 Hsiao-Ying Kuo
Hsiao-Ying Kuo Fu-Chin Liu
Fu-Chin Liu- 1Institute of Neuroscience, National Yang-Ming University, Taipei, Taiwan
- 2Brain Research Center, National Yang-Ming University, Taipei, Taiwan
Autism spectrum disorder (ASD) is a heterogeneous neurodevelopmental disorder with a high prevalence rate. The core symptoms of ASD patients are impaired social communication and repetitive behavior. Genetic and environmental factors contribute to pathophysiology of ASD. Regarding environmental risk factors, it is known that valproic acid (VPA) exposure during pregnancy increases the chance of ASD among offspring. Over a decade of animal model studies have shown that maternal treatment with VPA in rodents recapitulates ASD-like pathophysiology at a molecular, cellular and behavioral level. Here, we review the prevailing theories of ASD pathogenesis, including excitatory/inhibitory imbalance, neurotransmitter dysfunction, dysfunction of mTOR and endocannabinoid signaling pathways, neuroinflammation and epigenetic alterations that have been associated with ASD. We also describe the evidence linking neuropathological changes to ASD-like behavioral abnormalities in maternal VPA-treated rodents. In addition to obtaining an understanding of the neuropathological mechanisms, the VPA-induced ASD-like animal models also serve as a good platform for testing pharmacological reagents that might be use treating ASD. We therefore have summarized the various pharmacological studies that have targeted the classical neurotransmitter systems, the endocannabinoids, the Wnt signal pathway and neuroinflammation. These approaches have been shown to often be able to ameliorate the ASD-like phenotypes induced by maternal VPA treatments.
Introduction
Autism spectrum disorder (ASD) is a heterogeneous neurodevelopmental disorder with a high prevalence approximately 16.8 in 1,000; these individuals share the core symptoms of impaired social communication/interaction and restrictive/repetitive behavior (American Psychiatric Association, 2013; Baio et al., 2018). In addition to the core symptoms, ASD patients also show various physiological and psychiatric comorbid symptoms, including anxiety, intellectual disability, epilepsy, hypersensitivity, aggression, sleep disturbance, and gastrointestinal problems (Lai et al., 2014). Despite the high prevalence and significant social-economic burdens that ASD imposes, effective treatment(s) are not yet available. There are so far only two U.S. Food and Drug Administration (FDA)-approved drugs; namely risperidone and aripiprazole, that are available for treating irritability in ASD, but none that target the defective social communication (U.S. Food and Drug Administration, 2007, 2009).
Although ASD is a highly heritable disorder (Abrahams and Geschwind, 2008), environmental influences also play a significant role in the etiology of ASD. Risk factors have effects during critical periods in embryogenesis that may enhance susceptibility to ASD. Epidemiological studies have suggested that maternal infection, ethanol exposure and anti-epileptic drug treatment increase the risk of ASD in offspring (Arndt et al., 2005).
A prospective report including 632 subjects has shown that children exposed to valproic acid (VPA) during pregnancy have a significantly higher chance of developing ASD (Bromley et al., 2008). VPA is clinically prescribed as an anti-epileptic and mood-stabilizing drug. VPA is also considered to be a first-line prophylactic drug for migraine headaches. The chemical structure of VPA consists of a simple branched-chain fatty acid (2-propylpentanoic acid) that can indirectly inhibit the enzyme γ-aminobutyric acid (GABA) transferase; this leads to an increase in GABA levels in the brain. It is also important to note that VPA is also able to inhibit voltage-gated Na+ channels and this directly suppresses the high-frequency firing of neurons (Rosenberg, 2007). Interestingly, VPA is also known as a non-specific histone deacetylase (HDAC) inhibitor and has been shown to promote the differentiation of carcinoma cells (Földy et al., 2001), neural stem cells (Hao et al., 2004) and adult neural stem cells in the subgranular zone of the hippocampus (Yu et al., 2009). Therefore, VPA has dual pharmacological impacts on VPA-exposed offspring; one effect is reducing the excitability of neural circuits by inhibiting synaptic GABA uptake, the other is epigenetic regulation of chromosome remodeling by inhibiting HDAC activity.
Maternal administration of VPA during pregnancy in rodents results in lifelong abnormalities that recapitulate ASD-like clinical phenotypes. After VPA injection into pregnant rats at embryonic day (E) 12.5 or E13, their offspring exhibit delayed developmental milestones, stereotypic and self-injurious behaviors, and impaired social behavior (Schneider and Przewlocki, 2005; Wagner et al., 2006). These VPA-induced behavioral phenotypes are similar to the major symptoms found in human ASD patients. Subsequent studies have identified abnormal cellular and molecular changes resulting from maternal VPA exposure (Roullet et al., 2013; Nicolini and Fahnestock, 2018). Essentially, the validity of the VPA-induced ASD-like animal model has been discussed widely and it would seem that the biological changes in VPA-treated animals are similar to hypotheses suggested as the origins of ASD; furthermore, the pathological changes and therapeutic response in VPA-treated animals and human patients are similar (Mabunga et al., 2015; Nicolini and Fahnestock, 2018). An advantage of the VPA model is that the ASD-like phenotypes characterized in VPA animals resemble the clinical symptoms of ASD patients. Moreover, the VPA model provides an entry point for researchers when investigating how environmental risk factors can influence the neurodevelopmental processes underlying ASD pathogenesis. It should be noted that VPA exposure during prenatal critical time windows reliably recapitulates the human clinical findings and produces ASD-like phenotypes in rodents ranging from the behavioral level to the molecular level. The maternal VPA-induced ASD-like animal model therefore provides a paradigm by which the etiology of ASD can be experimentally investigated in order to develop potential treatments for ASD. Here, we review the pathological mechanisms of ASD, as well as exploring pharmacological approaches to relieving ASD-like phenotypes using maternal VPA-induced ASD-like animal models (Table 1).
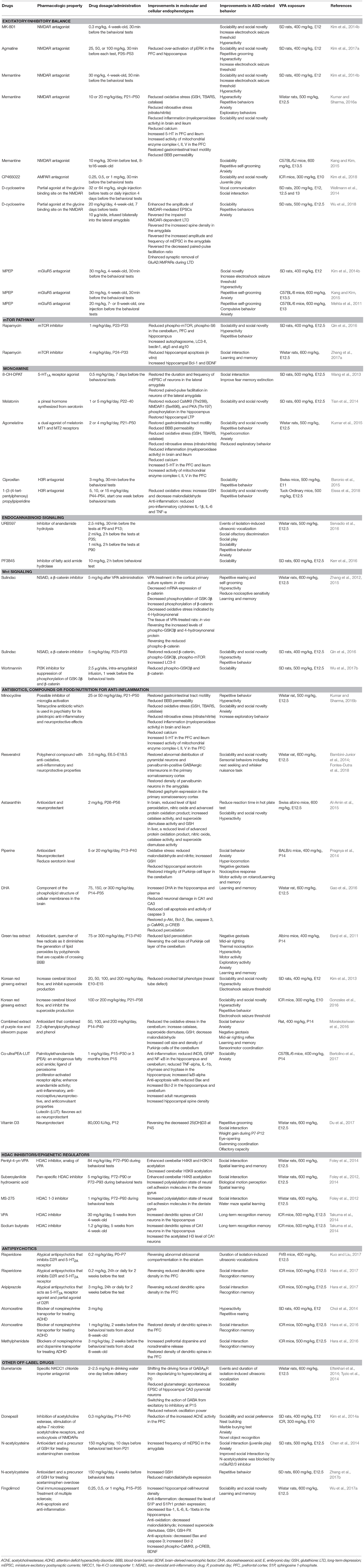
Table 1. Summary of the molecular, cellular and behavioral effects of pharmacological treatments in VPA-induced ASD-like animal models.
The Excitatory/Inhibitory Imbalance Hypothesis
An influential theory regarding psychiatric diseases over the last two decades is one that involves an imbalance affecting the excitatory/inhibitory (E/I) neural circuits (Rubenstein and Merzenich, 2003; Marín, 2012; Nelson and Valakh, 2015). Hussman (2001) first proposed that hypofunction of GABAergic neurotransmission as part of ASD pathophysiology (Hussman, 2001). Rubenstein and Merzenich (2003) further postulated that an increased E/I ratio might be a pathogenic mechanism for ASD. The E/I imbalance theory is based on the observation that there are higher incidences of epilepsy and abnormal GABAergic function in patients with ASD. The theory proposes that a loss of inhibitory control may cause elevated noise in the brain's networks and this alters sensory, emotional and social information processing, which in turn reduces the adaptive ability of ASD patients to process and respond to environmental stimulation (Rubenstein and Merzenich, 2003). Supporting evidence has come from clinical and genetic mouse model studies that have shown a disruption of E/I homeostasis may result from defective GABAergic inhibitory input, abnormal glutamatergic transmission and/or homeostatic compensation (Nelson and Valakh, 2015). In ASD models involving maternal VPA treatment, NMDA receptor (NMDAR)-mediated currents are increased in the medial prefrontal cortex (mPFC) in maternal VPA-treated rats before P16, but not between P30 and P50 (Rinaldi et al., 2007; Walcott et al., 2011). It is worth noting that the reduction in NMDAR function during adulthood may reflect a compensatory homeostasis for the presence of an E/I imbalance during development (Martin and Manzoni, 2014). Therefore, an E/I imbalance would seem to be involved in the pathogenesis of ASD-like phenotypes that are induced by VPA.
Based on the E/I imbalance theory, several VPA-induced ASD model studies have tested whether activity-dependent manipulation via glutamate receptors is beneficial and able to alleviate ASD-like phenotypes. Clinical studies have shown that genetic mutation of the GRIA2 and GRIA3 subunits of AMPA receptors (AMPAR), are associated with ASD (Ramanathan et al., 2004; Jacquemont et al., 2006). In VPA model systems, GluA1 protein levels and mEPSC amplitudes are increased in the mPFC of VPA-treated mice; furthermore, AMPA antagonist treatment is able to improve the social deficits present in VPA-exposed mice (Kim et al., 2018). Along with this, de novo mutation of GRIN2B, a subunit of the NMDAR, has also been found in patients with ASD (Tarabeux et al., 2011; O'Roak et al., 2012a). Consistent with findings observed in human patients, the GRIN2A and GRIN2B subunits of the NMDAR are upregulated in the primary somatosensory cortex of VPA-treated rats (Rinaldi et al., 2007). Interestingly, there is increased local connectivity and NMDAR-mediated long-term potentiation (LTP) but there is also reduced connective strength and excitability in the mPFC and the primary somatosensory cortex of rodents after treatment with VPA in the second postnatal week (Rinaldi et al., 2007, 2008). Based on these findings, NMDAR antagonists have been tested as potential drugs for alleviating the ASD-like phenotypes of VPA-treated rodent offspring. A reduction in the threshold for electronic shock seizure in VPA-treated offspring has been found and this mimics the higher susceptibility of epilepsy in ASD patients. The NMDAR blockers MK-801 and agmatine increase the threshold of electronic shock seizure of VPA-treated rats (Kim et al., 2014b, 2017a). NMDA antagonists, including MK-801, agmatine and memantine, relieve the hyperactivity, anxiety, social impairments and the repetitive behavior of VPA-treated rats (Kim et al., 2014b, 2017a; Kang and Kim, 2015; Kumar and Sharma, 2016a). Moreover, administration of donepezil, an acetylcholine esterase inhibitor that is able to induce endocytosis of the NMDAR, improves VPA-induced ASD-like phenotypes (Kim et al., 2014a). Interestingly, D-cycloserine, a partial NMDA agonist that acts on the glycine binding site of NMDAR, not only decreases NMDA function in the amygdala, but also alleviates anxiety, social and vocal communication in VPA-treated rats (Wellmann et al., 2014; Wu et al., 2018). D-cycloserine treatment also enhances the endocytosis of NMDAR at synaptic sites during long-term depression (LTD) induction in the amygdala of the VPA-treated rats. These findings suggest that D-cycloserine is able to regulate the E/I neuronal activity balance via modulation of NMDA function (Wu et al., 2018).
In addition to ionotropic glutamate receptors, metabotropic glutamate receptors (mGluR) have also been implicated in ASD pathogenesis. For example, hyperfunction and hypofunction of mGluR5 signaling is known to be involved in Fragile X syndrome and tuberous sclerosis complex (TSC), respectively (Auerbach et al., 2011). ASD-like phenotypes induced by the Fmr1 mutation, a gene responsible for Fragile X syndrome, are able to be alleviated by administration of mGluR antagonists in mouse models (Choi et al., 2011; Thomas et al., 2012). Furthermore, a recent study has shown that there is a higher prevalence of copy number variation of the gene RANBP1, which affects the mGluR and mGluR network, in ASD patients (Wenger et al., 2016). In the VPA model system, disruption of the E/I balance is accompanied by reduced levels of mGluR2/3 in the lateral amygdala of VPA-treated mouse offspring (Lin et al., 2013; Chen et al., 2014). Furthermore, a mGluR5 antagonist is capable of relieving ASD-like behaviors, including repetitive behavior and anxiety (Mehta et al., 2011; Kang and Kim, 2015).
ASD is highly heterogeneous and therefore the E/I imbalance observed in ASD brains may be a result of dysfunction of either or both the excitatory and inhibitory synapses, which suggests that there are aberrant developmental processes affecting multiple cell types contributing this whole complex of abnormalities. During early development, a high level of sodium-potassium-chloride cotransporter 1 (NKCC1) results in an increase in intracellular chloride and this then leads to the excitatory effects of GABA. After the end of the first week after birth, GABA's effects become inhibitory due to the gradual expression of chloride exporter potassium-chloride transporter 2 (Rivera et al., 1999; Valeeva et al., 2013). Oxytocin is known to induce a transient reduction in intracellular chloride levels and thus it can cause a temporary GABA switch during fetus delivery (Tyzio et al., 2006). However, this transient reduction in intracellular chloride level is absent in VPA-treated mouse offspring; this effect may be related to aberrant neonatal vocal communication, abnormal social behavior, and neuronal oscillation during adulthood (Eftekhari et al., 2014; Tyzio et al., 2014). These ASD-like phenotypes are able to be rescued by perinatal administration of an antagonist of NKCC1, which augments the transient reduction in intracellular chloride during fetus delivery (Eftekhari et al., 2014; Tyzio et al., 2014). These results not only help to elucidate GABA-mediated network maturation, but also provide evidence to support the hypothesis than an E/I imbalance can be a therapeutic target when treating ASD.
Abnormal mTOR Signaling As Part of ASD Pathophysiology
The presence of abnormal mammalian target of rapamycin (mTOR) and related mTOR-mediated signaling has been demonstrated in several syndromic ASD models, including TSC, Fragile X syndrome, Rett syndrome, and Angelman syndrome (Winden et al., 2018). mTOR is a core kinase of mTORC1 and mTORC2, which are regulated by Wnt signaling and PI3K pathway. The mTOR signal pathway integrates various cellular signals related to ATP-mediated energy metabolism, trophic factors, neurotransmitters and amino acids (Lipton and Sahin, 2014). Under physiological conditions, mTOR signaling controls cell survival, cell proliferation, the cytoskeleton, mRNA translation, protein synthesis, and autophagy. These mTOR-mediated basic cellular functions are involved in the control of extensive neurodevelopmental processes including neurogenesis, neuronal migration, axonogenesis, synaptogenesis, and circuity formation (Lipton and Sahin, 2014; Crino, 2016; Winden et al., 2018). The genes TSC, phosphatase and tensin homolog (PTEN) and neurofibromatosis type 1 (NF1), which are negatively regulated by mTOR kinase activity, are responsible for syndromic ASD pathogenesis in various diseases including TSC, PTEN hamartoma tumor syndrome, and RASopathies, respectively (Winden et al., 2018). Consistently, over-activation of mTOR signaling has been found in the cerebral cortex of human postmortem ASD brains (Tang et al., 2014). Rapamycin is an antibiotic that binds to the FKPB12-rapamycin binding site of mTOR and inhibits mTORC1, but not mTORC2, activity. Rapamycin was originally used as an immunosuppressant during tissue transplantation. In ASD transgenic mice models, rapamycin has been tested as a potential pharmacotherapic agent to treat ASD. For example, it has been shown that the abnormal social interaction and stereotypic behaviors are ameliorated by rapamycin treatment in heterozygotes carry one copy of the TSC knockout and in neuronal, astroglial or cerebellar Purkinje cell-specific conditional TSC knockout mice (Meikle et al., 2008; Zeng et al., 2008; Sato et al., 2012; Tsai et al., 2012; Tang et al., 2014). In addition to rescuing behavioral deficits, rapamycin also improves several endophenotypes of the TSC mutant models, including abnormal myelination, reduced spinogenesis and blockade of autophagy (Meikle et al., 2008; Sato et al., 2012; Tsai et al., 2012; Tang et al., 2014). In an ASD model in which Pten is conditionally knocked out in oligodendrocyte and Schwann cells, rapamycin treatment was found to reduce the hypertrophy of white matter (Goebbels et al., 2010). These findings support the hypothesis that the mTOR pathway is a potential intervention target for ASD.
Abnormal up-regulation of mTOR signaling is observed in the brains of VPA-treated rats (Qin et al., 2016; Zhang et al., 2017a). Increased phospho-mTOR and phospho-S6, a downstream target of mTOR, have been found in the mPFC, hippocampus and cerebellum of VPA-treated brains and these changes were associated with reduced autophagosome and increased apoptosis (Qin et al., 2016; Zhang et al., 2017a). Treating VPA mice with rapamycin not only is able to improve social impairment, hyperactivity, repetitive behavior, and learning/memory, but it also increases BDNF and Bcl-2 expression (Qin et al., 2016; Zhang et al., 2017a). Moreover, the impairment of autophagy is ameliorated by rapamycin treatment as a result of up-regulation of the components of autophagosomes in the cerebellum, mPFC and hippocampus; this was observed after chronic rapamycin treatment of VPA-treated rats (Qin et al., 2016). Taken the above findings together, a potential ASD therapeutic approach involving a reduction in the over-activated mTOR signal transduction is supported by investigations that have used a VPA rodent model.
Dysregulation of Monoamine Transmission In ASD
Abnormalities of the various monoaminergic systems, including serotonin, catecholamine and histamine, have been observed in ASD patients (Lake et al., 1977; Gabriele et al., 2014; Wright et al., 2017). VPA-treated animals also exhibit aberrant monoamine transmission (see references below). We discuss here the involvement of monoamines in the pathophysiology of ASD and current progress regarding therapeutic strategies that focus on the monoamines systems.
Serotonin
Serotonin (5-hydroxytryptamine, 5-HT) is a neurotransmitter known to be involved in regulating psychiatric function. 5-HT is synthesized from tryptophan by a two-step process. Tryptophan is first converted into 5-HTP by tryptophan hydroxylase (TPH). 5-HTP is then converted into 5-HT by aromatic acid decarboxylase (AADC). Serotonin is transported into synaptic vesicles by vesicular monoamine transporter (VMAT). When released, serotonin in the synaptic cleft is taken up by serotonin transporter (SERT) into the presynaptic terminal in order to terminate neurotransmission and to allow it to be recycled (Frazer and Hensler, 1999).
Hyperserotonemia was first detected in early onsets infantile ASD children, but six out of 23 of the ASD children with highest 5-HT levels were reported in this study to not show specifically correlated clinical signs (Schain and Freedman, 1961). Elevated levels of blood 5-HT have been reported by a meta-analysis study to be present in 25.4% of ASD patients (Gabriele et al., 2014). However, neuropathological studies of human ASD postmortem brains are not consistent with clinical studies. Azmitia et al. (2011) reported that the number of SERT-immunoreactive axons is increased in the 5-HT fibers innervating the cortex and forebrain of ASD brains (Azmitia et al., 2011). On the other hand, Oblak et al. (2013) showed that there were significant reductions in SERT, 5-HT receptor 1A (5-HT1A receptor) and 5-HT receptor 2A (5-HT2A receptor) in the posterior cingulate cortex and fusiform gyrus of human ASD postmortem brains; these two cortical regions are involved in social-emotional processing (Oblak et al., 2013). In addition, genetic linkage studies have identified variants of the SERT gene, SLC6A4, as being present in autistic patients, and that this was found to be correlated with impaired social communication by the ASD patients (Cook et al., 1997; Tordjman et al., 2001). A later study further indicates that such SLC6A4 variants are correlated with platelet hyperserotonemia in ASD individuals (Coutinho et al., 2004). Despite the presence of some inconsistencies, a meta-analysis study has shown that prenatally exposure to selective serotonin reuptake inhibitors (SSRI) leads to a higher odds ratio of having a offspring with ASD (Man et al., 2015). Notably, ASD patients with short-term depletion of tryptophan show deteriorated repetitive behavior and a poorer anxiety status (McDougle et al., 1996), which suggests that a dysregulation in serotonin function is a risk factor for ASD (Muller et al., 2016).
Serotonin is a precursor of melatonin, which is produced in the pineal gland and is involved in the regulation of circadian rhythms (Frazer and Hensler, 1999). Melatonin is relevant to ASD because there is a high prevalence of sleep disturbance among ASD patients (Rossignol and Frye, 2011; Tordjman et al., 2013). Abnormal melatonin biosynthesis and reduced levels of melatonin have been found in children and adults who are ASD patients (Nir et al., 1995; Kulman et al., 2000; Melke et al., 2008). Treatment with melatonin is able to reduce such sleep disturbance as well as autistic behavior among ASD individuals (Rossignol and Frye, 2011). It should be noted that sleep problems are also found in patients with other psychiatric diseases, such as schizophrenia, depression and anxiety (Krystal, 2012). The specificity, the mechanisms involved and the causality of the therapeutic effects of melatonin treatment of ASD need to be further clarified in future studies.
Catecholamine
Dopamine and norepinephrine, two major catecholamines in the brain, are both synthesized from tyrosine. Tyrosine hydroxylase first converts tyrosine into L-DOPA. Subsequent decarboxylation of L-DOPA leads to the production of dopamine. After release from presynaptic sites, dopamine binds to dopamine receptors within the postsynaptic sites. Dopamine can be cleared from synaptic clefts by uptake into the presynaptic terminals via the dopamine transporter (DAT) (Kuhar et al., 1999). Early evidence pointing to catecholamine dysfunction in ASD patients is based on the finding that in ASD patients there are increased plasma levels of norepinephrine and decreased plasma dopamine-β-hydroxylase activity levels, the latter being the enzyme that converts dopamine to norepinephrine (Lake et al., 1977). Genetic mutation of the regulators of dopaminergic neurotransmission have been identified as present in ASD patients (Nguyen et al., 2014), and these include DRD1 (Hettinger et al., 2008), DRD2 (Hettinger et al., 2012), DRD3 (de Krom et al., 2009; Staal et al., 2015), DRD4 (Gadow et al., 2010), and DAT (Hamilton et al., 2013). Notably, the midbrain dopamine systems seem to be involved in the pathology of autism. The dopaminergic mesolimbic and mesocortical pathways have been shown to participate in the reward circuits associated with social motivation and social interaction (Dichter et al., 2012; Gunaydin and Deisseroth, 2014). Reduced dopamine levels in the mPFC of medication-free ASD children, as measured by positron emission tomographic scanning, suggest that hypofunction of the dopamine reward system may be involved in the social communication deficits of ASD patients (Ernst et al., 1997). The dopaminergic mesostriatal pathways have been shown to control movement, and degeneration of the mesostriatal pathways is a hallmark of Parkinson's disease (Iversen and Iversen, 2007). Abnormalities affecting the mesostriatal pathways have been linked to repetitive behavior in ASD. This is based on the fact that drug-induced changes in the mesostriatal pathways result in altered stereotypic behaviors in rats (Iwamoto et al., 1976). Given the importance of the mesostriatal pathways to reward prediction, motivational control, and decision making (Matsumoto and Hikosaka, 2009; Ilango et al., 2014; Lerner et al., 2015), it seems highly likely that these pathological changes in the midbrain dopamine systems may be involved in ASD pathogenesis.
Histamine
The histaminergic system has been postulated to be involved in the pathophysiology of neurodevelopmental disorders, especially schizophrenia and ASD (Baronio et al., 2014; Wright et al., 2017). Histamine is derived from histidine by decarboxylation and is largely released by the histaminergic neurons present in the tuberomammillary nucleus of the hypothalamus. The extracellular histamine is inactivated and converted into tele-methylhistamine by neuronal histamine N-methyltransferase (HNMT) in the brain (Haas et al., 2008). The histaminergic system is believed to play an important role in regulating a variety of physiological function; these include the sleep/awake cycle, addiction, neuroinflammation, endocrine control, emotion, learning, and memory (Haas et al., 2008). Wright et al. (2017) found increased levels of HNMT and three types of histaminergic receptors (H1R-H3R) in postmortem dorsolateral prefrontal cortex of ASD patients' brains (Wright et al., 2017). It is noteworthy that Gi-coupled H3R is known to be able to indirectly regulate the other neurotransmitter systems that are involved in ASD pathophysiology owing to the fact that this protein can function as both an autoreceptor and a heteroreceptor. This means that it brings about an inhibition of histamine synthesis and the release of other neurotransmitters, respectively. These properties make H3R a potential therapeutic target for the treatment of related cognitive disorders (Esbenshade et al., 2008; Passani and Blandina, 2011).
The Presence of an Abnormal Monoamine System in Mice With a Maternal VPA-Induced ASD-Like Phenotype
Hyperserotonemia is found in ASD patients and an ASD mouse model where the mice had a variant of SLC6A4 that results in a SERT gain-of-function (Muller et al., 2016). Hyperserotonemia has also been found in VPA-treated animals. Maternal VPA administration not only results in an aberrant migration of serotonin neurons within the embryonic hindbrain, but also their abnormal distribution together with increased tryptophan immunoreactivity in the dorsal raphe nucleus of brains of the adult rats (Miyazaki et al., 2005; Kuwagata et al., 2009; Wang et al., 2013). Serotonin is well known to be involved in melatonin synthesis, and it is also well known that melatonin regulates circadian rhythms; thus the findings in VPA-treated rats of hyperserotonemia of the frontal cortex, hippocampus, cerebellum and blood plasma, when this is linked to the present of abnormal circadian rhythms, implies that there is a disrupted melatonin system in the VPA-treated brain (Narita et al., 2002; Tsujino et al., 2007). Moreover, increased SERT activity is found in the midbrain of SLC6A4 gain-of-function mice and in the amygdala of VPA-treated mice (Veenstra-VanderWeele et al., 2012; Wang et al., 2013). However, some studies have reported hyperserotonemia in the prefrontal cortex and hippocampus of VPA-treated animals (Dufour-Rainfray et al., 2010; Kumar et al., 2015). Interestingly, reduced levels of serotonin in the intestines of VPA-treated rodents, but increased levels of serotonin in the serum of VPA-treated mice, implies that the presence of defective brain-gut interactions that are associated with ASD pathology (de Theije et al., 2014a,b; Kumar et al., 2015; Lim et al., 2017).
Abnormal midbrain dopamine system has been detected in a VPA-treated ASD model. Dopamine levels and turnover rates in the frontal cortex are higher in VPA-treated rats, which suggests hyperfunctionality of the mesocortical system in these VPA-treated animals (Narita et al., 2002; Nakasato et al., 2008). Dopamine D2 receptor (D2R)-positive pyramidal projection neurons in the mPFC layer V in VPA-treated mice are abnormally activated during social interaction (Brumback et al., 2017). Prenatal VPA exposure reduces methamphetamine-induced increases in extracellular DA levels and also reduces the levels of Drd1 and Drd2 mRNAs in the prefrontal cortex; this occurs without alteration in extracellular 5-HT and norepinephrine levels (Hara et al., 2015). In the striatum, alterations in the mesostriatal dopamine pathways remain controversial. Cezar et al. (2018) have reported a reduction in the tyrosine hydroxylase level of the striatum of VPA-treated mice, which suggests hypofunctionality of the mesostriatal dopamine pathways (Cezar et al., 2018). However, Acosta et al. (2018) found that increased levels in the striatum of dopamine and its metabolites are correlated with abnormal timing accuracy and precision when these are measured in an operant lever-press conditioning experiment (Acosta et al., 2018).
Based on the above studies, pharmacological manipulation of the monoamine system has been tested as a strategy for treating ASD. Repression of the 5-HT system by systematic administration of a Gi-coupled 5-HT1A receptor agonist or a Gq-coupled 5-HT2A receptor antagonist has been found to improve ASD-like phenotypes. Chronic treatments with 8-OH-DPAT, a 5-HT1A receptor agonist, not only ameliorates fear memory extinction and social impairment, but also normalizes the E/I balance within the amygdala (Wang et al., 2013). Hara et al. (2017) found that aripiprazole (an atypical antipsychotics that acts as a 5-HT1A receptor agonist and a partial agonist of D2R in order to reduce DA transmission) and risperidone (an atypical antipsychotics that inhibits D2R and the 5-HT2A receptor), but not haloperidol, are able to alleviate reverse recognition memory deficits and social impairment of VPA-treated rats, as well as restoring the reduced number of dendritic spines in the mPFC of VPA-treated rats (Hara et al., 2017). Abnormal vocal communication and striatal compartmentation in VPA-treated mouse neonates are also partially rescued by chronic risperidone treatment after birth (Kuo and Liu, 2017). Notably, risperidone, and aripiprazole are the only two drugs that the FDA has approved for treating irritability among ASD patients (U.S. Food and Drug Administration, 2007, 2009). The pharmacological mechanisms and neurodevelopmental impacts of these pharmacological treatments on the various different brain regions obviously need further investigation.
Administration of melatonin or agomelatine (a dual agonist of melatonin type 1 and type 2 receptors) in VPA-treated rats was found to have beneficial effects, not only on ASD-like phenotypes, but also on the pathophysiology, including improvement in the impairment of CaMKII and PKA signaling in the hippocampus, an increase in blood-brain-barrier (BBB) permeability, a reduction in neuroinflammation, and a lowering of mitochondria dysfunction (Tian et al., 2014; Kumar et al., 2015).
Targeting the catecholamine system has been shown to be an effective way of treating VPA-exposed rodents. In addition to risperidone, which has high affinity to D2R, two other ADHD drugs, atomoxetine and methylphenidate, which act as inhibitors of norepinephrine transporter (NET) and DAT, are able to alleviate ASD-like phenotypes, abnormal catecholamine release, and aberrant dendritic spines in the prefrontal cortex of VPA-treated rats (Choi et al., 2014; Hara et al., 2016). Notably, Hara et al. (2016) found that inhibition of DAT by atomoxetine and methylphenidate has beneficial effects on VPA-induced ASD phenotypes, but not on NET (Hara et al., 2016). These findings are consistent with there being increased extracellular dopamine levels in the prefrontal cortex of VPA-induced ASD animal models after chronic risperidone treatments (Hara et al., 2017).
Notwithstanding the above, there is no evidence as yet to indicate that the histaminergic system in VPA-induced ASD rodents is altered. Nevertheless, administration of the H3R antagonists ciproxifan and 1-(3-(4-tert-pentylphenoxy)propyl)piperidine have been shown to relieve ASD-like behaviors in the VPA-treated mice, as well as reducing oxidative stress and lowering the level of pro-inflammatory cytokines (Baronio et al., 2015; Eissa et al., 2018). These results highlight the therapeutic potential of H3R antagonists as part of the pharmacological treatment of ASD.
Endocannabinoid Signaling
Alterations in neuromodulatory endocannabinoid signaling have been reported in human patients and in ASD animal studies, the latter including VPA-induced rodent models (Chakrabarti et al., 2015; Zamberletti et al., 2017). The presynaptic release of glutamate is able to lead to postsynaptic mGluR-mediated up-regulation of endocannabinoids, including anandamide (AEA), and 2-arachidonoylglycerol (2-AG). The endocannabinoids then pass through the cellular membrane, across the synaptic cleft, which finally leads to retrograde activation of presynaptic cannabinoid receptors; this subsequently induces LTD. This feedback inhibition of synaptic transmission occurs in many brain regions and is involved in the regulation of a diverse range of physiological and pathological functions (Heifets and Castillo, 2009). Alterations affecting the endocannabinoid system have been documented in neurodegenerative and neuropsychiatric diseases related to synaptic plasticity or neuroinflammation; these include Parkinson's diseases, drug addiction, and ASD (Heifets and Castillo, 2009; Mechoulam and Parker, 2013; Chakrabarti et al., 2015; Fernández-Ruiz et al., 2015). Clinical studies support the hypothesis that there is a disruption of endocannabinoid signaling in ASD patients. Down-regulation of cannabinoid receptor type I (CB1R) was first detected in the cerebellum of postmortem ASD patients (Purcell et al., 2001). Up-regulation of cannabinoid receptor type II (CB2R) has been found in the peripheral blood mononuclear cells of ASD children (Siniscalco et al., 2013). Moreover, CB1R gene variants in humans have been linked to the modulation of the striatal response and gaze duration, both in response to social stimulation (Chakrabarti et al., 2006; Chakrabarti and Baron-Cohen, 2011). These findings suggest an involvement of the endocannabinoid system in ASD symptoms. In genetic animal models of ASD, dysfunction of the endocannabinoid system has been found in Fmr1 and neuroligin 3 mutant mice (Földy et al., 2013). For example, elevated endocannabinoid-mediated LTD is found in Fmr1 knockout hippocampus and striatum, and this is correlated with enhanced activity of the 2-AG synthesis enzyme, diacylglycerol lipase (DAGL) (Maccarrone et al., 2010; Zhang and Alger, 2010). Moreover, in these Fmr1 knockout mice, impaired endocannabinoid-mediated LTD in the striatum and linked behavioral abnormalities are able to be rescued by an inhibition of monoacylglycerol lipase (MGL), an enzyme that degrades 2-AG, (Jung et al., 2012). Inhibition of fatty acid amide hydrolase (FAAH), an AEA hydrolase, is able to reverse the aversive memory and social interaction abnormalities presence in Fmr1 mutant mice (Qin et al., 2015; Wei et al., 2016). Taken together, the above findings support the use of endocannabinoid modulators as a potential approach to alleviating ASD symptoms in humans (Zamberletti et al., 2017).
Disturbed endocannabinoid signaling seem to contribute to the ASD-like phenotypes in VPA-induced ASD animal models. A reduction in the level of DAGLα in the cerebellum, a lower MGL level in the hippocampus and reduced endocannabinoid signal transduction in the frontal cortex and hippocampus have been found in VPA-exposed rats, despite unaltered levels of CB1R, AEA, and 2-AG (Kerr et al., 2013). Other studies of the brains of VPA-treated rats have found increased phosphorylation of CB1R in the amygdala, hippocampus and dorsal striatum, as well as reduced levels of the AEA synthesis enzyme (Servadio et al., 2016). Furthermore, Kerr et al. (2016) have reported that acute administration of a FAAH inhibitor was sufficient to attenuate social abnormalities, but not the repetitive behavior and defective exploratory behaviors, of VPA-exposed rats with a dimorphic sexual manner (Kerr et al., 2016). On the other hand, Servadio et al. (2016) carried out a modulation of 2-AG degradation by giving an MGL inhibitor and they found there to be an alleviation of social abnormalities, vocalization, repetitive behaviors, and anxiety in the VPA-treated group (Servadio et al., 2016). Deciphering the mechanisms linked to the various distinct elements found within the endocannabinoid system and related to ASD-like phenotypes requires further investigation.
Dysregulation of Canonical WNT Signaling and ASD Pathophysiology
Wnt signaling is highly conserved and is involved in many neuronal functions including patterning, neurogenesis, axon guidance and synaptogenesis (Rosso and Inestrosa, 2013). Genetic evidence suggests that some molecules that are part of Wnt signaling are also involved in ASD pathogenesis. For example, genetic mutation of the chromodomain helicase DNA binding protein 8 (CHD8, a negative regulator of Wnt signaling) and of CTNNB1 (the β-catenin gene) have been identified as present in ASD patients (O'Roak et al., 2012a,b). ASD has been proposed to involve a developmental disconnection disorder linked to synaptopathy. Several components of Wnt signaling, including the ligands, β-catenin, CHD8, and GSK3β in pre-synaptic and post-synaptic sites, are known to be involved in synaptic development and the regulation of synaptic transmission, both of which affect the E/I balance within the brain (Rosso and Inestrosa, 2013; Caracci et al., 2016). Moreover, Wnt signaling interacts with other signaling pathways that are known to be involved in ASD pathogenesis, such as the mTOR pathway through PTEN (Chen et al., 2015). In addition to synaptic function, Wnt signaling also is known to regulate neuroinflammation in the CNS via NFκB and various other inflammatory pathways (Marchetti and Pluchino, 2013; Ma and Hottiger, 2016). In the Fragile X syndrome genetic mouse model, elevated phosphorylation of GSK3β was found in the Fmr1 knockout mice, which suggests overactivation of Wnt signaling in this genetic ASD model (Min et al., 2009). Subsequent studies have shown that the ASD-related symptoms of Fmr1 knockout mice are able to be ameliorated by inhibition of GSK3β (Min et al., 2009; Mines et al., 2010; Guo et al., 2012; Franklin et al., 2014). Based on the versatile roles of Wnt signaling, the Wnt signal pathway has become a prospective target site for the pharmacological treatment of ASD.
Overactivation of Wnt signaling has been reported in rodents that have undergone maternal VPA treatments. Increased phospho-GSK3β and phospho-β-catenin have been found in in the prefrontal cortex, hippocampus, cerebellum and amygdala of these VPA-treated rats (Go et al., 2012; Zhang et al., 2012, 2017b; Qin et al., 2016; Wu et al., 2017b). Moreover, demethylation of the promoter regions, which led to increased mRNA levels of Wnt1 and Wnt2, has been detected in the prefrontal cortex and hippocampus of VPA-treated rats (Wang et al., 2010). Sulindac acts as non-steroidal anti-inflammatory drug (NSAID) and as an anti-metastasis drug and these effect involve the inhibition β-catenin. Prenatal or postnatal administration of sulindac alleviates ASD-like phenotypes in VPA-treated rats, including social abnormalities, repetitive behaviors, hyperactivity, hypersensitivity and learning/memory deficits (Zhang et al., 2012, 2015; Qin et al., 2016). Notably, sulindac treatment not only restores Wnt signaling, but also deactivates mTOR signaling, as well as increasing the number of autophagosomes in VPA-treated brains (Qin et al., 2016). These findings suggest the possibility that the beneficial effects of a Wnt signaling inhibitor may be partially mediated through a restoration of mTOR signaling. It should be noted that an infusion of wortmannin, an inhibitor of the PI3K-AKT-β-catenin pathway, into the amygdala is able to alleviate the social impairment of VPA-treated rats, and that this occurs via a reduction in the over-activation of Wnt signaling (Wu et al., 2017b).
Neuroinflammation and ASD Pathogenesis
Chronic neuroinflammation within the brain has been shown to have deleterious effects and to be associated with neuropsychiatric and neurodegenerative diseases (Najjar et al., 2013; Ransohoff, 2016). Clinical studies have reported evidence that there is neuroinflammation present in ASD brains, including activation of astrocytes and microglia, the presence of elevated levels of cytokines and higher levels of various other inflammatory biomakers (Kern et al., 2015). It is likely that neuroinflammation has a negative effect on ASD pathogenesis, and therefore the intravenous administration of immunoglobulin has been used to suppress the systematic inflammation that may be present in ASD patients (Gupta et al., 1996; Melamed et al., 2018). Behavioral assessments have shown there to be significant improvements in ASD symptoms after intravenous immunoglobulin infusion (Melamed et al., 2018). Maternal immune activation during pregnancy has been reported to significantly increase the probability of psychosis in offspring, both in humans and using animal models (Buka et al., 2001; Shi et al., 2005; Estes and McAllister, 2016; Careaga et al., 2017). Interestingly, two recent studies have shown that social impairment and repetitive behavior induced by maternal immune activation require the presence of segmented filamentous bacteria in the intestine that bring about the release of IL-17a by TH17 cells (Choi et al., 2016; Kim et al., 2017b). These studies seem to have uncovered the presence of complex interactions among neural development, the immune system and the gut microbiota. These findings highlight the essential role that the brain-gut axis and inflammation plays in ASD pathogenesis.
Neuroinflammation pathways have been shown to be altered in VPA-induced ASD animal models. Reactive oxygen species (ROS), apoptotic markers, the expression level of NFκB and the levels of pro-inflammatory cytokines are all increased in VPA-treated rodents, whereas the antioxidant glutathione and various anti-inflammatory cytokines are reduced (Tung and Winn, 2011). Moreover, increased BBB permeability and perioxidation, which are indicators of pro-inflammatory responses, have also been found in VPA-exposed animals (Banji et al., 2011; Pragnya et al., 2014; Al-Amin et al., 2015; Gao et al., 2016; Kumar and Sharma, 2016b; Morakotsriwan et al., 2016; Zhang et al., 2017b). Several substances have been tested as methods of reducing this inflammatory response and thus the related ASD-like phenotypes. For example, the antibiotic minocycline reduces not only oxidative stress, nitrosative stress, and BBB permeability in the brain, but also restores gastrointestinal tract motility and reduces excessive inflammation in the ileum (Kumar and Sharma, 2016b). Various natural compounds, including resveratrol, astaxanthin, piperine, docosahexaenoic acid, and palmitoylethanolamide/luteolin, have been shown to have neuroprotective and antioxidative effects using VPA-treated rodent models (Bambini-Junior et al., 2014; Pragnya et al., 2014; Al-Amin et al., 2015; Gao et al., 2016; Fontes-Dutra et al., 2018). Fingolimod and N-acetylcysteine, off-label drugs used to treat multiple sclerosis and acetaminophen overdose, respectively, have been reported to have anti-inflammation and anti-apoptotic effects in the VPA-treated rodent brains (Wu et al., 2017a; Zhang et al., 2017b). Nutrition and food extracts from green tea, Korean red ginseng, purple rice, and silkworm pupae have also been shown to reduce ASD-like phenotypes in VPA-treated rodent models, possibly via an anti-inflammatory mechanism (Banji et al., 2011; Kim et al., 2013; Gonzales et al., 2016; Morakotsriwan et al., 2016; Du et al., 2017). From the above findings, it is clear that further studies are needed to elucidate the various neuro-immune mechanisms underlying VPA-induced pathophysiology. Such mechanisms might include the effects of crosstalk between neurons and glia in the microenvironment of defective brain regions, which in turn might help with the development of treatments for ASD that involve anti-inflammation reagents.
Epigenetic Changes and ASD Pathogenesis
Epigenetic changes may occur as part of ASD pathophysiology and these changes often occur as a consequence of genetic alterations and/or environmental stimulation. Previous studies have shown that genes associated with ASD risk often undergo epigenetic modulation during neurodevelopment and these events may be involved in the pathogenesis of ASD (Loke et al., 2015). Genetic mutations of Methyl-CpG binding protein 2 (MECP2), which binds methylated CpG sites and represses gene expression, has been suggested to account for autistic Rett syndrome (Amir et al., 1999). Another example is an increased level of methylation of OXTR, which encodes the oxytocin receptor, which has also been found in ASD patients (Gregory et al., 2009). In response to external environmental stimulation including VPA, increased acetylated levels of histone H3 and H4 have been detected in VPA-treated brains at 6 h after VPA treatments at E12.5 (Kataoka et al., 2011). Moreover, VPA treatment has been shown to increase acetylation in the promoter region of Pax6 in the brain cortex (Kim et al., 2014b). Given that VPA is a potent HDAC inhibitor that is able to regulate gene expression, it was reasonable to test whether ASD-like phenotypes that are induced by maternal VPA treatment are able to be relieved by epigenetic regulators. Treatment with a HDAC inhibitor, including Pentyl-4-yn-VPA, suberoylanilide hydroxamic acid and sodium butyrate, during adulthood not only can relieve ASD-like phenotypes, but also can modulate the acetylation status of neurons in the cerebellum and hippocampus (Foley et al., 2012, 2014; Takuma et al., 2014). In addition to behavioral rescue, sodium butyrate and VPA treatments increase the dendritic spine in hippocampal neurons of prenatal VPA-treated mouse brains (Takuma et al., 2014). These studies suggest that the VPA-induced transient increases in acetylation that occur during the embryonic stages may lead to an abnormal epigenetic state that persists after birth. Genome-wide epigenetic analysis targeting specific regions of VPA-treated brains may thus help to identify the specific genes that are regulated by VPA in the pathogenesis of ASD.
Conclusion
Here we have reviewed current theories related to ASD pathophysiology and explored the potential treatments available for ASD that have used maternal VPA-induced ASD-like animal models. The findings outlined here should help to provide insights into the pathogenesis of ASD at both a cellular and a molecular level. We have summarized the potential therapeutic candidates identified using VPA-induced animal models of ASD. This type of experimental ASD model has been found to be quite useful clinically because the drugs often prescribed for ASD patients have been found to be able to reduce some ASD-like phenotypes in VPA-treated animals. Thus, we should take advantage of this animal model and use it to screen for new pharmacological compounds that can be used to treat ASD.
ASD is a complex neurodevelopmental disease that includes both genetic and epigenetic components. Abnormalities of the sensory, motor, and limbic systems in the brain are involved in ASD neuropathology. An interesting and important question that remains is how the neural deficits found in broad regions of the brain manifest themselves as ASD core symptoms and allow us to distinguish ASD from other psychiatric diseases. One possibility is that ASD pathology may affect specific cell types within neural circuits across different brain regions. Recent advances in systems neuroscience and single-cell RNA sequencing technology may enable us to address this key issue and help to untangle the complexities of ASD pathophysiology.
Author Contributions
H-YK and F-CL wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This review was supported by the Ministry of Science and Technology-Taiwan grants MOST107-2321-B-010-002, MOST107-2320-B-010-041-MY3, Featured Areas Research Center Program grant from Ministry of Education through Brain Research Center, National Yang-Ming University (F-CL), and Postdoctoral Fellowship grants MOST107-2811-B-010-011, MOST107-2321-B-010-010-MY3 (H-YK).
References
Abrahams, B. S., and Geschwind, D. H. (2008). Advances in autism genetics: on the threshold of a new neurobiology. Nat. Rev. Genet. 9, 341–355. doi: 10.1038/nrg2346
Acosta, J., Campolongo, M. A., Hocht, C., Depino, A. M., Golombek, D. A., and Agostino, P. V. (2018). Deficits in temporal processing in mice prenatally exposed to valproic acid. Eur. J. Neurosci. 47, 619–630. doi: 10.1111/ejn.13621
Al-Amin, M. M., Rahman, M. M., Khan, F. R., Zaman, F., and Mahmud Reza, H. (2015). Astaxanthin improves behavioral disorder and oxidative stress in prenatal valproic acid-induced mice model of autism. Behav. Brain Res. 286, 112–121. doi: 10.1016/j.bbr.2015.02.041
American Psychiatric Association, A. P. (2013). Diagnostic and Statistical Manual of Mental Disorders: DSM-5. Arlington, VA: American Psychiatric Publishing.
Amir, R. E., Van den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188. doi: 10.1038/13810
Arndt, T., Stodgell, C., and Rodier, P. M. (2005). The teratology of autism. Int. J. Dev. Neurosci. 23, 189–199. doi: 10.1016/j.ijdevneu.2004.11.001
Auerbach, B. D., Osterweil, E. K., and Bear, M. F. (2011). Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature 480, 63–68. doi: 10.1038/nature10658
Azmitia, E. C., Singh, J. S., and Whitaker-Azmitia, P. M. (2011). Increased serotonin axons (immunoreactive to 5-HT transporter) in postmortem brains from young autism donors. Neuropharmacology 60, 1347–1354. doi: 10.1016/j.neuropharm.2011.02.002
Baio, J., Wiggins, L., Christensen, D. L., Maenner, M. J., Daniels, J., Warren, Z., et al. (2018). Prevalence of autism spectrum disorder among children aged 8 years - autism and developmental disabilities monitoring network, 11 sites, United States, 2014. MMWR Surveill. Summ. 67, 1–23. doi: 10.15585/mmwr.ss6706a1
Bambini-Junior, V., Zanatta, G., Della Flora Nunes, G., Mueller de Melo, G., Michels, M., Fontes-Dutra, M., et al. (2014). Resveratrol prevents social deficits in animal model of autism induced by valproic acid. Neurosci. Lett. 583, 176–181. doi: 10.1016/j.neulet.2014.09.039
Banji, D., Banji, O. J., Abbagoni, S., Hayath, M. S., Kambam, S., and Chiluka, V. L. (2011). Amelioration of behavioral aberrations and oxidative markers by green tea extract in valproate induced autism in animals. Brain Res. 1410, 141–151. doi: 10.1016/j.brainres.2011.06.063
Baronio, D., Castro, K., Gonchoroski, T., de Melo, G. M., Nunes, G. D., Bambini-Junior, V., et al. (2015). Effects of an H3R antagonist on the animal model of autism induced by prenatal exposure to valproic acid. PLoS ONE 10:e0116363. doi: 10.1371/journal.pone.0116363
Baronio, D., Gonchoroski, T., Castro, K., Zanatta, G., Gottfried, C., and Riesgo, R. (2014). Histaminergic system in brain disorders: lessons from the translational approach and future perspectives. Ann. Gen. Psychiatry 13:34. doi: 10.1186/s12991-014-0034-y
Bertolino, B., Crupi, R., Impellizzeri, D., Bruschetta, G., Cordaro, M., Siracusa, R., et al. (2017). Beneficial effects of co-ultramicronized palmitoylethanolamide/luteolin in a mouse model of autism and in a case report of autism. CNS Neurosci. Ther. 23, 87–98. doi: 10.1111/cns.12648
Bromley, R. L., Mawer, G., Clayton-Smith, J., and Baker, G. A. (2008). Autism spectrum disorders following in utero exposure to antiepileptic drugs. Neurology 71, 1923–1924. doi: 10.1212/01.wnl.0000339399.64213.1a
Brumback, A. C., Ellwood, I. T., Kjaerby, C., Iafrati, J., Robinson, S., Lee, A. T., et al. (2017). Identifying specific prefrontal neurons that contribute to autism-associated abnormalities in physiology and social behavior. Mol. Psychiatry doi: 10.1038/mp.2017.213. [Epub ahead of print]
Buka, S. L., Tsuang, M. T., Torrey, E. F., Klebanoff, M. A., Bernstein, D., and Yolken, R. H. (2001). Maternal infections and subsequent psychosis among offspring. Arch. Gen. Psychiatry 58, 1032–1037. doi: 10.1001/archpsyc.58.11.1032
Caracci, M. O., Avila, M. E., and De Ferrari, G. V. (2016). Synaptic Wnt/GSK3beta signaling hub in autism. Neural Plast. 2016:9603751. doi: 10.1155/2016/9603751
Careaga, M., Murai, T., and Bauman, M. D. (2017). Maternal immune activation and autism spectrum disorder: from rodents to nonhuman and human primates. Biol. Psychiatry 81, 391–401. doi: 10.1016/j.biopsych.2016.10.020
Cezar, L. C., Kirsten, T. B., da Fonseca, C. C. N., de Lima, A. P. N., Bernardi, M. M., and Felicio, L. F. (2018). Zinc as a therapy in a rat model of autism prenatally induced by valproic acid. Prog. Neuropsychopharmacol. Biol. Psychiatry 84, 173–180. doi: 10.1016/j.pnpbp.2018.02.008
Chakrabarti, B., and Baron-Cohen, S. (2011). Variation in the human cannabinoid receptor CNR1 gene modulates gaze duration for happy faces. Mol. Autism 2:10. doi: 10.1186/2040-2392-2-10
Chakrabarti, B., Kent, L., Suckling, J., Bullmore, E., and Baron-Cohen, S. (2006). Variations in the human cannabinoid receptor (CNR1) gene modulate striatal responses to happy faces. Eur. J. Neurosci. 23, 1944–1948. doi: 10.1111/j.1460-9568.2006.04697.x
Chakrabarti, B., Persico, A., Battista, N., and Maccarrone, M. (2015). Endocannabinoid signaling in autism. Neurotherapeutics 12, 837–847. doi: 10.1007/s13311-015-0371-9
Chen, Y., Huang, W. C., Sejourne, J., Clipperton-Allen, A. E., and Page, D. T. (2015). Pten mutations alter brain growth trajectory and allocation of cell types through elevated beta-catenin signaling. J. Neurosci. 35, 10252–10267. doi: 10.1523/JNEUROSCI.5272-14.2015
Chen, Y. W., Lin, H. C., Ng, M. C., Hsiao, Y. H., Wang, C. C., Gean, P. W., et al. (2014). Activation of mGluR2/3 underlies the effects of N-acetylcystein on amygdala-associated autism-like phenotypes in a valproate-induced rat model of autism. Front. Behav. Neurosci. 8:219. doi: 10.3389/fnbeh.2014.00219
Choi, C. H., Schoenfeld, B. P., Bell, A. J., Hinchey, P., Kollaros, M., Gertner, M. J., et al. (2011). Pharmacological reversal of synaptic plasticity deficits in the mouse model of fragile X syndrome by group II mGluR antagonist or lithium treatment. Brain Res. 1380, 106–119. doi: 10.1016/j.brainres.2010.11.032
Choi, C. S., Hong, M., Kim, K. C., Kim, J. W., Yang, S. M., Seung, H., et al. (2014). Effects of atomoxetine on hyper-locomotive activity of the prenatally valproate-exposed rat offspring. Biomol. Ther. 22, 406–413. doi: 10.4062/biomolther.2014.027
Choi, G. B., Yim, Y. S., Wong, H., Kim, S., Kim, H., Kim, S. V., et al. (2016). The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science 351, 933–939. doi: 10.1126/science.aad0314
Cook, E. H. Jr., Courchesne, R., Lord, C., Cox, N. J., Yan, S., Lincoln, A., et al. (1997). Evidence of linkage between the serotonin transporter and autistic disorder. Mol. Psychiatry 2, 247–250.
Coutinho, A. M., Oliveira, G., Morgadinho, T., Fesel, C., Macedo, T. R., Bento, C., et al. (2004). Variants of the serotonin transporter gene (SLC6A4) significantly contribute to hyperserotonemia in autism. Mol. Psychiatry 9, 264–271. doi: 10.1038/sj.mp.4001409
Crino, P. B. (2016). The mTOR signalling cascade: paving new roads to cure neurological disease. Nat. Rev. Neurol. 12, 379–392. doi: 10.1038/nrneurol.2016.81
de Krom, M., Staal, W. G., Ophoff, R. A., Hendriks, J., Buitelaar, J., Franke, B., et al. (2009). A common variant in DRD3 receptor is associated with autism spectrum disorder. Biol. Psychiatry 65, 625–630. doi: 10.1016/j.biopsych.2008.09.035
de Theije, C. G., Koelink, P. J., Korte-Bouws, G. A., Lopes da Silva, S., Korte, S. M., Olivier, B., et al. (2014a). Intestinal inflammation in a murine model of autism spectrum disorders. Brain Behav. Immun. 37, 240–247. doi: 10.1016/j.bbi.2013.12.004
de Theije, C. G., Wopereis, H., Ramadan, M., van Eijndthoven, T., Lambert, J., Knol, J., et al. (2014b). Altered gut microbiota and activity in a murine model of autism spectrum disorders. Brain Behav. Immun. 37, 197–206. doi: 10.1016/j.bbi.2013.12.005
Dichter, G. S., Damiano, C. A., and Allen, J. A. (2012). Reward circuitry dysfunction in psychiatric and neurodevelopmental disorders and genetic syndromes: animal models and clinical findings. J. Neurodev. Disord. 4:19. doi: 10.1186/1866-1955-4-19
Du, L., Zhao, G., Duan, Z., and Li, F. (2017). Behavioral improvements in a valproic acid rat model of autism following vitamin D supplementation. Psychiatry Res. 253, 28–32. doi: 10.1016/j.psychres.2017.03.003
Dufour-Rainfray, D., Vourc'h, P., Le Guisquet, A. M., Garreau, L., Ternant, D., Bodard, S., et al. (2010). Behavior and serotonergic disorders in rats exposed prenatally to valproate: a model for autism. Neurosci. Lett. 470, 55–59. doi: 10.1016/j.neulet.2009.12.054
Eftekhari, S., Shahrokhi, A., Tsintsadze, V., Nardou, R., Brouchoud, C., Conesa, M., et al. (2014). Response to comment on “Oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring”. Science 346:176. doi: 10.1126/science.1256009
Eissa, N., Jayaprakash, P., Azimullah, S., Ojha, S. K., Al-Houqani, M., Jalal, F. Y., et al. (2018). The histamine H3R antagonist DL77 attenuates autistic behaviors in a prenatal valproic acid-induced mouse model of autism. Sci. Rep. 8:13077. doi: 10.1038/s41598-018-31385-7
Ernst, M., Zametkin, A. J., Matochik, J. A., Pascualvaca, D., and Cohen, R. M. (1997). Low medial prefrontal dopaminergic activity in autistic children. Lancet 350:638.
Esbenshade, T. A., Browman, K. E., Bitner, R. S., Strakhova, M., Cowart, M. D., and Brioni, J. D. (2008). The histamine H3 receptor: an attractive target for the treatment of cognitive disorders. Br. J. Pharmacol. 154, 1166–1181. doi: 10.1038/bjp.2008.147
Estes, M. L., and McAllister, A. K. (2016). Maternal immune activation: implications for neuropsychiatric disorders. Science 353, 772–777. doi: 10.1126/science.aag3194
Fernández-Ruiz, J., Romero, J., and Ramos, J. A. (2015). “Endocannabinoids and neurodegenerative disorders: parkinson's disease, huntington's chorea, alzheimer's disease, and others,” in Endocannabinoids, ed R.G. Pertwee (Cham: Springer International Publishing), 233–259.
Földy, C., Malenka, R. C., and Sudhof, T. C. (2013). Autism-associated neuroligin-3 mutations commonly disrupt tonic endocannabinoid signaling. Neuron 78, 498–509. doi: 10.1016/j.neuron.2013.02.036
Földy, C. M., Minucci, S., Zhu, P., Kramer, O. H., Schimpf, A., Giavara, S., et al. (2001). Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 20, 6969–6978. doi: 10.1093/emboj/20.24.6969
Foley, A. G., Cassidy, A. W., and Regan, C. M. (2014). Pentyl-4-yn-VPA, a histone deacetylase inhibitor, ameliorates deficits in social behavior and cognition in a rodent model of autism spectrum disorders. Eur. J. Pharmacol. 727, 80–86. doi: 10.1016/j.ejphar.2014.01.050
Foley, A. G., Gannon, S., Rombach-Mullan, N., Prendergast, A., Barry, C., Cassidy, A. W., et al. (2012). Class I histone deacetylase inhibition ameliorates social cognition and cell adhesion molecule plasticity deficits in a rodent model of autism spectrum disorder. Neuropharmacology 63, 750–760. doi: 10.1016/j.neuropharm.2012.05.042
Fontes-Dutra, M., Santos-Terra, J., Deckmann, I., Brum Schwingel, G., Della-Flora Nunes, G., Hirsch, M. M., et al. (2018). Resveratrol prevents cellular and behavioral sensory alterations in the animal model of autism induced by valproic acid. Front. Synaptic Neurosci. 10:9. doi: 10.3389/fnsyn.2018.00009
Franklin, A. V., King, M. K., Palomo, V., Martinez, A., McMahon, L. L., and Jope, R. S. (2014). Glycogen synthase kinase-3 inhibitors reverse deficits in long-term potentiation and cognition in fragile X mice. Biol. Psychiatry 75, 198–206. doi: 10.1016/j.biopsych.2013.08.003
Frazer, A., and Hensler, J. G. (1999). “Serotonin,” in Basic Neurochemistry: Molecular, Cellular and Medical Aspects, 6th Edn, eds R. Wayne Albers and S. G. Siegel (Philadelphia, PA: Lippincott-Raven).
Gabriele, S., Sacco, R., and Persico, A. M. (2014). Blood serotonin levels in autism spectrum disorder: a systematic review and meta-analysis. Eur. Neuropsychopharmacol. 24, 919–929. doi: 10.1016/j.euroneuro.2014.02.004
Gadow, K. D., Devincent, C. J., Olvet, D. M., Pisarevskaya, V., and Hatchwell, E. (2010). Association of DRD4 polymorphism with severity of oppositional defiant disorder, separation anxiety disorder and repetitive behaviors in children with autism spectrum disorder. Eur. J. Neurosci. 32, 1058–1065. doi: 10.1111/j.1460-9568.2010.07382.x
Gao, J., Wang, X., Sun, H., Cao, Y., Liang, S., Wang, H., et al. (2016). Neuroprotective effects of docosahexaenoic acid on hippocampal cell death and learning and memory impairments in a valproic acid-induced rat autism model. Int. J. Dev. Neurosci. 49, 67–78. doi: 10.1016/j.ijdevneu.2015.11.006
Go, H. S., Kim, K. C., Choi, C. S., Jeon, S. J., Kwon, K. J., Han, S. H., et al. (2012). Prenatal exposure to valproic acid increases the neural progenitor cell pool and induces macrocephaly in rat brain via a mechanism involving the GSK-3beta/beta-catenin pathway. Neuropharmacology 63, 1028–1041. doi: 10.1016/j.neuropharm.2012.07.028
Goebbels, S., Oltrogge, J. H., Kemper, R., Heilmann, I., Bormuth, I., Wolfer, S., et al. (2010). Elevated phosphatidylinositol 3,4,5-trisphosphate in glia triggers cell-autonomous membrane wrapping and myelination. J. Neurosci. 30, 8953–8964. doi: 10.1523/JNEUROSCI.0219-10.2010
Gonzales, E. L., Jang, J. H., Mabunga, D. F., Kim, J. W., Ko, M. J., Cho, K. S., et al. (2016). Supplementation of Korean red ginseng improves behavior deviations in animal models of autism. Food Nutr. Res. 60:29245. doi: 10.3402/fnr.v60.29245
Gregory, S. G., Connelly, J. J., Towers, A. J., Johnson, J., Biscocho, D., Markunas, C. A., et al. (2009). Genomic and epigenetic evidence for oxytocin receptor deficiency in autism. BMC Med. 7:62. doi: 10.1186/1741-7015-7-62
Gunaydin, L. A., and Deisseroth, K. (2014). Dopaminergic dynamics contributing to social behavior. Cold Spring Harb. Symp. Quant. Biol. 79, 221–227. doi: 10.1101/sqb.2014.79.024711
Guo, W., Murthy, A. C., Zhang, L., Johnson, E. B., Schaller, E. G., Allan, A. M., et al. (2012). Inhibition of GSK3beta improves hippocampus-dependent learning and rescues neurogenesis in a mouse model of fragile X syndrome. Hum. Mol. Genet. 21, 681–691. doi: 10.1093/hmg/ddr501
Gupta, S., Aggarwal, S., and Heads, C. (1996). Dysregulated immune system in children with autism: beneficial effects of intravenous immune globulin on autistic characteristics. J. Autism Dev. Disord. 26, 439–452.
Haas, H. L., Sergeeva, O. A., and Selbach, O. (2008). Histamine in the nervous system. Physiol. Rev. 88, 1183–1241. doi: 10.1152/physrev.00043.2007
Hamilton, P. J., Campbell, N. G., Sharma, S., Erreger, K., Herborg Hansen, F., Saunders, C., et al. (2013). De novo mutation in the dopamine transporter gene associates dopamine dysfunction with autism spectrum disorder. Mol. Psychiatry 18, 1315–1323. doi: 10.1038/mp.2013.102
Hao, Y., Creson, T., Zhang, L., Li, P., Du, F., Yuan, P., et al. (2004). Mood stabilizer valproate promotes ERK pathway-dependent cortical neuronal growth and neurogenesis. J. Neurosci. 24, 6590–6599. doi: 10.1523/JNEUROSCI.5747-03.2004
Hara, Y., Ago, Y., Taruta, A., Hasebe, S., Kawase, H., Tanabe, W., et al. (2017). Risperidone and aripiprazole alleviate prenatal valproic acid-induced abnormalities in behaviors and dendritic spine density in mice. Psychopharmacology 234, 3217–3228. doi: 10.1007/s00213-017-4703-9
Hara, Y., Ago, Y., Taruta, A., Katashiba, K., Hasebe, S., Takano, E., et al. (2016). Improvement by methylphenidate and atomoxetine of social interaction deficits and recognition memory impairment in a mouse model of valproic acid-induced autism. Autism Res. 9, 926–939. doi: 10.1002/aur.1596
Hara, Y., Takuma, K., Takano, E., Katashiba, K., Taruta, A., Higashino, K., et al. (2015). Reduced prefrontal dopaminergic activity in valproic acid-treated mouse autism model. Behav. Brain Res. 289, 39–47. doi: 10.1016/j.bbr.2015.04.022
Heifets, B. D., and Castillo, P. E. (2009). Endocannabinoid signaling and long-term synaptic plasticity. Annu. Rev. Physiol. 71, 283–306. doi: 10.1146/annurev.physiol.010908.163149
Hettinger, J. A., Liu, X., Hudson, M. L., Lee, A., Cohen, I. L., Michaelis, R. C., et al. (2012). DRD2 and PPP1R1B (DARPP-32) polymorphisms independently confer increased risk for autism spectrum disorders and additively predict affected status in male-only affected sib-pair families. Behav. Brain Funct. 8:19. doi: 10.1186/1744-9081-8-19
Hettinger, J. A., Liu, X., Schwartz, C. E., Michaelis, R. C., and Holden, J. J. (2008). A DRD1 haplotype is associated with risk for autism spectrum disorders in male-only affected sib-pair families. Am. J. Med. Genet. B Neuropsychiatr. Genet. 147, 628–636. doi: 10.1002/ajmg.b.30655
Hussman, J. P. (2001). Letters to the editor: suppressed GABAergic inhibition as a common factor in suspected etiologies of autism. J. Autism Dev. Disord. 31, 247–248. doi: 10.1023/A:1010715619091
Ilango, A., Kesner, A. J., Keller, K. L., Stuber, G. D., Bonci, A., and Ikemoto, S. (2014). Similar roles of substantia nigra and ventral tegmental dopamine neurons in reward and aversion. J. Neurosci. 34, 817–822. doi: 10.1523/JNEUROSCI.1703-13.2014
Iversen, S. D., and Iversen, L. L. (2007). Dopamine: 50 years in perspective. Trends Neurosci. 30, 188–193. doi: 10.1016/j.tins.2007.03.002
Iwamoto, E. T., Loh, H. H., and Way, E. L. (1976). Circling behavior in rats with 6-hydroxydopamine or electrolytic nigral lesions. Eur. J. Pharmacol. 37, 339–356.
Jacquemont, M. L., Sanlaville, D., Redon, R., Raoul, O., Cormier-Daire, V., Lyonnet, S., et al. (2006). Array-based comparative genomic hybridisation identifies high frequency of cryptic chromosomal rearrangements in patients with syndromic autism spectrum disorders. J. Med. Genet. 43, 843–849. doi: 10.1136/jmg.2006.043166
Jung, K. M., Sepers, M., Henstridge, C. M., Lassalle, O., Neuhofer, D., Martin, H., et al. (2012). Uncoupling of the endocannabinoid signalling complex in a mouse model of fragile X syndrome. Nat. Commun. 3:1080. doi: 10.1038/ncomms2045
Kang, J., and Kim, E. (2015). Suppression of NMDA receptor function in mice prenatally exposed to valproic acid improves social deficits and repetitive behaviors. Front. Mol. Neurosci. 8:17. doi: 10.3389/fnmol.2015.00017
Kataoka, S., Takuma, K., Hara, Y., Maeda, Y., Ago, Y., and Matsuda, T. (2011). Autism-like behaviours with transient histone hyperacetylation in mice treated prenatally with valproic acid. Int. J. Neuropsychopharmacol. 16, 91–103. doi: 10.1017/S1461145711001714
Kern, J. K., Geier, D. A., Sykes, L. K., and Geier, M. R. (2015). Relevance of neuroinflammation and encephalitis in autism. Front. Cell. Neurosci. 9:519. doi: 10.3389/fncel.2015.00519
Kerr, D. M., Downey, L., Conboy, M., Finn, D. P., and Roche, M. (2013). Alterations in the endocannabinoid system in the rat valproic acid model of autism. Behav. Brain Res. 249, 124–132. doi: 10.1016/j.bbr.2013.04.043
Kerr, D. M., Gilmartin, A., and Roche, M. (2016). Pharmacological inhibition of fatty acid amide hydrolase attenuates social behavioural deficits in male rats prenatally exposed to valproic acid. Pharmacol. Res. 113, 228–235. doi: 10.1016/j.phrs.2016.08.033
Kim, J. W., Park, K., Kang, R. J., Gonzales, E. L. T., Kim, D. G., Oh, H. A., et al. (2018). Pharmacological modulation of AMPA receptor rescues social impairments in animal models of autism. Neuropsychopharmacology doi: 10.1038/s41386-018-0098-5. [Epub ahead of print].
Kim, J. W., Seung, H., Kim, K. C., Gonzales, E. L. T., Oh, H. A., Yang, S. M., et al. (2017a). Agmatine rescues autistic behaviors in the valproic acid-induced animal model of autism. Neuropharmacology 113, 71–81. doi: 10.1016/j.neuropharm.2016.09.014
Kim, J. W., Seung, H., Kwon, K. J., Ko, M. J., Lee, E. J., Oh, H. A., et al. (2014a). Subchronic treatment of donepezil rescues impaired social, hyperactive, and stereotypic behavior in valproic acid-induced animal model of autism. PLoS ONE 9:e104927. doi: 10.1371/journal.pone.0104927
Kim, K. C., Lee, D. K., Go, H. S., Kim, P., Choi, C. S., Kim, J. W., et al. (2014b). Pax6-dependent cortical glutamatergic neuronal differentiation regulates autism-like behavior in prenatally valproic acid-exposed rat offspring. Mol. Neurobiol. 49, 512–528. doi: 10.1007/s12035-013-8535-2
Kim, P., Park, J. H., Kwon, K. J., Kim, K. C., Kim, H. J., Lee, J. M., et al. (2013). Effects of Korean red ginseng extracts on neural tube defects and impairment of social interaction induced by prenatal exposure to valproic acid. Food Chem. Toxicol. 51, 288–296. doi: 10.1016/j.fct.2012.10.011
Kim, S., Kim, H., Yim, Y. S., Ha, S., Atarashi, K., Tan, T. G., et al. (2017b). Maternal gut bacteria promote neurodevelopmental abnormalities in mouse offspring. Nature 549, 528–532. doi: 10.1038/nature23910
Krystal, A. D. (2012). Psychiatric disorders and sleep. Neurol. Clin. 30, 1389–1413. doi: 10.1016/j.ncl.2012.08.018
Kuhar, M. J., Couceyro, P. R., and Lambert, P. D. (1999). “Biosynthesis of catecholamines,” in Basic Neurochemistry: Molecular, Cellular and Medical Aspects, 6th Edn, eds G. J. Siegel, B. W. Agranoff, and R. W. Albers (Philadelphia, PA: Lippincott-Raven).
Kulman, G., Lissoni, P., Rovelli, F., Roselli, M. G., Brivio, F., and Sequeri, P. (2000). Evidence of pineal endocrine hypofunction in autistic children. Neuro Endocrinol. Lett. 21, 31–34.
Kumar, H., and Sharma, B. (2016a). Memantine ameliorates autistic behavior, biochemistry & blood brain barrier impairments in rats. Brain Res. Bull. 124, 27–39. doi: 10.1016/j.brainresbull.2016.03.013
Kumar, H., and Sharma, B. (2016b). Minocycline ameliorates prenatal valproic acid induced autistic behaviour, biochemistry and blood brain barrier impairments in rats. Brain Res. 1630, 83–97. doi: 10.1016/j.brainres.2015.10.052
Kumar, H., Sharma, B. M., and Sharma, B. (2015). Benefits of agomelatine in behavioral, neurochemical and blood brain barrier alterations in prenatal valproic acid induced autism spectrum disorder. Neurochem. Int. 91, 34–45. doi: 10.1016/j.neuint.2015.10.007
Kuo, H. Y., and Liu, F. C. (2017). Valproic acid induces aberrant development of striatal compartments and corticostriatal pathways in a mouse model of autism spectrum disorder. FASEB J. 31, 4458–4471. doi: 10.1096/fj.201700054R
Kuwagata, M., Ogawa, T., Shioda, S., and Nagata, T. (2009). Observation of fetal brain in a rat valproate-induced autism model: a developmental neurotoxicity study. Int. J. Dev. Neurosci. 27, 399–405. doi: 10.1016/j.ijdevneu.2009.01.006
Lai, M. C., Lombardo, M. V., and Baron-Cohen, S. (2014). Autism. Lancet 383, 896–910. doi: 10.1016/S0140-6736(13)61539-1
Lake, C. R., Ziegler, M. G., and Murphy, D. L. (1977). Increased norepinephrine levels and decreased dopamine-beta-hydroxylase activity in primary autism. Arch. Gen. Psychiatry 34, 553–556.
Lerner, T. N., Shilyansky, C., Davidson, T. J., Evans, K. E., Beier, K. T., Zalocusky, K. A., et al. (2015). Intact-brain analyses reveal distinct information carried by SNc dopamine subcircuits. Cell 162, 635–647. doi: 10.1016/j.cell.2015.07.014
Lim, J. S., Lim, M. Y., Choi, Y., and Ko, G. (2017). Modeling environmental risk factors of autism in mice induces IBD-related gut microbial dysbiosis and hyperserotonemia. Mol. Brain 10:14. doi: 10.1186/s13041-017-0292-0
Lin, H. C., Gean, P. W., Wang, C. C., Chan, Y. H., and Chen, P. S. (2013). The amygdala excitatory/inhibitory balance in a valproate-induced rat autism model. PLoS ONE 8:e55248. doi: 10.1371/journal.pone.0055248
Lipton, J. O., and Sahin, M. (2014). The neurology of mTOR. Neuron 84, 275–291. doi: 10.1016/j.neuron.2014.09.034
Loke, Y. J., Hannan, A. J., and Craig, J. M. (2015). The role of epigenetic change in autism spectrum disorders. Front. Neurol. 6:107. doi: 10.3389/fneur.2015.00107
Ma, B., and Hottiger, M. O. (2016). Crosstalk between Wnt/beta-catenin and NF-kappaB signaling pathway during inflammation. Front. Immunol. 7:378. doi: 10.3389/fimmu.2016.00378
Mabunga, D. F., Gonzales, E. L., Kim, J. W., Kim, K. C., and Shin, C. Y. (2015). Exploring the validity of valproic acid animal model of autism. Exp. Neurobiol. 24, 285–300. doi: 10.5607/en.2015.24.4.285
Maccarrone, M., Rossi, S., Bari, M., De Chiara, V., Rapino, C., Musella, A., et al. (2010). Abnormal mGlu 5 receptor/endocannabinoid coupling in mice lacking FMRP and BC1 RNA. Neuropsychopharmacology 35, 1500–1509. doi: 10.1038/npp.2010.19
Man, K. K., Tong, H. H., Wong, L. Y., Chan, E. W., Simonoff, E., and Wong, I. C. (2015). Exposure to selective serotonin reuptake inhibitors during pregnancy and risk of autism spectrum disorder in children: a systematic review and meta-analysis of observational studies. Neurosci. Biobehav. Rev. 49, 82–89. doi: 10.1016/j.neubiorev.2014.11.020
Marchetti, B., and Pluchino, S. (2013). Wnt your brain be inflamed? Yes, it Wnt! Trends Mol. Med. 19, 144–156. doi: 10.1016/j.molmed.2012.12.001
Marín, O. (2012). Interneuron dysfunction in psychiatric disorders. Nat. Rev. Neurosci. 13, 107–120. doi: 10.1038/nrn3155
Martin, H. G., and Manzoni, O. J. (2014). Late onset deficits in synaptic plasticity in the valproic acid rat model of autism. Front. Cell. Neurosci. 8:23. doi: 10.3389/fncel.2014.00023
Matsumoto, M., and Hikosaka, O. (2009). Two types of dopamine neuron distinctly convey positive and negative motivational signals. Nature 459, 837–841. doi: 10.1038/nature08028
McDougle, C. J., Naylor, S. T., Cohen, D. J., Aghajanian, G. K., Heninger, G. R., and Price, L. H. (1996). Effects of tryptophan depletion in drug-free adults with autistic disorder. Arch. Gen. Psychiatry 53, 993–1000.
Mechoulam, R., and Parker, L. A. (2013). The endocannabinoid system and the brain. Annu. Rev. Psychol. 64, 21–47. doi: 10.1146/annurev-psych-113011-143739
Mehta, M. V., Gandal, M. J., and Siegel, S. J. (2011). mGluR5-antagonist mediated reversal of elevated stereotyped, repetitive behaviors in the VPA model of autism. PLoS ONE 6:e26077. doi: 10.1371/journal.pone.0026077
Meikle, L., Pollizzi, K., Egnor, A., Kramvis, I., Lane, H., Sahin, M., et al. (2008). Response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: effects on mTORC1 and Akt signaling lead to improved survival and function. J. Neurosci. 28, 5422–5432. doi: 10.1523/JNEUROSCI.0955-08.2008
Melamed, I. R., Heffron, M., Testori, A., and Lipe, K. (2018). A pilot study of high-dose intravenous immunoglobulin 5% for autism: impact on autism spectrum and markers of neuroinflammation. Autism Res. 11, 421–433. doi: 10.1002/aur.1906
Melke, J., Goubran Botros, H., Chaste, P., Betancur, C., Nygren, G., Anckarsater, H., et al. (2008). Abnormal melatonin synthesis in autism spectrum disorders. Mol. Psychiatry 13, 90–98. doi: 10.1038/sj.mp.4002016
Min, W. W., Yuskaitis, C. J., Yan, Q., Sikorski, C., Chen, S., Jope, R. S., et al. (2009). Elevated glycogen synthase kinase-3 activity in Fragile X mice: key metabolic regulator with evidence for treatment potential. Neuropharmacology 56, 463–472. doi: 10.1016/j.neuropharm.2008.09.017
Mines, M. A., Yuskaitis, C. J., King, M. K., Beurel, E., and Jope, R. S. (2010). GSK3 influences social preference and anxiety-related behaviors during social interaction in a mouse model of fragile X syndrome and autism. PLoS ONE 5: e9706. doi: 10.1371/journal.pone.0009706
Miyazaki, K., Narita, N., and Narita, M. (2005). Maternal administration of thalidomide or valproic acid causes abnormal serotonergic neurons in the offspring: implication for pathogenesis of autism. Int. J. Dev. Neurosci. 23, 287–297. doi: 10.1016/j.ijdevneu.2004.05.004
Morakotsriwan, N., Wattanathorn, J., Kirisattayakul, W., and Chaisiwamongkol, K. (2016). Autistic-like behaviors, oxidative stress status, and histopathological changes in cerebellum of valproic acid rat model of autism are improved by the combined extract of purple rice and silkworm pupae. Oxid. Med. Cell. Longev. 2016:3206561. doi: 10.1155/2016/3206561
Muller, C. L., Anacker, A. M. J., and Veenstra-VanderWeele, J. (2016). The serotonin system in autism spectrum disorder: from biomarker to animal models. Neuroscience 321, 24–41. doi: 10.1016/j.neuroscience.2015.11.010
Najjar, S., Pearlman, D. M., Alper, K., Najjar, A., and Devinsky, O. (2013). Neuroinflammation and psychiatric illness. J. Neuroinflammation 10:43. doi: 10.1186/1742-2094-10-43
Nakasato, A., Nakatani, Y., Seki, Y., Tsujino, N., Umino, M., and Arita, H. (2008). Swim stress exaggerates the hyperactive mesocortical dopamine system in a rodent model of autism. Brain Res. 1193, 128–135. doi: 10.1016/j.brainres.2007.11.043
Narita, N., Kato, M., Tazoe, M., Miyazaki, K., Narita, M., and Okado, N. (2002). Increased monoamine concentration in the brain and blood of fetal thalidomide- and valproic acid-exposed rat: putative animal models for autism. Pediatr. Res. 52, 576–579. doi: 10.1203/00006450-200210000-00018
Nelson, S. B., and Valakh, V. (2015). Excitatory/inhibitory balance and circuit homeostasis in autism spectrum disorders. Neuron 87, 684–698. doi: 10.1016/j.neuron.2015.07.033
Nguyen, M., Roth, A., Kyzar, E. J., Poudel, M. K., Wong, K., Stewart, A. M., et al. (2014). Decoding the contribution of dopaminergic genes and pathways to autism spectrum disorder (ASD). Neurochem. Int. 66, 15–26. doi: 10.1016/j.neuint.2014.01.002
Nicolini, C., and Fahnestock, M. (2018). The valproic acid-induced rodent model of autism. Exp. Neurol. 299, 217–227. doi: 10.1016/j.expneurol.2017.04.017
Nir, I., Meir, D., Zilber, N., Knobler, H., Hadjez, J., and Lerner, Y. (1995). Brief report: circadian melatonin, thyroid-stimulating hormone, prolactin, and cortisol levels in serum of young adults with autism. J. Autism Dev. Disord. 25, 641–654.
Oblak, A., Gibbs, T. T., and Blatt, G. J. (2013). Reduced serotonin receptor subtypes in a limbic and a neocortical region in autism. Autism Res. 6, 571–583. doi: 10.1002/aur.1317
O'Roak, B. J., Vives, L., Fu, W., Egertson, J. D., Stanaway, I. B., Phelps, I. G., et al. (2012a). Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 338, 1619–1622. doi: 10.1126/science.1227764
O'Roak, B. J., Vives, L., Girirajan, S., Karakoc, E., Krumm, N., Coe, B. P., et al. (2012b). Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250. doi: 10.1038/nature10989
Passani, M. B., and Blandina, P. (2011). Histamine receptors in the CNS as targets for therapeutic intervention. Trends Pharmacol. Sci. 32, 242–249. doi: 10.1016/j.tips.2011.01.003
Pragnya, B., Kameshwari, J. S., and Veeresh, B. (2014). Ameliorating effect of piperine on behavioral abnormalities and oxidative markers in sodium valproate induced autism in BALB/C mice. Behav. Brain Res. 270, 86–94. doi: 10.1016/j.bbr.2014.04.045
Purcell, A. E., Jeon, O. H., Zimmerman, A. W., Blue, M. E., and Pevsner, J. (2001). Postmortem brain abnormalities of the glutamate neurotransmitter system in autism. Neurology 57, 1618–1628. doi: 10.1212/WNL.57.9.1618
Qin, L., Dai, X., and Yin, Y. (2016). Valproic acid exposure sequentially activates Wnt and mTOR pathways in rats. Mol. Cell. Neurosci. 75, 27–35. doi: 10.1016/j.mcn.2016.06.004
Qin, M., Zeidler, Z., Moulton, K., Krych, L., Xia, Z., and Smith, C. B. (2015). Endocannabinoid-mediated improvement on a test of aversive memory in a mouse model of fragile X syndrome. Behav. Brain Res. 291, 164–171. doi: 10.1016/j.bbr.2015.05.003
Ramanathan, S., Woodroffe, A., Flodman, P. L., Mays, L. Z., Hanouni, M., Modahl, C. B., et al. (2004). A case of autism with an interstitial deletion on 4q leading to hemizygosity for genes encoding for glutamine and glycine neurotransmitter receptor sub-units (AMPA 2, GLRA3, GLRB) and neuropeptide receptors NPY1R, NPY5R. BMC Med. Genet. 5:10. doi: 10.1186/1471-2350-5-10
Ransohoff, R. M. (2016). How neuroinflammation contributes to neurodegeneration. Science 353, 777–783. doi: 10.1126/science.aag2590
Rinaldi, T., Kulangara, K., Antoniello, K., and Markram, H. (2007). Elevated NMDA receptor levels and enhanced postsynaptic long-term potentiation induced by prenatal exposure to valproic acid. Proc. Natl. Acad. Sci. U.S.A. 104, 13501–13506. doi: 10.1073/pnas.0704391104
Rinaldi, T., Perrodin, C., and Markram, H. (2008). Hyper-connectivity and hyper-plasticity in the medial prefrontal cortex in the valproic acid animal model of autism. Front. Neural Circuits 2:4. doi: 10.3389/neuro.04.004.2008
Rivera, C., Voipio, J., Payne, J. A., Ruusuvuori, E., Lahtinen, H., Lamsa, K., et al. (1999). The K+/Cl- co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397, 251–255. doi: 10.1038/16697
Rosenberg, G. (2007). The mechanisms of action of valproate in neuropsychiatric disorders: can we see the forest for the trees? Cell. Mol. Life Sci. 64, 2090–2103. doi: 10.1007/s00018-007-7079-x
Rossignol, D. A., and Frye, R. E. (2011). Melatonin in autism spectrum disorders: a systematic review and meta-analysis. Dev. Med. Child Neurol. 53, 783–792. doi: 10.1111/j.1469-8749.2011.03980.x
Rosso, S. B., and Inestrosa, N. C. (2013). WNT signaling in neuronal maturation and synaptogenesis. Front. Cell. Neurosci. 7:103. doi: 10.3389/fncel.2013.00103
Roullet, F. I., Lai, J. K., and Foster, J. A. (2013). In utero exposure to valproic acid and autism–a current review of clinical and animal studies. Neurotoxicol. Teratol. 36, 47–56. doi: 10.1016/j.ntt.2013.01.004
Rubenstein, J. L., and Merzenich, M. M. (2003). Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2, 255–267. doi: 10.1034/j.1601-183X.2003.00037.x
Sato, A., Kasai, S., Kobayashi, T., Takamatsu, Y., Hino, O., Ikeda, K., et al. (2012). Rapamycin reverses impaired social interaction in mouse models of tuberous sclerosis complex. Nat. Commun. 3:1292. doi: 10.1038/ncomms2295
Schain, R. J., and Freedman, D. X. (1961). Studies on 5-hydroxyindole metabolism in autistic and other mentally retarded children. J. Pediatr. 58, 315–320.
Schneider, T., and Przewlocki, R. (2005). Behavioral alterations in rats prenatally exposed to valproic acid: animal model of autism. Neuropsychopharmacology 30, 80–89. doi: 10.1038/sj.npp.1300518
Servadio, M., Melancia, F., Manduca, A., di Masi, A., Schiavi, S., Cartocci, V., et al. (2016). Targeting anandamide metabolism rescues core and associated autistic-like symptoms in rats prenatally exposed to valproic acid. Transl. Psychiatry 6:e902. doi: 10.1038/tp.2016.182
Shi, L., Tu, N., and Patterson, P. H. (2005). Maternal influenza infection is likely to alter fetal brain development indirectly: the virus is not detected in the fetus. Int. J. Dev. Neurosci. 23, 299–305. doi: 10.1016/j.ijdevneu.2004.05.005
Siniscalco, D., Sapone, A., Giordano, C., Cirillo, A., de Magistris, L., Rossi, F., et al. (2013). Cannabinoid receptor type 2, but not type 1, is up-regulated in peripheral blood mononuclear cells of children affected by autistic disorders. J. Autism Dev. Disord. 43, 2686–2695. doi: 10.1007/s10803-013-1824-9
Staal, W. G., Langen, M., van Dijk, S., Mensen, V. T., and Durston, S. (2015). DRD3 gene and striatum in autism spectrum disorder. Br. J. Psychiatry 206, 431–432. doi: 10.1192/bjp.bp.114.148973
Takuma, K., Hara, Y., Kataoka, S., Kawanai, T., Maeda, Y., Watanabe, R., et al. (2014). Chronic treatment with valproic acid or sodium butyrate attenuates novel object recognition deficits and hippocampal dendritic spine loss in a mouse model of autism. Pharmacol. Biochem. Behav. 126, 43–49. doi: 10.1016/j.pbb.2014.08.013
Tang, G., Gudsnuk, K., Kuo, S. H., Cotrina, M. L., Rosoklija, G., Sosunov, A., et al. (2014). Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 83, 1131–1143. doi: 10.1016/j.neuron.2014.07.040
Tarabeux, J., Kebir, O., Gauthier, J., Hamdan, F., Xiong, L., Piton, A., et al. (2011). Rare mutations in N-methyl-D-aspartate glutamate receptors in autism spectrum disorders and schizophrenia. Transl. Psychiatry 1:e55. doi: 10.1038/tp.2011.52
Thomas, A. M., Bui, N., Perkins, J. R., Yuva-Paylor, L. A., and Paylor, R. (2012). Group I metabotropic glutamate receptor antagonists alter select behaviors in a mouse model for fragile X syndrome. Psychopharmacology 219, 47–58. doi: 10.1007/s00213-011-2375-4
Tian, Y., Yabuki, Y., Moriguchi, S., Fukunaga, K., Mao, P. J., Hong, L. J., et al. (2014). Melatonin reverses the decreases in hippocampal protein serine/threonine kinases observed in an animal model of autism. J. Pineal Res. 56, 1–11. doi: 10.1111/jpi.12081
Tordjman, S., Gutknecht, L., Carlier, M., Spitz, E., Antoine, C., Slama, F., et al. (2001). Role of the serotonin transporter gene in the behavioral expression of autism. Mol. Psychiatry 6, 434–439. doi: 10.1038/sj.mp.4000873
Tordjman, S., Najjar, I., Bellissant, E., Anderson, G. M., Barburoth, M., Cohen, D., et al. (2013). Advances in the research of melatonin in autism spectrum disorders: literature review and new perspectives. Int. J. Mol. Sci. 14, 20508–20542. doi: 10.3390/ijms141020508
Tsai, P. T., Hull, C., Chu, Y., Greene-Colozzi, E., Sadowski, A. R., Leech, J. M., et al. (2012). Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 488, 647–651. doi: 10.1038/nature11310
Tsujino, N., Nakatani, Y., Seki, Y., Nakasato, A., Nakamura, M., Sugawara, M., et al. (2007). Abnormality of circadian rhythm accompanied by an increase in frontal cortex serotonin in animal model of autism. Neurosci. Res. 57, 289–295. doi: 10.1016/j.neures.2006.10.018
Tung, E. W., and Winn, L. M. (2011). Valproic acid increases formation of reactive oxygen species and induces apoptosis in postimplantation embryos: a role for oxidative stress in valproic acid-induced neural tube defects. Mol. Pharmacol. 80, 979–987. doi: 10.1124/mol.111.072314
Tyzio, R., Cossart, R., Khalilov, I., Minlebaev, M., Hubner, C. A., Represa, A., et al. (2006). Maternal oxytocin triggers a transient inhibitory switch in GABA signaling in the fetal brain during delivery. Science 314, 1788–1792. doi: 10.1126/science.1133212
Tyzio, R., Nardou, R., Ferrari, D. C., Tsintsadze, T., Shahrokhi, A., Eftekhari, S., et al. (2014). Oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring. Science 343, 675–679. doi: 10.1126/science.1247190
U.S. Food and Drug Administration (2007). First drug to treat irritability associated with autism. FDA Consum. 41, 1–4.
U.S. Food and Drug Administration (2009). U.S. Food and Drug Administration Approves ABILIFY® (aripiprazole) for the treatment of irritability associated with autistic disorder in pediatric patients (Ages 6 to 17 Years). FDA Consum.
Valeeva, G., Valiullina, F., and Khazipov, R. (2013). Excitatory actions of GABA in the intact neonatal rodent hippocampus in vitro. Front. Cell. Neurosci. 7:20. doi: 10.3389/fncel.2013.00020
Veenstra-VanderWeele, J., Muller, C. L., Iwamoto, H., Sauer, J. E., Owens, W. A., Shah, C. R., et al. (2012). Autism gene variant causes hyperserotonemia, serotonin receptor hypersensitivity, social impairment and repetitive behavior. Proc. Natl. Acad. Sci. U.S.A. 109, 5469–5474. doi: 10.1073/pnas.1112345109
Wagner, G. C., Reuhl, K. R., Cheh, M., McRae, P., and Halladay, A. K. (2006). A new neurobehavioral model of autism in mice: pre- and postnatal exposure to sodium valproate. J. Autism Dev. Disord. 36, 779–793. doi: 10.1007/s10803-006-0117-y
Walcott, E. C., Higgins, E. A., and Desai, N. S. (2011). Synaptic and intrinsic balancing during postnatal development in rat pups exposed to valproic acid in utero. J. Neurosci. 31, 13097–13109. doi: 10.1523/JNEUROSCI.1341-11.2011
Wang, C. C., Lin, H. C., Chan, Y. H., Gean, P. W., Yang, Y. K., and Chen, P. S. (2013). 5-HT1A-receptor agonist modified amygdala activity and amygdala-associated social behavior in a valproate-induced rat autism model. Int. J. Neuropsychopharmacol. 16, 2027–2039. doi: 10.1017/S1461145713000473
Wang, Z., Xu, L., Zhu, X., Cui, W., Sun, Y., Nishijo, H., et al. (2010). Demethylation of specific Wnt/beta-catenin pathway genes and its upregulation in rat brain induced by prenatal valproate exposure. Anat. Rec. 293, 1947–1953. doi: 10.1002/ar.21232
Wei, D., Dinh, D., Lee, D., Li, D., Anguren, A., Moreno-Sanz, G., et al. (2016). Enhancement of anandamide-mediated endocannabinoid signaling corrects autism-related social impairment. Cannabis Cannabinoid Res. 1, 81–89. doi: 10.1089/can.2015.0008
Wellmann, K. A., Varlinskaya, E. I., and Mooney, S. M. (2014). D-Cycloserine ameliorates social alterations that result from prenatal exposure to valproic acid. Brain Res. Bull. 108, 1–9. doi: 10.1016/j.brainresbull.2014.08.001
Wenger, T. L., Kao, C., McDonald-McGinn, D. M., Zackai, E. H., Bailey, A., Schultz, R. T., et al. (2016). The role of mGluR copy number variation in genetic and environmental forms of syndromic autism spectrum disorder. Sci. Rep. 6:19372. doi: 10.1038/srep19372
Winden, K. D., Ebrahimi-Fakhari, D., and Sahin, M. (2018). Abnormal mTOR activation in autism. Annu. Rev. Neurosci. 41, 1–23. doi: 10.1146/annurev-neuro-080317-061747
Wright, C., Shin, J. H., Rajpurohit, A., Deep-Soboslay, A., Collado-Torres, L., Brandon, N. J., et al. (2017). Altered expression of histamine signaling genes in autism spectrum disorder. Transl. Psychiatry 7:e1126. doi: 10.1038/tp.2017.87
Wu, H., Wang, X., Gao, J., Liang, S., Hao, Y., Sun, C., et al. (2017a). Fingolimod (FTY720) attenuates social deficits, learning and memory impairments, neuronal loss and neuroinflammation in the rat model of autism. Life Sci. 173, 43–54. doi: 10.1016/j.lfs.2017.01.012
Wu, H. F., Chen, P. S., Chen, Y. J., Lee, C. W., Chen, I. T., and Lin, H. C. (2017b). Alleviation of N-methyl-D-aspartate receptor-dependent long-term depression via regulation of the glycogen synthase kinase-3beta pathway in the amygdala of a valproic acid-induced animal model of autism. Mol. Neurobiol. 54, 5264–5276. doi: 10.1007/s12035-016-0074-1
Wu, H. F., Chen, P. S., Hsu, Y. T., Lee, C. W., Wang, T. F., Chen, Y. J., et al. (2018). D-cycloserine ameliorates autism-like deficits by removing GluA2-containing AMPA receptors in a valproic acid-induced rat model. Mol. Neurobiol. 55, 4811–4824. doi: 10.1007/s12035-017-0685-1
Yu, I. T., Park, J. Y., Kim, S. H., Lee, J. S., Kim, Y. S., and Son, H. (2009). Valproic acid promotes neuronal differentiation by induction of proneural factors in association with H4 acetylation. Neuropharmacology 56, 473–480. doi: 10.1016/j.neuropharm.2008.09.019
Zamberletti, E., Gabaglio, M., and Parolaro, D. (2017). The endocannabinoid system and autism spectrum disorders: insights from animal models. Int. J. Mol. Sci. 18:E1916. doi: 10.3390/ijms18091916
Zeng, L. H., Xu, L., Gutmann, D. H., and Wong, M. (2008). Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann. Neurol. 63, 444–453. doi: 10.1002/ana.21331
Zhang, J., Liu, L. M., and Ni, J. F. (2017a). Rapamycin modulated brain-derived neurotrophic factor and B-cell lymphoma 2 to mitigate autism spectrum disorder in rats. Neuropsychiatr. Dis. Treat. 13, 835–842. doi: 10.2147/NDT.S125088
Zhang, L., and Alger, B. E. (2010). Enhanced endocannabinoid signaling elevates neuronal excitability in fragile X syndrome. J. Neurosci. 30, 5724–5729. doi: 10.1523/JNEUROSCI.0795-10.2010
Zhang, Y., Cui, W., Zhai, Q., Zhang, T., and Wen, X. (2017b). N-acetylcysteine ameliorates repetitive/stereotypic behavior due to its antioxidant properties without activation of the canonical Wnt pathway in a valproic acid-induced rat model of autism. Mol. Med. Rep. 16, 2233–2240. doi: 10.3892/mmr.2017.6787
Zhang, Y., Sun, Y., Wang, F., Wang, Z., Peng, Y., and Li, R. (2012). Downregulating the canonical Wnt/beta-catenin signaling pathway attenuates the susceptibility to autism-like phenotypes by decreasing oxidative stress. Neurochem. Res. 37, 1409–1419. doi: 10.1007/s11064-012-0724-2
Zhang, Y., Yang, C., Yuan, G., Wang, Z., Cui, W., and Li, R. (2015). Sulindac attenuates valproic acid-induced oxidative stress levels in primary cultured cortical neurons and ameliorates repetitive/stereotypic-like movement disorders in Wistar rats prenatally exposed to valproic acid. Int. J. Mol. Med. 35, 263–270. doi: 10.3892/ijmm.2014.1996
Keywords: autism, valproic acid, excitatory/inhibitory imbalance, endocannabinoid system, mTOR signaling, Wnt signaling, neuroinflammation, epigenetics
Citation: Kuo H-Y and Liu F-C (2018) Molecular Pathology and Pharmacological Treatment of Autism Spectrum Disorder-Like Phenotypes Using Rodent Models. Front. Cell. Neurosci. 12:422. doi: 10.3389/fncel.2018.00422
Received: 24 July 2018; Accepted: 29 October 2018;
Published: 20 November 2018.
Edited by:
Chang-hui Shen, College of Staten Island, United StatesReviewed by:
Yuriko Iwakura, Niigata University, JapanElisa L. Hill-Yardin, RMIT University, Australia
Copyright © 2018 Kuo and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fu-Chin Liu, ZnVjaGluQHltLmVkdS50dw==