 Aaron Mellesmoen
Aaron Mellesmoen Carrie Sheeler
Carrie Sheeler Austin Ferro
Austin Ferro Orion Rainwater
Orion Rainwater Marija Cvetanovic
Marija Cvetanovic- 1Department of Neuroscience, University of Minnesota, Minneapolis, MN, United States
- 2Department of Lab Medicine and Pathology, University of Minnesota, Minneapolis, MN, United States
- 3Institute for Translational Neuroscience, University of Minnesota, Minneapolis, MN, United States
Spinocerebellar ataxia type 1 (SCA1) is a fatal neurodegenerative disease caused by an abnormal expansion of CAG repeats in the Ataxin-1 (ATXN1) gene and characterized by motor deficits and cerebellar neurodegeneration. Even though mutant ATXN1 is expressed from an early age, disease onset usually occurs in patient’s mid-thirties, indicating the presence of compensatory factors that limit the toxic effects of mutant ATXN1 early in disease. Brain derived neurotrophic factor (BDNF) is a growth factor known to be important for the survival and function of cerebellar neurons. Using gene expression analysis, we observed altered BDNF expression in the cerebella of Purkinje neuron specific transgenic mouse model of SCA1, ATXN1[82Q] mice, with increased expression during the early stage and decreased expression in the late stage of disease. We therefore investigated the potentially protective role of BDNF in early stage SCA1 through intraventricular delivery of BDNF via ALZET osmotic pumps. Extrinsic BDNF delivery delayed onset of motor deficits and Purkinje neuron pathology in ATXN1[82Q] mice supporting its use as a novel therapeutic for SCA1.
Introduction
Spinocerebellar ataxia type 1 (SCA1) is a dominantly inherited and fatal neurodegenerative disease resulting from an overexpansion of CAG repeats within the Ataxin-1 (ATXN1) gene (Banfi et al., 1996; Zoghbi and Orr, 2009). SCA1 belongs to a group of polyglutamine (polyQ) disorders that also includes SCA2, 3, 6, 7, 17, spinobulbar muscular atrophy, Huntington’s disease (HD), and dentatorubropallidoluysian atrophy (Genis et al., 1995; Gusella and MacDonald, 2000; La Spada and Taylor, 2010). Clinical onset of SCA1 is characterized by ataxia, or loss of motor coordination and balance, which typically presents during patient’s mid-thirties (Genis et al., 1995; Orr and Zoghbi, 2007; Rüb et al., 2013; Matilla-Dueñas et al., 2014). Progressive degradation of motor function leads to death within 10–20 years following clinical onset (Rüb et al., 2013). Currently, no treatments exist for SCA1 (Paulson et al., 2017).
ATXN1 is expressed prior to birth in humans [Figure 1A, created using human developmental transcriptome data from BrainSpan (Miller et al., 2014)], yet the clinical phenotype of SCA1 emerges decades later. Similarly, in ATXN1[82Q] mice, Purkinje neuron specific transgenic mouse model of SCA1, mutant ATXN1 is expressed from postnatal day 10 (P10; Serra et al., 2006) yet they exhibit motor deficits and Purkinje neuron pathology earliest at 12 weeks of age, in our hands (Cvetanovic, 2015; Qu et al., 2017; Ferro et al., 2018). Neuroprotective factors may compensate for early mutant ATXN1-mediated neural toxicity, and gradual decline in compensation may contribute to late onset of disease. Indeed, pre-manifest carriers of SCA1 and other polyQ mutations demonstrate mild coordination deficits and brain abnormalities several years prior to estimated disease onset (Jacobi et al., 2013; Storey, 2013; Tabrizi et al., 2013; Espinoza et al., 2018; Joers et al., 2018). We and others have observed cellular and molecular alterations that may compensate for neuronal dysfunction during the early stages of disease in ATXN1[82Q] mice (Dell’Orco et al., 2015; Kim et al., 2018). Here, we propose that increased expression of brain derived neurotrophic factor (BDNF) by reactive astroglia may be a part of this compensatory mechanism to delay disease onset in ATXN1[82Q] mice.
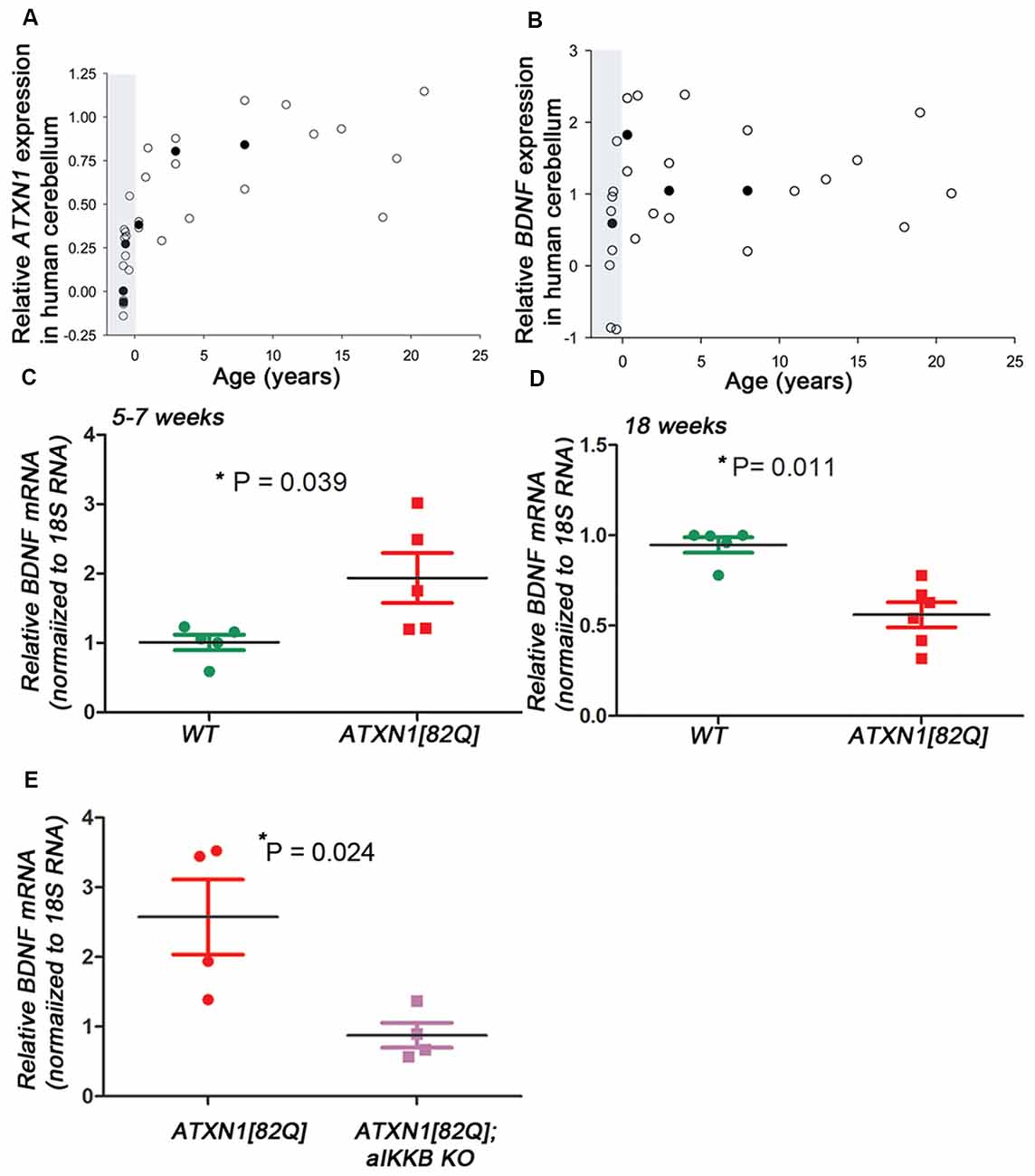
Figure 1. Brain derived neurotrophic factor (BDNF) expression in transgenic spinocerebellar ataxia type 1 (SCA1) mice. (A–B) Relative Ataxin-1 (ATXN1) (A) and BDNF (B) expression in the human cerebellar cortex [polyglutamine (polyQ) sequence/SCA1 status unknown] obtained from the transcriptome data at Allen Brain Institute. Raw reads per kilobase million (RPKM) values were normalized to the highest and lowest values and data was averaged when multiple patient samples fell within the same postnatal year or same post conception week. Open circles represent single patient samples and closed circles represent average expression (used when possible). Shaded areas mark pre-natal period. (C–E) Reverse transcription and quantitative polymerase chain reaction (RT-qPCR) using cerebellar extracts from (C) early stage (5–7 weeks) and (D) late stage (18–24 weeks old) ATXN1[82Q] mice. Results are normalized using 18S RNA and age-matched wild-type (WT) littermates. (E) RT-qPCR using cerebellar extracts from ATXN1[82Q] mice and mice in which astroglialnuclear factor κ-light-chain-enhancer of activated B cells (NF-κB) is inhibited early indisease [ATXN1[82Q]; aIKKβ KO]. Results are normalized using 18S RNA and age-matched WT littermates. For (C–E) error bars = SEM. Student’s t-test P values. Each dot represents a biological sample.
Materials and Methods
Human RNAseq
Reads per kilobase million (RPKM) RNAseq data was obtained from the BrainSpan database1 with permission (Miller et al., 2014). RPKM values were averaged over patients with equal ages and then normalized as follows:
y, youngest datapoint; o, oldest datapoint.
Mice
The creation of the ATXN1[82Q] mice was previously described (Clark et al., 1997). Equal number of male and female mice were randomly allocated to BDNF (20 μg of human recombinant BDNF (R&D Systems Cat. 248-BD-250/CF) in 100 μl per micropump, ~ 0.71μg/day) or control artificial cerebrospinal fluid (aCSF) groups. We surgically implanted ALZET pumps (Alzet Model 1004) into 8-week-old mice in a subcutaneous pocket in the back and the delivery cannula in the right lateral ventricle (A/P, 1.1; M/L, 0.5 D/V, −2.5 mm from Bregma) as we previously described (Cvetanovic et al., 2011).
In all the experiments investigators were blinded to the genotype/treatment.
Animal experimentation was approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Minnesota and was conducted in accordance with the National Institutes of Health’s (NIH) Principles of Laboratory Animal Care (86-23, revised 1985), and the American Physiological Society’s Guiding Principles in the Use of Animals.
Mouse RNA Sequencing
Mouse RNA sequencing was done as previously described (Ingram et al., 2016).
Rotarod Analysis
Mice were tested on rotatod (#47600; Ugo Basile) to evaluate motor deficits as described previously (Kim et al., 2018). Rotarod paradigm consisted of acceleration from 5 rpm to 40 rpm over 0–5 min, followed by 40 rpm constant speed from 5 min to 10 min. Latency to fall was recorded.
Immunofluorescent (IF) Staining
Immunofluorescent (IF) was performed on floating 45 μm brain sections using primary antibodies [Calbindin #C9848, Sigma-Aldrich; vesicular glutamate transporter 2 (VGLUT2) #MAB5504, Millipore, Burlington, MA, USA] as previously described (Cvetanovic et al., 2011).
Quantitative analysis was performed using ImageJ National Institutes of Health’s (NIH) as described previously. We quantified minimum of six different slices from each mouse. Per each slice we randomly chose two lobules and draw a line from the base of the Purkinje soma to the end of the dendrites to determine width of the molecular layer (length of the line) and average calbindin intensity. For assessing climbing fiber height, the distance from the Purkinje neuron soma to the end of VGLUT2 staining was measured (VGLUT2), and the extension of climbing fibers was depicted relative to the molecular layer thickness (VGLUT2/calbindin; Joers et al., 2018).
Reverse Transcription and Quantitative Polymerase Chain Reaction (RT-qPCR)
Total RNA was extracted from mouse cerebella using TRIzol (Life Technologies) and (reverse transcription and quantitative polymerase chain reaction (RT-qPCR) was performed as described previously (Kim et al., 2018).
Statistical Analysis
Wherever possible, sample sizes were calculated using power analyses based on the standard deviations from our previous studies, significance level of 5%, and power of 90%. Statistical tests were performed with GraphPad Prism. We have used one-way analyses of variance (ANOVA) followed by Bonferroni or Kruskal-Wallis post hoc tests (depending on the normality of the data) or two-tailed Student’s t-test.
Data Availability
All the data from this study are available from the authors.
Results
BDNF Expression Is Altered in ATXN1[82Q] Mice
We used unbiased RNA sequencing to examine gene expression changes during disease progression. We have found that expression of neuroprotective factor BDNF is increased early and decreased late in ATXN1[82Q] mice (Cvetanovic et al., 2015; Supplementary Table S1), 5-week-old ATXN1[82Q] mice 6.87 vs. 5.3 fragments per kilobase of exon per million reads mapped (FPKM) in wild-type (WT) littermates, p = 0.00005, adjusted q = 0.0165; 12-week-old ATXN1[82Q] mice 3.2 vs. 5.03 fragments per kilobase of exon per million reads mapped (FPKM) in WT littermates, p = 0.00015, adjusted q = 0.0021). Using RT-qPCR we have confirmed increased BDNF expression in the cerebella during early stage (Figure 1C, 1.934 ± 0.359 relative to WT littermates, N = 5 each, Student’s t-test P = 0.0394) and later decrease (Figure 1D, 0.56 ± 006 relative to WT, N = 6 ATXN1[82Q] and N = 5 WT mice, Student’s t-test P = 0.0014).
Hallmark features of astrogliosis, the process by which astroglia react to neuronal dysfunction, are detectable early in SCA1 mice (Cvetanovic et al., 2015). We demonstrated that reducing astrogliosis early in SCA1, through inducible genetic inhibition of nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB) signaling selectively in astroglia (astroglial IKKβ knock-out, aIKKβ KO), exacerbates motor deficits and neuropathology in ATXN1[82Q] mice (Kim et al., 2018). Inhibition of astroglial NF-κB prevented the early increase in BDNF expression (Figure 1E, 2.57 ± 0.11 increase in ATXN1[82Q] mice compared to 0.88 ± 0.17 in ATXN1[82Q];aIKKB KO, N = 4, t-test P = 0.024), indicating that astroglial NF-κB signaling may regulate increased BDNF expression early in SCA1 mice.
Delivery of BDNF Early in Disease Ameliorates SCA1
In our hands, ATXN1[82Q] mice demonstrate reproducible motor deficits and cerebellar pathology earliest at 12 weeks. However, as the onset of motor deficits has been observed earlier by others (Clark et al., 1997; Duvick et al., 2010; Hourez et al., 2011; Ibrahim et al., 2017), we refer to mice younger than 12 weeks as early stage instead of pre-symptomatic. To determine the neuroprotective effects of BDNF, we used osmotic ALZET pumps to deliver recombinant BDNF to 8-weeks-old SCA1 or WT littermate mice for a total of 4 weeks (~0.71 μg/day). This study consisted of four experimental groups: WT and ATXN1[82Q] mice with pumps delivering BDNF, and control WT and ATXN1[82Q] mice with pumps delivering aCSF. Mice were tested on rotarod at 12 weeks and pathology was examined at 14 weeks (Figure 2A).
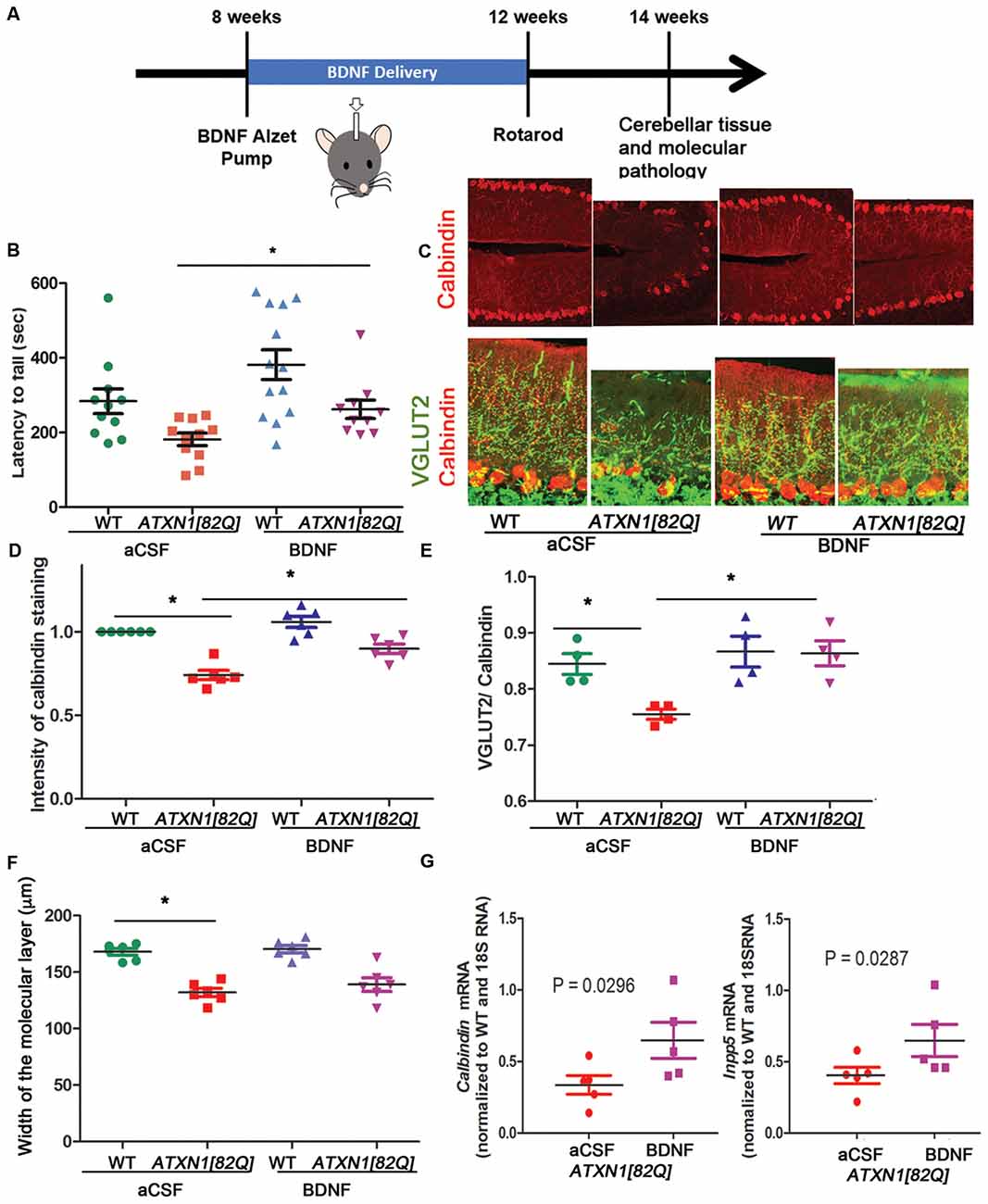
Figure 2. BDNF delivery delays disease pathogenesis in ATXN1[82Q] mice. (A) Experimental scheme to test the role of BDNF early in SCA1. (B) Rotarod performance of mice at 12 weeks. *P < 0.05 using one-way ANOVA with Kruskal-Wallis test. (C–F) Cerebellar slices from 14-week-old mice were stained with antibody specific for Purkinje neuron-marker calbindin and vesicular glutamate transporter 2 (VGLUT2) to label climbing fiber synapses on Purkinje neurons. ImageJ was used to quantify (D) calbindin intensity in the Purkinje neurons, (E) length of climbing fiber synapses (VGLUT2 puncta) on Purkinje neuron dendrites (determined as VGLUT2/calbindin ratio), and (F) width of the molecular layer. *P < 0.05 using one-way ANOVA with Bonferroni’s multiple comparison test. (G) RT-qPCR analysis of disease associated Purkinje neurons genes calbindin (left) and Inpp5 (right). Student’s t-test P values. For all error bars = SEM. Each dot represents a biological sample.
BDNF-treated ATXN1[82Q] mice performed significantly better on rotarod compared to control aCSF-treated ATXN1[82Q] mice (e.g., day 4 average latency to fall of BDNF-treated ATXN1[82Q] mice = 262.5 ± 24.3 s, N = 10; compared to aCSF-treated ATXN1[82Q] mice = 181 ± 16.73 s, N = 11, P < 0.05, one-way ANOVA with Kruskal-Wallis test H = 15.74, P = 0.0013; Figure 2B). Importantly, BDNF-treated ATXN1[82Q] mice were indistinguishable from BDNF- or aCSF-treated WT mice (aCSF- and BDNF- treated WT mice latency was 283 ± 36.7 s, N = 12, and 382 ± 39.78, N = 13, respectively, P > 0.05 for both comparisons one-way ANOVA with Kruskal-Wallis test).
We used immunofluorescence to determine whether BDNF ameliorates decreased calbindin expression and synaptic loss, two well-characterized hallmarks of Purkinje neuron pathology in SCA1 mice (Zu et al., 2004; Serra et al., 2006; Duvick et al., 2010; Barnes et al., 2011; Ebner et al., 2013; Ruegsegger et al., 2016; Ibrahim et al., 2017). BDNF-treated ATXN1[82Q] and WT mice were indistinguishable and both groups had significantly higher calbindin intensity of Purkinje neurons compared to aCSF-treated ATXN1[82Q] controls (Figures 2C,D, average relative intensity of aCSF-treated ATXN1[82Q] mice was 0.7417 ± 0.028 compared to 0.8989 ± 0.027 in BDNF-treated ATXN1[82Q] mice, N = 6 of each, P < 0.05 one-way ANOVA with post hoc Bonferroni test F(3,20) = 29.32, P < 0.0001). BDNF also prevented loss of climbing fibers synapses on Purkinje neuron dendrites in ATXN1[82Q] mice as quantified with VGLUT2 staining (Figures 2C,E, average VGLUT2/CAL ratio for aCSF-treated ATXN1[82Q] mice was 0.7553 ± 0.009 compared to 0.8638 ± 0.02 in BDNF–treated ATXN1[82Q] mice, N = 4 each, one-way ANOVA F(3,12) = 3.878 P = 0.0071, post hoc Bonferroni test P < 0.05). However, width of the molecular layer was not rescued (Figure 2F, average width for aCSF-treated ATXN1[82Q] mice was 132.1 ± 3.667 μm compared to 139 ± 6.1 in BDNF–treated ATXN1[82Q] mice, N = 6 each, one-way ANOVA with post hoc Bonferroni test P > 0.05).
We used RT-qPCR to demonstrate that BDNF treatment rescued expression of Purkinje neuron genes Calbindin (Ingram et al., 2016; Figure 2G left, aCSF-treated ATXN1[82Q] mice = 0.336 ± 0.065 compared to 0.648 ± 0.125 in BDNF-treated ATXN1[82Q] mice, N = 5, t-test, P = 0.0296) and INNP5 (Figure 2G right, aCSF-treated ATXN1[82Q] mice = 0.404 ± 0.057 compared to 0.648 ± 0.115 in BDNF-treated ATXN1[82Q] mice, t-test, P = 0.02872, N = 5). Together, these results indicate that BDNF may be neuroprotective early in SCA1.
Discussion
We report that early delivery of BDNF delays motor deficits and pathology of Purkinje neurons. In addition, we report an early increase in BDNF expression in SCA1 that may be regulated by astroglial NF-κB-signaling. Based on these results, we propose that early increase in BDNF expression may be neuroprotective and compensate for ATXN1 toxicity, thus delaying disease onset.
BDNF belongs to the family of neurotrophins (Barde et al., 1982) and is expressed in human cerebellum from early development (Figure 1B). Its cognate receptor, Tyrosine kinase B receptor (TrkB), is highly expressed on Purkinje neurons where BDNF signaling plays a host of neuroprotective and modulatory functions such as regulating dendritic branching and synaptic strength (Schwartz et al., 1997; Kafitz et al., 1999; Carter et al., 2002; Furutani et al., 2006; Huang et al., 2012). Purkinje neuron pathology in SCA1 includes shrinking of the dendritic arbor, loss of synapses, and altered evoked and spontaneous firing (Duvick et al., 2010; Cvetanovic et al., 2011; Hourez et al., 2011; Dell’Orco et al., 2015). It is possible that BDNF exerts its neuroprotective effect in SCA1 at least in part through prevention of these Purkinje neuron pathologies. Intriguingly, during postnatal cerebellar development BDNF signaling seems to promote elimination of climbing fiber synapses, and this may be regulated by temporal expression of short TrkB isoform (Bosman et al., 2006; Sherrard et al., 2009; Choo et al., 2017). Delivery of BDNF to WT mice at 8 weeks did not seem to affect climbing fiber synapses and these mice performed well on rotarod (Figure 2B) indicating that post-development extrinsic BDNF may not have obvious side effects on these tests. Since BDNF is delivered intracerebroventricularly, we cannot exclude the possibility of off-target BDNF effects. However, ATXN1[82Q] mice pathology is limited to cerebellum (Burright et al., 1995) due to Purkinje neuron selective expression of mutant ATXN1.
BDNF expression is decreased in many neuropsychiatric and neurodegenerative diseases, including depression, bipolar disease, schizophrenia, Alzheimer’s disease, HD, SCA6 and Parkinson’s disease (Ferrer et al., 2000; Zuccato and Cattaneo, 2007; Takahashi et al., 2012; He et al., 2013; Shin et al., 2015). Moreover, beneficial treatments for these diseases correlate with increased BDNF expression, and rescued BDNF expression ameliorates disease symptoms in mouse models (Willson et al., 2008; Nagahara et al., 2009; Giampà et al., 2013). While these studies underlie the importance of BDNF in brain pathology and pathophysiology, regulation of BDNF expression during disease is less understood. Recent studies indicate a link between BDNF and neuroinflammation (Lima Giacobbo et al., 2018), another phenomenon that is ubiquitously present in these diseases (Ilieva et al., 2009; Heneka et al., 2014; Pekny et al., 2016). NF-κB, a key regulator of neuroinflammation (Mincheva-Tasheva and Soler, 2013), modulates BDNF expression (Saha et al., 2006; Lima Giacobbo et al., 2018). Astroglia react to neuronal dysfunction by activating NF-κB (Barres, 2008), and our results implicate that astroglial NF-κB contributes to elevated BDNF levels early in SCA1 mice. Likewise, astroglia-derived BDNF delayed disease onset in a transgenic mouse model of HD (Giralt et al., 2011).
Previous studies indicate that reversibility of polyQ neurodegeneration diminishes with disease progression (Yamamoto and Lucas, 2000; Zu et al., 2004; Rubinsztein and Orr, 2016; Ibrahim et al., 2017), strongly supporting consideration of early stage treatments. Our results indicate that early BDNF treatment may delay disease onset in SCA1 carriers. We also observed decreased BDNF expression late in SCA1 that may contribute to disease severity (Hourez et al., 2011). Future work will examine the effects of late BDNF delivery on disease severity and progression in SCA1 mice.
Author Contributions
MC conceived the study. AM, MC and CS performed the experiments and analyzed the data. AF analyzed data from BrainSpan database linked from Allen brain atlas. All authors prepared the figures and wrote the manuscript.
Funding
MC is supported by the funding from the National Institutes of Health 1R01NS107387-01 and Minnesota Regenerative Medicine RMM 101617 TR 001.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Dr. Harry Orr for the generous gift of ATXN1[82Q] mice Katherine Hamel for help with data acquisition, and to all the members of Cvetanovic and Orr laboratories for suggestions.
Footnotes
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fncel.2018.00509/full#supplementary-material
TABLE S1 | BDNF expression from RNA sequencing of mouse cerebellar extracts.
References
Banfi, S., Servadio, A., Chung, M., Capozzoli, F., Duvick, L. A., Elde, R., et al. (1996). Cloning and developmental expression analysis of the murine homolog of the spinocerebellar ataxia type 1 gene (Sca1). Hum. Mol. Genet. 5, 33–40. doi: 10.1093/hmg/5.1.33
Barde, Y., Edgar, D., and Thoenen, H. (1982). Purification of a new neurotrophic factor from mammalian brain. EMBO J. 1, 549–553. doi: 10.1002/j.1460-2075.1982.tb01207.x
Barnes, J. A., Ebner, B. A., Duvick, L. A., Gao, W., Chen, G., Orr, H. T., et al. (2011). Abnormalities in the climbing fiber-Purkinje cell circuitry contribute to neuronal dysfunction in ATXN1[82Q] mice. J. Neurosci. 31, 12778–12789. doi: 10.1523/jneurosci.2579-11.2011
Barres, B. A. (2008). The mystery and magic of glia: a perspective on their roles in health and disease. Neuron 60, 430–440. doi: 10.1016/j.neuron.2008.10.013
Bosman, L. W. J., Hartmann, J., Barski, J. J., Lepier, A., Noll-Hussong, M., and Reichardt, L. F. (2006). Requirement of TrkB for synapse elimination in developing cerebellar Purkinje cells. Brain Cell Biol. 35, 87–101. doi: 10.1007/s11068-006-9002-z
Burright, E. N., Clark, H. B., Servadio, A., Matilla, T., Feddersen, R. M., Yunis, W. S., et al. (1995). SCA1 transgenic mice: a model for neurodegeneration caused by an expanded CAG trinucleotide repeat. Cell 82, 937–948. doi: 10.1016/0092-8674(95)90273-2
Carter, A. R., Chen, C., Schwartz, P. M., and Segal, R. A. (2002). Brain-derived neurotrophic factor modulates cerebellar plasticity and synaptic ultrastructure. J. Neurosci. 22, 1316–1327. doi: 10.1523/jneurosci.22-04-01316.2002
Choo, M., Miyazaki, T., Yamazaki, M., Kawamura, M., Nakazawa, T., Zhang, J., et al. (2017). Retrograde BDNF to TrkB signaling promotes synapse elimination in the developing cerebellum. Nat. Commun. 8:195. doi: 10.1038/s41467-017-00260-w
Clark, H. B., Burright, E. N., Yunis, W. S., Larson, S., Wilcox, C., Hartman, B., et al. (1997). Purkinje cell expression of a mutant allele of SCA1 in transgenic mice leads to disparate effects on motor behaviors, followed by a progressive cerebellar dysfunction and histological alterations. J. Neurosci. 17, 7385–7395. doi: 10.1523/JNEUROSCI.17-19-07385.1997
Cvetanovic, M. (2015). Decreased expression of glutamate transporter GLAST in bergmann glia is associated with the loss of Purkinje neurons in the spinocerebellar ataxia type 1. Cerebellum 14, 8–11. doi: 10.1007/s12311-014-0605-0
Cvetanovic, M., Ingram, M., Orr, H., and Opal, P. (2015). Early activation of microglia and astrocytes in mouse models of spinocerebellar ataxia type 1. Neuroscience 289, 289–299. doi: 10.1016/j.neuroscience.2015.01.003
Cvetanovic, M., Patel, J. M., Marti, H. H., Kini, A. R., and Opal, P. (2011). Vascular endothelial growth factor ameliorates the ataxic phenotype in a mouse model of spinocerebellar ataxia type 1. Nat. Med. 17, 1445–1447. doi: 10.1038/nm.2494
Dell’Orco, J. M., Wasserman, A. H., Chopra, R., Ingram, M. A. C., Hu, Y.-S., Singh, V., et al. (2015). Neuronal atrophy early in degenerative ataxia is a compensatory mechanism to regulate membrane excitability. J. Neurosci. 35, 11292–11307. doi: 10.1523/jneurosci.1357-15.2015
Duvick, L., Barnes, J., Ebner, B., Agrawal, S., Andresen, M., Lim, J., et al. (2010). SCA1-like disease in mice expressing wild-type Ataxin-1 with a serine to aspartic acid replacement at residue 776. Neuron 67, 929–935. doi: 10.1016/j.neuron.2010.08.022
Ebner, B. A., Ingram, M. A., Barnes, J. A., Duvick, L. A., Frisch, J. L., Clark, H. B., et al. (2013). Purkinje cell ataxin-1 modulates climbing fiber synaptic input in developing and adult mouse cerebellum. J. Neurosci. 33, 5806–5820. doi: 10.1523/jneurosci.6311-11.2013
Espinoza, F. A., Turner, J. A., Vergara, V. M., Miller, R. L., Mennigen, E., Liu, J., et al. (2018). Whole-brain connectivity in a large study of Huntington’s disease gene mutation carriers and healthy controls. Brain Connect 8, 166–178. doi: 10.1089/brain.2017.0538
Ferrer, I., Goutan, E., Marín, C., Rey, M. J., and Ribalta, T. (2000). Brain-derived neurotrophic factor in Huntington disease. Brain Res. 866, 257–261. doi: 10.1016/s0006-8993(00)02237-x
Ferro, A., Qu, W., Lukowicz, A., Svedberg, D., Johnson, A., and Cvetanovic, M. (2018). Inhibition of NF-κB signaling in IKKβF/F;LysM Cre mice causes motor deficits but does not alter pathogenesis of Spinocerebellar ataxia type 1. PLoS One 13:e0200013. doi: 10.1371/journal.pone.0200013
Furutani, K., Okubo, Y., Kakizawa, S., and Iino, M. (2006). Postsynaptic inositol 1,4,5-trisphosphate signaling maintains presynaptic function of parallel fiber—Purkinje cell synapses via BDNF. Proc. Natl. Acad. Sci. U S A 103, 8528–8533. doi: 10.1073/pnas.0600497103
Genis, D., Matilla, T., Volpini, V., Rosell, J., Dávalos, A., Ferrer, I., et al. (1995). Clinical, neuropathologic and genetic studies of a large spinocerebellar ataxia type 1 (SCA1) kindred: (CAG)n expansion and early premonitory signs and symptoms. Neurology 45, 24–30. doi: 10.1212/wnl.45.1.24
Giampà, C., Montagna, E., Dato, C., Melone, M. A. B., Bernardi, G., and Fusco, F. R. (2013). Systemic delivery of recombinant brain derived neurotrophic factor (BDNF) in the R6/2 mouse model of Huntington’s disease. PLoS One 8:e64037. doi: 10.1371/journal.pone.0064037
Giralt, A., Carretón, O., Lao-Peregrin, C., Martín, E. D., and Alberch, J. (2011). Conditional BDNF release under pathological conditions improves Huntington’s disease pathology by delaying neuronal dysfunction. Mol. Neurodegener. 6:71. doi: 10.1186/1750-1326-6-71
Gusella, J. F., and MacDonald, M. E. (2000). Molecular genetics: unmasking polyglutamine triggers in neurodegenerative disease. Nat. Rev. Neurosci. 1, 109–115. doi: 10.1038/nrd1077
He, Y. Y., Zhang, X. Y., Yung, W. H., Zhu, J.-N., and Wang, J.-J. (2013). Role of BDNF in central motor structures and motor diseases. Mol. Neurobiol. 48, 783–793. doi: 10.1007/s12035-013-8466-y
Heneka, M. T., Kummer, M. P., and Latz, E. (2014). Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 14, 463–477. doi: 10.1038/nri3705
Hourez, R., Servais, L., Orduz, D., Gall, D., Millard, I., and de Kerchove d’Exaerde, A. (2011). Aminopyridines correct early dysfunction and delay neurodegeneration in a mouse model of spinocerebellar ataxia type 1. J. Neurosci. 31, 11795–11807. doi: 10.1523/jneurosci.0905-11.2011
Huang, Y., Ko, H., Cheung, Z. H., Yung, K. K. L., Yao, T., Wang, J., et al. (2012). Dual actions of brain-derived neurotrophic factor on GABAergic transmission in cerebellar Purkinje neurons. Exp. Neurol. 233, 791–798. doi: 10.1016/j.expneurol.2011.11.043
Ibrahim, M. F., Power, E. M., Potapov, K., and Empson, R. M. (2017). Motor and cerebellar architectural abnormalities during the early progression of ataxia in a mouse model of SCA1 and how early prevention leads to a better outcome later in life. Front. Cell. Neurosci. 11:292. doi: 10.3389/fncel.2017.00292
Ilieva, H., Polymenidou, M., and Cleveland, D. W. (2009). Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol. 187, 761–772. doi: 10.1083/jcb.200908164
Ingram, M., Wozniak, E. A. L., Duvick, L., Yang, R., Bergmann, P., Carson, R., et al. (2016). Cerebellar transcriptome profiles of ATXN1 transgenic mice reveal SCA1 disease progression and protection pathways article cerebellar transcriptome profiles of ATXN1 transgenic mice reveal SCA1 disease progression and protection pathways. Neuron 89, 1194–1207. doi: 10.1016/j.neuron.2016.02.011
Jacobi, H., Reetz, K., du Montcel, S. T., Bauer, P., Mariotti, C., Nanetti, L., et al. (2013). Biological and clinical characteristics of individuals at risk for spinocerebellar ataxia types 1, 2, 3 and 6 in the longitudinal RISCA study: analysis of baseline data. Lancet Neurol. 12, 650–658. doi: 10.1016/S1474-4422(13)70104-2
Joers, J. M., Deelchand, D. K., Lyu, T., Emir, U. E., Hutter, D., Gomez, C. M., et al. (2018). Neurochemical abnormalities in premanifest and early spinocerebellar ataxias. Ann. Neurol. 83, 816–829. doi: 10.1002/ana.25212
Kafitz, K. W., Rose, C. R., Thoenen, H., and Konnerth, A. (1999). Neurotrophin-evoked rapid excitation through TrkB receptors. Nature 401, 918–921. doi: 10.1038/44847
Kim, J. H., Lukowicz, A., Qu, W., Johnson, A., and Cvetanovic, M. (2018). Astroglia contribute to the pathogenesis of spinocerebellar ataxia Type 1 (SCA1) in a biphasic, stage-of-disease specific manner. Glia 66, 1972–1987. doi: 10.1002/glia.23451
La Spada, A. R., and Taylor, J. P. (2010). Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat. Rev. Genet. 11, 247–258. doi: 10.1038/nrg2748
Lima Giacobbo, B., Doorduin, J., Klein, H. C., Dierckx, R. A. J. O., Bromberg, E., and de Vries, E. F. J. (2018). Brain-derived neurotrophic factor in brain disorders: focus on neuroinflammation. Mol. Neurobiol. doi: 10.1007/s12035-018-1283-6 [Epub ahead of print].
Matilla-Dueñas, A., Ashizawa, T., Brice, A., Magri, S., McFarland, K. N., Pandolfo, M., et al. (2014). Consensus paper: pathological mechanisms underlying neurodegeneration in spinocerebellar ataxias. Cerebellum 13, 269–302. doi: 10.1007/s12311-013-0539-y
Miller, J. A., Ding, S., Sunkin, S. M., Smith, K. A., Ng, L., Szafer, A., et al. (2014). Transcriptional landscape of the prenatal human brain. Nature 508, 199–206. doi: 10.1038/nature13185
Mincheva-Tasheva, S., and Soler, R. M. (2013). NF-κB signaling pathways: role in nervous system physiology and pathology. Neuroscientist 19, 175–194. doi: 10.1177/1073858412444007
Nagahara, A. H., Merrill, D. A., Coppola, G., Tsukada, S., Schroeder, B. E., Shaked, G. M., et al. (2009). Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat. Med. 15, 331–337. doi: 10.1038/nm.1912
Orr, H. T., and Zoghbi, H. Y. (2007). Trinucleotide repeat disorders. Annu. Rev. Neurosci. 30, 575–621. doi: 10.1146/annurev.neuro.29.051605.113042
Paulson, H. L., Shakkottai, V. G., Clark, H. B., and Orr, H. T. (2017). Polyglutamine spinocerebellar ataxias—from genes to potential treatments. Nat. Rev. Neurosci. 18, 613–626. doi: 10.1038/nrn.2017.92
Pekny, M., Pekna, M., Messing, A., Steinhäuser, C., Lee, J. M., Parpura, V., et al. (2016). Astrocytes: a central element in neurological diseases. Acta Neuropathol. 131, 323–345. doi: 10.1007/s00401-015-1513-1
Qu, W., Johnson, A., Kim, J. H., Lukowicz, A., Svedberg, D., and Cvetanovic, M. (2017). Inhibition of colony-stimulating factor 1 receptor early in disease ameliorates motor deficits in SCA1 mice. J. Neuroinflammation 14:107. doi: 10.1186/s12974-017-0880-z
Rüb, U., Schöls, L., Paulson, H., Auburger, G., Kermer, P., Jen, J. C., et al. (2013). Clinical features, neurogenetics and neuropathology of the polyglutamine spinocerebellar ataxias type 1, 2, 3, 6 and 7. Prog. Neurobiol. 104, 38–66. doi: 10.1016/j.pneurobio.2013.01.001
Rubinsztein, D. C., and Orr, H. T. (2016). Diminishing return for mechanistic therapeutics with neurodegenerative disease duration?: there may be a point in the course of a neurodegenerative condition where therapeutics targeting disease-causing mechanisms are futile. Bioessays 38, 977–980. doi: 10.1002/bies.201600048
Ruegsegger, C., Stucki, D. M., Steiner, S., Angliker, N., Radecke, J., Keller, E., et al. (2016). Impaired mTORC1-dependent expression of homer-3 influences SCA1 pathophysiology. Neuron 89, 129–146. doi: 10.1016/j.neuron.2015.11.033
Saha, R. N., Liu, X., and Pahan, K. (2006). Up-regulation of BDNF in astrocytes by TNF-α: a case for the neuroprotective role of cytokine. J. Neuroimmune Pharmacol. 1, 212–222. doi: 10.1007/s11481-006-9020-8
Schwartz, P. M., Borghesani, P. R., Levy, R. L., Pomeroy, S. L., and Segal, R. A. (1997). Abnormal cerebellar development and foliation in BDNF−/− mice reveals a role for neurotrophins in CNS Patterning. Neuron 19, 269–281. doi: 10.1016/s0896-6273(00)80938-1
Serra, H. G., Duvick, L., Zu, T., Carlson, K., Stevens, S., Jorgensen, N., et al. (2006). ROR α-mediated purkinje cell development determines disease severity in adult SCA1 mice. Cell 127, 697–708. doi: 10.1016/j.cell.2006.09.036
Sherrard, R. M., Dixon, K. J., Bakouche, J., Rodger, J., Lemaigre-dubreuil, Y., and Mariani, J. (2009). Differential expression of TrkB isoforms switches climbing fiber-purkinje cell synaptogenesis to selective synapse elimination. Dev. Neurobiol. 69, 647–662. doi: 10.1002/dneu.20730
Shin, H. W., Kim, H., and Lee, K. J. (2015). Differences in BDNF serum levels in patients with Alzheimer’s disease and mild cognitive impairment. J. Psychiatry 18:245. doi: 10.4172/psychiatry.1000245
Storey, E. (2013). Presymptomatic features of spinocerebellar ataxias. Lancet Neurol. 12, 625–626. doi: 10.1016/s1474-4422(13)70116-9
Tabrizi, S. J., Scahill, R. I., Owen, G., Durr, A., Leavitt, B. R., Roos, R. A., et al. (2013). Predictors of phenotypic progression and disease onset in premanifest and early-stage Huntington’s disease in the TRACK-HD study: analysis of 36-month observational data. Lancet Neurol. 12, 637–649. doi: 10.1016/s1474-4422(13)70088-7
Takahashi, M., Ishikawa, K., Sato, N., Obayashi, M., Niimi, Y., Ishiguro, T., et al. (2012). Reduced brain-derived neurotrophic factor (BDNF) mRNA expression and presence of BDNF-immunoreactive granules in the spinocerebellar ataxia type 6 (SCA6) cerebellum. Neuropathology 6, 595–603. doi: 10.1111/j.1440-1789.2012.01302.x
Willson, M. L., McElnea, C., Mariani, J., Lohof, A. M., and Sherrard, R. M. (2008). BDNF increases homotypic olivocerebellar reinnervation and associated fine motor and cognitive skill. Brain 131, 1099–1112. doi: 10.1093/brain/awn024
Yamamoto, A., and Lucas, J. (2000). Reversal of neuropathology and motor dysfunction in a conditional model of Huntington’s disease. Cell 101, 57–66. doi: 10.1016/s0092-8674(00)80623-6
Zoghbi, H. Y., and Orr, H. T. (2009). Pathogenic mechanisms of a polyglutamine-mediated neurodegenerative disease, spinocerebellar ataxia type 1. J. Biol. Chem. 284, 7425–7429. doi: 10.1074/jbc.R800041200
Zu, T., Duvick, L. A., Kaytor, M. D., Berlinger, M. S., Zoghbi, H. Y., Clark, H. B., et al. (2004). Recovery from polyglutamine-induced neurodegeneration in conditional SCA1 transgenic mice. J. Neurosci. 24, 8853–8861. doi: 10.1523/JNEUROSCI.2978-04.2004
Keywords: ATAXIN-1, BDNF, astroglia, neuroprotective, nuclear factor κb, cerebellum, neurodegeneration
Citation: Mellesmoen A, Sheeler C, Ferro A, Rainwater O and Cvetanovic M (2019) Brain Derived Neurotrophic Factor (BDNF) Delays Onset of Pathogenesis in Transgenic Mouse Model of Spinocerebellar Ataxia Type 1 (SCA1). Front. Cell. Neurosci. 12:509. doi: 10.3389/fncel.2018.00509
Received: 11 September 2018; Accepted: 10 December 2018;
Published: 21 January 2019.
Edited by:
Ying Shen, Zhejiang University, ChinaReviewed by:
Ruth M. Empson, University of Otago, New ZealandLaurens Bosman, Erasmus University Rotterdam, Netherlands
Hong Jiang, Central South University, China
Copyright © 2019 Mellesmoen, Sheeler, Ferro, Rainwater and Cvetanovic. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marija Cvetanovic, bWN2ZXRhbm9AdW1uLmVkdQ==