Abstract
Neurons depend on the highly dynamic microtubule (MT) cytoskeleton for many different processes during early embryonic development including cell division and migration, intracellular trafficking and signal transduction, as well as proper axon guidance and synapse formation. The coordination and support from MTs is crucial for newly formed neurons to migrate appropriately in order to establish neural connections. Once connections are made, MTs provide structural integrity and support to maintain neural connectivity throughout development. Abnormalities in neural migration and connectivity due to genetic mutations of MT-associated proteins can lead to detrimental developmental defects. Growing evidence suggests that these mutations are associated with many different neurodevelopmental disorders, including intellectual disabilities (ID) and autism spectrum disorders (ASD). In this review article, we highlight the crucial role of the MT cytoskeleton in the context of neurodevelopment and summarize genetic mutations of various MT related proteins that may underlie or contribute to neurodevelopmental disorders.
Introduction
The development of the central nervous system (CNS) and wiring of the brain is an extremely complex process, governed by the communication and careful coordination of the neuronal cytoskeleton, comprised of microtubule (MT), actin and intermediate filament networks (Menon and Gupton, 2016; Pacheco and Gallo, 2016; Kirkcaldie and Dwyer, 2017). Newly formed neurons face many challenges as they undergo dramatic changes in shape and migrate their way through the extracellular terrain in order to establish connections with other cells. Specifically, dynamic MTs play pivotal roles in creating cell polarity, as well as aiding in neural migration in order to establish appropriate neural connectivity throughout development and into adulthood. The elaborate MT network is integral to facilitate numerous morphological and functional processes during neurodevelopment, including cell proliferation, differentiation and migration, as well as accurate axon guidance and dendrite arborization. The organization and remodeling of the MT network is also essential for developing neurons to form axons, dendrites and assemble synapses. Moreover, in mature neurons, MTs continue to maintain the structure of axons and dendrites, and serve as tracks for intracellular trafficking, allowing motor proteins to deliver specific cargoes within the cell.
As brain development relies heavily on proper MT function, defects in the MT cytoskeleton can lead to detrimental effects on neural proliferation, migration and connectivity. Over the last several years, numerous studies have identified mutations within genes coding for proteins that interact with and directly modulate the structure and function of the MT cytoskeleton. Many of these MT-associated mutations have been linked to various neurodevelopmental disorders including lissencephaly, polymicrogyria, autism spectrum disorders (ASD) and intellectual disabilities (ID; Srivastava and Schwartz, 2014; Chakraborti et al., 2016; Stouffer et al., 2016). The regulation of the MT cytoskeleton during specific stages of brain development still remains an active topic of research. In this review article, we highlight various studies that illustrate important functions of the MT cytoskeleton that contribute to proper neural development and how genetic mutations within MT-related proteins can alter these crucial functions that may lead to disorders of neural development.
The Role of the Microtubule Cytoskeleton During Neural Development
MTs are one of the major cytoskeletal components present in all eukaryotic cell types. They are composed of α- and β-tubulin heterodimers, which bind to form 13 polarized linear protofilaments that associate laterally together to create the MT (Figure 1; Akhmanova and Steinmetz, 2008). MTs are extremely dynamic structures, existing in either a growing state (polymerization) or shrinking state (depolymerization). The plus ends of MTs can rapidly switch between these two states, going from growth to shrinkage (catastrophe), or from shrinkage to growth (rescue), a process called “dynamic instability” (Mitchison and Kirschner, 1984). Developing neurons depend on this stochastically dynamic nature of the MT cytoskeleton in order to remodel their shape, proliferate and migrate, as well as other processes during different phases of neural development, as described in more detail below.
Figure 1
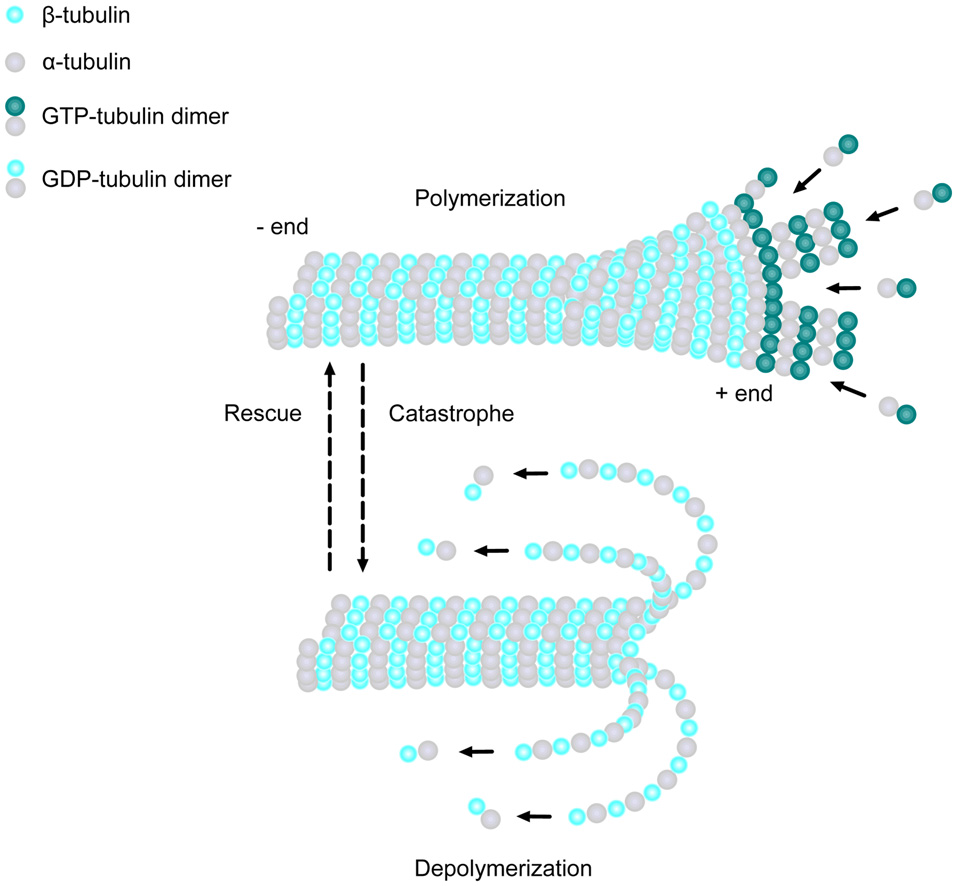
Microtubule (MT) basics. MTs are linear structures comprised of α-tubulin and β-tubulin heterodimers. MTs are extremely dynamic, existing in either a growing state (polymerization) or shrinking state (depolymerization), and can rapidly switch from growth to shrinkage (catastrophe) or from shrinkage to growth (rescue). Addition of new GTP-bound heterodimers occurs at the MT plus end during polymerization. Shortly thereafter, the tubulin subunits hydrolyze their bound GTP to GDP. When the addition of GTP-bound heterodimers slows and the MT lattice is composed of predominantly GDP-tubulin, the protofilaments splay apart and the MT depolymerizes.
Neurite Formation and Axon Specification
Neurons begin their development as spherical, unpolarized cells, with MTs emanating from MT organizing centers (MTOCs), such as the centrosome, and are nucleated by the γ-tubulin ring complex (γ-TuRC; Kuijpers and Hoogenraad, 2011). This structure acts as a template for the α- and β-tubulin dimers to begin polymerization and is the cap of the minus end as the MT plus end grows away from the MTOC (Kuijpers and Hoogenraad, 2011). Once differentiation occurs, neurons form multiple processes, termed neurites, which extend from the spherical cell body and elongate to form thin protrusions (Götz and Huttner, 2005). MTs are one of the major players which influence the formation of neurites by creating small bundles that invade lamellipodia in multiple directions (Götz and Huttner, 2005). It has been suggested that MT sliding may play a key role in initiating neurite formation. One study demonstrated that MT-associated protein 2c (MAP2c) could stabilize MT bundles in vitro, which then rapidly move toward the cell periphery, prompting protrusions via a dynein-driven force (Dehmelt et al., 2006). It has also been shown that neurite formation can be induced by the MT motor protein, kinesin-1, which drives MT sliding and generates a mechanical force on the cell membrane (Lu et al., 2013; Winding et al., 2016). Studies have also demonstrated that actin filaments contribute to membrane protrusions, working in combination with stable MTs to initiate neurite outgrowth (Dent et al., 2011; Sainath and Gallo, 2015). Together, these results suggest that the local increase in actin dynamics, as well as the mechanical forces produced by MT sliding, all play key roles in stimulating neurite formation.
After neurite extension, one of the multiple processes becomes the axon, while the others later develop into dendrites (Menon and Gupton, 2016). Stable MTs are essential in axon specification and actively determine the polarity of developing neurons (Conde and Cáceres, 2009; Hoogenraad and Bradke, 2009). Interestingly, the ratio of stable to dynamic MTs was found to be significantly higher in one specific neurite compared to other neurites during this stage of neural development (Witte et al., 2008). Furthermore, axon formation was induced in unpolarized neurons following the addition of a photoactivatable analog of the MT-stabilizing drug taxol, while the addition of low doses of taxol led to the formation of multiple axons (Witte et al., 2008). These results suggest that in unpolarized neurons, the stabilization of MTs within one specific neurite occurs before axon formation begins. Once neurons begin to establish a distinct polarity, the centrosome progressively loses its function as an MTOC, with MTs beginning to nucleate from non-centrosomal MTOCs, such as Gogli outposts (Stiess et al., 2010; Stiess and Bradke, 2011; Ori-McKenney et al., 2012; Yau et al., 2014). Moreover, the stabilization of non-centrosomal MTs by the minus-end binding protein, calmodulin-regulated spectrin-associated protein 2 (CAMSAP2), was shown to be required for the establishment of neuronal polarity and axon formation (Yau et al., 2014). Reduction of CAMSAP2 inhibited both proper polarization and axon formation in vitro, and led to defects in neural migration in vivo, providing further evidence that MT stabilization is critical for these processes during neural development (Yau et al., 2014). Increased MT stabilization may also provide tracks for selective targeting of MT motor proteins to aid in the transportation of various organelles and proteins that are necessary for the eventual formation of axonal segments (Kapitein and Hoogenraad, 2011). Kinesin-1 has been shown to preferentially bind to stabilized MTs and accumulate in the future axon, contributing to early polarized trafficking, suggesting that an increase of stable MTs may lead to increased kinesin-mediated transport, driving both neuronal polarization and eventual axon specification (Nakata and Hirokawa, 2003; Jacobson et al., 2006).
Axon Elongation and Branching
A major change that occurs, following axon specification, is the formation and enlargement of the growth cone, a dynamic structure at the tip of the growing axon responsible for driving axon elongation and branching (Kahn and Baas, 2016). Neuronal growth cones probe the extracellular environment and come into contact with various external stimuli, thus steering the axon in a particular direction (Lowery and Van Vactor, 2009). The growth cone uses the cytoskeletal machinery to progress through three distinct morphological stages, termed protrusion, engorgement and consolidation (Lowery and Van Vactor, 2009). To progress through these stages, an array of dynamic MTs penetrate into the central and peripheral domains of the growth cone, and can display different behaviors which include splaying, looping and bundling (Tanaka and Kirschner, 1991). This remodeling is essential for MTs to probe the growth cone periphery in search of guidance cues, which prompt growth cone advancement and turning. Additionally, growth cone formation and advancement require the interaction between actin filaments and dynamic MTs, which work in combination to promote the extension of lamellipodia and filopodia at the tip of the axon (Dent et al., 2011).
Cytoskeletal remodeling must also occur during axon branching, which involves an accumulation of actin filaments that will form axonal filopodia along the axon shaft (Mattila and Lappalainen, 2008). Shortly thereafter, MTs begin to invade the actin-rich filopodia with localized splaying of the normally bundled MT array (Dent et al., 1999; Gallo and Letourneau, 1999). This invasion by the axonal MTs into the filopodia allows their maturation into collateral branches as they continue extending. Early studies showed that there was an increase in the number of MTs at regions that eventually become axon branches, suggesting that MTs within the parent axon undergo fragmentation, and a portion of these fragments are then translocated to the developing branches (Yu et al., 1994; Kalil and Dent, 2014; Armijo-Weingart and Gallo, 2017). Thus, MT fragmentation and transportation appear to be key mechanisms which regulate the beginning of axon branch formation. Crosstalk between actin and MTs also seems to be required during the initial steps of axon branching (Dent and Kalil, 2001; Kalil and Dent, 2014; Gallo, 2016; Pacheco and Gallo, 2016). Dynamic MTs colocalize with F-actin in regions of axon branching, whereas stable MTs are excluded from these regions (Dent and Kalil, 2001). Moreover, when MT dynamics were dampened in neurons treated with either taxol or nocodazole, invasion of MTs into filopodia was reduced and they were unable to interact with actin-filament bundles, which resulted in decreased neurite formation (Dent et al., 2007). Recently, the cytoskeleton-associated protein, drebrin, was shown to regulate both actin filaments and MTs to initiate the formation of axon branches (Ketschek et al., 2016; Zhao et al., 2017). Endogenous drebrin was found to localize to axon actin patches in vivo, which eventually form axonal filopodia, suggesting that drebrin may contribute to the development of these precursor structures before axon branching (Ketschek et al., 2016). Additionally, reduction of drebrin severely inhibited axonal filopodia formation and the number of collateral branches in vitro, further demonstrating an essential role for drebrin in regulating these processes (Ketschek et al., 2016). Expression of drebrin also increased the number of axonal filopodia that contained end-binding protein 3 (EB3) comets, indicating that drebrin can promote the entry of dynamic MTs into axonal filopodia during the formation of axon branches (Ketschek et al., 2016). Thus, dynamic MTs and their interactions with actin filaments are crucial for the establishment and formation of collateral branches during neuronal development.
Dendritogenesis and Synapse Formation
Following axon formation, other neurites begin to develop into dendrites, prompting dramatic changes to the MT network. While dendrites branch more extensively than axons, the behavior of MTs during this process has not yet been fully elucidated. One of the most striking features that distinguishes dendrites from axons is the orientation of their MTs. Axons display uniform plus-end distal MTs, while dendrites harbor a population of MTs with mixed polarity, where both plus and minus ends are oriented towards the cell body (Kapitein and Hoogenraad, 2015; Tas et al., 2017). These distinct polarity patterns are essential for determining the directionality of intracellular cargo transport, which maintains both the composition and morphological differences between axons and dendrites. Several studies have suggested that the translocation of MTs of differing orientations within dendrites plays a key role in establishing their mixed polarity (Sharp et al., 1995, 1997; Yu et al., 2000; Zheng et al., 2008; Rao et al., 2017). When MT assembly was inhibited with low levels of vinblastine, dendritic elongation was reduced, however MT reorientation was not hindered, suggesting that transport of MTs from the cell body is a possible mechanism for creating the non-uniform orientation of dendritic MTs (Sharp et al., 1995). Moreover, several studies have shown that neurons use distinct motor proteins to engage in specific transportation events that may drive this difference in MT orientation between axons and dendrites (Sharp et al., 1997; Yu et al., 2000; Zheng et al., 2008; Rao et al., 2017). Reduction of the kinesin-related motor protein CHO1 (also known as KIF23) from cultured neurons inhibited the movement of minus-end distal MTs into nascent dendrites and these processes failed to differentiate (Sharp et al., 1997; Yu et al., 2000). Likewise, reduction of kinesin-12 (also known as KIF15) produced similar phenotypic results (Lin et al., 2012). The abnormalities observed with depletion of kinesin-6 could be rescued by overexpression of kinesin-12, indicating that these two motor proteins may share functional redundancy within dendrites (Lin et al., 2012). Similarly, neurons treated with the dynein inhibitor, Ciliobrevin D, displayed a decrease in MT transport and abnormal MT orientation in the axon, which could be rescued after Ciliobrevin D washout (Rao et al., 2017). Thus, active MT transport by specific motor proteins is essential for both establishment and maintenance of MT bi-directionality during dendritic development.
The next steps in neuronal development are the formation and maturation of dendritic spines, which represent postsynaptic sites of excitatory synapses. The connections between an axon and a dendrite of neighboring neurons is a process that continuously occurs throughout development and into adulthood, as the brain rewires these connections in response to novel stimuli. For some time, it was thought that dendritic spines were devoid of dynamic MTs, and that actin was the main regulator of spine morphology and dynamics associated with synaptic plasticity. However, within the last decade, the use of new visualization techniques has revealed that MT dynamics do play an essential role in dendritic spine development (Yau et al., 2016; Dent, 2017). In concert with MT transport, MT polymerization actively occurs and contributes to the development of the dendritic branches. MT assembly occurs within the cell body, and after transport, these newly formed MTs are incorporated into the dendrite (Dent, 2017). Dynamic MTs can penetrate into dendritic spines of different shapes, including mushroom, stubby and thin, and can modulate their morphology by interacting with F-actin via +TIPs, such as EB3 (Gu et al., 2008; Jaworski et al., 2009; Dent, 2017). Furthermore, reduction of EB3 impairs spine development, and pharmacological manipulation of MT dynamics severely decreases the total number of spines and inhibits the formation of spines that are induced by brain-derived neurotrophic factor (BDNF; Gu et al., 2008; Jaworski et al., 2009). Interestingly, N-methyl-D-aspartate receptor (NMDAR)-dependent synaptic activation in hippocampal cell cultures increases the proportion of dendritic spines containing dynamic MTs, which contributes to spine enlargement, and this increase in MT invasion is inhibited by either blocking action potential activity or by dampening MT dynamics (Merriam et al., 2011). Additionally, dendritic spines exhibiting elevations in calcium signaling contain increased amounts of F-actin, and these spines are preferentially targeted by dynamic MTs, which interact with F-actin in a drebrin-dependent manner (Merriam et al., 2013). Moreover, studies have shown that the invasion of dynamic MTs into dendritic spines provide tracks for MT-dependent motors to deliver specific cargoes that are essential for synaptic plasticity (McVicker et al., 2016). It has also been demonstrated that the severing of dynamic MTs by the MT-severing proteins, katanin and fidgetin, are essential for dendrite development and synapse formation (Mao et al., 2014; Leo et al., 2015), which will be discussed in greater detail in subsequent sections. Together, these results strongly indicate that dynamic MTs are key regulators of dendritic spine formation, maintenance and synaptic activity, and that +TIPs play a role in this dynamicity to modulate spine morphology during neural development.
Microtubules Guide Intracellular Transport During Axon and Dendrite Maintenance
MT-dependent cargo transport is an essential process that regulates the unique composition of both axons and dendrites and aids in maintaining their polarized morphology. This active transport mechanism is also necessary to accurately distribute specific cargoes and establish particular signaling pathways throughout the neuron. Neuronal intracellular transport is driven by the MT-dependent motors, kinesin and dynein, which move in opposite directions to deliver various cargo such as organelles, neurotransmitter receptors, cell signaling molecules and mRNAs, to the correct location within the cell (Hirokawa et al., 2010). Moreover, adaptor proteins and local signaling pathways regulate proper cargo delivery by aiding in cargo loading, anchoring and motility (Maday et al., 2014). The specific orientations of MTs are critical in determining the routes in which cargoes will travel throughout the neuron. For example, studies have demonstrated that dynein selectively transports cargoes along minus-end out oriented MTs found within dendrites (Kapitein et al., 2010; Tas et al., 2017). It has also been shown that kinesin-1 preferentially transports cargoes along MTs in axons, whereas kinesin-3 exhibits no selectivity, delivering cargoes to both axons and dendrites. It has been reported that specific post translational modifications of MTs are recognized by various kinesin superfamily members, which can regulate their localization and functions within neurons (Kapitein et al., 2010; Tas et al., 2017). As mentioned previously, it has been suggested that kinesin-1 preferentially binds to stabilized MTs that have either been acetylated or detyrosinated (Nakata et al., 2011), while kinesin-3 prefers to bind to tyrosinated MTs (Tas et al., 2017). However, the exact mechanisms which govern MT properties within axons and dendrites that subsequently guide specific motor proteins still remain unknown.
Regulators of the Neuronal Microtubule Network
The essential remodeling and organization of the MT cytoskeleton during neuronal morphogenesis relies on a vast array of MT-regulating proteins that have been identified over the last few decades (Figure 2). These proteins carry out specific functions to control MT dynamicity, fragmentation, stabilization, and intracellular transport. Many act directly on MTs to affect their nucleation, assembly, or stability, while others act indirectly by modulating tubulin levels or intracellular transport, producing downstream effects on neuronal differentiation. The combined efforts of MT-regulating proteins such as MT associated proteins (MAPs), +TIPs and MT motor proteins, provide the mechanisms by which the MT network reshapes its architecture during neuronal development. Here, we highlight several groups of MT regulators and their respective functions that control the assembly of new MTs and their dynamicity, as well as how they regulate MT stability, fragmentation and intracellular trafficking within neurons.
Figure 2
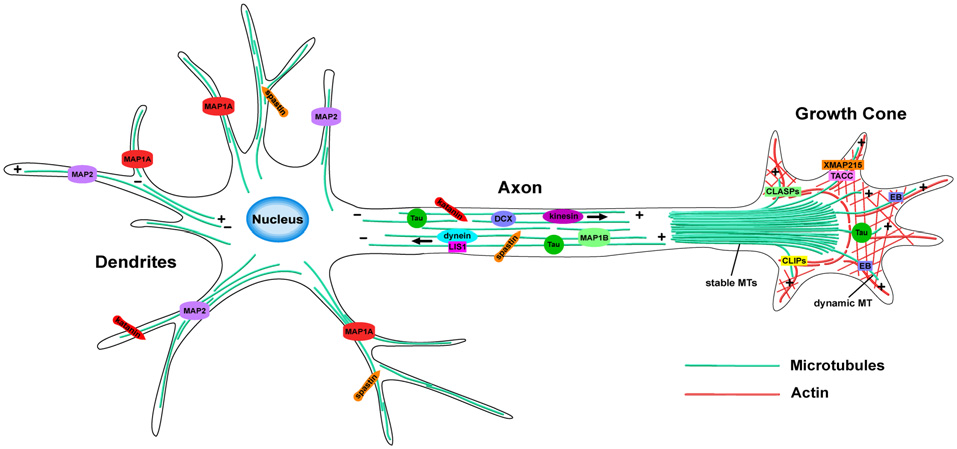
MT organization and MT-associated proteins (MAPs) in axons and dendrites. In axons, MTs form stable, polarized bundles, which provide structural integrity and serve as tracks to guide MT-dependent motor proteins. Axonal MTs are stabilized by several MAPs including Tau, MAP1B and DCX. The growth cone contains an array of both stable and dynamic MTs, which prompt growth cone advancement and turning. Various +TIPs accumulate at the growing MT plus ends in the growth cone, where they regulate MT dynamics during axon outgrowth and guidance. MTs of mixed polarity are located within dendrites where MAP1A and MAP2 aid in MT stabilization. The MT-severing proteins, katanin and spastin, are critical for reorganizing the MT network in both axons and dendrites.
Microtubule Plus End Tracking Proteins (+TIPs)
+TIPs are proteins which accumulate at growing MT plus-ends where they regulate MT dynamics, as well as facilitate their interactions with other proteins, organelles, and actin (Akhmanova and Steinmetz, 2008). Many families of +TIPs have been found to be crucial for different neurodevelopmental processes, specifically during neurite extension and axon outgrowth. For example, adenomatous polyposis coli (APC) is enriched in the nervous system where it plays a significant role in establishing neuronal polarity, as well as aiding in migration and axon navigation by stabilizing MTs (Shi et al., 2004; Koester et al., 2007; Eom et al., 2014). Other core regulators of the +TIP network involved in neural development are the EBs. EBs autonomously recognize the growing MT plus end, functioning as a scaffold for other +TIPs to associate and interact with the plus end, thereby regulating MT dynamics (Akhmanova and Steinmetz, 2008). Additionally, several studies suggest differential roles of EB proteins during neurodevelopment, demonstrating that the mechanisms underlying their regulation of the MT cytoskeleton are quite complex. For instance, as mentioned previously, EB3 expression is upregulated during neurodevelopment and is important for regulating MT dynamicity during dendritic spine development (Jaworski et al., 2008). Interestingly, this increase in EB3 expression coincides with a decrease in EB1 expression (Jaworski et al., 2008). However, EB1 is specifically upregulated during axon extension and has been shown to be critical for proper MT organization, as depletion of EB1 in Drosophila leads to abnormalities in MT architecture leading to aberrant axon outgrowth (Alves-Silva et al., 2012).
Cytoplasmic linker proteins (CLIPs) are another group of +TIPs that participate in several neural developmental processes including axon formation and outgrowth, as well as dendrite arborization (van de Willige et al., 2016). CLIPs are enriched in axonal growth cones, where they stabilize MTs invading the growth cone leading edge, and in dendritic spines, where they regulate the crosstalk between actin and dynamic MTs (Neukirchen and Bradke, 2011; Swiech et al., 2011). Additionally, mutations within CLIPs have been associated with ID (Larti et al., 2015), which will be discussed further in subsequent sections. Similar to CLIPs, cytoplasmic linker associated proteins (CLASPs) also regulate MT dynamics during axon outgrowth where they accumulate along MTs at the leading edge of the growth cone, suggesting a role in facilitating axon navigation during development (Lee et al., 2004; Marx et al., 2013). Additional +TIPs found to be important for regulating MT dynamics during neural development include the MT polymerase XMAP215 (Lowery et al., 2013), the XMAP215-interactor, TACC3 (Nwagbara et al., 2014; Bearce et al., 2015; Cammarata et al., 2016; Erdogan et al., 2017), as well as the other TACC family members, TACC1 and TACC2 (Lucaj et al., 2015; Rutherford et al., 2016).
Microtubule Lattice-Binding Proteins
Remodeling of MTs within neurons is essential for the establishment and maintenance of neuronal polarity, axon outgrowth and guidance, synapse formation and migration. There are numerous MAPs that can bind along the MT lattice and regulate MT dynamics in order to properly organize and remodel the MT cytoskeleton during neural development. The most widely studied and best-characterized group of MT lattice-binding proteins are structural MAPs, which are comprised of three distinct groups including MAP1, MAP2 and tau.
There are three members of the MAP1 family found in vertebrates, MAP1A, MAP1B and MAP1S. MAP1B is highly expressed during early neuronal development and is found to be enriched at growing axons, where it regulates axon outgrowth via its interaction with the MT cytoskeleton (Bouquet et al., 2004; Jayachandran et al., 2016). Neuronal subtypes derived from map1b−/− mice revealed several defects including higher collateral axon branching, improper growth cone turning, as well as abnormalities in synaptic vesicle fusion and synaptic plasticity, suggesting that MAP1B is necessary during these processes (Bouquet et al., 2004; Bodaleo et al., 2016). Furthermore, reduction of MAP1B was also shown to decrease MT growth speed in the proximal and distal axon shaft, suggesting that MAP1B may function as a regulator of MT dynamics during axon outgrowth (Tymanskyj et al., 2012). Less is currently known about MAP1A, but it appears to participate in dendritic remodeling and preferentially localizes to postsynaptic densities (PSD) where it interacts with PSD proteins to support synaptic function (Koleske, 2013; Takei et al., 2015). It has also been shown that expression of MAP1A increases MT stability in vitro (Vaillant et al., 1998) and Map1a mutant mouse Purkinje neurons were shown to have abnormal MT networks, demonstrating an important role for MAP1A in MT organization and stabilization (Liu et al., 2015).
Similar to MAP1A, the MAP2 family of proteins are also enriched within dendrites and they have been shown to increase MT stability by rescuing catastrophes (Dehmelt and Halpain, 2005). Furthermore, they are important for regulating MT organization in dendrites by participating in MT bundling, as well as creating the proper spacing between MTs (Teng et al., 2001; Dehmelt and Halpain, 2005). Moreover, it was recently demonstrated that MAP2 can interact with both kinesin-1 and kinesin-3 motors to regulate their distribution of specific cargoes throughout the neuron (Gumy et al., 2017). MAP2-deficient mice show no overt neurological abnormalities, though they do display a reduction in MT density and dendrite length (Harada et al., 2002). However, mice deficient in both MAP2 and MAP1B have severe cortical defects, suggesting there may be a compensatory mechanism between these two proteins (Teng et al., 2001).
Tau is the most widely studied MT lattice binding protein due to its implication in neurodegenerative diseases (Matamoros and Baas, 2016). It is expressed in the brain throughout development into adulthood and appears to have several different functions in relation to MTs. Tau has been shown to increase MT polymerization, prevent MT depolymerization, and regulate MT organization (Wang and Mandelkow, 2016). Additionally, it has been demonstrated that tau dynamically interacts with MTs through a kiss-and-hop mechanism, where it remains associated with a single MT filament for only milliseconds before rapidly dissociating (Janning et al., 2014). Moreover, axonal transport rates are not significantly affected by either the elimination or overexpression of tau, further supporting the kiss-and-hop mechanism, as MT-dependent motor proteins would be impeded by tau if it associated with MTs for longer periods of time (Yuan et al., 2008; Janning et al., 2014). Tau is also expressed within axonal growth cones where it co-localizes with both MTs and actin filaments, and it can influence the organization of dynamic MTs to facilitate axon outgrowth and turning mediated by guidance cues (Li et al., 2014; Biswas and Kalil, 2018). Furthermore, tau seems to be critical for neuronal migration during brain development, as reduction of tau impaired the radial migration of cortical neurons, in addition to causing morphological abnormalities of the leading edge in these cells (Sapir et al., 2012). Interestingly, there may be functional redundancy between tau and MAP1B, as both have been shown to act synergistically to promote axon outgrowth and neuronal migration (Takei et al., 2000).
Microtubule Severing Proteins
The reorganization of MTs depends on the process of MT severing, accomplished by enzymes such as katanin, spastin and fidgetin, which act to cut MTs into small pieces so that they can be transported to other locations within the cell in order to form a new MT array. Katanin consists of a P60 subunit, encoded by KATNA1, which functions to sever MTs, and a P80 subunit, encoded by KATNB1, which regulates the ATPase activity and localizes the protein primarily to centrosomes (McNally et al., 2000). In addition to the centrosome, katanin has been found to be highly expressed in growing axons (Karabay et al., 2004). Proper function of katanin within neurons seems to be dosage-dependent. Expression of a dominant-negative P60-katanin construct inhibited MT severing leading to abnormal axon outgrowth in cultured rat sympathetic neurons (Karabay et al., 2004). Conversely, overexpression of the P60-katanin construct led to increased MT severing, which was also detrimental to axon outgrowth (Karabay et al., 2004). More recently, katanin-mediated severing has been shown to regulate the development of dendrites and synapses at the neuromuscular junction (NMJ). The loss of the katanin P60 subunit in Drosophila resulted in abnormal synapse morphology at the NMJ, including an increased number of small boutons, more stable MT loops and dendritic overgrowth (Mao et al., 2014). Moreover, genetic mutations within several katanin-associated proteins have been linked to microcephaly, suggesting that proper function of katanin is important during early brain development (Bartholdi et al., 2014).
Similar to katanin, spastin is another MT severing protein that is highly enriched in developing axons and is thought to be involved in promoting axon outgrowth (Yu et al., 2008). Reduction of spastin function in zebrafish caused severe defects in motor axon outgrowth, as well as abnormalities in neuronal connectivity (Wood et al., 2006). Additionally, knockdown of spastin in cultured neurons reduced axonal branching, whereas overexpression increased the number of branches and filopodia, indicating that spastin also functions in a dosage-dependent manner (Yu et al., 2008). Furthermore, mutations within spastin cause autosomal dominant hereditary spastic paraplegia, which is a disease characterized by axonal degeneration that leads to a progressive gait disorder (Hazan et al., 1999). Therefore, the regulation of MT severing by spastin is vital for axon outgrowth that is necessary to establish proper neural connectivity.
Less is currently known about the precise functions of fidgetin, however it has been associated with various aspects of vertebrate embryonic development (Cox et al., 2000). It has been demonstrated that fidgetin can depolymerize and sever MTs in vitro, as well as regulate MT dynamics at mitotic centrosomes (Mukherjee et al., 2012). Moreover, knocking down fidgetin in Drosophila disrupts normal axon development by causing an increase in the number of synaptic boutons, as well as an increase in stable MTs (Leo et al., 2015). Interestingly, these results are similar to the phenotypes previously observed with the reduction of katanin, suggesting that fidgetin behaves in a similar fashion to other MT-severing enzymes (Mao et al., 2014). However, axons in cultured vertebrate neurons are significantly longer with more minor processes and contain an increase in labile MT mass when fidgetin is depleted, suggesting that fidgetin regulates axon elongation by severing MT labile domains during neurodevelopment (Leo et al., 2015).
The Major Players of Neuronal Movement: Microtubule-Associated Genes Linked to Disorders of Neuronal Migration
Development of the brain requires extensive neuronal proliferation and migration. This orchestrated movement of neurons to their final destination within the brain relies heavily on the support and coordination of the MT cytoskeleton, along with various MT-associated proteins. Abnormalities in proliferation or migration can lead to downstream defects in neural connectivity and various neurodevelopmental disorders. Human and mouse genetic studies have been instrumental in understanding the dynamic nature and function of the MT cytoskeleton and its associated regulators during neuronal migration. In the following sections, we discuss several well-characterized neuronal migration disorders and the various MT-associated genetic mutations that lead to these disorders during brain development (Table 1).
Table 1
| Disease | Gene | Major pathways/roles | Additional phenotypes | References |
|---|---|---|---|---|
| Intellectual disabilities (ID) | KIF1A | Kinesin; MT-dependent motor | Willemsen et al. (2014) | |
| KIF4A | Kinesin; MT-dependent motor | Kondo et al. (2012), Lee et al. (2015), Ohba et al. (2015) and McVicker et al. (2016) | ||
| CLIP1 | MT binding; +TIP; MT dynamics | Coquelle et al. (2002), Jaworski et al. (2008), Swiech et al. (2011) and Larti et al. (2015) | ||
| KATNAL1 | MT severing | Microcephaly | Bartholdi et al. (2014) and Banks et al. (2018) | |
| MID2 | Ubiquitin ligase; MT binding | Geetha et al. (2014) and Gholkar et al. (2016) | ||
| Autism spectrum disorders (ASD) | AUTS2 | Cytoskeletal remodeling | Kawauchi et al. (2003), Kalscheuer et al. (2007), Oksenberg et al. (2013) and Hori et al. (2014) | |
| ADNP | Chromatin remodeling | ID, developmental delay, motor delay | Mandel et al. (2007), Vulih-Shultzman et al. (2007), Oz et al. (2012), Helsmoortel et al. (2014), Schirer et al. (2014), Gozes et al. (2017a), Gozes et al. (2017b), Ivashko-Pachima et al. (2017), Gozes et al. (2018) and Van Dijck et al. (2018) | |
| JAKMIP1 | MT-associated kinase; MT dynamics; GABA receptor trafficking | Couve et al. (2004), Steindler et al. (2004), Vidal et al. (2007), Kaminsky et al. (2011), Hedges et al. (2012), Poultney et al. (2013) and Berg et al. (2015) | ||
| MARK1 | MT-associated kinase; MT dynamics; mitochondrial trafficking | Drewes et al. (1997), Mandelkow et al. (2004), Maussion et al. (2008) and Sapir et al. (2008) | ||
| Microcephaly | ASPM | Mitotic spindle protein; cell division | ID, speech delay, seizures, short stature | Kouprina et al. (2005), Jiang et al. (2017) and Duerinckx and Abramowicz (2018) |
| MCPH1 | Mitotic spindle protein; DNA damage response; chromosome condensation | ID, seizures, short stature | Trimborn et al. (2004) and Gruber et al. (2011) | |
| STIL | Mitotic spindle checkpoint protein; centriole amplification | ID, seizures, short stature | Kumar et al. (2009), Kitagawa et al. (2011) and Vulprecht et al. (2012) | |
| CDK5RAP2 | Centrosome integrity; spindle pole morphology | ID | Bond et al. (2005), Choi et al. (2010) and Lizarraga et al. (2010) | |
| CENPJ | Centrosome integrity; spindle pole morphology | ID, seizures | Kitagawa et al. (2011) and Garcez et al. (2015) | |
| PRUNE1 | Cell motility; MT dynamics | Zollo et al. (2017) | ||
| KIF20B | Kinesin; MT-dependent motor; cell polarity; cytokinesis | McNeely et al. (2017) | ||
| Polymicrogyria (PMG) | TUBA8 | MT component | Abdollahi et al. (2009), Jaglin et al. (2009) and Poirier et al. (2010) | |
| TUBB2B | MT component | ID, epilepsy | Abdollahi et al. (2009), Jaglin et al. (2009), Jaglin and Chelly (2009), Poirier et al. (2010), Cushion et al. (2013) and Stouffer et al. (2016) | |
| TUBB3 | MT component | Abdollahi et al. (2009), Jaglin et al. (2009), Poirier et al. (2010), Tischfield et al. (2010) and Whitman et al. (2016) | ||
| KIF5C | Kinesin; MT-dependent motor | ID, seizures | Kanai et al. (2000), Homma et al. (2003), Poirier et al. (2013a) and Willemsen et al. (2014) | |
| KIF2A | Kinesin; MT-dependent motor | ID, epilepsy, developmental delay | Kanai et al. (2000), Homma et al. (2003) and Poirier et al. (2013a) | |
| DYNC1H1 | Dynein; MT-dependent motor | Hafezparast et al. (2003), Shu et al. (2004), Ori-McKenney and Vallee (2011), Poirier et al. (2013a) and Zhao et al. (2016) | ||
| Lissencephaly | LIS1 (PAFAH1B1) | MT binding; dynein binding; MT stability; neuronal migration | ID, epilepsy | Reiner et al. (1993), Sapir et al. (1997), Hirotsune et al. (1998), Paylor et al. (1999), Smith et al. (2000), Fleck et al. (2000), Gambello et al. (2003), Shu et al. (2004), Li et al. (2005), Rehberg et al. (2005), Tsai et al. (2007), Bi et al. (2009), Youn et al. (2009), Egan et al. (2012), Moughamian et al. (2013), Vagnoni et al. (2016) and DeSantis et al. (2017) |
| DCX | MT stability; promotes growth cone formation; neuronal migration | ID, epilepsy | Gleeson et al. (1998), des Portes et al. (1998), Pilz et al. (1998), Burgess and Reiner (2000), Corbo et al. (2002), Bai et al. (2003), Moores et al. (2004), Deuel et al. (2006), Tanaka et al. (2006), Bechstedt and Brouhard (2012) and Stouffer et al. (2016) | |
| TUBA1A | MT component | ID, PMG, epilepsy, motor delay | Poirier et al. (2007), Keays et al. (2007), Abdollahi et al. (2009), Jaglin et al. (2009), Jaglin and Chelly (2009), Poirier et al. (2010), Cushion et al. (2013), Fry et al. (2014), Bahi-Buisson et al. (2014) and Stouffer et al. (2016)⊥rule |
Microtubule (MT)-associated genes linked to neurodevelopmental diseases.
Type I Lissencephaly
One of the most widely known neuronal migration disorders is type I lissencephaly (“smooth brain”), also known as classic lissencephaly. This malformation is characterized by a spectrum of cortical abnormalities, which are defined by the absence or reduction of cortical folds (gyri) and grooves (sulci), making parts of the surface of the brain appear smooth. More severe forms of type I lissencephaly result in the complete loss of cortical folds (agyria), while milder forms result in the reduction of cortical folds (pachygyria), or areas of heterotopic bands of gray matter within the cortex (subcortical band heterotopia (SBH); Forman et al., 2005; Barkovich et al., 2012). Agyria and pachygyria arise from post-mitotic neurons failing to reach their proper positions, leading to a disorganized and thickened four-layer cortex instead of the normal six-layered cortex. In SBH, neurons migrate abnormally and form an extra layer of cells underneath the gray matter within the cortex. Type I lissencephaly is typically diagnosed in children within the first few months of life and patients often have many other symptoms including epilepsy, ID, developmental delays and motor function impairments.
Over the last few decades, studies of families with one or multiple individuals affected by type I lissencephaly led to the discovery of several mutations associated with MT-related genes (Kerjan and Gleeson, 2007). The first two genes to be identified were lissencephaly 1 (LIS1; Reiner et al., 1993), located on the short arm of chromosome 17, and doublecortin (DCX; Gleeson et al., 1998; des Portes et al., 1998), located on the X-chromosome. Human mutations of LIS1 (also known as PAFAH1B1, platelet-activating factor acetylhydrolase 1b, regulatory subunit 1) are heterozygous, affecting both males and females, while mutations of DCX are hemizygous, occurring on the X-chromosome, and largely affect only males. Mutations within LIS1 tend to be associated with a more severe phenotype in posterior brain regions, whereas mutations within DCX have the reverse effect with a more severe phenotype in frontal brain regions (Pilz et al., 1998). Both of these genes account for most cases of type I lissencephaly and code for MAPs that are necessary for neuronal differentiation and migration due to their essential interactions with the MT cytoskeleton (Pilz et al., 1998; Stouffer et al., 2016).
In migrating neurons, LIS1 predominately localizes to the centrosome where it aids in nuclear movement and acts as an adaptor protein to stabilize MTs by minimizing catastrophe (Sapir et al., 1997; Shu et al., 2004). LIS1 has also been shown to regulate the MT minus end directed motor, dynein (Smith et al., 2000; Rehberg et al., 2005), by increasing its localization and stabilization at MT plus ends (Li et al., 2005; DeSantis et al., 2017), and assisting in its transport of cargo along the MT (Egan et al., 2012; Moughamian et al., 2013; Vagnoni et al., 2016). DCX has been shown to be highly expressed in the growing axon, where it directly nucleates and stabilizes MTs by supporting their 13 protofilament structure and participates in promoting growth cone formation (Burgess and Reiner, 2000; Tint et al., 2009; Bechstedt and Brouhard, 2012; Jean et al., 2012). The interactions of LIS1 and DCX with MTs are regulated by numerous phosphorylation events (Sapir et al., 1999; Schaar et al., 2004). However, the specific sites of phosphorylation, as well as the kinases and phosphatases which regulate this activity, have still not been fully elucidated in relation to classic lissencephaly. It is possible that genetic mutations at specific phosphorylation sites may alter the interaction between LIS1 and DCX with the MT cytoskeleton, thereby changing their functions and producing variable phenotypic outcomes.
The creation of several mouse models of classic lissencephaly have been instrumental in investigating the cellular and molecular irregularities that occur due to the implicated genetic mutations of this disorder. Homozygous Lis1 knockout (KO) mice die prenatally, indicating that LIS1 is absolutely essential for normal embryonic development (Hirotsune et al., 1998). Lis1-null heterozygous mice display motor coordination and cognition deficits, as well as severe disorganization of the cortical layers, hippocampus, and other areas within the brain (Paylor et al., 1999; Fleck et al., 2000; Youn et al., 2009). Moreover, overexpression of LIS1 caused severe structural brain abnormalities, including smaller brain size, distorted cellular organization and an increase in apoptotic cells, suggesting proper function of LIS1 is required in a dose-dependent manner (Bi et al., 2009). Inhibition of dynein results in a similar phenotype, further supporting the role of LIS1 in dynein function (Shu et al., 2004; Grabham et al., 2007; Tsai et al., 2007). Additional studies using histological analysis and BrdU labeling experiments have also provided strong evidence towards in vivo migrational defects in mice with reduced Lis1 dosage, demonstrating that LIS1 is necessary for proper neuronal migration (Hirotsune et al., 1998; Gambello et al., 2003). Interestingly, mice with mutations in Dcx only display mild disordered neuronal migration phenotypes, mostly occurring in the hippocampus of both heterozygous females and hemizygous males (Corbo et al., 2002). In contrast, RNAi-directed depletion of Dcx showed abnormal radial migration of neurons in rat neocortex (Bai et al., 2003). It is possible that this variability of phenotypic severity may be due to compensation by other members of the DCX superfamily. For example, doublecortin-like kinase-1 (DCLK1) is a closely related gene to DCX, and Dcx/Dckl1 KO mice display a more severe phenotype of cortical development, supporting the idea of genetic redundancy (Deuel et al., 2006; Tanaka et al., 2006). Together, in vivo and in vitro studies have confirmed that these two proteins are critical regulators of proper neuronal migration by working in combination, potentially in similar molecular pathways, to stabilize the MT cytoskeleton throughout neurodevelopment.
More recently, another genetic mutation linked to classic lissencephaly has been identified, occurring in the TUBA1A gene locus (Poirier et al., 2007). TUBA1A is an α-tubulin isotype specifically expressed in the developing nervous system and is required for proper MT structure and function (Keays et al., 2007; Poirier et al., 2007; Fry et al., 2014). Mice with heterozygous mutations of TubA1A were shown to have abnormal neuronal migration and lamination defects similar to the human phenotype (Keays et al., 2007, 2010). Subsequent patient studies revealed several TUBA1A mutations in locations that are predicted to interfere with the interactions between known binding partners, like DCX and other tubulins, suggesting that this may be a possible mechanism to explain how the mutations lead to cortical migration defects (Poirier et al., 2007; Bahi-Buisson et al., 2014), however, the molecular basis of these alterations still remains unclear. In addition to causing symptoms associated with classic lissencephaly, mutations within this gene have also been shown to cause a wide range of other cortical malformations, which will be discussed further in subsequent sections.
Polymicrogyria
Another well-known neuronal migration disorder is Polymicrogyria (PMG), which is a spectrum of disorders characterized by excessive cerebral cortex folding and malformations of cortical layering. It has been described that hypoxia, congenital infections, inflammation of the microvasculature, as well as mitochondrial diseases are among the non-genetic causes that may lead to the cortical abnormalities related to PMG during early embryonic development (Gressens, 2000; Stutterd and Leventer, 2014). Clinical manifestations of PMG are heterogeneous and cause a wide range of developmental disabilities, making a uniform classification of this disorder difficult. Both environmental and genetic causes have been implicated in PMG; however, our current understanding of this cortical malformation still remains incomplete. Defining features of PMG are controversial, as some sources debate whether this disorder is truly due to a neuronal migration defect or a post-migrational defect, with abnormalities occurring after neurons are properly positioned to form the cortical layers (Judkins et al., 2011; Barkovich et al., 2012). These ambiguities make the need for more pathological and molecular studies essential in order to determine the mechanisms that lead to this developmental disorder.
Genetic studies have implicated numerous candidate genes associated with PMG, including transcription factors, signaling molecules and various cytoskeletal components, such as multiple tubulin isotypes and kinesin family members. The α-tubulin genes, TUBA1A and TUBA8, as well as the β-tubulin genes, TUBB2B and TUBB3, have all been identified in connection with PMG (Abdollahi et al., 2009; Jaglin et al., 2009; Poirier et al., 2010, 2013b). Mutations within these neuronally-expressed genes occur in a heterozygous fashion, with the exception of TUBA8 mutations, which are homozygous. Alterations within any of these genes can lead to structural and functional defects of the MT cytoskeleton, as well as interfere with the interactions between the MT cytoskeleton and other MT-related proteins. The abnormalities which arise from mutations of these tubulin isotypes are subtle yet distinct, indicating that they each play a unique role in regulating the MT cytoskeleton. For example, PMG patients harboring a mutation within TUBA8 have been shown to also have optic nerve hypoplasia and callosal dysgenesis (Abdollahi et al., 2009), whereas patients with a mutation in TUBB2B have asymmetrical PMG, dysmorphic basal ganglia, as well as heterotopic neuronal cells in the white matter areas of the cortex (Jaglin et al., 2009; Cushion et al., 2013). Additionally, axonal defects are present in all known TUBB3 mutations, which create abnormalities in axon targeting of the oculomotor muscles, leading to eye movement disorders such as congenital fibrosis of the external ocular muscles (CFEOM; Tischfield et al., 2010; Whitman et al., 2016). PMG patients with either TUBA1A or TUBB2B mutations have been found to display overlapping cortical malformations, possibly due to their similar roles in the formation of tubulin heterodimers, suggesting that MT stability may underlie some of the clinical phenotypes (Jaglin and Chelly, 2009; Cushion et al., 2013; Stouffer et al., 2016).
Within the last several years, studies have identified additional PMG-associated mutations within genes encoding several MT motor proteins including kinesin family members KIF5C and KIF2A, and a dynein-associated protein, DYNC1H1 (Poirier et al., 2013a; Fiorillo et al., 2014). Each mutation results in varying phenotypes, however, all of these genes play pivotal roles in regulating the MT cytoskeleton within neurons. KIF5C and KIF2A encode members of the kinesin superfamily, both of which are highly expressed in the developing nervous system and are involved in the intracellular transport of cargo along MTs (Kanai et al., 2000; Homma et al., 2003). Kif2a KO mice die shortly after birth and display numerous brain abnormalities including aberrant axon outgrowth and collateral branching, as well as delayed neuronal migration (Homma et al., 2003). Additionally, the MT-depolymerizing activity in neuronal growth cones was found to be reduced, indicating that KIF2A mechanistically regulates the growth cone via its interactions with the MT cytoskeleton during axon outgrowth. In contrast, Kif5C KO mice are viable and do not display any gross malformations within the CNS, aside from smaller brain size and a reduction of motor neurons (Kanai et al., 2000). This drastic difference in phenotype could be due to compensation by other closely related genes, suggesting that there may be functional redundancy among the kinesin family members. DYNC1H1 is a large subunit of the cytoplasmic dynein complex and various mouse models indicate that this gene is also vital for cortical development. Inactivation of the mouse homolog causes embryonic lethality, and N-ethyl-N-nitrosourea (ENU)-induced heterozygous missense mutations result in neurodegenerative diseases, as well as abnormal neuronal migration and retrograde axonal transport (Hafezparast et al., 2003; Ori-McKenney and Vallee, 2011; Zhao et al., 2016). Moreover, RNAi-directed reduction of Dync1h1 similarly resulted in impaired neuronal migration (Shu et al., 2004). Taken together, these results highlight the importance of MT-dependent intracellular trafficking during early neural development.
Microcephaly
Primary microcephaly (MCPH) is a neurodevelopmental disorder characterized by a smaller-than-normal head size arising from abnormal prenatal brain growth. This reduced head size occurs due to insufficient proliferation or increased apoptosis of neural stem cells, leading to a reduction in the number of neurons and impaired neurogenesis during development (Barkovich et al., 2012). Often, individuals with this disorder also present with ID, poor motor function, abnormal craniofacial features and seizures. A wide range of factors have been linked to cases of microcephaly such as genetic mutations, chromosomal abnormalities, vertically transmitted infections and other environmental factors (Barkovich et al., 2012).
Several genes have been discovered to be associated with MCPH, many of which code for proteins that localize to the centrosome and play a significant role in regulating MT dynamics. Mutations in abnormal spindle-like microcephaly-associated (ASPM) have been found to be the most common cause of MCPH (Duerinckx and Abramowicz, 2018). ASPM is essential for normal function and organization of mitotic spindle poles specifically within the developing brain (Kouprina et al., 2005). A recent study showed that ASPM can also recruit Katanin to promote MT severing and disassembly, suggesting that misregulation of this process may lead to microcephaly (Jiang et al., 2017). Likewise, microcephalin 1 (MCPH1), a gene which regulates DNA-damage responses, has also been found to be crucial for proper mitotic spindle alignment within neuroprogenitors (Gruber et al., 2011). Reduction of MCPH1 results in an imbalance between mitosis and the centrosome cycle, causing asymmetric cell division and dysregulation of chromosome condensation (Trimborn et al., 2004; Gruber et al., 2011). Another gene, SCL-TAL1 interrupting locus (STIL), encodes a cytoplasmic centriole duplication factor that is required for centriole formation and proper spindle positioning during embryonic brain development (Kumar et al., 2009; Kitagawa et al., 2011). Depletion of STIL results in a loss of centriole amplification, whereas overexpression leads to excess centriole formation (Vulprecht et al., 2012). CDK5 regulatory subunit associated protein 2 (CDK5RAP2) also codes for a protein that localizes to the centrosome and is involved in centrosome function and MT nucleation (Bond et al., 2005; Choi et al., 2010). Mutations of this gene result in cells displaying mitotic delay with abnormal spindle pole number and orientation (Lizarraga et al., 2010). Centromere protein J (CENPJ) is also involved in maintaining centrosome integrity and normal spindle pole morphology. Downregulation or loss of CENPJ leads to centrosome duplication abnormalities which contribute to spindle orientation defects, as well as improper neuronal migration and morphology (Kitagawa et al., 2011; Garcez et al., 2015). Together, these genes code for proteins which all play pivotal roles in regulating centrosome and spindle pole related functions, as well as MT dynamics. Disruptions within any of these genes may lead to defects in the cell cycle, MT nucleation and reduced proliferation of neurons, thus leading to microcephaly. The overlapping functions of these genes in relation to the centrosome are striking and provides strong evidence towards a link between proper centrosome function and MT dynamics during neurogenesis.
More recently, two other genes, PRUNE1 and KIF20B, have been identified in relation to MCPH. Prune exopolyphosphatase 1 (PRUNE1) encodes a member of the DHH (Asp-His-His) phosphoesterase protein superfamily important for cell motility. Genetic studies conducted by Zollo et al. (2017) identified biallelic mutations of PRUNE1 in 13 different individuals with microcephaly and developmental delay. Mutations in PRUNE1 impaired MT polymerization, cell migration and proliferation, suggesting that PRUNE1 may have a fundamental role in regulating these processes throughout cortical development. Kinesin Family Member 20B (KIF20B) is a MT plus end-directed motor that is required for the completion of cytokinesis and regulates cell polarity in neurons (McNeely et al., 2017). Loss of Kif20b disrupts cerebral cortex growth and cell polarization, as well as neurite outgrowth and branching (McNeely et al., 2017). Authors suggest that KIF20B may act to stabilize or bundle MTs in neurites to allow for proper polarization and outgrowth during brain development (McNeely et al., 2017).
Microtubule-Associated Genes Linked to Intellectual Disabilities and Autism Spectrum Disorders
Intellectual Disabilities
ID are complex neurodevelopmental disorders that affect a significant portion of the general population and are an immense health issue. ID is defined by an IQ score under 70 and is characterized by impaired intellectual and adaptive functioning that affects everyday living. ID may occur in isolation or it can be accompanied with other medical or behavioral symptoms such as seizures, craniofacial abnormalities, and microcephaly. ID can be caused by both genetic and environmental factors, with genetic causes representing up to 50% of all ID cases (Kaufman et al., 2010). Single gene mutations, as well as pathogenic copy number variants (CNVs), have been associated with ID, several of the implicated genes are involved in MT function.
Whole exome sequencing and next generation sequencing (NGS) studies have identified various mutations within kinesin superfamily members linked to ID. Willemsen et al. (2014) discovered pathogenic mutations in KIF4A and KIF5C in individuals from two different families. In vivo studies further confirmed the link between these genes and ID. Knockdown of both KIF4A and KIF5C disrupted the balance between excitatory and inhibitory synaptic activity, which may be a contributing factor that leads to ID (Willemsen et al., 2014). Furthermore, de novo mutations in KIF1A were found in several patients that had a range of cognitive and motor defects, including ID (Lee et al., 2015; Ohba et al., 2015). KIF1A, an anterograde motor protein, transports membranous organelles along MTs within axons and dendrites, and has been shown to be involved in synaptogenesis, as well as learning and memory (Kondo et al., 2012; McVicker et al., 2016). Reduction or loss of this protein results in abnormal interactions with MTs and disruptions in axonal and dendritic transport. Together, these results highlight the importance of the kinesin superfamily members during neural development and how alterations of their MT-dependent functions can lead to a spectrum of neurological defects.
A recent NGS study of large consanguineous Iranian families affected by ID identified a novel mutation in CAP-Gly domain containing linker protein 1 (CLIP1), which encodes a +TIP, CLIP-170, that localizes to the ends of growing MTs (Larti et al., 2015). CLIP1 regulates MT behavior and participates in MT-mediated transport in neurons (Jaworski et al., 2008; Swiech et al., 2011). The protein encoded by CLIP1 was absent from cell lines derived from these ID patients, suggesting that loss of CLIP1 function can lead to cognitive impairments. It has also been shown that CLIP-170 may interact with LIS1 to mediate the recruitment of dynein to MTs and regulate MT dynamics (Coquelle et al., 2002). It is possible that the interaction between CLIP1 and LIS1 may be important for proper neuronal migration during brain development.
Microdeletions on chromosome 13 encompassing several genes, including katanin catalytic subunit A1 like 1 (KATNAL1), were found to result in ID and microcephaly (Bartholdi et al., 2014). KATNAL1 encodes a MT-severing enzyme that regulates the remodeling of cellular MT arrays. Mouse lines carrying a loss-of-function allele in Katnal1 display defects in learning and memory, as well as abnormal neuronal migration and morphology (Banks et al., 2018). However, there are genetic discrepancies between humans and mice. Human patients are heterozygous for the KATNAL1 deletion, whereas heterozygous mice show no overt phenotypes, suggesting that further investigations are required to understand the causative mechanisms in regards to this genetic mutation (Bartholdi et al., 2014; Banks et al., 2018). Nevertheless, it is evident that KATNAL1 plays an important role during several neuronal processes, and that perturbations of KATNAL1 function can lead to various defects which may eventually contribute to neurodevelopmental disorders.
X-linked ID have been associated with mutations in midline 2 (MID2), a gene that codes for an ubiquitin ligase, which localizes to MTs and regulates their activity during neural tube closure (Geetha et al., 2014). Mid2 was shown to localize and ubiquitinate Astrin, which is a MT organizing protein that regulates MTs during cell division. Loss of Mid2 led to the stabilization of Astrin, causing defects in MT organization, cytokinesis and cell death (Gholkar et al., 2016). This suggests that ubiquitination of Astrin by Mid2 is essential for regulating MT function during cell division and could explain how mutations of MID2 lead to X-linked ID.
Autism Spectrum Disorders
ASD are a heterogeneous group of disorders characterized by a wide range of symptoms and disabilities that can vary in severity. These symptoms include verbal and non-verbal communication deficits, difficulties with social interactions, repetitive behaviors and restrictive interests. Individuals with ASD can also present with additional medical conditions such as epilepsy, motor function impairments, ID, anxiety and sleep disorders. The pathogenesis of ASD is not yet fully understood and a majority of ASD cases do not have a specific known cause. However, increased research efforts have identified various genetic mutations which have been linked to ASD, including several MT-associated genes (Pinto et al., 2014).
One of these genes is autism susceptibility candidate 2 (AUTS2), however the exact functions of AUTS2 have not yet been entirely characterized, though studies suggest that it may play a significant role during early brain development (Kalscheuer et al., 2007). AUTS2 has been found to be highly expressed in the developing brain of zebrafish, and knockdown of this gene resulted in microcephaly along with a reduction in the total number of neurons (Oksenberg et al., 2013). Further studies found that the protein functioned as a regulator of Rac1, a Rho-family GTPase that is crucial for coordinating cytoskeletal rearrangements (Hori et al., 2014). The reduction or loss of Auts2 in mice caused abnormal morphologies of embryonic neurons and impaired their migration (Hori et al., 2014). Knockdown of Auts2 also suppressed the activation of c-Jun N-terminal kinase (JNK). JNK is regulated by Rac1 and is involved in MT formation, as well as MT dynamics at leading processes of migrating neurons (Kawauchi et al., 2003). Taken together, alterations in AUTS2 may inhibit its regulation of Rac1, which then has downstream effects on subsequent target molecules, like JNK, that can lead to defects in cytoskeletal remodeling in migrating neurons.
Another gene implicated in ASD is activity-dependent neuroprotective protein (ADNP; Helsmoortel et al., 2014). This gene is part of the SWI/SNF chromatin remodeling complex and is vital for brain development (Mandel et al., 2007; Helsmoortel et al., 2014). Adnp−/− mice die prenatally due to failure of neural tube closure, and Adnp+/− mice have increased neuronal death along with abnormal cognitive functioning (Vulih-Shultzman et al., 2007). ADNP was found to be associated with tau mRNA splicing (Schirer et al., 2014), and also participates in the recruitment of Tau to MTs, possibly to prevent free Tau accumulation that eventually leads to neurodegenerative disorders (Oz et al., 2012). Furthermore, ADNP has been shown to directly interact with MT EBs, EB1 and EB3, to promote neurite outgrowth and dendritic spine formation (Oz et al., 2014; Ivashko-Pachima et al., 2017). Together, these findings indicate that mutations of ADNP may alter its interactions with several MT-associated proteins, which have negative downstream effects that alter MT dynamics and hinder different neuronal processes during early development.
Dysregulation of several MT-associated kinases have also been linked to ASD. Janus kinase and MT interacting protein 1 (JAKMIP1) is an RNA binding protein that is highly expressed in glutamatergic neurons (Couve et al., 2004), and has been shown to modify MT polymers and influence MT dynamics (Steindler et al., 2004). Rare deletions of this gene have been found in individuals with ASD (Kaminsky et al., 2011; Hedges et al., 2012; Poultney et al., 2013), and loss of Jakmip1 in mice results in autistic-like behavior, possibly by affecting the expression of downstream mRNA targets during synaptogenesis (Berg et al., 2015). It has also been suggested that JAKMIP1 modulates intracellular trafficking of GABA receptors via its interaction with the MT cytoskeleton (Vidal et al., 2007). Given these results, it is possible that mutations of JAKMIP1 cause defects in synapse formation, specifically within glutamatergic neurons, that may eventually lead to ASD. Another MT-associated kinase, MT affinity regulating kinase 1 (MARK1), was shown to have altered transcript and protein levels in postmortem brains from patients with ASD (Maussion et al., 2008). MARK1 is a kinase which phosphorylates several MAPs, causing them to dissociate from MTs (Drewes et al., 1997). MARK1 also participates in the regulation of mitochondrial trafficking along MTs in both axons and dendrites and plays a significant role during neuronal polarization and migration (Mandelkow et al., 2004; Maussion et al., 2008). The reduction or overexpression of MARK1 has been shown to cause defects in synaptic function as well as cell migration (Maussion et al., 2008; Sapir et al., 2008). It is possible that mutations within MARK1 modulate its phosphorylation activity of MAPs, leading to aberrant alterations of MT dynamics that disrupt proper neural development.
Conclusion
The elaborate MT cytoskeletal network plays many instrumental roles during development of the nervous system. Young neurons rely on the dynamic properties of MTs in order to proliferate and migrate to their final destinations. Moreover, MTs are vital for the formation and extension of both axons and dendrites, which enables the neuron to navigate through the extracellular terrain to create synapses and connections with neighboring cells. Elucidating the molecular mechanisms responsible for regulating the MT cytoskeleton throughout specific stages of neural development still remain an important area of research. In recent years, genetic studies have uncovered numerous mutations within genes that negatively affect the MT cytoskeleton, causing abnormalities in neural migration, proliferation and connectivity. The discovery of mutations occurring within MT-associated genes that lead to neurodevelopmental disorders has provided an opportunity to investigate the functions of MTs at the cellular and molecular level during brain development. Future studies must continue to focus on understanding how these mutations contribute to altered MT function underlying various brain malformations, which will allow insight into the essential roles MTs and their associated proteins play during neural development.
Statements
Author contributions
ML and LAL conceived of the manuscript. ML, JT and LAL wrote and edited the manuscript.
Funding
LAL is funded by National Institutes of Health (NIH; grant no. R01 MH109651) and ML by NIH grant no. R01 GM121907.
Acknowledgments
We thank members of the Lowery lab for helpful discussions and suggestions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
AbdollahiM. R.MorrisonE.SireyT.MolnárZ.HaywardB. E.CarrI. M.et al. (2009). Mutation of the variant α-tubulin TUBA8 results in polymicrogyria with optic nerve hypoplasia. Am. J. Hum. Genet.85, 737–744. 10.1016/j.ajhg.2009.10.007
2
AkhmanovaA.SteinmetzM. O. (2008). Tracking the ends: a dynamic protein network controls the fate of microtubule tips. Nat. Rev. Mol. Cell Biol.9, 309–322. 10.1038/nrm2369
3
Alves-SilvaJ.Sánchez-SorianoN.BeavenR.KleinM.ParkinJ.MillardT. H.et al. (2012). Spectraplakins promote microtubule-mediated axonal growth by functioning as structural microtubule-associated proteins and EB1-dependent +TIPs (tip interacting proteins). J. Neurosci.32, 9143–9158. 10.1523/JNEUROSCI.0416-12.2012
4
Armijo-WeingartL.GalloG. (2017). It takes a village to raise a branch: cellular mechanisms of the initiation of axon collateral branches. Mol. Cell. Neurosci.84, 36–47. 10.1016/j.mcn.2017.03.007
5
Bahi-BuissonN.PoirierK.FourniolF.SaillourY.ValenceS.LebrunN.et al. (2014). The wide spectrum of tubulinopathies: what are the key features for the diagnosis?Brain137, 1676–1700. 10.1093/brain/awu082
6
BaiJ.RamosR. L.AckmanJ. B.ThomasA. M.LeeR. V.LoTurcoJ. J. (2003). RNAi reveals doublecortin is required for radial migration in rat neocortex. Nat. Neurosci.6, 1277–1283. 10.1038/nn1153
7
BanksG.LassiG.Hoerder-SuabedissenA.TinarelliF.SimonM. M.WilcoxA.et al. (2018). A missense mutation in Katnal1 underlies behavioural, neurological and ciliary anomalies. Mol. Psychiatry23, 713–722. 10.1038/mp.2017.54
8
BarkovichA. J.GuerriniR.KuznieckyR. I.JacksonG. D.DobynsW. B. (2012). A developmental and genetic classification for malformations of cortical development: update 2012. Brain135, 1348–1369. 10.1093/brain/aws019
9
BartholdiD.Stray-PedersenA.Azzarello-BurriS.KibaekM.KirchhoffM.OnedaB.et al. (2014). A newly recognized 13q12.3 microdeletion syndrome characterized by intellectual disability, microcephaly and eczema/atopic dermatitis encompassing the HMGB1 and KATNAL1 genes. Am. J. Med. Genet. A164A, 1277–1283. 10.1002/ajmg.a.36439
10
BearceE. A.ErdoganB.LoweryL. A. (2015). TIPsy tour guides: how microtubule plus-end tracking proteins (+TIPs) facilitate axon guidance. Front. Cell. Neurosci.9:241. 10.3389/fncel.2015.00241
11
BechstedtS.BrouhardG. J. (2012). Doublecortin recognizes the 13-protofilament microtubule cooperatively and tracks microtubule ends. Dev. Cell23, 181–192. 10.1016/j.devcel.2012.05.006
12
BergJ. M.LeeC.ChenL.GalvanL.CepedaC.ChenJ. Y.et al. (2015). JAKMIP1, a novel regulator of neuronal translation, modulates synaptic function and autistic-like behaviors in mouse. Neuron88, 1173–1191. 10.1016/j.neuron.2015.10.031
13
BiW.SapirT.ShchelochkovO. A.ZhangF.WithersM. A.HunterJ. V.et al. (2009). Increased LIS1 expression affects human and mouse brain development. Nat. Genet.41, 168–177. 10.1038/ng.302
14
BiswasS.KalilK. (2018). The microtubule-associated protein tau mediates the organization of microtubules and their dynamic exploration of actin-rich lamellipodia and filopodia of cortical growth cones. J. Neurosci.38, 291–307. 10.1523/JNEUROSCI.2281-17.2017
15
BodaleoF. J.Montenegro-VenegasC.HenríquezD. R.CourtF. A.González-BillaultC. (2016). Microtubule-associated protein 1B (MAP1B)-deficient neurons show structural presynaptic deficiencies in vitro and altered presynaptic physiology. Sci. Rep.6:30069. 10.1038/srep32275
16
BondJ.RobertsE.SpringellK.LizarragaS. B.LizarragaS.ScottS.et al. (2005). A centrosomal mechanism involving CDK5RAP2 and CENPJ controls brain size. Nat. Genet.37, 353–355. 10.1038/ng1539
17
BouquetC.SoaresS.von BoxbergY.Ravaille-VeronM.PropstF.NothiasF. (2004). Microtubule-associated protein 1B controls directionality of growth cone migration and axonal branching in regeneration of adult dorsal root ganglia neurons. J. Neurosci.24, 7204–7213. 10.1523/JNEUROSCI.2254-04.2004
18
BurgessH. A.ReinerO. (2000). Doublecortin-like kinase is associated with microtubules in neuronal growth cones. Mol. Cell. Neurosci.16, 529–541. 10.1006/mcne.2000.0891
19
CammarataG. M.BearceE. A.LoweryL. A. (2016). Cytoskeletal social networking in the growth cone: how +TIPs mediate microtubule-actin cross-linking to drive axon outgrowth and guidance. Cytoskeleton73, 461–476. 10.1002/cm.21272
20
ChakrabortiS.NatarajanK.CurielJ.JankeC.LiuJ. (2016). The emerging role of the tubulin code: from the tubulin molecule to neuronal function and disease. Cytoskeleton73, 521–550. 10.1002/cm.21290
21
ChoiY.-K.LiuP.SzeS. K.DaiC.QiR. Z. (2010). CDK5RAP2 stimulates microtubule nucleation by the γ-tubulin ring complex. J. Cell Biol.191, 1089–1095. 10.1083/jcb.201007030
22
CondeC.CáceresA. (2009). Microtubule assembly, organization and dynamics in axons and dendrites. Nat. Rev. Neurosci.10, 319–332. 10.1038/nrn2631
23
CoquelleF. M.CaspiM.CordelièresF. P.DompierreJ. P.DujardinD. L.KoifmanC.et al. (2002). LIS1, CLIP-170’s key to the dynein/dynactin pathway. Mol. Cell. Biol.22, 3089–3102. 10.1128/mcb.22.9.3089-3102.2002
24
CorboJ. C.DeuelT. A.LongJ. M.LaPorteP.TsaiE.Wynshaw-BorisA.et al. (2002). Doublecortin is required in mice for lamination of the hippocampus but not the neocortex. J. Neurosci.22, 7548–7557. 10.1523/JNEUROSCI.22-17-07548.2002
25
CouveA.RestituitoS.BrandonJ. M.CharlesK. J.BawaganH.FreemanK. B.et al. (2004). Marlin-1, a novel RNA-binding protein associates with GABA receptors. J. Biol. Chem.279, 13934–13943. 10.1074/jbc.M311737200
26
CoxG. A.MahaffeyC. L.NystuenA.LettsV. A.FrankelW. N. (2000). The mouse fidgetin gene defines a new role for AAA family proteins in mammalian development. Nat. Genet.26, 198–202. 10.1038/79923
27
CushionT. D.DobynsW. B.MullinsJ. G. L.StoodleyN.ChungS.-K.FryA. E.et al. (2013). Overlapping cortical malformations and mutations in TUBB2B and TUBA1A. Brain136, 536–548. 10.1093/brain/aws338
28
DehmeltL.HalpainS. (2005). The MAP2/Tau family of microtubule-associated proteins. Genome Biol.6:204. 10.1186/gb-2004-6-1-204
29
DehmeltL.NalbantP.SteffenW.HalpainS. (2006). A microtubule-based, dynein-dependent force induces local cell protrusions: implications for neurite initiation. Brain Cell Biol.35, 39–56. 10.1007/s11068-006-9001-0
30
DentE. W. (2017). Of microtubules and memory: implications for microtubule dynamics in dendrites and spines. Mol. Biol. Cell28, 1–8. 10.1091/mbc.E15-11-0769
31
DentE. W.CallawayJ. L.SzebenyiG.BaasP. W.KalilK. (1999). Reorganization and movement of microtubules in axonal growth cones and developing interstitial branches. J. Neurosci.19, 8894–8908. 10.1523/JNEUROSCI.19-20-08894.1999
32
DentE. W.GuptonS. L.GertlerF. B. (2011). The growth cone cytoskeleton in axon outgrowth and guidance. Cold Spring Harb. Perspect. Biol.3:a001800. 10.1101/cshperspect.a001800
33
DentE. W.KalilK. (2001). Axon branching requires interactions between dynamic microtubules and actin filaments. J. Neurosci.21, 9757–9769. 10.1523/JNEUROSCI.21-24-09757.2001
34
DentE. W.KwiatkowskiA. V.MebaneL. M.PhilipparU.BarzikM.RubinsonD. A.et al. (2007). Filopodia are required for cortical neurite initiation. Nat. Cell Biol.9, 1347–1359. 10.1038/ncb1654
35
DeSantisM. E.CianfroccoM. A.HtetZ. M.TranP. T.Reck-PetersonS. L.LeschzinerA. E. (2017). Lis1 has two opposing modes of regulating cytoplasmic dynein. Cell170, 1197.e12–1208.e12. 10.1016/j.cell.2017.08.037
36
des PortesV.FrancisF.PinardJ. M.DesguerreI.MoutardM. L.SnoeckI.et al. (1998). Doublecortin is the major gene causing X-linked subcortical laminar heterotopia (SCLH). Hum. Mol. Genet.7, 1063–1070. 10.1093/hmg/7.7.1063
37
DeuelT. A. S.LiuJ. S.CorboJ. C.YooS.-Y.Rorke-AdamsL. B.WalshC. A. (2006). Genetic interactions between doublecortin and doublecortin-like kinase in neuronal migration and axon outgrowth. Neuron49, 41–53. 10.1016/j.neuron.2005.10.038
38
DrewesG.EbnethA.PreussU.MandelkowE. M.MandelkowE. (1997). MARK, a novel family of protein kinases that phosphorylate microtubule-associated proteins and trigger microtubule disruption. Cell89, 297–308. 10.1016/s0092-8674(00)80208-1
39
DuerinckxS.AbramowiczM. (2018). The genetics of congenitally small brains. Semin. Cell Dev. Biol.76, 76–85. 10.1016/j.semcdb.2017.09.015
40
EganM. J.TanK.Reck-PetersonS. L. (2012). Lis1 is an initiation factor for dynein-driven organelle transport. J. Cell Biol.197, 971–982. 10.1083/jcb.201112101
41
EomT.-Y.StancoA.GuoJ.WilkinsG.DeslauriersD.YanJ.et al. (2014). Differential regulation of microtubule severing by APC underlies distinct patterns of projection neuron and interneuron migration. Dev. Cell31, 677–689. 10.1016/j.devcel.2014.11.022
42
ErdoganB.CammarataG. M.LeeE. J.PrattB. C.FranclA. F.RutherfordE. L.et al. (2017). The microtubule plus-end-tracking protein TACC3 promotes persistent axon outgrowth and mediates responses to axon guidance signals during development. Neural Dev.12:3. 10.1186/s13064-017-0080-7
43
FiorilloC.MoroF.YiJ.WeilS.BriscaG.AstreaG.et al. (2014). Novel dynein DYNC1H1 neck and motor domain mutations link distal spinal muscular atrophy and abnormal cortical development. Hum. Mutat.35, 298–302. 10.1002/humu.22491
44
FleckM. W.HirotsuneS.GambelloM. J.Phillips-TanseyE.SuaresG.MervisR. F.et al. (2000). Hippocampal abnormalities and enhanced excitability in a murine model of human lissencephaly. J. Neurosci.20, 2439–2450. 10.1523/JNEUROSCI.20-07-02439.2000
45
FormanM. S.SquierW.DobynsW. B.GoldenJ. A. (2005). Genotypically defined lissencephalies show distinct pathologies. J. Neuropathol. Exp. Neurol.64, 847–857. 10.1097/01.jnen.0000182978.56612.41
46
FryA. E.CushionT. D.PilzD. T. (2014). The genetics of lissencephaly. Am. J. Med. Genet. C Semin Med. Genet.166C, 198–210. 10.1002/ajmg.c.31402
47
GötzM.HuttnerW. B. (2005). The cell biology of neurogenesis. Nat. Rev. Mol. Cell Biol.6, 777–788. 10.1038/nrm1739
48
GalloG. (2016). Coordination of the axonal cytoskeleton during the emergence of axon collateral branches. Neural Regen. Res.11, 709–711. 10.4103/1673-5374.182684
49
GalloG.LetourneauP. C. (1999). Different contributions of microtubule dynamics and transport to the growth of axons and collateral sprouts. J. Neurosci.19, 3860–3873. 10.1523/JNEUROSCI.19-10-03860.1999
50
GambelloM. J.DarlingD. L.YinglingJ.TanakaT.GleesonJ. G.Wynshaw-BorisA. (2003). Multiple dose-dependent effects of Lis1 on cerebral cortical development. J. Neurosci.23, 1719–1729. 10.1523/JNEUROSCI.23-05-01719.2003
51
GarcezP. P.Diaz-AlonsoJ.Crespo-EnriquezI.CastroD.BellD.GuillemotF. (2015). Cenpj/CPAP regulates progenitor divisions and neuronal migration in the cerebral cortex downstream of Ascl1. Nat. Commun.6:6474. 10.1038/ncomms7474
52
GeethaT. S.MichealrajK. A.KabraM.KaurG.JuyalR. C.ThelmaB. K. (2014). Targeted deep resequencing identifies MID2 mutation for X-linked intellectual disability with varied disease severity in a large kindred from India. Hum. Mutat.35, 41–44. 10.1002/humu.22453
53
GholkarA. A.SeneseS.LoY.-C.VidesE.ContrerasE.HodaraE.et al. (2016). The X-linked-intellectual-disability-associated ubiquitin ligase Mid2 interacts with astrin and regulates astrin levels to promote cell division. Cell Rep.14, 180–188. 10.1016/j.celrep.2015.12.035
54
GleesonJ. G.AllenK. M.FoxJ. W.LampertiE. D.BerkovicS.SchefferI.et al. (1998). Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell92, 63–72. 10.1016/s0092-8674(00)80899-5
55
GozesI.HelsmoortelC.VandeweyerG.Van der AaN.KooyF.Bedrosian-SermoneS. (2018). The compassionate side of neuroscience: tony sermone’s undiagnosed genetic journey—ADNP mutation. J. Mol. Neurosci.56, 751–757. 10.1007/s12031-018-1028-z
56
GozesI.PattersonM. C.Van DijckA.KooyR. F.PeedenJ. N.EichenbergerJ. A.et al. (2017a). The eight and a half year journey of undiagnosed AD: gene sequencing and funding of advanced genetic testing has led to hope and new beginnings. Front. Endocrinol.8:107. 10.3389/fendo.2017.00107
57
GozesI.Van DijckA.Hacohen-KleimanG.GriggI.KarmonG.GiladiE.et al. (2017b). Premature primary tooth eruption in cognitive/motor-delayed ADNP-mutated children. Transl. Psychiatry7:e1043. 10.1038/tp.2017.128
58
GrabhamP. W.SealeG. E.BennecibM.GoldbergD. J.ValleeR. B. (2007). Cytoplasmic dynein and LIS1 are required for microtubule advance during growth cone remodeling and fast axonal outgrowth. J. Neurosci.27, 5823–5834. 10.1523/JNEUROSCI.1135-07.2007
59
GressensP. (2000). Mechanisms and disturbances of neuronal migration. Pediatr. Res.48, 725–730. 10.1203/00006450-200012000-00004
60
GruberR.ZhouZ.SukchevM.JoerssT.FrappartP.-O.WangZ.-Q. (2011). MCPH1 regulates the neuroprogenitor division mode by coupling the centrosomal cycle with mitotic entry through the Chk1-Cdc25 pathway. Nat. Cell Biol.13, 1325–1334. 10.1038/ncb2342
61
GuJ.FiresteinB. L.ZhengJ. Q. (2008). Microtubules in dendritic spine development. J. Neurosci.28, 12120–12124. 10.1523/JNEUROSCI.2509-08.2008
62
GumyL. F.KatrukhaE. A.GrigorievI.JaarsmaD.KapiteinL. C.AkhmanovaA.et al. (2017). MAP2 defines a pre-axonal filtering zone to regulate KIF1- versus KIF5-dependent cargo transport in sensory neurons. Neuron94, 347.e7–362.e7. 10.1016/j.neuron.2017.03.046
63
HafezparastM.KlockeR.RuhrbergC.MarquardtA.Ahmad-AnnuarA.BowenS.et al. (2003). Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science300, 808–812. 10.1126/science.1083129
64
HaradaA.TengJ.TakeiY.OguchiK.HirokawaN. (2002). MAP2 is required for dendrite elongation, PKA anchoring in dendrites, and proper PKA signal transduction. J. Cell Biol.158, 541–549. 10.1083/jcb.200110134
65
HazanJ.FonknechtenN.MavelD.PaternotteC.SamsonD.ArtiguenaveF.et al. (1999). Spastin, a new AAA protein, is altered in the most frequent form of autosomal dominant spastic paraplegia. Nat. Genet.23, 296–303. 10.1038/15472
66
HedgesD. J.Hamilton-NelsonK. L.SacharowS. J.NationsL.BeechamG. W.KozhekbaevaZ. M.et al. (2012). Evidence of novel fine-scale structural variation at autism spectrum disorder candidate loci. Mol. Autism3:2. 10.1186/2040-2392-3-2
67
HelsmoortelC.Vulto-van SilfhoutA. T.CoeB. P.VandeweyerG.RoomsL.van den EndeJ.et al. (2014). A SWI/SNF-related autism syndrome caused by de novo mutations in ADNP. Nat. Genet.46, 380–384. 10.1038/ng.2899
68
HirokawaN.NiwaS.TanakaY. (2010). Molecular motors in neurons: transport mechanisms and roles in brain function, development and disease. Neuron68, 610–638. 10.1016/j.neuron.2010.09.039
69
HirotsuneS.FleckM. W.GambelloM. J.BixG. J.ChenA.ClarkG. D.et al. (1998). Graded reduction of Pafah1b1 (Lis1) activity results in neuronal migration defects and early embryonic lethality. Nat. Genet.19, 333–339. 10.1038/1221
70
HommaN.TakeiY.TanakaY.NakataT.TeradaS.KikkawaM.et al. (2003). Kinesin superfamily protein 2A (KIF2A) functions in suppression of collateral branch extension. Cell114, 229–239. 10.1016/s0092-8674(03)00522-1
71
HoogenraadC. C.BradkeF. (2009). Control of neuronal polarity and plasticity—a renaissance for microtubules?Trends Cell Biol.19, 669–676. 10.1016/j.tcb.2009.08.006
72
HoriK.NagaiT.ShanW.SakamotoA.TayaS.HashimotoR.et al. (2014). Cytoskeletal regulation by AUTS2 in neuronal migration and neuritogenesis. Cell Rep.9, 2166–2179. 10.1016/j.celrep.2014.11.045
73
Ivashko-PachimaY.SayasC. L.MalishkevichA.GozesI. (2017). ADNP/NAP dramatically increase microtubule end-binding protein-Tau interaction: a novel avenue for protection against tauopathy. Mol. Psychiatry22, 1335–1344. 10.1038/mp.2016.255
74
JacobsonC.SchnappB.BankerG. A. (2006). A change in the selective translocation of the Kinesin-1 motor domain marks the initial specification of the axon. Neuron49, 797–804. 10.1016/j.neuron.2006.02.005
75
JaglinX. H.ChellyJ. (2009). Tubulin-related cortical dysgeneses: microtubule dysfunction underlying neuronal migration defects. Trends Genet.25, 555–566. 10.1016/j.tig.2009.10.003
76
JaglinX. H.PoirierK.SaillourY.BuhlerE.TianG.Bahi-BuissonN.et al. (2009). Mutations in the β-tubulin gene TUBB2B result in asymmetrical polymicrogyria. Nat. Genet.41, 746–752. 10.1038/ng.380
77
JanningD.IgaevM.SündermannF.BrühmannJ.BeutelO.HeinischJ. J.et al. (2014). Single-molecule tracking of tau reveals fast kiss-and-hop interaction with microtubules in living neurons. Mol. Biol. Cell25, 3541–3551. 10.1091/mbc.E14-06-1099
78
JaworskiJ.HoogenraadC. C.AkhmanovaA. (2008). Microtubule plus-end tracking proteins in differentiated mammalian cells. Int. J. Biochem. Cell Biol.40, 619–637. 10.1016/j.biocel.2007.10.015
79
JaworskiJ.KapiteinL. C.GouveiaS. M.DortlandB. R.WulfP. S.GrigorievI.et al. (2009). Dynamic microtubules regulate dendritic spine morphology and synaptic plasticity. Neuron61, 85–100. 10.1016/j.neuron.2008.11.013
80
JayachandranP.OlmoV. N.SanchezS. P.McFarlandR. J.VitalE.WernerJ. M.et al. (2016). Microtubule-associated protein 1b is required for shaping the neural tube. Neural Dev.11:1. 10.1186/s13064-015-0056-4
81
JeanD. C.BaasP. W.BlackM. M. (2012). A novel role for doublecortin and doublecortin-like kinase in regulating growth cone microtubules. Hum. Mol. Genet.21, 5511–5527. 10.1093/hmg/dds395
82
JiangK.RezabkovaL.HuaS.LiuQ.CapitaniG.AltelaarA. F. M.et al. (2017). Microtubule minus-end regulation at spindle poles by an ASPM-katanin complex. Nat. Cell Biol.19, 480–492. 10.1038/ncb3511
83
JudkinsA. R.MartinezD.FerreiraP.DobynsW. B.GoldenJ. A. (2011). Polymicrogyria includes fusion of the molecular layer and decreased neuronal populations but normal cortical laminar organization. J. Neuropathol. Exp. Neurol.70, 438–443. 10.1097/NEN.0b013e31821ccf1c
84
KahnO. I.BaasP. W. (2016). Microtubules and growth cones: motors drive the turn. Trends Neurosci.39, 433–440. 10.1016/j.tins.2016.04.009
85
KalilK.DentE. W. (2014). Branch management: mechanisms of axon branching in the developing vertebrate CNS. Nat. Rev. Neurosci.15, 7–18. 10.1038/nrn3650
86
KalscheuerV. M.FitzPatrickD.TommerupN.BuggeM.NiebuhrE.NeumannL. M.et al. (2007). Mutations in autism susceptibility candidate 2 (AUTS2) in patients with mental retardation. Hum. Genet.121, 501–509. 10.1007/s00439-006-0284-0
87
KaminskyE. B.KaulV.PaschallJ.ChurchD. M.BunkeB.KunigD.et al. (2011). An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Genet. Med.13, 777–784. 10.1097/GIM.0b013e31822c79f9
88
KanaiY.OkadaY.TanakaY.HaradaA.TeradaS.HirokawaN. (2000). KIF5C, a novel neuronal kinesin enriched in motor neurons. J. Neurosci.20, 6374–6384. 10.1523/JNEUROSCI.20-17-06374.2000
89
KapiteinL. C.HoogenraadC. C. (2011). Which way to go? Cytoskeletal organization and polarized transport in neurons. Mol. Cell. Neurosci.46, 9–20. 10.1016/j.mcn.2010.08.015
90
KapiteinL. C.HoogenraadC. C. (2015). Building the neuronal microtubule cytoskeleton. Neuron87, 492–506. 10.1016/j.neuron.2015.05.046
91
KapiteinL. C.SchlagerM. A.KuijpersM.WulfP. S.van SpronsenM.MacKintoshF. C.et al. (2010). Mixed microtubules steer dynein-driven cargo transport into dendrites. Curr. Biol.20, 290–299. 10.1016/j.cub.2009.12.052
92
KarabayA.YuW.SolowskaJ. M.BairdD. H.BaasP. W. (2004). Axonal growth is sensitive to the levels of katanin, a protein that severs microtubules. J. Neurosci.24, 5778–5788. 10.1523/JNEUROSCI.1382-04.2004
93
KaufmanL.AyubM.VincentJ. B. (2010). The genetic basis of non-syndromic intellectual disability: a review. J. Neurodev. Disord.2, 182–209. 10.1007/s11689-010-9055-2
94
KawauchiT.ChihamaK.NabeshimaY.-I.HoshinoM. (2003). The in vivo roles of STEF/Tiam1, Rac1 and JNK in cortical neuronal migration. EMBO J.22, 4190–4201. 10.1093/emboj/cdg413
95
KeaysD. A.CleakJ.HuangG.-J.EdwardsA.BraunA.TreiberC. D.et al. (2010). The role of Tuba1a in adult hippocampal neurogenesis and the formation of the dentate gyrus. Dev. Neurosci.32, 268–277. 10.1159/000319663
96
KeaysD. A.TianG.PoirierK.HuangG.-J.SieboldC.CleakJ.et al. (2007). Mutations in α-tubulin cause abnormal neuronal migration in mice and lissencephaly in humans. Cell128, 45–57. 10.1016/j.cell.2006.12.017
97
KerjanG.GleesonJ. G. (2007). Genetic mechanisms underlying abnormal neuronal migration in classical lissencephaly. Trends Genet.23, 623–630. 10.1016/j.tig.2007.09.003
98
KetschekA.SpillaneM.DunX.-P.HardyH.ChiltonJ.GalloG. (2016). Drebrin coordinates the actin and microtubule cytoskeleton during the initiation of axon collateral branches. Dev. Neurobiol.76, 1092–1110. 10.1002/dneu.22377
99
KirkcaldieM. T. K.DwyerS. T. (2017). The third wave: intermediate filaments in the maturing nervous system. Mol. Cell. Neurosci.84, 68–76. 10.1016/j.mcn.2017.05.010
100
KitagawaD.KohlmaierG.KellerD.StrnadP.BalestraF. R.FlückigerI.et al. (2011). Spindle positioning in human cells relies on proper centriole formation and on the microcephaly proteins CPAP and STIL. J. Cell. Sci.124, 3884–3893. 10.1242/jcs.089888
101
KoesterM. P.MüllerO.PollerbergG. E. (2007). Adenomatous polyposis coli is differentially distributed in growth cones and modulates their steering. J. Neurosci.27, 12590–12600. 10.1523/JNEUROSCI.2250-07.2007
102
KoleskeA. J. (2013). Molecular mechanisms of dendrite stability. Nat. Rev. Neurosci.14, 536–550. 10.1038/nrn3486
103
KondoM.TakeiY.HirokawaN. (2012). Motor protein KIF1A is essential for hippocampal synaptogenesis and learning enhancement in an enriched environment. Neuron73, 743–757. 10.1016/j.neuron.2011.12.020
104
KouprinaN.PavlicekA.CollinsN. K.NakanoM.NoskovV. N.OhzekiJ.-I.et al. (2005). The microcephaly ASPM gene is expressed in proliferating tissues and encodes for a mitotic spindle protein. Hum. Mol. Genet.14, 2155–2165. 10.1093/hmg/ddi220
105
KuijpersM.HoogenraadC. C. (2011). Centrosomes, microtubules and neuronal development. Mol. Cell. Neurosci.48, 349–358. 10.1016/j.mcn.2011.05.004
106
KumarA.GirimajiS. C.DuvvariM. R.BlantonS. H. (2009). Mutations in STIL, encoding a pericentriolar and centrosomal protein, cause primary microcephaly. Am. J. Hum. Genet.84, 286–290. 10.1016/j.ajhg.2009.01.017
107
LartiF.KahriziK.MusanteL.HuH.PapariE.FattahiZ.et al. (2015). A defect in the CLIP1 gene (CLIP-170) can cause autosomal recessive intellectual disability. Eur. J. Hum. Genet.23, 331–336. 10.1038/ejhg.2014.13
108
LeeH.EngelU.RuschJ.ScherrerS.SheardK.Van VactorD. (2004). The microtubule plus end tracking protein Orbit/MAST/CLASP acts downstream of the tyrosine kinase Abl in mediating axon guidance. Neuron42, 913–926. 10.1016/j.neuron.2004.05.020
109
LeeJ.-R.SrourM.KimD.HamdanF. F.LimS.-H.Brunel-GuittonC.et al. (2015). De novo mutations in the motor domain of KIF1A cause cognitive impairment, spastic paraparesis, axonal neuropathy, and cerebellar atrophy. Hum. Mutat.36, 69–78. 10.1002/humu.22709
110
LeoL.YuW.D’RozarioM.WaddellE. A.MarendaD. R.BairdM. A.et al. (2015). Vertebrate fidgetin restrains axonal growth by severing labile domains of microtubules. Cell Rep.12, 1723–1730. 10.1016/j.celrep.2015.08.017
111
LiL.FothergillT.HutchinsB. I.DentE. W.KalilK. (2014). Wnt5a evokes cortical axon outgrowth and repulsive guidance by tau mediated reorganization of dynamic microtubules. Dev. Neurobiol.74, 797–817. 10.1002/dneu.22102
112
LiJ.LeeW.-L.CooperJ. A. (2005). NudEL targets dynein to microtubule ends through LIS1. Nat. Cell Biol.7, 686–690. 10.1038/ncb1273
113
LinS.LiuM.MozgovaO. I.YuW.BaasP. W. (2012). Mitotic motors coregulate microtubule patterns in axons and dendrites. J. Neurosci.32, 14033–14049. 10.1523/JNEUROSCI.3070-12.2012
114
LiuY.LeeJ. W.AckermanS. L. (2015). Mutations in the microtubule-associated protein 1A (Map1a) gene cause Purkinje cell degeneration. J. Neurosci.35, 4587–4598. 10.1523/JNEUROSCI.2757-14.2015
115
LizarragaS. B.MargossianS. P.HarrisM. H.CampagnaD. R.HanA.-P.BlevinsS.et al. (2010). Cdk5rap2 regulates centrosome function and chromosome segregation in neuronal progenitors. Development137, 1907–1917. 10.1242/dev.040410
116
LoweryL. A.StoutA.FarisA. E.DingL.BairdM. A.DavidsonM. W.et al. (2013). Growth cone-specific functions of XMAP215 in restricting microtubule dynamics and promoting axonal outgrowth. Neural Dev.8:22. 10.1186/1749-8104-8-22
117
LoweryL. A.Van VactorD. (2009). The trip of the tip: understanding the growth cone machinery. Nat. Rev. Mol. Cell Biol.10, 332–343. 10.1038/nrm2679
118
LuW.FoxP.LakonishokM.DavidsonM. W.GelfandV. I. (2013). Initial neurite outgrowth in Drosophila neurons is driven by kinesin-powered microtubule sliding. Curr. Biol.23, 1018–1023. 10.1016/j.cub.2013.04.050
119
LucajC. M.EvansM. F.NwagbaraB. U.EbbertP. T.BakerC. C.VolkJ. G.et al. (2015). Xenopus TACC1 is a microtubule plus-end tracking protein that can regulate microtubule dynamics during embryonic development. Cytoskeleton72, 225–234. 10.1002/cm.21224
120
MadayS.TwelvetreesA. E.MoughamianA. J.HolzbaurE. L. F. (2014). Axonal transport: cargo-specific mechanisms of motility and regulation. Neuron84, 292–309. 10.1016/j.neuron.2014.10.019
121
MandelS.RechaviG.GozesI. (2007). Activity-dependent neuroprotective protein (ADNP) differentially interacts with chromatin to regulate genes essential for embryogenesis. Dev. Biol.303, 814–824. 10.1016/j.ydbio.2006.11.039
122
MandelkowE.-M.ThiesE.TrinczekB.BiernatJ.MandelkowE. (2004). MARK/PAR1 kinase is a regulator of microtubule-dependent transport in axons. J. Cell Biol.167, 99–110. 10.1083/jcb.200401085
123
MaoC.-X.XiongY.XiongZ.WangQ.ZhangY. Q.JinS. (2014). Microtubule-severing protein Katanin regulates neuromuscular junction development and dendritic elaboration in Drosophila. Development141, 1064–1074. 10.1242/dev.097774
124
MarxA.GodinezW. J.TsimashchukV.BankheadP.RohrK.EngelU. (2013). Xenopus cytoplasmic linker-associated protein 1 (XCLASP1) promotes axon elongation and advance of pioneer microtubules. Mol. Biol. Cell24, 1544–1558. 10.1091/mbc.E12-08-0573
125
MatamorosA. J.BaasP. W. (2016). Microtubules in health and degenerative disease of the nervous system. Brain Res. Bull.126, 217–225. 10.1016/j.brainresbull.2016.06.016
126
MattilaP. K.LappalainenP. (2008). Filopodia: molecular architecture and cellular functions. Nat. Rev. Mol. Cell Biol.9, 446–454. 10.1038/nrm2406
127
MaussionG.CarayolJ.Lepagnol-BestelA.-M.ToresF.Loe-MieY.MilbretaU.et al. (2008). Convergent evidence identifying MAP/microtubule affinity-regulating kinase 1 (MARK1) as a susceptibility gene for autism. Hum. Mol. Genet.17, 2541–2551. 10.1093/hmg/ddn154
128
McNallyK. P.BazirganO. A.McNallyF. J. (2000). Two domains of p80 katanin regulate microtubule severing and spindle pole targeting by p60 katanin. J. Cell. Sci.113, 1623–1633. Available online at: https://www.ncbi.nlm.nih.gov/pubmed/?term=Two+domains+of+p80+katanin+regulate+microtubule+severing+and+spindle+pole+targeting+by+p60+katanin
129
McNeelyK. C.CuppT. D.LittleJ. N.JanischK. M.ShresthaA.DwyerN. D. (2017). Mutation of Kinesin-6 Kif20b causes defects in cortical neuron polarization and morphogenesis. Neural Dev.12:5. 10.1186/s13064-017-0082-5
130
McVickerD. P.AweA. M.RichtersK. E.WilsonR. L.CowdreyD. A.HuX.et al. (2016). Transport of a kinesin-cargo pair along microtubules into dendritic spines undergoing synaptic plasticity. Nat. Commun.7:12741. 10.1038/ncomms12741
131
MenonS.GuptonS. L. (2016). Building blocks of functioning brain: cytoskeletal dynamics in neuronal development. Int. Rev. Cell Mol. Biol.322, 183–245. 10.1016/bs.ircmb.2015.10.002
132
MerriamE. B.LumbardD. C.ViesselmannC.BallwegJ.StevensonM.PietilaL.et al. (2011). Dynamic microtubules promote synaptic NMDA receptor-dependent spine enlargement. PLoS One6:e27688. 10.1371/journal.pone.0027688
133
MerriamE. B.MilletteM.LumbardD. C.SaengsawangW.FothergillT.HuX.et al. (2013). Synaptic regulation of microtubule dynamics in dendritic spines by calcium, F-actin, and drebrin. J. Neurosci.33, 16471–16482. 10.1523/JNEUROSCI.0661-13.2013
134
MitchisonT.KirschnerM. (1984). Dynamic instability of microtubule growth. Nature312, 237–242. 10.1038/312237a0
135
MoughamianA. J.OsbornG. E.LazarusJ. E.MadayS.HolzbaurE. L. F. (2013). Ordered recruitment of dynactin to the microtubule plus-end is required for efficient initiation of retrograde axonal transport. J. Neurosci.33, 13190–13203. 10.1523/JNEUROSCI.0935-13.2013
136
MooresC. A.PerdeerisetM.FrancisF.ChellyJ.HoudusseA.MilliganR. A.et al. (2004). Mechanism of microtubule stabilization by doublecortin. Mol. Cell14, 833–839. 10.1016/j.molcel.2004.06.009
137
MukherjeeS.Diaz ValenciaJ. D.StewmanS.MetzJ.MonnierS.RathU.et al. (2012). Human Fidgetin is a microtubule severing the enzyme and minus-end depolymerase that regulates mitosis. Cell Cycle11, 2359–2366. 10.4161/cc.20849
138
NakataT.HirokawaN. (2003). Microtubules provide directional cues for polarized axonal transport through interaction with kinesin motor head. J. Cell Biol.162, 1045–1055. 10.1083/jcb.200302175
139
NakataT.NiwaS.OkadaY.PerezF.HirokawaN. (2011). Preferential binding of a kinesin-1 motor to GTP-tubulin-rich microtubules underlies polarized vesicle transport. J. Cell Biol.194, 245–255. 10.1083/jcb.201104034
140
NeukirchenD.BradkeF. (2011). Cytoplasmic linker proteins regulate neuronal polarization through microtubule and growth cone dynamics. J. Neurosci.31, 1528–1538. 10.1523/JNEUROSCI.3983-10.2011
141
NwagbaraB. U.FarisA. E.BearceE. A.ErdoganB.EbbertP. T.EvansM. F.et al. (2014). TACC3 is a microtubule plus end-tracking protein that promotes axon elongation and also regulates microtubule plus end dynamics in multiple embryonic cell types. Mol. Biol. Cell25, 3350–3362. 10.1091/mbc.E14-06-1121
142
OhbaC.HaginoyaK.OsakaH.KubotaK.IshiyamaA.HiraideT.et al. (2015). De novo KIF1A mutations cause intellectual deficit, cerebellar atrophy, lower limb spasticity and visual disturbance. J. Hum. Genet.60, 739–742. 10.1038/jhg.2015.108
143
OksenbergN.StevisonL.WallJ. D.AhituvN. (2013). Function and regulation of AUTS2, a gene implicated in autism and human evolution. PLoS Genet.9:e1003221. 10.1371/journal.pgen.1003221
144
Ori-McKenneyK. M.JanL. Y.JanY. N. (2012). Golgi outposts shape dendrite morphology by functioning as sites of acentrosomal microtubule nucleation in neurons. Neuron76, 921–930. 10.1016/j.neuron.2012.10.008
145
Ori-McKenneyK. M.ValleeR. B. (2011). Neuronal migration defects in the Loa dynein mutant mouse. Neural Dev.6:26. 10.1186/1749-8104-6-26
146
OzS.Ivashko-PachimaY.GozesI. (2012). The ADNP derived peptide, NAP modulates the tubulin pool: implication for neurotrophic and neuroprotective activities. PLoS One7:e51458. 10.1371/journal.pone.0051458
147
OzS.KapitanskyO.Ivashco-PachimaY.MalishkevichA.GiladiE.SkalkaN.et al. (2014). The NAP motif of activity-dependent neuroprotective protein (ADNP) regulates dendritic spines through microtubule end binding proteins. Mol. Psychiatry19, 1115–1124. 10.1038/mp.2014.97
148
PachecoA.GalloG. (2016). Actin filament-microtubule interactions in axon initiation and branching. Brain Res. Bull.126, 300–310. 10.1016/j.brainresbull.2016.07.013
149
PaylorR.HirotsuneS.GambelloM. J.Yuva-PaylorL.CrawleyJ. N.Wynshaw-BorisA. (1999). Impaired learning and motor behavior in heterozygous Pafah1b1 (Lis1) mutant mice. Learn. Mem.6, 521–537. 10.1101/lm.6.5.521
150
PilzD. T.MatsumotoN.MinnerathS.MillsP.GleesonJ. G.AllenK. M.et al. (1998). LIS1 and XLIS (DCX) mutations cause most classical lissencephaly, but different patterns of malformation. Hum. Mol. Genet.7, 2029–2037. 10.1093/hmg/7.13.2029
151
PintoD.DelabyE.MericoD.BarbosaM.MerikangasA.KleiL.et al. (2014). Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet.94, 677–694. 10.1016/j.ajhg.2014.03.018
152
PoirierK.KeaysD. A.FrancisF.SaillourY.BahiN.ManouvrierS.et al. (2007). Large spectrum of lissencephaly and pachygyria phenotypes resulting from de novo missense mutations in tubulin α 1A (TUBA1A). Hum. Mutat.28, 1055–1064. 10.1002/humu.20572
153
PoirierK.LebrunN.BroixL.TianG.SaillourY.BoscheronC.et al. (2013a). Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat. Genet.45, 639–647. 10.1038/ng.2613
154
PoirierK.SaillourY.FourniolF.FrancisF.SouvilleI.ValenceS.et al. (2013b). Expanding the spectrum of TUBA1A-related cortical dysgenesis to Polymicrogyria. Eur. J. Hum. Genet.21, 381–385. 10.1038/ejhg.2012.195
155
PoirierK.SaillourY.Bahi-BuissonN.JaglinX. H.Fallet-BiancoC.NabboutR.et al. (2010). Mutations in the neuronal ß-tubulin subunit TUBB3 result in malformation of cortical development and neuronal migration defects. Hum. Mol. Genet.19, 4462–4473. 10.1093/hmg/ddq377
156
PoultneyC. S.GoldbergA. P.DrapeauE.KouY.Harony-NicolasH.KajiwaraY.et al. (2013). Identification of small exonic CNV from whole-exome sequence data and application to autism spectrum disorder. Am. J. Hum. Genet.93, 607–619. 10.1016/j.ajhg.2013.09.001
157
RaoA. N.PatilA.BlackM. M.CraigE. M.MyersK. A.YeungH. T.et al. (2017). Cytoplasmic dynein transports axonal microtubules in a polarity-sorting manner. Cell Rep.19, 2210–2219. 10.1016/j.celrep.2017.05.064
158
RehbergM.Kleylein-SohnJ.FaixJ.HoT.-H.SchulzI.GräfR. (2005). Dictyostelium LIS1 is a centrosomal protein required for microtubule/cell cortex interactions, nucleus/centrosome linkage, and actin dynamics. Mol. Biol. Cell16, 2759–2771. 10.1091/mbc.E05-01-0069
159
ReinerO.CarrozzoR.ShenY.WehnertM.FaustinellaF.DobynsW. B.et al. (1993). Isolation of a Miller-Dieker lissencephaly gene containing G protein β-subunit-like repeats. Nature364, 717–721. 10.1038/364717a0
160
RutherfordE. L.CarandangL.EbbertP. T.MillsA. N.BowersJ. T.LoweryL. A. (2016). Xenopus TACC2 is a microtubule plus end-tracking protein that can promote microtubule polymerization during embryonic development. Mol. Biol. Cell27, 3013–3020. 10.1091/mbc.E16-03-0198
161
SainathR.GalloG. (2015). Cytoskeletal and signaling mechanisms of neurite formation. Cell Tissue Res.359, 267–278. 10.1007/s00441-014-1955-0
162
SapirT.CahanaA.SegerR.NekhaiS.ReinerO. (1999). LIS1 is a microtubule-associated phosphoprotein. Eur. J. Biochem.265, 181–188. 10.1046/j.1432-1327.1999.00711.x
163
SapirT.ElbaumM.ReinerO. (1997). Reduction of microtubule catastrophe events by LIS1, platelet-activating factor acetylhydrolase subunit. EMBO J.16, 6977–6984. 10.1093/emboj/16.23.6977
164
SapirT.FrotscherM.LevyT.MandelkowE.-M.ReinerO. (2012). Tau’s role in the developing brain: implications for intellectual disability. Hum. Mol. Genet.21, 1681–1692. 10.1093/hmg/ddr603
165
SapirT.SapoznikS.LevyT.FinkelshteinD.ShmueliA.TimmT.et al. (2008). Accurate balance of the polarity kinase MARK2/Par-1 is required for proper cortical neuronal migration. J. Neurosci.28, 5710–5720. 10.1523/JNEUROSCI.0911-08.2008
166
SchaarB. T.KinoshitaK.McConnellS. K. (2004). Doublecortin microtubule affinity is regulated by a balance of kinase and phosphatase activity at the leading edge of migrating neurons. Neuron41, 203–213. 10.1016/s0896-6273(03)00843-2
167
SchirerY.MalishkevichA.OphirY.LewisJ.GiladiE.GozesI. (2014). Novel marker for the onset of frontotemporal dementia: early increase in activity-dependent neuroprotective protein (ADNP) in the face of Tau mutation. PLoS One9:e87383. 10.1371/journal.pone.0087383
168
SharpD. J.YuW.BaasP. W. (1995). Transport of dendritic microtubules establishes their nonuniform polarity orientation. J. Cell Biol.130, 93–103. 10.1083/jcb.130.1.93
169
SharpD. J.YuW.FerhatL.KuriyamaR.RuegerD. C.BaasP. W. (1997). Identification of a microtubule-associated motor protein essential for dendritic differentiation. J. Cell Biol.138, 833–843. 10.1083/jcb.138.4.833
170
ShiS.-H.ChengT.JanL. Y.JanY. N. (2004). APC and GSK-3β are involved in mPar3 targeting to the nascent axon and establishment of neuronal polarity. Curr. Biol.14, 2025–2032. 10.1016/j.cub.2004.11.009
171
ShuT.AyalaR.NguyenM.-D.XieZ.GleesonJ. G.TsaiL.-H. (2004). Ndel1 operates in a common pathway with LIS1 and cytoplasmic dynein to regulate cortical neuronal positioning. Neuron44, 263–277. 10.1016/j.neuron.2004.09.030
172
SmithD. S.NiethammerM.AyalaR.ZhouY.GambelloM. J.Wynshaw-BorisA.et al. (2000). Regulation of cytoplasmic dynein behaviour and microtubule organization by mammalian Lis1. Nat. Cell Biol.2, 767–775. 10.1038/35041000
173
SrivastavaA. K.SchwartzC. E. (2014). Intellectual disability and autism spectrum disorders: causal genes and molecular mechanisms. Neurosci. Biobehav. Rev.46, 161–174. 10.1016/j.neubiorev.2014.02.015
174
SteindlerC.LiZ.AlgartéM.AlcoverA.LibriV.RagimbeauJ.et al. (2004). Jamip1 (marlin-1) defines a family of proteins interacting with janus kinases and microtubules. J. Biol. Chem.279, 43168–43177. 10.1074/jbc.M401915200
175
StiessM.BradkeF. (2011). Neuronal polarization: the cytoskeleton leads the way. Dev. Neurobiol.71, 430–444. 10.1002/dneu.20849
176
StiessM.MaghelliN.KapiteinL. C.Gomis-RüthS.Wilsch-BräuningerM.HoogenraadC. C.et al. (2010). Axon extension occurs independently of centrosomal microtubule nucleation. Science327, 704–707. 10.1126/science.1182179
177
StoufferM. A.GoldenJ. A.FrancisF. (2016). Neuronal migration disorders: focus on the cytoskeleton and epilepsy. Neurobiol. Dis.92, 18–45. 10.1016/j.nbd.2015.08.003
178
StutterdC. A.LeventerR. J. (2014). Polymicrogyria: a common and heterogeneous malformation of cortical development. Am. J. Med. Genet. C Semin Med. Genet.166C, 227–239. 10.1002/ajmg.c.31399
179
SwiechL.BlazejczykM.UrbanskaM.PietruszkaP.DortlandB. R.MalikA. R.et al. (2011). CLIP-170 and IQGAP1 cooperatively regulate dendrite morphology. J. Neurosci.31, 4555–4568. 10.1523/JNEUROSCI.6582-10.2011
180
TakeiY.KikkawaY. S.AtapourN.HenschT. K.HirokawaN. (2015). Defects in synaptic plasticity, reduced NMDA-receptor transport and instability of postsynaptic density proteins in mice lacking microtubule-associated protein 1A. J. Neurosci.35, 15539–15554. 10.1523/JNEUROSCI.2671-15.2015
181
TakeiY.TengJ.HaradaA.HirokawaN. (2000). Defects in axonal elongation and neuronal migration in mice with disrupted tau and map1b genes. J. Cell Biol.150, 989–1000. 10.1083/jcb.150.5.989
182
TanakaE. M.KirschnerM. W. (1991). Microtubule behavior in the growth cones of living neurons during axon elongation. J. Cell Biol.115, 345–363. 10.1083/jcb.115.2.345
183
TanakaT.KoizumiH.GleesonJ. G. (2006). The doublecortin and doublecortin-like kinase 1 genes cooperate in murine hippocampal development. Cereb. Cortex16, i69–i73. 10.1093/cercor/bhk005
184
TasR. P.ChazeauA.CloinB. M. C.LambersM. L. A.HoogenraadC. C.KapiteinL. C. (2017). Differentiation between oppositely oriented microtubules controls polarized neuronal transport. Neuron96, 1264.e5–1271.e5. 10.1016/j.neuron.2017.11.018
185
TengJ.TakeiY.HaradaA.NakataT.ChenJ.HirokawaN. (2001). Synergistic effects of MAP2 and MAP1B knockout in neuronal migration, dendritic outgrowth, and microtubule organization. J. Cell Biol.155, 65–76. 10.1083/jcb.200106025
186
TintI.JeanD.BaasP. W.BlackM. M. (2009). Doublecortin associates with microtubules preferentially in regions of the axon displaying actin-rich protrusive structures. J. Neurosci.29, 10995–11010. 10.1523/JNEUROSCI.3399-09.2009
187
TischfieldM. A.BarisH. N.WuC.RudolphG.Van MaldergemL.HeW.et al. (2010). Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell140, 74–87. 10.1016/j.cell.2009.12.011
188
TrimbornM.BellS. M.FelixC.RashidY.JafriH.GriffithsP. D.et al. (2004). Mutations in microcephalin cause aberrant regulation of chromosome condensation. Am. J. Hum. Genet.75, 261–266. 10.1086/422855
189
TsaiJ.-W.BremnerK. H.ValleeR. B. (2007). Dual subcellular roles for LIS1 and dynein in radial neuronal migration in live brain tissue. Nat. Neurosci.10, 970–979. 10.1038/nn1934
190
TymanskyjS. R.ScalesT. M. E.Gordon-WeeksP. R. (2012). MAP1B enhances microtubule assembly rates and axon extension rates in developing neurons. Mol. Cell. Neurosci.49, 110–119. 10.1016/j.mcn.2011.10.003
191
VagnoniA.HoffmannP. C.BullockS. L. (2016). Reducing Lissencephaly-1 levels augments mitochondrial transport and has a protective effect in adult Drosophila neurons. J. Cell. Sci.129, 178–190. 10.1242/jcs.179184
192
VaillantA. R.MüllerR.LangkopfA.BrownD. L. (1998). Characterization of the microtubule-binding domain of microtubule-associated protein 1A and its effects on microtubule dynamics. J. Biol. Chem.273, 13973–13981. 10.1074/jbc.273.22.13973
193
van de WilligeD.HoogenraadC. C.AkhmanovaA. (2016). Microtubule plus-end tracking proteins in neuronal development. Cell. Mol. Life Sci.73, 2053–2077. 10.1007/s00018-016-2168-3
194
Van DijckA.Vulto-van SilfhoutA. T.CappuynsE.van der WerfI. M.ManciniG. M.TzschachA.et al. (2018). Clinical presentation of a complex neurodevelopmental disorder caused by mutations in ADNP. Biol. Psychiatry [Epub ahead of print]. 10.1016/j.biopsych.2018.02.1173
195
VidalR. L.RamírezO. A.SandovalL.Koenig-RobertR.HärtelS.CouveA. (2007). Marlin-1 and conventional kinesin link GABAB receptors to the cytoskeleton and regulate receptor transport. Mol. Cell. Neurosci.35, 501–512. 10.1016/j.mcn.2007.04.008
196
Vulih-ShultzmanI.PinhasovA.MandelS.GrigoriadisN.TouloumiO.PittelZ.et al. (2007). Activity-dependent neuroprotective protein snippet NAP reduces tau hyperphosphorylation and enhances learning in a novel transgenic mouse model. J. Pharmacol. Exp. Ther.323, 438–449. 10.1124/jpet.107.129551
197
VulprechtJ.DavidA.TibeliusA.CastielA.KonotopG.LiuF.et al. (2012). STIL is required for centriole duplication in human cells. J. Cell. Sci.125, 1353–1362. 10.1242/jcs.104109
198
WangY.MandelkowE. (2016). Tau in physiology and pathology. Nat. Rev. Neurosci.17, 22–35. 10.1038/nrn.2015.1
199
WhitmanM. C.AndrewsC.ChanW.-M.TischfieldM. A.StasheffS. F.BrancatiF.et al. (2016). Two unique TUBB3 mutations cause both CFEOM3 and malformations of cortical development. Am. J. Med. Genet. A170A, 297–305. 10.1002/ajmg.a.37362
200
WillemsenM. H.BaW.Wissink-LindhoutW. M.de BrouwerA. P. M.HaasS. A.BienekM.et al. (2014). Involvement of the kinesin family members KIF4A and KIF5C in intellectual disability and synaptic function. J. Med. Genet.51, 487–494. 10.1136/jmedgenet-2013-102182
201
WindingM.KelliherM. T.LuW.WildongerJ.GelfandV. I. (2016). Role of kinesin-1-based microtubule sliding in Drosophila nervous system development. Proc. Natl. Acad. Sci. U S A113, E4985–E4994. 10.1126/science.353.6302.883-a
202
WitteH.NeukirchenD.BradkeF. (2008). Microtubule stabilization specifies initial neuronal polarization. J. Cell Biol.180, 619–632. 10.1083/jcb.200707042
203
WoodJ. D.LandersJ. A.BingleyM.McDermottC. J.Thomas-McArthurV.GleadallL. J.et al. (2006). The microtubule-severing protein Spastin is essential for axon outgrowth in the zebrafish embryo. Hum. Mol. Genet.15, 2763–2771. 10.1093/hmg/ddl212
204
YauK. W.SchätzleP.TortosaE.PagèsS.HoltmaatA.KapiteinL. C.et al. (2016). Dendrites in vitro and in vivo contain microtubules of opposite polarity and axon formation correlates with uniform plus-end-out microtubule orientation. J. Neurosci.36, 1071–1085. 10.1523/JNEUROSCI.2430-15.2016
205
YauK. W.van BeuningenS. F. B.Cunha-FerreiraI.CloinB. M. C.van BattumE. Y.WillL.et al. (2014). Microtubule minus-end binding protein CAMSAP2 controls axon specification and dendrite development. Neuron82, 1058–1073. 10.1016/j.neuron.2014.04.019
206
YounY. H.PramparoT.HirotsuneS.Wynshaw-BorisA. (2009). Distinct dose-dependent cortical neuronal migration and neurite extension defects in Lis1 and Ndel1 mutant mice. J. Neurosci.29, 15520–15530. 10.1523/JNEUROSCI.4630-09.2009
207
YuW.AhmadF. J.BaasP. W. (1994). Microtubule fragmentation and partitioning in the axon during collateral branch formation. J. Neurosci.14, 5872–5884. 10.1523/JNEUROSCI.14-10-05872.1994
208
YuW.CookC.SauterC.KuriyamaR.KaplanP. L.BaasP. W. (2000). Depletion of a microtubule-associated motor protein induces the loss of dendritic identity. J. Neurosci.20, 5782–5791. 10.1523/JNEUROSCI.20-15-05782.2000
209
YuW.QiangL.SolowskaJ. M.KarabayA.KoruluS.BaasP. W. (2008). The microtubule-severing proteins spastin and katanin participate differently in the formation of axonal branches. Mol. Biol. Cell19, 1485–1498. 10.1091/mbc.E07-09-0878
210
YuanA.KumarA.PeterhoffC.DuffK.NixonR. A. (2008). Axonal transport rates in vivo are unaffected by tau deletion or overexpression in mice. Mol. Hum. Reprod.28, 1682–1687. 10.1523/JNEUROSCI.5242-07.2008
211
ZhaoB.MekaD. P.ScharrenbergR.KönigT.SchwankeB.KoblerO.et al. (2017). Microtubules modulate F-actin dynamics during neuronal polarization. Sci. Rep.7:9583. 10.1038/s41598-017-09832-8
212
ZhaoJ.WangY.XuH.FuY.QianT.BoD.et al. (2016). Dync1h1 mutation causes proprioceptive sensory neuron loss and impaired retrograde axonal transport of dorsal root ganglion neurons. CNS Neurosci. Ther.22, 593–601. 10.1111/cns.12552
213
ZhengY.WildongerJ.YeB.ZhangY.KitaA.YoungerS. H.et al. (2008). Dynein is required for polarized dendritic transport and uniform microtubule orientation in axons. Nat. Cell Biol.10, 1172–1180. 10.1038/ncb1777
214
ZolloM.AhmedM.FerrucciV.SalpietroV.AsadzadehF.CarotenutoM.et al. (2017). PRUNE is crucial for normal brain development and mutated in microcephaly with neurodevelopmental impairment. Brain140, 940–952. 10.1093/brain/awx014
Summary
Keywords
cytoskeleton, microtubule dynamics, MAPs, neuronal migration, neurodevelopmental disorders
Citation
Lasser M, Tiber J and Lowery LA (2018) The Role of the Microtubule Cytoskeleton in Neurodevelopmental Disorders. Front. Cell. Neurosci. 12:165. doi: 10.3389/fncel.2018.00165
Received
23 February 2018
Accepted
28 May 2018
Published
14 June 2018
Volume
12 - 2018
Edited by
C. Laura Sayas, Universidad de La Laguna, Spain
Reviewed by
Marina Mikhaylova, Universitätsklinikum Hamburg-Eppendorf, Germany; Roland Brandt, University of Osnabrück, Germany; Illana Gozes, Tel Aviv University, Israel
Updates
Copyright
© 2018 Lasser, Tiber and Lowery.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura Anne Lowery laura.lowery@bc.edu
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.