Abstract
Postnatally derived cultures of ventral mesencephalic neurons offer several crucial advantages over embryonic ventral mesencephalic cultures, including a higher content of TH-positive cells and the ability to derive cells from the substantia nigra, which contains the neurons most vulnerable to Parkinson’s disease. On the other hand, these cultures are more challenging to produce consistently. Here, we provide an easy-to-implement protocol for culturing postnatal ventral mesencephalic cells from the substantia nigra (SN) and the ventral tegmental area using commercially available media, dishes, and general lab equipment, avoiding extensive material and equipment purchases. The protocol can be completed in about 5 h and provides ventral midbrain neuron cultures on cortex glia feeder layers in three weeks’ time. The protocol uses an optimized protease digestion, tissue storage in Hibernate A during dissection and purification of neurons on an OptiPrep density gradient.
Introduction
When neuronal cell lines are not sufficient to study the aspects of neuron physiology and pathophysiology, primary neuronal cultures can often be used instead. Primary neuronal cultures can be derived either from embryonic or postnatal tissue and both have their advantages and disadvantages depending on the scientific question at hand. Embryonic ventral midbrain (VM) neuronal cultures are easier to prepare, however they have the drawback of only containing 1–2% of Tyrosine hydroxylase (TH) -positive cells, limiting quantitative and single cell analyses. Conversely, primary postnatal cultures are more difficult to derive, but contain a substantial amount of TH-positive cells. Furthermore, neurons from either the substantia nigra (SN) or the ventral tegmental area (VTA) can be isolated separately, as shown and characterized in previous papers (Masuko et al., 1992; Burke et al., 1998; Mosharov et al., 2009), enabling studies comparing cells, which are either more vulnerable (SN) or less vulnerable (VTA) in patients suffering from Parkinson’s disease (PD).
Postnatal rodent midbrain cultures were introduced 25 years ago (Rayport et al., 1992) and have been extensively employed since. It was observed that cell survival drops significantly if the pups are older than 2 days postpartum, limiting the time at which neurons can be obtained for culturing. Nevertheless, we and others have demonstrated several important physiological characteristics of cultured ventral mesencephalic neurons, including (1) stimulation-dependent dopamine exocytosis (Pothos et al., 2000; Staal et al., 2004; Mosharov et al., 2009), (2) the presence of ‘adult’ plasma membrane channels (Rayport et al., 1992; Sulzer et al., 1998; Dryanovski et al., 2013), and (3) morphological differences between SN and VTA neurons (Dryanovski et al., 2013; Pacelli et al., 2015). Previously, we demonstrated that whereas total VM (SN+VTA) cultures contain ∼50% of TH-positive cells that are also calbindin-positive, this proportion is ∼25% in SN and ∼85% in VTA cultures, mirroring the in situ distribution of calbindin positive neurons (Thompson, 2005).
One of the drawbacks of both early (Masuko et al., 1992; Rayport et al., 1992; Cardozo, 1993) and newer protocols (Smeyne and Smeyne, 2002; Fasano et al., 2008; Frank et al., 2008) for preparing postnatal ventral mesencephalic cultures is that in general they are more elaborate in terms of media and reagent set up compared to protocols for embryonic cultures. A simple PubMed query shows that embryonic dopaminergic neuron cultures are preferentially used (1819 publications) and postnatal dopaminergic neuron protocols have not become a general tool (484 publications). This paper aims to provide an easy-to-implement protocol for the culture of postnatal ventral mesencephalic neurons by using standard media for adult neuronal cultures, minimizing reagent setup and not having the need of oxygenation, thus encouraging the transition from embryonic to postnatal ventral mesencephalic cultures. Applications for this protocol mainly lie in the field of PD, for example enabling the study of differential toxicity of MPP+ (1-methyl-4-phenylpyridinium) in SN versus VTA neurons or the effects of cytosolic dopamine, calcium and alpha-synuclein levels. However, this protocol is also interesting for the understanding of physiological properties of ventral mesencephalic neurons such as the mechanism of dopamine release (Staal et al., 2004; Mosharov et al., 2009; Dryanovski et al., 2013; Pacelli et al., 2015; Lieberman et al., 2017).
Experimental Design
The protocol describes a culture system where ventral mesencephalic cells are maintained on a glia feeder layer. Thus, cortex glia cultures need to be obtained first (day 1), while ventral mesencephalic neurons are obtained in a second dissection and plated onto by then confluent glia feeder layers (day 14). Cultures can be obtained from rat as well as mouse brain (P0-P2). Figure 1 presents the flow chart of the protocol.
FIGURE 1

Flow chart diagram of experimental design.
Glia Feeder Layers
First, the brain needs to be removed from the skull, which takes about 30 min, with less than 2 min per brain (12–15 rat pups per litter). Then the cortex tissue for glia feeder layers is dissected (1 h, Figure 2). The tissue can be triturated directly without enzymatic digestion, cells are plated on poly-L-lysine coated glass-bottom dishes (30 min). Critical steps during maintaining glia cultures include a rough wash the day after plating to remove non-attached cells and a mitotic inhibition after reaching confluency.
FIGURE 2
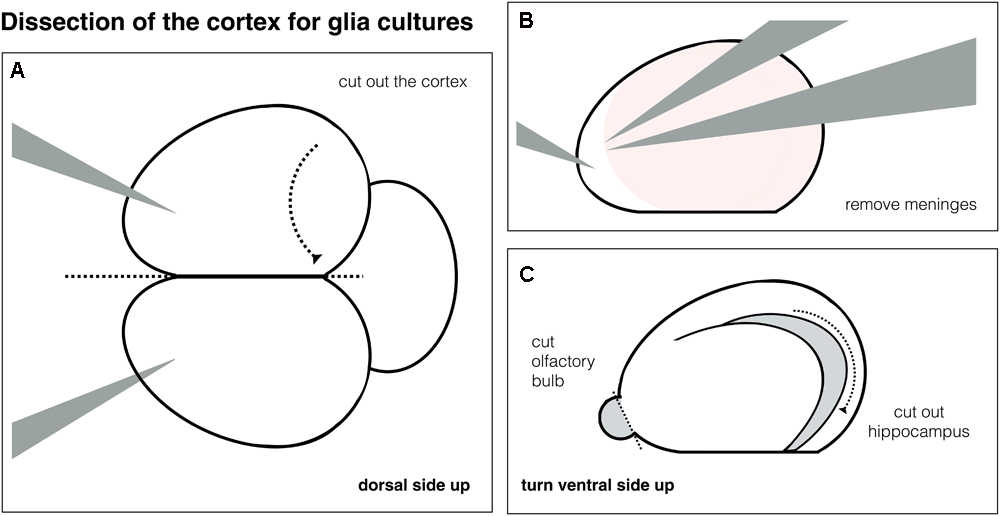
Schematic of cortex dissection. (A) Whole brain and dissection of one cortex. (B) Removal of meninges from the cortex. (C) Removal of olfactory bulb and the hippocampus.
Ventral Mesencephalic Cultures
One or 2 days before the dissection of VM neurons, confluent glia feeder layers receive postnatal neuron medium to condition the media before the neurons are plated on top. The dissection of the VM takes about 1 h, with less than 5 min per brain (Figure 3). The tissue is enzymatically digested using papain and triturated afterward to obtain a single cell suspension (30 min). The cell suspension is centrifuged on an OptiPrep density gradient to purify neurons and to remove cell debris. Neurons are resuspended and plated in MatTek dishes with glia feeder layers and postnatal neuronal medium. GDNF (glial cell derived neurotrophic factor) is added as a growth factor (altogether 1 h). The same brains can be used to obtain new glia cultures from cortex if a second VM dissection is planned.
FIGURE 3
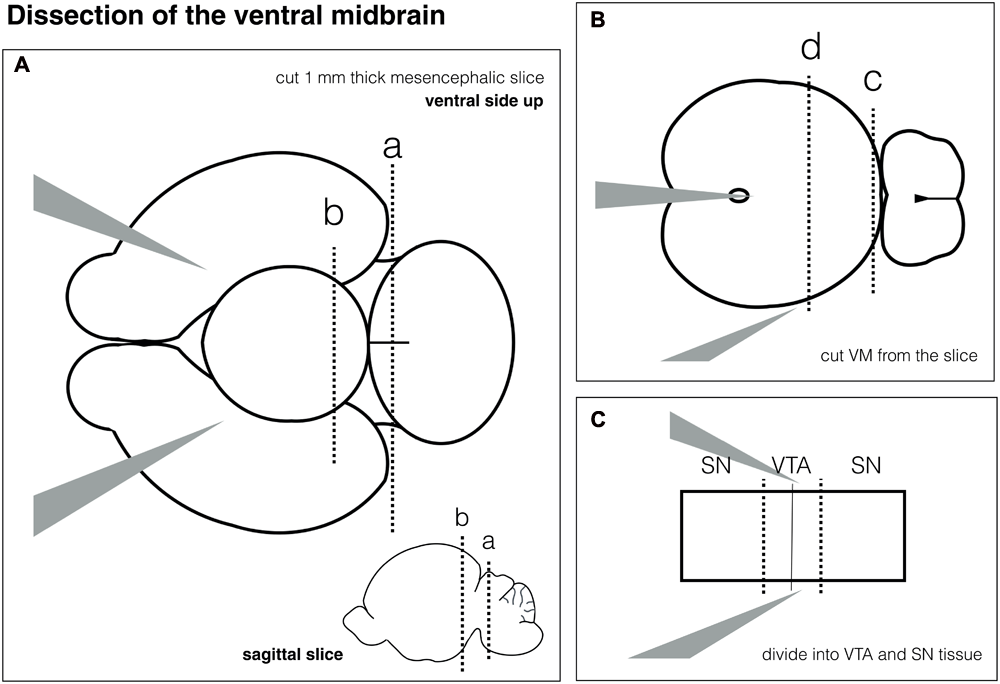
Schematic for ventral midbrain (VM) dissection. (A) Dissection of mesencephalic slice from the whole brain. (B) Dissection of the VM. (C) Separation of Substantia nigra (SN) and ventral tegmental area (VTA).
Materials and Equipment
Materials and Reagents
All materials and reagents needed for the coating process, the dissection itself and for the neuron and glia media are given in Table 1.
Table 1
| Product code | Price in USD | |
|---|---|---|
| Coating process | ||
| MatTek dishes | MatTek Cor., P35G-1.5-14-C, 200 units | 335 |
| Poly-L-lysine | Sigma–Aldrich®, P4707-50ML | 85 |
| Sterile water | GElifesciences, SH30221.17, 500 mL | 15 |
| Dissection | ||
| Minisart filter 0.2 μm | Sigma–Aldrich®, 16534K, 50 units | 161 |
| 10 mL Syringe | BD Plastipak, 302188, 100 units | 9 |
| Sylgard® 184 kit | Dow Corning®, 184S1.1, 1.1 KG | 122 |
| Sterile PBS | Gibco, 14190250, 10x 500 mL | 174 |
| 45% Glucose solution | Sigma–Aldrich®, G8769-100ML | 30 |
| Hibernate® A minus Calcium | BrainBits UK, HA-Ca, 500 mL | 110 |
| Papain suspension | Worthington Bio. Cor., LS003126, 100 mg | 70 |
| DNase I | Sigma–Aldrich®, DN25-100MG | 75 |
| OptiPrep Density Gradient | Sigma–Aldrich®, D1556-250ML | 257 |
| Pasteur pipets 230 mm | Fisher Scientific, 11566963, 1000 units | 52 |
| Slide rings 5 mm × 18 mm 12/CT | Fisher Scientific, NC0639576, 12 units | 50 |
| Neuron media | ||
| Neurobasal-A medium | Life Technologies, 10888022, 500 mL | 65 |
| B-27® Supplement 50x | Life Technologies, 17504044, 10 mL | 90 |
| GlutaMAXTM Supplement 100x | Life Technologies, 35050038, 100 mL | 30 |
| Gentamycin 10 mg/mL | Life Technologies, 15710049, 50 mL | 137 |
| GDNF | Merck Millipore, GF030, 10 μg | 359 |
| Glia media | ||
| DMEM – high glucose | Sigma–Aldrich®, D6546-500ML | 30 |
| FBS | Life Technologies, 10270106, 500 mL | 61 |
| Antibiotics | Gibco, 15240062, 100 mL | 36 |
| Cytarabine | Abcam, ab141924, 100 mg | 39 |
Overview of materials and reagents.
Equipment
Standard cell culture lab equipment, including:
- •
Laminar Flow Cabinet, CO2 cell incubator, inverted microscope.
- •
Hemocytometer, 0.4% Trypan Blue stain.
- •
Dissection stereomicroscope, Petri dishes, a flat cool pack.
- •
Swinging bucket centrifuge with 15 mL tube inlets.
Dissection Instruments
- •
Lister Scissors, serrated 14 cm (Fine Science Tools, cat no. 14131-14), $100.75.
- •
Tissue Forceps, 1x2 Teeth 15.5 cm (Fine Science Tools, cat no. 11021-15), $27.00.
- •
Alternatively, for mice: 1x2 Teeth 12 cm (Fine Science Tools, cat no. 11021-12), $22.00.
- •
Standard Pattern Forceps, curved 12 cm (Fine Science Tools, cat no. 11001-12), $24.00.
- •
Spring Scissors, Angl. 10 mm (Fine Science Tools, cat no. 15006-09), $308.00.
- •
Spatula (VWR, cat no. BOCH3340), $8.10.
- •
2x Dumont #5 Forceps (Fine Science Tools, cat no. 11253-20), 2x $40.50.
- •
1x Dumont # 5A Forceps (Fine Science Tools, cat no. 11253-21), $38.25.
- •
No. 3 Scalpel handle (Swann-Morton®, No 3G S/S), $3.73.
- •
Blades No. 11 (Swann-Morton®, 0303100), $15.20.
NOTE! Instruments have to be cleaned and sterilized on a regular basis to preserve functionality and avoid contamination.
Stock Solution and Media Setup
Cytarabine 100x Stock Solution at 10 mM
- •
Dissolve 100 mg cytarabine with 41.115 mL sterile water (cytarabine is a chemotherapeutic agent, use chemical hood during dissolving procedure, see MSDS for further information).
- •
Filter sterilize and store in aliquots of 5 mL at -20°C.
DNase 100x Stock Solution
- •
Dissolve 100 mg DNase in 2 mL of sterile water.
- •
Filter sterilize and store as 20 μL aliquots at -20°C.
GDNF 20 μg/mL Stock Solution
- •
Dissolve 10 μg GDNF in 500 μL sterile water, store as 10 μL aliquots at -20°C.
- •
Dilute 1:2000 in Postnatal neuron medium for a final concentration of 10 ng/mL.
Glia Plating Medium – DMEM With 10% FBS
- •
Add 5 mL of fetal bovine serum (FBS) to 45 mL of DMEM high glucose medium.
Glia Stopping Medium – DMEM With 5% FBS and 1:100 Cytarabine
- •
Add 2.5 mL of FBS and 500 μL of the cytarabine stock solution to 47 mL of DMEM high glucose medium.
Glia Maintaining Medium – DMEM With 5% FBS
- •
Add 2.5 mL of FBS to 47.5 mL of DMEM high glucose medium.
OptiPrep Gradient 6.2%
∗OptiPrepTM is commonly used to separate cells on a density gradient. In the same way it is able to separate intact cells from cell debris which is critical to obtain healthy cultures.
- •
Add 930 μL OptiPrep to 8 mL Hibernate A® minus Calcium and store at 4°C, protect from light.
Papain Dissociation Solution – 20 Units/mL
- •
Add appropriate volume of the papain suspension to 3 mL Hibernate A® minus Calcium to achieve a concentration of 20 units/mL.
NOTE! The amount of papain is batch specific and has to be calculated from the activity per mg protein (u/mgP) and the concentration of protein per mL (mgP/mL).
- •
Activate in the incubator at 37°C for 30 min, then filter sterilize into a new Falcon tube.
CRITICAL STEP! The papain solution has to be made up freshly for each dissection. The solution is cloudy but should get clear after activation – see Troubleshooting point – Papain solution.
PBS Dissection Buffer
- •
Add 650 μL of 45% glucose solution to 49.35 mL sterile PBS without CaCl2 and MgCl2.
- •
Solution needs to be cooled to 4°C.
- •
You will need 1x 50 mL Falcon tube per dissection, some spare solution for the glia dissociation and 2x half-filled 50 mL Falcon tubes if you need to transport pup heads.
Postnatal Neuron Medium – Neurobasal A With 2% B27, 0.5 mM GlutaMAX and 10 μg/mL Gentamycin
- •
Add 200 μL B27, 25 μL GlutaMAX, and 10 μL Gentamycin to 9.8 mL of Neurobasal A medium.
CRITICAL STEP! Postnatal neuron medium has to be added to the dishes with glia cultures 1 or 2 days before neurons are plated so that media can be conditioned.
Procedure
Preparations
∗The Sylgard is used to make flat pads which function as a bumper when cutting the brain tissue during dissection. Sylgard pads, rings and Pasteur pipets need to be autoclaved before usage.
- •
Mix 1 g curing agent and 10 g elastomer, pour in a 12 well plate so that the liquid level is about 1–2 mm in height. Leave at room temperature for 2 days until solid.
- •
Wrap single pads in aluminum foil, autoclave and keep until dissection.
- •
Wrap single slide rings in aluminum foil, autoclave and keep until dissection.
- •
Put Pasteur pipets in steel canister and autoclave.
∗For appropriate cell attachment, glass-bottom dishes (MatTek dishes, glass culture dishes from Electron Microscopy Science (70760), or custom made glass bottom dishes) have to be coated, Poly-L-lysine has been proven to be sufficient. The coating can be done the day before, if done longer in advance feeder layers may take longer to achieve confluency.
- •
Put a drop of about 200 μL Poly-L-lysine solution onto the glass surface.
- •
Leave for 1 h, wash 3x times with sterile water and let dry in the Laminar Flow Cabinet.
∗Meanwhile, you can prepare the Postnatal neuron medium, which has to be added to the dishes with the pre-established glia feeder layers. Glia plating media can be prepared in advance as well. Please refer to STOCK SOLUTIONS AND MEDIA for recipes.
Removing the Brain – 30 min
∗Please note that all animal experiments underlie the respective national authorities. Animals used were bred and supplied by Charles River UK, Ltd., Scientific, Breeding and Supplying Establishment, registered under Animals (Scientific Procedures) Act 1986, and AAALAC International accredited. This study was carried out in accordance with the recommendations of the Animals (Scientific Procedures) Act 1986 provided by the UK Home Office. The protocol was approved by the NACWO and University of Cambridge Ethics Board.
- •
Decapitate rat pups (P0-P2) using the Listor scissors. The heads can be used fresh, or stored in PBS dissection buffer for up to 2 h.
- •
Put heads onto a 9–10 cm Petri dish lid, hold the heads using the teeth forceps grabbing at the level of the eyes.
- •
Remove the skin from the skull using the curved forceps grabbing a large skin fold and pulling lightly cranial, then caudal.
- •
Cut round the skull with the spring scissors. First cut the half facing away from you, pointing the scissors away from you. Then do the same for the side facing toward you, scissors pointing toward you. Make sure to cut a full circle including the sutura sagittalis.
- •
The skullcap can now be removed easily in one piece using the fine forceps No. 5.
- •
Remove the brain using the small spoon of the spatula.
TIP! Set up a 9–10 cm Petri dish under the dissection stereomicroscope, stick the Sylgard pad with the concave side facing up onto the bottom and fill with 50 mL cooled PBS dissection buffer prior to removing the brain. Make sure not to touch the liquid with unsterile parts. An ice pad underneath helps to cool the solution during the dissection.
Dissection of the Cortex – 1 h
∗The cortex tissue is needed to obtain glia feeder layers for the neuronal cultures. For cortex dissection see Figure 2 (The cortex can also be obtained from brains used for VM dissection before).
- •
Put the brain onto the Sylgard pad, with the dorsal side facing up. Make sure the brains are fully submerged in PBS.
- •
Cut the cortex by inserting the scalpel between the two hemispheres, then carefully divide the cortex from the diencephalon – see Figure 2A.
- •
Remove the meninges using the forceps 5A and 5, one holding the tissue, one gently pulling – see Figure 2B, check dorsal and ventral side are free of meninges.
- •
Remove the olfactory bulb and the hippocampus – Figure 2C.
- •
Store tissue in PBS dissection buffer, put on ice. Pause point – tissue can be kept for up to 2–3 h before trituration (if both VM and glia cultures are done at the same day, VM tissue can be obtained and triturated first, while cortex tissue is maintained on ice).
Trituration of Glia Cells – 30 min
∗The glia cells can be triturated with a simpler approach than for neurons, as neuronal viability is not intended at this point. From one half of the pup cortex, you can obtain about 2 million cells. Cells reach confluence after about 7–11 days in vitro.
- •
Put 5 mL of the PBS dissection buffer in a 50 mL Falcon tube and transfer the cortex tissue into the Falcon tube.
- •
Triturate 2x 15 times with a FBS-coated 1000 μL pipet tip.
- •
Mix 10 μL of cell suspension with 10 μL Trypan blue stain and load 9 μL onto the Hemocytometer, count unstained cells.
- •
Let tissue chunks sediment, take 5 mL of single cell suspension in a new Falcon tube.
- •
Centrifuge at 750 g for 5 min and resuspend cell pellet in 1 mL of Glia plating media.
- •
Plate 650,000 cells per MatTek dish.
TIP! You can already prepare the dishes with Glia plating medium (10% FBS in DMEM), then putting the cells in the middle on the coverslip. Let them sediment briefly for 1 or 2 min, then swirl cells gently around and put dishes into the incubator.
Maintenance of Glia Cultures
∗There are some critical steps in the maintenance of the glia cells to get high quality monolayers. Cells must be washed rigorously and cell division has to be stopped at the right time point. The media changes can be done using media at 4°C, as this helps to retain cultures free from neurons.
- •
The day after dissection glia cultures must be washed to remove non-attached cells.
TIP! Use a 5 mL stripette with a pipetboy for this washing step. Take up the old medium and rigorously pipette the media directly onto the cells on the cover slip, repeat five times. If you have done this properly, only a few attached cells should be left on the coverslip now.
- •
Remove the old medium and add 2 mL of fresh Glia plating medium.
- •
Glia medium needs to be changed twice a week using Glia plating medium (10% FBS in DMEM).
TIP! Watch your cells carefully before each media change. With confluency single glia cells take an angular shape within the monolayer (see Figure 6). If feeder layers are stopped too late, microglia, small round cells with high phase contrast, might enrich on top of the monolayer.
- •
Upon reaching confluence (normally between DIV 7 and 11) cell division has to be halted by changing media to Glia stopping medium (5% FBS in DMEM plus cytarabine).
- •
With the next regular media change, wash cells carefully and put Glia maintaining medium.
TIP! Before removing glia stopping medium, use a 1000 μL pipet tip to swirl medium around, repeat five times. This will help to remove microglia, which are loose after the treatment with Glia stopping medium. Discard media according to national waste disposal guidelines as it contains cytarabine.
- •
The twice weekly media change should be done with Glia maintaining medium (5% FBS in DMEM) from now on.
- •
Please also see Troubleshooting point – Maintenance of glia cultures.
Dissection of the Ventral Midbrain (VM) for Neuronal Cultures – 1 h, See Figure 3
∗This section describes how to dissect the ventral midbrain from postnatal rodent brain. The VM is isolated using a slice cutting approach and can be separated into VTA and SN. Figure 3 gives a schematic overview of individual steps.
- •
Put the brain onto the Sylgard pad with the ventral side facing up to see the midbrain flexure.
- •
Hold the brain by inserting the fine forceps No. 5A into both hemispheres.
- •
Gently remove the meninges over the hindbrain using the fine forceps No. 5.
- •
Make a vertical cut with the scalpel at the level of the hindbrain. see Figure 3A, dotted line a.
- •
Then make another vertical cut cranial to the midbrain flexure – see Figure 3A, dotted line b.
- •
You will have an approximately 1 mm thick coronal brain slice of the mesencephalon. The IV ventricle should be visible on the caudal side of the slice.
- •
Put the fine forceps No. 5A into the ventricle to hold the slice – Figure 3B.
- •
Make a vertical cut between midbrain and hindbrain – see Figure 3B, dotted line c.
- •
Then make another cut half-way between the 1st cut and the ventricle – see Figure 3B, dotted line d.
CRITICAL STEP! Sometimes there can be some meninges left. Make sure to remove them.
- •
Divide the VM into SN and VTA by cutting 1/3, 1/3, 1/3. See dotted lines in Figure 3C.
- •
Put tissue chunks in a 1.5 mL Eppendorf tubes containing cooled Hibernate A® minus Calcium, using a transfer pipette, put back on ice.
- •
Repeat all steps with the next brain.
- •
Pause point – tissue can be kept for about an hour in Hibernate A® minus Calcium before papain digestion is started.
Papain Digestion of VM Tissue – 30 min
∗The dissociation of VM tissue is done using a mild papain digestion. For dissociation, a standard 1000 μL pipet tip coated with FBS is used. The papain can be prepared before starting the dissection. Filtered solution can be kept at 37°C for up to 3 h, otherwise store at 4°C.
- •
Remove Hibernate A® minus Calcium from the tube with the tissue chunks.
- •
Add 1 mL of the activated and filtered papain solution.
- •
Incubate for 10 min in the incubator at 37°C.
- •
In the meantime, prepare the washing solution – add 20 μl of the DNase stock solution to 2 mL Hibernate A® minus Calcium, prepare twice if you do SN and VTA separately.
- •
After 10 min take your cells from the incubator and remove the papain solution, rinse 4x with 500 μL of washing solution each time.
- •
Add 1 mL of Hibernate A® minus Calcium.
- •
Triturate with a FBS-coated 1000 μL pipet tip.
- •
About 30–40 trituration steps in total should be sufficient to reach a single cell suspension, let tissue chunks settle after every 10 steps.
TIP! Use a FBS coated pipet tip. For this, pipette up and down several times in a small volume of FBS before triturating papain-digested tissue chunks.
OptiPrep Purification and Plating of VM Cells – 1 h
∗The OptiPrep purification is a critical step to obtain healthy neuron cultures. Neurons can survive without this step but cell debris can interfere with neuronal sprouting and development.
- •
Put 1 mL of cell suspension in a 15 mL Falcon tube and add 3 mL of Hibernate A® minus Calcium.
- •
Use the long Pasteur pipet to add a sublayer of 1–1.5 mL of the 6.2% OptiPrep solution.
CRITICAL STEP! Make sure to obtain two separate layers. Take up the OptiPrep solution with a long Pasteur pipet, carefully go to the bottom of the Falcon tube and slowly release the OptiPrep solution.
- •
Centrifuge for 15 min at 500 g, do not use the brake to avoid disturbing the layers.
- •
After centrifugation remove about 3 mL from the top containing cell debris.
- •
Collect cells by harvesting the turbid band just in between the two layers, put these 1–2 mL in a new 15 mL Falcon tube.
- •
Add the same volume of Hibernate A® minus Calcium to your cells and centrifuge for 3 min at 500 g, no brake.
- •
Re-suspend your pellet in 1 mL Hibernate A® minus Calcium.
- •
Count your cells by loading 9 μL of the cell suspension onto the Hemocytometer.
- •
According to the experimental setup plate 50,000 to 100,000 cells in the middle of each MatTek dish with the pre-established glia feeder layers and the pre-conditioned Postnatal neuron medium, let cells settle for 10–15 min.
- •
Add 1 μL of GDNF stock solution to each dish with 2 mL media (10 ng/mL) (Burke et al., 1998).
- •
The next day add 2 μL of the cytarabine stock solution to 2 mL media to avoid glia growth.
TIP! Slide rings can be used to keep the cells in the middle of the dish. Here, cell numbers should be reduced. Rings can be reused, wash with plenty of water, spray with ethanol and autoclave again.
- •
Put one of the autoclaved slide rings in the middle of each MatTek dish with the pre-established glia feeder layers and the pre-conditioned Postnatal neuron medium.
- •
Plate your cells in the middle of the slide ring.
- •
Remove the slide rings the next day. Use forceps covered by sterile non-filter pipet tips to avoid contamination.
The cells can be used at DIV 7-14, but also maintenance up to DIV 48 is possible.
TIP! Try to keep your cells without media change, they are healthier with conditioned media. If you must change media, keep half of the old media and add half of fresh preconditioned media.
Results
Postnatal ventral mesencephalic cultures obtained with this protocol are shown in Figure 4A. The cultures show 40.86 ± 6.24% and 34.10 ± 2.40% of TH-positive cells for VTA and SN cultures respectively, as determined by beta-III tubulin (ab7751, Abcam, Cambridge, United Kingdom)/tyrosine hydroxylase (ab41528, Abcam) co-staining (Figures 4B,C). The presence of synaptic connections is shown by co-labeling for the presynaptic marker bassoon (ab82958, Abcam) and beta-III tubulin (ab7751, Abcam, Cambridge, United Kingdom) (Figure 4D). TH-positive ventral mesencephalic cells are highly connected within the neuronal network, showing multiple synaptic inputs (Figure 5A) as well as TH-positive axon terminals co-labeled with bassoon (Figure 5B, arrowheads). Further experiments, including electrophysiology, are required to fully characterize these cultures.
FIGURE 4

Rat ventral midbrain cultures. (A) Brightfield image of live cultures. (B,C) β-III tubulin and tyrosine hydroxylase co-staining for VTA and SN cultures. (D) β-III tubulin and bassoon co-staining showing the development of synapses. Scale bars represent 30 μm.
FIGURE 5
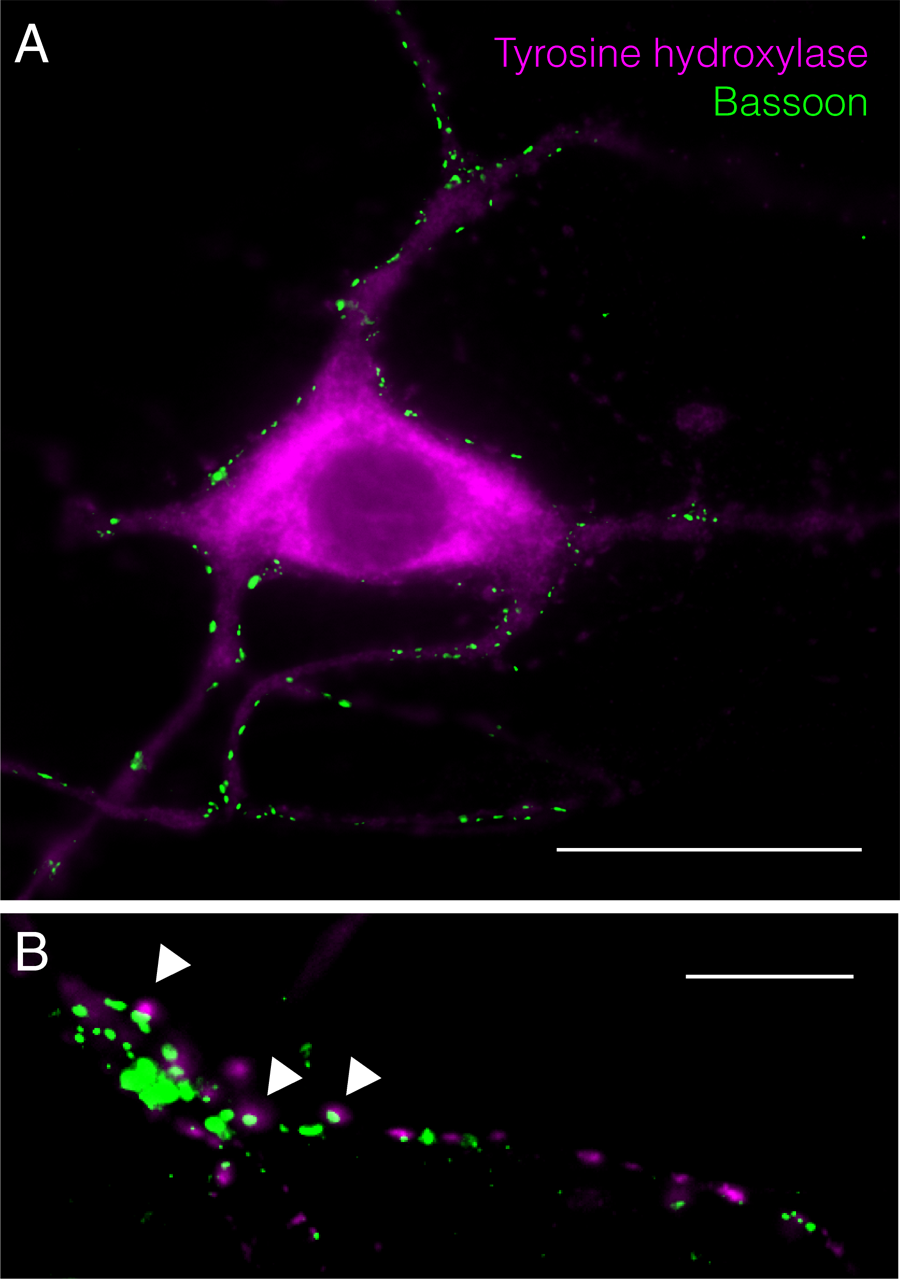
Tyrosine hydroxylase and bassoon co-staining for VM cultures. (A) Synaptic inputs on TH-positive cell. Scale bar represent 30 μm. (B) TH-positive axon terminals with bassoon co-labeling. Scale bar represent 10 μm.
Conclusion
Cultures of postnatal ventral mesencephalic neurons have a wide range of applications, in the study of PD as well as in studying physiologic properties of neuronal subtypes. The culture protocol we present here uses standard media for postnatal neuron cultures and avoids extensive media and equipment setup, thus giving an easy start up protocol for everyone aiming to implement postnatal ventral mesencephalic cultures.
Troubleshooting
Troubleshooting Point – Papain Solution
The papain solution should be clear after activation at 37°C. If this is not the case, the papain is not adequately dissolved and crystals can rupture cells membranes. The protocol from Worthington Biochemical Corporation recommends adding EDTA, 2-mercaptoethanol and cysteine HCl to dissolve papain (see papain datasheet for details). Please note that in this case the solution needs to be pH adjusted afterward. In general, the papain suspension should dissolve in Hibernate A® minus Calcium, if this is not the case the medium might have lost its oxide reductase activity.
Troubleshooting Point – Maintenance of Glia Cultures
It is critical to stop glia cells from dividing in order to achieve good feeder layers, which provide optimal support for neuronal cultures afterward. For this, check your glia cultures regularly. In general, they need Glia stopping medium between DIV 7-11, however, timing may vary depending on the seeding density and quality of the coating. Optimal glia feeder layers show an angular shape of single cells when they are closely packed (Figure 6). If cell division is stopped too early, glia cells do not form a continuous monolayer, however if cell division is stopped too late, microglia settle on top of the feeder layer and can have neurotoxic effects. Microglia may be removed with the media change after stopping with cytarabine using rough washing.
FIGURE 6
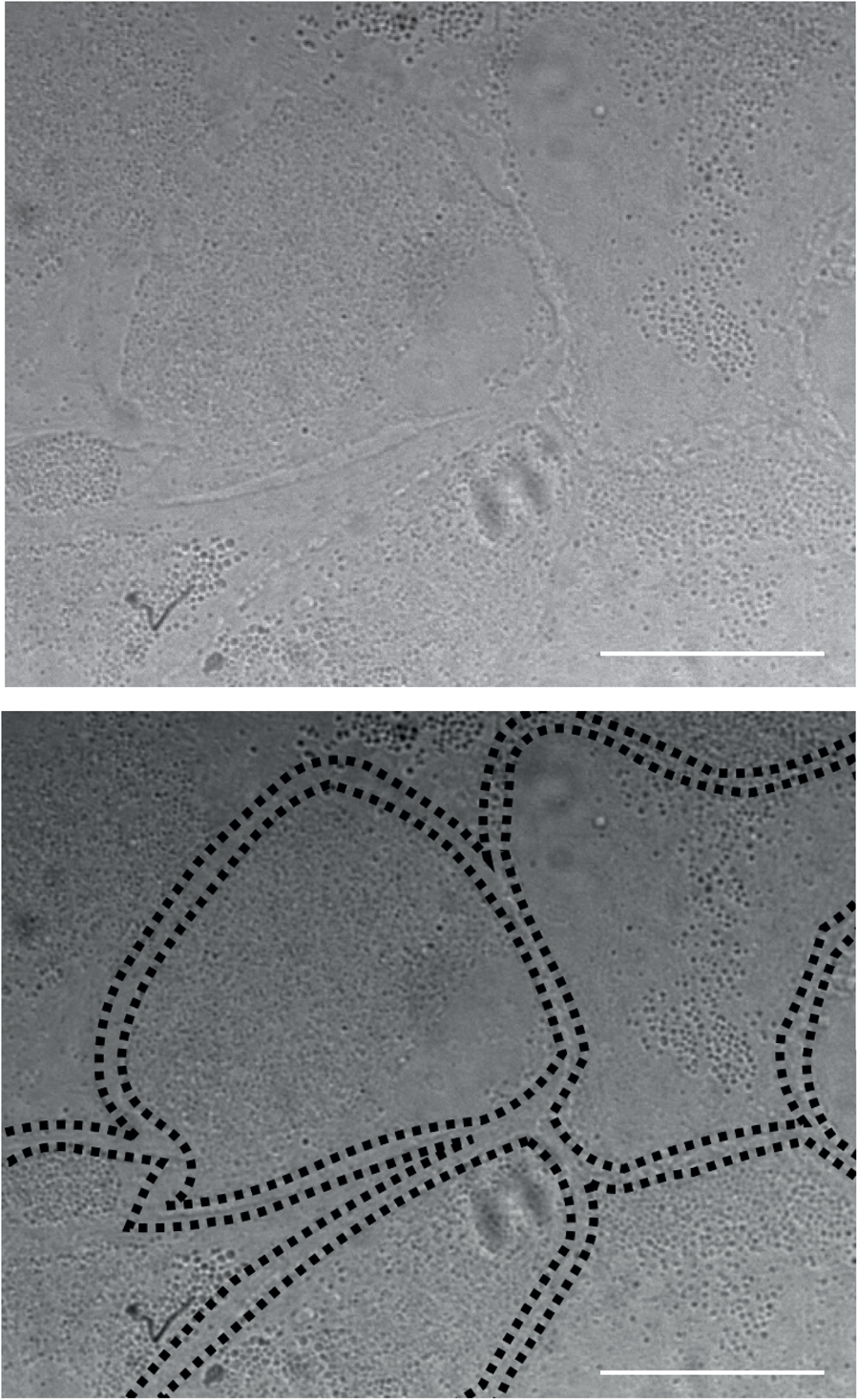
Maintenance of glia feeder layers. Optimal glia feeder layer with closely packed cells in an angular shape. Scale bars represent 30 μm.
The glia culture protocol used here was adapted from (Pébay et al., 2001; Rouach et al., 2006), however, other protocols using papain digestion of cortical tissue or passaging of glia in cell culture flasks can give similar outcomes and can substituted if favored.
Troubleshooting Point – Neurons Don’t Survive
- 1.
When implementing cultures, it is good to start with mixed VM cultures, as this will give you an idea if the protocol works before going to the more vulnerable SN cultures. If you make mixed VM cultures, cut tissue into small chunks to ensure appropriate digestion during papain incubation.
- 2.
In general, cultures should be handled with care. Avoid shaking and extensive times out of the incubator. Do not place dishes on cold steel surfaces, but rather use a heating pad or pre-warmed plastic support underneath.
- 3.
Make sure to have good feeder layers. If glia cells are too sparse, the neuronal cultures are not sufficiently supported, on the other hand if the glia feeder layers are too dense, the postnatal neuron medium is exhausted too quickly and you need to change media.
- 4.
The B27 is a critical supplement in the neuronal medium, however, it can vary from batch to batch. It is therefore, good practice to try the new batch in comparison with the old one before running out.
- 5.
Additionally acetylcysteine, desferoxamine, and kynurenic acid have been shown to have beneficial effect on postnatal VM neuron cultures and can be tried to improve culture conditions (Mena et al., 1997; Sulzer et al., 2000; Petersen et al., 2001).
Statements
Author contributions
JL carried out the experiments. JL wrote the manuscript with support from EM, EK, DS, and GKS. DS and GKS helped supervise the project. All authors were involved in the conception of the work, discussed the results, approved the final manuscript, and agreed to be accountable for all aspects of the work.
Funding
JL was supported by a research fellowship from the Deutsche Forschungsgemeinschaft (DFG; award LA 3609/2-1). GKS acknowledges funding from the Wellcome Trust, the UK Medical Research Council (MRC), Alzheimer’s Research UK (ARUK), and Infinitus China Company, Ltd. EM was supported from an R01 grant from NINDS (NS075222).
Acknowledgments
We thank Dr. Colin Hockings for proofreading the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
BurkeR. E.AntonelliM.SulzerD. (1998). Glial cell line-derived neurotrophic growth factor inhibits apoptotic death of postnatal substantia nigra dopamine neurons in primary culture.J. Neurochem.71517–525. 10.1046/j.1471-4159.1998.71020517.x
2
CardozoD. L. (1993). Midbrain dopaminergic neurons from postnatal rat in long-term primary culture.Neuroscience56409–421. 10.1016/0306-4522(93)90342-D
3
DryanovskiD. I.GuzmanJ. N.XieZ.GalteriD. J.Volpicelli-daleyL. A.LeeV. M.et al (2013). Calcium entry and alpha-synuclein inclusions elevate dendritic mitochondrial oxidant stress in dopaminergic neurons.J. Neurosci.3310154–10164. 10.1523/JNEUROSCI.5311-12.2013
4
FasanoC.ThibaultD.TrudeauL. É (2008). Culture of postnatal mesencephalic dopamine neurons on an astrocyte monolayer.Curr. Protoc. Neurosci.443.21.1–3.21.19. 10.1002/0471142301.ns0321s44
5
FrankL. E.Caldera-SiuA. D.PothosE. N. (2008). Primary dissociated midbrain dopamine cell cultures from rodent neonates.J. Vis. Exp.21:820. 10.3791/820
6
LiebermanO. J.ChoiS. J.KanterE.SaverchenkoA.FrierM. D.FioreG. M.et al (2017). Alpha-Synuclein dependent calcium entry underlies differential sensitivity of cultured SN and VTA dopaminergic neurons to a Parkinsonian neurotoxin effect of MPP on SN and VTA neurons.eNeuro4:ENEURO.0167-17.2017. 10.1523/ENEURO.0167-17.2017
7
MasukoS.NakajimaS.NakajimaY. (1992). Dissociated high-purity dopaminergic neuron cultures from the substantia nigra and the ventral tegmental area of the postnatal rat.Neuroscience49347–364. 10.1016/0306-4522(92)90101-7
8
MenaM. A.DavilaV.SulzerD. (1997). Neurotrophic effects of L-DOPA in postnatal midbrain dopamine neuron/cortical astrocyte cocultures.J. Neurochem.691398–1408. 10.1046/j.1471-4159.1997.69041398.x
9
MosharovE. V.LarsenK. E.KanterE.PhillipsK. A.WilsonK.SchmitzY.et al (2009). Interplay between cytosolic dopamine, calcium, and α-synuclein causes selective death of substantia nigra neurons.Neuron62218–229. 10.1016/j.neuron.2009.01.033
10
PacelliC.GiguèreN.BourqueM. J.LévesqueM.SlackR. S.TrudeauL. É (2015). Elevated mitochondrial bioenergetics and axonal arborization size are key contributors to the vulnerability of dopamine neurons.Curr. Biol.252349–2360. 10.1016/j.cub.2015.07.050
11
PébayA.ToutantM.PrémontJ.CalvoC. F.VenanceL.CordierJ.et al (2001). Sphingosine-1-phosphate induces proliferation of astrocytes: regulation by intracellular signalling cascades.Eur. J. Neurosci.132067–2076. 10.1046/j.0953-816x.2001.01585.x
12
PetersenK. E.BehrG. G.RomeroN.PrzedborskiS.BrundinP.SulzerD. (2001). Brain-derived neurotrophic factor inhibits apoptosis and dopamine-induced free radical production in striatal neurons but does not prevent cell death.Brain Res. Bull.56331–335. 10.1016/S0361-9230(01)00580-9
13
PothosE. N.LarsenK. E.KrantzD. E.LiuY.HaycockJ. W.SetlikW.et al (2000). Synaptic vesicle transporter expression regulates vesicle phenotype and quantal size.J. Neurosci.207297–7306.
14
RayportS.SulzerD.ShiW. X.SawasdikosolS.MonacoJ.BatsonD.et al (1992). Identified postnatal mesolimbic dopamine neurons in culture: morphology and electrophysiology.J. Neurosci.124264–4280.
15
RouachN.PébayA.MêmeW.CordierJ.EzanP.EtienneE.et al (2006). S1P inhibits gap junctions in astrocytes: involvement of Gi and Rho GTPase/ROCK.Eur. J. Neurosci.231453–1464. 10.1111/j.1460-9568.2006.04671.x
16
SmeyneM.SmeyneR. J. (2002). Method for culturing postnatal substantia nigra as an in vitro model of experimental Parkinson’s disease.Brain Res. Brain Res. Protoc.9105–111. 10.1016/S1385-299X(02)00117-4
17
StaalR. G. W.MosharovE. V.SulzerD. (2004). Dopamine neurons release transmitter via a flickering fusion pore.Nat. Neurosci.7341–346. 10.1038/nn1205
18
SulzerD.BogulavskyJ.LarsenK. E.BehrG.KaratekinE.KleinmanM. H.et al (2000). Neuromelanin biosynthesis is driven by excess cytosolic catecholamines not accumulated by synaptic vesicles.Proc. Natl. Acad. Sci. U.S.A.9711869–11874. 10.1073/pnas.97.22.11869
19
SulzerD.JoyceM. P.LinL.GeldwertD.HaberS. N.HattoriT.et al (1998). Dopamine neurons make glutamatergic synapses in vitro.J. Neurosci.184588–4602.
20
ThompsonL. (2005). Identification of dopaminergic neurons of nigral and ventral tegmental area subtypes in grafts of fetal ventral mesencephalon based on cell morphology, protein expression, and efferent projections.J. Neurosci.256467–6477.
Summary
Keywords
ventral mesencephalic neurons, dopaminergic neurons, postnatal neuron cultures, substantia nigra, ventral tegmental area, postnatal neurons, protocol, Parkinson’s disease
Citation
Lautenschläger J, Mosharov EV, Kanter E, Sulzer D and Kaminski Schierle GS (2018) An Easy-to-Implement Protocol for Preparing Postnatal Ventral Mesencephalic Cultures. Front. Cell. Neurosci. 12:44. doi: 10.3389/fncel.2018.00044
Received
26 October 2017
Accepted
02 February 2018
Published
02 March 2018
Volume
12 - 2018
Edited by
Pier Giorgio Mastroberardino, Erasmus University Rotterdam, Netherlands
Reviewed by
Louis-Eric Trudeau, Université de Montréal, Canada; Ezia Guatteo, Università degli Studi di Napoli Parthenope, Italy
Updates
Copyright
© 2018 Lautenschläger, Mosharov, Kanter, Sulzer and Kaminski Schierle.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Janin Lautenschläger, jl865@cam.ac.uk; janin.lautenschlaeger@gmail.com Gabriele S. Kaminski Schierle, gsk20@cam.ac.uk
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.