Abstract
Migraine is one of the most prevalent and disabling neurovascular disorders worldwide. However, despite the increase in awareness and research, the understanding of migraine pathophysiology and treatment options remain limited. For centuries, migraine was considered to be a vascular disorder. In fact, the throbbing, pulsating quality of the headache is thought to be caused by mechanical changes in vessels. Moreover, the most successful migraine treatments act on the vasculature and induction of migraine can be accomplished with vasoactive agents. However, over the past 20 years, the emphasis has shifted to the neural imbalances associated with migraine, and vascular changes have generally been viewed as an epiphenomenon that is neither sufficient nor necessary to induce migraine. With the clinical success of peripherally-acting antibodies that target calcitonin gene-related peptide (CGRP) and its receptor for preventing migraine, this neurocentric view warrants a critical re-evaluation. This review will highlight the likely importance of the vasculature in migraine.
Introduction
Migraine is a highly prevalent and complex, neurovascular disorder that has been recognized since ancient times. It afflicts approximately 1 in 10 people worldwide and causes a significant socioeconomic burden on society (Smitherman et al., 2013; Burch et al., 2015; Woldeamanuel and Cowan, 2017). Most migraines are diagnosed as episodic, however, approximately 5% of migraineurs experience chronic migraine (>15 headache days monthly; Couch, 2011). As a result, migraine has become a $13 billion dollar industry in direct healthcare costs (Hawkins et al., 2008). The presentation of migraine varies between each individual patient, but a common diagnostic feature is a severe headache with a pulsating quality. The pulsating or throbbing pain underlies migraine being thought of as a vascular disease. The most common treatments, the triptans, are effective in only ~60% of patients and little advancement in effective therapy has occurred since their development (Loder, 2010). However, the promise of monoclonal antibodies for prevention of migraine has reignited the theory of a peripheral input, which most likely involves a vascular contribution. Here, we revisit the vascular basis of migraine.
Theories of migraine pathophysiology have been debated for quite some time. In particular, whether migraine genesis and/or maintenance involves a primary role for the vasculature remains controversial (Goadsby, 2009; Messlinger, 2009; Shevel, 2011). The vascular theory was first articulated by Galen in the second century and later re-proposed by Thomas Willis in the late 17th century (Isler, 1992). However, it wasn’t until the early 1940’s that Harold Wolff first showed that intensity of migraine was closely linked to the pulsating branches of the external carotid arteries and decreasing the amplitude of the pulsations decreased the intensity of the headache (Tfelt-Hansen and Koehler, 2008). In the same study, ergots were used to induce vasoconstriction of temporal and middle meningeal arteries. The vasoconstriction of these vessels reduced throbbing while coincidently diminishing the intensity of the headache. These observations were the genesis of what would later be known as the vascular theory of migraine. However, Ahn reported a study that opposed the observations of Wolff (Ahn, 2010). In a cohort of 20 migraine patients he did not find a temporal relationship between throbbing migraine pain and arterial pulsation.
It seems possible that both intracranial and extracranial arteries play a nociceptive role in migraine (Asghar et al., 2010, 2011; Shevel, 2011). Intracranial vessel dilation has been implicated in migraine, specifically meningeal arteries. In one study, during experimental induction of migraine, dilation of both middle meningeal and middle cerebral arteries was observed (Asghar et al., 2011). During spontaneous migraine, intracranial arteries have been reported to be more dilated during attacks (Amin et al., 2013). For extracranial arteries, the superficial temporal arteries have been reported to be wider on the pain side of the head, and compression of the artery relieved pain in ~30% of patients (Blau and Dexter, 1981; Drummond and Lance, 1983). Additionally, Elkind et al. (1964) concluded that during unilateral headache, blood flow was increased only on the painful side and treatment with ergotamine reduced blood flow and headache in many of the patients. However, it is not clear if ergotamine affected both extracranial and/or intracranial vessels in this study. Other studies used ultrasonography to measure blood flow velocity, a marker for increased artery lumen, during migraine attacks (Thomsen et al., 1995). More recently, studies using magnetic resonance angiography to observe the circumference of extracranial arteries during attacks reached an opposing conclusion that dilation of extracranial arteries was not associated with migraine pain, although there was slight intracranial dilation of the vessels (Amin et al., 2013). Interestingly, both intracranial and extracranial arteries can potentially be innervated by collaterals of the same trigeminal nerve that transverse the skull (Kosaras et al., 2009). Further investigation of both intracranial and extracranial vessels in migraine are necessary.
Induction of Migraine by Vasoactive Compounds
Using the vascular hypothesis as inspiration, several studies have shown that migraine attacks were associated with release of vasoactive peptides. The vasodilatory peptides calcitonin gene-related peptide (CGRP) and pituitary adenylate cyclase-activating polypeptide (PACAP-38), as well as the neurotransmitter nitric oxide (NO), are all potent vasodilators implicated in migraine pathophysiology (Brain et al., 1985; Moncada et al., 1991; Messlinger et al., 2012; Kaiser and Russo, 2013; Russo, 2015, 2017; Jansen-Olesen and Hougaard Pedersen, 2018).
CGRP is a multifunctional neuropeptide found on sensory nerve fibers (Russell et al., 2014). These fibers innervate vessels where CGRP receptor activation can cause both smooth muscle-dependent and endothelium-dependent activation (Wang et al., 1991; Raddino et al., 1997; Brain and Grant, 2004). Infusion of CGRP in migraine patients results in a delayed migraine-like headache, occurring around 1–5 h after treatment. However, this study also reported a decrease in blood pressure following infusion of CGRP that returned to levels similar to baseline within 60 min of administration (Lassen et al., 2002). To understand the mechanisms of migraine, several studies have induced migraine-like behavior via administration of CGRP (Marquez de Prado et al., 2009; Recober et al., 2009, 2010; Kaiser et al., 2012; Mason et al., 2017; Yao et al., 2017). We have previously reported that overexpression of CGRP receptors in the nervous system enhanced photophobia following central, but not peripheral, administration of CGRP (Mason et al., 2017). These studies suggest that peripheral CGRP had targets outside of nervous tissue that may be relevant in migraine, which is consistent with a possible vascular site of action. However, not all data fit the hypothesis that vasodilation can cause migraine pain. Levy et al. (2005) reported that CGRP-evoked vasodilation failed to induce a nociceptive effect in meningeal nociceptors. Since data focused on vasodilation-induced nociception are conflicting, future studies that dissect whether vasodilation can sensitize dural nociceptors in vivo are warranted.
Similar to CGRP, PACAP-38 and NO also can induce migraine. PACAP-38 is present in sensory neurons and on the vascular smooth muscle cells of vessels (Uddman et al., 1993; Mulder et al., 1994; Fahrenkrug and Hannibal, 1998; Tajti et al., 1999; Vaudry et al., 2009). Reports also show that intravenous administration of PACAP-38 can elicit a migraine-like attack in patients (Schytz et al., 2009) and cause sustained meningeal vasodilation and migraine-like photophobia in mice (Markovics et al., 2012). Additionally, both CGRP and PACAP cause vasodilation of the middle meningeal artery (Asghar et al., 2010; Amin et al., 2013; Kaiser and Russo, 2013; Jansen-Olesen and Hougaard Pedersen, 2018). Nitroglycerin (glycerol trinitrate, GTN) is another vasoactive compound implicated in migraine. GTN is enzymatically reduced to NO and/or NO precursors that cause substantial vasodilation (Hill et al., 1992; Millar et al., 1998; Chen et al., 2005; Bonini et al., 2008). GTN administration also causes an immediate headache and a delayed migraine-like headache in migraineurs and a less severe headache in control subjects (Iversen et al., 1989; Thomsen et al., 1994). For these studies, it was reported that during the actual headache phase vasodilation no longer occurs. This is consistent with studies in 1981 by Olesen reporting that the pain was manifested only after vasodilation had subsided (Olesen et al., 1981), although the prolonged initial vasodilatory phase could be important in the pain process. Finally, an important consideration is that vascular agents related to migraine also have neural and/or immune activity (Coleman, 2001; Delgado et al., 2003; Cury et al., 2011; Kaiser and Russo, 2013).
Perhaps the most convincing argument that vasodilation may be an epiphenomenon and not a causative factor in migraine came from a study showing that vasoactive intestinal peptide (VIP) induces vasodilation, but only produced a mild headache, not a migraine (Rahmann et al., 2008). However, the conclusion that vasodilation is an epiphenomenon based on the inability of VIP to induce migraine, may be a premature conclusion. CGRP, PACAP-38 and GTN all cause sustained vasodilation following infusion (Brain et al., 1985; Iversen et al., 1989; Bhatt et al., 2014). In contrast, VIP-induced vasodilation is transitory compared to PACAP-38 (Amin et al., 2014; Edvinsson et al., 2018). Additionally, It was recently reported that PACAP, but not VIP, can dilate the middle meningeal artery (Jansen-Olesen and Hougaard Pedersen, 2018). Hence, there are differences in kinetics and possibly vascular targets of VIP compared to PACAP. Moreover, CGRP, PACAP and GTN cause pro-inflammatory molecule release(Reuter et al., 2001; Raddant and Russo, 2011; Jansen-Olesen et al., 2014; Jansen-Olesen and Hougaard Pedersen, 2018), while VIP is well known for its immuno-protective role and its anti-inflammatory effect (Delgado et al., 2002). Along this line, VIP may also inhibit mast cell degranulation whereas PACAP induces dural mast cell degranulation (Tunçel et al., 2000; Baun et al., 2012). Therefore, it is possible that: (1) increasing VIP infusion time could unveil an ability of VIP to induce migraine-like headaches or (2) the anti-inflammatory properties of VIP play a role in its inability to induce migraine. Future studies with prolonged VIP infusion in migraineurs and examination of cephalic mast cells after infusion of VIP in rodents are warranted.
Although most studies focus on vasodilatory agents implicated in migraine, the role of vasoconstrictors must not be ruled out. There have been observations that plasma levels of endothelin-1 (ET-1), a potent vasoconstrictor, were increased during early stages of a migraine attack, but rapidly decreased at the onset of the headache (Kallela et al., 1998). ET-1 is an important regulator of cerebral blood flow and its receptors are found in endothelium and vascular smooth muscle cells of the arterial system and throughout the CNS (Arai et al., 1990; Sakurai et al., 1990). Kallela et al. (1998) observed that even though ET-1 was elevated during early phases of the migraine attack, the cubital vein blood pressure measurements were unchanged. ET-1 levels rapidly decline approximately 3–4 h after the initiation of the attack, which coincides with reports of headache onset (Kallela et al., 1998). However, the authors did not report the actual blood pressure values and the observation time points are unclear. It is important to note that ET-1 has the ability to either induce vasoconstriction or an initial vasodilation followed by vasoconstriction depending on whether it is activating endothelin type A receptor (ETA) or endothelin type B (ETB). ETA activation causes sustained vasoconstriction via smooth muscle and can inhibit NO synthesis (Arai et al., 1990; Ikeda et al., 1997) however, ETB activation initially increases release of NO and prostacyclin, which are known vasodilators, followed by sustained vasoconstriction via endothelial cells (de Nucci et al., 1988; Hoffman et al., 1989; Winquist et al., 1989; Sakurai et al., 1990). Furthermore, the non-specific endothelin receptor (A/B) antagonist bosentan inhibits neurogenic inflammation but not vasoconstriction and is not effective for the treatment of migraine (May et al., 1996). However, the ability of bosentan to act on the initial vasodilation that occurs with ETB activation was not assessed. This, along with other studies showing inhibition of neurogenic inflammation is not enough to abort migraine, supports a possible vascular role in migraine. Future studies dissecting the temporal relationship among the release of vasoactive agents such as CGRP, NO and ET-1 in migraine patients are necessary.
Non-vasodilatory Role of the Vasculature
The vascular aspects of migraine have largely focused on changes in vascular tone, however a non-vasodilatory role of the vasculature has recently been suggested (Jacobs and Dussor, 2016). Vascular inflammation is a mechanism that may contribute to migraine pathogenesis. Dural vessels are thought to contribute to neurogenic inflammation, an event that activates sensory neurons and is characterized by vasodilation, plasma extravasation, and release of pro-inflammatory molecules from mast cells (Raddant and Russo, 2011). These non-vasomotor roles may involve all three layers of vessels: the inner endothelium layer, the middle smooth muscle layer and the outermost adventitia layer of fibroblasts and connective tissue.
The endothelium can both send and respond to signals via release of vasoactive substances to maintain vessel homeostasis (Tomiyama and Yamashina, 2010; Jacobs and Dussor, 2016). For example, when perturbed, cells of the vasculature can release ATP, consequently activate purinergic receptors, stimulate release of NO and pro-inflammatory mediators from endothelial cells (Burnstock, 2016; Jacobs and Dussor, 2016). Endothelium-induced NO release is then capable of sensitizing nearby afferents and possibly contributing to pain experienced during migraine. Two studies reported migraineurs have a decreased count of circulating endothelial progenitor cells (Hill et al., 2003; Rodríguez-Osorio et al., 2012). These cells are a marker of endothelium integrity and function, and a reduction suggest endothelial cell dysfunction (Hill et al., 2003). Additionally, there is mounting evidence of circulating endothelial microparticles in female migraine patients, particularly those diagnosed with migraine with aura (Liman et al., 2015). Tietjen et al. (2009) concluded that decreased concentrations of urinary NO stable metabolites in migraineurs in between migraine attacks compared to control subjects was indicative of endothelial cell dysfunction. Reports from the Levine lab suggest that vascular endothelial cells play a role in enhanced peripheral hyperalgesia via endothelin-1 and both β-adrenergic antagonist ICI-118551 and sumatriptan, both which have receptors on endothelial cells, attenuated endothelin-induced enhancement of hyperalgesia (Joseph et al., 2013). These findings suggest that anti-migraine drugs can produce anti-nociceptive effects by actions on endothelial cells. Conversely, Napoli et al. (2009) concluded that endothelial cells were properly functioning in migraineurs, however, smooth muscle cells failed to function properly following a diminished response to NO. Thus, there is evidence of endothelial dysfunction in migraine, although it is not without controversy.
Smooth muscle cells are of particular interest in migraine although studies that focus on their non-vasomotor contributions are limited. However, one promising area is the ability of NO to activate soluble guanylyl cyclase (sGC) in vascular smooth muscle cells. Recently sGC has been implicated in migraine pathogenesis (Ben Aissa et al., 2017). sGC is a major NO receptor and has been reported as a mediator of nitroglycerin-induced migraine pain (Ben Aissa et al., 2017). While NO induction of sGC causes vasodilation, it can influence dural nociceptors via the NO-cGMP pathway (Levy and Strassman, 2004). More recently Zhang et al. (2013) showed that this NTG infusion has been shown to cause delayed meningeal inflammation via vascular phosphorylated ERK expression. These data, though few, warrant more comprehensive studies to determine the role of smooth muscle activation in migraine models.
Finally, an unexpected contribution of the fibroblasts has recently been suggested by the Dussor lab (Wei et al., 2014). Cultured fibroblasts from the dura are capable of releasing mediators that sensitize dural afferents and induce-migraine-like behavior in rodents (Wei et al., 2014). These cells release IL-6 which is elevated during migraine attacks (Fidan et al., 2006; Sarchielli et al., 2006). It is important to note the fibroblasts used in this study were not cultured from dural vessels and cytokine release was induced by lipopolysaccharide. Future studies that dissect whether adventitial fibroblasts from dural vessels play a role in migraine and a whether a spontaneous mechanism can induce cytokine release are warranted.
Mast Cell, Neurons and Vessel Cross Talk
The mechanisms that underlie aberrant nociceptor activation are still poorly understood, but are believed to involve changes in the meningeal environment, especially in the dura mater (Zhang et al., 2007, 2010; Zhang and Levy, 2008; Levy, 2009, 2010). The dura mater is a highly vascularized membrane that is heavily innervated by pain fibers and has a dense population of immune cells (Fricke et al., 2001; Jacobs and Dussor, 2016). One theory is that neurogenic inflammation caused by activated mast cells can sensitize nociceptors and thus trigger headache (Theoharides et al., 2005; Waeber and Moskowitz, 2005; Levy, 2009). While direct evidence is lacking, clinical studies have reported increased circulating intracranial inflammatory mediators during an attack (Sarchielli et al., 2006; Goadsby and Edvinsson, 1993). Moreover, activated mast cells release histamine, prostaglandins and a host of pro-inflammatory peptides (Roberts et al., 1979; Heatley et al., 1982; Lewis et al., 1982; Tetlow et al., 1998; Theoharides et al., 2005; Aich et al., 2015). Specifically, tryptase and histamine release have been reported to release neuropeptides from proximal nerve endings and contribute to hyperalgesia (Kleij and Bienenstock, 2005; Aich et al., 2015). This along with the ability of mast cells to increase pERK, cfos, and excitation of meningeal nociceptors provide insight into a mechanism of how mast cells might contribute to peripheral sensitization.
Histamine has been reported to be increased in plasma levels during a migraine attack (Heatley et al., 1982; Moskowitz, 1993; Theoharides et al., 2005). Also, infusion of histamine in migraineurs causes a severe pulsating headache compared to controls (Krabbe and Olesen, 1980). Although with some debate, anti-histamines have been effective in treating migraine in some clinical studies and their potential role in migraine is nicely summarized by Silberstein (Yuan and Silberstein, 2018). Histamine causes dilation of cranial arteries via activation of endothelial histamine receptor H1 and inducing formation of NO (Toda, 1990; Ottosson et al., 1991). Moreover, histamine disrupts endothelial barrier formation by altering vascular endothelial cadherin and inducing dilation of vessels (Ashina et al., 2015).
There is an overwhelming abundance of evidence that suggest inflammatory pain states can alter blood brain barrier (BBB) permeability. The BBB is a selective barrier that limits paracellular diffusion via tight junctions between endothelial cells (DosSantos et al., 2014). Given that migraine has increased release of pro-inflammatory peptides, it could involve BBB disruption, although this is still controversial. In a mouse model of cortical spreading depression, the detection of brain edema, plasma extravasation, and altered metalloprotease and matrix proteins were indicative of BBB dysfunction (Gursoy-Ozdemir et al., 2004). In addition to barrier dysfunction, alterations in gap junctions may play a role in migraine. Gap junctions are specialized regions of the plasma membrane that connect cytoplasms of adjacent cells. Tonabersat, a gap junction inhibitor that binds to connexin 43, has been shown to be effective in a subset of migraine patients with aura (Sarrouilhe et al., 2014). Of particular interest, connexin 43 is found on neuronal cells and is one of the connexin proteins associated with cells of the cardiovascular system (Figueroa and Duling, 2009). Given these data, the efficacy of tonabersat suggest a possible role for dysfunction of gap junctions in migraine.
Based on these observations, we propose a possible mechanism for how an altered trigeminovascular microenvironment may initiate vascular-neural cross talk (Figure 1). The meninges are densely vascularized and the layers are innervated by sensory fibers that relay information from the periphery to higher order neurons in the brain. Distention of intracranial blood vessels, possibly from the dura, mechanically activates trigeminal perivascular afferents (Davis and Dostrovsky, 1986; Buzzi et al., 1995). Those activated neurons can release molecules that cause mast cell activation and vasodilation of the nearby vessels in a feed-back loop (Figure 1). In this model, mast cell activation increases vascular permeability and/or causes neuronal activation and neuropeptide release, which causes subsequent release of inflammatory mediators from the vessels that modulate sensory input. Most studies focus on the effect of mast cells on neurons in migraine. Future studies that examine the effect of mast cells on vasculature in translational models of migraine could reveal a role for mast cells in vascular-neural coupling.
Figure 1
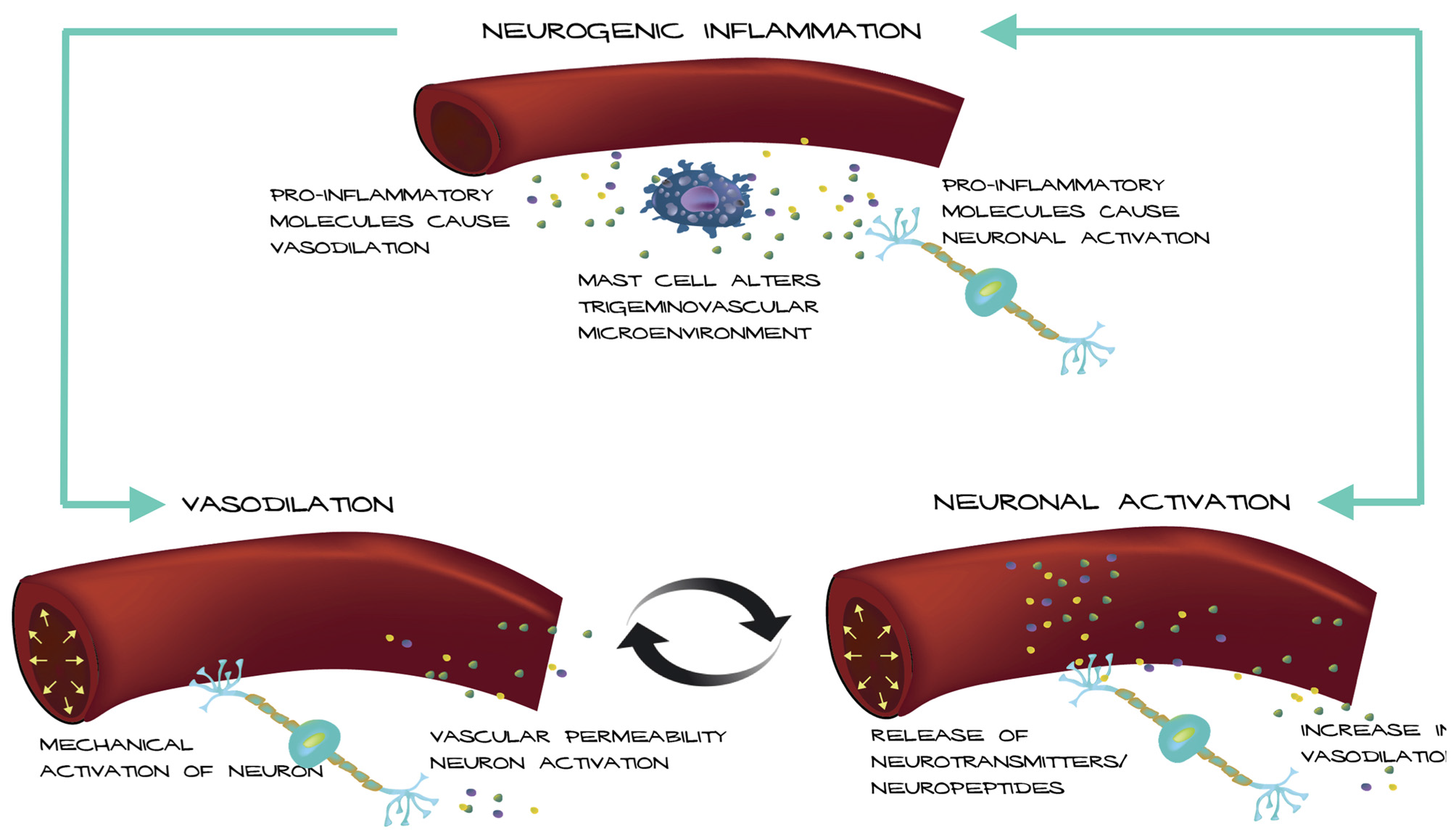
Model of vascular-neural coupling. Mast cell activation and degranulation alter the trigeminovascular microenvironment via release of inflammatory molecules. Inflammatory molecules can mediate vasodilation of nearby vessels and cause nociceptor activation. Vessels can activate trigeminal neurons mechanically or by release of inflammatory mediators due to increased vascular permeability causing a positive feedback loop.
Migraine Treatments and Their Actions on Vessels
Based on the theory that cranial vasodilation was the sole cause of migraine and intravenous infusion of serotonin could successfully treat migraine, a new evidence-based target for migraine therapy was born, the triptans (Humphrey et al., 1989). The triptans have now become the gold-standard in migraine treatment (Ferrari et al., 2001).
Despite the wide use and tolerability of the triptans, the mechanism of action has never been entirely defined. It is known that triptans are 5-HT1B/D/F serotonin receptor agonists that can inhibit the release of neuropeptides involved in migraine and act as vasoconstrictors (Jansen et al., 1992; Nozaki et al., 1992; Williamson et al., 1997; Knight et al., 2001; Wackenfors et al., 2005). Moreover, sumatriptan treatment reduced plasma levels of CGRP in humans (Goadsby and Edvinsson, 1993; Juhasz et al., 2003) and animal models (Buzzi and Moskowitz, 1990; Buzzi et al., 1991; Nozaki et al., 1992). Vascular studies of triptans in people have given insight into its mechanism of action and the roles vessels might play in migraine. In fact, a study using single-photon emission computed tomography combined with Doppler sonography showed that if sumatriptan is infused in people, only the abnormally dilated vessels were reversed back to normal (Asghar et al., 2010, 2011). This observation suggests that triptans only cause significant vasoconstriction on dilated vessels during a migraine. Furthermore, it is interesting to note that triptans work best if used within the first 2 h of the attack (Linde et al., 2006). This coincides with the vasodilatory period following CGRP administration (Lassen et al., 2002). In addition, sumatriptan does not appear to be effective for relieving pain in other disorders (Ahn and Basbaum, 2005), which may be due to the vascular events in migraine. These observations suggest that the vasoconstrictor activity of triptans should not be ignored.
CGRP receptors are found throughout the cranial vasculature. Olcegepant is a non-peptide CGRP antagonist that has high specificity for human CGRP receptors and reported to be efficacious in migraine (Edvinsson, 2008, 2015). Olcegepant blocks dilation of the middle meningeal and extracranial temporal arteries. Similarly, the CGRP receptor antagonist telcagepant has also been shown to inhibit vasodilation of cultured human cerebral and meningeal arteries (Edvinsson et al., 2010). These observations leave open the possibility that some of the CGRP receptor antagonist efficacy might involve the vasculature.
Association of Migraine With Cardiovascular Disease
Several studies have linked migraine with increased risk of cardiovascular disease. It is reported that migraineurs with aura have a two-fold increased risk for ischemic stroke (Schürks et al., 2009; Sacco and Kurth, 2014). Moreover, patients that experience high frequency migraine have a further increased risk of stroke (Kurth et al., 2009). More to the point, in the largest ever meta-analysis that included over 1 million subjects, migraine has been confirmed to be associated with a higher long-term risk of both ischemic and hemorrhagic stroke and myocardial infarction (Mahmoud et al., 2018). Another study that corroborates a role in migraine is a genome wide association meta-analysis in 2016 by Gormley et al. (2016) that identified 38 susceptibility loci that were enriched for genes associated with arterial tissue. Moreover, several of these genes are associated with smooth muscle dysfunction and cardiovascular disorders linked to migraine as a comorbidity.
There is debate on whether hypertension and/or hypotension have a relationship with migraine (Hagen et al., 2002; Low and Merikangas, 2003; Hamed et al., 2010). It has been suggested that migraine and hypertension have a high prevalence of co-morbidity. Reports that there is a high incidence of migraine in patients with hypertension go as far back as 1913 (Janeway, 1913). Additionally, in a retrospective study, Grebe et al. (2001) found 61% of patients that had developed medicine overuse headache from the treatment of migraine also had hypertension. The same group also found that migraineurs with aura had increased systolic pressure compared to control subjects.
Migraine has a sex disparity and affects a substantial number of women in their reproductive years (Sacco et al., 2012; Finocchi and Strada, 2014). Preeclampsia is a vascular disorder of pregnancy and is characterized by the sudden onset of hypertension and occurrence of vasospasm. It is the leading cause of death among pregnant women (MacKay et al., 2001). In 1959, Rotton et al. (1959) published the first study correlating migraine and preeclampsia and eclampsia in pregnant women. This study noted that a large population of women whose migraine attacks were exacerbated during pregnancy, were also found to have preeclampsia. Moreover, Adeney and Williams used a retrospective approach to examine the association of migraine and preeclampsia and found that 8 out of 10 studies showed a correlation between the two disorders (Adeney and Williams, 2006).
The renin-angiotensin system (RAS), which is involved in hypertension, has been thought to be involved in migraine pathogenesis (Ba’albaki and Rapoport, 2008). Indeed, the efficacy of angiotensin converting enzyme inhibitors in migraine treatment is indicative of a link between migraine and hypertension (Tronvik et al., 2003). Additionally, blood pressure homeostasis is maintained by close communication between the RAS and natriuretic peptides. It has been reported that brain natriuretic peptide (BNP), which is produced by cardiac cells, is elevated in migraine (Uzar et al., 2011). However, BNP is reported to negatively regulate sensory neuron excitability (Vilotti et al., 2013). Further investigations are necessary to elucidate the role of BNP, RAS and hypertension in migraine. Conversely, several studies have also found no correlation between migraine and hypertension (Hagen et al., 2002; Wiehe et al., 2002; Tzourio et al., 2003). In fact, one study suggested that individuals with migraine-like episodes had a higher correlation with lower blood pressure than individuals without headache (Seçil et al., 2010). Furthermore, Seçil et al. (2010) detected diastolic hypotension in normotensive patients at the beginning, during, and up to 1 h following a migraine attack. These observations point to a possible link, but more comprehensive studies are needed to determine if hypertension or hypotension contribute to a sub-population of migraine attacks.
Conclusion
The ability of vasoactive substances to induce migraine, effective drugs to have a vascular site of action, and the associated correlation of migraine and cardiovascular disease convey that vascular contributions should not be considered an epiphenomenon, but more so a causative component in migraine. Yet, clearly many lines of evidence establish that migraine is a neural disorder. We suggest that the vascular and neural theories can be linked by vascular activation of the nervous system (Figure 1). Understanding the communication between blood vessels, neurons and possibly mast cells will be integral in unraveling the pathophysiology of migraine and future studies should focus on dissecting this intersection of vascular and neural actions in migraine.
Statements
Author contributions
The manuscript was written by BM and edited by AR.
Funding
This study was supported by National Institutes of Health (NIH) NRSA F31NS098825 to BM and NIH (NS075599), U.S. Department of Veterans Affairs Medical Center (1I01RX002101), and U.S. Department of Defense USAMRAA (W81XWH-16-1-0071 and W81XWH-16-1-0211) to AR.
Conflict of interest
AR is a consultant to Alder BioPharmaceuticals, Inc. and has served as a consultant to Eli Lilly and Amgen/Novartis. The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
AdeneyK. L.WilliamsM. A. (2006). Migraine headaches and preeclampsia: an epidemiologic review. Headache46, 794–803. 10.1111/j.1526-4610.2006.00432.x
2
AhnA. H. (2010). On the temporal relationship between throbbing migraine pain and arterial pulse. Headache50, 1507–1510. 10.1111/j.1526-4610.2010.01765.x
3
AhnA. H.BasbaumA. I. (2005). Where do triptans act in the treatment of migraine?Pain115, 1–4. 10.1016/j.pain.2005.03.008
4
AichA.AfrinL. B.GuptaK. (2015). Mast cell-mediated mechanisms of nociception. Int. J. Mol. Sci.16, 29069–29092. 10.3390/ijms161226151
5
AminF. M.AsgharM. S.HougaardA.HansenA. E.LarsenV. A.de KoningP. J.et al. (2013). Magnetic resonance angiography of intracranial and extracranial arteries in patients with spontaneous migraine without aura: a cross-sectional study. Lancet Neurol.12, 454–461. 10.1016/s1474-4422(13)70067-x
6
AminF. M.HougaardA.SchytzH. W.AsgharM. S.LundholmE.ParvaizA. I.et al. (2014). Investigation of the pathophysiological mechanisms of migraine attacks induced by pituitary adenylate cyclase-activating polypeptide-38. Brain137, 779–794. 10.1093/brain/awt369
7
AraiH.HoriS.AramoriI.OhkuboH.NakanishiS. (1990). Cloning and expression of a cDNA encoding an endothelin receptor. Nature348, 730–732. 10.1038/348730a0
8
AsgharM. S.HansenA. E.AminF. M.van der GeestR. J.KoningP.LarssonH. B.et al. (2011). Evidence for a vascular factor in migraine. Ann. Neurol.69, 635–645. 10.1002/ana.22292
9
AsgharM. S.HansenA. E.KapijimpangaT.van der GeestR. J.van der KoningP.LarssonH. B.et al. (2010). Dilation by CGRP of middle meningeal artery and reversal by sumatriptan in normal volunteers. Neurology75, 1520–1526. 10.1212/WNL.0b013e3181f9626a
10
AshinaK.TsubosakaY.NakamuraT.OmoriK.KobayashiK.HoriM.et al. (2015). Histamine induces vascular hyperpermeability by increasing blood flow and endothelial barrier disruption in vivo. PLoS One10:e0132367. 10.1371/journal.pone.0132367
11
Ba’albakiH.RapoportA. (2008). Mast cells activate the renin angiotensin system and contribute to migraine: a hypothesis. Headache48, 1499–1505. 10.1111/j.1526-4610.2008.00852.x
12
BaunM.PedersenM. H.OlesenJ.Jansen-OlesenI. (2012). Dural mast cell degranulation is a putative mechanism for headache induced by PACAP-38. Cephalalgia32, 337–345. 10.1177/0333102412439354
13
Ben AissaM.TiptonA. F.BertelsZ.GandhiR.MoyeL. S.NovackM.et al. (2017). Soluble guanylyl cyclase is a critical regulator of migraine-associated pain. Cephalalgia [Epub ahead of print]. 10.1177/0333102417737778
14
BhattD. K.GuptaS.OlesenJ.Jansen-OlesenI. (2014). PACAP-38 infusion causes sustained vasodilation of the middle meningeal artery in the rat: possible involvement of mast cells. Cephalalgia34, 877–886. 10.1177/0333102414523846
15
BlauJ. N.DexterS. L. (1981). The site of pain origin during migraine attacks. Cephalalgia1, 143–147. 10.1046/j.1468-2982.1981.0103143.x
16
BoniniM. G.StadlerK.SilvaS. O.CorbettJ.DoreM.PetrankaJ.et al. (2008). Constitutive nitric oxide synthase activation is a significant route for nitroglycerin-mediated vasodilation. Proc. Natl. Acad. Sci. U S A105, 8569–8574. 10.1073/pnas.0708615105
17
BrainS. D.GrantA. D. (2004). Vascular actions of calcitonin gene-related peptide and adrenomedullin. Physiol. Rev.84, 903–934. 10.1152/physrev.00037.2003
18
BrainS. D.WilliamsT. J.TippinsJ. R.MorrisH. R.MacIntyreI. (1985). Calcitonin gene-related peptide is a potent vasodilator. Nature313, 54–56. 10.1038/313054a0
19
BurchR. C.LoderS.LoderE.SmithermanT. A. (2015). The prevalence and burden of migraine and severe headache in the United States: updated statistics from government health surveillance studies. Headache55, 21–34. 10.1111/head.12482
20
BurnstockG. (2016). Purinergic signalling and endothelium. Curr. Vasc. Pharmacol.14, 130–145. 10.2174/1570161114666151202204948
21
BuzziM. G.MoskowitzM. A. (1990). The antimigraine drug, sumatriptan (GR43175), selectively blocks neurogenic plasma extravasation from blood vessels in dura mater. Br. J. Pharmacol.99, 202–206. 10.1111/j.1476-5381.1990.tb14679.x
22
BuzziM. G.BonaminiM.MoskowitzM. A. (1995). Neurogenic model of migraine. Cephalalgia15, 277–280. 10.1046/j.1468-29821995.1504277.x
23
BuzziM. G.CarterW. B.ShimizuT.HeathH.III.MoskowitzM. A. (1991). Dihydroergotamine and sumatriptan attenuate levels of CGRP in plasma in rat superior sagittal sinus during electrical stimulation of the trigeminal ganglion. Neuropharmacology30, 1193–1200. 10.1016/0028-3908(91)90165-8
24
ChenZ.FosterM. W.ZhangJ.MaoL.RockmanH. A.KawamotoT.et al. (2005). An essential role for mitochondrial aldehyde dehydrogenase in nitroglycerin bioactivation. Proc. Natl. Acad. Sci. U S A102, 12159–12164. 10.1073/pnas.0503723102
25
ColemanJ. W. (2001). Nitric oxide in immunity and inflammation. Int. Immunopharmacol.1, 1397–1406. 10.1016/s1567-5769(01)00086-8
26
CouchJ. R. (2011). Update on chronic daily headache. Curr. Treat. Options Neurol.13, 41–55. 10.1007/s11940-010-0104-7
27
CuryY.PicoloG.GutierrezV. P.FerreiraS. H. (2011). Pain and analgesia: the dual effect of nitric oxide in the nociceptive system. Nitric Oxide25, 243–254. 10.1016/j.niox.2011.06.004
28
DavisK. D.DostrovskyJ. O. (1986). Activation of trigeminal brain-stem nociceptive neurons by dural artery stimulation. Pain25, 395–401. 10.1016/0304-3959(86)90244-7
29
de NucciG.ThomasR.D’Orleans-JusteP.AntunesE.WalderC.WarnerT. D.et al. (1988). Pressor effects of circulating endothelin are limited by its removal in the pulmonary circulation and by the release of prostacyclin and endothelium-derived relaxing factor. Proc. Natl. Acad. Sci. U S A85, 9797–9800. 10.1073/pnas.85.24.9797
30
DelgadoM.AbadC.MartinezM.ArranzA.GomarizR. (2002). Vasoactive intestinal peptide in the immune system: potential therapeutic role of inflammatory and autoimmune disease. J. Mol. Med.80, 16–24. 10.1007/s00109-001-0291-5
31
DelgadoM.AbadC.MartinezC.JuarranzM. G.LecetaJ.GaneaD.et al. (2003). PACAP in immunity and inflammation. Ann. N Y Acad. Sci.992, 141–157. 10.1111/j.1749-6632.2003.tb03145.x
32
DosSantosM. F.Holanda-AfonsoR. C.LimaR. L.DaSilvaA. F.Moura-NetoV. (2014). The role of the blood-brain barrier in the development and treatment of migraine and other pain disorders. Front. Cell. Neurosci.8:302. 10.3389/fncel.2014.00302
33
DrummondP. D.LanceJ. W. (1983). Extracranial vascular changes and the source of pain in migraine headache. Ann. Neurol.13, 32–37. 10.1002/ana.410130108
34
EdvinssonL. (2008). CGRP blockers in migraine therapy: where do they act?Br. J. Pharmacol.155, 967–969. 10.1038/bjp.2008.346
35
EdvinssonL. (2015). CGRP receptor antagonists and antibodies against CGRP and its receptor in migraine treatment. Br. J. Clin. Pharmacol.80, 193–199. 10.1111/bcp.12618
36
EdvinssonL.ChanK. Y.EftekhariS.NilssonE.de VriesR.SavelandH.et al. (2010). Effect of the calcitonin gene-related peptide (CGRP) receptor antagonist telcagepant in human cranial arteries. Cephalalgia30, 1233–1240. 10.1177/0333102410362122
37
EdvinssonL.TajtiJ.SzalárdyL.VécseiL. (2018). PACAP and its role in primary headaches. J. Headache Pain19:21. 10.1186/s10194-018-0852-4
38
ElkindA. H.FriedmanA. P.GrossmanJ. (1964). Cutaneous blood flow in vascular headaches of the migraine type. Neurology14, 24–30. 10.1212/WNL.14.1.24
39
FahrenkrugJ.HannibalJ. (1998). Pituitary adenylate cyclase activating polypeptide immunoreactivity in capsaicin-sensitive nerve fibres supplying the rat urinary tract. Neuroscience83, 1261–1272. 10.1016/s0306-4522(97)00474-0
40
FerrariM. D.RoonK. I.LiptonR. B.GoadsbyP. J. (2001). Oral triptans (serotonin 5-HT1B/1D agonists) in acute migraine treatment: a meta-analysis of 53 trials. Lancet358, 1668–1675. 10.1016/s0140-6736(01)06711-3
41
FidanI.YükselS.YmirT.IrkeçC.AksakalF. N. (2006). The importance of cytokines, chemokines and nitric oxide in pathophysiology of migraine. J. Neuroimmunol.171, 184–188. 10.1016/j.jneuroim.2005.10.005
42
FigueroaX. F.DulingB. R. (2009). Gap junctions in the control of vascular function. Antioxid. Redox Signal.11, 251–266. 10.1089/ars.2008.2117
43
FinocchiC.StradaL. (2014). Sex-related differences in migraine. Neurol. Sci.35, 207–213. 10.1007/s10072-014-1772-y
44
FrickeB.AndresK. H.Von DüringM. (2001). Nerve fibers innervating the cranial and spinal meninges: morphology of nerve fiber terminals and their structural integration. Microsc. Res. Tech.53, 96–105. 10.1002/jemt.1074
45
GoadsbyP. J. (2009). The vascular theory of migraine—a great story wrecked by the facts. Brain132, 6–7. 10.1093/brain/awn321
46
GoadsbyP. J.EdvinssonL. (1993). The trigeminovascular system and migraine: studies characterizing cerebrovascular and neuropeptide changes seen in humans and cats. Ann. Neurol.33, 48–56. 10.1002/ana.410330109
47
GormleyP.AnttilaV.WinsvoldB. S.PaltaP.EskoT.PersT. H.et al. (2016). Meta-analysis of 375,000 individuals identifies 38 susceptibility loci for migraine. Nat. Genet.48, 856–866. 10.1038/ng.3598
48
GrebeH. P.Nunes da SilvaM. J.Diogo-SousaL. (2001). [Role of arterial hypertension in comorbidity of chronic headaches]. Rev. Neurol.33, 119–122.
49
Gursoy-OzdemirY.QiuJ.MatsuokaN.BolayH.BermpohlD.JinH.et al. (2004). Cortical spreading depression activates and upregulates MMP-9. J. Clin. Invest.113, 1447–1455. 10.1172/jci21227
50
HagenK.StovnerL. J.VattenL.HolmenJ.ZwartJ. A.BovimG. (2002). Blood pressure and risk of headache: a prospective study of 22 685 adults in Norway. J. Neurol. Neurosurg. Psychiatry72, 463–466. 10.1136/jnnp.72.4.463
51
HamedS. A.HamedE. A.Ezz EldinA. M.MahmoudN. M. (2010). Vascular risk factors, endothelial function, and carotid thickness in patients with migraine: relationship to atherosclerosis. J. Stroke Cerebrovasc. Dis.19, 92–103. 10.1016/j.jstrokecerebrovasdis.2009.04.007
52
HawkinsK.WangS.RupnowM. (2008). Direct cost burden among insured US employees with migraine. Headache48, 553–563. 10.1111/j.1526-4610.2007.00990.x
53
HeatleyR. V.DenburgJ. A.BayerN.BienenstockJ. (1982). Increased plasma histamine levels in migraine patients. Clin. Allergy12, 145–149. 10.1111/j.1365-2222.1982.tb01633.x
54
HillJ. M.ZalosG.HalcoxJ. P.SchenkeW. H.WaclawiwM. A.QuyyumiA. A.et al. (2003). Circulating endothelial progenitor cells, vascular function and cardiovascular risk. N. Engl. J. Med.348, 593–600. 10.1056/nejmoa022287
55
HillK. E.HuntR. W.Jr.JonesR.HooverR. L.BurkR. F. (1992). Metabolism of nitroglycerin by smooth muscle cells. Involvement of glutathione and glutathione S-transferase. Biochem. Pharmacol.43, 561–566. 10.1016/0006-2952(92)90579-8
56
HoffmanA.GrossmanE.OhmanK. P.MarksE.KeiserH. R. (1989). Endothelin induces an initial increase in cardiac output associated with selective vasodilation in rats. Life Sci.45, 249–255. 10.1016/0024-3205(89)90257-9
57
HumphreyP. P.FeniukW.PerrenM. J.ConnorH. E.OxfordA. W. (1989). The pharmacology of the novel 5-HT1-like receptor agonist, GR43175. Cephalalgia9, 23–33.
58
IkedaU.YamamotoK.MaedaY.ShimpoM.KanbeT.ShimadaK. (1997). Endothelin-1 inhibits nitric oxide synthesis in vascular smooth muscle cells. Hypertension29, 65–69. 10.1161/01.hyp.29.1.65
59
IslerH. (1992). The Galenic tradition and migraine. J. Hist. Neurosci.1, 227–233. 10.1080/09647049209525535
60
IversenH. K.OlesenJ.Tfelt-HansenP. (1989). Intravenous nitroglycerin as an experimental model of vascular headache. Basic characteristics. Pain38, 17–24. 10.1016/0304-3959(89)90067-5
61
JacobsB.DussorG. (2016). Neurovascular contributions to migraine: moving beyond vasodilation. Neuroscience338, 130–144. 10.1016/j.neuroscience.2016.06.012
62
JanewayH. H. (1913). Intratracheal anaesthesia: A. By nitrous oxide and oxygen. B. By nitrous oxide and oxygen under conditions of differential pressure. Ann. Surg.58, 927–933. 10.1097/00000658-191312000-00025
63
JansenI.EdvinssonL.MortensenA.OlesenJ. (1992). Sumatriptan is a potent vasoconstrictor of human dural arteries via a 5-HT1-like receptor. Cephalalgia12, 202–205. 10.1046/j.1468-2982.1992.1204202.x
64
Jansen-OlesenI.Hougaard PedersenS. (2018). PACAP and its receptors in cranial arteries and mast cells. J. Headache Pain19:16. 10.1186/s10194-017-0822-2
65
Jansen-OlesenI.BaunM.AmrutkarD. V.RamachandranR.ChristophersenD. V.OlesenJ. (2014). PACAP-38 but not VIP induces release of CGRP from trigeminal nucleus caudalis via a receptor distinct from the PAC receptor. Cephalalgia48, 53–64. 10.1016/j.npep.2014.01.004
66
JosephE. K.GreenP. G.BogenO.AlvarezP.LevineJ. D. (2013). Vascular endothelial cells mediate mechanical stimulation-induced enhancement of endothelin hyperalgesia via activation of P2X2/3 receptors on nociceptors. J. Neurosci.33, 2849–2859. 10.1523/JNEUROSCI.3229-12.2013
67
JuhaszG.ZsombokT.ModosE. A.OlajosS.JakabB.NemethJ.et al. (2003). NO-induced migraine attack: strong increase in plasma calcitonin gene-related peptide (CGRP) concentration and negative correlation with platelet serotonin release. Pain106, 461–470. 10.1016/j.pain.2003.09.008
68
KaiserE. A.RussoA. F. (2013). CGRP and migraine: could PACAP play a role too?Neuropeptides47, 451–461. 10.1016/j.npep.2013.10.010
69
KaiserE. A.KuburasA.RecoberA.RussoA. F. (2012). Modulation of CGRP-induced light aversion in wild-type mice by a 5-HT1B/D agonist. J. Neurosci.32, 15439–15449. 10.1523/JNEUROSCI.3265-12.2012
70
KallelaM.FärkkiläM.SaijonmaaO.FyhrquistF. (1998). Endothelin in migraine patients. Cephalalgia18, 329–332. 10.1046/j.1468-2982.1998.1806303-3.x
71
KleijH. P.BienenstockJ. (2005). Significance of conversation between mast cells and nerves. Allergy Asthma Clin. Immunol.1, 65–80. 10.1186/1710-1492-1-2-65
72
KnightY. E.EdvinssonL.GoadsbyP. J. (2001). 4991W93 inhibits release of calcitonin gene-related peptide in the cat but only at doses with 5HT1B/1D receptor agonist activity?Neuropharmacology40, 520–525. 10.1016/s0028-3908(00)00187-8
73
KosarasB.JakubowskiM.KainzV.BursteinR. (2009). Sensory innervation of the calvarial bones of the mouse. J. Comp. Neurol.515, 331–348. 10.1002/cne.22049
74
KrabbeA. A.OlesenJ. (1980). Headache provocation by continuous intravenous infusion of histamine. Clinical results and receptor mechanisms. Pain8, 253–259. 10.1016/0304-3959(88)90012-7
75
KurthT.SchurksM.LogroscinoG.BuringJ. E. (2009). Migraine frequency and risk of cardiovascular disease in women. Neurology73, 581–588. 10.1212/WNL.0b013e3181ab2c20
76
LassenL. H.HaderslevP. A.JacobsenV. B.IversenH. K.SperlingB.OlesenJ. (2002). CGRP may play a causative role in migraine. Cephalalgia22, 54–61. 10.1046/j.1468-2982.2002.00310.x
77
LevyD. (2009). Migraine pain, meningeal inflammation and mast cells. Curr. Pain Headache Rep.13, 237–240. 10.1007/s11916-009-0040-y
78
LevyD. (2010). Migraine pain and nociceptor activation—where do we stand?Headache50, 909–916. 10.1111/j.1526-4610.2010.01670.x
79
LevyD.StrassmanA. M. (2004). Modulation of dural nociceptor mechanosensitivity by the nitric oxide-cyclic GMP signaling cascade. J. Neurophysiol.92, 766–772. 10.1152/jn.00058.2004
80
LevyD.BursteinR.StrassmanA. M. (2005). Calcitonin gene-related peptide does not excite or sensitize meningeal nociceptors: implications for the pathophysiology of migraine. Ann. Neurol.58, 698–705. 10.1002/ana.20619
81
LewisR. A.SoterN. A.DiamondP. T.AustenK. F.OatesJ. A.RobertsL. J.II. (1982). Prostaglandin D2 generation after activation of rat and human mast cells with anti-IgE. J. Immunol.129, 1627–1631.
82
LimanT. G.Bachelier-WalentaK.NeebL.RosinskiJ.ReuterU.BohmM.et al. (2015). Circulating endothelial microparticles in female migraineurs with aura. Cephalalgia35, 88–94. 10.1177/0333102414529671
83
LindeM.MellbergA.DahlöfC. (2006). Subcutaneous sumatriptan provides symptomatic relief at any pain intensity or time during the migraine attack. Cephalalgia26, 113–121. 10.1111/j.1468-2982.2005.00999.x
84
LoderE. (2010). Triptan therapy in migraine. N. Engl. J. Med.363, 63–70. 10.1056/NEJMct0910887
85
LowN. C.MerikangasK. R. (2003). The comorbidity of migraine. CNS Spectr.8, 433–434, 437–444. 10.1017/S1092852900018745
86
MacKayA. P.BergC. J.AtrashH. K. (2001). Pregnancy-related mortality from preeclampsia and eclampsia. Obstet. Gynecol.97, 533–538. 10.1097/00006250-200104000-00011
87
MahmoudA. N.MentiasA.ElgendyA. Y.QaziA.BarakatA. F.SaadM.et al. (2018). Migraine and the risk of cardiovascular and cerebrovascular events: a meta-analysis of 16 cohort studies including 1 152 407 subjects. BMJ Open8:e020498. 10.1136/bmjopen-2017-020498
88
MarkovicsA.KormosV.GasznerB.LashgararaA.SzokeE.SandorK.et al. (2012). Pituitary adenylate cyclase-activating polypeptide plays a key role in nitroglycerol-induced trigeminovascular activation in mice. Neurobiol. Dis.45, 633–644. 10.1016/j.nbd.2011.10.010
89
Marquez de PradoB.HammondD. L.RussoA. F. (2009). Genetic enhancement of calcitonin gene-related peptide-induced central sensitization to mechanical stimuli in mice. J. Pain10, 992–1000. 10.1016/j.jpain.2009.03.018
90
MasonB. N.KaiserE. A.KuburasA.LoomisM. M.LathamJ. A.Garcia-MartinezL. F.et al. (2017). Induction of migraine-like photophobic behavior in mice by both peripheral and central CGRP mechanisms. J. Neurosci.37, 204–216. 10.1523/JNEUROSCI.2967-16.2016
91
MayA.GijsmanH. J.WallnoferA.JonesR.DienerH. C.FerrariM. D. (1996). Endothelin antagonist bosentan blocks neurogenic inflammation, but is not effective in aborting migraine attacks. Pain67, 375–378. 10.1016/0304-3959(96)03137-5
92
MesslingerK. (2009). Migraine: where and how does the pain originate?Exp. Brain Res.196, 179–193. 10.1007/s00221-009-1756-y
93
MesslingerK.LennerzJ. K.EberhardtM.FischerM. J. (2012). CGRP and NO in the trigeminal system: mechanisms and role in headache generation. Headache52, 1411–1427. 10.1111/j.1526-4610.2012.02212.x
94
MillarT. M.StevensC. R.BenjaminN.EisenthalR.HarrisonR.BlakeD. R. (1998). Xanthine oxidoreductase catalyses the reduction of nitrates and nitrite to nitric oxide under hypoxic conditions. FEBS Lett.427, 225–228. 10.1016/s0014-5793(98)00430-x
95
MoncadaS.PalmerR. M.HiggsE. A. (1991). Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol. Rev.43, 109–142.
96
MoskowitzM. A. (1993). Neurogenic inflammation in the pathophysiology and treatment of migraine. Neurology43, S16–S20.
97
MulderH.UddmanR.MollerK.ZhangY. Z.EkbladE.AlumetsJ.et al. (1994). Pituitary adenylate cyclase activating polypeptide expression in sensory neurons. Neuroscience63, 307–312. 10.1016/0306-4522(94)90025-6
98
NapoliR.GuardasoleV.ZarraE.MatarazzoM.D’AnnaC.SaccaF.et al. (2009). Vascular smooth muscle cell dysfunction in patients with migraine. Neurology72, 2111–2114. 10.1212/WNL.0b013e3181aa53ce
99
NozakiK.MoskowitzM. A.BoccaliniP. (1992). CP-93,129, sumatriptan, dihydroergotamine block c-fos expression within rat trigeminal nucleus caudalis caused by chemical stimulation of the meninges. Br. J. Pharmacol.106, 409–415. 10.1111/j.1476-5381.1992.tb14348.x
100
OlesenJ.LarsenB.LauritzenM. (1981). Focal hyperemia followed by spreading oligemia and impaired activation of rCBF in classic migraine. Ann. Neurol.9, 344–352. 10.1002/ana.410090406
101
OttossonA. L.JansenI.LangemarkM.OlesenJ.EdvinssonL. (1991). Histamine receptors in the isolated human middle meningeal artery. A comparison with cerebral and temporal arteries. Cephalalgia11, 183–188. 10.1046/j.1468-2982.1991.1104183.x
102
RaddantA. C.RussoA. F. (2011). Calcitonin gene-related peptide in migraine: intersection of peripheral inflammation and central modulation. Expert Rev. Mol. Med.13:e36. 10.1017/s1462399411002067
103
RaddinoR.PelàG.MancaC.BarbagalloM.D’AloiaA.PasseriM.et al. (1997). Mechanism of action of human calcitonin gene-related peptide in rabbit heart and in human mammary arteries. J. Cardiovasc. Pharmacol.29, 463–470. 10.1097/00005344-199704000-00006
104
RahmannA.WieneckeT.HansenJ. M.FahrenkrugJ.OlesenJ.AshinaM. (2008). Vasoactive intestinal peptide causes marked cephalic vasodilation, but does not induce migraine. Cephalalgia28, 226–236. 10.1111/j.1468-2982.2007.01497.x
105
RecoberA.KaiserE. A.KuburasA.RussoA. F. (2010). Induction of multiple photophobic behaviors in a transgenic mouse sensitized to CGRP. Neuropharmacology58, 156–165. 10.1016/j.neuropharm.2009.07.009
106
RecoberA.KuburasA.ZhangZ.WemmieJ. A.AndersonM. G.RussoA. F. (2009). Role of calcitonin gene-related peptide in light aversive behavior: implications for migraine. J. Neurosci.29, 8798–8804. 10.1523/JNEUROSCI.1727-09.2009
107
ReuterU.BolayH.Jansen-OlesenI.ChiarugiA.Sanchez del RioM.LetourneauR.et al. (2001). Delayed inflammation in rat meninges: implications for migraine pathophysiology. Brain124, 2490–2502. 10.1093/brain/124.12.2490
108
RobertsL. J.II.LewisR. A.OatesJ. A.AustenK. F. (1979). Prostaglandin thromboxane and 12-hydroxy-5,8,10,14-eicosatetraenoic acid production by ionophore-stimulated rat serosal mast cells. Biochim. Biophys. Acta575, 185–192. 10.1016/0005-2760(79)90020-1
109
Rodríguez-OsorioX.SobrinoT.BreaD.MartínezF.CastilloJ.LeiraR. (2012). Endothelial progenitor cells: a new key for endothelial dysfunction in migraine. Neurology79, 474–479. 10.1212/wnl.0b013e31826170ce
110
RottonW. N.SachtlebenM. R.FriedmanE. A. (1959). Migraine and eclampsia. Obstet. Gynecol.14, 322–330.
111
RussellF. A.KingR.SmillieS. J.KodjiX.BrainS. D. (2014). Calcitonin gene-related peptide: physiology and pathophysiology. Physiol. Rev.94, 1099–1142. 10.1152/physrev.00034.2013
112
RussoA. F. (2015). CGRP as a neuropeptide in migraine: lessons from mice. Br. J. Clin. Pharmacol.80, 403–414. 10.1111/bcp.12686
113
RussoA. F. (2017). Overview of neuropeptides: awakening the senses?Headache57, 37–46. 10.1111/head.13084
114
SaccoS.KurthT. (2014). Migraine and the risk for stroke and cardiovascular disease. Curr. Cardiol. Rep.16:524. 10.1007/s11886-014-0524-1
115
SaccoS.RicciS.DeganD.CaroleiA. (2012). Migraine in women: the role of hormones and their impact on vascular diseases. J. Headache Pain13, 177–189. 10.1007/s10194-012-0424-y
116
SakuraiT.YanagisawaM.TakuwaY.MiyazakiH.KimuraS.GotoK.et al. (1990). Cloning of a cDNA encoding a non-isopeptide-selective subtype of the endothelin receptor. Nature348, 732–735. 10.1038/348732a0
117
SarchielliP.AlbertiA.BaldiA.CoppolaF.RossiC.PierguidiL.et al. (2006). Proinflammatory cytokines, adhesion molecules and lymphocyte integrin expression in the internal jugular blood of migraine patients without aura assessed ictally. Headache46, 200–207. 10.1111/j.1526-4610.2006.00337.x
118
SarrouilheD.DejeanC.MesnilM. (2014). Involvement of gap junction channels in the pathophysiology of migraine with aura. Front. Physiol.5:78. 10.3389/fphys.2014.00078
119
SchürksM.RistP. M.BigalM. E.BuringJ. E.LiptonR. B.KurthT. (2009). Migraine and cardiovascular disease: systematic review and meta-analysis. BMJ339:b3914. 10.1136/bmj.b3914
120
SchytzH. W.BirkS.WieneckeT.KruuseC.OlesenJ.AshinaM. (2009). PACAP38 induces migraine-like attacks in patients with migraine without aura. Brain132, 16–25. 10.1093/brain/awn307
121
SeçilY.UndeC.BeckmannY. Y.BozkayaY. T.OzerkanF.BasogluM. (2010). Blood pressure changes in migraine patients before, during and after migraine attacks. Pain Pract.10, 222–227. 10.1111/j.1533-2500.2009.00349.x
122
ShevelE. (2011). The extracranial vascular theory of migraine—a great story confirmed by the facts. Headache51, 409–417. 10.1111/j.1526-4610.2011.01844.x
123
SmithermanT. A.BurchR.SheikhH.LoderE. (2013). The prevalence, impact and treatment of migraine and severe headaches in the United States: a review of statistics from national surveillance studies. Headache53, 427–436. 10.1111/head.12074
124
TajtiJ.UddmanR.MöllerS.SundlerF.EdvinssonL. (1999). Messenger molecules and receptor mRNA in the human trigeminal ganglion. J. Auton. Nerv. Syst.76, 176–183. 10.1016/s0165-1838(99)00024-7
125
TetlowL. C.HarperN.DunninghamT.MorrisM. A.BertfieldH.WoolleyD. E. (1998). Effects of induced mast cell activation on prostaglandin E and metalloproteinase production by rheumatoid synovial tissue in vitro. Ann. Rheum. Dis.57, 25–32. 10.1136/ard.57.1.25
126
Tfelt-HansenP. C.KoehlerP. J. (2008). History of the use of ergotamine and dihydroergotamine in migraine from 1906 and onward. Cephalalgia28, 877–886. 10.1111/j.1468-2982.2008.01578.x
127
TheoharidesT. C.DonelanJ.Kandere-GrzybowskaK.KonstantinidouA. (2005). The role of mast cells in migraine pathophysiology. Brain Res. Rev.49, 65–76. 10.1016/j.brainresrev.2004.11.006
128
ThomsenL. L.IversenH. K.OlesenJ. (1995). Cerebral blood flow velocities are reduced during attacks of unilateral migraine without aura. Cephalalgia15, 109–116. 10.1046/j.1468-2982.1995.015002109.x
129
ThomsenL. L.KruuseC.IversenH. K.OlesenJ. (1994). A nitric oxide donor (nitroglycerin) triggers genuine migraine attacks. Eur. J. Neurol.1, 73–80. 10.1111/j.1468-1331.1994.tb00053.x
130
TietjenG. E.HerialN. A.WhiteL.UtleyC.KosmynaJ. M.KhuderS. A. (2009). Migraine and biomarkers of endothelial activation in young women. Stroke40, 2977–2982. 10.1161/strokeaha.109.547901
131
TodaN. (1990). Mechanism underlying responses to histamine of isolated monkey and human cerebral arteries. Am. J. Physiol.258, H311–H317. 10.1152/ajpheart.1990.258.2.h311
132
TomiyamaH.YamashinaA. (2010). Non-invasive vascular function tests: their pathophysiological background and clinical application. Circ. J.74, 24–33. 10.1253/circj.cj-09-0534
133
TronvikE.StovnerL. J.HeldeG.SandT.BovimG. (2003). Prophylactic treatment of migraine with an angiotensin II receptor blocker: a randomized controlled trial. JAMA289, 65–69. 10.1001/jama.289.1.65
134
TunçelN.TöreF.SahintürkV.AkD.TunçelM. (2000). Vasoactive intestinal peptide inhibits degranulation and changes granular content of mast cells: a potential therapeutic strategy in controlling septic shock. Peptides21, 81–89. 10.1016/s0196-9781(99)00177-1
135
TzourioC.GagnièreB.El AmraniM.AlpérovitchA.BousserM. G. (2003). Relationship between migraine, blood pressure and carotid thickness. A population-based study in the elderly. Cephalalgia23, 914–920. 10.1046/j.1468-2982.2003.00613.x
136
UddmanR.GoadsbyP. J.JansenI.EdvinssonL. (1993). PACAP, a VIP-like peptide: immunohistochemical localization and effect upon cat pial arteries and cerebral blood flow. J. Cereb. Blood Flow Metab.13, 291–297. 10.1038/jcbfm.1993.36
137
UzarE.EvliyaogluO.YucelY.Ugur CevikM.AcarA.GuzelI.et al. (2011). Serum cytokine and pro-brain natriuretic peptide (BNP) levels in patients with migraine. Eur. Rev. Med. Pharmacol. Sci.15, 1111–1116.
138
VaudryD.Falluel-MorelA.BourgaultS.BasilleM.BurelD.WurtzO.et al. (2009). Pituitary adenylate cyclase-activating polypeptide and its receptors: 20 years after the discovery. Pharmacol. Rev.61, 283–357. 10.1124/pr.109.001370
139
VilottiS.MarchenkovaA.NtamatiN.NistriA. (2013). B-type natriuretic peptide-induced delayed modulation of TRPV1 and P2X3 receptors of mouse trigeminal sensory neurons. PLoS One8:e81138. 10.1371/journal.pone.0081138
140
WackenforsA.JarviusM.IngemanssonR.EdvinssonL.MalmsjoM. (2005). Triptans induce vasoconstriction of human arteries and veins from the thoracic wall. J. Cardiovasc. Pharmacol.45, 476–484. 10.1097/01.fjc.0000159639.59770.36
141
WaeberC.MoskowitzM. A. (2005). Migraine as an inflammatory disorder. Neurology64, S9–S15. 10.1212/wnl.64.10_suppl_2.s9
142
WangX.HanC.FiscusR. R. (1991). Calcitonin gene-related peptide (CGRP) causes endothelium-dependent cyclic AMP, cyclic GMP and vasorelaxant responses in rat abdominal aorta. Neuropeptides20, 115–124. 10.1016/0143-4179(91)90061-m
143
WeiX.MelemedjianO. K.AhnD. D.WeinsteinN.DussorG. (2014). Dural fibroblasts play a potential role in headache pathophysiology. Pain155, 1238–1244. 10.1016/j.pain.2014.03.013
144
WieheM.FuchsS. C.MoreiraL. B.MoraesR. S.FuchsF. D. (2002). Migraine is more frequent in individuals with optimal and normal blood pressure: a population-based study. J. Hypertens.20, 1303–1306. 10.1097/00004872-200207000-00016
145
WilliamsonD. J.HargreavesR. J.HillR. G.ShepheardS. L. (1997). Sumatriptan inhibits neurogenic vasodilation of dural blood vessels in the anaesthetized rat—intravital microscope studies. Cephalalgia17, 525–531. 10.1046/j.1468-2982.1997.1704525.x
146
WinquistR. J.BuntingP. B.GarskyV. M.LummaP. K.SchofieldT. L. (1989). Prominent depressor response to endothelin in spontaneously hypertensive rats. Eur. J. Pharmacol.163, 199–203. 10.1016/0014-2999(89)90420-2
147
WoldeamanuelY. W.CowanR. P. (2017). Migraine affects 1 in 10 people worldwide featuring recent rise: a systematic review and meta-analysis of community-based studies involving 6 million participants. J. Neurol. Sci.372, 307–315. 10.1016/j.jns.2016.11.071
148
YaoG.HuangQ.WangM.YangC. L.LiuC. F.YuT. M. (2017). Behavioral study of a rat model of migraine induced by CGRP. Neurosci. Lett.651, 134–139. 10.1016/j.neulet.2017.04.059
149
YuanH.SilbersteinS. D. (2018). Histamine and migraine. Headache58, 184–193. 10.1111/head.13164
150
ZhangX.KainzV.ZhaoJ.StrassmanA. M.LevyD. (2013). Vascular extracellular signal-regulated kinase mediates migraine-related sensitization of meningeal nociceptors. Ann. Neurol.73, 741–750. 10.1002/ana.23873
151
ZhangX.LevyD.NosedaR.KainzV.JakubowskiM.BursteinR. (2010). Activation of meningeal nociceptors by cortical spreading depression: implications for migraine with aura. J. Neurosci.30, 8807–8814. 10.1523/JNEUROSCI.0511-10.2010
152
ZhangX. C.LevyD. (2008). Modulation of meningeal nociceptors mechanosensitivity by peripheral proteinase-activated receptor-2: the role of mast cells. Cephalalgia28, 276–284. 10.1111/j.1468-2982.2007.01523.x
153
ZhangX. C.StrassmanA. M.BursteinR.LevyD. (2007). Sensitization and activation of intracranial meningeal nociceptors by mast cell mediators. J. Pharmacol. Exp. Ther.322, 806–812. 10.1124/jpet.107.123745
Summary
Keywords
vascular, migraine, vasodilation, vasoconstriction, inflammation, CGRP
Citation
Mason BN and Russo AF (2018) Vascular Contributions to Migraine: Time to Revisit?. Front. Cell. Neurosci. 12:233. doi: 10.3389/fncel.2018.00233
Received
02 May 2018
Accepted
16 July 2018
Published
03 August 2018
Volume
12 - 2018
Edited by
Greg Dussor, The University of Texas, Dallas, United States
Reviewed by
Dan Levy, Beth Israel Deaconess Medical Center, Harvard Medical School, United States; Elsa Fabbretti, University of Trieste, Italy
Updates
Copyright
© 2018 Mason and Russo.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrew F. Russo andrew-russo@uiowa.edu
†Present address: Bianca N. Mason, School of Behavioral and Brain Sciences, The University of Texas, Dallas, Richardson, TX, United States
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.