 Geoffrey Canet1†
Geoffrey Canet1† Chloé Dias2†
Chloé Dias2† Audrey Gabelle3
Audrey Gabelle3 Yannick Simonin2
Yannick Simonin2 Fabien Gosselet4
Fabien Gosselet4 Nicola Marchi5
Nicola Marchi5 Alain Makinson6Edouard Tuaillon2,7
Alain Makinson6Edouard Tuaillon2,7 Philippe Van de Perre2,7
Philippe Van de Perre2,7 Laurent Givalois1*
Laurent Givalois1* Sara Salinas2*
Sara Salinas2*- 1Molecular Mechanisms in Neurodegenerative Dementia, INSERM, University of Montpellier/EPHE, Montpellier, France
- 2Pathogenesis and Control of Chronic Infections, INSERM, University of Montpellier, Etablissement français du Sang, Montpellier, France
- 3Memory Research and Resources Center, CHU Montpellier, University of Montpellier, Montpellier, France
- 4Laboratoire de la Barrière Hémato-Encéphalique, Université d’Artois, Lens, France
- 5Cerebrovascular Mechanisms of Brain Disorders, Department of Neuroscience, Institute of Functional Genomics, CNRS, INSERM, University of Montpellier, Montpellier, France
- 6Department of Infectious Diseases CHU Montpellier, INSERM, IRD, University of Montpellier, Montpellier, France
- 7Pathogenesis and Control of Chronic Infections, INSERM, University of Montpellier, Etablissement français du Sang, CHU Montpellier, Montpellier, France
Environmental factors such as chemicals, stress and pathogens are now widely believed to play important roles in the onset of some brain diseases, as they are associated with neuronal impairment and acute or chronic inflammation. Alzheimer’s disease (AD) is characterized by progressive synaptic dysfunction and neurodegeneration that ultimately lead to dementia. Neuroinflammation also plays a prominent role in AD and possible links to viruses have been proposed. In particular, the human immunodeficiency virus (HIV) can pass the blood-brain barrier and cause neuronal dysfunction leading to cognitive dysfunctions called HIV-associated neurocognitive disorders (HAND). Similarities between HAND and HIV exist as numerous factors involved in AD such as members of the amyloid and Tau pathways, as well as stress-related pathways or blood brain barrier (BBB) regulators, seem to be modulated by HIV brain infection, leading to the accumulation of amyloid plaques or neurofibrillary tangles (NFT) in some patients. Here, we summarize findings regarding how HIV and some of its proteins such as Tat and gp120 modulate signaling and cellular pathways also impaired in AD, suggesting similarities and convergences of these two pathologies.
Introduction
Neurodegenerative disorders, whether they are sporadic, genetic or due to pathogens, are caused by the specific impairment (cell death or cell dysfunction) of different neural cell types and represent a major health issue in developed and developing countries. Central nervous system (CNS) impairment can have tremendous effects on day-to-day activities, inducing a loss of autonomy and the need for continuous care and support. Indeed, cognitive processes, as well as locomotor and sensory functions can be severely affected. In this context, the World Health Organization estimated that close to 2 million persons died in 2012 from a neurological disorder (WHO, 2014) and Alzheimer’s disease (AD) International reported that more than 46 millions of people suffered from AD dementia worldwide in 2015 (Prince et al., 2015).
Aging is certainly the primary risk factor for neurodegenerative disorders such as AD and Parkinson’s disease (PD). In AD, the risk doubles every 5 years after the age of 65, and reaches 50% after 85. Genetic factors, often involved in the regulation of neuronal activity or glial function, are also found associated with several types of pathologies such as motor neuron diseases (MND), AD, PD or Huntington’s disease (HD). However, many of these disorders can be sporadic and emphasize the role of the environment in their etiology. For example, in AD, familial forms due to genetic mutations represent less than 5% of cases, whereas sporadic forms are predominant (Selkoe, 2001; Fratiglioni et al., 1993). In these forms multifactorial factors have been identified such as environmental agents that may increase the probability of developing AD (Hayden et al., 2010; Yan et al., 2016). Among such environmental factors, neurotropic infectious agents such as bacteria, parasites or viruses can cause neuronal dysfunction: some pathogens have been selected throughout evolution for their ability to reach the CNS using different strategies such as crossing the blood brain barrier (BBB) or by axonal transport (Smith et al., 2001; Samuel et al., 2007; Salinas et al., 2010; Roe et al., 2014). Once in the CNS, they can trigger a cascade of events directly or indirectly due to their replication cycle, which will lead to neuronal defects, and in some cases, host death. Because some pathophysiological mechanisms involved in neuronal infection and neurodegenerative diseases are relatively similar, and sometimes overlapping, links have been proposed between the onset of certain brain diseases and prior encounters with neurotropic pathogens (Mattson, 2004; De Chiara et al., 2012). For example, some studies proposed potential links between herpes simplex virus (HSV) and/or cytomegalovirus (CMV) infection and the etiology of AD (Itzhaki et al., 1997; Lurain et al., 2013; Itzhaki, 2014). In particular, the presence of HSV in patients carrying susceptibility genes (e.g., APOE-e4 allele) is associated with the disease (Itzhaki et al., 1997). In the same light, CMV infection has been proposed to have a role in AD by modulating inflammatory responses (Lurain et al., 2013). In this context, accumulating evidence show that neuroinflammation and cerebrovascular permeability are key mechanisms in the etiology of many of these diseases including AD, MND and multiple sclerosis (MS; Hong H. et al., 2016). In the brain, the resident cells involved in inflammation are the glial cells, namely astrocytes and microglia. Interestingly, links between glial-mediated immune response and regulation of cognitive processes are emerging. For example, the pro-inflammatory cytokine tumor necrosis factor tumor necrosis factor-α (TNF-α) can trigger an astrocyte-dependent response that will ultimately lead to excitatory synapses impairment (Habbas et al., 2015). Along the same line, microglia cells are also key regulators of synaptic function and could be responsible for some cognitive deficits after synaptic impairment following activation by viral infections (Vasek et al., 2016), in some brain diseases such as Oculoleptomeningeal amyloidosis (Azevedo et al., 2013) or in AD (Hong S. et al., 2016; Rajendran and Paolicelli, 2018).
One physiological system regulating inflammation is the hypothalamo-pituitary-adrenal (HPA) axis, a major neuroendocrine system. This axis is highly involved in stress responses and triggers the adrenal cortex to release glucocorticoids (GC; cortisol in humans and corticosterone in rodents). These steroid hormones readily cross the BBB and bind to low affinity glucocorticoid receptors (GR) and high affinity mineralocorticoid receptors (MR; Reul and de Kloet, 1985). These receptors are necessary for normal cellular activity, inflammatory and stress responses, and crucial for many CNS functions, including learning and memory (Roozendaal, 2000; Chen et al., 2012). GC via their receptors, increase the transcription of anti-inflammatory genes but also inhibit the expression of multiple inflammatory genes (cytokines, enzymes, receptors and adhesion molecules; Coutinho and Chapman, 2011; Van Bogaert et al., 2011). Interestingly, pathogens can activate the HPA axis and induce subsequent secretion of GC, directly by their structural and genetic components and indirectly by the immune response involving cytokines and inflammatory mediators secreted from activated immune cells and infected tissues (Givalois et al., 1994; Kino, 2000). However, in case of excessive secretion due to chronic stress, GC become unable to exert their effects on target tissues, and trigger the syndrome of GC resistance (Chrousos et al., 1993; Charmandari et al., 2005). Thus, prolonged stimulation of GC secretion can induce or potentiate neuroinflammation, but also excitotoxicity and oxidative stress via synergistic effects with excitatory amino acids such as glutamate (Takahashi et al., 2002; McEwen, 2008).
As yet, there is no definitive and conclusive data showing a causative link between the human immunodeficiency virus (HIV) neuroinfection and the onset of AD. However, accumulating evidence suggests that common pathways and factors are modulated in the brains of HIV+ and AD patients, thus pointing out similarities and convergence in these two pathologies. In particular, neuroinflammation is strongly associated with both diseases and is emerging as a major player in the onset and progression of neuropathologies. Understanding the potential link between molecular mechanisms such as neuroinflammation, viral CNS infection, HPA axis deregulation and neurodegenerative disorders is therefore more than pertinent. Here, we summarize existing data, drawing parallels between HIV-associated neurocognitive disorders (HAND) and AD where similar cellular pathways are impaired, and propose potential new links between HIV, HPA pathways and AD that were not previously reported or addressed.
HIV Neuroinfection
HIV is a retrovirus that depletes CD4+ cells and strongly impairs the immune response, thus opening up the way for opportunistic infections that cause the acquired immunodeficiency syndrome (AIDS). However, the immune system is not the only target. HIV is sometimes classified as a neurotropic virus, even though it cannot directly infect neurons, due to their lack of expression of its main receptor CD4 (Peudenier et al., 1991). Nonetheless, the virus can access the brain very early on during primary infection (within the first 2 weeks) where it can locally replicate and become compartmentalized, as determined by recent phylogenetic analyses (Sturdevant et al., 2015). The virus then leads to neurotoxicity that is associated with motor, sensory and cognitive impairment in around 50% of HIV+ patients (Becker et al., 2009; Thakur et al., 2018). These neuronal impairments are collectively named HAND (González-Scarano and Martín-García, 2005; Clifford and Ances, 2013; Thakur et al., 2018). Depending on the severity of the symptoms, these conditions are classified into three groups: asymptomatic neurocognitive impairment (ANI), mild neurocognitive disorder (MND) and HIV-associated dementia (HAD) (Antinori et al., 2007). Clinically, patients can display a range of symptoms from cognitive deficits (memory, attention, language, behavior), motor and sensory impairment, mood changes to dementia. Even though asymptomatic, ANI HIV+ patients present higher risk for developing cognitive impairments compared to controls and have been proposed to mirror early phases of AD (Ellis et al., 2007; McArthur et al., 2010).
With the introduction of successful combination antiretroviral therapy (cART), the incidence of HAND has decreased (Maschke et al., 2000; González-Scarano and Martín-García, 2005). However, its prevalence is increasing, mainly due to the increased life expectancy of patients, cardiovascular risks factors, exposure hazards, and ongoing nervous system inflammation despite cART. Patients diagnosed with HAND, with mild or severe cognitive impairments, have a lower quality of life and shorter lifespan (Heaton et al., 2011). Before the establishment of cART, HAD could be found in up to 15%–20% of HIV+ individuals and was one of the main risk factor (McArthur et al., 1993, 2003). In the post-cART era, the total proportion of patients with HAND did not vary but the distribution of the classes changed with a decrease in HAD and an increase in MND and ANI (González-Scarano and Martín-García, 2005). Moreover, neuronal disorders are becoming more frequent in the aging HIV+ population (Thakur et al., 2018). Prospective trials have found poor prediction on the effect of cART on the patient’s cognitive impairment as the BBB limits the penetration of the drugs into the brain and some antiretrovirals (ARV) can show neurotoxicity (Soontornniyomkij et al., 2018; Thakur et al., 2018). In this context, HAD has also been proposed as the most common form of dementia in people under 40 years of age (Janssen, 1992).
HIV enters the CNS by a mechanism called “Trojan horse” that consists of the migration of infected monocytes through the BBB (Williams et al., 2015; Zhang et al., 2015). Recent work showed that CD14+CD16+ monocytes are able to efficiently transmigrate through the BBB and are found in high numbers in HIV-infected individuals (Williams et al., 2015). Once in the brain, HIV can infect various cell types expressing the CD4 receptor including microglia (Cosenza et al., 2002), perivascular macrophages and potentially adult neural precursors (Rothenaigner et al., 2007). Viral replication is also seen in a restrictive manner in astrocytes (Eugenin et al., 2011). These observations have led to the classification of the brain as a reservoir and a sanctuary for HIV (Hellmuth et al., 2015). In the brain, direct and indirect effects of HIV infection will lead to neuronal dysfunction (Figure 1): infected astrocytes and microglia release factors inducing neurotoxicity such as cytokines, chemokine and reactive oxygen species (ROS) (González-Scarano and Martín-García, 2005). Later, this may also contribute to the disruption of the BBB and result in further entry/exit of virions and viral proteins (see below). Some HIV proteins (glycoprotein120 (gp120), transactivator of transcription (Tat), viral protein R (Vpr) and negative regulatory factor (Nef)) can have direct effects on neurons and trigger signaling cascades leading to neuronal impairment. These proteins can be released from infected non-neuronal cells or shed from virions (Nath, 2002; Churchill et al., 2015). Consistently, some viral proteins such as Tat and Vpr are found in the cerebrospinal fluid (CSF; Levy et al., 1994; Hudson et al., 2000; Nath, 2002). Numerous studies using ex vivo and in vivo models show a plethora of neurotoxic effects: the envelope protein gp120 was shown to promote the release of IL-1β and TNFα, as well as neurotoxic factors such as glutamate, which in turn triggers neuronal apoptosis. (Garden et al., 2002; Bachis et al., 2009). Tat potentiates glutamate overactivation of NMDA receptors and release of cytokines through astrocytes (Haughey et al., 2001; King et al., 2006) and triggers neuronal apoptosis (Kruman et al., 1998). Tat and gp120-mediated apoptosis also triggers an increase in intracellular calcium levels which is classically associated with excitotoxicity processes and is induced by glutamate accumulation in the extracellular space. HIV+ patients show elevated CSF glutamate levels that correlate with dementia severity and the degree of brain atrophy (Ferrarese et al., 2001). Similarly, the protein Nef can trigger cytotoxic effects (Sami Saribas et al., 2017). Vpr has been proposed to induce mitochondrial neuronal accumulation and impaired axonal transport by modulating microtubule stability (Wang et al., 2017).
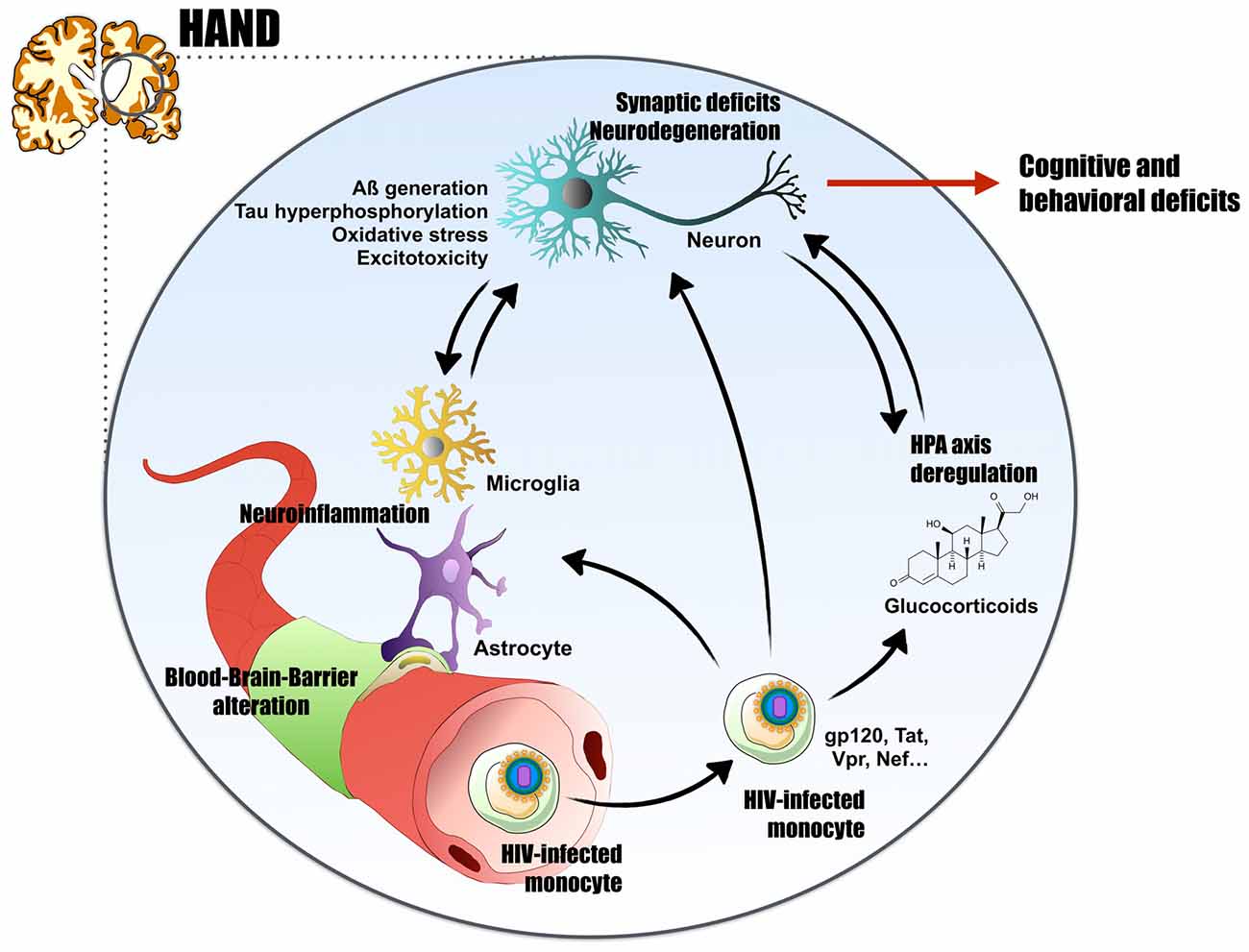
Figure 1. Proposed mechanism for human immunodeficiency virus (HIV)-associated neurocognitive disorders (HAND). HIV-1, through infected monocytes, can cross the blood brain barrier (BBB) by the Trojan horse mechanism. In the central nervous system (CNS), neuroinflammation triggered directly by viral replication or by HIV-viral proteins (glycoprotein 120 (gp120), transactivator of transcription (Tat), viral protein R (Vpr), negative regulatory factor (Nef)…) exert neurotoxic effects. They impact neurons integrity and lead to Alzheimer’s disease (AD)-like characteristics such as A generation, abnormal Tau phosphorylation, oxidative stress and excitotoxicity. The virus can induces neuroinflammation by several mechanisms: direct infection of astrocytes, BBB impairment and peripheral macrophages invasion, or massive gliosis and cytokines release. Finally, HIV+ patients present high glucocorticoids (cortisol) levels, characteristic of a hypothalamic–pituitary–adrenal (HPA) axis deregulation. Glucocorticoids and their receptors are highly involved in the etiology of AD. By these numerous pathways, HIV-1 induces synaptic deficits and neurodegeneration, thus leads to cognitive and behavioral deficits, and could explain the establishment of HAND in HIV+ patients and potentially the onset of AD.
Ultimately, if the viral load is not controlled, neuronal loss will occur. Interestingly, similarly as during the early stages of AD (Bateman et al., 2012; Marcello et al., 2012), HIV-induced neurodegeneration does not correlate completely with cognitive impairments. Cognitive deficits in patients with HAD have been shown to better correlate with synaptic impairment than neurodegeneration as cell death is also accompanied by axonal degeneration, astrocytosis and synaptic loss (Adle-Biassette et al., 1999; Avdoshina et al., 2013). Moreover, some studies aimed to decipher whether neurocognitive deficits are still found in populations where the viral load is well controlled. While some cohorts showed that cognitive functions are generally not affected in individuals with no detectable viremia (Lopardo et al., 2009), others suggest that there is still a high prevalence of HAND in HIV+ aviremic patients (Cysique et al., 2006; McArthur et al., 2010). This could be explained by several non-exclusive mechanisms such as: (i) poor cART regime penetration of the BBB; (ii) toxicity of anti-retrovirals; (iii) prolonged lifespan of infected individuals; (iv) local low-noise viral replication; and (v) chronic neuroinflammation. Continuous inflammatory injuries could pave the way for some neurodegenerative disorders. Potential links have therefore been suggested between HIV and amyotrophic lateral sclerosis (ALS; Alfahad and Nath, 2013) and AD (Chakradhar, 2018; see below).
Key Features of AD
AD is a devastating neurodegenerative disorder and the most common cause of neurodegenerative dementia in the elderly. It is characterized by a progressive impairment of cognitive functions, associated with synaptic and neuronal loss, as well as senile plaques and neurofibrillary tangles (NFT) in the brain (Selkoe, 2001; Cummings, 2004). Plaques are composed of insoluble extracellular aggregates consisting mainly of amyloid-β (Aβ) peptides, whereas NFT are the result of hyper- and abnormal phosphorylation on the intracellular microtubule-stabilizing protein Tau (Selkoe, 2001). Aβ production is dependent of the amyloidogenic pathway induction, and results from a pathological cleavage of the amyloid precursor protein (APP) by β-secretase (BACE1), which releases the C99 fragment, and a second cleavage by γ-secretase releasing Aβ into the extracellular medium (Cummings, 2004; Flammang et al., 2012). This generation of Aβ, in synergy with Tau, induces a cascade of neurodegenerative mechanisms such as cholinergic neurons degeneration, synaptic deficit, neuroinflammation, oxidative stress, apoptosis and autophagy, in part responsible for cognitive and behavioral deficits (Selkoe, 2001; Cummings, 2004). Moreover, neurotoxicity in AD is correlated with the total level of brain circulating oligomeric Aβ peptides, mainly consisting of Aβ1–40 and Aβ1–42 peptides (Selkoe, 2001; Cummings, 2004; Cleary et al., 2005; Blennow et al., 2010).
Common Mechanisms Between HIV Neuroinfection and AD
Because AIDS became a chronic disease with the advent of cART, an important proportion of HIV+ patient population is now over 50 years of age and also facing age-related disorders (Milanini and Valcour, 2017). In addition, accelerated ageing, including immunosenescence, is a constitutive part of the natural history of HIV infection. In particular, HAND is weakening an already age-targeted organ and could favor the occurrence of neurodegenerative diseases. Regarding AD, questions have risen about potential links with HIV CNS infection from observations and studies demonstrating modulation of common pathways/mechanism (such as the amyloid and Tau pathways). In this context, several symptoms associated with AD physiopathology were reported in HIV+ patients and murine in vivo and in vitro models of neuroAIDS (Table 1).
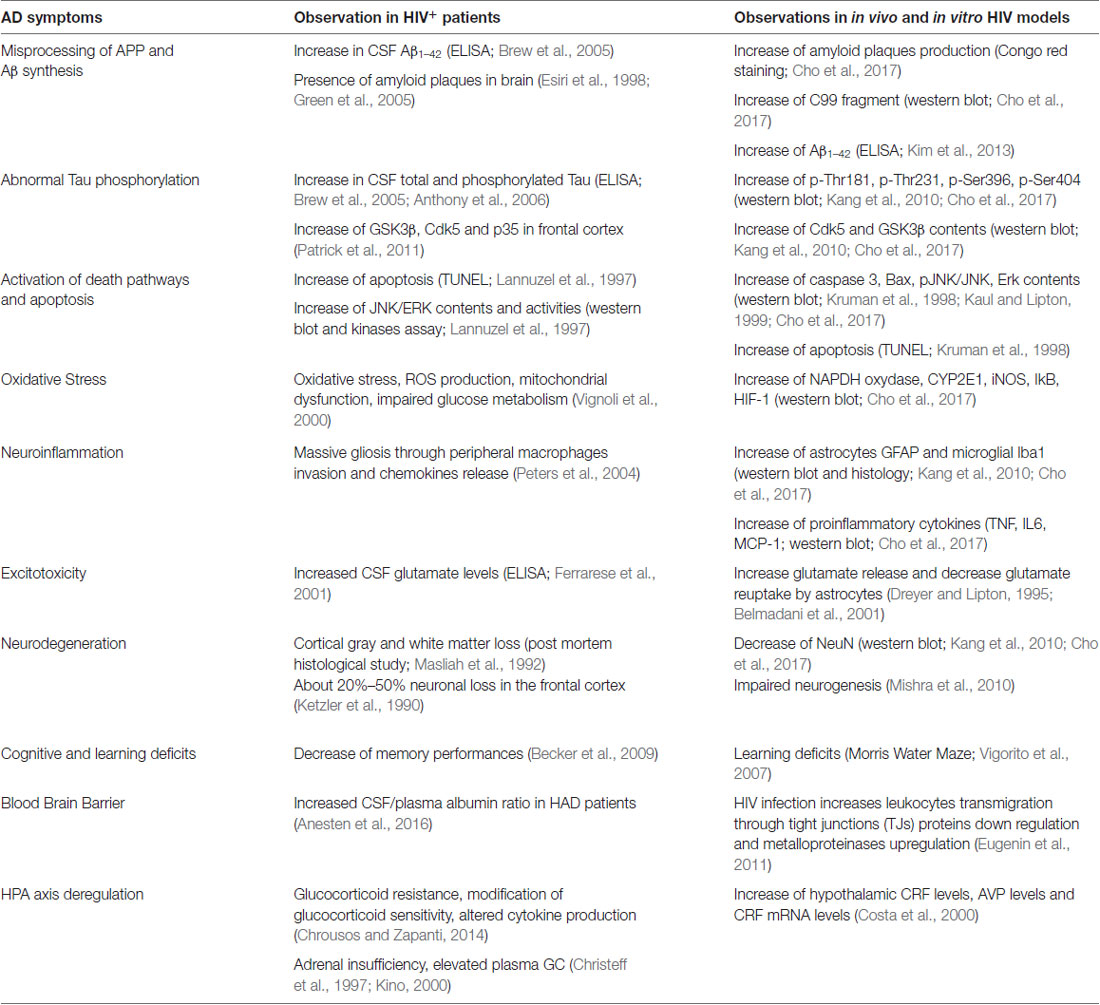
Table 1. Common neurotoxicity pathways between Alzheimer’s disease (AD) and human immunodeficiency virus (HIV).
HIV+ patients presenting HAND show CSF features very similar to early and late stages of AD (Table 1). For instance, biomarkers such as Aβ1–42 are found to be dysregulated in the CSF of HAND patients (Clifford et al., 2009). When comparing CSF from age-matched controls, HAND and late-stage AD patients, a similar trend for decreased Aβ1–42 levels was found in HIV+ individuals suffering from neuronal disorders (522 pg/ml for HAND and 421 pg/ml for AD, compared to 722 pg/ml for controls; Clifford et al., 2009). Notably, HIV+ patients without neurological symptoms had levels of Aβ1–42 in the same range as non-dementia controls (Clifford et al., 2009). Consequently, plaques caused by extracellular amyloid peptide accumulation can be seen in patients, particularly before the cART era (Esiri et al., 1998; Green et al., 2005).
Emerging evidence suggests that brain exposure to HIV particles and HIV proteins can directly or indirectly modulate the amyloid and Tau pathways (Chen et al., 2013; Ortega and Ances, 2014; Cho et al., 2017; Hategan et al., 2017). In HIV murine models (HIV-1 transgenic rats and gp120 transgenic mice) neurodegeneration is observed and associated with apoptosis, gliosis, oxidative stress, Aβ synthesis and increases in Tau phosphorylation (Table 1). Tat can affect Aβ synthesis through several mechanisms. It can increases Aβ production by modulating endolysosomal structure and function (Chen et al., 2013). Conversely, Tat leads to an increase of Aβ accumulation by inhibiting its degradation by neprilysin (Daily et al., 2006), and increasing BACE1 activity and synthesis of the C99 fragment (Chen et al., 2013; Cho et al., 2017). This increase in BACE1, which is also found elevated in AD, was recently confirmed in HIV+ patients (Stern et al., 2018). Similarly, treatment of primary hippocampal cell cultures with recombinant gp120 promotes Aβ1–42 secretion (Aksenov et al., 2010). In addition, when lentiviral vector-derived Tat is expressed in the hippocampus of APP/PS1 transgenic mice (a widely used mouse model of AD), it potentiates Aβ1–42 synthesis and increases the size of amyloid plaques (Kim et al., 2013). More recently, it was demonstrated that in primary hippocampal neurons, Tat interacts with Aβ peptides and forms complexes that increase damage likely through membrane pore formation (Hategan et al., 2017). In HIV-1 transgenic rats (which express seven of the nine HIV-1 viral proteins including gp120, Nef and Tat), the number and size of amyloid plaques were significantly elevated in the cerebral cortex compared to wild type (WT) animals (Cho et al., 2017). This was accompanied by an increase of amyloid C-terminal fragment C99 levels (>5-fold) in brains of HIV-1 transgenic rats (Cho et al., 2017). Similarly, The HIV-1 matrix protein p17 released from HIV-1 infected cells, participates in amyloid deposits toxicity by its ability to misfold and aggregate, even in the presence of protease inhibitors (Zeinolabediny et al., 2017). When injected into the mouse hippocampus, p17 colocalizes with phospho-Tau, plaque and fibril-like structures, where it increases Aβ expression and plaque-like development (Zeinolabediny et al., 2017). This led to cognitive impairment, as measured by recognition and Morris water maze tests (Zeinolabediny et al., 2017). Recently, APP metabolism in macrophage and microglia was reported to be modulated by HIV-1 Gag protein (Chai et al., 2017). The Gag polyprotein was shown to increase Aβ production and associated neurotoxicity by the activation of secretases. APP on the other hand, was reported to act as an antiviral factor by sequestering Gag in lipid rafts and restricting HIV-1 release (Chai et al., 2017). The balance between these two mechanisms (restriction and evasion), as well as the impact on Aβ peptide genesis will need further investigation.
The causative role of Tau and/or hyperphosphorylation of Tau in HAND is still poorly established. However, in 10 month-old gp120 transgenic mice, cognitive abnormalities, which are associated with an increase in neuronal death and gliosis, are also linked with an increase in Tau hyperphosphorylation (Kang et al., 2010). This effect is concomitant with an over-activation of GSK3-β, the main enzyme involved in Tau phosphorylation (Kang et al., 2010). In HIV-1 transgenic rats, levels of phosphorylated-Tau (p-Thr181, p-Thr231 and p-Ser396) were markedly elevated in the hippocampus and associated with an increase in Cdk5 activity, the other main enzyme involved in Tau phosphorylation (Cho et al., 2017). These animal model observations are in accordance with significantly elevated levels of phospho-Tau and abnormal NFT in HIV+ patients with HAND (Brew et al., 2005; Anthony et al., 2006; Kang et al., 2010).
HIV, AD and the Blood Brain Barrier
Neurodegenerative disorders, including HAND, are often associated with BBB impairment (Atluri et al., 2015; Zhao et al., 2015). Endothelial microvascular cells, pericytes, neurons and astrocytes form and regulate the BBB neurovascular unit (NVU), a tightly regulated endothelium that separates the brain from the systemic circulation. The BBB is also a metabolic barrier because endothelial cells express several enzymes and efflux pumps impeding the entry of xenobiotics and cells into the CNS (Cecchelli et al., 2014). When the modulation of the microenvironment by inflammation or cell damage occurs, BBB integrity can be perturbed. In AD, the damage of microvessels is believed to be associated with the progression of the disease (Johnson et al., 2005; Marchesi, 2011; Rosenberg, 2014). In mouse models and in AD patients, an alteration of the BBB physiology exists and is linked with amyloid deposits (Gosselet et al., 2013; Montagne et al., 2017; Yamazaki and Kanekiyo, 2017) Moreover, Aβ peptides are normally cleared from the brain by specific transport across the BBB. Receptors and transporters expressed by NVU cells like LRP1, the P-glycoprotein (P-gp) and Breast Cancer Resistance Protein (BCRP), play an important role in Aβ transport through the BBB (Gosselet et al., 2013; Storck et al., 2016). LRP1 may also be involved in Aβ endocytosis in endothelial cells for degradation via the lysosomal pathway (Nazer et al., 2008). Other receptors such as the receptor for advanced glycation end products (RAGE) are involved in Aβ entry into the CNS (Candela et al., 2010; Bu et al., 2017). Downregulations of LRP1, P-gp and BCRP or upregulation of RAGE during AD potentiate the Aβ accumulation (Gosselet et al., 2013; Yamazaki and Kanekiyo, 2017). In addition, Aβ can lead to a decrease of tight junction (TJ) proteins (occludin, claudin-5 and ZO-1) in endothelial cells and reduction of microvessel coverage by pericytes, which induces higher BBB permeability (Gosselet et al., 2013; Yamazaki and Kanekiyo, 2017).
During HIV-1 infection, impairment of the BBB occurs and is probably responsible for the spread of virions from the vascular compartment, enhances immune cell recruitment, and may allow brain infection by other opportunistic pathogens (Atluri et al., 2015; Zhang et al., 2015; Anesten et al., 2016). The interaction between HIV-1 and the BBB occurs with all of the NVU cells and often involves viral proteins. HIV-infected astrocytes can directly impair BBB integrity by dysregulating gap junctions (Eugenin et al., 2011). Tat, gp120, Vpr and Nef, have shown cellular alteration, dysregulation of molecular pathways and an inhibition of repair mechanisms resulting in impairment of the BBB (Zhang et al., 2015). Tat has been shown to directly modulate the endothelium through several cellular pathways, including the inhibition of the RhoA/ROCK or Ras pathways, which lead to the downregulation of TJ proteins expression, resulting in the BBB impairment (Chen et al., 2012, 2016; Jiang et al., 2017). These effects are accompanied by an accumulation of Aβ peptide in the brain, suggesting a direct role for HIV proteins in Aβ-BBB interaction. Interestingly, Tat can also modulate the expression of receptors and transporters of Aβ, which are involved in bi-directional transport of peptides across the BBB. Extracellular Tat can upregulate RAGE expression resulting in Ras/MAPK signaling pathway activation and in fine Aβ accumulation (András and Toborek, 2013; Chen et al., 2016). Tat can also reduce Aβ clearance across endothelial cells in inhibiting the synthesis of LRP-1 (Chen et al., 2016). Similarly, gp120 has been shown to impair BBB integrity through the PKC and JAK/STAT pathways and to enhance monocyte migration, a process that can also increase the number of HIV-infected monocytes that cross the BBB to get to the CNS (Kanmogne et al., 2007; Yang et al., 2009; Zhang et al., 2015). Conversely, rats injected with recombinant gp120 in the caudate putamen showed lesion in brain microvessels, suggesting that gp120 could directly impair brain endothelial cell physiology and therefore BBB integrity (Louboutin et al., 2010).
Ultimately, these mechanisms could disturb Aβ clearance through the interstitial fluid, resulting in an increase in Aβ deposit and accumulation. In this context, it is more than pertinent to consider the role of the BBB in AD and HAND pathogenesis (Yamazaki and Kanekiyo, 2017).
HIV, HPA Axis Dysregulation and AD: GC as a Link Between HIV Infection and AD?
In HIV+ patients treated with cART, the adrenal gland is frequently affected, resulting in higher serum cortisol levels both in early stages and severely affected patients (Sellmeyer and Grunfeld, 1996; Christeff et al., 1997; Collazos et al., 2003; Langerak et al., 2015). In addition, an excess of circulating GC can also dysregulate numerous brain functions. As GC act in synergy with glutamate, a deregulation of the HPA axis activity or a modification of GR function can be toxic, especially in the hippocampus by inducing excitotoxicity, neuronal damage (neuronal death by apoptosis and synaptic deficits), neuroinflammation, oxidative stress and cognitive decline (Magariños and McEwen, 1995; de Kloet et al., 2005). In fact, it appears that plasma cortisol levels correlate with the severity of hippocampal atrophy and therefore could contribute to the cognitive decline and psychological symptoms that occur in neurodegenerative pathologies and particularly in AD (Lupien et al., 1998). In AD, this view is particularly sustained by the fact that cognitive deficits and psychological symptoms are associated with an early deregulation of the HPA axis, as well as elevated levels of GC in plasma and CSF (Csernansky et al., 2006; Hoogendijk et al., 2006). Moreover, GC and GR can directly trigger APP misprocessing and Aβ pathway through the direct transcription of APP and BACE1 genes (Lahiri, 2004). Along this line, GC and stress appear to induce abnormal Tau hyperphosphorylation and accumulation (Sotiropoulos et al., 2011). This suggests that dysregulations of the HPA axis would likely increase Aβ pathology and subsequent Tau accumulation and hyperphosphorylation, resulting inexorably in a vicious circle whereby the pathology increases the secretion of GC, which further increases the pathology (Green et al., 2006; Brureau et al., 2013; Pineau et al., 2016).
In HAND, viral proteins such as Tat and gp120, together with GC, could have synergistic effects on excitotoxicity and oxidative stress, by decreasing glutamate uptake by different but complementary processes, as previously mentioned. This is illustrated by the fact that combined treatment of GC and gp120 directly increases calcium mobilization, ATP depletion, decrease of mitochondrial potential, ROS production and neurotoxicity (Brooke and Sapolsky, 2003). Basal levels of GC enhanced the disruptive effects of gp120 on metabolism in the hippocampus and in the cortex. Moreover, raising GC concentration exacerbated the ability of gp120 to mobilize cytosolic calcium in hippocampal cells (Yusim et al., 2000; Brooke and Sapolsky, 2003). Acute exposure to GC initiated the endocytosis of glucose transporter in neurons and glia, whereas chronic exposure to GC could directly inhibit its transcription, resulting in energy depletion (Brooke and Sapolsky, 2003). The decrease of glucose transporter is responsible for a lack of ATP production, necessary to supply sodium/potassium pumps that are part of the glutamate transporter system (EAATs; Virgin et al., 1991; Chan et al., 1996). Gp120 and Tat act on astrocyte, microglia and neurons. Especially, gp120 induces arachidonic acid and superoxide release that in turn inhibits glutamate uptake by glia cells (Dreyer and Lipton, 1995; Belmadani et al., 2001). So, taken together, GC, Tat and gp120 strongly block glutamate uptake (Wang et al., 2004; Cheung et al., 2008; Potter et al., 2013), which could potentiate excitotoxicity and neuronal loss observed in HAND patients. Such mechanisms were already described in AD, and potentially due to GC excess (Virgin et al., 1991; Goodman et al., 1996). Even if this view is speculative and needs further investigation, GC and HPA axis deregulation could participate in neuronal dysfunction in HIV patients. Excessive GC secretion, in synergy with viral proteins, could trigger and/or accelerate the onset of the pathology by altering APP processing, Tau phosphorylation and BBB integrity. In addition, synergetic action between GC and HIV could potentiate oxidative stress induction, excitotoxicity and neuroinflammation and, finally, cognitive decline (Figure 2).
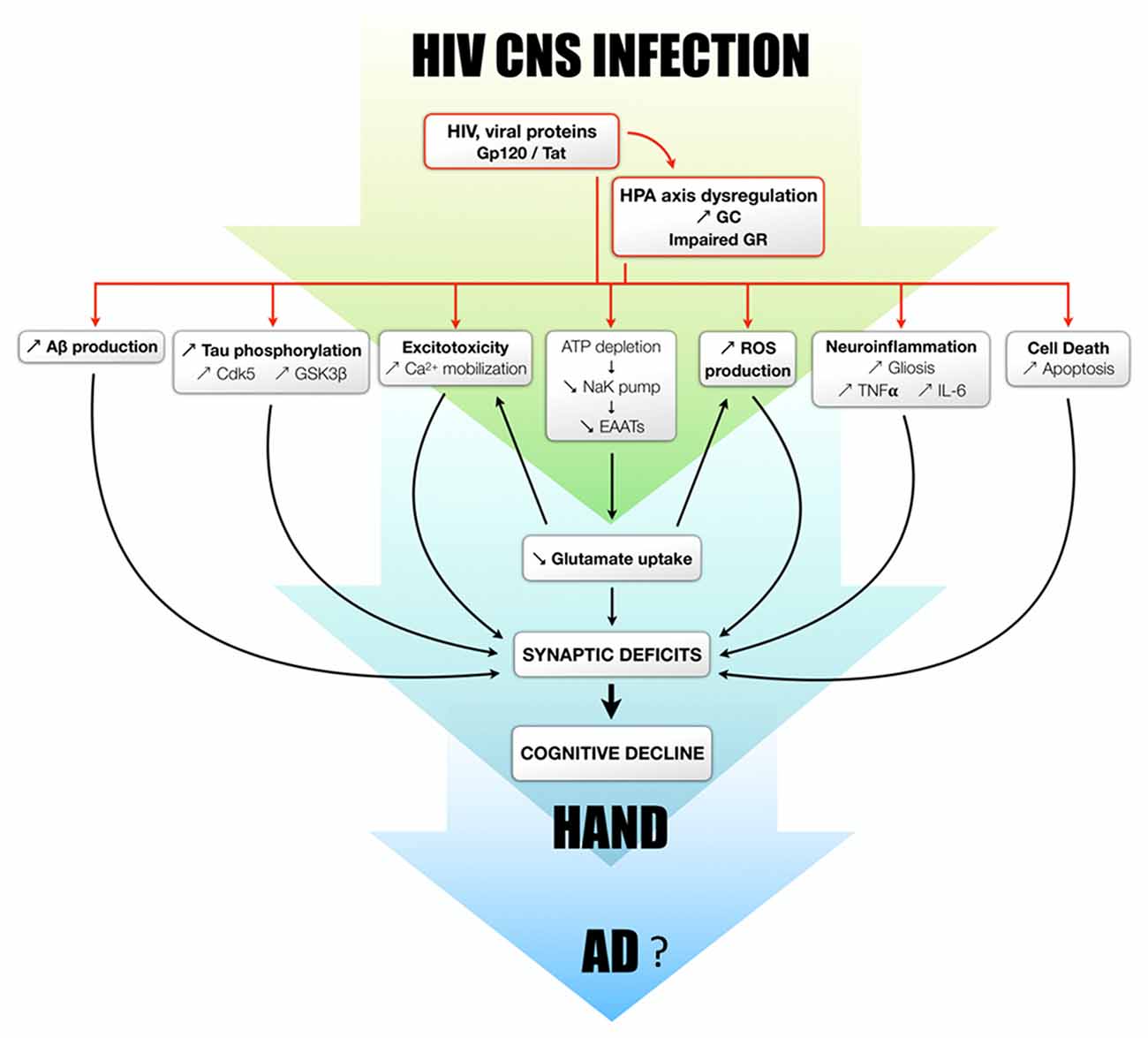
Figure 2. Proposed mechanisms by which glucocorticoids and their receptors modulate/potentiate the development of HAND and potentially AD. The dysregulation of the HPA axis is observed both in HIV patients and rodent models. GC overexposure, in combination with viral proteins or not, is able to induce the increase of Aβ production, Tau phosphorylation, excitotoxicity, oxidative stress, neuroinflammation and apoptosis. It should be also mentioned that Aβ itself can trigger Tau phosphorylation, excitotoxicity, oxidative stress, neuroinflammation and cell death. All these processes lead to neurodegeneration and synaptic deficits and potentially responsible for cognitive decline observed in HAND patients, all of which could progressively favor to the development of AD.
Perspectives and Conclusion
In recent years, a consensus has emerged regarding the etiology of neurological disorders, which is now believed to be complex and multifactorial, even in the case of familial (genetic) forms. A concept that is clearly emerging though is that acute and chronic (neuro) inflammation may pave the way to such disorders and neurotropic pathogens may therefore represent likely candidates among environmental factors promoting this inflammatory/causative state. Inflammation, in particular in the brain, is a clear hallmark of HIV infection, and can also be found in virally suppressed individuals (Edén et al., 2007). in vitro and in vivo studies show neurotoxic effects of some HIV proteins. Direct and indirect effects, coupled to the potential toxicity of some anti-HIV drugs could provide a weakened environment where the onset of neurological disorders (e.g., AD), could be favored.
HIV neuroinfection shares many pathways and characteristics of familial and sporadic forms of AD, but whether HIV patients will have more chance to develop bona fide AD is not yet fully understood and will have to be determined in the following years. The HIV+ population is aging and starting to face age-related disorders. However, there is still little information on their susceptibility to neuronal disorders besides what qualifies as HAND. Although large epidemiological studies are still needed to definitively conclude that aging HIV+ patients are more at risk of developing AD, there are however key physiological findings that suggest that this could be the case, though debate between neurologists exists (Ortega and Ances, 2014; Chakradhar, 2018). In this context, it was reported that HIV-1 modulates Aβ metabolism differently from what is observed in AD (in terms of Aβ deposition patterns), which suggests unique features for HAD and HAND (Ortega and Ances, 2014). This also suggests that the onset of dementia in aged HIV+ patients could be due to either HIV but also to AD or a combination of both. Identification of HIV+ patients with AD are starting to be reported and may provide more clinical data regarding how these two diseases may or may not be connected (Mäkitalo et al., 2015; Turner et al., 2016). Because of the similarities between HAND and AD one of the challenges facing neurologists will be to discriminate between advanced cognitive impairment caused by HIV (HAD) and AD (Xu and Ikezu, 2009). Distinctive CSF biomarker profiles, coupled to appropriate imaging techniques are keys to understanding and diagnosing these pathologies. In this light, the study by Mäkitalo et al. (2015) is of particular importance as it reports the CSF pattern of a HIV+ patient having developed AD, possibly because of HIV CSF escape.
In conclusion, the causes behind sporadic forms of AD are still poorly characterized, but environmental factors, in combination or not with genetic triggers, are emerging as key players in their onset. In particular, the dysregulation of the HPA axis that can associated with HIV infection could favor an environment where oxidative stress, neuroinflammation, excitotoxicity, BBB disruption and amyloid-β load are exacerbated and thus, combined with other (genetic or environmental) factors pave the way for the establishment of brain diseases such as AD. It is therefore not unlikely that HIV infection may represent a risk factor for AD and other related neuronal disorders.
Author Contributions
SS and LG participated in manuscript design, writing and coordination. GC, CD, AG, YS, FG, NM, AM, ET and PP participated in manuscript writing and editing. All authors read and approved the final manuscript.
Funding
This work was supported by La Région Languedoc Roussillon (SS), France Alzheimer (LG), Fédération pour la Recherche sur le Cerveau (LG, NM), ANR-Epicyte (NM), ANR Hepatobrain (NM). GC is supported by a Ph.D. Grant from the University of Montpellier, France (CBS2 Ph.D. program).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank members of PCCI and MMDN laboratories for their help and support and Vasiliki Kalatzis for proof reading. LG is a CNRS fellow and SS is an Institut National de la Santé et de la Recherche Médicale (INSERM) research fellow.
Abbreviations
AD, Alzheimer’s disease; AIDS, acquired immunodeficiency syndrome; ALCAM, activated leukocyte cell adhesion molecule; ALS, amyotrophic lateral sclerosis; ANI, asymptomatic neurocognitive impairment; APP, amyloid precursor protein; Aβ, amyloid-β peptides; BACE1, β-secretase converting enzyme 1; BBB, blood brain barrier; BCRP, breast cancer resistance protein; cART, combination antiretroviral therapy; CMV, cytomegalovirus; CNS, central nervous system; CSF, cerebrospinal fluid; GC, glucocorticoids; gp120, glycoprotein 120; GR, glucocorticoid receptors; HAD, HIV-associated dementia; HAND, HIV-associated neurocognitive disorders; HD, Huntington disease; HIV, human immunodeficiency virus; HPA axis, hypothalamo-pituitary-adrenal axis; HSV, Herpes simplex virus; JAM-A, junctional adhesion molecule A; LPR, low-density lipoprotein receptor-related protein; MND, mild neurocognitive disorder; MND, motor neuron diseases; MR, mineralocorticoid receptors; MS, multiple sclerosis; Nef, negative regulatory factor; NFT, neurofibrillary tangles; NVU, neurovascular unit; P-gp, P-glycoprotein; PD, Parkinson’s disease; RAGE, receptor for advanced glycation end products; ROS, reactive oxygen species; Tat, transactivator of transcription; TJ, tight junctions; TNF-α, tumor necrosis factor-α; Vpr, viral protein R.
References
Adle-Biassette, H., Chrétien, F., Wingertsmann, L., Héry, C., Ereau, T., Scaravilli, F., et al. (1999). Neuronal apoptosis does not correlate with dementia in HIV infection but is related to microglial activation and axonal damage. Neuropathol. Appl. Neurobiol. 25, 123–133. doi: 10.1046/j.1365-2990.1999.00167.x
Aksenov, M. Y., Aksenova, M. V., Mactutus, C. F., and Booze, R. M. (2010). HIV-1 protein-mediated amyloidogenesis in rat hippocampal cell cultures. Neurosci. Lett. 475, 174–178. doi: 10.1016/j.neulet.2010.03.073
Alfahad, T., and Nath, A. (2013). Retroviruses and amyotrophic lateral sclerosis. Antiviral Res. 99, 180–187. doi: 10.1016/j.antiviral.2013.05.006
András, I. E., and Toborek, M. (2013). Amyloid beta accumulation in HIV-1-infected brain: the role of the blood brain barrier. IUBMB Life 65, 43–49. doi: 10.1002/iub.1106
Anesten, B., Yilmaz, A., Hagberg, L., Zetterberg, H., Nilsson, S., Brew, B. J., et al. (2016). Blood-brain barrier integrity, intrathecal immunoactivation and neuronal injury in HIV. Neurol. Neuroimmunol. Neuroinflamm. 3:e300. doi: 10.1212/nxi.0000000000000300
Anthony, I. C., Ramage, S. N., Carnie, F. W., Simmonds, P., and Bell, J. E. (2006). Accelerated Tau deposition in the brains of individuals infected with human immunodeficiency virus-1 before and after the advent of highly active anti-retroviral therapy. Acta Neuropathol. 111, 529–538. doi: 10.1007/s00401-006-0037-0
Antinori, A., Arendt, G., Becker, J. T., Brew, B. J., Byrd, D. A., Cherner, M., et al. (2007). Updated research nosology for HIV-associated neurocognitive disorders. Neurology 69, 1789–1799. doi: 10.1212/01.WNL.0000287431.88658.8b
Atluri, V. S. R., Hidalgo, M., Samikkannu, T., Kurapati, K. R. V., Jayant, R. D., Sagar, V., et al. (2015). Effect of human immunodeficiency virus on blood-brain barrier integrity and function: an update. Front. Cell. Neurosci. 9:212. doi: 10.3389/fncel.2015.00212
Avdoshina, V., Bachis, A., and Mocchetti, I. (2013). Synaptic dysfunction in human immunodeficiency virus type-1-positive subjects: inflammation or impaired neuronal plasticity? J. Intern. Med. 273, 454–465. doi: 10.1111/joim.12050
Azevedo, E. P., Ledo, J. H., Barbosa, G., Sobrinho, M., Diniz, L., Fonseca, A. C. C., et al. (2013). Activated microglia mediate synapse loss and short-term memory deficits in a mouse model of transthyretin-related oculoleptomeningeal amyloidosis. Cell Death Dis. 4:e789. doi: 10.1038/cddis.2013.325
Bachis, A., Biggio, F., Major, E. O., and Mocchetti, I. (2009). M- and T-tropic HIVs promote apoptosis in rat neurons. J. Neuroimmune Pharmacol. 4, 150–160. doi: 10.1007/s11481-008-9141-3
Bateman, R. J., Xiong, C., Benzinger, T. L. S., Fagan, A. M., Goate, A., Fox, N. C., et al. (2012). Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 367, 795–804. doi: 10.1056/NEJMoa1202753
Becker, J. T., Kingsley, L., Mullen, J., Cohen, B., Martin, E., Miller, E. N., et al. (2009). Vascular risk factors, HIV serostatus and cognitive dysfunction in gay and bisexual men. Neurology 73, 1292–1299. doi: 10.1212/wnl.0b013e3181bd10e7
Belmadani, A., Zou, J. Y., Schipma, M. J., Neafsey, E. J., and Collins, M. A. (2001). Ethanol pre-exposure suppresses HIV-1 glycoprotein 120-induced neuronal degeneration by abrogating endogenous glutamate/Ca2+-mediated neurotoxicity. Neuroscience 104, 769–781. doi: 10.1016/s0306-4522(01)00139-7
Blennow, K., Hampel, H., Weiner, M., and Zetterberg, H. (2010). Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat. Rev. Neurol. 6, 131–144. doi: 10.1038/nrneurol.2010.4
Brew, B. J., Pemberton, L., Blennow, K., Wallin, A., and Hagberg, L. (2005). CSF amyloid β42 and tau levels correlate with AIDS dementia complex. Neurology 65, 1490–1492. doi: 10.1212/01.wnl.0000183293.95787.b7
Brooke, S. M., and Sapolsky, R. M. (2003). Effects of glucocorticoids in the gp120-induced inhibition of glutamate uptake in hippocampal cultures. Brain Res. 972, 137–141. doi: 10.1016/s0006-8993(03)02517-4
Brureau, A., Zussy, C., Delair, B., Ogier, C., Ixart, G., Maurice, T., et al. (2013). Deregulation of hypothalamic-pituitary-adrenal axis functions in an Alzheimer’s disease rat model. Neurobiol. Aging 34, 1426–1439. doi: 10.1016/j.neurobiolaging.2012.11.015
Bu, X.-L., Xiang, Y., Jin, W.-S., Wang, J., Shen, L.-L., Huang, Z.-L., et al. (2017). Blood-derived amyloid-β protein induces Alzheimer’s disease pathologies. Mol. Psychiatry doi: 10.1038/mp.2017.204 [Epub ahead of print].
Candela, P., Gosselet, F., Saint-Pol, J., Sevin, E., Boucau, M.-C., Boulanger, E., et al. (2010). Apical-to-basolateral transport of amyloid-β peptides through blood-brain barrier cells is mediated by the receptor for advanced glycation end-products and is restricted by P-glycoprotein. J. Alzheimers Dis. 22, 849–859. doi: 10.3233/jad-2010-100462
Cecchelli, R., Aday, S., Sevin, E., Almeida, C., Culot, M., Dehouck, L., et al. (2014). A stable and reproducible human blood-brain barrier model derived from hematopoietic stem cells. PLoS One 9:e99733. doi: 10.1371/journal.pone.0099733
Chai, Q., Jovasevic, V., Malikov, V., Sabo, Y., Morham, S., Walsh, D., et al. (2017). HIV-1 counteracts an innate restriction by amyloid precursor protein resulting in neurodegeneration. Nat. Commun. 8:1522. doi: 10.1038/s41467-017-01795-8
Chakradhar, S. (2018). A tale of two diseases: aging HIV patients inspire a closer look at Alzheimer’s disease. Nat. Med. 24, 376–377. doi: 10.1038/nm0418-376
Chan, R. S., Huey, E. D., Maecker, H. L., Cortopassi, K. M., Howard, S. A., Iyer, A. M., et al. (1996). Endocrine modulators of necrotic neuron death. Brain Pathol. 6, 481–491. doi: 10.1111/j.1750-3639.1996.tb00877.x
Charmandari, E., Raji, A., Kino, T., Ichijo, T., Tiulpakov, A., Zachman, K., et al. (2005). A novel point mutation in the ligand-binding domain (LBD) of the human glucocorticoid receptor (hGR) causing generalized glucocorticoid resistance: the importance of the C terminus of hGR LBD in conferring transactivational activity. J. Clin. Endocrinol. Metab. 90, 3696–3705. doi: 10.1210/jc.2004-1920
Chen, D. Y., Bambah-Mukku, D., Pollonini, G., and Alberini, C. M. (2012). Glucocorticoid receptors recruit the CaMKIIα-BDNF-CREB pathways to mediate memory consolidation. Nat. Neurosci. 15, 1707–1714. doi: 10.1038/nn.3266
Chen, L., Choi, J. J., Choi, Y. J., Hennig, B., and Toborek, M. (2012). HIV-1 Tat-induced cerebrovascular toxicity is enhanced in mice with amyloid deposits. Neurobiol. Aging 33, 1579–1590. doi: 10.1016/j.neurobiolaging.2011.06.004
Chen, Y., Huang, W., Jiang, W., Wu, X., Ye, B., and Zhou, X. (2016). HIV-1 Tat regulates occludin and Aβ transfer receptor expression in brain endothelial cells via Rho/ROCK signaling pathway. Oxid. Med. Cell. Longev. 2016:4196572. doi: 10.1155/2016/4196572
Chen, X., Hui, L., Geiger, N. H., Haughey, N. J., and Geiger, J. D. (2013). Endolysosome involvement in HIV-1 transactivator protein-induced neuronal amyloid beta production. Neurobiol. Aging 34, 2370–2378. doi: 10.1016/j.neurobiolaging.2013.04.015
Cheung, R., Ravyn, V., Wang, L., Ptasznik, A., and Collman, R. G. (2008). Signaling mechanism of HIV-1 gp120 and virion-induced IL-1β release in primary human macrophages. J. Immunol. 180, 6675–6684. doi: 10.4049/jimmunol.180.10.6675
Cho, Y.-E., Lee, M.-H., and Song, B.-J. (2017). Neuronal cell death and degeneration through increased nitroxidative stress and tau phosphorylation in HIV-1 transgenic rats. PLoS One 12:e0169945. doi: 10.1371/journal.pone.0169945
Christeff, N., Gherbi, N., Mammes, O., Dalle, M. T., Gharakhanian, S., Lortholary, O., et al. (1997). Serum cortisol and DHEA concentrations during HIV infection. Psychoneuroendocrinology 22, S11–S18. doi: 10.1016/s0306-4530(97)00015-2
Chrousos, G. P., Detera-Wadleigh, S. D., and Karl, M. (1993). Syndromes of glucocorticoid resistance. Ann. Intern. Med. 119, 1113–1124. doi: 10.7326/0003-4819-119-11-199312010-00009
Chrousos, G. P., and Zapanti, E. D. (2014). Hypothalamic-pituitary-adrenal axis in HIV infection and disease. Endocrinol. Metab. Clin. North Am. 43, 791–806. doi: 10.1016/j.ecl.2014.06.002
Churchill, M. J., Cowley, D. J., Wesselingh, S. L., Gorry, P. R., and Gray, L. R. (2015). HIV-1 transcriptional regulation in the central nervous system and implications for HIV cure research. J. Neurovirol. 21, 290–300. doi: 10.1007/s13365-014-0271-5
Cleary, J. P., Walsh, D. M., Hofmeister, J. J., Shankar, G. M., Kuskowski, M. A., Selkoe, D. J., et al. (2005). Natural oligomers of the amyloid-β protein specifically disrupt cognitive function. Nat. Neurosci. 8, 79–84. doi: 10.1038/nn1372
Clifford, D. B., and Ances, B. M. (2013). HIV-associated neurocognitive disorder. Lancet Infect. Dis. 13, 976–986. doi: 10.1016/S1473-3099(13)70269-X
Clifford, D. B., Fagan, A. M., Holtzman, D. M., Morris, J. C., Teshome, M., Shah, A. R., et al. (2009). CSF biomarkers of Alzheimer disease in HIV-associated neurologic disease. Neurology 73, 1982–1987. doi: 10.1212/WNL.0b013e3181c5b445
Collazos, J., Mayo, J., Martínez, E., and Ibarra, S. (2003). Serum cortisol in HIV-infected patients with and without highly active antiretroviral therapy. AIDS 17, 123–126. doi: 10.1097/01.aids.0000042598.93174.d2
Cosenza, M. A., Zhao, M.-L., Si, Q., and Lee, S. C. (2002). Human brain parenchymal microglia express CD14 and CD45 and are productively infected by HIV-1 in HIV-1 encephalitis. Brain Pathol. 12, 442–455. doi: 10.1111/j.1750-3639.2002.tb00461.x
Costa, A., Nappi, R. E., Polatti, F., Poma, A., Grossman, A. B., and Nappi, G. (2000). Stimulating effect of HIV-1 coat protein gp120 on corticotropin-releasing hormone and arginine vasopressin in the rat hypothalamus: involvement of nitric oxide. Exp. Neurol. 166, 376–384. doi: 10.1006/exnr.2000.7502
Coutinho, A. E., and Chapman, K. E. (2011). The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol. Cell. Endocrinol. 335, 2–13. doi: 10.1016/j.mce.2010.04.005
Csernansky, J. G., Dong, H., Fagan, A. M., Wang, L., Xiong, C., Holtzman, D. M., et al. (2006). Plasma cortisol and progression of dementia in subjects with Alzheimer-type dementia. Am. J. Psychiatry 163, 2164–2169. doi: 10.1176/appi.ajp.163.12.2164
Cysique, L. A. J., Maruff, P., and Brew, B. J. (2006). Variable benefit in neuropsychological function in HIV-infected HAART-treated patients. Neurology 66, 1447–1450. doi: 10.1212/01.wnl.0000210477.63851.d3
Daily, A., Nath, A., and Hersh, L. B. (2006). Tat peptides inhibit neprilysin. J. Neurovirol. 12, 153–160. doi: 10.1080/13550280600760677
De Chiara, G., Marcocci, M. E., Sgarbanti, R., Civitelli, L., Ripoli, C., Piacentini, R., et al. (2012). Infectious agents and neurodegeneration. Mol. Neurobiol. 46, 614–638. doi: 10.1007/s12035-012-8320-7
de Kloet, E. R., Joëls, M., and Holsboer, F. (2005). Stress and the brain: from adaptation to disease. Nat. Rev. Neurosci. 6, 463–475. doi: 10.1038/nrn1683
Dreyer, E. B., and Lipton, S. A. (1995). The coat protein gp120 of HIV-1 inhibits astrocyte uptake of excitatory amino acids via macrophage arachidonic acid. Eur. J. Neurosci. 7, 2502–2507. doi: 10.1111/j.1460-9568.1995.tb01048.x
Edén, A., Price, R. W., Spudich, S., Fuchs, D., Hagberg, L., and Gisslén, M. (2007). Immune activation of the central nervous system is still present after >4 years of effective highly active antiretroviral therapy. J. Infect. Dis. 196, 1779–1783. doi: 10.1086/523648
Ellis, R., Langford, D., and Masliah, E. (2007). HIV and antiretroviral therapy in the brain: neuronal injury and repair. Nat. Rev. Neurosci. 8, 33–44. doi: 10.1038/nrn2040
Esiri, M. M., Biddolph, S. C., and Morris, C. S. (1998). Prevalence of Alzheimer plaques in AIDS. J. Neurol. Neurosurg. Psychiatry 65, 29–33. doi: 10.1136/jnnp.65.1.29
Eugenin, E. A., Clements, J. E., Zink, M. C., and Berman, J. W. (2011). Human immunodeficiency virus infection of human astrocytes disrupts blood-brain barrier integrity by a gap junction-dependent mechanism. J. Neurosci. 31, 9456–9465. doi: 10.1523/JNEUROSCI.1460-11.2011
Ferrarese, C., Aliprandi, A., Tremolizzo, L., Stanzani, L., De Micheli, A., Dolara, A., et al. (2001). Increased glutamate in CSF and plasma of patients with HIV dementia. Neurology 57, 671–675. doi: 10.1212/wnl.57.4.671
Flammang, B., Pardossi-Piquard, R., Sevalle, J., Debayle, D., Dabert-Gay, A.-S., Thévenet, A., et al. (2012). Evidence that the amyloid-β protein precursor intracellular domain, AICD, derives from β-secretase-generated C-terminal fragment. J. Alzheimers Dis. 30, 145–153. doi: 10.3233/JAD-2012-112186
Fratiglioni, L., Ahlbom, A., Viitanen, M., and Winblad, B. (1993). Risk factors for late- onset Alzheimer’s disease: a population- based, case-control study. Ann. Neurol. 33, 258–266. doi: 10.1002/ana.410330306
Garden, G. A., Budd, S. L., Tsai, E., Hanson, L., Kaul, M., D’Emilia, D. M., et al. (2002). Caspase cascades in human immunodeficiency virus-associated neurodegeneration. J. Neurosci. 22, 4015–4024. doi: 10.1523/jneurosci.22-10-04015.2002
Givalois, L., Dornand, J., Mekaouche, M., Solier, M. D., Bristow, A. F., Ixart, G., et al. (1994). Temporal cascade of plasma level surges in ACTH, corticosterone and cytokines in endotoxin-challenged rats. Am. J. Physiol. 267, R164–R170. doi: 10.1152/ajpregu.1994.267.1.r164
González-Scarano, F., and Martín-García, J. (2005). The neuropathogenesis of AIDS. Nat. Rev. Immunol. 5, 69–81. doi: 10.1038/nri1527
Goodman, Y., Bruce, A. J., Cheng, B., and Mattson, M. P. (1996). Estrogens attenuate and corticosterone exacerbates excitotoxicity, oxidative injury and amyloid β-peptide toxicity in hippocampal neurons. J. Neurochem. 66, 1836–1844. doi: 10.1046/j.1471-4159.1996.66051836.x
Gosselet, F., Saint-Pol, J., Candela, P., and Fenart, L. (2013). Amyloid-β peptides, Alzheimer’s disease and the blood-brain barrier. Curr. Alzheimer Res. 10, 1015–1033. doi: 10.2174/15672050113106660174
Green, K. N., Billings, L. M., Roozendaal, B., McGaugh, J. L., and LaFerla, F. M. (2006). Glucocorticoids increase amyloid-β and tau pathology in a mouse model of Alzheimer’s disease. J. Neurosci. 26, 9047–9056. doi: 10.1523/jneurosci.2797-06.2006
Green, D. A., Masliah, E., Vinters, H. V., Beizai, P., Moore, D. J., and Achim, C. L. (2005). Brain deposition of beta-amyloid is a common pathologic feature in HIV positive patients. AIDS 19, 407–411. doi: 10.1097/01.aids.0000161770.06158.5c
Habbas, S., Santello, M., Becker, D., Stubbe, H., Zappia, G., Liaudet, N., et al. (2015). Neuroinflammatory TNFα impairs memory via astrocyte signaling. Cell 163, 1730–1741. doi: 10.1016/j.cell.2015.11.023
Hategan, A., Bianchet, M. A., Steiner, J., Karnaukhova, E., Masliah, E., Fields, A., et al. (2017). HIV Tat protein and amyloid-β peptide form multifibrillar structures that cause neurotoxicity. Nat. Struct. Mol. Biol. 24, 379–386. doi: 10.1038/nsmb.3379
Haughey, N. J., Nath, A., Mattson, M. P., Slevin, J. T., and Geiger, J. D. (2001). HIV-1 Tat through phosphorylation of NMDA receptors potentiates glutamate excitotoxicity. J. Neurochem. 78, 457–467. doi: 10.1046/j.1471-4159.2001.00396.x
Hayden, K. M., Norton, M. C., Darcey, D., Ostbye, T., Zandi, P. P., Breitner, J. C. S., et al. (2010). Occupational exposure to pesticides increases the risk of incident AD: the Cache County study. Neurology 74, 1524–1530. doi: 10.1212/wnl.0b013e3181dd4423
Heaton, R. K., Franklin, D. R., Ellis, R. J., McCutchan, J. A., Letendre, S. L., Leblanc, S., et al. (2011). HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. J. Neurovirol. 17, 3–16. doi: 10.1007/s13365-010-0006-1
Hellmuth, J., Valcour, V., and Spudich, S. (2015). CNS reservoirs for HIV: implications for eradication. J. Virus Erad. 1, 67–71.
Hong, S., Beja-Glasser, V. F., Nfonoyim, B. M., Frouin, A., Li, S., Ramakrishnan, S., et al. (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. doi: 10.1126/science.aad8373
Hong, H., Kim, B. S., and Im, H. (2016). Pathophysiological role of neuroinflammation in neurodegenerative diseases and psychiatric disorders. Int. Neurourol. J. 20, S2–S7. doi: 10.5213/inj.1632604.302
Hoogendijk, W. J. G., Meynen, G., Endert, E., Hofman, M. A., and Swaab, D. F. (2006). Increased cerebrospinal fluid cortisol level in Alzheimer’s disease is not related to depression. Neurobiol. Aging 27, 780.e1–780.e2. doi: 10.1016/j.neurobiolaging.2005.07.017
Hudson, L., Liu, J., Nath, A., Jones, M., Raghavan, R., Narayan, O., et al. (2000). Detection of the human immunodeficiency virus regulatory protein tat in CNS tissues. J. Neurovirol. 6, 145–155. doi: 10.3109/13550280009013158
Itzhaki, R. F. (2014). Herpes simplex virus type 1 and Alzheimer’s disease: increasing evidence for a major role of the virus. Front. Aging Neurosci. 6:202. doi: 10.3389/fnagi.2014.00202
Itzhaki, R. F., Lin, W.-R., Shang, D., Wilcock, G. K., Faragher, B., and Jamieson, G. A. (1997). Herpes simplex virus type 1 in brain and risk of Alzheimer’s disease. Lancet 349, 241–244. doi: 10.1016/S0140-6736(96)10149-5
Janssen, R. (1992). Epidemiology of human immunodeficiency virus infection and the neurologic complications of the infection. Semin. Neurol. 12, 10–17. doi: 10.1055/s-2008-1041152
Jiang, W., Huang, W., Chen, Y., Zou, M., Peng, D., and Chen, D. (2017). HIV-1 transactivator protein induces ZO-1 and neprilysin dysfunction in brain endothelial cells via the ras signaling pathway. Oxid. Med. Cell. Longev. 2017:3160360. doi: 10.1155/2017/3160360
Johnson, N. A., Jahng, G.-H., Weiner, M. W., Miller, B. L., Chui, H. C., Jagust, W. J., et al. (2005). Pattern of cerebral hypoperfusion in Alzheimer disease and mild cognitive impairment measured with arterial spin-labeling MR imaging: initial experience. Radiology 234, 851–859. doi: 10.1148/radiol.2343040197
Kang, Y.-J., Digicaylioglu, M., Russo, R., Kaul, M., Achim, C. L., Fletcher, L., et al. (2010). Erythropoietin plus insulin-like growth factor-I protects against neuronal damage in a murine model of human immunodeficiency virus-associated neurocognitive disorders. Ann. Neurol. 68, 342–352. doi: 10.1002/ana.22070
Kanmogne, G. D., Schall, K., Leibhart, J., Knipe, B., Gendelman, H. E., and Persidsky, Y. (2007). HIV-1 gp120 compromises blood-brain barrier integrity and enhance monocyte migration across blood-brain barrier: implication for viral neuropathogenesis. J. Cereb. Blood Flow Metab. 27, 123–134. doi: 10.1038/sj.jcbfm.9600330
Kaul, M., and Lipton, S. A. (1999). Chemokines and activated macrophages in HIV gp120-induced neuronal apoptosis. Proc. Natl. Acad. Sci. U S A 96, 8212–8216. doi: 10.1073/pnas.96.14.8212
Ketzler, S., Weis, S., Haug, H., and Budka, H. (1990). Loss of neurons in the frontal cortex in AIDS brains. Acta Neuropathol. 80, 92–94. doi: 10.1007/bf00294228
Kim, J., Yoon, J.-H., and Kim, Y.-S. (2013). HIV-1 Tat interacts with and regulates the localization and processing of amyloid precursor protein. PLoS One 8:e77972. doi: 10.1371/journal.pone.0077972
King, J. E., Eugenin, E. A., Buckner, C. M., and Berman, J. W. (2006). HIV tat and neurotoxicity. Microbes Infect. 8, 1347–1357. doi: 10.1016/j.micinf.2005.11.014
Kino, T. (2000). “AIDS/HPA axis,” in Endotext [Internet]. eds L. J. De Groot, G. Chrousos and K. Dungan (South Dartmouth, MA: MDText.com Inc.) Available online at: www.endotext.org
Kruman, I. I., Nath, A., and Mattson, M. P. (1998). HIV-1 protein Tat induces apoptosis of hippocampal neurons by a mechanism involving caspase activation, calcium overload and oxidative stress. Exp. Neurol. 154, 276–288. doi: 10.1006/exnr.1998.6958
Lahiri, D. K. (2004). Functional characterization of amyloid β precursor protein regulatory elements: rationale for the identification of genetic polymorphism. Ann. N Y Acad. Sci. 1030, 282–288. doi: 10.1196/annals.1329.035
Langerak, T., van den Dries, L. W. J., Wester, V. L., Staufenbiel, S. M., Manenschijn, L., van Rossum, E. F. C., et al. (2015). The relation between long-term cortisol levels and the metabolic syndrome in HIV-infected patients. Clin. Endocrinol. 83, 167–172. doi: 10.1111/cen.12790
Lannuzel, A., Barnier, J. V., Hery, C., Huynh, V. T., Guibert, B., Gray, F., et al. (1997). Human immunodeficiency virus type 1 and its coat protein gp120 induce apoptosis and activate JNK and ERK mitogen-activated protein kinases in human neurons. Ann. Neurol. 42, 847–856. doi: 10.1002/ana.410420605
Levy, D. N., Refaeli, Y., MacGregort, R. O. B. R., and Weiner, D. B. (1994). Serum Vpr regulates productive infection and latency of human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. U S A 91, 10873–10877. doi: 10.1073/pnas.91.23.10873
Lopardo, G. D., Bissio, E., Iannella Mdel, C., Crespo, A. D., Garone, D. B., and Cassetti, L. I. (2009). Good neurocognitive performance measured by the international HIV dementia scale in early HIV-1 infection. J. Acquir. Immune Defic. Syndr. 52, 488–492. doi: 10.1097/QAI.0b013e3181b06348
Louboutin, J.-P., Reyes, B. A. S., Agrawal, L., Maxwell, C. R., Van Bockstaele, E. J., and Strayer, D. S. (2010). Blood-brain barrier abnormalities caused by exposure to HIV-1 gp120–protection by gene delivery of antioxidant enzymes. Neurobiol. Dis. 38, 313–325. doi: 10.1016/j.nbd.2010.02.007
Lupien, S. J., de Leon, M., de Santi, S., Convit, A., Tarshish, C., Nair, N. P. V., et al. (1998). Cortisol levels during human aging predict hippocampal atrophy and memory deficits. Nat. Neurosci. 1, 69–73. doi: 10.1038/271
Lurain, N. S., Hanson, B. A., Martinson, J., Leurgans, S. E., Landay, A. L., Bennett, D. A., et al. (2013). Virological and immunological characteristics of human cytomegalovirus infection associated with Alzheimer disease. J. Infect. Dis. 208, 564–572. doi: 10.1093/infdis/jit210
Magariños, A. M., and McEwen, B. S. (1995). Stress-induced atrophy of apical dendrites of hippocampal CA3c neurons: involvement of glucocorticoid secretion and excitatory amino acid receptors. Neuroscience 69, 89–98. doi: 10.1016/0306-4522(95)00259-l
Mäkitalo, S., Mellgren, Å., Borgh, E., Kilander, L., Skillbäck, T., Zetterberg, H., et al. (2015). The cerebrospinal fluid biomarker profile in an HIV-infected subject with Alzheimer’s disease. AIDS Res. Ther. 12:23. doi: 10.1186/s12981-015-0063-x
Marcello, E., Epis, R., Saraceno, C., and Di Luca, M. (2012). Synaptic dysfunction in Alzheimer’s disease. Adv. Exp. Med. Biol. 970, 573–601. doi: 10.1007/978-3-7091-0932-8_25
Marchesi, V. T. (2011). Alzheimer’s dementia begins as a disease of small blood vessels, damaged by oxidative-induced inflammation and dysregulated amyloid metabolism: implications for early detection and therapy. FASEB J. 25, 5–13. doi: 10.1096/fj.11-0102ufm
Maschke, M., Kastrup, O., Esser, S., Ross, B., Hengge, U., and Hufnagel, A. (2000). Incidence and prevalence of neurological disorders associated with HIV since the introduction of highly active antiretroviral therapy (HAART). J. Neurol. Neurosurg. Psychiatry 69, 376–380. doi: 10.1136/jnnp.69.3.376
Masliah, E., Achim, C. L., Ge, N., DeTeresa, R., Terry, R. D., and Wiley, C. A. (1992). Spectrum of human immunodeficiency virus-associated neocortical damage. Ann. Neurol. 32, 321–329. doi: 10.1002/ana.410320304
Mattson, M. P. (2004). Infectious agents and age-related neurodegenerative disorders. Ageing Res. Rev. 3, 105–120. doi: 10.1016/j.arr.2003.08.005
McArthur, J. C., Haughey, N., Gartner, S., Conant, K., Pardo, C., Nath, A., et al. (2003). Human immunodeficiency virus-associated dementia: an evolving disease. J. Neurovirol. 9, 205–221. doi: 10.1080/13550280390194109
McArthur, J. C., Hoover, D. R., Bacellar, H., Miller, E. N., Cohen, B. A., Becker, J. T., et al. (1993). Dementia in AIDS patients: incidence and risk factors. Multicenter AIDS Cohort study. Neurology 43, 2245–2252. doi: 10.1212/wnl.43.11.2245
McArthur, J. C., Steiner, J., Sacktor, N., and Nath, A. (2010). Human immunodeficiency virus-associated neurocognitive disorders: mind the gap. Ann. Neurol. 67, 699–714. doi: 10.1002/ana.22053
McEwen, B. S. (2008). Central effects of stress hormones in health and disease: understanding the protective and damaging effects of stress and stress mediators. Eur. J. Pharmacol. 583, 174–185. doi: 10.1016/j.ejphar.2007.11.071
Milanini, B., and Valcour, V. (2017). Differentiating HIV-associated neurocognitive disorders from Alzheimer’s disease: an emerging issue in geriatric NeuroHIV. Curr. HIV/AIDS Rep. 14, 123–132. doi: 10.1007/s11904-017-0361-0
Mishra, M., Taneja, M., Malik, S., Khalique, H., and Seth, P. (2010). Human immunodeficiency virus type 1 Tat modulates proliferation and differentiation of human neural precursor cells: implication in NeuroAIDS. J. Neurovirol. 16, 355–367. doi: 10.3109/13550284.2010.513028
Montagne, A., Zhao, Z., and Zlokovic, B. V. (2017). Alzheimer’s disease: a matter of blood-brain barrier dysfunction? J. Exp. Med. 214, 3151–3169. doi: 10.1084/jem.20171406
Nath, A. (2002). Human immunodeficiency virus (HIV) proteins in neuropathogenesis of HIV dementia. J. Infect. Dis. 7609, 193–198. doi: 10.1086/344528
Nazer, B., Hong, S., and Selkoe, D. J. (2008). LRP promotes endocytosis and degradation, but not transcytosis, of the amyloid-β peptide in a blood-brain barrier in vitro model. Neurobiol. Dis. 30, 94–102. doi: 10.1016/j.nbd.2007.12.005
Ortega, M., and Ances, B. M. (2014). Role of HIV in amyloid metabolism. J. Neuroimmune Pharmacol. 9, 483–491. doi: 10.1007/s11481-014-9546-0
Patrick, C., Crews, L., Desplats, P., Dumaop, W., Rockenstein, E., Achim, C. L., et al. (2011). Increased CDK5 expression in HIV encephalitis contributes to neurodegeneration via tau phosphorylation and is reversed with Roscovitine. Am. J. Pathol. 178, 1646–1661. doi: 10.1016/j.ajpath.2010.12.033
Peters, P. J., Bhattacharya, J., Hibbitts, S., Dittmar, M. T., Simmons, G., Bell, J., et al. (2004). Biological analysis of human immunodeficiency virus type 1 R5 envelopes amplified from brain and lymph node tissues of AIDS patients with neuropathology reveals two distinct tropism phenotypes and identifies envelopes in the brain that confer an enhanced tropism and fusigenicity for macrophages. J. Virol. 78, 6915–6926. doi: 10.1128/JVI.78.13.6915-6926.2004
Peudenier, S., Héry, C., Ng, K. H., and Tardieu, M. (1991). HIV receptors within the brain: a study of CD4 and MHC-II on human neurons, astrocytes and microglial cells. Res. Virol. 142, 145–149. doi: 10.1016/0923-2516(91)90051-4
Pineau, F., Canet, G., Desrumaux, C., Hunt, H., Chevallier, N., Ollivier, M., et al. (2016). New selective glucocorticoid receptor modulators reverse amyloid-β peptide-induced hippocampus toxicity. Neurobiol. Aging 45, 109–122. doi: 10.1016/j.neurobiolaging.2016.05.018
Potter, M. C., Figuera-Losada, M., Rojas, C., and Slusher, B. S. (2013). Targeting the glutamatergic system for the treatment of HIV-associated neurocognitive disorders. J. Neuroimmune Pharmacol. 8, 594–607. doi: 10.1007/s11481-013-9442-z
Prince, M., Wimo, A., Guerchet, M., Gemma-Claire, A., Wu, Y.-T., and Prina, M. (2015). World Alzheimer report 2015: the global impact of dementia–an analysis of prevalence, incidence, cost and trends. Alzheimers Dis. Int. 84.
Rajendran, L., and Paolicelli, R. C. (2018). Microglia-Mediated synapse loss in Alzheimer’s disease. J. Neurosci. 38, 2911–2919. doi: 10.1523/JNEUROSCI.1136-17.2017
Reul, J. M., and de Kloet, E. R. (1985). Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology 117, 2505–2511. doi: 10.1210/endo-117-6-2505
Roe, K., Orillo, B., and Verma, S. (2014). West nile virus-induced cell adhesion molecules on human brain microvascular endothelial cells regulate leukocyte adhesion and modulate permeability of the in vitro blood-brain barrier model. PLoS One 9:e102598. doi: 10.1371/journal.pone.0102598
Roozendaal, B. (2000). Glucocorticoids and the regulation of memory consolidation. Psychoneuroendocrinology 25, 213–238. doi: 10.1016/s0306-4530(99)00058-x
Rosenberg, G. A. (2014). Blood-brain barrier permeability in aging and Alzheimers disease. J. Prev. Alzheimers Dis. 1, 138–139. doi: 10.14283/jpad.2014.25
Rothenaigner, I., Kramer, S., Ziegler, M., Wolff, H., Kleinschmidt, A., and Brack-werner, R. (2007). Long-term HIV-1 infection of neural progenitor populations. AIDS 21, 2271–2281. doi: 10.1097/QAD.0b013e3282f12f27
Salinas, S., Schiavo, G., and Kremer, E. J. (2010). A hitchhiker’s guide to the nervous system: the complex journey of viruses and toxins. Nat. Rev. Microbiol. 8, 645–655. doi: 10.1038/nrmicro2395
Sami Saribas, A., Cicalese, S., Ahooyi, T. M., Khalili, K., Amini, S., and Sariyer, I. K. (2017). HIV-1 Nef is released in extracellular vesicles derived from astrocytes: evidence for Nef-mediated neurotoxicity. Cell Death Dis. 8:e2542. doi: 10.1038/cddis.2016.467
Samuel, M. A., Wang, H., Siddharthan, V., Morrey, J. D., and Diamond, M. S. (2007). Axonal transport mediates West Nile virus entry into the central nervous system and induces acute flaccid paralysis. Proc. Natl. Acad. Sci. U S A 104, 17140–17145. doi: 10.1073/pnas.0705837104
Selkoe, D. J. (2001). Alzheimer’s disease: genes, proteins and therapy. Physiol. Rev. 81, 741–766. doi: 10.1152/physrev.2001.81.2.741
Sellmeyer, D. E., and Grunfeld, C. (1996). Endocrine and metabolic disturbances in human immunodeficiency virus infection and the acquired immune deficiency syndrome. Endocr. Rev. 17, 518–532. doi: 10.1210/edrv-17-5-518
Smith, G. A., Gross, S. P., and Enquist, L. W. (2001). Herpesviruses use bidirectional fast-axonal transport to spread in sensory neurons. Proc. Natl. Acad. Sci. U S A 98, 3466–3470. doi: 10.1073/pnas.061029798
Soontornniyomkij, V., Umlauf, A., Soontornniyomkij, B., Gouaux, B., Ellis, R. J., Levine, A. J., et al. (2018). Association of antiretroviral therapy with brain aging changes among HIV-infected adults. AIDS doi: 10.1097/QAD.0000000000001927 [Epub ahead of print].
Sotiropoulos, I., Catania, C., Pinto, L. G., Silva, R., Pollerberg, G. E., Takashima, A., et al. (2011). Stress acts cumulatively to precipitate Alzheimer’s disease-like tau pathology and cognitive deficits. J. Neurosci. 31, 7840–7847. doi: 10.1523/JNEUROSCI.0730-11.2011
Stern, A. L., Ghura, S., Gannon, P. J., Akay-espinoza, C., Phan, X. M., Yee, X. A. C., et al. (2018). BACE1 mediates HIV-associated and excitotoxic neuronal damage through an APP-dependent mechanism. J. Neurosci. 38, 4288–4300. doi: 10.1523/JNEUROSCI.1280-17.2018
Storck, S. E., Meister, S., Nahrath, J., Meißner, J. N., Schubert, N., Di Spiezio, A., et al. (2016). Endothelial LRP1 transports amyloid- β1-42 across the blood-brain barrier. J. Clin. Invest. 126, 123–136. doi: 10.1172/jci81108
Sturdevant, C. B., Joseph, S. B., Schnell, G., Price, R. W., Swanstrom, R., and Spudich, S. (2015). Compartmentalized replication of R5 T cell-tropic HIV-1 in the central nervous system early in the course of infection. PLoS Pathog. 11:e1004720. doi: 10.1371/journal.ppat.1004720
Takahashi, T., Kimoto, T., Tanabe, N., Hattori, T., Yasumatsu, N., and Kawato, S. (2002). Corticosterone acutely prolonged N-methyl-d-aspartate receptor-mediated Ca2+ elevation in cultured rat hippocampal neurons. J. Neurochem. 83, 1441–1451. doi: 10.1046/j.1471-4159.2002.01251.x
Thakur, K. T., Boubour, A., Saylor, D., Das, M., Bearden, D. R., and Birbeck, G. L. (2018). Global HIV neurology: a comprehensive review. AIDS doi: 10.1097/QAD.0000000000001796 [Epub ahead of print].
Turner, R. S., Chadwick, M., Horton, W. A., Simon, G. L., Jiang, X., and Esposito, G. (2016). An individual with human immunodeficiency virus, dementia and central nervous system amyloid deposition. Alzheimers Dement. 4, 1–5. doi: 10.1016/j.dadm.2016.03.009
Van Bogaert, T., Vandevyver, S., Dejager, L., Van Hauwermeiren, F., Pinheiro, I., Petta, I., et al. (2011). Tumor necrosis factor inhibits glucocorticoid receptor function in mice: a strong signal toward lethal shock. J. Biol. Chem. 286, 26555–26567. doi: 10.1074/jbc.M110.212365
Vasek, M. J., Garber, C., Dorsey, D., Durrant, D. M., Bollman, B., Soung, A., et al. (2016). A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature 534, 538–543. doi: 10.1038/nature18283
Vignoli, A. L., Martini, I., Haglid, K. G., Silvestroni, L., Augusti-Tocco, G., and Biagioni, S. (2000). Neuronal glycolytic pathway impairment induced by HIV envelope glycoprotein gp120. Mol. Cell. Biochem. 215, 73–80. doi: 10.1023/A:1026590916661
Vigorito, M., LaShomb, A. L., and Chang, S. L. (2007). Spatial learning and memory in HIV-1 transgenic rats. J. Neuroimmune Pharmacol. 2, 319–328. doi: 10.1007/s11481-007-9078-y
Virgin, C. E., Ha, T. P., Packan, D. R., Tombaugh, G. C., Yang, S. H., Horner, H. C., et al. (1991). Glucocorticoids inhibit glucose transport and glutamate uptake in hippocampal astrocytes: implications for glucocorticoid neurotoxicity. J. Neurochem. 57, 1422–1428. doi: 10.1111/j.1471-4159.1991.tb08309.x
Wang, Y., Santerre, M., Tempera, I., Martin, K., Mukerjee, R., and Sawaya, B. E. (2017). HIV-1 Vpr disrupts mitochondria axonal transport and accelerates neuronal aging. Neuropharmacology 117, 364–375. doi: 10.1016/j.neuropharm.2017.02.008
Wang, Z., Trillo-Pazos, G., Kim, S.-Y., Canki, M., Morgello, S., Sharer, L. R., et al. (2004). Effects of human immunodeficiency virus type 1 on astrocyte gene expression and function: potential role in neuropathogenesis. J. Neurovirol. 10, 25–32. doi: 10.1080/753312749
WHO. (2014). Estimates for 2000–2012 [WWW Document]. Available online at: http://www.who.int/healthinfo/global_burden_disease/estimates/en/index2.html [Accessed on February 8, 2016].
Williams, D. W., Anastos, K., Morgello, S., and Berman, J. W. (2015). JAM-A and ALCAM are therapeutic targets to inhibit diapedesis across the BBB of CD14+CD16+ monocytes in HIV-infected individuals. J. Leukoc. Biol. 97, 401–412. doi: 10.1189/jlb.5A0714-347R
Xu, J., and Ikezu, T. (2009). The comorbidity of HIV-associated neurocognitive disorders and Alzheimer’s disease: a foreseeable medical challenge in post-HAART era. J. Neuroimmune Pharmacol. 4, 200–212. doi: 10.1007/s11481-008-9136-0
Yamazaki, Y., and Kanekiyo, T. (2017). Blood-brain barrier dysfunction and the pathogenesis of Alzheimer’s disease. Int. J. Mol. Sci. 18:1965. doi: 10.3390/ijms18091965
Yan, D., Zhang, Y., Liu, L., and Yan, H. (2016). Pesticide exposure and risk of Alzheimer’s disease: a systematic review and meta-analysis. Sci. Rep. 6:32222. doi: 10.1038/srep32222
Yang, B., Akhter, S., Chaudhuri, A., and Kanmogne, G. D. (2009). HIV-1 gp120 induces cytokine expression, leukocyte adhesion and transmigration across the blood-brain barrier: modulatory effects of STAT1 signaling. Microvasc. Res. 77, 212–219. doi: 10.1016/j.mvr.2008.11.003
Yusim, A., Franklin, L., Brooke, S., Ajilore, O., and Sapolsky, R. (2000). Glucocorticoids exacerbate the deleterious effects of gp120 in hippocampal and cortical explants. J. Neurochem. 74, 1000–1007. doi: 10.1046/j.1471-4159.2000.0741000.x
Zeinolabediny, Y., Caccuri, F., Colombo, L., Morelli, F., Romeo, M., Rossi, A., et al. (2017). HIV-1 matrix protein p17 misfolding forms toxic amyloidogenic assemblies that induce neurocognitive disorders. Sci. Rep. 7:10313. doi: 10.1038/s41598-017-10875-0
Zhang, Y. L., Ouyang, Y. B., Liu, L. G., and Chen, D. X. (2015). Blood-brain barrier and neuro-AIDS. Eur. Rev. Med. Pharmacol. Sci. 19, 4927–4939.
Keywords: HIV-associated neurocognitive disorders, neuroinflammation, viral neuroinfection, Alzheimer’s disease, hypothalamo-pituitary-adrenal axis
Citation: Canet G, Dias C, Gabelle A, Simonin Y, Gosselet F, Marchi N, Makinson A, Tuaillon E, Van de Perre P, Givalois L and Salinas S (2018) HIV Neuroinfection and Alzheimer’s Disease: Similarities and Potential Links? Front. Cell. Neurosci. 12:307. doi: 10.3389/fncel.2018.00307
Received: 21 June 2018; Accepted: 23 August 2018;
Published: 11 September 2018.
Edited by:
Rena Li, Roskamp Institute, United StatesReviewed by:
Marcella Reale, Università degli Studi G. d’Annunzio Chieti e Pescara, ItalyResham Chhabra, School Johns Hopkins University, United States
Copyright © 2018 Canet, Dias, Gabelle, Simonin, Gosselet, Marchi, Makinson, Tuaillon, Van de Perre, Givalois and Salinas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laurent Givalois, bGF1cmVudC5naXZhbG9pc0B1bW9udHBlbGxpZXIuZnI=
Sara Salinas, c2FyYS5zYWxpbmFzQGluc2VybS5mcg==
† These authors have contributed equally to this work