 Linlin Wan
Linlin Wan Keqin Xu
Keqin Xu Zhao Chen
Zhao Chen Beisha Tang1,2,3,4,5,6,7
Beisha Tang1,2,3,4,5,6,7 Hong Jiang
Hong Jiang- 1Department of Neurology, Xiangya Hospital, Central South University, Changsha, China
- 2National Clinical Research Center for Geriatric Diseases, Central South University, Changsha, China
- 3Key Laboratory of Hunan Province in Neurodegenerative Disorders, Central South University, Changsha, China
- 4Laboratory of Medical Genetics, Central South University, Changsha, China
- 5Parkinson’s Disease Center of Beijing Institute for Brain Disorders, Beijing, China
- 6Collaborative Innovation Center for Brain Science, Shanghai, China
- 7Collaborative Innovation Center for Genetics and Development, Shanghai, China
- 8Department of Neurology, Xinjiang Medical University, Ürümqi, China
Post-translational modifications (PTMs), including phosphorylation, acetylation, ubiquitination, SUMOylation, etc., of proteins can modulate protein properties such as intracellular distribution, activity, stability, aggregation, and interactions. Therefore, PTMs are vital regulatory mechanisms for multiple cellular processes. Spinocerebellar ataxias (SCAs) are hereditary, heterogeneous, neurodegenerative diseases for which the primary manifestation involves ataxia. Because the pathogenesis of most SCAs is correlated with mutant proteins directly or indirectly, the PTMs of disease-related proteins might functionally affect SCA development and represent potential therapeutic interventions. Here, we review multiple PTMs related to disease-causing proteins in SCAs pathogenesis and their effects. Furthermore, we discuss these PTMs as potential targets for treating SCAs and describe translational therapies targeting PTMs that have been published.
Introduction
Post-translational modifications (PTMs) of proteins are crucial regulators of protein properties which can generate various types of proteins with different functions (Ratovitski et al., 2017). These modifications mostly arise from covalently attaching functional groups to specific amino acid residues including phosphate, acetate, and other chemical moieties, while another modification called proteolytic cleavage results from removing or separating specific portions of proteins (Karve and Cheema, 2011; Matos et al., 2017) (Table 1). PTMs, such as phosphorylation, acetylation, ubiquitination, SUMOylation, and proteolytic cleavage, can modulate the turnover, localisation, activity, and interaction of proteins (Ju et al., 2014). Thus, PTMs play pivotal roles in regulating multiple cellular pathways to further modulate pathogenesis in a number of diseases. For example, PTMs have been shown to modulate pathogenesis in cardiovascular diseases, neurological diseases, diabetes, liver cirrhosis, and cancer (Mendez et al., 2010; Modol et al., 2011; Orr, 2012; Xie et al., 2017; Zhang et al., 2017). Studying the roles of PTMs in disease processes can facilitate a better understanding of pathogenesis and uncover therapeutic targets for these diseases.
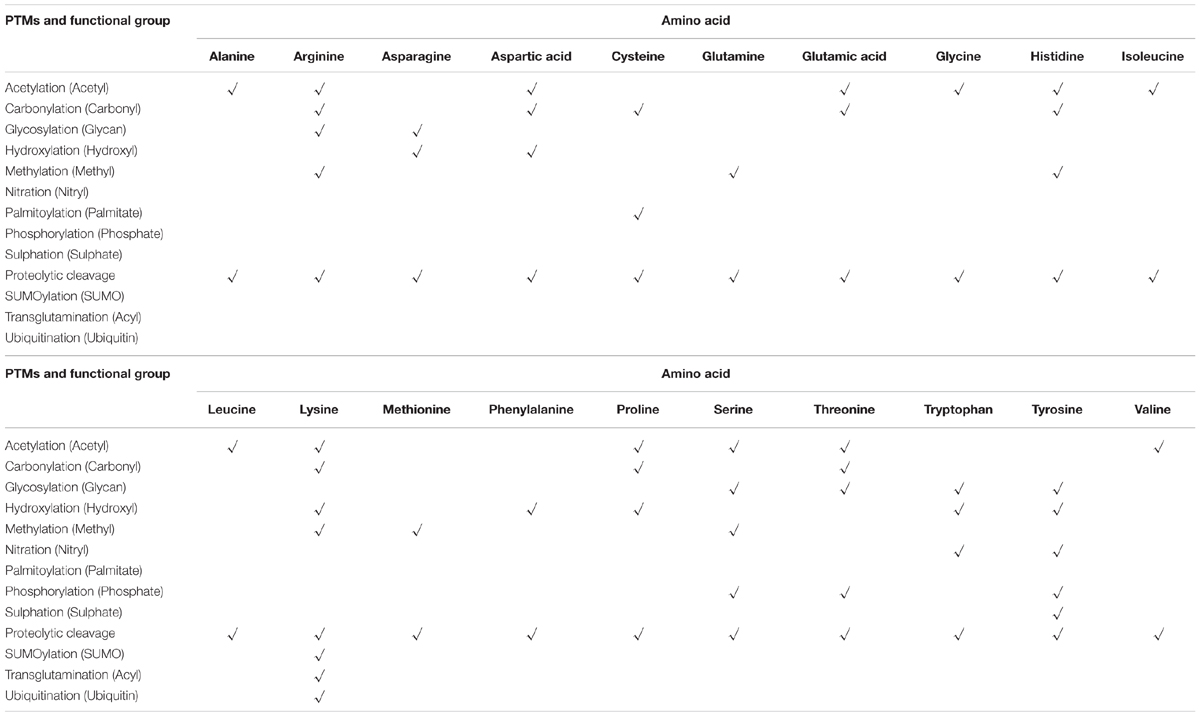
TABLE 1. Summary of common PTMs, their functional groups and possible amino acid sites (Klemm, 1984; Violante et al., 2001; Karve and Cheema, 2011; Hickey et al., 2012).
Spinocerebellar ataxias (SCAs) are a group of heterogeneously neurodegenerative diseases characterised by the progressive disequilibrium of motor coordination, cerebellar atrophy and other various non-ataxia manifestations (Rossi et al., 2014). To date, more than 40 subtypes of SCAs have been reported1, including dentatorubral-pallidoluysian atrophy (DRPLA), which is classified as a specific subtype (Durr, 2010). The most common SCAs are caused by (CAG)n expansions in a specific gene that encode expanded polyglutamine (polyQ) tracts. Some SCAs arise from a non-coding repeat expansion, and others result from point mutations or insertions/deletions in their respective gene (Pandolfo and Manto, 2013). The pathogenesis of SCAs is most commonly correlated with aggregation of mutant proteins containing polyQ, such as SCA1, SCA2, SCA3, SCA6, SCA7, SCA17, and DRPLA. In addition, some specific proteins are also implicated in non-polyQ SCAs, such as mutant KCNC3 in SCA13 (Gallego-Iradi et al., 2014). Recent studies have revealed that the PTMs of proteins correlated with neurodegenerative diseases modulate their properties, including intracellular distribution, activity, stability, aggregation, protein interaction network and clearance (Sambataro and Pennuto, 2017). Therefore, investigation into the relationship between SCAs and PTMs may shed light on SCA pathogenesis. For instance, one characteristic feature of polyQ diseases is selective neurodegeneration, yet the mechanism remains unclear. Two possible factors contributing to selective neuronal impairment are the abnormal subcellular localisation of polyQ proteins and the change in their folding and function (Sambataro and Pennuto, 2012). PTMs are shown to regulate protein properties including their intracellular localisation and functions (Sambataro and Pennuto, 2017). Therefore, understanding the PTMs in polyQ SCAs may yield important clues into mechanisms behind the selective neuronal damage. What’s more, these PTMs involved in SCA disease processes can help us discover novel therapeutic targets. Till now, many studies have investigated this topic.
Here, we review PTMs that have been proven to serve regulative roles in SCA pathogenesis; these PTMs include phosphorylation, ubiquitination, SUMOylation, proteolytic cleavage, transglutamination, acetylation and N-glycosylation. We describe how these PTMs might modulate protein characteristics and further affect the disease process (Table 2). The potential and limitation of these PTMs as therapeutic targets for SCAs are also discussed.
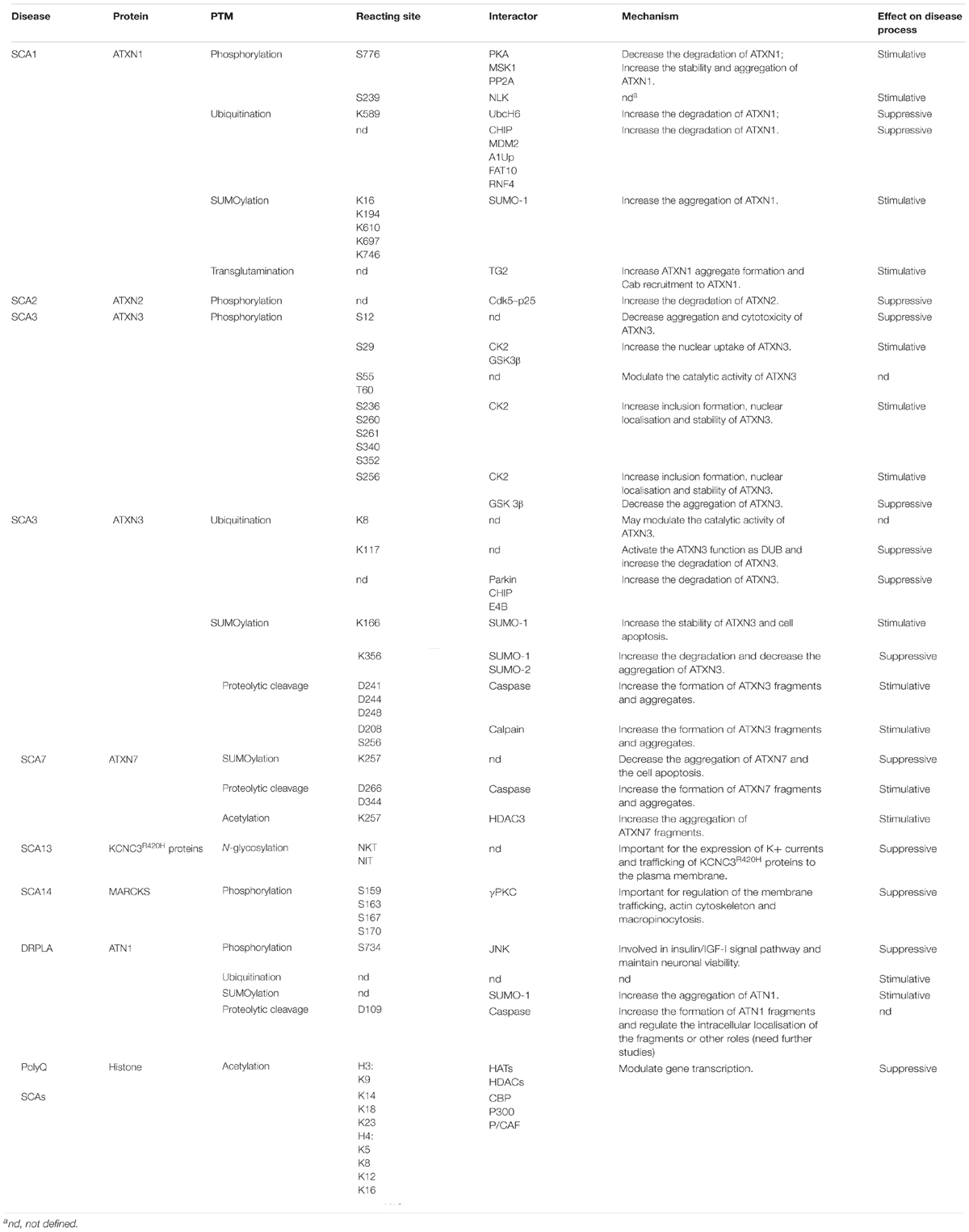
TABLE 2. Summary of post-translational modifications (PTMs) in proteins related to SCAs.
Phosphorylation
Phosphorylation is a vital PTM in which a phosphate group is added to an amino acid. This PTM can regulate protein–protein interactions, cellular metabolism, protein degradation, and enzyme reactions (Karve and Cheema, 2011). If the activity and function of the disease-causing protein are controlled by phosphorylation, phosphorylation might play a significant role in the disease.
SCA1 is caused by a (CAG)n expansion in the ataxin1 (ATXN1)-encoding gene, which results in a polyQ expansion in ATXN1. ATXN1 is a DNA-binding protein incorporating the expanded polyQ stretch, the globular ataxin1/HBP1 domain (AXH) and a nuclear localisation sequence (NLS) (Figure 1A). In addition, wild-type ATXN1 contains from 6 to 44 polyQ repeats, while the repeat number is from 39 to 83 in mutant ATXN1 (Zoghbi and Orr, 2009). At least seven phosphorylation sites have been identified in vivo in ATXN1 (Huttlin et al., 2010), and phosphorylation at serine residues 776 (S776) and 239 (S239) is believed to significantly affect SCA1 pathogenesis (Figure 1A) (Ju et al., 2013; Park et al., 2013). Phosphorylation at S776 of polyQ-expanded ATXN1 might stabilise ATXN1 by blocking its proteolysis (Orr, 2012). In addition, it also regulates the interaction between ATXN1 and other cellular proteins. S776 phosphorylation promotes binding to the cellular signalling protein family 14-3-3, which prevents pS776 dephosphorylation and protects ATXN1 from proteolysis. In addition, S776 phosphorylation inhibits ATXN1 localisation to the nucleus by masking the nuclear localisation signal, which also prevents dephosphorylation (Lai et al., 2011). S776 phosphorylation is also essential for the formation of the ATXN1-CIC complex, which is vital for ATXN1 maintenance and stabilisation; thus, this complex might be relevant to SCA1 disease progression (Ju et al., 2014). Binding to U2AF65, a constitutive component of the spliceosome, is impeded but not eliminated by the phosphorylation of S776, and this reduces ATXN1 binding to the large spliceosome machinery, leaving ATXN1 conformationally extended and vulnerable to self-association and aggregation (Menon et al., 2012). In addition, the phosphorylation status of S776 is critical to the interaction between ATXN1 and RBM17 (splicing factor RNA-binding motif protein 17), an interaction that contributes to the neuropathology of SCA1 (Lim et al., 2008). For the regulation of phosphorylation, cyclic AMP-dependent protein kinase (PKA) and mitogen- and stress-activated protein kinase-1 (MSK1) phosphorylate ATXN1 at S776 (Jorgensen et al., 2009; Park et al., 2013). In addition, the RAS-MAPK-MSK1 signalling pathway, which includes the components ERK1, ERK2, MEK2, MEK3, and MEK6, serves as an important regulator of S776 phosphorylation and ATXN1 stability (Park et al., 2013). Conversely, protein phosphatase 2A (PP2A) can dephosphorylate ATXN1-pS776, playing an important role in regulating S776 phosphorylation (Lai et al., 2011). One study suggested that ATXN1 could be phosphorylated at S239 by Nemo-like kinase (NLK), an ATXN1 interactor that modulates the toxicity of SCA1; this hypothesis is supported by NLK overexpression in a Drosophila model, which increased ATXN1 disease-related phenotypes (Ju et al., 2013). However, the specific role of S239 phosphorylation in the pathogenesis of SCA1 requires further studies.
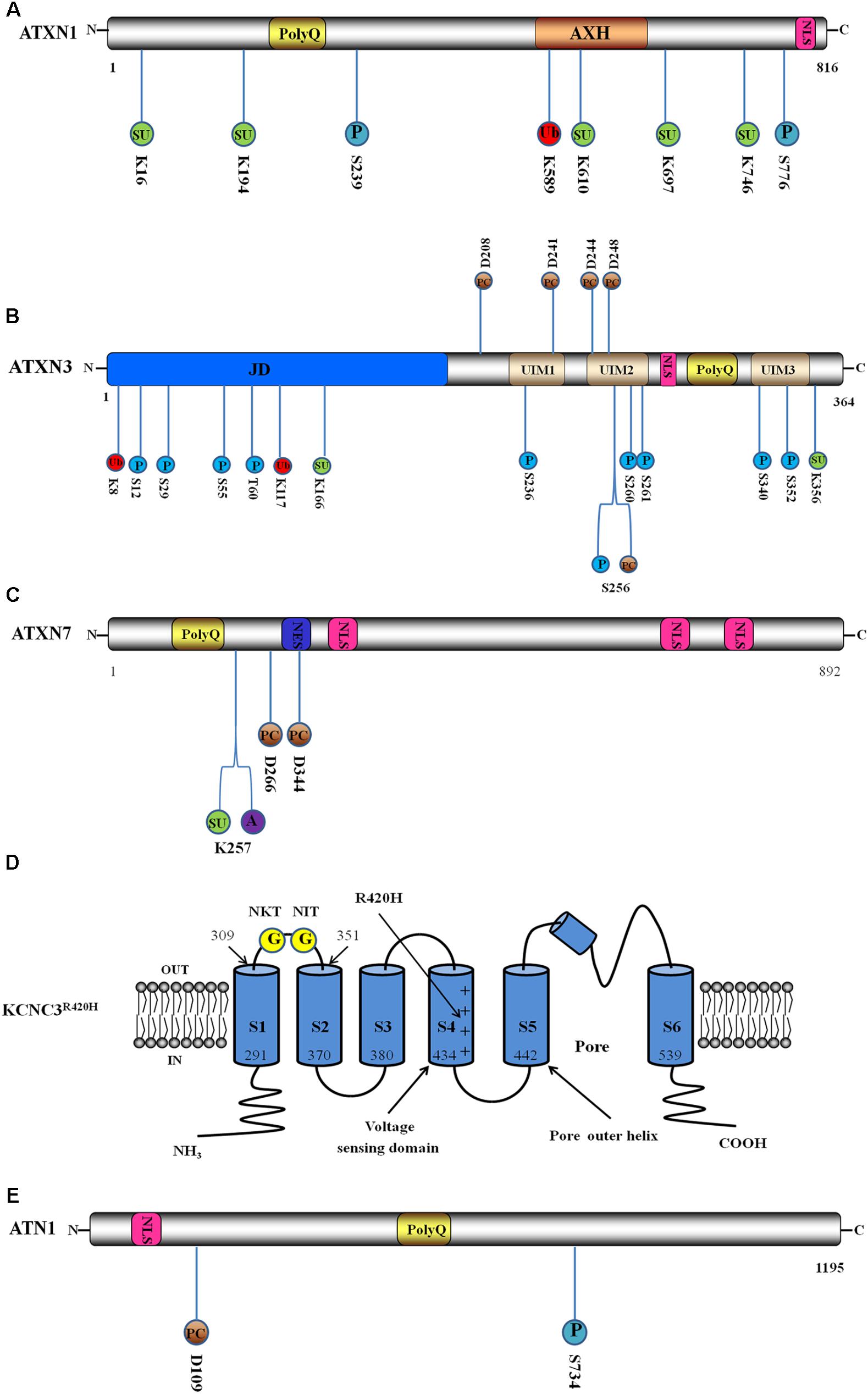
FIGURE 1. Schematic interpretation of direct pathogenesis-related mutant protein constructs and post-translational modification (PTM) sites. Blue circle stands for phosphorylation site, red circle stands for ubiquitination site, green circle stands for SUMOylation site, brown circle represents proteolytic cleavage site, purple circle represents acetylation site, and yellow circle represents N-glycosylation site. The number below or above the circle represents the position of PTM sites. (A) Domain architecture and PTM sites of ATXN1: ATXN1 incorporates the expanded polyQ stretch, the globular ataxin1/HBP1 domain (AXH) and a nuclear localisation sequence (NLS). Here are illustrated two phosphorylation sites, a ubiquitination site and five SUMOylation sites. (B) Domain structure and PTM sites of ATXN3: ATXN3 incorporates the globular Josephin domain (JD), a flexible C-terminal tail containing three ubiquitin interaction motifs (UIMs), an NLS and the expanded polyQ. Here are illustrated ten phosphorylation sites, two ubiquitination sites, two SUMOylation sites and five proteolytic cleavage sites. (C) Domain structure and PTM sites of ATXN7: ATXN7 incorporates the expanded polyQ, a nuclear export signal (NES) and three NLS. Here are shown a SUMOylation site, an acetylation site and two proteolytic cleavage sites. (D) Schematic interpretation of the Kv3.3 channel in R420H variant (KCNC3R420H) and PTM sites. Here are illustrated the six transmembrane segments and pore re-entrant loop. The numbers show the positions of the ending and beginning amino acids of S1 and S2 domains. Location of R420H-mutation is indicated by the long arrow. The two N-glycosylation sites are located in the S1–S2 extracytoplasmic loop. Adapted from Waters et al. (2006). (E) Domain structure and PTM sites of ATN1. ATN1 incorporates an NLS and the expanded polyQ. Here are shown a phosphorylation site and a proteolytic cleavage site.
SCA2, one of the nine polyQ neurodegenerative diseases, is caused by aberrant expansion of the CAG repeat in the ATXN2-encoding gene. ATXN2 is a member of the Like-Sm (LSm) protein family and contains the expanded polyQ stretch, the LSm domain, the LSm-associated domain, proline-rich domains and the PAM2 domain. ATXN2 contains from 13 to 31 polyQ stretches in healthy individuals, but SCA2 patients possess more than 31 repetitions (Magana et al., 2013). SCA2 pathogenesis is linked to ATXN2 phosphorylation by cyclin-dependent kinase 5-p25 (Cdk5–p25), which induces ATXN2 degradation (Asada et al., 2014). Thus, Cdk5–p25 may play a protective role in SCA2 pathogenesis.
SCA3 (also called Machado-Joseph Disease, MJD), is caused by CAG repeat expansion in the ATXN3-encoding gene. ATXN3 is a deubiquitinating enzyme (DUB) consisting of the globular Josephin domain (JD), a flexible C-terminal tail containing three ubiquitin interaction motifs (UIMs), an NLS and the expanded polyQ stretch. The range of polyQ is from 12 to 44 in healthy individuals but from approximately 55 to 80 in MJD patients (Li et al., 2015) (Figure 1B). Several phosphorylation sites have been identified (S12, S29, S55, T60, S236, S256, S260, S261, S340, and S352) in ATXN3: S12, S29, S55, and T60 are located in the catalytic JD (aa 8–168), which is the N-terminal domain of ATXN3; S236 is located in the first UIM; S256, S260, and S261 are located in the second UIM; and S340 and S352 are located in the third UIM (Figure 1B) (Fei et al., 2007; Tao et al., 2008; Mueller et al., 2009; Pastori et al., 2010; Matos et al., 2016; Kristensen et al., 2017). A recent study showed that phosphorylation at S12 decreased the DUB activity, aggregation and cytotoxicity of ATXN3 (Matos et al., 2016) and that this site might be a novel therapeutic target for SCA3. Protein casein kinase 2 (CK2) and glycogen synthase kinase 3β (GSK3β) phosphorylate ATXN3 at S29, which promotes ATXN3 nuclear localisation and thus contributes to the pathogenesis of SCA3 (Pastori et al., 2010). Phosphorylation of the remaining sites in the JD, S55, and T60, may modulate catalytic activity. Increased phosphorylation of S55 has been detected in polyQ-expanded ATXN3, which may contribute to disease processes; however, more studies are needed to determine a specific effect. In addition to S29, CK2 can also phosphorylate sites S236, 256, 260, 261, 340, and 352 in ATXN3, altering the inclusion formation, nuclear localisation and stability of ATXN3 (Tao et al., 2008; Mueller et al., 2009), which indicates that CK2-dependent phosphorylation stimulates SCA3 pathogenesis. However, phosphorylation at S256 by GSK 3β inhibits ATXN3 aggregation, which plays a protective role in SCA3 pathophysiology (Fei et al., 2007). We conclude that phosphorylation of the same sites by different kinases produces different effects on protein activities.
SCA14 is reported to arise from missense mutations in the gene encoding protein kinase Cγ (γPKC) (Chen et al., 2005), which can be activated by TPA (12-O-tetradecanoylphorbol 13-acetate) and regulates membrane trafficking, the actin cytoskeleton and macropinocytosis. A mutant form of γPKC that is linked to SCA14 cannot perform these regulatory activities because of reduced phosphorylation of its plasma membrane substrate MARCKS (myristoylated alanine-rich C-kinase substrate) (Yamamoto et al., 2014). MARCKS (Figure 2A) contains three conserved regions: a myristoylated N-terminal domain, an MH2 domain and a phosphorylation site domain (PSD) with four phosphorylation sites (S159, S163, S167, and S170) (Fong et al., 2017). Aberrant phosphorylation of MARCKS may be involved in the pathogenesis of SCA14, and further studies are required.
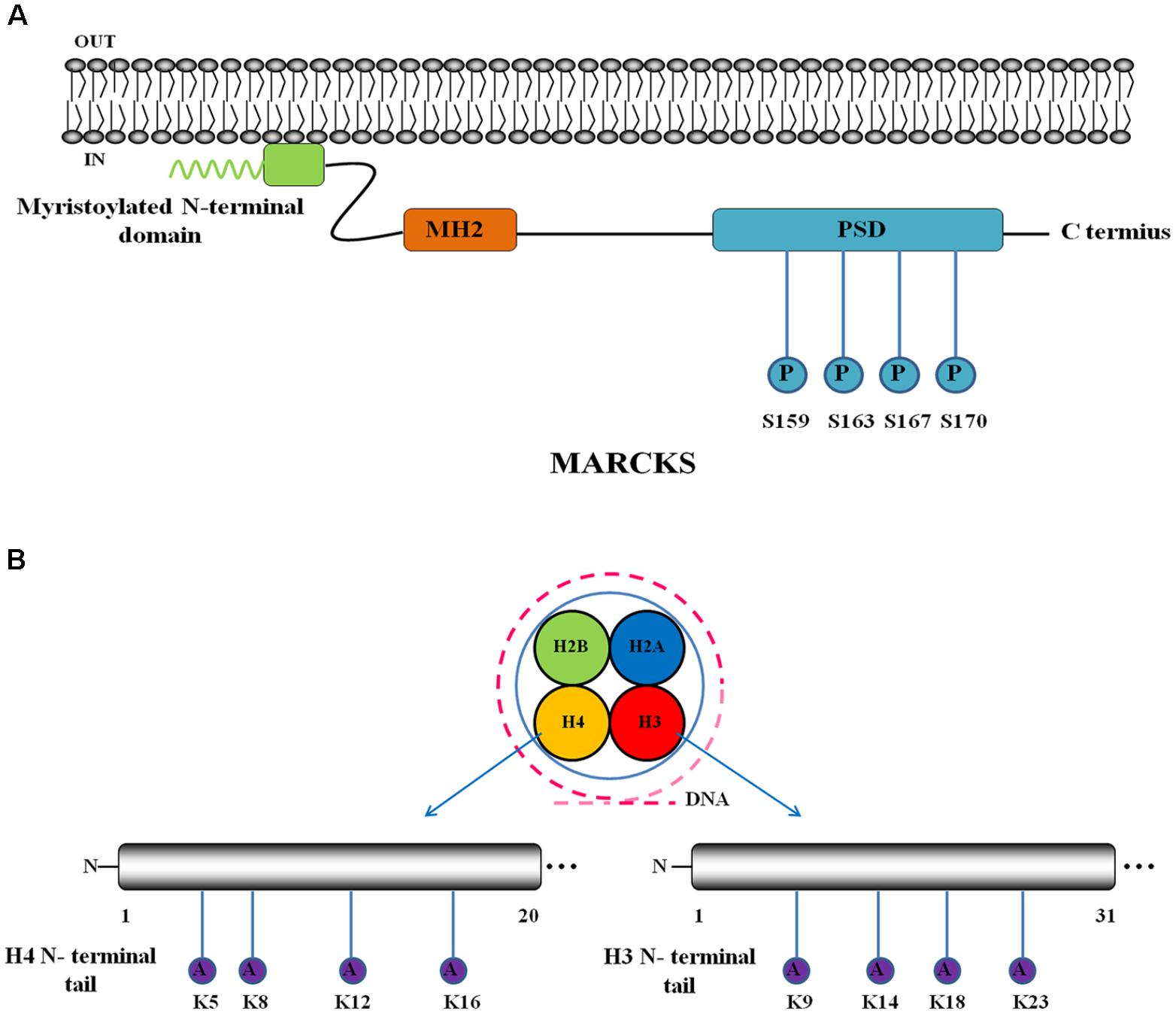
FIGURE 2. Schematic interpretation of non-direct pathogenesis-related protein constructs and post-translational modification (PTM) sites. Blue circle stands for phosphorylation site and purple circle represents acetylation. (A) Domain structure and PTM sites of MARCKS: MARCKS incorporates a myristoylated N-terminal domain, an MH2 domain and a phosphorylation site domain (PSD) with four phosphorylation sites. Adapted from Fong et al. (2017). (B) Schematic interpretation of PTM sites in histone N-terminal tails: H3 and H4 have four acetylation sites in their N-terminal tails respectively.
Dentatorubral-pallidoluysian atrophy is specific subtype of SCA caused by an expansion of the CAG trinucleotide repeat in the atrophin-1 (ATN1) gene; the number of repeats ranges from 3 to 36 in healthy individuals to 49 to 88 copies in DRPLA patients (Schols et al., 2004; Durr, 2010). ATN1 incorporates an NLS and the expanded polyQ tract (Okamura-Oho et al., 1999) (Figure 1E). In vitro experiments provided evidence of ATN1 phosphorylation, a process primarily mediated by c-Jun NH2-terminal kinase (JNK) at S734 (Figure 1E). However, expanded polyQ might inhibit the affinity of JNK for ATN1 and cause that ATN1 is slowly phosphorylated in DRPLA patients. Aberrant phosphorylation of ATN1 might delay the insulin/IGF-I signalling pathway, a pathway which ATN1 is involved in and is vital to maintaining neuronal viability (Okamura-Oho et al., 1999; Okamura-Oho et al., 2003).
In conclusion, phosphorylation can alter the stability, degradation, aggregation or nuclear uptake of mutant proteins and thus may be protective or toxic for SCA pathogenesis. In addition, the phosphorylation status of some specific proteins involved in SCAs pathogenesis is critical to pivotal pathways; for example, phosphorylation of MARCKS is essential for membrane trafficking, actin cytoskeleton regulation and micropinocytosis. Based on such roles, some exploratory translational therapies related to phosphorylation in SCAs have been examined. Park et al. (2013) found that downregulating some components of the RAS-MAPK-MSK1 signalling pathway, such as RAS, ERK1, ERK2, and MAPK, suppresses ATXN1 levels and neurotoxicity in Drosophila and mouse models. Hearst et al. (2014) designed a therapeutic polypeptide, Synb1-ELP-PKI, which contains a thermally responsive elastin-like peptide (ELP) carrier, a PKA inhibitory peptide (PKI) and a cell-penetrating peptide (Synb1), to inhibit PKA based on phosphorylation at S776 by PKA. They found that treatment with Synb1-ELP-PKI decreased phosphorylation on S776 and suppressed the formation of intranuclear inclusions containing mutant ATXN1 in cell culture. In addition, this peptide was verified to improve SCA1 Purkinje cell morphology in cerebellar slice cultures, and the validity of intranasal delivery of the peptide to mouse brains has been demonstrated. However, most of these explorations are still in primary in vitro-level or model-level stages, and clinical drug trials are required before these therapeutic explorations become reliable. In addition, how to ensure accurate regulation on a specific residue is still a challenge due to the diverse effects of phosphorylation on different residues.
Ubiquitination
Ubiquitination is a reversible PTM that occurs by forming an isopeptide bond between the K residues of a target protein and the C-terminal glycine of ubiquitin (Faggiano et al., 2015) and is regulated by numerous related enzymes, including the ubiquitin-activating enzyme (E1), the ubiquitin-conjugating enzyme (E2), the ubiquitin ligase (E3), and DUBs. An additional ubiquitylation enzyme has been recently identified (Koegl et al., 1999): E4, a novel conjugation factor known as yeast Ufd2, is required for polyubiquitin chain assembly in conjunction with E1, E2, and E3. Ubiquitination labels target proteins for degradation primarily by the ubiquitin proteasome system (UPS) (Hershko and Ciechanover, 1998) and autophagy, which have important regulatory functions in protein homeostasis. Because impaired protein degradation is always involved in SCA pathogenesis, ubiquitination is also significant in SCA pathogenesis.
A proteasome inhibition study (Cummings et al., 1999) confirmed the role of the UPS in ATXN1 degradation; inhibiting proteasome increases ATXN1 aggregation, and mutant ATXN1 is more resistant to ubiquitin-mediated degradation. UbcH6, a class III ubiquitin-conjugating enzyme (E2), ubiquitinates polyQ-mutated ATXN1 and enhances its subsequent degradation, which can uniquely be accomplished in the absence of E3 ligases (Hong et al., 2008), whereas most E3 ligases are indispensable for the ubiquitination function of the E2 conjugating enzyme. UbcH6 interacts with ATXN1 at K589 (Figure 1A) in the AXH domain of ATXN1, and a lysine 589 to arginine substitution impairs degradation, thus enhancing ATXN1 aggregation (Kang et al., 2015). Furthermore, the function of ATXN1 as a transcriptional repressor was suppressed by UbcH6 by modulating ATXN1 degradation (Lee et al., 2008). Because the repression activity of ATXN1 has a vital role in SCA1 pathogenesis, the effect of UbcH6 on transcriptional repression might be a potential therapeutic target. CHIP (C-terminal of Hsc70 interacting protein), which is also known as the STIP1 homology and U-box containing protein 1 (STUB1) (Shi et al., 2013), acts as a molecular co-chaperone through the TPR domain and as an E3 ubiquitin ligase through the U-box domain, linking the cellular folding-refolding machinery with the UPS (McDonough and Patterson, 2003). CHIP interacts with the molecular chaperone Hsp70, promoting Hsp70 induction and removing excess Hsp70 (Qian et al., 2006). This interaction enhances the ubiquitylation and degradation of misfolded proteins through the UPS (McDonough and Patterson, 2003). CHIP directly interacts with and localises to ATXN1 in human SCA1 neurons, where it ubiquitinates mutant ATXN1 in a chaperone-dependent manner, inducing degradation. These results demonstrate the therapeutic potential of CHIP for SCA1. The Notch intracellular domain (NICD) might also enhance the degradation of ATXN1 through MDM2-mediated ubiquitination (Kang et al., 2017). In addition, some ubiquitin-like proteins, such as A1Up and FAT10, potentially regulate the proteasomal degradation of mutant ATXN1 (Davidson et al., 2000; Hipp et al., 2005), but the specific mechanism still requires further exploration. One study (Guo et al., 2014) has shown that PQC systems, including PML (promyelocytic leukaemia protein) and RNF4 (a ubiquitin ligase), mediate the degradation of misfolded proteins including ATXN1. In this system, PML binds to the misfolded protein and conjugates it to poly-SUMO2/3 chains, and RNF4 ubiquitinates SUMOylated misfolded proteins, targeting them to the UPS. This system serves as another potential therapeutic target for neurodegenerative diseases, although its role in other SCAs still requires further verification.
ATXN3, a DUB containing the ubiquitin-binding UIM and a ubiquitin protease domain, suppresses polyQ neurotoxicity through degradation, although this function is partially impaired in pathogenic ATXN3 (Warrick et al., 2005). Ubiquitination has been reported to promote the DUB activity of ATXN3, which further contributes to Ub-dependent homoeostasis and neuroprotection in SCA3 (Todi et al., 2009). Parkin, an E3 ubiquitin ligase, promotes the ubiquitination and degradation of ATXN3, which is enhanced by Hsp70. Furthermore, parkin improves proteasome activity after impairment by misfolded polyQ proteins (Tsai et al., 2003), indicating that parkin might serve a protective role in SCA3. CHIP also promotes the degradation of ATXN3 by enhancing its ubiquitination, which would be increased by Hsp70 (Jana et al., 2005). Another ubiquitin-related protein is the ubiquitin chain assembly factor E4B. E4B also promotes the degradation of polyQ-expanded ATXN3 by enhancing ATXN3 polyubiquitination and conjugation to VCP (an AAA-type ATPase) to modulate a shift between ATXN3 and the proteasome. Consequently, the targeted expression of parkin, CHIP and E4B could be a novel therapeutic strategy for SCA3. To date, several sites on ATXN3 have been found to be ubiquitinated in mammalian cells (K8, K117, K190, K200, K206, and K29, with K117 and K200 being the primary sites) (Figure 1B), and there are no significant differences between the ubiquitination of normal and mutant ATXN3 except that ubiquitination on K8 is enhanced on mutant ATXN3, which might affect catalytic activity. In addition, the ubiquitination of K200 was not detected on mutant ATXN3 (Todi et al., 2010; Kristensen et al., 2017). The ubiquitination of ATXN3, primarily at site K117, activates the DUB function of ATXN3 through a conformational switch, which improves the editing of Ub chains on substrates. This activates Ub-dependent pathways, including the proteasomal degradation of misfolded proteins, and improves neuroprotective activity (Todi et al., 2010; Faggiano et al., 2015). Interestingly, Blount et al. (2014) found that ubiquitination is not absolutely necessary for ATXN3 proteasomal turnover, which is modulated by ubiquitin-binding Site 2 (UbS2) on the amino terminus. UbS2 might promote the interaction between ATXN3 and Rad23A/B, the proteasome-associated proteins, and this interaction might block ATXN3 turnover. Thus, occupation of the UbS2 binding site, preventing an interaction with Rad23A/B, would promote the proteasomal degradation of ATXN3 and further suppress neurotoxicity, suggesting a novel target for SCA3 therapy.
As a common process in polyQ neurodegenerative disorders, ubiquitination is commonly believed to be involved in DRPLA pathogenesis. In DRPLA brains, ATN1 forms an abnormal complex that is pathologically ubiquitinated to form neuronal cytoplasmic inclusions. In addition, this ubiquitination is discovered selectively in affected lesions (Yazawa et al., 1999). Through immunoblots of brain tissues from DRPLA patients, pathological ubiquitination is found to correlate with the size of an expanded glutamine repeat in ATN1 and the onset of symptoms. Thus, pathological ubiquitination of the ATN1 complex plays an important role in DRPLA pathogenesis. Interestingly, this abnormal complex is insoluble in water but soluble in SDS and reducing agents, unlike the characteristic solubility of abnormally ubiquitinated complexes in Alzheimer’s disease. This study reveals a unique pathological mechanism for DRPLA in which an insoluble complex is formed by spontaneous accumulation rather than by a qualitative change in water solubility (Yazawa, 1999).
Overall, an in-depth study of the aberrant ubiquitination in SCAs can facilitate a better understanding of the pathogenesis since UPS is the main pathway for protein clearance. Besides, targeting the various chaperons involved in ubiquitination to upregulate the ubiquitination of mutant proteins in SCAs may be a protective therapeutic strategy. Al-Ramahi et al. (2006) have demonstrated that CHIP overexpression lessens the toxicity induced by mutant ATXN1 in Drosophila models of SCA1 due to upregulation of ATXN1 ubiquitination. E4B has been verified to suppress the neurotoxicity of polyQ-expanded ATXN3 in Drosophila models of SCA3 by promoting the ubiquitination and degradation of mutant ATXN3 (Matsumoto et al., 2004). The targeted expression of specific ubiquitination interactors such as CHIP and E4B suggests potential gene therapy routes to treat SCAs.
SUMOylation
SUMOylation which is a vital regulator of the proteostasis is the binding of SUMO (small ubiquitin-like modifier) proteins, including SUMO-1, SUMO-2, SUMO-3, and SUMO-4, to the lysine (K) residues of target proteins. SUMOylation can modulate proteostasis independently and usually enhances protein stability; however, the interaction between SUMOylation and ubiquitination can be cooperative or competitive and is a primary regulator of proteostasis (Liebelt and Vertegaal, 2016). Protein SUMOylation mediates the pathogenesis of multiple diseases, including neurodegenerative diseases (Yang et al., 2017); for example, the SUMOylation of HTT protein by SUMO-1 enhances its stability and contributes to the neurodegeneration observed in Huntington’s disease (Steffan et al., 2004). Here, we review the role of SUMOylation in the pathogenesis of SCAs.
In ATXN1, there are five lysine residues that are SUMOylated: K16, K194, K610, K697, and K746 (Figure 1A). The SUMOylation of ATXN1 is likely modulated by multiple factors, such as phosphorylation of Ser776, polyQ length, nuclear localisation and the self-association region (Riley et al., 2005). The SUMOylation of ATXN1 by SUMO-1 promotes ATXN1 aggregation, thus enhancing neurodegeneration in SCA1. Furthermore, incubation with hydrogen peroxide to stimulate oxidative stress accelerates SUMO conjugation to ATXN1 and enhances aggregation as a result, which could be modulated by the JNK pathway (Ryu et al., 2010).
In SCA3, ATXN3 can be SUMOylated at site K166 (Zhou et al., 2013) (Figure 1B) by SUMO-1, which would enhance mutant ATXN3 stability without affecting aggregate formation. In addition, ATXN3 SUMOylation by SUMO-1 on site K166 also increases apoptosis in SCA3; therefore, SUMOylation by SUMO-1 might stimulate SCA3 pathogenesis through both effects described above. In addition to K166, ATXN3 can also be SUMOylated at site K356 (Figure 1B) by SUMO-1 and SUMO-2. SUMOylation at K356 (Almeida et al., 2015) reduces the formation of ATXN3 aggregates and strengthens the association with the AAA+ ATPase p97, which can promote p97-mediated ER-associated protein degradation. Consequently, SUMOylation at K356 impedes ATXN3 aggregation through two pathways, further exerting a protective influence during SCA3 pathogenesis. We observed different effects when different ATXN3 sites were SUMOylated.
SCA7 is caused by the aberrant expansion of CAG repeat in the first exon of the ATXN7-encoding gene (David et al., 1997). ATXN7 is a subunit of the DUB module in the Spt-AdaGcn5-acetyltransferase (SAGA) complex, and this complex can regulate histone acetylation and ubiquitination to modulate gene expression (Koutelou et al., 2010). ATXN7 incorporates the expanded polyQ, a nuclear export signal (NES) and three NLS, in which the range of polyQ repeats is from 37 to 130 in SCA7 patients but from 7 to 35 in healthy individuals (David et al., 1998; Young et al., 2007) (Figure 1C). SUMOylation on K257 (Figure 1C) reduces the toxic aggregate production and apoptosis caused by expanded polyQ-ATXN7 without affecting ATXN7 subcellular localisation (Janer et al., 2010). Therefore, changing the SUMOylation status of SCA7 might reduce SCA7 pathogenesis.
SUMO-1 has been verified to co-localise with polyQ aggregates in both the DRPLA cellular model and brain tissue. Moreover, co-transfection of SUMO-1 with polyQ ATN1 dramatically enhances the number of intranuclear inclusions (NIs) and, consequently, neuronal apoptosis, whereas a non-conjugating mutant of SUMO-1 shows the opposite effect. These results reveal that SUMO-1 is involved in the DRPLA process and promotes polyQ aggregate formation and cell death (Terashima et al., 2002). Further exploration is needed to elucidate the specific mechanisms involved in the association between SUMOylation and DRPLA.
Serving as a critical regulator of proteostasis, SUMOylation can change the aggregation formation and degradation of mutant proteins in SCAs and whether the regulation is promotion or inhibition is related to the different amino acid residues. What’s more, SUMOylation has many more roles, such as regulating genome organisation, protein-DNA binding and DNA repair (Hickey et al., 2012). Whether these roles are involved in SCA pathogenesis still needs further investigation, and this complexity poses a barrier to transferring SUMOylation roles into therapeutic targets.
Proteolytic Cleavage
Proteolytic cleavage is a type of PTM with important roles in neurodegenerative diseases. This process is catalysed by special proteases, such as the caspase and calpain families, and removes or separates the specific parts of protein sequences, turning long proteins into short fragments. Increasing evidence has suggested that fragments containing polyQ expansions are more likely to aggregate and cause toxicity than full-length polyQ-expanded proteins; this is known as the toxic fragment hypothesis (Matos et al., 2017). Here, we primarily discuss SCA3, SCA7, and DRPLA, in which stable proteolytic fragments are observed.
Schmidt et al. (1998) discovered that NIs in neuronal SCA3 patient cells are created by fragments of pathogenic proteins, implying that ATXN3 undergoes proteolytic cleavage prior to its transport into the nucleus. ATXN3, a protein with a molecular weight of approximately 42 kDa, is proteolytically modified by caspases and calpains (Berke et al., 2004; Weber et al., 2017). D241, D244, and/or D248 are the three most relevant caspase cleavage sites in ATXN3 (Evers et al., 2014), and the two major calpain cleavage sites occur at amino acids D208 and S256 (Weber et al., 2017) (Figure 1B). Short polyQ-expanded fragments of ATXN3 that aggregate are essential factors in the pathogenesis of SCA3 (Schaffar et al., 2004). The toxic polyQ-expanded fragments led to increased mitochondrial fission, decreased mitochondrial membrane potential and increased reactive oxygen species, causing cell death (Hsu et al., 2017). In addition, the structure of full-length ATXN3 can be distorted by polyQ-expanded fragments, causing them to interact with each other and form co-aggregates (Haacke et al., 2006). These observations led to the current consensus that ATXN3 fragments are more toxic than full-length ATXN3. Caspases involved in proteolytic cleavage are initially activated after signalling events. Then, the activated caspases cleave their target substrates, such as full-length ATXN3, after an aspartate residue (McIlwain et al., 2015). One study has shown that inducing caspase activity leads to increased aggregation and cleavage of a C-terminal 28 kDa ATXN3 fragment in cells expressing mutant ATXN3, which facilitates neurodegeneration (Liman et al., 2014). However, studies suggest that calpains have a greater influence on aggregate formation than caspases (Koch et al., 2011; Evers et al., 2014). When the endogenous calpain inhibitor calpastatin was knocked out to enhance calpain activity in SCA3 mice, increased aggregation and accelerated neurodegeneration were observed in the cerebellum (Hubener et al., 2013). Similarly, a study overexpressing the endogenous calpain inhibitor calpastatin to inhibit calpain activity in SCA3 mice showed decreased aggregation and nuclear toxicity (Simoes et al., 2012). Oral administration of the calpain inhibitor BDA-410 has the same outcome (Simoes et al., 2014). Thus, we conclude that calpains contribute significantly to the ATXN3 cleavage pathway.
ATXN7, a member of the STAGA complex, is involved in transcriptional regulation (Helmlinger et al., 2004). The specificity of neuronal cell death in SCAs may be correlated with caspase expression (Hermel et al., 2004). ATXN7 is responsible for the recruitment and activation of caspase-7 in the nucleus, which, in turn, cleaves ATXN7, contributing to the formation of Nis (Young et al., 2007). PolyQ-expanded proteins can activate caspase-7 in vivo, which cleaves ATXN7 at the D266 and D344 sites (Figure 1C) and generates an ATXN7 fragment that is more toxic than the full-length protein (Young et al., 2007; Guyenet et al., 2015). In addition to the toxic fragments arising from the polyQ-expanded protein, caspase-3 cleavage of amyloid precursor-like protein 2 (APLP2) may also contribute to SCA7 pathogenesis by producing intracellular C-terminal domains that might enhance aberrant transcriptional regulation (Takahashi-Fujigasaki et al., 2011). Thus, we conclude that caspase cleavage is a critical event in ATXN7 neurotoxicity and the pathogenesis of SCA7. In addition, the inhibition of caspase cleavage may provide a positive avenue for SCA7 treatment.
Multiple studies have shown that the caspase family is associated with DRPLA protein cleavage. The C-terminal polyQ-expanded fragment produced by proteolytic caspase-3 cleavage at D109 of ATN1 (Figure 1E) in vitro is responsible for the formation of aggregates and neurodegeneration (Miyashita et al., 1998; Ellerby et al., 1999). In DRPLA disease models and patient brain tissue, it has been demonstrated that ATN1 proteolytic processing modulates the intracellular localisation of fragments, which are also involved in DRPLA pathogenesis (Suzuki and Yazawa, 2011). Interestingly, the selective accumulation of the C-terminal ATN1 fragment is enhanced by the inhibition of caspases in COS-7 cells. This may arise from additional roles of caspases; for example, they are involved in the regulation of ATN1 (Suzuki et al., 2010). Further exploration is required to determine the specific mechanisms of caspases in DRPLA pathogenesis.
In brief, proteolytic cleavage could generate short polyQ-expanded fragments, causing more aggregation and toxicity in cells. Based on the existing studies, several therapeutic strategies could be considered. For one thing, altering the specific cleavage site of disease proteins could offer opportunities to reduce toxicity, thus affecting the neurodegenerative process. In SCA3, the identification of the ATXN3 cleavage sites D208 and D256 provided a potential technique by skipping exons containing the calpain recognition motif (Weber et al., 2017). For another, treatment with caspase and calpain inhibitors may also demonstrate their therapeutic potential. Calpastatin, a calpain inhibitor, decreases the nuclear uptake and aggregation of mutant ATXN3, delaying neurodegeneration in SCA3 mouse models (Simoes et al., 2012). However, this result does not coincide with the result of caspase inhibitors used in DRPLA disease models, suggesting that another signal pathway of caspases and calpains is involved.
Transglutamination
Transglutamination is a PTM that results in an acyl shift between the γ-carboxamide group of a polypeptide-bound glutamine and the ε-amino group of a lysine residue in another protein, which is catalysed by transglutaminases (TGs) (Violante et al., 2001). TG families include three types of TGs (Lorand and Graham, 2003): papain-like TGs, bacterial toxin TGs and protein disulfide isomerase-like TGs. The primary function of mammalian TG is to modulate the formation of cross links between proteins for the stabilisation of biological structures (Aeschlimann and Thomazy, 2000). Because expanded polyQ tracts, which provide substrates for TGs, account for most common SCAs, TGs may play important roles in SCA pathogenesis.
Tissue-specific TGs have been demonstrated to exist widely in the rat and human CNS, such as in the brain, the cerebellum and in different classes of neurons (Maggio et al., 2001). TGs have been reported to facilitate mutant protein aggregation in SCAs, and the integration of polyQ AR with TG contributes to proteasome dysfunction (Kahlem et al., 1998; Mandrusiak et al., 2003); thus, TG can promote neurodegeneration in SCAs. ATXN1 has been demonstrated to be a substrate of TG type 2 (TG2), and SCA1 transgenic mice have showed elevated nuclear TG2 in the Purkinje cells of the cerebellum (D’Souza et al., 2006). In addition, TG2 mediates the recruitment of the calcium binding protein calbindin-D28k (CaB) to ATXN1 (Vig et al., 2007). The interaction between CaB and myoinositol monophosphatase (IMPase) is necessary for inositol-1, 4,5-trisphosphate (IP3)-mediated Ca2+ signalling, which is significantly involved in synaptic integration and plasticity in the peripheral nervous system (Schmidt et al., 2005). The recruitment of CaB to ATXN1 might decrease CaB levels and impede IP3 signalling, which contributes to SCA1 neurodegeneration.
To conclude, transglutamination is an important driver of SCA1 pathogenesis, possibly by modulating aggregate formation and CaB recruitment. Given the enzyme-substrate relationship between TG2 and ATXN1, further exploration on specific mechanism is needed as well as the effects of transglutamination in other SCAs.
Acetylation
Acetylation is the process of covalently binding an acetyl group to a lysine residue on a protein and is regulated by two groups of enzymes: histone deacetylases (HDACs) and histone acetyltransferases (HATs) (Park et al., 2015). There have been 18 HDAC subtypes identified in humans, which include Class I HDACs (HDAC1, 2, 3 and 8), class IIa HDACs (HDAC4, 5, 7 and 9), class IIb HDACs (HDAC6 and 10), class III HDACs and class IV HDACs (Xu et al., 2007). Many studies show that acetylation plays diverse roles in neurodegenerative diseases.
Epigenetic dysregulation is an important characteristic of polyQ diseases (Cohen-Carmon and Meshorer, 2012). Nucleosome is the basic unit of chromatin, which incorporates 147 base pairs of DNA enwinding an octamer of core histones (H2A, H2B, H3, and H4) (Quina et al., 2006). The equilibrium between the acetylation and deacetylation of histones, which occurs at the N-terminal tails of histones [H3 is acetylated at K9, K14, K18, and K23; H4 is acetylated at K5, K8, K12, and K16 (Carrozza et al., 2003)], modulates gene transcription by changing the interaction between histones and DNA as well as nuclear proteins (Figure 2B) (Kornberg and Lorch, 1999). The mutant proteins containing expanded polyQ can suppress the histone acetylase activities of some acetylation-related interactors, such as CREB-binding protein (CBP), p300 and p300/CBP-associated factor (P/CAF), through direct interaction with their acetyltransferase domains, which further inhibits histone acetylation and leads to epigenetic dysregulation (Hughes et al., 2001; Steffan et al., 2001). Aberrant histone acetylation and epigenetic dysregulation have been verified in a variety of SCAs. Among them, ATXN1 is connected to leucine-rich acidic nuclear protein (LANP), an ATXN1-binding co-repressor, and can inhibit the activity of the histone acetyltransferase CBP (Cvetanovic et al., 2012). ATXN3 has been verified to inhibit the acetylase activities of CBP and p300 in vitro through protein–protein interactions (Li et al., 2002). ATXN7 is a constituent of the SAGA complex, which acetylates targeted histones. Furthermore, mutant ATXN7 disrupts structural integrity and interferes with the substrate targeting of the SAGA complex, further affecting histone acetylation (McCullough and Grant, 2010). Therefore, reversing histone hypoacetylation by treating patients with HDAC inhibitors might alleviate neurodegeneration symptoms.
In addition to the acetylation of histones, acetylation at the K257 residue of ATXN7 (Figure 1C) enhances the accumulation of the caspase-7 cleavage product by inhibiting protein turnover, and accumulation of this fragment promotes SCA7 pathogenesis (Mookerjee et al., 2009). HDAC3 increases K257 acetylation through a direct protein–protein interaction with ATXN3 instead of through deacetylase activity, which contributes to SCA7 disease progression (Duncan et al., 2013). Thus, inhibiting the acetylation of ATXN7 by inhibiting HDAC3 may slow SCA7 pathogenesis.
Taken together, abnormal histone acetylation might partially contribute to the pathogenesis of SCAs. The major mechanism is that polyQ expanded proteins suppress histone acetylase activities, leading to histone hypoacetylation. Therefore, altering the decreased expression of acetylated histones could be an effective therapeutic target in SCAs. In-depth studies on HDAC inhibitors have developed. There are several HDAC inhibitors, including sodium butyrate (SB), suberoylanilide hydroxamic acid (SAHA), and valproic acid (VPA), which have been shown to reverse histone hypoacetylation and improve motor performance in DRPLA, HD, and SCA3 mouse models (Ying et al., 2006; Mielcarek et al., 2011; Yi et al., 2013; Long et al., 2014). The safety and validity of VPA have been verified in SCA3 patients in a clinical trial, which showed that treatment with VPA improved patient locomotor function at a dose that patients could tolerate (Lei et al., 2016). However, the complex roles of acetylation in different proteins in SCA pathogenesis should be considered; thus, the targeted accuracy is truly essential.
N-Glycosylation
N-Glycosylation, one of many glycosylation patterns, is another PTM related to SCA disease progression. The N-linked protein glycosylation process contains two phases, which occur in the ER and the Golgi apparatus (Marquardt and Denecke, 2003). The first phase is the transfer of a preassembled oligosaccharide to the asparagine of the motif Asn-X-Ser/Thr [the common glycosylation site, X, can be any amino acid but proline (Gavel and von Heijne, 1990)] of a protein in the ER, and this process is accomplished by oligosaccharyl transferase (OST), a multimeric enzyme complex. In the second phase, the removal of several monosaccharides and the addition of several units, including GlcNAc, sialic acid, galactose and fucose residues, occur in the ER and Golgi. A variety of enzymes are involved in the second phase, including mannosidases (I, II, III), galactosyltransferase, and sialyltransferase. N-linked protein glycosylation affects multiple cellular pathways, such as protein folding, quality control, degradation, and secretion (Marquardt and Denecke, 2003; Freeze and Aebi, 2005).
SCA13 is an infrequent autosomal dominant cerebellar ataxia caused by point mutations in KCNC3, which encodes the Kv3.3 voltage-gated potassium channel, and its symptoms include intellectual disability, seizure and ataxia. The Kv3.3 channel, a 757-amino-acid protein, contains six transmembrane segments and a pore re-entrant loop. Among these segments, the first four transmembrane segments (S1–S4) form the voltage sensor domain and the last two segments (S5–S6) compose the ion-selective pore (Figure 1D). In addition, this channel could facilitate the firing of vast action potentials per unit time in neurons (Waters et al., 2006). To date, several variants have been described, including the R420H variant, the R423H variant and the F448L variant (Figueroa et al., 2010). Two conserved N-glycosylation sites have been confirmed on neural Kv3.3 channels and are located in the S1–S2 extra-cytoplasmic loop: the NKT amino acid residues, located from 321 to 323; and the NIT amino acid residues, located from 337 to 339 (Figure 1D). The N-glycosylation status of Kv3.3 channels is thought to affect K+ currents at the neuronal surface, which may affect neurodegeneration (Cartwright et al., 2007). Gallego-Iradi et al. (2014) detected aberrant N-glycosylation of the Kv3.3 channel in the R420H variant (KCNC3R420H). They also found that KCNC3R420H proteins were retained in the Golgi and exhibited limited trafficking to the plasma membrane because protein glycosylation is essential for transmission through the ER and Golgi. Aberrant Golgi and cellular morphology arise from this, which might be a cellular manifestation of SCA13 pathogenesis.
Overall, the aberrant N-glycosylation of KCNC3R420H can result in the abnormal K+ currents of neurons and the reduced membrane expression of KCNC3R420H along with the abnormal Golgi and cellular morphology. Studying N-glycosylation may help to explain the molecular mechanism behind SCA13 manifestations.
Conclusion
We reviewed multiple PTMs that affect the pathogenesis of SCAs through various mechanisms (Table 2). By regulating protein homeostasis, including protein interactions and the subcellular distribution, aggregation, stability and clearance of proteins, PTMs play significant and complicated roles in the regulation of SCA disease development. For instance, PTMs reducing the stability and aggregation of mutant proteins or modulating gene transcription, can generate protective influences on pathogenesis, such as the phosphorylation of ATXN2 by Cdk5–p25 and the ubiquitination of ATXN1. On the other hand, some PTMs contribute to SCA pathogenesis by promoting the stability and aggregation of mutant proteins. Phosphorylation at S776 of polyQ-expanded ATXN1 and proteolytic cleavage at D266 and D344 of ATXN7 could serve as examples. Due to the diversity of roles in SCAs, the regulation of PTMs uncovers novel potential therapeutic targets. PTMs are regulated through multiple cellular signalling pathways involving various molecules (Ju et al., 2014), such as kinases, ubiquitin ligases, phosphatases, transcription and splicing factors; therefore, therapies targeting PTMs by interfering with these pathways could potentially relieve neurodegeneration in SCAs. Massive efforts have been put into achieving this aim by silencing the pathways that induce PTMs stimulating SCA pathogenesis or activating the pathways responsible for PTMs that suppress SCA pathogenesis. For instance, inhibiting the phosphorylation-relevant pathways by downregulating the interactors RAS, ERK1, ERK2, and MAPK is proven to suppress ATXN1 levels and neurotoxicity in Drosophila and mouse models (Park et al., 2013). VPA, an HDAC inhibitor that reverses histone hypoacetylation, has been reported as a well-known therapeutic candidate in SCA3 (Lei et al., 2016). Although substantial studies have verified the validity of these therapeutic strategies in cells and animal models, which yield hope for clinical application, the clinical safety and efficacy of these agents might still be a huge problem due to the inconsistency among in vitro, in vivo, and clinical trials. In addition, due to the possible different outcomes arising from the same PTM at different sites, how to ensure target accuracy is still a big challenge. Furthermore, inhibiting or activating the distinct molecules responsible for PTMs mainly aim to regulate different downstream pathways, playing a destructive or protective role in SCAs. Therefore, none of them can relieve the SCAs from genome levels radically, such as gene editing. This means that even if the feasibility of these therapies is validated in clinical trials, patients with SCAs will have to take lifelong medication. Nevertheless, the deeper understanding of PTMs mechanisms in SCAs could provide us more efficient therapeutic avenues to treat SCAs, and the therapies at PTM levels are promising as well as challenging and thus are worth pursuing further.
Author Contributions
LW and KX made substantial contributions to conception and design of the review article and drafted the manuscript. ZC, BT, and HJ revised the manuscript. HJ agreed to be accountable for all aspects of the work. All authors read and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (Nos. 81471156 and 81771231 to HJ; Nos. 81430023 and 81130021 to BT), the National Key Research and Development Program of China (Nos. 2016YFC0901504 and 2016YFC0905100 to HJ; 2016YFC1306000 to BT), Clinical Research Funds of Xiangya Hospital (No. 2014L03 to HJ), and the Clinical and Rehabilitation Research Foundation of Xiangya hospital – Beidaweiming (No. xywm2015I10 to HJ).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
- ^Neuromuscular disease center [Internet]. Saint Louis: Washington University in St. Louis; [updated 2018 June]. Hereditary ataxias: dominant; [updated 2018 May 25; cited 2018 Jul 1]; [about 5 screens]. Available from: https://neuromuscular.wustl.edu/ataxia/domatax.html
References
Aeschlimann, D., and Thomazy, V. (2000). Protein crosslinking in assembly and remodelling of extracellular matrices: the role of transglutaminases. Connect. Tissue Res. 41, 1–27. doi: 10.3109/03008200009005638
Almeida, B., Abreu, I. A., Matos, C. A., Fraga, J. S., Fernandes, S., Macedo, M. G., et al. (2015). SUMOylation of the brain-predominant Ataxin-3 isoform modulates its interaction with p97. Biochim. Biophys. Acta 1852, 1950–1959. doi: 10.1016/j.bbadis.2015.06.010
Al-Ramahi, I., Lam, Y. C., Chen, H. K., de Gouyon, B., Zhang, M., Perez, A. M., et al. (2006). CHIP protects from the neurotoxicity of expanded and wild-type ataxin-1 and promotes their ubiquitination and degradation. J. Biol. Chem. 281, 26714–26724. doi: 10.1074/jbc.M601603200
Asada, A., Yamazaki, R., Kino, Y., Saito, T., Kimura, T., Miyake, M., et al. (2014). Cyclin-dependent kinase 5 phosphorylates and induces the degradation of ataxin-2. Neurosci. Lett. 563, 112–117. doi: 10.1016/j.neulet.2014.01.046
Berke, S. J., Schmied, F. A., Brunt, E. R., Ellerby, L. M., and Paulson, H. L. (2004). Caspase-mediated proteolysis of the polyglutamine disease protein ataxin-3. J. Neurochem. 89, 908–918. doi: 10.1111/j.1471-4159.2004.02369.x
Blount, J. R., Tsou, W. L., Ristic, G., Burr, A. A., Ouyang, M., Galante, H., et al. (2014). Ubiquitin-binding site 2 of ataxin-3 prevents its proteasomal degradation by interacting with Rad23. Nat. Commun. 5:4638. doi: 10.1038/ncomms5638
Carrozza, M. J., Utley, R. T., Workman, J. L., and Cote, J. (2003). The diverse functions of histone acetyltransferase complexes. Trends Genet. 19, 321–329. doi: 10.1016/s0168-9525(03)00115-x
Cartwright, T. A., Corey, M. J., and Schwalbe, R. A. (2007). Complex oligosaccharides are N-linked to Kv3 voltage-gated K+ channels in rat brain. Biochim. Biophys. Acta 1770, 666–671. doi: 10.1016/j.bbagen.2006.11.013
Chen, D. H., Cimino, P. J., Ranum, L. P., Zoghbi, H. Y., Yabe, I., Schut, L., et al. (2005). The clinical and genetic spectrum of spinocerebellar ataxia 14. Neurology 64, 1258–1260. doi: 10.1212/01.wnl.0000156801.64549.6b
Cohen-Carmon, D., and Meshorer, E. (2012). Polyglutamine (polyQ) disorders: the chromatin connection. Nucleus 3, 433–441. doi: 10.4161/nucl.21481
Cummings, C. J., Reinstein, E., Sun, Y., Antalffy, B., Jiang, Y., Ciechanover, A., et al. (1999). Mutation of the E6-AP ubiquitin ligase reduces nuclear inclusion frequency while accelerating polyglutamine-induced pathology in SCA1 mice. Neuron 24, 879–892. doi: 10.1016/S0896-6273(00)81035-1
Cvetanovic, M., Kular, R. K., and Opal, P. (2012). LANP mediates neuritic pathology in Spinocerebellar ataxia type 1. Neurobiol. Dis. 48, 526–532. doi: 10.1016/j.nbd.2012.07.024
David, G., Abbas, N., Stevanin, G., Durr, A., Yvert, G., Cancel, G., et al. (1997). Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat. Genet. 17, 65–70. doi: 10.1038/ng0997-65
David, G., Durr, A., Stevanin, G., Cancel, G., Abbas, N., Benomar, A., et al. (1998). Molecular and clinical correlations in autonomic dominant cerebellar ataxia with progressive macular dystrophy (SCA7). Hum. Mol. Genet. 7, 165–170. doi: 10.1093/hmg/7.2.165
Davidson, J. D., Riley, B., Burright, E. N., Duvick, L. A., Zoghbi, H. Y., and Orr, H. T. (2000). Identification and characterization of an ataxin-1-interacting protein: a1Up, a ubiquitin-like nuclear protein. Hum. Mol. Genet. 9, 2305–2312. doi: 10.1093/oxfordjournals.hmg.a018922
D’Souza, D. R., Wei, J., Shao, Q., Hebert, M. D., Subramony, S. H., and Vig, P. J. (2006). Tissue transglutaminase crosslinks ataxin-1: possible role in SCA1 pathogenesis. Neurosci. Lett. 409, 5–9. doi: 10.1016/j.neulet.2006.08.003
Duncan, C. E., An, M. C., Papanikolaou, T., Rugani, C., Vitelli, C., and Ellerby, L. M. (2013). Histone deacetylase-3 interacts with ataxin-7 and is altered in a spinocerebellar ataxia type 7 mouse model. Mol. Neurodegener. 8:42. doi: 10.1186/1750-1326-8-42
Durr, A. (2010). Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol. 9, 885–894. doi: 10.1016/s1474-4422(10)70183-6
Ellerby, L. M., Andrusiak, R. L., Wellington, C. L., Hackam, A. S., Propp, S. S., Wood, J. D., et al. (1999). Cleavage of atrophin-1 at caspase site aspartic acid 109 modulates cytotoxicity. J. Biol. Chem. 274, 8730–8736. doi: 10.1074/jbc.274.13.8730
Evers, M. M., Toonen, L. J., and van Roon-Mom, W. M. (2014). Ataxin-3 protein and RNA toxicity in spinocerebellar ataxia type 3: current insights and emerging therapeutic strategies. Mol. Neurobiol. 49, 1513–1531. doi: 10.1007/s12035-013-8596-2
Faggiano, S., Menon, R. P., Kelly, G. P., Todi, S. V., Scaglione, K. M., Konarev, P. V., et al. (2015). Allosteric regulation of deubiquitylase activity through ubiquitination. Front. Mol. Biosci. 2:2. doi: 10.3389/fmolb.2015.00002
Fei, E., Jia, N., Zhang, T., Ma, X., Wang, H., Liu, C., et al. (2007). Phosphorylation of ataxin-3 by glycogen synthase kinase 3beta at serine 256 regulates the aggregation of ataxin-3. Biochem. Biophys. Res. Commun. 357, 487–492. doi: 10.1016/j.bbrc.2007.03.160
Figueroa, K. P., Minassian, N. A., Stevanin, G., Waters, M., Garibyan, V., Forlani, S., et al. (2010). KCNC3: phenotype, mutations, channel biophysics-a study of 260 familial ataxia patients. Hum. Mutat. 31, 191–196. doi: 10.1002/humu.21165
Fong, L. W. R., Yang, D. C., and Chen, C. H. (2017). Myristoylated alanine-rich C kinase substrate (MARCKS): a multirole signaling protein in cancers. Cancer Metastasis Rev. 36, 737–747. doi: 10.1007/s10555-017-9709-6
Freeze, H. H., and Aebi, M. (2005). Altered glycan structures: the molecular basis of congenital disorders of glycosylation. Curr. Opin. Struct. Biol. 15, 490–498. doi: 10.1016/j.sbi.2005.08.010
Gallego-Iradi, C., Bickford, J. S., Khare, S., Hall, A., Nick, J. A., Salmasinia, D., et al. (2014). KCNC3(R420H), a K(+) channel mutation causative in spinocerebellar ataxia 13 displays aberrant intracellular trafficking. Neurobiol. Dis. 71, 270–279. doi: 10.1016/j.nbd.2014.08.020
Gavel, Y., and von Heijne, G. (1990). Sequence differences between glycosylated and non-glycosylated Asn-X-Thr/Ser acceptor sites: implications for protein engineering. Protein Eng. 3, 433–442. doi: 10.1093/protein/3.5.433
Guo, L., Giasson, B. I., Glavis-Bloom, A., Brewer, M. D., Shorter, J., Gitler, A. D., et al. (2014). A cellular system that degrades misfolded proteins and protects against neurodegeneration. Mol. Cell. 55, 15–30. doi: 10.1016/j.molcel.2014.04.030
Guyenet, S. J., Mookerjee, S. S., Lin, A., Custer, S. K., Chen, S. F., Sopher, B. L., et al. (2015). Proteolytic cleavage of ataxin-7 promotes SCA7 retinal degeneration and neurological dysfunction. Hum. Mol. Genet. 24, 3908–3917. doi: 10.1093/hmg/ddv121
Haacke, A., Broadley, S. A., Boteva, R., Tzvetkov, N., Hartl, F. U., and Breuer, P. (2006). Proteolytic cleavage of polyglutamine-expanded ataxin-3 is critical for aggregation and sequestration of non-expanded ataxin-3. Hum. Mol. Genet. 15, 555–568. doi: 10.1093/hmg/ddi472
Hearst, S. M., Shao, Q., Lopez, M., Raucher, D., and Vig, P. J. (2014). The design and delivery of a PKA inhibitory polypeptide to treat SCA1. J. Neurochem. 131, 101-114. doi: 10.1111/jnc.12782
Helmlinger, D., Hardy, S., Sasorith, S., Klein, F., Robert, F., Weber, C., et al. (2004). Ataxin-7 is a subunit of GCN5 histone acetyltransferase-containing complexes. Hum. Mol. Genet. 13, 1257–1265. doi: 10.1093/hmg/ddh139
Hermel, E., Gafni, J., Propp, S. S., Leavitt, B. R., Wellington, C. L., Young, J. E., et al. (2004). Specific caspase interactions and amplification are involved in selective neuronal vulnerability in Huntington’s disease. Cell Death Differ. 11, 424–438. doi: 10.1038/sj.cdd.4401358
Hershko, A., and Ciechanover, A. (1998). The ubiquitin system. Annu. Rev. Biochem. 67, 425–479. doi: 10.1146/annurev.biochem.67.1.425
Hickey, C. M., Wilson, N. R., and Hochstrasser, M. (2012). Function and regulation of SUMO proteases. Nat. Rev. Mol. Cell Biol. 13, 755–766. doi: 10.1038/nrm3478
Hipp, M. S., Kalveram, B., Raasi, S., Groettrup, M., and Schmidtke, G. (2005). FAT10, a ubiquitin-independent signal for proteasomal degradation. Mol. Cell. Biol. 25, 3483–3491. doi: 10.1128/mcb.25.9.3483-3491.2005
Hong, S., Lee, S., Cho, S. G., and Kang, S. (2008). UbcH6 interacts with and ubiquitinates the SCA1 gene product ataxin-1. Biochem. Biophys. Res. Commun. 371, 256–260. doi: 10.1016/j.bbrc.2008.04.066
Hsu, J. Y., Jhang, Y. L., Cheng, P. H., Chang, Y. F., Mao, S. H., Yang, H. I., et al. (2017). The truncated C-terminal fragment of mutant ATXN3 disrupts mitochondria dynamics in Spinocerebellar Ataxia type 3 models. Front. Mol. Neurosci. 10:196. doi: 10.3389/fnmol.2017.00196
Hubener, J., Weber, J. J., Richter, C., Honold, L., Weiss, A., Murad, F., et al. (2013). Calpain-mediated ataxin-3 cleavage in the molecular pathogenesis of spinocerebellar ataxia type 3 (SCA3). Hum. Mol. Genet. 22, 508–518. doi: 10.1093/hmg/dds449
Hughes, R. E., Lo, R. S., Davis, C., Strand, A. D., Neal, C. L., Olson, J. M., et al. (2001). Altered transcription in yeast expressing expanded polyglutamine. Proc. Natl. Acad. Sci. U.S.A. 98, 13201–13206. doi: 10.1073/pnas.191498198
Huttlin, E. L., Jedrychowski, M. P., Elias, J. E., Goswami, T., Rad, R., Beausoleil, S. A., et al. (2010). A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189. doi: 10.1016/j.cell.2010.12.001
Jana, N. R., Dikshit, P., Goswami, A., Kotliarova, S., Murata, S., Tanaka, K., et al. (2005). Co-chaperone CHIP associates with expanded polyglutamine protein and promotes their degradation by proteasomes. J. Biol. Chem. 280, 11635–11640. doi: 10.1074/jbc.M412042200
Janer, A., Werner, A., Takahashi-Fujigasaki, J., Daret, A., Fujigasaki, H., Takada, K., et al. (2010). SUMOylation attenuates the aggregation propensity and cellular toxicity of the polyglutamine expanded ataxin-7. Hum. Mol. Genet. 19, 181–195. doi: 10.1093/hmg/ddp478
Jorgensen, N. D., Andresen, J. M., Lagalwar, S., Armstrong, B., Stevens, S., Byam, C. E., et al. (2009). Phosphorylation of ATXN1 at Ser776 in the cerebellum. J. Neurochem. 110, 675–686. doi: 10.1111/j.1471-4159.2009.06164.x
Ju, H., Kokubu, H., and Lim, J. (2014). Beyond the glutamine expansion: influence of posttranslational modifications of ataxin-1 in the pathogenesis of spinocerebellar ataxia type 1. Mol. Neurobiol. 50, 866–874. doi: 10.1007/s12035-014-8703-z
Ju, H., Kokubu, H., Todd, T. W., Kahle, J. J., Kim, S., Richman, R., et al. (2013). Polyglutamine disease toxicity is regulated by Nemo-like kinase in spinocerebellar ataxia type 1. J. Neurosci. 33, 9328–9336. doi: 10.1523/jneurosci.3465-12.2013
Kahlem, P., Green, H., and Djian, P. (1998). Transglutaminase action imitates Huntington’s disease: selective polymerization of Huntingtin containing expanded polyglutamine. Mol. Cell 1, 595–601. doi: 10.1016/S1097-2765(00)80059-3
Kang, A. R., An, H. T., Ko, J., and Kang, S. (2017). Ataxin-1 regulates epithelial-mesenchymal transition of cervical cancer cells. Oncotarget 8, 18248–18259. doi: 10.18632/oncotarget.15319
Kang, A. R., Park, S. H., Lee, S., Choi, D. Y., Kim, K. P., Song, H. K., et al. (2015). A key lysine residue in the AXH domain of ataxin-1 is essential for its ubiquitylation. Biochim. Biophys. Acta 1854, 356–364. doi: 10.1016/j.bbapap.2015.01.012
Karve, T. M., and Cheema, A. K. (2011). Small changes huge impact: the role of protein posttranslational modifications in cellular homeostasis and disease. J. Amino Acids 2011:207691. doi: 10.4061/2011/207691
Klemm, P. (1984). Carboxy-terminal sequence determination of proteins and peptides with carboxypeptidase y. Methods Mol. Biol. 1, 255–259. doi: 10.1385/0-89603-062-8:255
Koch, P., Breuer, P., Peitz, M., Jungverdorben, J., Kesavan, J., Poppe, D., et al. (2011). Excitation-induced ataxin-3 aggregation in neurons from patients with Machado-Joseph disease. Nature 480, 543–546. doi: 10.1038/nature10671
Koegl, M., Hoppe, T., Schlenker, S., Ulrich, H. D., Mayer, T. U., and Jentsch, S. (1999). A novel ubiquitination factor, E4, is involved in multiubiquitin chain assembly. Cell 96, 635–644. doi: 10.1016/S0092-8674(00)80574-7
Kornberg, R. D., and Lorch, Y. (1999). Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell 98, 285–294. doi: 10.1016/S0092-8674(00)81958-3
Koutelou, E., Hirsch, C. L., and Dent, S. Y. (2010). Multiple faces of the SAGA complex. Curr. Opin. Cell Biol. 22, 374–382. doi: 10.1016/j.ceb.2010.03.005
Kristensen, L. V., Oppermann, F. S., Rauen, M. J., Hartmann-Petersen, R., and Thirstrup, K. (2017). Polyglutamine expansion of ataxin-3 alters its degree of ubiquitination and phosphorylation at specific sites. Neurochem. Int. 105, 42–50. doi: 10.1016/j.neuint.2016.12.019
Lai, S., O’Callaghan, B., Zoghbi, H. Y., and Orr, H. T. (2011). 14-3-3 Binding to ataxin-1(ATXN1) regulates its dephosphorylation at Ser-776 and transport to the nucleus. J. Biol. Chem. 286, 34606–34616. doi: 10.1074/jbc.M111.238527
Lee, S., Hong, S., and Kang, S. (2008). The ubiquitin-conjugating enzyme UbcH6 regulates the transcriptional repression activity of the SCA1 gene product ataxin-1. Biochem. Biophys. Res. Commun. 372, 735–740. doi: 10.1016/j.bbrc.2008.05.125
Lei, L. F., Yang, G. P., Wang, J. L., Chuang, D. M., Song, W. H., Tang, B. S., et al. (2016). Safety and efficacy of valproic acid treatment in SCA3/MJD patients. Parkinsonism. Relat. Disord. 26, 55-61. doi: 10.1016/j.parkreldis.2016.03.005
Li, F., Macfarlan, T., Pittman, R. N., and Chakravarti, D. (2002). Ataxin-3 is a histone-binding protein with two independent transcriptional corepressor activities. J. Biol. Chem. 277, 45004–45012. doi: 10.1074/jbc.M205259200
Li, X., Liu, H., Fischhaber, P. L., and Tang, T. S. (2015). Toward therapeutic targets for SCA3: insight into the role of Machado-Joseph disease protein ataxin-3 in misfolded proteins clearance. Prog. Neurobiol. 132, 34–58. doi: 10.1016/j.pneurobio.2015.06.004
Liebelt, F., and Vertegaal, A. C. (2016). Ubiquitin-dependent and independent roles of SUMO in proteostasis. Am. J. Physiol. Cell Physiol. 311, C284–C296. doi: 10.1152/ajpcell.00091.2016
Lim, J., Crespo-Barreto, J., Jafar-Nejad, P., Bowman, A. B., Richman, R., Hill, D. E., et al. (2008). Opposing effects of polyglutamine expansion on native protein complexes contribute to SCA1. Nature 452, 713–718. doi: 10.1038/nature06731
Liman, J., Deeg, S., Voigt, A., Vossfeldt, H., Dohm, C. P., Karch, A., et al. (2014). CDK5 protects from caspase-induced Ataxin-3 cleavage and neurodegeneration. J. Neurochem. 129, 1013–1023. doi: 10.1111/jnc.12684
Long, Z., Tang, B., and Jiang, H. (2014). Alleviating neurodegeneration in Drosophila models of PolyQ diseases. Cerebellum Ataxias 1:9. doi: 10.1186/2053-8871-1-9
Lorand, L., and Graham, R. M. (2003). Transglutaminases: crosslinking enzymes with pleiotropic functions. Nat. Rev. Mol. Cell Biol. 4, 140–156. doi: 10.1038/nrm1014
Magana, J. J., Velazquez-Perez, L., and Cisneros, B. (2013). Spinocerebellar ataxia type 2: clinical presentation, molecular mechanisms, and therapeutic perspectives. Mol. Neurobiol. 47, 90–104. doi: 10.1007/s12035-012-8348-8
Maggio, N., Sellitti, S., Capano, C. P., and Papa, M. (2001). Tissue-transglutaminase in rat and human brain: light and electron immunocytochemical analysis and in situ hybridization study. Brain Res. Bull. 56, 173–182. doi: 10.1016/S0361-9230(01)00649-9
Mandrusiak, L. M., Beitel, L. K., Wang, X., Scanlon, T. C., Chevalier-Larsen, E., Merry, D. E., et al. (2003). Transglutaminase potentiates ligand-dependent proteasome dysfunction induced by polyglutamine-expanded androgen receptor. Hum. Mol. Genet. 12, 1497–1506. doi: 10.1093/hmg/ddg161
Marquardt, T., and Denecke, J. (2003). Congenital disorders of glycosylation: review of their molecular bases, clinical presentations and specific therapies. Eur. J. Pediatr. 162, 359–379. doi: 10.1007/s00431-002-1136-0
Matos, C. A., Almeida, L. P., and Nobrega, C. (2017). Proteolytic cleavage of polyglutamine disease-causing proteins: revisiting the toxic fragment hypothesis. Curr. Pharm. Des. 23, 753–775. doi: 10.2174/1381612822666161227121912
Matos, C. A., Nobrega, C., Louros, S. R., Almeida, B., Ferreiro, E., Valero, J., et al. (2016). Ataxin-3 phosphorylation decreases neuronal defects in spinocerebellar ataxia type 3 models. J. Cell Biol. 212, 465–480. doi: 10.1083/jcb.201506025
Matsumoto, M., Yada, M., Hatakeyama, S., Ishimoto, H., Tanimura, T., Tsuji, S., et al. (2004). Molecular clearance of ataxin-3 is regulated by a mammalian E4. EMBO J. 23, 659–669. doi: 10.1038/sj.emboj.7600081
McCullough, S. D., and Grant, P. A. (2010). Histone acetylation, acetyltransferases, and ataxia–alteration of histone acetylation and chromatin dynamics is implicated in the pathogenesis of polyglutamine-expansion disorders. Adv. Protein Chem. Struct. Biol. 79, 165–203. doi: 10.1016/s1876-1623(10)79005-2
McDonough, H., and Patterson, C. (2003). CHIP: a link between the chaperone and proteasome systems. Cell Stress Chaperones 8, 303–308. doi: 10.1379/1466-1268(2003)008<0303:CALBTC>2.0.CO;2
McIlwain, D. R., Berger, T., and Mak, T. W. (2015). Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 5:a008656. doi: 10.1101/cshperspect.a026716.
Mendez, J. D., Xie, J., Aguilar-Hernandez, M., and Mendez-Valenzuela, V. (2010). Molecular susceptibility to glycation and its implication in diabetes mellitus and related diseases. Mol. Cell. Biochem. 344, 185–193. doi: 10.1007/s11010-010-0541-3
Menon, R. P., Soong, D., de Chiara, C., Holt, M. R., Anilkumar, N., and Pastore, A. (2012). The importance of serine 776 in Ataxin-1 partner selection: a FRET analysis. Sci. Rep. 2:919. doi: 10.1038/srep00919
Mielcarek, M., Benn, C. L., Franklin, S. A., Smith, D. L., Woodman, B., Marks, P. A., et al. (2011). SAHA decreases HDAC 2 and 4 levels in vivo and improves molecular phenotypes in the R6/2 mouse model of Huntington’s disease. PLoS One 6:e27746. doi: 10.1371/journal.pone.0027746
Miyashita, T., Nagao, K., Ohmi, K., Yanagisawa, H., Okamura-Oho, Y., and Yamada, M. (1998). Intracellular aggregate formation of dentatorubral-pallidoluysian atrophy (DRPLA) protein with the extended polyglutamine. Biochem. Biophys. Res. Commun. 249, 96–102. doi: 10.1006/bbrc.1998.9096
Modol, T., Natal, C., Perez de Obanos, M. P., Domingo de Miguel, E., Iraburu, M. J., and Lopez-Zabalza, M. J. (2011). Apoptosis of hepatic stellate cells mediated by specific protein nitration. Biochem. Pharmacol. 81, 451–458. doi: 10.1016/j.bcp.2010.10.017
Mookerjee, S., Papanikolaou, T., Guyenet, S. J., Sampath, V., Lin, A., Vitelli, C., et al. (2009). Posttranslational modification of ataxin-7 at lysine 257 prevents autophagy-mediated turnover of an N-terminal caspase-7 cleavage fragment. J. Neurosci. 29, 15134–15144. doi: 10.1523/jneurosci.4720-09.2009
Mueller, T., Breuer, P., Schmitt, I., Walter, J., Evert, B. O., and Wullner, U. (2009). CK2-dependent phosphorylation determines cellular localization and stability of ataxin-3. Hum. Mol. Genet. 18, 3334–3343. doi: 10.1093/hmg/ddp274
Okamura-Oho, Y., Miyashita, T., Nagao, K., Shima, S., Ogata, Y., Katada, T., et al. (2003). Dentatorubral-pallidoluysian atrophy protein is phosphorylated by c-Jun NH2-terminal kinase. Hum. Mol. Genet. 12, 1535–1542. doi: 10.1093/hmg/ddg168
Okamura-Oho, Y., Miyashita, T., Ohmi, K., and Yamada, M. (1999). Dentatorubral-pallidoluysian atrophy protein interacts through a proline-rich region near polyglutamine with the SH3 domain of an insulin receptor tyrosine kinase substrate. Hum. Mol. Genet. 8, 947–957. doi: 10.1093/hmg/8.6.947
Orr, H. T. (2012). SCA1-phosphorylation, a regulator of Ataxin-1 function and pathogenesis. Prog. Neurobiol. 99, 179–185. doi: 10.1016/j.pneurobio.2012.04.003
Pandolfo, M., and Manto, M. (2013). Cerebellar and afferent ataxias. Continuum 19, 1312–1343. doi: 10.1212/01.con.0000436158.39285.22
Park, J., Al-Ramahi, I., Tan, Q., Mollema, N., Diaz-Garcia, J. R., Gallego-Flores, T., et al. (2013). RAS-MAPK-MSK1 pathway modulates ataxin 1 protein levels and toxicity in SCA1. Nature 498, 325–331. doi: 10.1038/nature12204
Park, J. M., Jo, S. H., Kim, M. Y., Kim, T. H., and Ahn, Y. H. (2015). Role of transcription factor acetylation in the regulation of metabolic homeostasis. Protein Cell 6, 804–813. doi: 10.1007/s13238-015-0204-y
Pastori, V., Sangalli, E., Coccetti, P., Pozzi, C., Nonnis, S., Tedeschi, G., et al. (2010). CK2 and GSK3 phosphorylation on S29 controls wild-type ATXN3 nuclear uptake. Biochim. Biophys. Acta 1802, 583–592. doi: 10.1016/j.bbadis.2010.03.007
Qian, S. B., McDonough, H., Boellmann, F., Cyr, D. M., and Patterson, C. (2006). CHIP-mediated stress recovery by sequential ubiquitination of substrates and Hsp70. Nature 440, 551–555. doi: 10.1038/nature04600
Quina, A. S., Buschbeck, M., and Di Croce, L. (2006). Chromatin structure and epigenetics. Biochem. Pharmacol. 72, 1563–1569. doi: 10.1016/j.bcp.2006.06.016
Ratovitski, T., O’Meally, R. N., Jiang, M., Chaerkady, R., Chighladze, E., Stewart, J. C., et al. (2017). Post-translational modifications (PTMS), identified on endogenous huntingtin, cluster within proteolytic domains between HEAT repeats. J. Proteome Res. 16, 2692–2708. doi: 10.1021/acs.jproteome.6b00991
Riley, B. E., Zoghbi, H. Y., and Orr, H. T. (2005). SUMOylation of the polyglutamine repeat protein, ataxin-1, is dependent on a functional nuclear localization signal. J. Biol. Chem. 280, 21942–21948. doi: 10.1074/jbc.M501677200
Rossi, M., Perez-Lloret, S., Doldan, L., Cerquetti, D., Balej, J., Millar Vernetti, P., et al. (2014). Autosomal dominant cerebellar ataxias: a systematic review of clinical features. Eur. J. Neurol. 21, 607–615. doi: 10.1111/ene.12350
Ryu, J., Cho, S., Park, B. C., and Lee, D. H. (2010). Oxidative stress-enhanced SUMOylation and aggregation of ataxin-1: implication of JNK pathway. Biochem. Biophys. Res. Commun. 393, 280–285. doi: 10.1016/j.bbrc.2010.01.122
Sambataro, F., and Pennuto, M. (2012). Cell-autonomous and non-cell-autonomous toxicity in polyglutamine diseases. Prog. Neurobiol. 97, 152–172. doi: 10.1016/j.pneurobio.2011.10.003
Sambataro, F., and Pennuto, M. (2017). Post-translational modifications and protein quality control in motor neuron and polyglutamine diseases. Front. Mol. Neurosci. 10:82. doi: 10.3389/fnmol.2017.00082
Schaffar, G., Breuer, P., Boteva, R., Behrends, C., Tzvetkov, N., Strippel, N., et al. (2004). Cellular toxicity of polyglutamine expansion proteins: mechanism of transcription factor deactivation. Mol. Cell 15, 95–105. doi: 10.1016/j.molcel.2004.06.029
Schmidt, H., Schwaller, B., and Eilers, J. (2005). Calbindin D28k targets myo-inositol monophosphatase in spines and dendrites of cerebellar Purkinje neurons. Proc. Natl. Acad. Sci. U.S.A. 102, 5850–5855. doi: 10.1073/pnas.0407855102
Schmidt, T., Landwehrmeyer, G. B., Schmitt, I., Trottier, Y., Auburger, G., Laccone, F., et al. (1998). An isoform of ataxin-3 accumulates in the nucleus of neuronal cells in affected brain regions of SCA3 patients. Brain Pathol. 8, 669–679. doi: 10.1111/j.1750-3639.1998.tb00193.x
Schols, L., Bauer, P., Schmidt, T., Schulte, T., and Riess, O. (2004). Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. Lancet Neurol. 3, 291–304. doi: 10.1016/s1474-4422(04)00737-9
Shi, Y., Wang, J., Li, J. D., Ren, H., Guan, W., He, M., et al. (2013). Identification of CHIP as a novel causative gene for autosomal recessive cerebellar ataxia. PLoS One 8:e81884. doi: 10.1371/journal.pone.0081884
Simoes, A. T., Goncalves, N., Koeppen, A., Deglon, N., Kugler, S., Duarte, C. B., et al. (2012). Calpastatin-mediated inhibition of calpains in the mouse brain prevents mutant ataxin 3 proteolysis, nuclear localization and aggregation, relieving Machado-Joseph disease. Brain 135, 2428–2439. doi: 10.1093/brain/aws177
Simoes, A. T., Goncalves, N., Nobre, R. J., Duarte, C. B., and Pereira de Almeida, L. (2014). Calpain inhibition reduces ataxin-3 cleavage alleviating neuropathology and motor impairments in mouse models of Machado-Joseph disease. Hum. Mol. Genet. 23, 4932–4944. doi: 10.1093/hmg/ddu209
Steffan, J. S., Agrawal, N., Pallos, J., Rockabrand, E., Trotman, L. C., Slepko, N., et al. (2004). SUMO modification of Huntingtin and Huntington’s disease pathology. Science 304, 100–104. doi: 10.1126/science.1092194
Steffan, J. S., Bodai, L., Pallos, J., Poelman, M., McCampbell, A., Apostol, B. L., et al. (2001). Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature 413, 739–743. doi: 10.1038/35099568
Suzuki, Y., Nakayama, K., Hashimoto, N., and Yazawa, I. (2010). Proteolytic processing regulates pathological accumulation in dentatorubral-pallidoluysian atrophy. FEBS J. 277, 4873–4887. doi: 10.1111/j.1742-4658.2010.07893.x
Suzuki, Y., and Yazawa, I. (2011). Pathological accumulation of atrophin-1 in dentatorubralpallidoluysian atrophy. Int. J. Clin. Exp. Pathol. 4, 378–384.
Takahashi-Fujigasaki, J., Breidert, T., Fujigasaki, H., Duyckaerts, C., Camonis, J. H., Brice, A., et al. (2011). Amyloid precursor-like protein 2 cleavage contributes to neuronal intranuclear inclusions and cytotoxicity in spinocerebellar ataxia-7 (SCA7). Neurobiol. Dis. 41, 33–42. doi: 10.1016/j.nbd.2010.08.016
Tao, R. S., Fei, E. K., Ying, Z., Wang, H. F., and Wang, G. H. (2008). Casein kinase 2 interacts with and phosphorylates ataxin-3. Neurosci. Bull. 24, 271–277. doi: 10.1007/s12264-008-0605-5
Terashima, T., Kawai, H., Fujitani, M., Maeda, K., and Yasuda, H. (2002). SUMO-1 co-localized with mutant atrophin-1 with expanded polyglutamines accelerates intranuclear aggregation and cell death. Neuroreport 13, 2359–2364. doi: 10.1097/01.wnr.0000045009.30898.94
Todi, S. V., Scaglione, K. M., Blount, J. R., Basrur, V., Conlon, K. P., Pastore, A., et al. (2010). Activity and cellular functions of the deubiquitinating enzyme and polyglutamine disease protein ataxin-3 are regulated by ubiquitination at lysine 117. J. Biol. Chem. 285, 39303–39313. doi: 10.1074/jbc.M110.181610
Todi, S. V., Winborn, B. J., Scaglione, K. M., Blount, J. R., Travis, S. M., and Paulson, H. L. (2009). Ubiquitination directly enhances activity of the deubiquitinating enzyme ataxin-3. EMBO J. 28, 372–382. doi: 10.1038/emboj.2008.289
Tsai, Y. C., Fishman, P. S., Thakor, N. V., and Oyler, G. A. (2003). Parkin facilitates the elimination of expanded polyglutamine proteins and leads to preservation of proteasome function. J. Biol. Chem. 278, 22044–22055. doi: 10.1074/jbc.M212235200
Vig, P. J., Wei, J., Shao, Q., Hebert, M. D., Subramony, S. H., and Sutton, L. T. (2007). Role of tissue transglutaminase type 2 in calbindin-D28k interaction with ataxin-1. Neurosci. Lett. 420, 53–57. doi: 10.1016/j.neulet.2007.04.005
Violante, V., Luongo, A., Pepe, I., Annunziata, S., and Gentile, V. (2001). Transglutaminase-dependent formation of protein aggregates as possible biochemical mechanism for polyglutamine diseases. Brain Res. Bull. 56, 169–172. doi: 10.1016/S0361-9230(01)00576-7
Warrick, J. M., Morabito, L. M., Bilen, J., Gordesky-Gold, B., Faust, L. Z., Paulson, H. L., et al. (2005). Ataxin-3 suppresses polyglutamine neurodegeneration in Drosophila by a ubiquitin-associated mechanism. Mol. Cell 18, 37–48. doi: 10.1016/j.molcel.2005.02.030
Waters, M. F., Minassian, N. A., Stevanin, G., Figueroa, K. P., Bannister, J. P., Nolte, D., et al. (2006). Mutations in voltage-gated potassium channel KCNC3 cause degenerative and developmental central nervous system phenotypes. Nat. Genet. 38, 447–451. doi: 10.1038/ng1758
Weber, J. J., Golla, M., Guaitoli, G., Wanichawan, P., Hayer, S. N., Hauser, S., et al. (2017). A combinatorial approach to identify calpain cleavage sites in the Machado-Joseph disease protein ataxin-3. Brain 140, 1280–1299. doi: 10.1093/brain/awx039
Xie, F., Jin, K., Shao, L., Fan, Y., Tu, Y., Li, Y., et al. (2017). FAF1 phosphorylation by AKT accumulates TGF-beta type II receptor and drives breast cancer metastasis. Nat. Commun. 8:15021. doi: 10.1038/ncomms15021
Xu, W. S., Parmigiani, R. B., and Marks, P. A. (2007). Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene 26, 5541–5552. doi: 10.1038/sj.onc.1210620
Yamamoto, K., Seki, T., Yamamoto, H., Adachi, N., Tanaka, S., Hide, I., et al. (2014). Deregulation of the actin cytoskeleton and macropinocytosis in response to phorbol ester by the mutant protein kinase C gamma that causes spinocerebellar ataxia type 14. Front. Physiol. 5:126. doi: 10.3389/fphys.2014.00126
Yang, Y., He, Y., Wang, X., Liang, Z., He, G., Zhang, P., et al. (2017). Protein SUMOylation modification and its associations with disease. Open Biol. 7:170167. doi: 10.1098/rsob.170167
Yazawa, I. (1999). Unique characteristics of ubiquitin-bonded complex play a pathological role in dentatorubral-pallidoluysian atrophy. Biochem. Biophys. Res. Commun. 264, 37–41. doi: 10.1006/bbrc.1999.1386
Yazawa, I., Nakase, H., and Kurisaki, H. (1999). Abnormal dentatorubral-pallidoluysian atrophy (DRPLA) protein complex is pathologically ubiquitinated in DRPLA brains. Biochem. Biophys. Res. Commun. 260, 133–138. doi: 10.1006/bbrc.1999.0839
Yi, J., Zhang, L., Tang, B., Han, W., Zhou, Y., Chen, Z., et al. (2013). Sodium valproate alleviates neurodegeneration in SCA3/MJD via suppressing apoptosis and rescuing the hypoacetylation levels of histone H3 and H4. PLoS One 8:e54792. doi: 10.1371/journal.pone.0054792
Ying, M., Xu, R., Wu, X., Zhu, H., Zhuang, Y., Han, M., et al. (2006). Sodium butyrate ameliorates histone hypoacetylation and neurodegenerative phenotypes in a mouse model for DRPLA. J. Biol. Chem. 281, 12580–12586. doi: 10.1074/jbc.M511677200
Young, J. E., Gouw, L., Propp, S., Sopher, B. L., Taylor, J., Lin, A., et al. (2007). Proteolytic cleavage of ataxin-7 by caspase-7 modulates cellular toxicity and transcriptional dysregulation. J. Biol. Chem. 282, 30150–30160. doi: 10.1074/jbc.M705265200
Zhang, L., Yang, T. H., and Li, D. W. (2017). Roles of SUMOylation in heart development and cardiovascular diseases. Curr. Mol. Med. 16, 877–884. doi: 10.2174/1566524016666161223110407
Zhou, Y. F., Liao, S. S., Luo, Y. Y., Tang, J. G., Wang, J. L., Lei, L. F., et al. (2013). SUMO-1 modification on K166 of polyQ-expanded ataxin-3 strengthens its stability and increases its cytotoxicity. PLoS One 8:e54214. doi: 10.1371/journal.pone.0054214
Keywords: post-translational modification, spinocerebellar ataxias, protein, pathogenesis, therapy
Citation: Wan L, Xu K, Chen Z, Tang B and Jiang H (2018) Roles of Post-translational Modifications in Spinocerebellar Ataxias. Front. Cell. Neurosci. 12:290. doi: 10.3389/fncel.2018.00290
Received: 12 May 2018; Accepted: 13 August 2018;
Published: 19 September 2018.
Edited by:
Antonio Gambardella, Università degli studi Magna Græcia di Catanzaro, ItalyReviewed by:
Clevio Nobrega, University of Algarve, PortugalMarie-Christine Galas, Institut National de la Santé et de la Recherche Médicale (INSERM), France
Copyright © 2018 Wan, Xu, Chen, Tang and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hong Jiang, amlhbmdob25nNzM4NjhAMTI2LmNvbQ==
†These authors have contributed equally to this work