominant isoform in cortex and hippocampus (Berna-Erro et al., 2009; Steinbeck et al., 2011), while STIM1 protein expression is higher in cerebellum (Lein et al., 2007; Hartmann et al., 2014; Kraft, 2015). Therefore, ORAI1, TRPC4 and STIM1 and 2, are probably responsible for SOC entry in mouse cortical neurons.
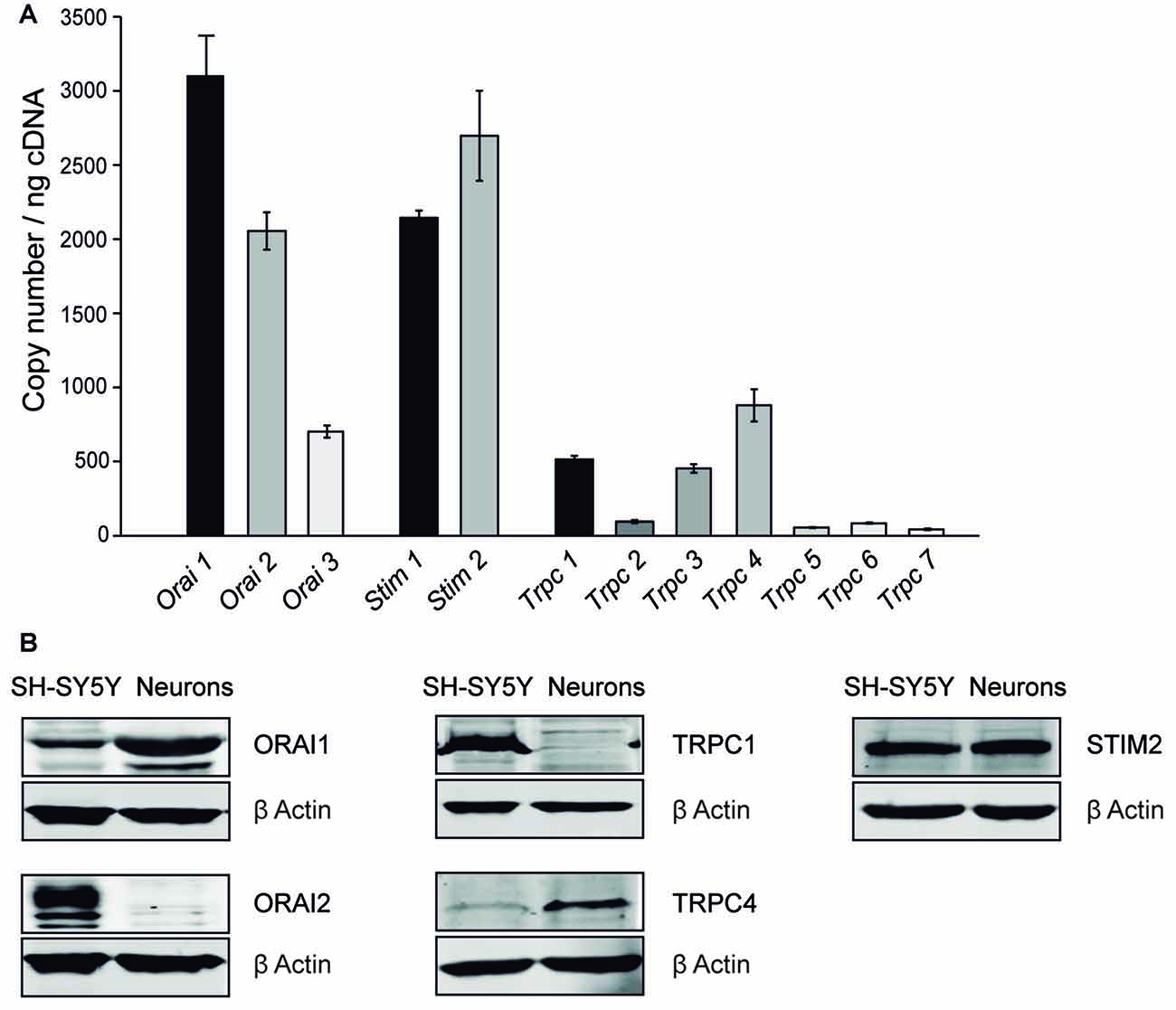
Figure 2. SOC channel proteins in mouse cortical neurons. (A) Real time PCR (RT-PCR) of DNA complementary from primary cortical neurons obtained from E-15 wild-type mouse embryo with Orai 1–3, Stim 1, 2 and Trpc 1–7 specific primers. The experiment was repeated four times with similar results. (B) Western Blot analysis of ORAI 1, ORAI 2, STIM 2, TRPC 1 and TRPC 4 levels. Protein extracts were obtained from SH-SY5Y cells and 8–9 day in vitro (DIV) cortical neurons. Primary antibodies used were α-Orai1, α-Orai2, α-Trpc1, α-Trpc4, α-Stim2 and α -β Actin as a control.
SOCE Is Involved in mGluR-Driven Ca2+ Signals
After determining that ER-Ca2+ depletion in neurons triggers Ca2+ influx into the cytosol by SOCE, we sought to study the activation of this pathway by physiological agonists, particularly glutamate. Activation of group I mGluRs (mGluRs I), formed by mGluR1 and 5, is coupled to a variety of signaling pathways. The Gαq/Gα11 protein/PLC/inositol-3,4,5-triphosphate (IP3) signal cascades have been considered as the canonical pathway, which leads to Ca2+ release from ER and activation of PKC. The decrease of ER-Ca2+ levels could activate SOCE in the PM and play a role in mGluR-driven Ca2+ signaling.
The experiments were performed at 10–12 DIV in a medium with 1 μM TTX in order to eliminate spontaneous activity. Application of 200 μM DHPG, a selective agonist of group I mGluRs, evoked a rapid increase in cytosolic Ca2+ followed by a more moderate but sustained Ca2+ signal that did not reach baseline during the recording period (Figure 3A, black circles). To determine the contribution of SOCE to the Ca2+ response to DHPG, we applied the SOCE blockers together with the agonist of mGluR I, as in previous experiments with Tg (Figure 1). Interestingly, we found that the cytosolic Ca2+ transient rapidly returned to baseline when 10 μM YM-58483 or 50 μM 2-APB were added together with DHPG (Figures 3A,B). This result indicates that SOCE is responsible for the sustained increase in Ca2+ levels above baseline in response to DHPG. On the other hand, the presence of YM-58483 did not affect the initial Ca2+ peak evoked by mGluRs, suggesting that SOC channels open secondarily to mGluRs activation and that the first peak involves the ER-Ca2+ mobilization by PLC pathway. In contrast, the addition of 2-APB decreased the initial Ca2+ peak, consistent with the effect of this compound on IP3 signaling (Prakriya and Lewis, 2015).
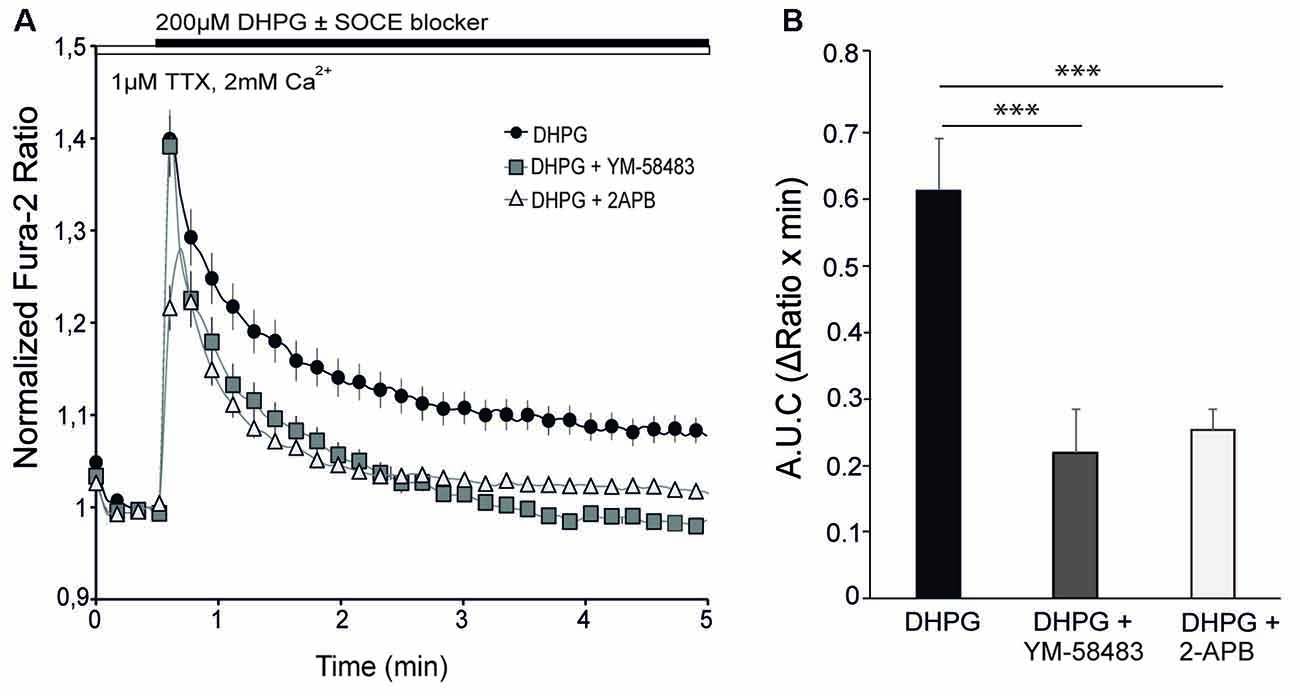
Figure 3. SOCE is involved in metabotropic glutamate receptors (mGluRs)-driven Ca2+ signal. (A) Fura-2 [Ca2+]i signals in cortical neurons in HCSS medium containing 2 mM CaCl2 and 1 μM tetrodotoxin (TTX) upon addition of 200 μM (S)-3,5-dihydroxyphenylglycine (DHPG) ± 10 μM YM-58483 or 50 μM 2-APB were indicated. (B) Quantification of area under the curve (ΔRatio·min) after DHPG ± YM-58483 or 2-APB addition. Data were obtained from three independent experiments (n = 3), and in each one a total of 25–30 cells were analyzed. All data are expressed as mean ± SEM and means were compared using one-way ANOVA, ***p < 0.001, post hoc Bonferroni test.
SOCE Inhibition Impairs DHPG-LTD
Finally, we wished to determine whether SOCE-dependent calcium signal in response to mGluR activation had physiological consequences for neuronal function. Stimulation of group I mGluRs can lead to a decrease in synaptic strength at many synapses, referred to as long-term depression (mGluR-LTD; Lüscher and Huber, 2010). This form of LTD can be induced by the application of the group I selective mGluR agonist DHPG (so called DHPG-LTD; Xiao et al., 2001; Jo et al., 2008), but the underlying molecular mechanism is still poorly understood and appears to depend on the specific neuronal type. Having shown that SOCE was activated after stimulation of group I mGluRs and was involved in the maintenance of cytosolic Ca2+ signal, we investigated whether the activation of SOCE may be involved in DHPG-LTD in cortical neurons. To this end, we first examined the effect of DHPG on synaptic transmission in primary cortical neurons. We found that bath application of 200 μM DHPG (5 min) caused a long lasting reduction of the mEPSC amplitude at 15 min after mGluR stimulation (Figures 4A,B,I). In contrast, we did not find any significant effect on the frequency of mEPSCs (Figures 4A,C,J), suggesting that this type of LTD is predominantly expressed via a postsynaptic mechanism. This interpretation is also consistent with an increase in the decay time constant of mEPSCs after DHPG application (Figures 4D,K). To study the contribution of SOCE to this form of synaptic plasticity, we evaluated the effect of the SOCE blocker YM-58483 (10 μM) applied together with DHPG. Interestingly, the long lasting decrease of mEPSCs amplitude was significantly attenuated (7.7 ± 2.9%; Figures 4E,F,I), as well as the effect on mEPSC kinetics (Figures 4H,K). The frequency of mEPSCs was not affected by DHPG with or without YM-58483 (Figures 4G,J). Importantly, bath application of 10 μM YM-58483 did not have any effect on its own on the amplitude, frequency or kinetics of mEPSCs (Figures 4I–K). All these results strongly suggest that the calcium influx via SOCE after group I mGluRs stimulation is a necessary step for the development of DHPG-LTD in cortical neurons.
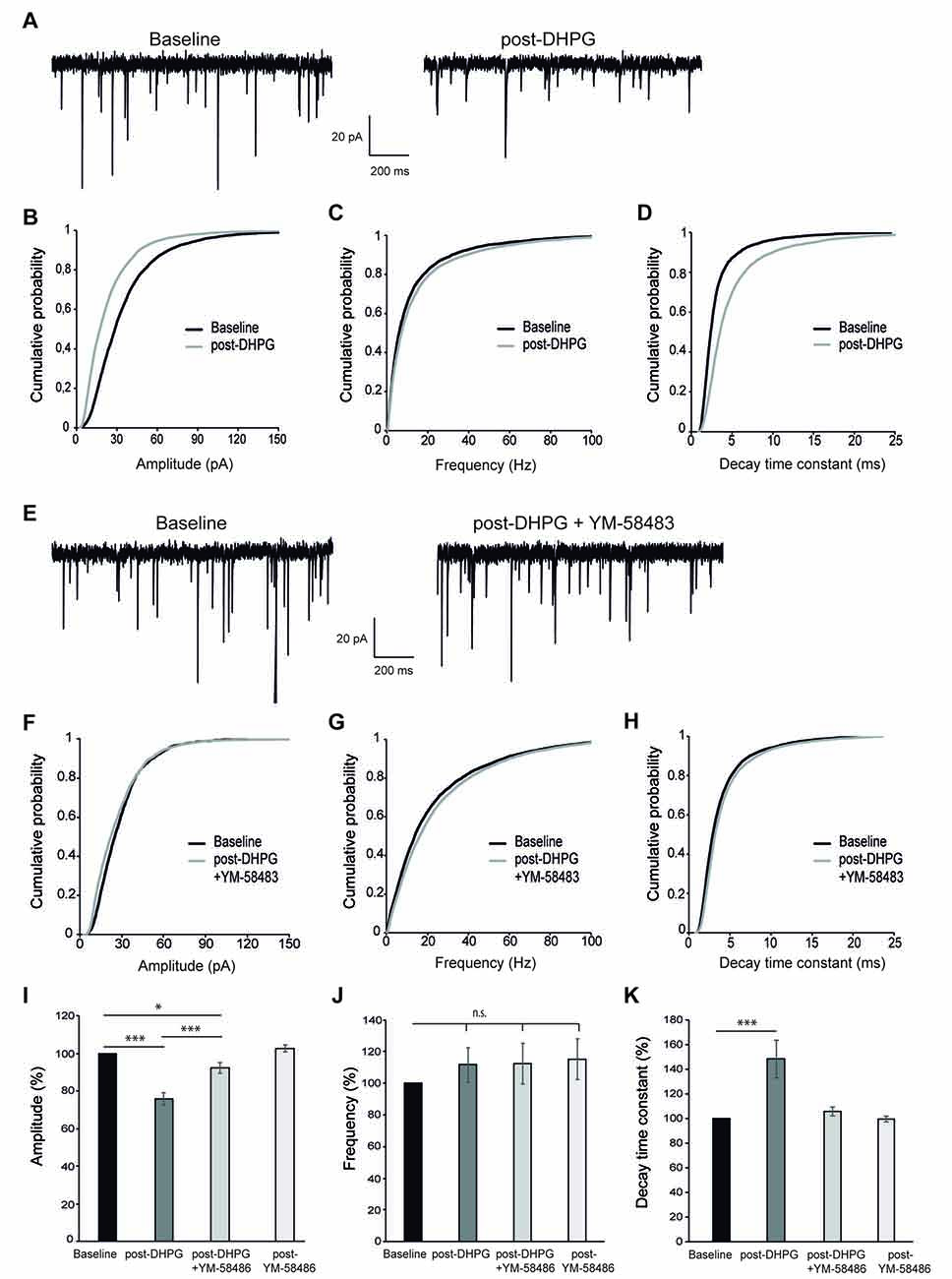
Figure 4. DHPG-long-term depression (LTD) is impaired by SOCE inhibition. (A) Representative miniature excitatory postsynaptic current (mEPSC) recordings from a cortical neuron before (left, “baseline”) and 15 min after 200 μM DHPG application (right, “post-DHPG”). (B–D) Cumulative probability graphs for amplitude (B), frequency (C) and decay time constant (D) of mEPSCs, showing baseline (2 min) and a 2 min interval beginning 15 min after DHPG application. (E) Representative mEPSC recordings from a cortical neuron before and 15 min after 200 μM DHPG + 10 μM YM-58483 application. (F–H) Cumulative probability graphs for amplitude (F), frequency (G) and decay time constant (H) of mEPSCs, showing baseline (2 min) and a 2 min interval beginning 15 min after DHPG + YM-58483 application. (I–K) Mean of mEPSCs amplitude (%) (I), frequency (%) (J) and decay time constant (K) comparing baseline (100%) to values obtained 15 min after application of 200 μM DHPG, 200 μM DHPG + 10 μM YM-58483 and 10 μM YM-58483 respectively. Data are expressed as mean ± SEM and were obtained from 14 independent experiments for results of DHPG addition, 13 for those of DHPG addition along with YM-58483 and six for those of YM-58483 application alone. Due to non-normal distribution of mEPSCs parameters, statistics were performed using the Wilcoxon matched pairs test, *p < 0.05, ***p < 0.001.
Discussion
SOCE is the main mechanism to replenish intracellular calcium stores in non-excitable cells but its existence in neurons is under debate (Putney, 2003; Lu and Fivaz, 2016), as neurons exhibit other major Ca2+ influx pathways through VGCCs and ionotropic glutamate receptors (Grienberger and Konnerth, 2012). Whereas Ca2+ entry through SOC channels has been reported in different neuronal populations (Emptage et al., 2001; Berna-Erro et al., 2009; Gemes et al., 2011; Alkhani et al., 2014; Sun et al., 2014; Xia et al., 2014), other groups did not find a clear SOCE response in neurons (Park et al., 2010; Garcia-Alvarez et al., 2015a). One of the possible reasons for this discrepancy could stem from the commonly used Ca2+ addback protocol to study SOCE, in which neuronal stores are emptied in a Ca2+-free medium, and then Ca2+ is added to the external medium. Neurons express multiple Ca2+ channels that can be activated in response to changes in extracellular calcium. In the present study, we investigated the existence of SOCE in cortical neurons in relatively mature state, in a medium containing physiological calcium concentrations. Our results clearly showed a small but maintained increase in cytosolic Ca2+ after Tg application, which rapidly returned to basal levels in the presence of SOCE blockers, and was unaltered after a cocktail of channel/receptor inhibitors, i.e., all conditions consistent with a bona fide Tg-induced SOCE. The analysis of the molecular players of SOCE in mouse cortical neurons, showed that STIM2 and probably STIM1 are highly expressed at the mRNA and protein levels, in agreement with previous findings (Berna-Erro et al., 2009; Gruszczynska-Biegala et al., 2011; Guner et al., 2017). In the PM, ORAI1 seems to form the store-operated pore, and to a lesser extent, TRPC4, as reported for cortical and hippocampal neurons (Klejman et al., 2009; Korkotian et al., 2017).
A second finding of this study was that a physiological stimulus, group I mGluR activation, induces SOCE in mouse cortical neurons. Group I metabotropic receptors are linked to activation of PLC, IP3 generation and release of Ca2+ from intracellular stores (Niswender and Conn, 2010). Here, we show that store depletion after mGluR I stimulation using the specific agonist DHPG activates SOCE in primary cortical neurons, and SOCE was necessary to maintain the mGluR-driven cytosolic Ca2+ signal. This result is in agreement with a previous report in which stimulation of mGluR I triggered STIM1 oligomerization and migration to PM (Ng et al., 2011), the first step needed in SOCE induction (Lewis, 2007). In fact, more recent findings point to a central role of STIM proteins as one of the intracellular links between mGluR I and its downstream effectors (Hartmann et al., 2014; Hou et al., 2015).
mGluRs are widely distributed throughout the CNS and play relevant roles for synaptic transmission, activity-dependent synaptic plasticity and higher cognitive functions (Niswender and Conn, 2010). Particularly, group I mGluRs induce a specific form of synaptic plasticity leading to long-term depression (LTD) of excitatory synaptic strength (mGluR LTD). In this work, we have observed that SOCE inhibition caused a strong impairment of DHPG-LTD in primary cortical neurons. In relation with SOCE components, STIM1 has been found to control mGluR1-dependent slow excitatory potentials in Purkinje neurons through its action in maintaining ER-Ca2+ levels (Hartmann et al., 2014), and both STIM2 knockout (Yap et al., 2017) and STIM1/STIM2 double knockout mice (Garcia-Alvarez et al., 2015b) have impaired LTP and LTD.
mGluR-LTD has been broadly studied in hippocampus and cerebellum (Jörntell and Hansel, 2006; Gladding et al., 2009; Lüscher and Huber, 2010), but less is known about other brain regions. It is still debated whether the expression mechanism of DHPG-LTD relies on presynaptic (neurotransmitter release) or postsynaptic (AMPA receptor internalization) changes (Anwyl, 2006). Moreover, in hippocampal neurons, the expression mechanism of DHPG-LTD depends on the developmental state of the synapse, being presynaptic in immature neurons but postsynaptic after neuronal maturation (Nosyreva and Huber, 2005). Our results show a clear reduction in the amplitude, but not frequency, of mEPSCs 15 min after DHPG application, which suggests a postsynaptic mechanism of expression. This agrees with the high maturation state of our neuronal cultures which has been previously documented when the BrainPhys Neuronal Medium was described (Bardy et al., 2015).
In cortical neurons, the strong impairment of DHPG-LTD and the failure to maintain DHPG-driven cytosolic Ca2+ signals in the presence of the SOCE blocker YM-58483 suggest that SOCE-mediated Ca2+ signal is essential to activate signaling pathways downstream mGluRs stimulation. It is particularly interesting that SOCE was required specifically for the maintenance of the calcium signal after mGluR activation, suggesting that this sustained signal is responsible for the long-term synaptic effects. SOCE may be acting directly by extracellular calcium influx through SOCs channels or by SOCE’s known function in maintaining the ER-Ca2+ levels (Samtleben et al., 2015), which are reduced by mGluRs stimulation. According to this, it has been demonstrated that ER-Ca2+ release from internal stores via IP3 generation after mGluR stimulation is required to induced LTD in cerebellum (Miyata et al., 2000; Kano et al., 2008). In the perirhinal cortex the induction of DHPG-LTD depends on interactions between the neuronal Ca2+ sensor protein (NCS-1) and protein interacting with C kinase (PICK1) in a Ca2+-dependent manner (Jo et al., 2008). However, it is relevant to note that the molecular mechanisms seem to differ between brain regions. Indeed, in hippocampus, DHPG-LTD is considered to be independent of PLC/IP3 pathway and postsynaptic Ca2+ (Schnabel et al., 1999; Fitzjohn et al., 2001; Mockett et al., 2011; Kim et al., 2015), but there is also evidence of IP3 mediated ER-Ca2+ release from dendritic spines and calmodulin activation for group I mGluR LTD (Holbro et al., 2009; Sethna et al., 2016).
Although Ca2+ influx through SOCs channels has been implicated in several functions in dendritic spines (Brini et al., 2014; Sun et al., 2014; Korkotian and Segal, 2017; Korkotian et al., 2017), SOCE proteins may also play roles outside the canonical SOCE pathway. A SOCE-independent activation of STIM proteins was first described in non-excitable cells (Lefkimmiatis et al., 2009; Tian et al., 2012; Shinde et al., 2013), and recently, this behavior has been also found in neurons. It has been shown that, in hippocampal neurons, STIM2 mediated PKA-dependent phosphorylation and trafficking of AMPA receptors is associated with an increase in cAMP levels (Garcia-Alvarez et al., 2015a). A recent study found that STIM1 overexpression in hippocampus impairs both synaptically and chemically induced mGluR-LTD, without affecting SOCE, among other calcium parameters (Majewski et al., 2017). As to the SOCE blocker used in this study to perform the experiments of DHPG-LTD, YM-58483, it seems to exert its action on the channel itself, without interfering with STIM proteins (He et al., 2005). Therefore, our results suggest a relevant role of Ca2+ entry through SOC channels themselves in DHPG-LTD.
In conclusion, our findings reveal a close relationship between mGluRs and SOCE in cortical neurons, and show that SOCE activation is a necessary step in the development of DHPG-driven cytosolic Ca2+ signals. Blocking SOCE activation upon mGluR stimulation prevented DHPG-LTD. Altogether the results suggest that, contrary to what has been observed in hippocampus, in cortical neurons SOCE-mediated Ca2+ signal could be relevant in generating downstream effects to evoke mGluR-LTD.
Author Contributions
PG-S: designed and performed the experiments, analyzed the data and wrote the manuscript. AdA: designed and performed the experiments. JAE and JS: designed the study and wrote the manuscript. All authors reviewed the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work has been funded by grants from the Spanish Ministry of Science and Innovation (Ministerio de Ciencia e Innovación) and BFU2011-30456-C02-01/BMC and SAF2014-56929 (to JS) and SAF2014-57233-R and PCIN-2016-095 (to JAE), the Consejería de Educación, Juventud y Deporte, Comunidad de Madrid S2010/BMD-2402 MITOLAB-CM (to JS), by an institutional grant from the Fundación Ramón Areces to the Centro de Biología Molecular Severo Ochoa. This work has also been funded by the CIBERER, an initiative from the Instituto de Salud Carlos III. PG-S is a recipient of a Formacion Personal Investigador-fellowship from Universidad Autónoma de Madrid (FPI-UAM). We thank Isabel Manso for technical support and Marta Navarrete and Jonathan Draffin for their help with data analysis.
Footnotes
References
Abe, T., Sugihara, H., Nawa, H., Shigemoto, R., Mizuno, N., and Nakanishi, S. (1992). Molecular characterization of a novel metabotropic glutamate receptor mGluR5 coupled to inositol phosphate/Ca2+ signal transduction. J. Biol. Chem. 267, 13361–13368.
Alkhani, H., Ase, A. R., Grant, R., O’Donnell, D., Groschner, K., and Séguéla, P. (2014). Contribution of TRPC3 to store-operated calcium entry and inflammatory transductions in primary nociceptors. Mol. Pain 10:43. doi: 10.1186/1744-8069-10-43
Anwyl, R. (2006). Induction and expression mechanisms of postsynaptic NMDA receptor-independent homosynaptic long-term depression. Prog. Neurobiol. 78, 17–37. doi: 10.1016/j.pneurobio.2005.12.001
Aramori, I., and Nakanishi, S. (1992). Signal transduction and pharmacological characteristics of a metabotropic glutamate receptor, mGluR1, in transfected CHO cells. Neuron 8, 757–765. doi: 10.1016/0896-6273(92)90096-v
Baba, A., Yasui, T., Fujisawa, S., Yamada, R. X., Yamada, M. K., Nishiyama, N., et al. (2003). Activity-evoked capacitative Ca2+ entry: implications in synaptic plasticity. J. Neurosci. 23, 7737–7741.
Bardo, S., Cavazzini, M. G., and Emptage, N. (2006). The role of the endoplasmic reticulum Ca2+ store in the plasticity of central neurons. Trends Pharmacol. Sci. 27, 78–84. doi: 10.1016/j.tips.2005.12.008
Bardy, C., van den Hurk, M., Eames, T., Marchand, C., Hernandez, R. V., Kellogg, M., et al. (2015). Neuronal medium that supports basic synaptic functions and activity of human neurons in vitro. Proc. Natl. Acad. Sci. U S A 112, E2725–E2734. doi: 10.1073/pnas.1504393112
Berna-Erro, A., Braun, A., Kraft, R., Kleinschnitz, C., Schuhmann, M. K., Stegner, D., et al. (2009). STIM2 regulates capacitive Ca2+ entry in neurons and plays a key role in hypoxic neuronal cell death. Sci. Signal. 2:ra67. doi: 10.1126/scisignal.2000522
Brini, M., Cali, T., Ottolini, D., and Carafoli, E. (2013). Intracellular calcium homeostasis and signaling. Met. Ions Life Sci. 12, 119–168. doi: 10.1007/978-94-007-5561-1_5
Brini, M., Cali, T., Ottolini, D., and Carafoli, E. (2014). Neuronal calcium signaling: function and dysfunction. Cell. Mol. Life Sci. 71, 2787–2814. doi: 10.1007/s00018-013-1550-7
Cavazzini, M., Bliss, T., and Emptage, N. (2005). Ca2+ and synaptic plasticity. Cell Calcium 38, 355–367. doi: 10.1016/j.ceca.2005.06.013
Dittmer, P. J., Wild, A. R., Dell’Acqua, M. L., and Sather, W. A. (2017). STIM1 Ca2+ sensor control of L-type Ca2+-channel-dependent dendritic spine structural plasticity and nuclear signaling. Cell Rep. 19, 321–334. doi: 10.1016/j.celrep.2017.03.056
Emptage, N. J., Reid, C. A., and Fine, A. (2001). Calcium stores in hippocampal synaptic boutons mediate short-term plasticity, store-operated Ca2+ entry, and spontaneous transmitter release. Neuron 29, 197–208. doi: 10.1016/s0896-6273(01)00190-8
Feske, S., Prakriya, M., Rao, A., and Lewis, R. S. (2005). A severe defect in CRAC Ca2+ channel activation and altered K+ channel gating in T cells from immunodeficient patients. J. Exp. Med. 202, 651–662. doi: 10.1084/jem.20050687
Fitzjohn, S. M., Palmer, M. J., May, J. E., Neeson, A., Morris, S. A., and Collingridge, G. L. (2001). A characterisation of long-term depression induced by metabotropic glutamate receptor activation in the rat hippocampus in vitro. J. Physiol. 537, 421–430. doi: 10.1111/j.1469-7793.2001.00421.x
Garcia-Alvarez, G., Lu, B., Yap, K. A., Wong, L. C., Thevathasan, J. V., Lim, L., et al. (2015a). STIM2 regulates PKA-dependent phosphorylation and trafficking of AMPARs. Mol. Biol. Cell 26, 1141–1159. doi: 10.1091/mbc.e14-07-1222
Garcia-Alvarez, G., Shetty, M. S., Lu, B., Yap, K. A., Oh-Hora, M., Sajikumar, S., et al. (2015b). Impaired spatial memory and enhanced long-term potentiation in mice with forebrain-specific ablation of the stim genes. Front. Behav. Neurosci. 9:180. doi: 10.3389/fnbeh.2015.00180
Gemes, G., Bangaru, M. L., Wu, H. E., Tang, Q., Weihrauch, D., Koopmeiners, A. S., et al. (2011). Store-operated Ca2+ entry in sensory neurons: functional role and the effect of painful nerve injury. J. Neurosci. 31, 3536–3549. doi: 10.1523/jneurosci.5053-10.2011
Gladding, C. M., Fitzjohn, S. M., and Molnár, E. (2009). Metabotropic glutamate receptor-mediated long-term depression: molecular mechanisms. Pharmacol. Rev. 61, 395–412. doi: 10.1124/pr.109.001735
González-Sánchez, P., Pla-Martín, D., Martinez-Valero, P., Rueda, C. B., Calpena, E., Del Arco, A., et al. (2017). CMT-linked loss-of-function mutations in GDAP1 impair store-operated Ca2+ entry-stimulated respiration. Sci. Rep. 7:42993. doi: 10.1038/srep42993
Grienberger, C., and Konnerth, A. (2012). Imaging calcium in neurons. Neuron 73, 862–885. doi: 10.1016/j.neuron.2012.02.011
Gruszczynska-Biegala, J., Pomorski, P., Wisniewska, M. B., and Kuznicki, J. (2011). Differential roles for STIM1 and STIM2 in store-operated calcium entry in rat neurons. PLoS One 6:e19285. doi: 10.1371/journal.pone.0019285
Gruszczynska-Biegala, J., Sladowska, M., and Kuznicki, J. (2016). AMPA receptors are involved in store-operated calcium entry and interact with STIM proteins in rat primary cortical neurons. Front. Cell. Neurosci. 10:251. doi: 10.3389/fncel.2016.00251
Guner, G., Guzelsoy, G., Isleyen, F. S., Sahin, G. S., Akkaya, C., Bayam, E., et al. (2017). NEUROD2 regulates Stim1 expression and store-operated calcium entry in cortical neurons. eNeuro 4:ENEURO.0255-16.2017. doi: 10.1523/eneuro.0255-16.2017
Gwack, Y., Srikanth, S., Feske, S., Cruz-Guilloty, F., Oh-hora, M., Neems, D. S., et al. (2007). Biochemical and functional characterization of Orai proteins. J. Biol. Chem. 282, 16232–16243. doi: 10.1074/jbc.M609630200
Hartmann, J., Karl, R. M., Alexander, R. P., Adelsberger, H., Brill, M. S., Rühlmann, C., et al. (2014). STIM1 controls neuronal Ca2+ signaling, mGluR1-dependent synaptic transmission and cerebellar motor behavior. Neuron 82, 635–644. doi: 10.1016/j.neuron.2014.03.027
He, L. P., Hewavitharana, T., Soboloff, J., Spassova, M. A., and Gill, D. L. (2005). A functional link between store-operated and TRPC channels revealed by the 3,5-bis(trifluoromethyl)pyrazole derivative, BTP2. J. Biol. Chem. 280, 10997–11006. doi: 10.1074/jbc.m411797200
Holbro, N., Grunditz, A., and Oertner, T. G. (2009). Differential distribution of endoplasmic reticulum controls metabotropic signaling and plasticity at hippocampal synapses. Proc. Natl. Acad. Sci. U S A 106, 15055–15060. doi: 10.1073/pnas.0905110106
Hou, P.-F., Liu, Z.-H., Li, N., Cheng, W.-J., and Guo, S.-W. (2015). Knockdown of STIM1 improves neuronal survival after traumatic neuronal injury through regulating mGluR1-dependent Ca2+ signaling in mouse cortical neurons. Cell. Mol. Neurobiol. 35, 283–292. doi: 10.1007/s10571-014-0123-0
Jo, J., Heon, S., Kim, M. J., Son, G. H., Park, Y., Henley, J. M., et al. (2008). Metabotropic glutamate receptor-mediated LTD involves two interacting Ca2+ sensors, NCS-1 and PICK1. Neuron 60, 1095–1111. doi: 10.1016/j.neuron.2008.10.050
Jörntell, H., and Hansel, C. (2006). Synaptic memories upside down: bidirectional plasticity at cerebellar parallel fiber-Purkinje cell synapses. Neuron 52, 227–238. doi: 10.1016/j.neuron.2006.10.015
Kann, O., Taubenberger, N., Huchzermeyer, C., Papageorgiou, I. E., Benninger, F., Heinemann, U., et al. (2012). Muscarinic receptor activation determines the effects of store-operated Ca2+-entry on excitability and energy metabolism in pyramidal neurons. Cell Calcium 51, 40–50. doi: 10.1016/j.ceca.2011.10.004
Kano, M., Hashimoto, K., and Tabata, T. (2008). Type-1 metabotropic glutamate receptor in cerebellar Purkinje cells: a key molecule responsible for long-term depression, endocannabinoid signalling and synapse elimination. Philos. Trans. R. Soc. Lond B Biol. Sci. 363, 2173–2186. doi: 10.1098/rstb.2008.2270
Kim, H. H., Lee, K. H., Lee, D., Han, Y. E., Lee, S. H., Sohn, J. W., et al. (2015). Costimulation of AMPA and metabotropic glutamate receptors underlies phospholipase C activation by glutamate in hippocampus. J. Neurosci. 35, 6401–6412. doi: 10.1523/jneurosci.4208-14.2015
Klejman, M. E., Gruszczynska-Biegala, J., Skibinska-Kijek, A., Wisniewska, M. B., Misztal, K., Blazejczyk, M., et al. (2009). Expression of STIM1 in brain and puncta-like co-localization of STIM1 and ORAI1 upon depletion of Ca2+ store in neurons. Neurochem. Int. 54, 49–55. doi: 10.1016/j.neuint.2008.10.005
Korkotian, E., Oni-Biton, E., and Segal, M. (2017). The role of the store-operated calcium entry channel Orai1 in cultured rat hippocampal synapse formation and plasticity. J. Physiol. 595, 125–140. doi: 10.1113/jp272645
Korkotian, E., and Segal, M. (2017). Orai1 regulates calcium entry into dendritic spines. Channels (Austin) 11, 99–100. doi: 10.1080/19336950.2016.1247528
Kraft, R. (2015). STIM and ORAI proteins in the nervous system. Channels (Austin) 9, 245–252. doi: 10.1080/19336950.2015.1071747
Lalonde, J., Saia, G., and Gill, G. (2014). Store-operated calcium entry promotes the degradation of the transcription factor Sp4 in resting neurons. Sci. Signal. 7:ra51. doi: 10.1126/scisignal.2005242
Lefkimmiatis, K., Srikanthan, M., Maiellaro, I., Moyer, M. P., Curci, S., and Hofer, A. M. (2009). Store-operated cyclic AMP signalling mediated by STIM1. Nat. Cell Biol. 11, 433–442. doi: 10.1038/ncb1850
Lein, E. S., Hawrylycz, M. J., Ao, N., Ayres, M., Bensinger, A., Bernard, A., et al. (2007). Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176. doi: 10.1038/nature05453
Lewis, R. S. (2007). The molecular choreography of a store-operated calcium channel. Nature 446, 284–287. doi: 10.1038/nature05637
Liou, J., Kim, M. L., Heo, W. D., Jones, J. T., Myers, J. W., Ferrell, J. E., et al. (2005). STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 15, 1235–1241. doi: 10.1016/j.cub.2005.05.055
Lu, B., and Fivaz, M. (2016). Neuronal SOCE: myth or reality? Trends Cell Biol. 26, 890–893. doi: 10.1016/j.tcb.2016.09.008
Lüscher, C., and Huber, K. M. (2010). Group 1 mGluR-dependent synaptic long-term depression: mechanisms and implications for circuitry and disease. Neuron 65, 445–459. doi: 10.1016/j.neuron.2010.01.016
Majewski, L., and Kuznicki, J. (2015). SOCE in neurons: signaling or just refilling? Biochim. Biophys. Acta 1853, 1940–1952. doi: 10.1016/j.bbamcr.2015.01.019
Majewski, L., Maciag, F., Boguszewski, P. M., Wasilewska, I., Wiera, G., Wojtowicz, T., et al. (2017). Overexpression of STIM1 in neurons in mouse brain improves contextual learning and impairs long-term depression. Biochim. Biophys. Acta 1864, 1071–1087. doi: 10.1016/j.bbamcr.2016.11.025
Miyata, M., Finch, E. A., Khiroug, L., Hashimoto, K., Hayasaka, S., Oda, S. I., et al. (2000). Local calcium release in dendritic spines required for long-term synaptic depression. Neuron 28, 233–244. doi: 10.1016/s0896-6273(00)00099-4
Moccia, F., Zuccolo, E., Soda, T., Tanzi, F., Guerra, G., Mapelli, L., et al. (2015). Stim and Orai proteins in neuronal Ca2+ signaling and excitability. Front. Cell. Neurosci. 9:153. doi: 10.3389/fncel.2015.00153
Mockett, B. G., Guévremont, D., Wutte, M., Hulme, S. R., Williams, J. M., and Abraham, W. C. (2011). Calcium/calmodulin-dependent protein kinase II mediates group I metabotropic glutamate receptor-dependent protein synthesis and long-term depression in rat hippocampus. J. Neurosci. 31, 7380–7391. doi: 10.1523/jneurosci.6656-10.2011
Ng, A. N., Krogh, M., and Toresson, H. (2011). Dendritic EGFP-STIM1 activation after type I metabotropic glutamate and muscarinic acetylcholine receptor stimulation in hippocampal neuron. J. Neurosci. Res. 89, 1235–1244. doi: 10.1002/jnr.22648
Niswender, C. M., and Conn, P. J. (2010). Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 50, 295–322. doi: 10.1146/annurev.pharmtox.011008.145533
Nosyreva, E. D., and Huber, K. M. (2005). Developmental switch in synaptic mechanisms of hippocampal metabotropic glutamate receptor-dependent long-term depression. J. Neurosci. 25, 2992–3001. doi: 10.1523/jneurosci.3652-04.2005
Olianas, M. C., Dedoni, S., and Onali, P. (2014). Involvement of store-operated Ca2+ entry in activation of AMP-activated protein kinase and stimulation of glucose uptake by M3 muscarinic acetylcholine receptors in human neuroblastoma cells. Biochim. Biophys. Acta 1843, 3004–3017. doi: 10.1016/j.bbamcr.2014.09.012
Pardo, B., Contreras, L., Serrano, A., Ramos, M., Kobayashi, K., Iijima, M., et al. (2006). Essential role of aralar in the transduction of small Ca2+ signals to neuronal mitochondria. J. Biol. Chem. 281, 1039–1047. doi: 10.1074/jbc.m507270200
Parekh, A. B., and Putney, J. W. Jr. (2005). Store-operated calcium channels. Physiol. Rev. 85, 757–810. doi: 10.1152/physrev.00057.2003
Park, C. Y., Shcheglovitov, A., and Dolmetsch, R. (2010). The CRAC channel activator STIM1 binds and inhibits L-type voltage-gated calcium channels. Science 330, 101–105. doi: 10.1126/science.1191027
Pla-Martín, D., Rueda, C. B., Estela, A., Sánchez-Piris, M., González-Sánchez, P., Traba, J., et al. (2013). Silencing of the Charcot–Marie–Tooth disease-associated gene GDAP1 induces abnormal mitochondrial distribution and affects Ca2+ homeostasis by reducing store-operated Ca2+ entry. Neurobiol. Dis. 55, 140–151. doi: 10.1016/j.nbd.2013.03.010
Prakriya, M., and Lewis, R. S. (2015). Store-Operated calcium channels. Physiol. Rev. 95, 1383–1436. doi: 10.1152/physrev.00020.2014
Putney, J. W. Jr. (1986). A model for receptor-regulated calcium entry. Cell Calcium 7, 1–12. doi: 10.1016/0143-4160(86)90026-6
Putney, J. W. Jr. (2003). Capacitative calcium entry in the nervous system. Cell Calcium 34, 339–344. doi: 10.1016/s0143-4160(03)00143-x
Ramos, M., del Arco, A., Pardo, B., Martínez-Serrano, A., Martínez-Morales, J. R., Kobayashi, K., et al. (2003). Developmental changes in the Ca2+-regulated mitochondrial aspartate-glutamate carrier aralar1 in brain and prominent expression in the spinal cord. Dev. Brain Res. 143, 33–46. doi: 10.1016/s0165-3806(03)00097-x
Roos, J., DiGregorio, P. J., Yeromin, A. V., Ohlsen, K., Lioudyno, M., Zhang, S., et al. (2005). STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 169, 435–445. doi: 10.1083/jcb.200502019
Ruiz, F., Alvarez, G., Pereira, R., Hernandez, M., Villalba, M., Cruz, F., et al. (1998). Protection by pyruvate and malate against glutamate-mediated neurotoxicity. Neuroreport 9, 1277–1282.
Samtleben, S., Wachter, B., and Blum, R. (2015). Store-operated calcium entry compensates fast ER calcium loss in resting hippocampal neurons. Cell Calcium 58, 147–159. doi: 10.1016/j.ceca.2015.04.002
Schnabel, R., Kilpatrick, I. C., and Collingridge, G. L. (1999). An investigation into signal transduction mechanisms involved in DHPG-induced LTD in the CA1 region of the hippocampus. Neuropharmacology 38, 1585–1596. doi: 10.1016/s0028-3908(99)00062-3
Segal, M., and Korkotian, E. (2016). Roles of calcium stores and store-operated channels in plasticity of dendritic spines. Neuroscientist 22, 477–485. doi: 10.1177/1073858415613277
Sethna, F., Zhang, M., Kaphzan, H., Klann, E., Autio, D., Cox, C. L., et al. (2016). Calmodulin activity regulates group I metabotropic glutamate receptor-mediated signal transduction and synaptic depression. J. Neurosci. Res. 94, 401–408. doi: 10.1002/jnr.23719
Shinde, A. V., Motiani, R. K., Zhang, X., Abdullaev, I. F., Adam, A. P., Gonzalez-Cobos, J. C., et al. (2013). STIM1 controls endothelial barrier function independently of Orai1 and Ca2+ entry. Sci. Signal. 6:ra18. doi: 10.1126/scisignal.2003425
Steinbeck, J. A., Henke, N., Opatz, J., Gruszczynska-Biegala, J., Schneider, L., Theiss, S., et al. (2011). Store-operated calcium entry modulates neuronal network activity in a model of chronic epilepsy. Exp. Neurol. 232, 185–194. doi: 10.1016/j.expneurol.2011.08.022
Stutzmann, G. E., and Mattson, M. P. (2011). Endoplasmic reticulum Ca2+ handling in excitable cells in health and disease. Pharmacol. Rev. 63, 700–727. doi: 10.1124/pr.110.003814
Südhof, T. C. (2012). Calcium control of neurotransmitter release. Cold Spring Harb. Perspect. Biol. 4:a011353. doi: 10.1101/cshperspect.a011353
Sun, S., Zhang, H., Liu, J., Popugaeva, E., Xu, N. J., Feske, S., et al. (2014). Reduced synaptic STIM2 expression and impaired store-operated calcium entry cause destabilization of mature spines in mutant presenilin mice. Neuron 82, 79–93. doi: 10.1016/j.neuron.2014.02.019
Tian, G., Tepikin, A. V., Tengholm, A., and Gylfe, E. (2012). cAMP induces stromal interaction molecule 1 (STIM1) puncta but neither Orai1 protein clustering nor store-operated Ca2+ entry (SOCE) in islet cells. J. Biol. Chem. 287, 9862–9872. doi: 10.1074/jbc.m111.292854
Verkhratsky, A. (2005). Physiology and pathophysiology of the calcium store in the endoplasmic reticulum of neurons. Physiol. Rev. 85, 201–279. doi: 10.1152/physrev.00004.2004
Wang, Y., Deng, X., Mancarella, S., Hendron, E., Eguchi, S., Soboloff, J., et al. (2010). The calcium store sensor, STIM1, reciprocally controls Orai and CaV1.2 channels. Science 330, 105–109. doi: 10.1126/science.1191086
Xia, J., Pan, R., Gao, X., Meucci, O., and Hu, H. (2014). Native store-operated calcium channels are functionally expressed in mouse spinal cord dorsal horn neurons and regulate resting calcium homeostasis. J. Physiol. 592, 3443–3461. doi: 10.1113/jphysiol.2014.275065
Xiao, M. Y., Zhou, Q., and Nicoll, R. A. (2001). Metabotropic glutamate receptor activation causes a rapid redistribution of AMPA receptors. Neuropharmacology 41, 664–671. doi: 10.1016/s0028-3908(01)00134-4
Yap, K. A., Shetty, M. S., Garcia-Alvarez, G., Lu, B., Alagappan, D., Oh-Hora, M., et al. (2017). STIM2 regulates AMPA receptor trafficking and plasticity at hippocampal synapses. Neurobiol. Learn. Mem. 138, 54–61. doi: 10.1016/j.nlm.2016.08.007
Keywords: store-operated calcium entry (SOCE), metabotropic glutamate receptors (mGluRs), long-term depression (LTD), calcium signaling, synaptic plasticity
Citation: González-Sánchez P, del Arco A, Esteban JA and Satrústegui J (2017) Store-Operated Calcium Entry Is Required for mGluR-Dependent Long Term Depression in Cortical Neurons. Front. Cell. Neurosci. 11:363. doi: 10.3389/fncel.2017.00363
Received: 04 September 2017; Accepted: 03 November 2017;
Published: 14 December 2017.
Edited by:
Alessandro Tozzi, University of Perugia, ItalyReviewed by:
Giuseppe Sciamanna, Università degli Studi di Roma Tor Vergata, ItalyFrancesco Lodola, Fondazione Istituto Italiano di Technologia, Italy
Copyright © 2017 González-Sánchez, del Arco, Esteban and Satrústegui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: José A. Esteban, amFlc3RlYmFuQGNibS5jc2ljLmVz
Jorgina Satrústegui, anNhdHJ1c3RlZ3VpQGNibS5jc2ljLmVz