 Satoshi Goto1,2*
Satoshi Goto1,2*- 1Department of Neurodegenerative Disorders Research, Institute of Biomedical Sciences, Graduate School of Medical Sciences, Tokushima University, Tokushima, Japan
- 2Parkinson’s Disease and Dystonia Research Center, Tokushima University Hospital, Tokushima, Japan
The motor symptoms of Parkinson’s disease (PD) result from striatal dopamine (DA) deficiency due to a progressive degeneration of nigral dopaminergic cells. Although DA replacement therapy is the mainstay to treat parkinsonian symptoms, a long-term daily administration of levodopa often develops levodopa-induced dyskinesia (LID). LID is closely linked to the dysregulation of cyclic adenosine monophosphate (cAMP) signaling cascades in the medium spiny neurons (MSNs), the principal neurons of the striatum, which are roughly halved with striatonigral MSNs by striatopallidal MSNs. The olfactory type G-protein α subunit (Gαolf) represents an important regulator of the cAMP signal activities in the striatum, where it positively couples with D1-type dopamine receptor (D1R) and adenosine A2A receptor (A2AR) to increase cAMP production in the MSNs. Notably, D1Rs are primarily expressed in striatonigral MSNs, whereas D2Rs and A2ARs are expressed in striatopallidal MSNs. Based on the evidence obtained from parkinsonian mice, we hypothesized that in the DA-denervated striatum with D1R hypersensitivity, a repeated and pulsatile exposure to levodopa might cause a usage-induced degradation of Gαolf proteins in striatal MSNs, resulting in increased and decreased levels of Gαolf protein in the striatonigral and striatopallidal MSNs, respectively. As a principal cause for generating LID, this might lead to an increased responsiveness to levodopa exposure in both striatonigral and striatopallidal MSNs. Our hypothesis reinforces the long-standing concept that LID might result from the reduced activity of the striatopallidal pathway and has important clinical implications.
Introduction
By transducing extracellular signals carried by neuromodulators, the cyclic adenosine monophosphate (cAMP) signaling plays a crucial role in the regulation of neuronal activities in the brain. Multiple guanine nucleotide-binding protein (G-protein)-coupled receptor (GPCR) cascades regulate the intracellular levels of cAMP, which activates its key effector protein kinase A. Seven-transmembrane domain receptors can transmit extracellular signals to the intracellular signaling cascades through the activation of heterotrimeric G-proteins, which are composed of the guanine nucleotide-binding Gα subunit and the dimeric βγ subunits (Pierce et al., 2002). Gαs is the predominant stimulatory G-protein subunit in the brain. However, in the striatum, Gαs is replaced by the olfactory type G protein α subunit (Gαolf), which is encoded by the GNAL gene (Jones and Reed, 1989). Cellular Gαolf/cAMP signaling pathway represents a principal regulator for the striatal functions in normal physiological processes and pathological conditions (Hervé, 2011). It is worth noting that mutations in the GNAL gene have been identified as a cause for generating dystonia (Fuchs et al., 2013; Pelosi et al., 2017), suggesting that the Gαolf function might participate in the brain circuit involving motor control.
The motor symptoms of Parkinson’s disease (PD) are caused by striatal dopamine (DA) deficiency, predominantly in the putamen, resulting from a progressive degeneration of nigrostriatal DA-producing cells (Kish et al., 1988; Goto et al., 1989). Although the DA replacement therapy remains the mainstay to treat PD symptoms, long-term exposure to dopaminergic drugs, particularly to the DA precursor levodopa, eventually causes adverse effects such as motor fluctuations and levodopa-induced dyskinesia (LID; Jenner, 2008; Calabresi et al., 2010; Bastide et al., 2015). LID is a major cause of disability in patients with PD, and occurs in approximately 80% of patients after 5 years of treatment with a daily administration of levodopa (Obeso et al., 1989; Luquin et al., 1992; Rascol et al., 2000). Importantly, once LID has been primed (or established), its severity progressively increases despite even when the used dosage of dopaminergic drugs is not increased (Brotchie, 2005). LID is known to be closely linked to the altered function of the DA signaling pathways in the striatum (Brotchie, 2005; Jenner, 2008; Bastide et al., 2015; Calabresi et al., 2016). It has also been suggested that LID is associated with the hypersensitivity of striatal MSNs to DA receptor stimulation and with ongoing deregulation of corticostriatal inputs, which activate striatal glutamate receptors, such as N-methyl-D-aspartate (NMDA) receptors (Brotchie, 2005; Jenner, 2008; Bastide et al., 2015; Calabresi et al., 2016). In this hypothesis article, we primarily considered the levodopa-induced changes in cellular Gαolf protein levels in the DA-denervated striatum as the key mechanism to increase the striatal responsiveness to DA receptor stimulation in LID.
Gαolf Regulates The Agonist-Induced cAMP Production in Striatal Cells
As being innervated by massive dopaminergic afferents originating from the midbrain, the striatum is highly enriched in DA receptors, which belong to a superfamily of GPCRs and are classified into two subtypes, D1- and D2-type receptors. Through their specific targeting of G proteins, the D1-type receptors (D1Rs) elicit the adenylyl cyclase type (AC) to increase the cAMP production, whereas the D2-type dopamine receptors (D2Rs) inhibit the cAMP production (Kebabian and Calne, 1979; Missale et al., 1998). Medium spiny neurons (MSNs) constitute more than 90% of the neuronal types in the striatum (Graybiel, 2008; Kreitzer, 2009; Gerfen and Surmeier, 2011). Anatomically, they are roughly halved with the MSN group to form the “direct” striatonigral pathway by the MSN group to from the “indirect” striatopallidal pathway (Crittenden and Graybiel, 2011; Gerfen and Surmeier, 2011; Calabresi et al., 2014). The striatonigral and striatopallidal MSNs express D1Rs and D2Rs, respectively. Moreover, the striatopallidal MSNs, but not the striatonigral MSNs, are enriched in adenosine A2A receptors (A2ARs), which are prototypical Gs-coupled receptors that elicit AC to increase cAMP production (Svenningsson et al., 1999; Schwarzschild et al., 2006; Fuxe et al., 2007). Figure 1 depicts the cell-type specific localization of Gαolf, D1R and A2AR among the striatal MSNs that constitute the basic circuits of the basal ganglia.
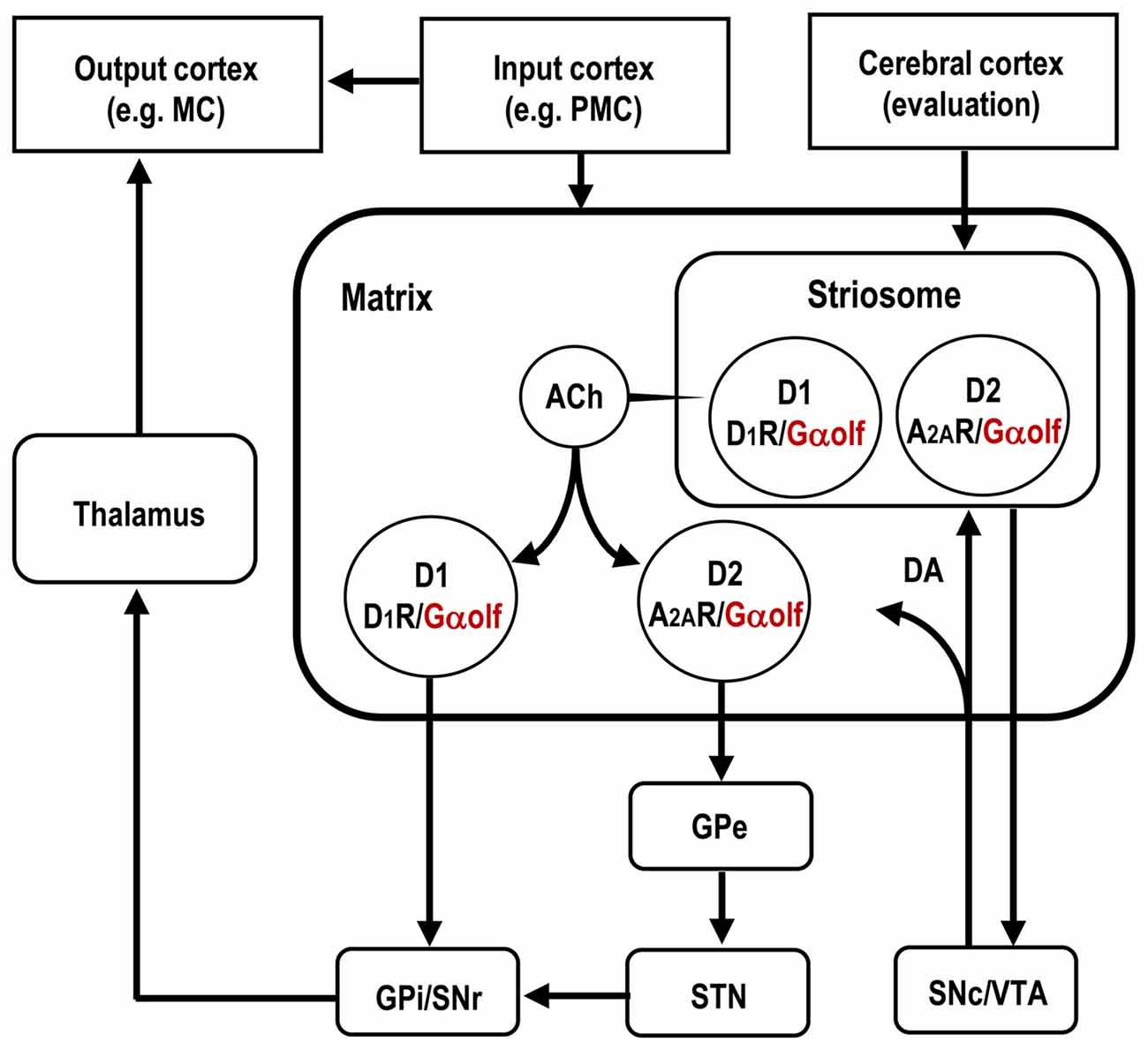
Figure 1. Distributional pattern of Gαolf proteins in striatal medium spiny neurons (MSNs) that form the basal ganglia circuit. Gαolf proteins are colocalized with DA D1 receptors (D1Rs) in the striatonigral MSNs (D1-cells), and with adenosine A2A receptors (A2ARs) in striatopallidal MSNs expressing DA D2 receptors (D2Rs; D2-cells). The striatonigral and striatopallidal pathways arising from the striosome are omitted in this scheme. ACh, acetylcholine; DA, dopamine; GPe, globus pallidus externa; GPi, globus pallidus interna; MC, motor cortex; PMC, premotor cortex; SNr, substantia nigra pars reticulata; SNc, substantia nigra pars compacta; STN, subthalamic nucleus; VTA, ventral tegmental area.
Gαolf is highly expressed in all striatal MSNs including those expressing the D1Rs and A2ARs (Kull et al., 2000; Hervé, 2011; Morigaki et al., 2017; see Figure 2). As Gαolf positively couples with D1R and A2AR to activate the AC type 5 (AC5) and, thereby, increase the intracellular cAMP levels, it serves as the rate-limiting factor for both the D1R- and A2AR-dependent cAMP production in striatal MSNs (Kull et al., 2000; Corvol et al., 2001). The Gαolf protein level plays a key role in regulating the D1R/cAMP- and A2AR/cAMP-signal activities of striatonigral and striatopallidal MSNs, respectively. The D1R/Gαolf-mediated increases in the cAMP levels cause the activation of the striatonigral MSNs (Hervé, 2011). On one hand, as D2R activation inhibits AC5 through Gi/o proteins but A2AR activation elicits AC5 through Gs/olf proteins (Kull et al., 2000), the A2AR/Gαolf-signal stimulation functionally opposes the actions of D2Rs on the striatopallidal MSNs (Schwarzschild et al., 2006; Fuxe et al., 2007).
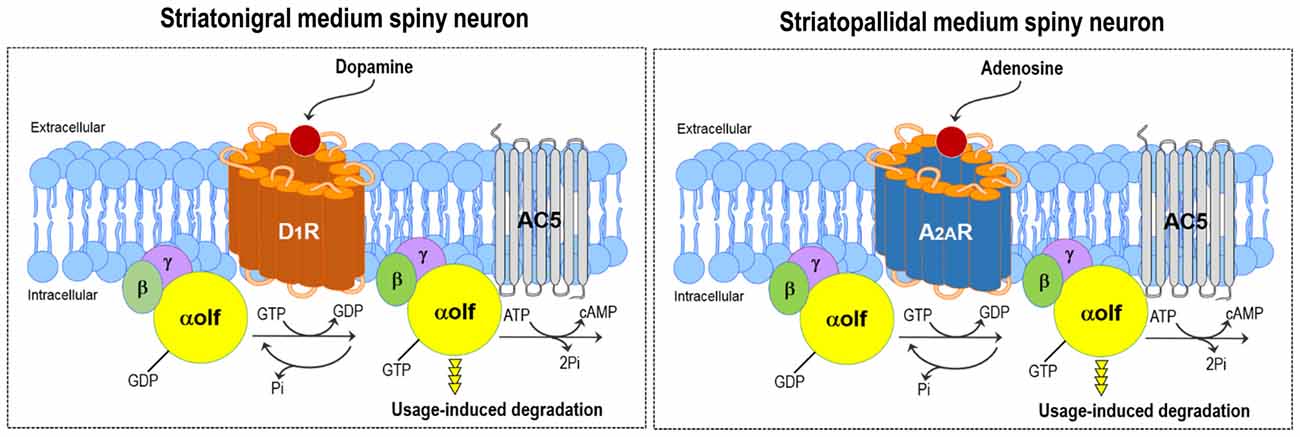
Figure 2. DA and adenosine-induced cAMP production in striatal MSNs. Gαolf positively couples with the DA D1 receptor (D1R) and adenosine A2A receptor (A2AR) to activate adenylyl cyclase type 5 (AC5) and subsequently increase cAMP production in striatonigral (left) and striatopallidal (right) MSNs, respectively. Thus, the DA-induced activation of D1R or adenosine-induced activation of A2AR leads to the degradation of the Gαolf protein through the usage-dependent mechanism in striatonigral or striatopallidal MSN, respectively.
Subdivisional and Compartmental Localization of Gαolf in The Striatum
Quantitative immunohistochemistry (IHC) has shown that the Gαolf protein is unevenly distributed within the mouse striatum, where it is highly concentrated in the dorsolateral striatum (Morigaki et al., 2017). Since the dorsolateral portion of the mouse striatum corresponds to the motor territory in rodents and is analogous to the putamen in primates (Graybiel, 2008), this strategic expression of Gαolf protein indicates that Gαolf may function as the stimulatory G protein that has a tight link to the basal ganglia “motor” circuit (Alexander and Crutcher, 1990) at the striatal level. With respect to the striatal compartments, there was a differential localization of Gαolf with higher densities of Gαolf proteins in the striosomes relative to the matrix compartment (Sako et al., 2010; Ruiz-DeDiego et al., 2015; Morigaki et al., 2017). This suggests that Gαolf may be a key molecule that determines differential responses between the striosome and matrix compartments to the D1R or A2AR activation in the striatum at maturity.
Homeostatic Regulation of The Cellular Gαolf Protein Levels in The Striatum
Rodent animal models for PD (Iderberg et al., 2012; Francardo and Cenci, 2014) have so far been used to elucidate the regulatory mechanism for the striatal expression of Gαolf. In line with the evidence that there is a significant increase in Gαolf protein levels in the putamen of patients with PD (Corvol et al., 2004), a dramatic increase in Gαolf protein levels has been identified in the DA-depleted striatum of rats (Hervé et al., 1993; Marcotte et al., 1994; Penit-Soria et al., 1997; Corvol et al., 2004; Rangel-Barajas et al., 2011) and mice (Alcacer et al., 2012; Ruiz-DeDiego et al., 2015; Morigaki et al., 2017) with nigrostriatal 6-hydroxydopamine lesions. However, this upregulation of the Gαolf protein levels is not associated with a parallel increase of the Gαolf mRNA expression. Accordingly, the homeostatic regulation of Gαolf protein levels is thought to occur through post-translational mechanisms in the striatum, where the altered expression of the Gαolf protein depends directly on its usage rate (Hervé, 2011). The persistent lack in the use of D1R and Gαolf could lower the Gαolf degradation rate and thereby result in the accumulation of Gαolf protein in the DA-denervated striatum of PD models. In agreement with this hypothesis, a total lack of D1Rs by D1R gene targeting induces a significant increase of the Gαolf protein levels without any changed expression of Gαolf mRNAs in the striatum of mutant mice (Hervé et al., 2001). In contrast, the decreased levels of striatal Gαolf proteins were found in mutant mice lacking the DA transporter (Hervé et al., 2001), which exhibit a marked increase in the extracellular DA levels leading to persistent activation of D1Rs in the striatum (Giros et al., 1996). Importantly, the lack of A2ARs in homozygous A2AR knock-out mice (Ledent et al., 1997) also results in an upregulation of Gαolf proteins with no obvious changes in the levels of Gαolf transcripts (Hervé et al., 2001). Collectively, the agonist-induced activation of D1Rs (Hervé et al., 2001; Corvol et al., 2004, 2007; Alcacer et al., 2012; Ruiz-DeDiego et al., 2015) or A2ARs (Hervé et al., 2001) might lead to the degradation of Gαolf proteins in striatal MSNs through posttranslational usage-dependent mechanism (see Figure 2).
Gαolf Protein Levels in Striatonigral and Striatopallidal MSNs in LID
On the hypothesis that the upregulation of the Gαolf protein levels results from the disuse of the D1Rs in the DA-depleted striatum in rodent models for PD, several studies with IHC and western blot analyses revealed that the Gαolf could be returned to normal levels by DA replacement with a daily exposure to levodopa in rodent models for PD with LID (Corvol et al., 2004; Rangel-Barajas et al., 2011; Ruiz-DeDiego et al., 2015; Morigaki et al., 2017). With respect to the striosome-matrix system, IHC studies revealed that the Gαolf levels were normally found in both the striosome and matrix compartments in PD with LID, although they were markedly increased in the matrix compartment, but not or only mildly increased in the striosome compartment, in PD (Ruiz-DeDiego et al., 2015; Morigaki et al., 2017). This novel finding indicates that there is a difference in the dopaminergic regulation of the Gαolf expression between the striosome and matrix compartments.
In situ proximity ligation assay (PLA) for dual-antigen recognition disclosed cell-type specific changes in the Gαolf levels in the DA-depleted striatum of mice with and without LID (Morigaki et al., 2017). The in situ PLA technique can indicate the presence of the Gαolf protein in close proximity to the D1R protein (D1R-Gαolf) or A2AR protein (A2AR-Gαolf). Quantitative in situ PLA showed that DA depletion caused a marked (~90%) increase in the striatal levels of D1R-Gαolf PLA signals, which were downregulated by a daily administration of levodopa. However, there remained a significant (~50%) increase in the striatal D1R-Gαolf PLA signals in mice with LID when compared with normal controls. On one hand, quantitative in situ PLA also disclosed that a daily exposure to levodopa, but not DA depletion per se, caused a significant (~40%) decrease in the striatal A2AR-Gαolf PLA signals in the DA-depleted striatum of mice with LID. These findings indicate that, in the DA-depleted striatum, DA replacement could induce the downregulation of the Gαolf protein levels not only in the striatonigral MSNs but also in the striatopallidal MSNs.
An intriguing question is how the Gαolf protein levels are decreased in the striatopallidal MSNs in LID. In animal models with nigrostriatal 6-OHDA-lesions, persistent (chronic) DA depletion per se has been shown to cause no apparent changes (Ballarin et al., 1987; Herrera-Marschitz et al., 1994; Nomoto et al., 2000) or mild decrease (Pinna et al., 2002) in the extracellular levels of adenosine in the DA-denervated striatum. However, evidence shows that the striatal adenosine levels are elevated by the activation of NMDA receptors (Delaney and Geiger, 1998; Delaney et al., 1998), which can be enhanced by D1R activation (Cepeda and Levine, 2012; Morigaki and Goto, 2015; see Figure 3). Interestingly, a pulsatile exposure to the D1R agonist reportedly facilitated the NMDA receptor-evoked increase in the extracellular adenosine release in the rat striatum (Harvey and Lacey, 1997). This evidence suggests that, in the DA-depleted striatum with D1R hypersensitivity, a repeated administration of levodopa may exert a pulsatile activation of D1Rs, which subsequently facilitates the NMDA receptor-evoked increase in the extracellular adenosine levels. Moreover, in the DA-depleted striatum, the activation of NMDA receptor could lead to a marked increase in the extracellular adenosine levels and, then, indirectly activate A2ARs (Nash and Brotchie, 2000). Thus, it is likely that the downregulation of the Gαolf levels in striatopallidal MSNs in LID might result from an increased usage of Gαolf proteins through the A2AR activation subsequent to the daily pulsatile activation of striatal D1Rs. This notion also suggests that the striatal D1R signals might play a critical role in the regulation of the Gαolf protein levels not only in the striatonigral MSNs, but also in the striatopallidal MSNs in the DA-denervated striatum. This consideration may corroborate the general concept that increased activities of striatal D1Rs are requisite for the genesis of LID (Westin et al., 2007; Darmopil et al., 2009; Alcacer et al., 2012).
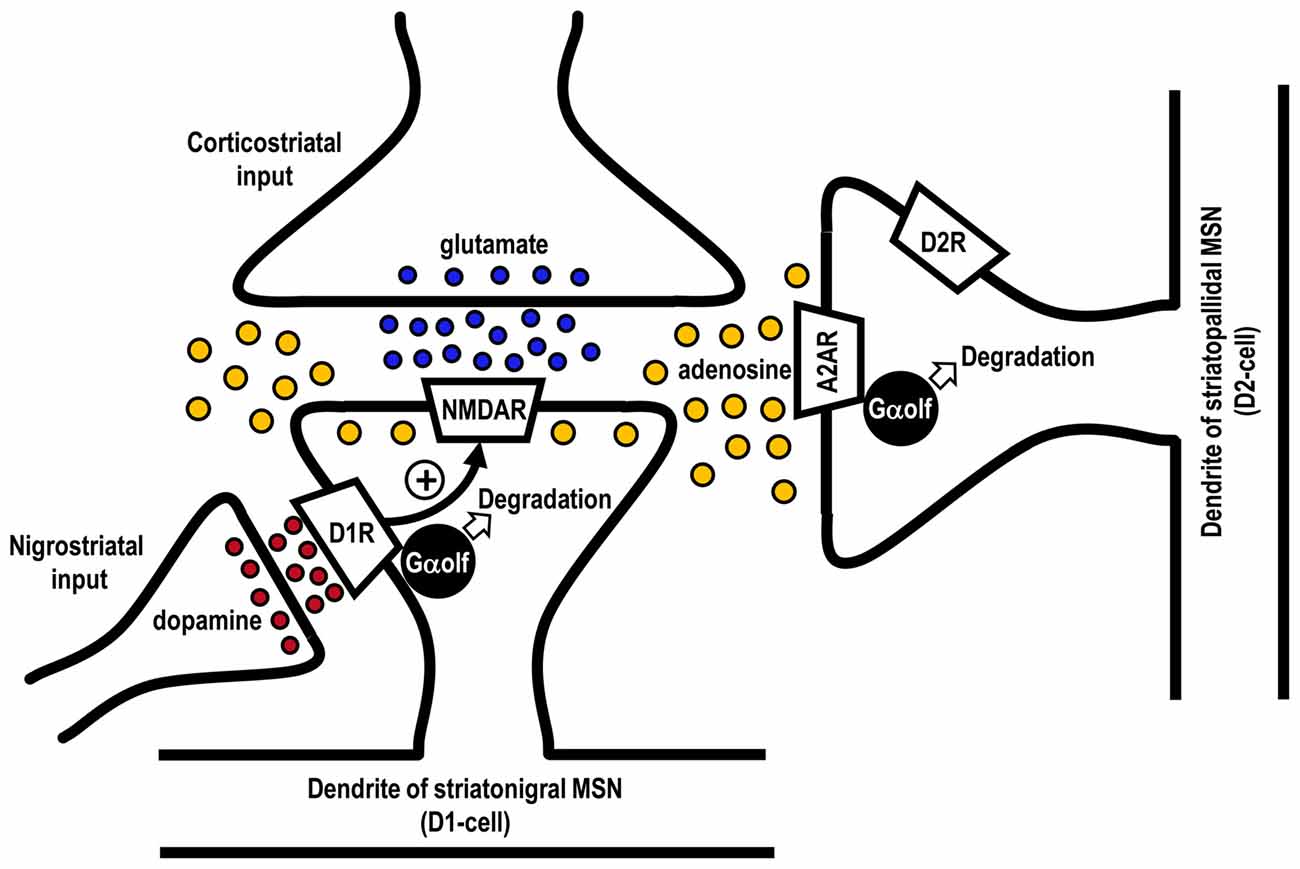
Figure 3. Possible mechanism for agonist-induced degradation of Gαolf proteins in striatonigral and striatopallidal MSNs. Glutamate released from the corticostriatal afferents could activate postsynaptic N-methyl-D-aspartate (NMDA) receptors (NMDARs) to increase the extracellular adenosine levels in the striatum. Repeated exposure to levodopa might cause a pulsatile release of DA from the nigrostriatal afferents to activate DA D1 receptors (D1Rs) in striatonigral MSNs (D1-cells). This might facilitate the NMDAR-evoked increase in extracellular adenosine release and, thereby, indirectly activate the adenosine A2A receptors (A2ARs) in striatopallidal MSNs expressing DA D2 receptors (D2Rs; D2-cells). Thus, a usage-induced downregulation of Gαolf protein levels could occur not only in the striatonigral MSNs but also in striatopallidal MSNs.
Striatal Gαolf/cAMP Signal-Dependent Mechanism for Generating LID
Figure 4 shows the hypothetical representation of the Gαolf protein levels in striatonigral and striatopallidal MSNs in the DA-denervated striatum under the conditions of both PD with and without LID. In PD, there is a dramatic increase in the Gαolf protein levels in the striatonigral MSNs, but not in the striatopallidal MSNs. Because of no apparent changes in the striatal D1R levels (Shinotoh et al., 1993; Turjanski et al., 1997; Hurley et al., 2001) and other principal mediators of the D1R signaling cascades (Girault et al., 1989; Nishino et al., 1993) in patients with PD, the marked increase in the Gαolf protein levels in the striatonigral MSNs may be a principal cause for generating striatal D1R hypersensitivity to levodopa exposure in PD. This notion corroborates the evidence that there is a marked increase in the responsiveness of the striatonigral MSNs to D1R activation in PD, as determined by the fos induction experiments (Engber et al., 1989; Asin et al., 1995; Kashihara et al., 2000; Xu et al., 2003; Morigaki et al., 2017).
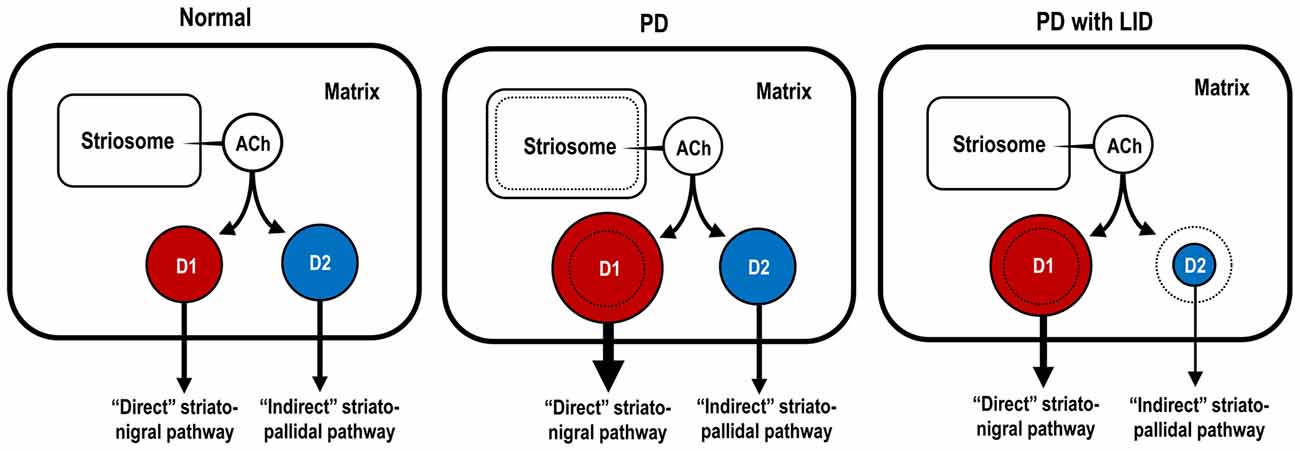
Figure 4. Hypothetical diagram for dopaminergic regulation of Gαolf protein levels in striatonigral and striatopallidal MSNs. The sizes of the circles, colored in red and blue, indicate the abundance of Gαolf proteins in striatonigral MSNs expressing dopamine D1 receptors (D1Rs; D1-cells; red) and in striatopallidal MSNs expressing dopamine D2 receptors (D2Rs; D2-cells; blue), respectively. In the conditions of Parkinson’s disease (PD), D1-cells, but not D2-cells, might exhibit a DA D1 hypersensitivity caused by a dramatic increase in their Gαolf levels. In the conditions of PD with levodopa-induced dyskinesia (LID), D1-cells might show an increase in their Gαolf levels, while D2-cells might show a decrease in their Gαolf levels, which might result in an enhanced responsiveness to D2R activation. ACh, acetylcholine; D1-cell, striatonigral medium spiny neuron expressing DA D1 receptor; D2-cell, striatopallidal medium spiny neuron expressing DA D2 receptor; PD, Parkinson’s disease; PD with LID, Parkinson’s disease with levodopa-induced dyskinesia.
In PD with LID, there is an important decrease in the Gαolf protein levels in the striatopallidal MSNs after a prolonged and pulsatile administration of levodopa. This leads to the facilitation of the effects of DA on striatopallidal MSNs by reducing the A2AR/Gαolf signal-mediated cAMP production and subsequently to the increase in the responsiveness of striatopallidal MSNs to D2R activation. Indeed, it was importantly noted that, during the increasing phase of dyskinesias, an abnormal lowering of intracellular cAMP levels transiently occurred in the DA-denervated striatum in rat model of LID (Sancesario et al., 2014). These novel findings parallel the evidence that a repeated exposure to levodopa results in a significant increase in the responsiveness of striatopallidal MSNs to dopaminergic stimulation, as determined by fos induction experiments (Engber et al., 1989; Asin et al., 1995; Kashihara et al., 2000; Xu et al., 2003; Morigaki et al., 2017). In addition, there is a significant increase in the Gαolf protein levels in striatonigral MSNs in PD with LID as compared to normal controls. Because Gαolf is the regulator of cAMP signal-dependent activities in the striatum, an increase in the responsiveness of both striatonigral and striatopallidal MSNs to levodopa exposure, which depends on the Gαolf protein levels, serves as a principal cause for generating LID.
Concluding Remarks
Since the intracellular cAMP signaling cascades serve as a determinant of striatal cell activities (Girault, 2012), maladaptive change in Gαolf protein levels is thought to be closely linked to the pathophysiology of PD (Hervé, 2011). Here, we hypothesized that DA depletion might cause a marked upregulation of the Gαolf protein levels in striatonigral MSNs, which results in a crucial hypersensitivity of the striatum to D1R stimulation in PD. A prolonged and pulsatile exposure to levodopa might lead to a usage-dependent decrease in the Gαolf protein levels not only in the nigrostriatal MSNs but also in the striatopallidal MSNs in PD with LID. This levodopa-induced decrease in Gαolf protein levels, which might be due to a pulsatile activation of postsynaptic D1Rs and NMDA receptors, could result in reduced A2AR/Gαolf/cAMP signal levels in striatopallidal MSNs. This might cause an increase in the responsiveness of striatopallidal MSNs to D2R activation, and thereby develop LID in PD. Our hypothesis corroborates the long-lasting concept that LIDs are associated with a decreased activity of the “indirect” striatopallidal pathway (Crossman, 1990; DeLong, 1990; Brotchie, 2005).
As an important cellular mechanism to regulate the activities of striatal MSNs, the recurrent collateral connections between the MSNs have also been identified (Bolam et al., 1983; Yung et al., 1996). The activities of striatopallidal MSNs can be inhibited by the GABAergic collateral axon branches from neighboring MSNs (Taverna et al., 2008; Lalchandani et al., 2013; Dobbs et al., 2016; Wei et al., 2017). Thus striatal D1 hypersensitivity could lead to an increased responsiveness of striatopallidal MSNs to D2R activation in the conditions of PD with and without LID, although only a small population of the striatonigral MSNs has been found to form collateral axon connections with striatopallidal MSNs in the mouse striatum (Taverna et al., 2008). However, this notion per se could not explain the progressive increase in the severity of LID, which occurs in the PD patients treated with unaltered dosages of given dopaminergic drugs (Brotchie, 2005), because there is an ongoing decline in striatal responsiveness to D1R activation along a repeated exposure to levodopa under the conditions of PD, as determined by fos induction experiments (Saka et al., 1999; Kashihara et al., 2000; Xu et al., 2003; Morigaki et al., 2017).
Finally, we suggest that the pharmacological concomitant therapy to increase Gαolf protein levels in the striatum might be useful in the management of LID and motor fluctuations in patients with PD treated with DA replacement therapy. The normalization of the decreased Gαolf protein levels in the striatopallidal MSNs might suppress LID. On one hand, the elevation of the Gαolf protein levels in the striatonigral MSNs could increase the striatal responsiveness to D1R activation and, thereby, facilitate the therapeutic efficacy of dopaminergic drugs. In considering the possible involvement of the activated NMDA receptors in lowering striatal Gαolf levels in LID, NMDA receptor antagonists (e.g., amantadine or memantine) might attenuate LID, as already shown in clinical practice (Rascol et al., 2015). Because A2AR activation, which could reduce the Gαolf protein levels in the striatopallidal MSNs leading to LID, might be required for the “priming” of LID (Brotchie, 2005; Xiao et al., 2006), it is suggested that A2AR antagonists (e.g., istradefylline) might be effective in dampening the “priming” of LID. However, after the establishment of LID, the adjunct use of A2AR antagonists might exacerbate the dyskinetic symptoms as shown in clinical practice (Kondo and Mizuno, 2015).
Author Contributions
SG wrote the manuscript.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author wishes to thank Dr. Ryoma Morigaki and Dr. Shinya Okita for their experimental supports. This work was supported in part by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan (grants-in-aid for Scientific Research no. 24390223, 26461272, 26430054 and 16k10788), Japan Agency for Medical Research and Development (AMED; No. 16ek0109182h0001) and the Research Cluster of Tokushima University (No. 1702004).
References
Alcacer, C., Santini, E., Valjent, E., Gaven, F., Girault, J.-A., and Hervé, D. (2012). Gαolf mutation allows parsing the role of cAMP-dependent and extracellular signal-regulated kinase-dependent signaling in L-3,4,-dihydroxyphenylalanine-induced dyskinesia. J. Neurosci. 32, 5900–5910. doi: 10.1523/JNEUROSCI.0837-12.2012
Alexander, G. E., and Crutcher, M. D. (1990). Functional architecture of basal ganglia circuits: neural substrates of parallel processing. Trends Neurosci. 13, 266–271. doi: 10.1016/0166-2236(90)90107-l
Asin, K. E., Bednarz, L., Nikkel, A., and Perner, R. (1995). Rotation and striatal c-fos expression after repeated, daily treatment with selective dopamine receptor agonists and levodopa. J. Pharmacol. Exp. Ther. 273, 1483–1490.
Ballarin, M., Herrera-Marschitz, M., Casas, M., and Ungerstedt, U. (1987). Striatal adenosine levels measured ‘in vivo’ by microdialysis in rats with unilateral dopamine denervation. Neurosci. Lett. 83, 338–344. doi: 10.1016/0304-3940(87)90111-x
Bastide, M. F., Meissner, W. G., Picconi, B., Fasano, S., Fernagut, P. O., Feyder, M., et al. (2015). Pathophysiology of L-dopa-induced motor and non-motor complications in Parkinson’s disease. Prog. Neurobiol. 132, 96–168. doi: 10.1016/j.pneurobio.2015.07.002
Bolam, J. P., Somogyi, P., Takagi, H., Fodor, I., and Smith, A. D. (1983). Localization of substance P-like immunoreactivity in neurons and nerve terminals in the neostriatum of the rat: a correlated light and electron microscopic study. J. Neurocytol. 12, 325–344. doi: 10.1007/bf01148468
Brotchie, J. M. (2005). Nondopaminergic mechanisms in levodopa-induced dyskinesia. Mov. Disord. 20, 919–931. doi: 10.1002/mds.20612
Calabresi, P., Di Filippo, M., Ghiglieri, V., Tambasco, N., and Picconi, B. (2010). Levodopa-induced dyskinesias in patients with Parkinson’s disease: filling the bench-to-bedside gap. Lancet Neurol. 9, 1106–1117. doi: 10.1016/S1474-4422(10)70218-0
Calabresi, P., Picconi, B., Tozzi, A., Ghiglieri, V., and Di Filippo, M. (2014). Direct and indirect pathways of basal ganglia: a critical reappraisal. Nat. Neurosci. 17, 1022–1030. doi: 10.1038/nn.3743
Calabresi, P., Pisani, A., Rothwell, J., Ghiglieri, V., Obeso, J. A., and Picconi, B. (2016). Hyperkinetic disorders and loss of synaptic downscaling. Nat. Neurosci. 19, 868–875. doi: 10.1038/nn.4306
Cepeda, C., and Levine, M. S. (2012). Dopamine-NMDA receptor interations: twenty years later. Dev. Neurosci. 34, 2–4. doi: 10.1159/000338590
Corvol, J. C., Muriel, M.-P., Valjent, E., Féger, J., Hanoun, N., Girault, J.-A., et al. (2004). Persistent increase in olfactory type G-protein α subunit levels may underlie D1 receptor functional hypersensitivity in Parkinson’s disease. J. Neurosci. 24, 7007–7014. doi: 10.1523/jneurosci.0676-04.2004
Corvol, J. C., Studler, J. M., Schonn, J. S., Girault, J. A., and Hervé, D. (2001). Gαolf is necessary for coupling D1 and A2a receptors to adenylyl cyclase in the striatum. J. Neurochem. 76, 1585–1588. doi: 10.1046/j.1471-4159.2001.00201.x
Corvol, J. C., Valjent, E., Pascoli, V., Robin, A., Stipanovich, A., Luedtke, R. R., et al. (2007). Quantitative changes in Gαolf protein levels, but not D1 receptor, alter specifically acute responses to psychostimulants. Neuropsychopharmacology 32, 1109–1121. doi: 10.1038/sj.npp.1301230
Crittenden, J. R., and Graybiel, A. M. (2011). Basal ganglia disorders associated with imbalances in the striatal striosome and matrix compartments. Front. Neuroanat. 5:59. doi: 10.3389/fnana.2011.00059
Crossman, A. R. (1990). A hypothesis on the pathophysiological mechanisms that underlie levodopa- or dopamine agonist-induced dyskinesia in Parkinson’s disease: implications for future strategies in treatment. Mov. Disord. 5, 100–108. doi: 10.1002/mds.870050203
Darmopil, S., Martín, A. B., De Diego, I. R., Ares, S., and Moratalla, R. (2009). Genetic inactivation of dopamine D1 but not D2 receptors inhibits L-DOPA-induced dyskinesia and histone activation. Biol. Psychiatry 66, 603–613. doi: 10.1016/j.biopsych.2009.04.025
Delaney, S. M., and Geiger, J. D. (1998). Levels of endogenous adenosine in rat striatum. II. Regulation of basal and N-methyl-D-aspartate-induced levels by inhibitors of adenosine transport and metabolism. J. Pharmacol. Exp. Ther. 285, 568–572.
Delaney, S. M., Shepel, P. N., and Geiger, J. D. (1998). Levels of endogenous adenosine in rat striatum I. Regulation by ionotropic glutamate receptors, nitric oxide and free radicals. J. Pharmacol. Exp. Ther. 285, 561–567.
DeLong, M. R. (1990). Primate models of movement disorders of basal ganglia origin. Trends Neurosci. 13, 281–285. doi: 10.1016/0166-2236(90)90110-v
Dobbs, L. K., Kaplan, A. R., Lemos, J. C., Matsui, A., Rubinstein, M., and Alvarez, V. A. (2016). Dopamine regulation of lateral inhibition between striatal neurons gates the stimulant actions of cocaine. Neuron 90, 1100–1113. doi: 10.1016/j.neuron.2016.04.031
Engber, T. M., Susel, Z., Juncos, J. L., and Chase, T. N. (1989). Continuous and intermittent levodopa differentially affect rotation induced by D-1 and D-2 dopamine agonists. Eur. J. Pharmacol. 168, 291–298. doi: 10.1016/0014-2999(89)90790-5
Francardo, V., and Cenci, M. A. (2014). Investigating the molecular mechanisms of L-DOPA-induced dyskinesia in the mouse. Parkinsonism Relat. Disord. 20, S20–S22. doi: 10.1016/s1353-8020(13)70008-7
Fuchs, T., Saunders-Pullman, R., Masuho, I., Luciano, M. S., Raymond, D., Factor, S., et al. (2013). Mutations in GNAL cause primary torsion dystonia. Nat. Genet. 45, 88–92. doi: 10.1038/ng.2496
Fuxe, K., Marcellino, D., Genedani, S., and Agnati, L. (2007). Adenosine A2A receptors, dopamine D2 receptors and their interactions in Parkinson’s disease. Mov. Disord. 22, 1990–2017. doi: 10.1002/mds.21440
Gerfen, C. R., and Surmeier, D. J. (2011). Modulation of striatal projection systems by dopamine. Annu. Rev. Neurosci. 34, 441–466. doi: 10.1146/annurev-neuro-061010-113641
Girault, J. A. (2012). Integrating neurotransmission in striatal medium spiny neurons. Adv. Exp. Med. Biol. 970, 407–429. doi: 10.1007/978-3-7091-0932-8_18
Girault, J. A., Raisman-Vozari, R., Agid, Y., and Greengard, P. (1989). Striatal phosphoproteins in Parkinson disease and progressive supranuclear palsy. Proc. Natl. Acad. Sci. U S A 86, 2493–2497. doi: 10.1073/pnas.86.7.2493
Giros, B., Jaber, M., Jones, S. R., Wightman, R. M., and Caron, M. G. (1996). Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature 379, 606–612. doi: 10.1038/379606a0
Goto, S., Hirano, A., and Matsumoto, S. (1989). Subdivisional involvement of nigrostriatal loop in idiopathic Parkinson’s disease and striatonigral degeneration. Ann. Neurol. 26, 766–770. doi: 10.1002/ana.410260613
Graybiel, A. M. (2008). Habits, rituals, and the evaluative brain. Annu. Rev. Neurosci. 31, 359–387. doi: 10.1146/annurev.neuro.29.051605.112851
Harvey, J., and Lacey, M. G. (1997). A postsynaptic interaction between dopamine D1 and NMDA receptors promotes presynaptic inhibition in the rat nucleus accumbens via adenosine release. J. Neurosci. 17, 5271–5280.
Herrera-Marschitz, M., Luthman, J., and Ferré, S. (1994). Unilateral neonatal intracerebroventricular 6-hydroxydopamine administration in rats: II. Effects on extracellular monoamine, acetylcholine and adenosine levels monitored with in vivo microdialysis. Psychopharmacology 116, 451–456. doi: 10.1007/bf02247477
Hervé, D. (2011). Identification of a specific assembly of the G protein Golf as a critical and regulated module of dopamine and adenosine-activated cAMP pathways in the striatum. Front. Neuroanat. 5:48. doi: 10.3389/fnana.2011.00048
Hervé, D., Le Moine, C., Corvol, J. C., Belluscio, L., Ledent, C., Fienberg, A. A., et al. (2001). Gα(olf) levels are regulated by receptor usage and control dopamine and adenosine action in the striatum. J. Neurosci. 21, 4390–4399.
Hervé, D., Lévi-Strauss, M., Marey-Semper, I., Verney, C., Tassin, J. P., Glowinski, J., et al. (1993). Golf and Gs in rat basal ganglia: possible involvement of Golf in the coupling of dopamine D1 receptor with adenylyl cyclase. J. Neurosci. 13, 2237–2248.
Hurley, M. J., Mash, D. C., and Jenner, P. (2001). Dopamine D1 receptor expression in human basal ganglia and changes in Parkinson’s disease. Mol. Brain Res. 87, 271–279. doi: 10.1016/s0169-328x(01)00022-5
Iderberg, H., Francardo, V., and Pioli, E. Y. (2012). Animal models of L-DOPA-induced dyskinesia: an update on the current options. Neuroscience 211, 13–27. doi: 10.1016/j.neuroscience.2012.03.023
Jenner, P. (2008). Molecular mechanisms of L-DOPA-induced dyskinesia. Nat. Rev. Neurosci. 9, 665–677. doi: 10.1038/nrn2471
Jones, D. T., and Reed, R. R. (1989). Golf: an olfactory neuron specific-G protein involved in odorant signal transduction. Science 244, 790–795. doi: 10.1126/science.2499043
Kashihara, K., Manabe, Y., Shiro, Y., Warita, H., and Abe, K. (2000). Effects of repeated methyl levodopa administration on apomorphine sensitivity of rotational behavior and striatal Fos expression of rats with unilateral 6-OHDA lesions. Neurosci. Res. 38, 273–279. doi: 10.1016/s0168-0102(00)00167-x
Kebabian, J. W., and Calne, D. B. (1979). Multiple receptors for dopamine. Nature 277, 93–96. doi: 10.1038/277093a0
Kish, S. J., Shannak, K., and Hornykiewicz, O. (1988). Uneven pattern of dopamine loss in the striatum of patients with idiopathic Parkinson’s disease. N. Engl. J. Med. 318, 876–880. doi: 10.1056/nejm198804073181402
Kondo, T., Mizuno, Y., and Japanese Istradefylline Study Group. (2015). A long-term study of istradefylline safety and efficacy in patients with Parkinson disease. Clin. Neuropharmacol. 38, 41–46. doi: 10.1097/WNF.0000000000000073
Kreitzer, A. C. (2009). Physiology and pharmacology of striatal neurons. Annu. Rev. Neurosci. 32, 127–147. doi: 10.1146/annurev.neuro.051508.135422
Kull, B., Svenningsson, P., and Fredholm, B. B. (2000). Adenosine A2A receptors are colocalized with and activate Golf in rat striatum. Mol. Pharmacol. 58, 771–777. doi: 10.1124/mol.58.4.771
Lalchandani, R. R., van der Goes, M. S., Partridge, J. G., and Vicini, S. (2013). Dopamine D2 receptors regulate collateral inhibition between striatal medium spiny neurons. J. Neurosci. 33, 14075–14086. doi: 10.1523/JNEUROSCI.0692-13.2013
Ledent, C., Vaugeois, J. M., Schiffmann, S. N., Pedrazzi, T., El Yacoubi, M., Vanderhaeghen, J. J., et al. (1997). Agressiveness, hypoalgesia and high blood pressure in mice lacking the adenosine A2a receptor. Nature 388, 674–678. doi: 10.1038/41771
Luquin, M. R., Scipioni, O., Vaamonde, J., Gershanik, O., and Obeso, J. A. (1992). Levodopa-induced dyskinesias in Parkinson’s disease: clinical and pharmacological classification. Mov. Disord. 7, 117–124. doi: 10.1002/mds.870070204
Marcotte, E. R., Sullivan, R. M., and Mishra, R. K. (1994). Striatal G-proteins: effects of unilateral 6-hydroxydopamine lesions. Neurosci. Lett. 169, 195–198. doi: 10.1016/0304-3940(94)90390-5
Missale, C., Nash, S. R., Robinson, S. W., Jaber, M., and Caron, M. G. (1998). Dopamine receptors: from structure to function. Physiol. Rev. 78, 189–225.
Morigaki, R., and Goto, S. (2015). Postsynaptic density protein 95 in the striosome and matrix compartments of the human neostriatum. Front. Neuroanat. 9:154. doi: 10.3389/fnana.2015.00154
Morigaki, R., Okita, S., and Goto, S. (2017). Dopamine-induced changes in Gαolf protein levels in striatonigral and striatopallidal medium spiny neurons underlie the genesis of L-DOPA-induced dyskinesia in parkinsonian mice. Front. Cell. Neurosci. 11:26. doi: 10.3389/fncel.2017.00026
Nash, J. E., and Brotchie, J. M. (2000). A common signaling pathway for striatal NMDA and adenosine A2a receptors: implications for the treatment of Parkinson’s disease. J. Neurosci. 20, 7782–7789.
Nishino, N., Kitamura, N., Hashimoto, T., and Tanaka, C. (1993). Transmembrane signaling systems in the brain of patients with Parkinson’s disease. Rev. Neurosci. 4, 213–222. doi: 10.1515/REVNEURO.1993.4.2.213
Nomoto, M., Kaseda, S., Iwata, S., Shimizu, T., Fukuda, T., and Nakagawa, S. (2000). The metabolic rate and vulnerability of dopaminergic neurons and adenosine dynamics in the cerebral cortex, nucleus accumbens, caudate nucleus, and putamen of the common marmoset. J. Neurol. 247, V16–V22. doi: 10.1007/pl00007779
Obeso, J. A., Grandas, F., Vaamonde, J., Luquin, M. R., Artieda, J., Lera, G., et al. (1989). Motor complications associated with chronic levodopa therapy in Parkinson’s disease. Neurology 39, 11–19.
Pelosi, A., Menardy, F., Popa, D., Girault, J. A., and Hervé, D. (2017). Heterozygous GNAL mice are a novel animal model with which to study dystonia pathophysiology. J. Neurosci. 37, 6253–6267. doi: 10.1523/JNEUROSCI.1529-16.2017
Penit-Soria, J., Durand, C., Besson, M. J., and Hervé, D. (1997). Levels of stimulatory G protein are increased in the rat striatum after neonatal lesion of dopamine neurons. Neuroreport 8, 829–833. doi: 10.1097/00001756-199703030-00005
Pierce, K. L., Premont, R. T., and Lefkowitz, R. J. (2002). Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 3, 639–650. doi: 10.1038/nrm908
Pinna, A., Corsi, C., Carta, A. R., Valentini, V., Pedata, F., and Morelli, M. (2002). Modification of adenosine extracellular levels and adenosine A2A receptor mRNA by dopamine denervation. Eur. J. Pharmacol. 446, 75–82. doi: 10.1016/s0014-2999(02)01818-6
Rangel-Barajas, C., Silva, I., Lopéz-Santiago, L. M., Aceves, J., Erlij, D., and Florán, B. (2011). L-DOPA-induced dyskinesia in hemiparkinsonian rats is associated with up-regulation of adenylyl cyclase type V/VI and increased GABA release in the substantia nigra reticulata. Neurobiol. Dis. 41, 51–61. doi: 10.1016/j.nbd.2010.08.018
Rascol, O., Brooks, D. J., Korczyn, A. D., De Deyn, P. P., Clarke, C. E., and Lang, A. E. (2000). A five-year study of the incidence of dyskinesia in patients with early Parkinson’s disease who were treated with ropinirole or levodopa. Mov. Disord. 342, 1484–1491. doi: 10.1056/NEJM200005183422004
Rascol, O., Perez-Lloret, S., and Ferreira, J. J. (2015). New treatments for levodopa-induced motor complications. Mov. Disord. 30, 1451–1460. doi: 10.1002/mds.26362
Ruiz-DeDiego, I., Naranjo, J. R., Hervé, D., and Moratalla, R. (2015). Dopaminergic regulation of olfactory type G-protein α subunit expression in the striatum. Mov. Disord. 30, 1039–1049. doi: 10.1002/mds.26197
Saka, E., Elibol, B., Erdem, S., and Dalkara, T. (1999). Compartmental changes in expression of c-Fos and FosB proteins in intact and dopamine-depleted striatum after chronic apomorphine treatment. Brain Res. 825, 104–114. doi: 10.1016/s0006-8993(99)01231-7
Sako, W., Morigaki, R., Nagahiro, S., Kaji, R., and Goto, S. (2010). Olfactory type G-protein α subunit in striosome-matrix dopamine systems in adult mice. Neuroscience 170, 497–502. doi: 10.1016/j.neuroscience.2010.06.072
Sancesario, G., Morrone, L. A., D’Angelo, V., Castelli, V., Ferrazzoli, D., Sica, F., et al. (2014). Levodopa-induced dyskinesias are associated with transient down-regulation of cAMP and cGMP in the caudate-putamen of hemiparkinsonian rats: reduced synthesis or increased catabolism? Neurochem. Int. 79, 44–56. doi: 10.1016/j.neuint.2014.10.004
Schwarzschild, M. A., Agnati, L., Fuxe, K., Chen, J. F., and Morelli, M. (2006). Targeting adenosine A2A receptors in Parkinson’s disease. Trends Neurosci. 29, 647–654. doi: 10.1016/j.tins.2006.09.004
Shinotoh, H., Inoue, O., Hirayama, K., Aotsuka, A., Asahina, M., Suhara, T., et al. (1993). Dopamine D1 receptors in Parkinson’s disease and striatonigral degeneration: a positron emission tomography study. J. Neurol. Neurosurg. Psychiatry 56, 467–472. doi: 10.1136/jnnp.56.5.467
Svenningsson, P., Le Moine, C., Fisone, G., and Fredholm, B. B. (1999). Distribution, biochemistry and function of striatal adenosine A2A receptors. Prog. Neurobiol. 59, 355–396. doi: 10.1016/s0301-0082(99)00011-8
Taverna, S., Ilijic, E., and Surmeier, D. J. (2008). Recurrent collateral connections of striatal medium spiny neurons are disrupted in models of Parkinson’s disease. J. Neurosci. 28, 5504–5512. doi: 10.1523/jneurosci.5493-07.2008
Turjanski, N., Lees, A. J., and Brooks, D. J. (1997). In vivo studies on striatal dopamine D1 and D2 site binding in L-dopa-treated Parkinson’s disease patients with and without dyskinesias. Neurology 49, 717–723. doi: 10.1212/wnl.49.3.717
Wei, W., Ding, S., and Zhou, F. M. (2017). Dopaminergic treatment weakens medium spiny neuron collateral inhibition in the parkinsonian striatum. J. Neurophysiol. 117, 987–999. doi: 10.1152/jn.00683.2016
Westin, J. E., Vercammen, L., Strome, E. M., Konradi, C., and Cenci, M. A. (2007). Spatiotemporal pattern of striatal ERK1/2 phosphorylation in a rat model of L-DOPA-induced dyskinesia and the role of dopamine D1 receptors. Biol. Psychiatry 62, 800–810. doi: 10.1016/j.biopsych.2006.11.032
Xiao, D., Bastia, E., Xu, Y. H., Benn, C. L., Cha, J. H., Peterson, T. S., et al. (2006). Forebrain adenosine A2A receptors contribute to L-3,4-dihydroxyphenylalanine-induced dyskinesia in hemiparkinsonian mice. J. Neurosci. 26, 13548–13555. doi: 10.1523/jneurosci.3554-06.2006
Xu, Y., Sun, S., and Cao, X. (2003). Effect of levodopa chronic administration on behavioral changes and fos expression in basal ganglia in rat model of PD. J. Huazhong Univ. Sci. Technolog. Med. Sci. 23, 258–262. doi: 10.1007/bf02829507
Yung, K. K., Smith, A. D., Levey, A. I., and Bolam, J. P. (1996). Synaptic connections between spiny neurons of the direct and indirect pathways in the neostriatum of the rat: evidence from dopamine receptor and neuropeptide immunostaining. Eur. J. Neurosci. 8, 861–869. doi: 10.1111/j.1460-9568.1996.tb01573.x
Keywords: olfactory type G-protein α subunit, levodopa-induced dyskinesia, Parkinson’s disease, dopamine, striatum
Citation: Goto S (2017) Striatal Gαolf/cAMP Signal-Dependent Mechanism to Generate Levodopa-Induced Dyskinesia in Parkinson’s Disease. Front. Cell. Neurosci. 11:364. doi: 10.3389/fncel.2017.00364
Received: 01 September 2017; Accepted: 06 November 2017;
Published: 21 November 2017.
Edited by:
Alessandro Tozzi, University of Perugia, ItalyReviewed by:
Yu-Wei Wu, Stanford University, United StatesGiuseppe Gangarossa, Paris Diderot University, France
Copyright © 2017 Goto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Satoshi Goto, c2dvdG9AdG9rdXNoaW1hLXUuYWMuanA=